
Abstract
Mutations in two highly related chromatin-remodeling complexes, BRG1/BRM-associated factor (BAF) and Polybromo-associated BAF (PBAF), cause various neurodevelopmental conditions. Although BAF activity is required at multiple steps of nervous system development, the specific neural functions of PBAF remain largely unexplored. Through an unbiased genetic screen in C. elegans, we identify here critical roles for PBAF in postmitotic neuronal identity. In nerve cord motor neurons, PBAF represses gene expression by antagonizing the terminal selector-type transcription factor UNC-3 (EBF1-4). In contrast, PBAF activates gene expression in caudal motor neurons. This dichotomy in PBAF function generates distinct motor neuron identities. PBAF disruption leads to locomotion defects. Genetic, genomic, and biochemical data suggest that the cell type-specific functions of PBAF in different motor neuron groups arise via its recruitment to specific gene loci by conserved transcription factors, such as MAB-9/TBX20. Altogether, our findings provide a conceptual framework to understand specific neuronal defects that arise in neurological conditions caused by mutations in broadly expressed chromatin-remodeling complexes.
Similar content being viewed by others

Introduction
Nervous systems are strikingly complex structures composed of numerous types of neurons and glia. Neuronal types have been traditionally defined based on anatomical and electrophysiological criteria1,2. Single-cell RNA sequencing (scRNA-Seq) has recently revolutionized neuronal classification efforts by generating molecular profiles of individual neurons3,4,5,6,7. Studies in model organisms (e.g., nematodes, fruit flies, zebrafish, and mice) have revealed a pervasive organizational principle: the existence of distinct neuronal types that share a number of traits (e.g., neurotransmitter usage) but can be further subdivided into subtypes based on anatomical, functional, or molecular criteria. In C. elegans, anatomically defined neuronal types can be molecularly subdivided into distinct subtypes5,8, and this is the case in every systematically studied component of animal nervous systems, such as the D. melanogaster optic lobe9,10, the mouse cortex11,12 and the mouse retina13. Given the ubiquity of neuronal subtype diversity, probing its underlying mechanisms is a key step in understanding neuronal development, function, and evolution.
To date, known molecular mechanisms underlying neuronal subtype diversification primarily involve transcription factors (TFs). At the level of neural progenitor cells, studies in the fly ventral nerve cord and optic lobe uncovered the concept of temporal TFs, whose sequential activation and cross-repressive interactions diversify neural progenitor cells, and thereby their postmitotic neuronal progeny14,15. TF-based mechanisms also operate in the vertebrate neural tube, where morphogen gradients determine TF expression in neural progenitors, ultimately generating distinct neuronal types and subtypes16,17,18,19. While studies like these enrich our understanding of diversification at the level of neural progenitors, much less is known about the transcriptional mechanisms that operate in post-mitotic neurons to diversify neuronal fate.
Terminal selectors are TFs that act in post-mitotic neurons20,21. They control the identity of individual neuron types and subtypes (e.g., sensory, inter-, and motor neurons) by directly activating the transcription of key effector genes, termed “terminal identity genes”. These encode proteins essential for neuronal function, such as neurotransmitter biosynthesis proteins, ion channels, and neuropeptides. To date, terminal selectors have been described in nematodes (C. elegans), fruit flies (Drosophila), cnidarians (Nematostella vectensis), marine chordates (Ciona robusta), and mice (Mus musculus), indicating a deeply conserved role for these critical regulators of neuronal identity20,21. For example, in the mouse brainstem, the terminal selector Pet-1 controls the identity of serotonergic neurons by activating scores of terminal identity genes22,23. Similarly, in the mouse midbrain, the terminal selector Nurr1 controls the identity of dopaminergic neurons24. As with most neuron types, serotonergic and dopaminergic neurons are heterogeneous populations of cells that are further subdivided into distinct subtypes based on anatomical, functional, and molecular criteria25,26. However, the precise mechanisms through which Pet-1 and Nurr1, respectively, control the identity of individual subtypes of serotonergic and dopaminergic neurons remain unclear. These two examples highlight a conceptual problem broadly applicable to most neuron types: a common TF (e.g., terminal selector) specifies a group of neurons with shared features, but additional gene regulatory mechanisms must exist to diversify those neurons into subtypes (Fig. 1a). Understanding these mechanisms is biomedically relevant because mutations and variants in dozens of human terminal selector orthologs (e.g., EBF3, OTX2, FEV, and NURR1) cause neurodevelopmental and neurodegenerative disorders27,28,29,30,31,32.

a A conceptual model for neuronal subtype diversification. A1 and A2: activator TFs, R1 and R2: repressor TFs. b Schematic of C. elegans MNs divided by region: retrovesicular ganglion (RVG), ventral nerve cord (VNC), preanal ganglion (PAG). 78 MNs are divided into 9 classes and 37 subclasses based on anatomical and molecular profile (bottom). Columns indicate molecular profile of MN subclasses based on scRNA-seq data. Rows indicate terminal identity genes (c) and transcription factors (d) expressed in MNs. Bold gene names indicate those analyzed in this study.
The inherent complexity of vertebrate nervous systems poses a significant challenge in efforts to uncover molecular mechanisms that control neuronal diversification. On the other hand, unbiased genetic screens in more tractable organisms, such as fruit flies and nematodes, have revealed a broadly applicable gene regulatory principle: TFs with either activator or repressor activities are expressed in specific neuronal subtypes to either collaborate with or counteract a common TF (terminal selector) shared by all subtypes of a given neuronal class, thereby generating unique subtype identities (Fig. 1a). For example, the terminal selector CHE-1 controls the identity of a bilateral pair of chemosensory neurons (ASEL/R) in C. elegans33,34. CHE-1 directly activates scores of terminal identity genes expressed in both ASE neurons. However, ASE diversification into ASEL and ASER subtypes occurs via the activity of distinct, subtype-specific TFs that can act as activators or repressors of subtype-specific terminal identity genes35. Similar TFs that either counteract or collaborate with a terminal selector have been identified in C. elegans touch receptors and motor neurons36,37,38, as well as in D. melanogaster photoreceptors39,40. Altogether, our current knowledge of neuronal diversification principles that operate in post-mitotic neurons is primarily based on two types of TFs: (a) terminal selectors that act in all subtypes of a given neuron type, and (b) additional TFs that act either as activators or repressors in select subtypes. However, the roles of epigenetic mechanisms, such as DNA methylation, histone modifications, and chromatin remodeling, in generating neuronal subtype diversity remain poorly understood.
In this study, we investigate the mechanisms of neuronal diversification in C. elegans cholinergic motor neurons (MNs) that control locomotion. We recently showed that these MNs are a heterogeneous population of cells subdivided molecularly into at least 22 subtypes, referred to as “subclasses” herein (Fig. 1b–d)8. These MNs also share several features, such as connectivity to muscle and use of the same neurotransmitter (acetylcholine, ACh) (Fig. 1b). They also share the same terminal selector, UNC-3, the sole C. elegans ortholog of the Collier/Olf/Ebf (COE) family of TFs41,42. Mutations in EBF3, the human ortholog of UNC-3, cause a neurodevelopmental syndrome characterized by intellectual disability and motor developmental delay30,31,32,43. It is known that UNC-3 acts directly to activate expression of terminal identity genes that are shared among all MN subclasses (e.g., those encoding ACh biosynthesis proteins), as well as terminal identity genes with subclass-specific expression (e.g., encoding ion channels and neuropeptides) (Fig. 1c)44. However, UNC-3 does not operate alone. At the level of post-mitotic MNs, additional conserved TFs have been identified to enable cholinergic MN diversification. These fall into two categories: (a) TFs that collaborate with UNC-3, such as the HOX proteins LIN-39, MAB-5, and EGL-5, to co-activate subclass-specific terminal identity genes (Fig. 1d)44, and (b) repressor TFs, such as the T-box ortholog MAB-9 and ARID3 ortholog CFI-1, that antagonize the ability of UNC-3 to broadly activate terminal identity genes in all MN subclasses (Fig. 1d)37,38. Altogether, the terminal selector UNC-3 and additional TFs (activators and repressors) act directly by recognizing their cognate DNA binding sites at the cis-regulatory regions of specific genes, and thereby generate distinct MN subclass identities (Fig. 1a, d).
Using an unbiased genetic screen in C. elegans, we identified here a crucial role for the polybromo-associated BRG1-associated factor (PBAF) chromatin-remodeling complex in MN diversification. PBAF belongs to the highly conserved SWI/SNF family of multi-subunit chromatin-remodeling complexes, mutations in which cause cancer and neurodevelopmental disorders such as Coffin–Siris syndrome, Nicolaides-Baraitser syndrome, Kleefstra syndrome, and autism spectrum disorders45,46. Despite its broad expression across tissues, PBAF exerts specific and strikingly different effects on post-mitotic MNs depending on cell body position. In nerve cord MNs, PBAF antagonizes the terminal selector UNC-3 by repressing UNC-3-dependent terminal identity genes. In posteriorly located (caudal) MNs, PBAF collaborates with UNC-3 to activate gene expression and thereby diversify these cells from their nerve cord counterparts. Mechanistically, we propose that the different effects of PBAF are due to the activity of distinct TFs in nerve cord (MAB-9/TBX20) and caudal (EGL-5/HOX) MNs. Altogether, our findings illuminate the epigenetic mechanisms that drive neuronal diversification by uncovering cell type-specific roles for an ancient and broadly expressed chromatin-remodeling complex.
Results
Defining MN diversity in the C. elegans ventral nerve cord
Based on distinct axodendritic morphologies, the MNs that populate the C. elegans ventral nerve cord (VNC) and its flanking ganglia (retrovesicular ganglion [RVG], preanal ganglion [PAG]) are divided into nine cardinal classes (SAB, DA, DB, DD, VA, VB, VC, VD, and AS) in hermaphrodite animals47. The number of neurons for each class ranges from 3 (SAB) to 13 (VD), totaling 78 cells (SAB = 3 neurons, DA = 9, DB = 7, DD = 6, VA = 12, VB = 11, VC = 6, VD = 13, AS = 11) (Fig. 1b). These classes differ in cell body position and muscle innervation: (a) SAB neurons innervate head muscle, (b) VC neurons innervate vulva muscle essential for egg laying, and (c) MNs of the remaining seven classes (AS, DA, DB, DD, VA, VB, and VD) innervate body wall muscles essential for sinusoidal locomotion. Except for the SAB and VC neurons, cell bodies of the remaining seven classes (DA, DB, DD, VA, VB, VD, and AS) intermingle along the VNC and its flanking ganglia (RVG, PAG) (Fig. 1b).
Based on neurotransmitter usage, seven MN classes are cholinergic (SAB, DA, DB, VA, VB, VC, and AS) and two are GABAergic (DD and VD)47. The seven cholinergic classes share a number of molecular features, such as expression of acetylcholine (ACh) biosynthesis genes (unc-17/VAChT and cho-1/ChT), neurotransmitter receptors (e.g., acr-2/CHRNA3/6), and ion channels (e.g., slo-2) (Fig. 1c). These features are not shared by the two GABAergic classes (DD, VD), which also express distinct sets of genes (e.g., unc-25/GAD and unc-47/VGAT)48. Through single-cell RNA-Sequencing (scRNA-Seq), we recently uncovered a striking degree of molecular diversity within cardinal MN classes in the adult8. For example, the five cholinergic MN classes (DA, DB, VA, VB, and AS), which control locomotion and express the terminal selector unc-3/EBF, can be further subdivided into 22 subclasses based on gene expression (DA subclasses: DA1, DA2-5, DA6-8, and DA9; DB subclasses: DB1, DB2, and DB3-7; VA subclasses: VA1, VA2, VA3-8, VA9-10, VA11, and VA12; VB subclasses: VB1, VB2, VB3, VB4-9, and VB10-11; AS subclasses: AS2-3, AS4-8, AS9-10, and AS11) (Fig. 1c). For the current study, we re-analyzed our scRNA-Seq dataset and found that the head muscle-innervating SAB neurons, which also require unc-3/EBF for their terminal identity, display a distinct molecular signature, albeit their limited representation in the dataset prevented possible identification of SAB subclasses (Fig. 1c, Supplementary Fig. 1). Altogether, both anatomical and molecular studies have firmly established that C. elegans MNs are remarkably diverse, offering a powerful platform to uncover molecular mechanisms of neuronal subclass diversification.
A genetic screen for regulators of MN diversity
The terminal selector UNC-3/EBF is expressed in six classes (SAB, DA, DB, VA, VB, and AS) of cholinergic MNs (Fig. 1d). Mechanistically, UNC-3 acts directly to activate expression of shared (e.g., unc-17/VAChT and cho-1/ChT) and subclass-specific terminal identity genes42,49. Our previous work showed that additional TFs act either as activators (e.g., LIN-39/Hox and MAB-5/Hox) or repressors (e.g., CFI-1/ARID3 and MAB-9/TBX20), thereby generating distinct signatures of terminal identity gene expression in individual MN subclasses37,50 (Fig. 1b–d). However, it remains unknown whether and how epigenetic mechanisms (e.g., DNA or histone modifications, chromatin remodeling) influence the activity of UNC-3, thereby dictating MN diversity.
To address this knowledge gap, we leveraged an available genetic tool that specifically labels a subset of unc-3-expressing MNs41. A transgenic red fluorescent reporter for glr-4 (glr-4prom::tagRFP), ortholog of human GRIK4 (glutamate receptor, ionotropic, and kainite 4) is expressed in SAB neurons (RVG ganglion) and the DA9 subclass (PAG ganglion) in an unc-3-dependent manner (Fig. 2a)41, partially recapitulating endogenous glr-4 expression (Fig. 1c)38. Our scRNA-Seq data show that glr-4 transcripts are detected at high levels in MNs located outside the ventral nerve cord (e.g., SAB and DA9) (Fig. 1c). Within the ventral nerve cord, glr-4 transcripts are normally expressed at low (basal) levels, or are undetectable in MNs. This pattern of expression is consistent with our recent study, where we analyzed the expression of the endogenous mScarlet::glr-4 reporter with single-cell resolution38. Nevertheless, the restricted expression pattern of the glr-4prom::tagRFP reporter offers an opportunity to conduct a genetic screen for mutants that display ectopic glr-4prom::tagRFP expression in MN subclasses located in the VNC, and thereby identify—in an unbiased manner—regulatory factors that prevent UNC-3 from activating glr-4 in VNC subtypes (Fig. 2a)38.

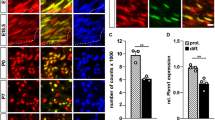
a Images show expression of glr-4prom::tagRFP in SAB MNs (magenta asterisk), VNC MNs (white arrowheads), and DA9 (blue asterisk) in WT and pbrm-1 mutants. Worm schematic represents glr-4prom::tagRFP expression in WT animals. Quantification of a showing number or percentage (%) of MNs expressing glr-4prom::tagRFP in VNC (b), SAB (c), and DA9 (d). Animals imaged at L4 stage. WT n = 35; ot792 n = 18; tm415 n = 20; ot792/tm415 n = 22. e Diagram of the pbrm-1 gene locus in C. elegans showing the location of mutant alleles, RNAi targets and reporter constructs. f Protein domains of PBRM-1 long and short isoforms in C. elegans and human (bottom): bromodomains (Bromo), bromo-adjacent homology (BAH), high mobility group (HMG) box. g Representative images of pbrm-1(ot792) animals injected with pbrm-1prom::pbrm-1::GFP (control–top). h Quantification showing number of MNs expressing glr-4prom::tagRFP in VNC. Animals imaged at L4 stage. Control n = 10; pbrm-1 rescue n = 15. i Representative images of L4 animals expressing glr-4prom::tagRFP in MNs of WT, pbrm-1(syb10332), and pbrm-1 (syb10440) animals. Quantification on the right. n = 15. For all quantifications, unpaired two-sided Welch’s t-test was used, p < .05 = *; p < .01 = **; p < .001 = ***; p < .0001 = ****. Box and whisker plots were used; all data points presented. Box boundaries indicate the 25th and 75th percentile. The limits indicate minima and maxima values. Center values (mean) are highlighted with a black horizontal line. j Summary of UNC-3 and PBRM-1 effects on glr-4 in SAB, nerve cord, and DA9 neurons. Source data are provided as a Source Data file.
Following random ethyl methanesulfonate [EMS] mutagenesis of the P0 generation, we screened F2 progeny (~1500 haploid genomes) for homozygous recessive mutants displaying altered glr-4prom::tagRFP expression in MNs at larval stage 4 (L4) (Fig. 2a). We retrieved 3 mutant alleles: two alleles (ot785 and ot786), which affect two TFs (CFI-1, LIN-13), have been previously described elsewhere37,38. This study focuses on the third allele, ot792. In homozygous ot792 mutant animals, we observed ectopic glr-4prom::tagRFP expression in MNs of the nerve cord, but no effect on SAB and DA9 neurons (Fig. 2a–d). Of the 58 MNs that normally reside in the nerve cord, 11.6 ± 2.2 cells express glr-4prom::tagRFP in ot792 mutant animals compared to 0.6 ± 0.9 cells in control animals (Fig. 2b). We obtained similar results with an endogenous mScarlet::glr-4 reporter; both the number of MNs expressing mScarlet::glr-4 and its fluorescent intensity are increased in ot792 mutants (Supplementary Fig. 2a–c)38. Hence, ot792 mutants display increased glr-4 expression in a subset of the 58 nerve cord MNs. Next, we used a previously published strain that does not carry the ot792 mutation but selectively overexpresses glr-4 in nerve cord MNs due to mutated binding sites for CFI-1, a TF that normally represses glr-438. We found defects in thrashing behavior of this strain (Supplementary Fig. 3a), indicating that increased glr-4 levels in MNs are detrimental to C. elegans motility. Therefore, the ot792 mutants offer an opportunity to identify a mechanism that tunes down (limits) glr-4 expression in nerve cord MNs.
PBRM-1 (PBRM1/BAF180) represses glr-4/GRIK4 in MNs
Upon single nucleotide polymorphism (SNP) mapping and whole genome sequencing51(Supplementary Fig. 4), we identified the molecular lesion in ot792 mutants: a C to T nucleotide substitution (at position +7115 bp from ATG) that generates a premature stop (TAG, Amber) codon in the 12th exon of pbrm-1, ortholog of human polybromo 1 (PBRM1/BAF180) (Fig. 2e). This point mutation of the ot792 allele affects all known pbrm-1 transcripts, C26C6.1a.1 and C26C6.1b.1-3 (Fig. 2e). We also characterized animals carrying an independent pbrm-1 deletion allele (tm415), which selectively affects a single transcript (C26C6.1a.1) (Fig. 2e). Again, we observed ectopic glr-4prom::tagRFP in MNs of the VNC, although the effect is weaker compared to pbrm-1(ot792) (Fig. 2b–d).
At the protein level, the C26C6.1a.1 transcript produces the long PBRM-1 isoform a, whereas transcripts C26C6.1b.1-3 produce the short PBRM-1 isoform b (Fig. 2e). Six bromodomains (BrD) and two bromo-adjacent homology (BAH) domains are present in the long PBRM-1 isoform but absent from the short one (Fig. 2e, f). Both short and long pbrm-1 isoforms contain the high mobility group (HMG) domain (Fig. 2e). All these domains are present in human PBRM1 (BAF180) (Fig. 2f). Next, we employed CRISPR/Cas9 mutagenesis to generate isoform-specific pbrm-1 mutant alleles. Mutation of the start ATG (Met) codon of the short isoform (C26C6.1b.1) into ATA (Ile) (pbrm-1(10332) allele) had no effect on glr-4 expression in nerve cord MNs (Fig. 2e, i). However, mutation of the start ATG (Met) codon of the long isoform (C26C6.1a.1) into ATA (Ile) (pbrm-1(10440) allele) led to ectopic glr-4 expression in MNs (Fig. 2e, i). Moreover, transgenic supply of the long pbrm-1 isoform driven by its endogenous promoter (pbrm-1prom::pbrm-1 cDNA) completely rescued the glr-4 phenotype in pbrm-1(ot792) mutants (Fig. 2g, h). Hence, the long isoform of PBRM-1 is specifically required for glr-4 repression.
Further, complementation testing showed that trans-heterozygote animals carrying both alleles (ot792 and tm415) also displayed ectopic expression of glr-4 in nerve cord MNs (7.7 ± 1.2 cells, p < 0.0001), corroborating that pbrm-1 gene activity is necessary to repress glr-4 (Fig. 2b). Altogether, our genetic screen identified pbrm-1 (PBRM1/BAF180) as a negative regulator of glr-4 expression in a subset of nerve cord MNs (Fig. 2i), implicating the long PBRM-1 isoform in the process of neuronal subclass diversification. As pbrm-1 is the signature subunit of the polybromo-associated BRG1/BRM-associated factor (PBAF) chromatin remodeling complex52, these results implicate PBAF-mediated epigenetic regulation in the process of MN diversification.
pbrm-1/PBRM1 acts in nerve cord MNs to repress glr-4/GRIK4
Due to its specific effect on glr-4/GRIK4 expression in a subset of nerve cord MNs, we wondered whether pbrm-1 expression is restricted to these cells. Analysis of available scRNA-Seq data of adult MNs8 revealed that pbrm-1 transcripts are present broadly in all MN subclasses (both cholinergic and GABAergic) (Fig. 3a). At the protein level, an endogenous C-terminal GFP reporter (PBRM-1::TY1::EGFP::3xFLAG, allele st12226) that captures expression of all PBRM-1 isoforms (Fig. 2e) is expressed broadly in most (if not all) C. elegans neurons and other cell types (Fig. 3b and Supplementary Fig. 5a), consistent with a recent study53.

a scRNA-seq data showing UNC-3 and PBAF/BAF component expression across MN subclasses. b pbrm-1::TY1::EGFP::3xFlag endogenous reporter shows broad expression. MN marker cho-1 labels cholinergic MNs (middle). Merged image reveals pbrm-1 expression in cholinergic MNs (white arrowheads) (bottom). Animals imaged at L4 stage. n = 15. c Left – Images of pbrm-1 RNAi animals using lin-39 and cho-1 promoters, targeting long (bottom) and all pbrm-1 isoforms (control – left, experimental – right). Right – Quantification showing number of MNs expressing glr-4prom::tagRFP in VNC. Animals imaged at L4 stage. All isoform knock-down: lin-39prom control n = 10; lin-39prom RNAi n = 20; cho-1prom control n = 9; cho-1prom RNAi n = 13. Long isoform knock-down: lin-39prom control n = 10; lin-39prom RNAi n = 15; cho-1prom control n = 9; cho-1prom RNAi n = 15. d Left – Quantification of PBRM-1::GFP expression without (left) and with (right) cho-1prom::GFPnano::ZIF-1. Animals imaged at L4 stage. The cho-1prom::GFPnano::ZIF-1 line carries a pharyngeal (myo-2::gfp) marker. GFPnano::ZIF-1(-) n = 7; GFPnano::ZIF-1(+) n = 8. Middle – Images showing glr-4prom::tagRFP expression in cho-1prom::GFPnano::ZIF-1 (top), cho-1prom::GFPnano::ZIF-1; pbrm-1::TY1::EGFP::3xFlag (middle), and pbrm-1::TY1::EGFP::3xFlag (bottom) animals. Right – Quantification showing number of MNs expressing glr-4prom::tagRFP in VNC. Animals imaged at L4 stage. Cho-1prom::GFPnano::ZIF-1 n = 8; pbrm-1(st12226); cho-1prom::GFPnano::ZIF-1 n = 11; pbrm-1(st12226) n = 8. For all quantifications, unpaired two-sided Welch’s t-test was used, p < .05 = *; p < .01 = **; p < .001 = ***; p < .0001 = ****. Box and whisker plots were used; all data points presented. Box boundaries indicate the 25th and 75th percentile. The limits indicate minima and maxima values. Center values (mean) are highlighted with a black horizontal line. Source data are provided as a Source Data file.
This broad expression suggests that pbrm-1 could act either in post-mitotic MNs to control glr-4 or in other cells that indirectly influence glr-4 expression in MNs. To address this question, we conducted MN-specific RNAi against pbrm-154. We used two cis-regulatory elements, from lin-39 and cho-1, known to drive expression in post-mitotic nerve cord MNs42,50. RNAi against all isoforms, or just the long isoform resulted in ectopic glr-4 expression in nerve cord MNs (Figs. 3c and 2e). Hence, we conclude that pbrm-1 is necessary to repress glr-4 in post-mitotic MNs.
In addition to RNAi, we used ZIF-1-mediated protein degradation to selectively deplete PBRM-1 in post-mitotic MNs55(Fig. 3d). We generated a transgenic line that expresses the GFP nanobody::ZIF-1 protein fusion in nerve cord MNs. We crossed it to PBRM-1::TY1::GFP::3xFLAG animals (st12226 allele) and observed efficient PBRM-1::GFP depletion in nerve cord MNs, accompanied by ectopic glr-4 expression (Fig. 3d). Altogether, our MN-specific RNAi and protein degradation experiments demonstrate a cell-autonomous role for PBRM-1 in repressing glr-4 in post-mitotic MNs.
PBAF-specific subunits are essential for glr-4 repression
PBRM-1/BAF180 is the chromatin targeting subunit of the highly conserved SWI/SNF (SWItch/Sucrose Non-Fermentable) ATP-dependent chromatin-remodeling complex PBAF (Polybromo, BRG1-Associated Factor)56. This complex is composed of four PBAF-specific subunits (PBRM-1/BAF180, SWSN-9/BRD7/9, SWSN-7/BAF200, and PHF-10/BAF45a). In addition, a core set of proteins (SWSN-1/BAF155/170, SWSN-4/BRG1, and SWSN-5/BAF47) together with accessory subunits (SWSN-2.1-2/BAF60a-c, SWSN-3/BAF57, and SWSN-6/BAF53a-b) are shared between PBAF and the highly related BAF complex (Fig. 4a)45,56. Like pbrm-1, transcripts of PBAF-specific-, BAF-specific-, and core component-encoding genes are present in all subclasses of nerve cord MNs (Fig. 3a). Consistently, GFP protein fusion reporters for SWSN-7 and SWSN-9 showed broad expression in MNs and other cell types (Supplementary Fig. 5b, c).

a Cartoon of the PBAF and BAF chromatin remodeling complexes. Complexes share a core ATPase (dark gray) and its accessory components (light gray), with PBAF specific subunits (orange) and BAF specific subunits (green). b Images showing glr-4prom::tagRFP expression in PBAF mutants (swsn-9[ok1354], phf-10[rd21]) and RNAi (swsn-7). c Quantification of (b) showing number of MNs expressing glr-4prom::tagRFP in VNC. Animals imaged at L4 stage. WT n = 35; swsn-9(−) n = 20; phf-10(−) n = 20; Plin-39::swsn-7 RNAi Line 1 (−) n = 10, (+) n = 15; Plin-39::swsn-7 RNAi Line 2 (−) n = 10, (+) n = 15; Plin-39::swsn-7 RNAi Line 3 (−) n = 9, (+) n = 15. d Quantification of BAF analysis (dpff-1[tm4287], let-526 RNAi) showing number of MNs expressing glr-4 in VNC. Animals imaged at L4 stage. WT n = 35; dpff-1 n = 15; Plin-39::let-526 RNAi (−) n = 10, (+) n = 11. e Quantification of PBAF/BAF analysis (swsn-1[os22], swsn-3[tm3647], swsn-4[os13], swsn-5[ok622], swsn-4 RNAi, swsn-6 RNAi) showing number of MNs expressing glr-4 in VNC. Animals imaged at L4 stage. WT n = 35; swsn-1 n = 21; swsn-3 n = 20; swsn-4 n = 15; swsn-5 n = 19; Plin-39::swsn-4 RNAi (−) n = 10, (+) n = 15; Plin-39::swsn-6 RNAi (−) n = 10, (+) n = 15. For all quantifications, unpaired two-sided Welch’s t-test, p < .05 = *; p < .01 = **; p < .001 = ***; p < .0001 = ****. f, g Representative images of L4 animals expressing glr-4::tagRFP in WT and swsn-4(K564)/nT1 background. The number of nerve cord MNs is quantified (N = 22 animals). Unpaired two-sided Welch’s t test). P = 4.28e-9. Box and whisker plots were used; all data points presented. Box boundaries indicate the 25th and 75th percentile. The limits indicate minima and maxima values. Center values (mean) are highlighted with a black horizontal line. Source data are provided as a Source Data file.
To elucidate whether the effect on glr-4 expression in nerve cord MNs depends on pbrm-1 specifically or the PBAF complex as a whole, we focused on the remaining three PBAF-specific components (SWSN-9, SWSN-7, PHF-10). Similar to pbrm-1 mutants, we observed ectopic glr-4::tagRFP expression in nerve cord MNs of mutant swsn-9(-) or phf-10(-) animals (Fig. 4b, c) carrying previously described loss-of-function57 alleles (swsn-9: ok1354; phf-10: rd21) (Supplementary Fig. 6)53,58. We obtained similar results in phf-10(-) mutants with the endogenous mScarlet::glr-4 reporter (Supplementary Fig. 2a, c). Further, transgenic supply of swsn-9 cDNA driven by its endogenous promoter rescued the glr-4 phenotype of swsn-9 (-) mutants (Supplementary Fig. 7c). Because animals carrying LOF alleles for pbrm-1, swsn-9, or phf-10 genes produce viable progeny, we were able to characterize the effect of PBAF-specific mutations at L4, a time point by which most C. elegans MNs have terminally differentiated. However, null mutants for swsn-7 are maternal effect embryonic lethal59. Therefore, we conducted MN-specific RNAi against swsn-7 and again observed the glr-4 expression phenotype, similar to pbrm-1, swsn-9, and phf-10 mutants (Fig. 4b, c). Next, we conducted a similar analysis for the two BAF-specific components, DPFF-1/BAF45b-d and LET-526/BAF250a-b (Fig. 4a). However, we observed no effect on glr-4 expression in nerve cord MNs in dpff-1(tm4287) mutant animals or upon MN-specific RNAi against let-526 (Fig. 4d; Supplementary Fig. 6 and 7a). In summary, genetic and RNAi evidence shows that each PBAF-specific component is required to repress glr-4 expression in nerve cord MNs, whereas disruption of BAF-specific components has no effect.
The ATPase activity of PBAF is needed for glr-4/GRIK4 repression
Through ATP hydrolysis, the PBAF complex facilitates ejection, sliding, and/or swapping of nucleosomes, thereby altering the chromatin state to affect gene transcription in a positive or negative manner60. We therefore asked whether SWSN-4, the sole SWI/SNF ATPase in C. elegans59, is necessary for glr-4 repression. Because complete elimination of swsn-4 gene activity leads to early lethality59, we used a hypomorphic, temperature-sensitive swsn-4(os13) allele and MN-specific RNAi. In both cases, we observed ectopic expression of glr-4 in nerve cord MNs at the restrictive temperature (20 °C); the effect of RNAi was stronger than that of the swsn-4(os13) allele (Fig. 4e, Supplementary Fig. 6-7). To assess whether the effect on glr-4 is due to ATP-dependent chromatin remodeling, we used CRISPR/Cas9 gene editing to generate an ATPase-dead version of SWSN-4 by introducing a lysine (K)-to-arginine (R) mutation at a conserved residue (K564) essential for ATP hydrolysis61,62,63,64. Although homozygous swsn4K564R/K564R animals are embryonic lethal consistent with a previous study64, heterozygous swsn4K564R/+ animals are viable and fertile. Compared to controls, we found ectopic glr-4::tagRFP expression in MNs of swsn4K564R/+ animals (Fig. 4f, g). This finding not only indicates that ATP-dependent chromatin remodeling is required for glr-4 repression but also suggests that the PBAF effect on glr-4 is dosage-dependent.
Next, we conducted a similar analysis for four other shared subunits of BAF and PBAF (SWSN-1, SWSN-3, SWSN-5, SWSN-6) using available mutant alleles or RNAi (Supplementary Fig. 6). We observed ectopic glr-4 expression in nerve cord MNs of swsn-1(os22) and swsn-5(ok622) single mutants and in animals with MN-specific RNAi against swsn-6 (Fig. 4e and Supplementary Fig. 7). In shared subunit mutants, glr-4 is ectopically expressed in fewer MNs when compared to PBAF-specific mutants (Fig. 4c, e). Further, we observed no ectopic glr-4 expression in MNs of swsn-3 LOF mutants (Fig. 4e and Supplementary Fig. 7). These weaker or absent effects on glr-4 are likely due to the weak nature of LOF mutant alleles for these shared subunit-encoding genes, which do not affect organismal survival (Supplementary Fig. 6). Strong LOF alleles for genes encoding shared core BAF/PBAF subunits are lethal59,65, and thereby not examined herein. In conclusion, several shared subunits of PBAF and BAF, as well as the activity of the ATPase SWSN-4/BRG1, are needed for glr-4 repression in nerve cord MNs.
PBAF antagonizes the terminal selector UNC-3/EBF in MNs
The terminal selector UNC-3/EBF acts directly to activate multiple effector target genes (including glr-4) in subclasses of cholinergic MNs41,42. In prior work with the endogenous mScarlet::glr-4 reporter38, we showed that UNC-3 is required to activate basal/low levels of glr-4 in nerve cord MNs. Here, we show that expression of the transgenic glr-4prom::tagRFP reporter in SAB and DA9 neurons is completely lost in unc-3 (n3435) null mutant animals (Fig. 5a–d), consistent with the known role of UNC-3 as transcriptional activator41,44. We therefore hypothesized that, in nerve cord MNs, unc-3 gene activity is necessary for the increased glr-4 expression we observe in PBAF mutants (Fig. 4c). Through double mutant analysis, we found this to be the case. Expression of glr-4 in nerve cord MNs is present in single pbrm-1(-) or swsn-9(-) mutants, but completely lost in unc-3(-); pbrm-1(-) and unc-3(-); swsn-9(-) double mutants (Fig. 5a, c). Next, we over-expressed UNC-3 specifically in nerve cord MNs and witnessed a modest increase in glr-4prom::tagRFP expression. However, UNC-3 over-expression in a PBAF (swsn-9) mutant was able to substantially increase glr-4prom::tagRFP in MNs (Fig. 5e), suggesting that perturbation of the PBAF complex enables UNC-3 to robustly activate glr-4 transcription. Altogether, our genetic analysis suggests that the PBAF complex represses glr-4 in nerve cord MNs by antagonizing the transcriptional activator UNC-3/EBF, albeit the precise molecular mechanism remains elusive (Fig. 5f). Despite the broad expression of PBAF, this effect is remarkably specific to a subset of nerve cord MNs. Outside the nerve cord, no change was observed in glr-4 expression in SAB and DA9 neurons of pbrm-1 or swsn-9 mutants (Fig. 5a, b, d, f).

a glr-4 expression in unc-3 and unc-3; PBAF double mutants. glr-4 expression is lost completely in RVG, VNC, and PAG MNs in all unc-3 mutants. Encircled neurons are head ganglia; asterisks (magenta, blue) and arrows indicate SAB, DA9, and VNC MNs, respectively. Quantification of a showing number of MNs expressing glr-4 in SAB (b), VNC (c), and DA9 (d). Animals imaged at L4 stage. WT n = 35; unc-3(n3435) n = 15; pbrm-1(tm415) n = 20; pbrm-1(tm415); unc-3(n3435) n = 15; swsn-9(ok1354) n = 20; swsn-9(ok1354); unc-3(n3435) n = 10. e Quantification of number of MNs expressing glr-4prom::tagRFP in WT and swsn-9(−) with or without UNC-3 overexpression (Punc-3::unc-3-CDNA). Animals analyzed at L4. n = 15. For all quantifications, unpaired two-sided Welch’s t-test, p < .05 = *; p < 0.01 = **; p < 0.001 = ***; p < 0.0001 = ****. f Schematic showing effect of UNC-3 and PBAF on glr-4 in SAB (left), VNC (middle), and DA9 (right). Box and whisker plots were used; all data points presented. Box boundaries indicate the 25th and 75th percentile. The limits indicate minima and maxima values. Center values (mean) are highlighted with a black horizontal line. Source data are provided as a Source Data file.
PBAF represses glr-4/GRIK4 in specific MN subclasses
Which MN subclasses show glr-4 expression in PBAF mutants? Our genetic analysis showed up to 14 ± 1 (mean ± STDV) nerve cord MNs ectopically express glr-4prom::tagRFP in PBAF mutants (Fig. 4c), indicating PBAF represses glr-4 in a subset of the 58 MNs that populate the VNC (Fig. 1a). Using a combination of criteria (stereotypic cell body position, birth order, and fluorescent MN reporters; see Methods) (Fig. 6a), we identified with single-cell resolution the specific MN subclasses that ectopically express glr-4 in three PBAF-specific mutants (pbrm-1, swsn-9, and phf-10). We found that PBAF represses glr-4prom::tagRFP primarily in subclasses of DA, VA, and AS neurons located specifically in the VNC (DA2-5, DA6-8, VA2, VA3-8, VA9-10, AS2-3, AS4-8, and AS9-10) (Fig. 6b, c). Compared to DA and VA, this effect is stronger in AS subclasses, where the percentage of animals ectopically expressing glr-4prom::tagRFP approaches 100% (Fig. 6b, c). We obtained similar results with MN-specific RNAi against swsn-7, which encodes the PBAF-specific subunit SWSN-7 (BAF200) (Fig. 6b, c). Because glr-4 transcripts are detected at low levels in AS and VA neurons (Fig. 1c), we propose that PBAF tunes down glr-4 to low levels in these neurons (Fig. 6d). On the other hand, glr-4 transcripts are undetectable in DA neurons (Fig. 1c) due to PBAF’s repressive activity (Fig. 6d).

a Single-cell analysis for phf-10(rd21) mutant animals: glr-4prom::tagRFP (top), acr-2::GFP (middle), merged (bottom). AS MNs labeled with white arrowheads. Animals imaged at L4 stage. b, c Results by MN subclass of single-cell analysis shown in (a) for all PBAF mutants: DA, VA, AS, DB, and VB. WT n = 20; pbrm-1(ot792) n = 16; swsn-9(ok1354) n = 16; phf-10(rd21) n = 18; swsn-7(RNAi) n = 20. Two-sided chi-square test, p < .05 = *; p < .01 = **; p < .001 = ***; p < .0001 = ****. d Schematic showing effects of UNC-3 and PBAF on glr-4 across MN subclasses. Source data are provided as a Source Data file.
Next, we wondered whether the molecular identities of the DA, VA, and AS subclasses are broadly affected in PBAF mutants. To test for this possibility, we evaluated expression of seven available terminal identity markers of DA, VA, and/or AS neurons (unc-17/VAChT, dbl-1/BMP7, del-1/SCNN1B/D/G, slo-2/KCNT1/2, acr-2/CHRNA3/6, unc-53/NAV1, and unc-129/GDF10). However, we found no changes in the number of MNs expressing each marker in multiple PBAF mutants (Supplementary Fig. 8). In addition, these findings show that the total number of MNs is not affected by lack of PBAF activity, indicating that PBAF does not affect MN generation in the C. elegans nerve cord. In summary, the PBAF chromatin remodeling complex selectively represses glr-4, and likely additional yet-to-be identified target genes, in DA, VA, and AS MN subclasses (Fig. 6d).
MAB-9/TBX20 represses glr-4/GRIK4 in AS subclasses of MNs
The specificity of the observed PBAF effect on glr-4 in distinct subclasses of DA, VA, and AS neurons is striking, given that PBAF subunit-encoding genes (pbrm-1, swsn-7, swsn-9, and phf-10) are broadly expressed in neurons and other C. elegans cell types (Fig. 7a). What is the molecular basis for this specificity? We hypothesized that in DA, VA, and AS subclasses, transcription factors with repressor activity recognize specific DNA sequences at the glr-4 locus and subsequently recruit the PBAF complex, resulting in glr-4 transcriptional repression.

a scRNA-seq data showing UNC-3, MAB-9, CFI-1, and PBAF/BAF component expression across MN subclasses. b Images show glr-4prom::tagRFP expression in WT and mab-9(e2410) mutant animals. c Quantification of b showing number of MNs expressing glr-4prom::tagRFP in VNC. WT: n = 35, mab-9(-): n = 17. Animals imaged at L4 stage. d ChIP-seq tracks for UNC-3, MAB-9, PBRM-1, and SWSN-7 at the glr-4 locus. PBRM-1 and MAB-9 tracks are represented as fold change over control and using a log scale. The SWSN-7 track is represented as control normalized signal and using a log scale. ChIP-seq for PBRM-1, SWSN-7, and MAB-9 were performed at L1 stage animals148, but for UNC-3 at L2 stage animals49. Diagram of glr-4 locus. MAB-9 and UNC-3 binding sites are indicated. Known DNA binding sites for UNC-3 and MAB-9/Tbx20 (right). e Quantification of MAB-9 site mutagenesis (deletion). Animals imaged at L4. N = 15. Three transgenic lines per construct were analyzed. f Images of MAB-9 site #6 animals and controls. AS motor neurons were identified based on their axonal morphology. g Quantification of MN number and fluorescence intensity in animals expressing mScarlet::glr-4 in WT and MAB-9 binding site mutants. L4 stage. n = 15. For all quantifications, unpaired two-sided Welch’s t-test was performed, p < .05 = *; p < .01 = **; p < .001 = ***; p < .0001 = ****. Box and whisker plots were used; all data points presented. Box boundaries indicate the 25th and 75th percentile. The limits indicate minima and maxima values. Center values (mean) are highlighted with a black horizontal line. h Proposed model: MAB-9 (TBX20) recognizes multiple cognate sites and acts as a PBAF recruiter in AS subclasses. Source data are provided as a Source Data file.
To test this idea, we focused on AS subclasses (AS2-3, AS4-8, and AS9-10) because the PBAF effect on glr-4 is stronger compared to DA and VA subclasses (Fig. 6b–d). The transcription factor MAB-9 (ortholog of human Tbx20) is expressed in AS neurons (Fig. 7a) and acts as a transcriptional repressor37,66. If our hypothesis is correct, we would expect mab-9 (e2410) LOF mutants to exhibit a similar phenotype as PBAF mutant animals, expressing high levels of glr-4 in nerve cord AS subclasses. We found this to be the case using two transgenic reporters (glr-4prom::tagRFP, glr-4 4.9kb prom::GFP) and one endogenous (mScarlet::glr-4) reporter for glr-4 (Fig. 7b, c and Supplementary Fig. 9a, c–e). Further, we found the expression of a mab-9 reporter to be unaffected in MNs of various PBAF mutants (Supplementary Fig. 9f, g). We conclude that mab-9 is required to tune down glr-4 expression in AS subclasses of nerve cord MNs.
MAB-9/TBX20 and PBAF act directly to repress glr-4/GRIK4
If MAB-9 recruits PBAF to the glr-4 locus, which is normally activated by UNC-3 in various MNs, our hypothesis predicts that in WT animals, MAB-9, PBAF, and UNC-3 directly bind to cis-regulatory elements of the glr-4 locus. We therefore interrogated available chromatin immunoprecipitation followed by sequencing (ChIP-Seq) whole-animal data for MAB-9, two PBAF proteins (PBRM-1 and SWSN-7) and UNC-349,67, and found that all four proteins bind to the glr-4 locus (Fig. 7d). Interestingly, MAB-9 and PBAF (PBRM-1 and SWSN-7) binding coincides prominently within the glr-4 locus and modestly upstream of glr-4, supporting the possibility of MAB-9 recruiting PBAF (Fig. 7d). Further, binding of the glr-4 repressor (MAB-9) is mostly excluded from regions bound by the glr-4 activator (UNC-3) (Fig. 7d).
To test whether MAB-9 binding on the glr-4 locus is functional, we first followed a bioinformatic approach (see “Methods”) and identified 10 predicted MAB-9 binding sites upstream and within the glr-4 locus (Fig. 7d and Supplementary Table 1). Next, we individually deleted the three most proximal MAB-9 sites (#5 - #7) in the context of our transgenic glr-4538bp prom::tagRFP animals (Fig. 7e, f). Deletion (Δ) of either site #5 or #7 yielded no effect on glr-4prom::tagRFP expression in nerve cord MNs (Fig. 7e, f). However, deletion of site #6 (Δ site #6) led to ectopic glr-4prom::tagRFP expression in AS subclasses in the VNC (Fig. 7e, f), suggesting that MAB-9 binds directly to site #6 to repress glr-4 transcription. Interestingly, MAB-9 site #6 lies next to UNC-3 site #1, which is necessary for driving basal levels of endogenous mScarlet::glr-4 expression in nerve cord MNs38.
Next, we used CRISPR/Cas9 to mutate site #6 by introducing nucleotide substitutions (WT: CCTTTTCTCCT, Mutated: CCAAAAAACT) in the context of the endogenous mScarlet::glr-4 reporter (Supplementary Table 1), but this manipulation had no effect on mScarlet::glr-4 expression in MNs (Fig. 7g). The discrepancy between the results obtained when MAB-9 site #6 was deleted in the transgenic glr-4538bp prom::tagRFP versus the endogenous mScarlet::glr-4 context suggests redundancy among the 10 MAB-9 sites. To test this idea, we simultaneously mutated site #6 and two other sites (#8 and #9)(Supplementary Table 1), but observed no effect on endogenous mScarlet::glr-4 expression in MNs (Fig. 7g), raising the possibility that one or more of the remaining seven MAB-9 sites are involved in glr-4 repression. This is consistent with previous work where simultaneous mutation of 11 binding sites for the repressor TF CFI-1 led to increased glr-4 expression in C. elegans MNs38. Altogether, genetic and biochemical (ChIP-Seq) evidence suggests that MAB-9/Tbx20 and PBAF act directly to repress glr-4/GRIK4 transcription in AS neuron subclasses (AS2-3, AS4-8, and AS9-10) (Fig. 7h).
To expand the repertoire of PBAF target genes, we identified 1000 genes that—like glr-4—are expressed at low levels in AS neurons based on available sc-RNA-Seq data5. Specifically, we included 1000 genes with expression values (transcripts per million, TPM) centered around glr-4 (TPM: 120.5) (see Methods). Next, we found that—like glr-4—247 of these 1000 genes are co-bound by UNC-3, MAB-9 and SWSN-7 (Supplementary Fig. 9b and Supplementary Data 1). Although validation of these 247 genes is beyond the scope of this work, this analysis nevertheless offers a list of putative target genes that are repressed by MAB-9 and PBAF in AS neurons.
Prediction/validation of MAB-9/TBX20 and PBAF interactions
A plausible mechanism by which MAB-9 could provide specificity to PBAF to repress glr-4 in AS neurons is through direct PBAF recruitment via protein-protein interactions. This model is consistent with the prominent co-binding of MAB-9, SWSN-7 and PBRM-1 within the glr-4 locus (Fig. 7d). Because co-immunoprecipitation experiments on isolated C. elegans MNs are not feasible, we tested this model via an in silico interaction screen using the LocalColabFold implementation of AlphaFold2, a powerful tool for accurate prediction of protein-protein interactions68,69,70,71. We screened for pairwise interactions between MAB-9 and the PBAF-specific subunits (SWSN-7, SWSN-9, PBRM-1, and PHF-10), as well as other core/accessory subunits (SWSN-2.2, SWSN-4, SWSN-5, SWSN-3, SWSN-1, SWSN-6, and SWSN-2.1). Following stringent criteria established in a previous study71, we assessed prediction quality using an interface predicted template modeling (ipTM) score cut-off of 0.3 for the majority (at least 3 out of 5) of the predicted models for each pairwise interaction (Fig. 8a and Supplementary Fig. 10). We further assessed the proposed interaction using the predicted alignment error plots (Fig. 8b and Supplementary Fig. 10). With this analysis, we identified the PBAF-specific subunit SWSN-7 and the accessory subunit SWSN-6 as predicted interactors with MAB-9. Predicted contacts of the top-ranked structures for both dimers were assessed using PDBsum and visualized with PyMOL (Fig. 8c, d). The MAB-9-SWSN-7 interface had an area of 1947 Å2 and 2069 Å2 for each protein, respectively. The MAB-9-SWSN-7 interaction consisted of 3 salt bridges, 9 hydrogen bonds, and 486 non-bonded contacts. The MAB-9-SWSN-6 interface had an area of 2252 Å2 and 2720 Å2 for each protein, respectively. The MAB-9-SWSN-6 interaction consisted of 1 salt bridge, 18 hydrogen bonds, and 1820 non-bonded contacts. These predicted interactions support a model of PBAF recruitment via direct protein interactions with MAB-9.

a ColabFold confidence metrics of predicted structures for MAB-9 and SWSN-7, and MAB-9 and SWSN-6 complexes, with 5 models generated for each pairwise interaction. pTM (red) and ipTM (blue) values for the 5 top-ranked structures for each complex are plotted. pTM: predicted template modelling—confidence of overall complex prediction. ipTM: interface predicted template modelling—confidence of relative subunit placement. Gray dashed line: 0.3 ipTM cutoff determined as warranting future investigation71. b Predicted Alignment Error plots for 5 top ranked models of MAB-9 and SWSN-7, and MAB-9 and SWSN-6, complexes. Heat map color corresponds to expected positional error at a residue in angstroms. Within a plot, top left represents MAB-9 monomer confidence, bottom right represents SWSN-7 (top row) or SWSN-6 (bottom row) monomer confidence, and off-diagonal boxes represent placement confidence of residues between subunits (interaction confidence). c Structural visualization of the top-ranked MAB-9/SWSN-7 complex prediction. Light orange: MAB-9. Dark orange: MAB-9 DNA binding domain (DBD). Blue: SWSN-7. Magenta dashed lines: inter-chain polar contacts predicted by PyMOL. Inset: zoomed in view of the interface region highlighting polar contacts. Side-chains involved in polar contacts shown as licorice-sticks. d Structural visualization of the top-ranked MAB-9/SWSN-6 complex prediction. Light orange: MAB-9. Dark orange: MAB-9 DNA binding domain (DBD). Blue: SWSN-6. Magenta dashed lines: inter-chain polar contacts predicted by PyMOL. Inset: zoomed in view of the interface region highlighting polar contacts. Side-chains involved in polar contacts shown as licorice-sticks. e Western blots (anti-HA) of co-immunoprecipitation (co-IP) experiments in HEK293 cells show binding between MAB-9 and both SWSN-6 and SWSN-7. Left: Western blot shows presence of MAB-9::HA in protein lysate when SWSN-6::3XFLAG and SWSN-7::3XFLAG are pulled down. Right: Western blot shows presence of SWSN-6::HA and SWSN-7::HA in protein lysate when MAB-9::Myc is pulled down. Red asterisks indicate target protein in pulldown lanes. Negative controls (lanes 5–8) show an absence of target protein in pulldown lanes. Negative controls include: (1) MAB-9::HA or MAB-9::Myc co-transfected with pcDNA3.1_3xFLAG::BAP (BAP: bacterial alkaline phosphatase) and (2) pcDNA3.1_3xFLAG::BAP alone. Bottom: Western blots show beta-Actin protein loading control. These reciprocal co-IP experiments were repeated once. Source data are provided as a Source Data file.
To experimentally test this model, we conducted co-immunoprecipitation assays in HEK293 cells co-transfected with epitope-tagged versions of MAB-9 and SWSN-6, or MAB-9 and SWSN-7. Following pull-down of MAB-9::Myc with anti-c-Myc magnetic beads, we detected SWSN-6::HA (49.5 kDa) or SWSN-7:HA (140.2 kDa) with western blotting against the HA epitope (Fig. 8e). Conversely, following pull-down of either SWSN-6::3XFLAG or SWSN-7:3XFLAG with anti-FLAG M2 magnetic beads, we detected MAB-9::HA (40.5 kDa) with anti-HA western blotting (Fig. 8e). Together, these results validate the AlphaFold2 predictions, supporting a model where MAB-9/TBX20 physically interacts with both SWSN-6 and SWSN-7 to recruit PBAF and thereby influence gene expression in AS neurons.
UNC-3 and PBAF control caudal MN terminal identity
In the PAG ganglion immediately posterior to the VNC, there are five subclasses of caudal MNs that are cholinergic (DA8, DA9, VA11, VA12, and AS11) (Fig. 9a); these co-express UNC-3 and PBAF components (Fig. 7a). UNC-3 activates glr-4 in DA9, but loss of PBAF has no effect on glr-4 expression in DA9 (Fig. 2d). We therefore asked whether other unc-3-dependent terminal identity markers for caudal MN subclasses are affected in PBAF mutant animals. Indeed, we found that PBAF gene activity (pbrm-1, swsn-9, phf-10) is required for the expression of itr-1/ITPR1 in DA9 (Fig. 9b, f), avr-15/GLRA1-3 in VA11 and AS11 (Fig. 9c, g), ser-2/HTR1A in DA8 (Fig. 9d, h), and glr-5/GRIK1/3 in DA8 and VA12 (Fig. 9e, i). Additional markers (flp-18, sra-36, nlp-7, and flp-7) for caudal MN subclasses are not affected by loss of PBAF gene activity (Supplementary Fig. 11). These findings point to a different role for PBAF in caudal MN subclasses: instead of a role in gene repression, which we observed in nerve cord MN subclasses, PBAF is required to activate expression of various terminal identity markers in caudal MN subclasses. Taken together, our results indicate a dichotomy in PBAF function in C. elegans cholinergic MNs. In nerve cord MN subclasses, PBAF antagonizes UNC-3, and thereby acts as a repressor of glr-4. In caudal MN subclasses, PBAF and UNC-3 are both required to activate expression of several terminal identity genes (e.g., itr-1, avr-15, ser-2, and glr-5).

a Schematic of C. elegans caudal MNs. b Images showing itr-1::GFP expression (DA9) in WT and PBAF mutants. c Images showing avr-15::GFP expression (VA11, AS11) in WT and PBAF mutants. d Images showing ser-2::GFP expression (DA8, VD12, and VD13) in WT and PBAF mutants. e Images showing glr-5::GFP expression (DA8, VA12) in WT and PBAF mutants. f Quantification of fluorescence intensity of itr-1::GFP in DA9 of WT and PBAF mutants. L4 stage. WT n = 21; pbrm-1(ot792) n = 17; swsn-9(ok1354) n = 20; phf-10(rd21) n = 19. g Quantification of fluorescence intensity of avr-15:GFP in AS11 of WT and PBAF mutants. Unpaired Welch’s t-test, p < .05 = *; p < .01 = **; p < .001 = ***; p < .0001 = ****. h Quantification of fluorescence intensity of ser-2::GFP in DA8 as a proxy for expression levels in WT and swsn-9(ok1354) mutants. L4 stage. WT n = 15; swsn-9(ok1354) n = 7. i Quantification of fluorescence intensity of glr-5::GFP in DA8 and VA12 of WT and swsn-9(ok1354) mutants. L4 stage. WT n = 13; swsn-9(ok1354) n = 9. Unpaired, two-sided Welch’s t-test, p < .05 = *; p < .01 = **; p < .001 = ***; p < .0001 = ****. Box and whisker plots were used; all data points presented. Box boundaries indicate the 25th and 75th percentile. The limits indicate minima and maxima values. Center values (mean) are highlighted with a black horizontal line. j Schematic summarizing the hypothesis that EGL-5 recruits PBAF to activate terminal identity genes in caudal MNs (DA9, VA12). k ChIP-seq tracks for UNC-3, PBRM-1, and SWSN-7 at the avr-15, itr-1, ser-2, and glr-5 loci. ChIP-seq data for UNC-3 (L2 stage), PBRM-1 (L1) and SWSN-7 (L1). Source data are provided as a Source Data file.
Loss of pbrm-1 leads to locomotion defects
Because our molecular analyses revealed gene expression defects in MNs, we wondered whether PBAF mutant animals display locomotion defects. To test this, we performed high-resolution behavioral analysis of freely moving adult (day 1) pbrm-1 mutant animals using automated multi-worm tracking72,73. This analysis can quantitate multiple features related to C. elegans locomotion (e.g., speed, crawling amplitude, curvature, pause, forward and backward locomotion) and each feature can be localized to a specific part of the nematode’s body (e.g., head, mid-body, tail). We assessed homozygous pbrm-1 mutants carrying either the tm415 or ot792 alleles (Fig. 2e). Hierarchical clustering analysis showed that pbrm-1(tm415) animals cluster more closely to control (N2) animals, whereas pbrm-1(ot792) animals displayed strong locomotion defects (Fig. 10a). These behavioral effects are consistent with the stronger effects we observed on glr-4 expression in pbrm-1(ot792) compared to pbrm-1(tm415) animals (Fig. 2b). Because the PBAF effect on glr-4 is stronger in AS neurons, which are known to control body wave propagation74, we asked whether this particular feature is disrupted in pbrm-1 mutants. Indeed, we found significant changes in the body wave, reflected in curvature features and the time derivative of curvature features (d_curvature) in pbrm-1(ot792) mutants (Fig. 10c–g). Further, median midbody speed is affected in pbrm-1(ot792) mutants (Fig. 10b). Altogether, this analysis shows that loss of pbrm-1 leads to significant defects in posture (curvature) and motion (speed), consistent with defective body wave propagation.

a Heatmap showing the phenotypic relationship between strains. Each column shows values of different behavioral features. Data are z-normalized and rows and columns are ordered using hierarchical clustering (Euclidean distance, mean linkage). pbrm-1(tm415) is more similar to control (N2) strain than pbrm-1(ot792) in terms of their distance in phenotype space which leads to N2 clustering with pbrm-1(tm415) in the dendrogram. The color shows the z-score of each feature (red shows values greater than the mean across strains while blue shows values less than the mean). b–g Examples of locomotion features (e.g., speed and curvature) that are significantly affected in pbrm-1(ot792) animals, but mildly affected in pbrm-1(tm415) animals compared to WT (N2). Parenthesis: unit of measurement. n = 15. Box and whisker plots were used. Box boundaries indicate the 25th and 75th percentile. The limits indicate minima and maxima values. Center values (mean) are highlighted with a black vertical line. Unpaired two-sided Welch’s t-tests independently for each feature were performed. The false discovery rate was controlled at 5% across all strain and feature comparisons using the Benjamini–Yekutieli procedure.
Last, we conducted thrashing assays on adult animals, in which we knocked-down PBRM-1 specifically in motor neurons with either cell-specific RNAi or ZIF-1 technology using the exact same worm strains shown in Fig. 3c, d. However, we were unable to identify any thrashing defects (Supplementary Fig. 12), likely due to partial decrease of PBMR-1 with RNAi or ZIF-1. In fact, we only achieve a modest decrease in the number of motor neurons expressing PBRM-1::GFP with ZIF-1-mediated degradation (Fig. 3d). We conclude that although our RNAi and ZIF-1 approaches lead to ectopic glr-4 expression in motor neurons (Fig. 3c, d), their partial knock-down effect on PBRM-1 is insufficient to lead to detectable behavioral effects. Alternatively, we cannot discard that the pbrm-1(ot792) locomotory defects are caused by non-cell autonomous effects in other neurons or non-neuronal cells.
Discussion
Our current understanding of neuronal diversification is primarily based on developmental mechanisms that involve TFs acting at the level of progenitor or postmitotic neuronal cells. By recognizing specific DNA sequences, TFs control gene expression and thereby generate distinct molecular profiles for individual neuronal types and subtypes. However, epigenetic mechanisms such as DNA methylation, histone modifications, and ATP-dependent chromatin remodeling are also known to affect gene expression in neurons. Yet, our understanding of their involvement in neuronal diversification remains incomplete. In this study, we identify key roles for the PBAF chromatin-remodeling complex of the SWI/SNF family in C. elegans MN diversification. Because the PBAF complex is expressed in animal nervous systems and implicated in human neurodevelopmental conditions, the PBAF-mediated mechanisms of neuronal diversification described herein may have biomedical implications.
The ancient SWI/SNF family of chromatin remodelers includes the BAF and PBAF complexes. A large body of work has uncovered essential functions for the BAF complex at various stages of neural development45,46. In mammals, distinct BAF complex subunit compositions are found in embryonic stem cells, neural progenitors and postmitotic neurons75,76. Neural progenitor-specific BAF, termed npBAF, controls cell proliferation during neurogenesis at various regions of the mammalian brain (forebrain, cortex, olfactory bulb)77,78,79,80, as well as the developmental switch between neurogenesis and gliogenesis81,82. On the other hand, neuronal BAF (nBAF) is necessary for dendritic morphogenesis and synaptic plasticity in flies and mice83,84,85,86. Further, the BAF complex controls axonal pathfinding in the mouse olfactory system87. In C. elegans, the BAF subunit SWSN-2.1 (human BAF60a-c) is not only required for axonal pathfinding, but also for serotonergic neuron differentiation88. Hence, it is well attested that the BAF complex plays crucial and conserved roles in nervous system development. In contrast, the closely related PBAF complex is understudied in the nervous system. Currently, our limited understanding of neural PBAF functions stems from a handful of studies that focused on individual PBAF-specific subunits and showed that: (a) PHF10/BAF45a (C. elegans phf-10) is essential for neural progenitor proliferation75, (b) PBRM1/BAF180 (C. elegans pbrm-1) controls the transition from progenitors to pre-myelinating oligodendrocytes89, and (c) BRD7 (C. elegans SWSN-9) controls dendritic morphogenesis in the prefrontal cortex90. Thus, unlike BAF, the function and target genes of PBAF in postmitotic neurons remain largely unknown.
In this study, we demonstrate that PBAF acts in C. elegans postmitotic MNs. Upon a comprehensive genetic and RNAi analysis of all four PBAF-specific components (PBRM-1, PHF-10, SWSN-7, SWSN-9) and several core subunits, we uncovered five terminal identity genes as targets of PBAF in MNs. These include the glutamate receptor subunit genes glr-4 and glr-5 (orthologs of human GRIK4), the chloride channel subunit gene avr-15 (human GLRA2/3), the serotonin receptor gene ser-2 (human HTR1D), and the inositol triphosphate receptor gene itr-1 (human ITPR1). Each of these genes is expressed continuously, from embryo to adult, in subsets of C. elegans MNs, defining their identity and function8. Altogether, our findings indicate that the PBAF complex, as a whole, acts in postmitotic MNs to control their terminal identity, critically extending the repertoire of known PBAF functions beyond neural progenitor cells75,89.
It is important to note that the composition of PBAF and BAF complexes in C. elegans are inferred from homology to yeast and mammalian SWI/SNF subunits, as no biochemical studies to date have examined the BAF/PBAF complexes in nematodes. Nevertheless, our genetic findings are consistent with previous studies supporting the existence of distinct PBAF and BAF complexes in C. elegans53,58,64,91.
Terminal selectors are conserved TFs that control the identity of individual neuron types21. An emerging picture in terminal selector biology is that these TFs often act in combinations with one another in specific neuron types. To date, combinations of up to six TFs that act as terminal selectors have been described in ~80 of 118 C. elegans neuron types20,92,93,94. Studies in the fly and mouse nervous systems further support the principle that neuron type identity is controlled by specific combinations of TFs that act as terminal selectors9,24,39,95,96,97,98,99,100. In striking contrast to the plethora of terminal selectors identified to date, the epigenetic mechanisms that influence terminal selector activity and thereby neuron type identity are poorly defined. Genetic studies in model organisms have begun to fill this knowledge gap. The MYST histone acetyltransferase complex and the terminal selector gene die-1 are both required to initiate and maintain the identity of a C. elegans chemosensory neuron subtype (ASEL)101. The Polycomb Repressive Complex 1 (PRC1) controls the terminal identity of a C. elegans head interneuron type (AIY) by controlling expression of two terminal selector genes102. Further, our previous work showed that H3K9 methylation, the epigenetic hallmark of heterochromatin103, is necessary for terminal identity gene repression in a subset of C. elegans nerve cord MNs that express the terminal selector UNC-338. In the adult mouse brain, the PRC2 complex is required to maintain the identity and function of dopaminergic and serotonergic neurons104, whereas acetylation of lysine 27 in H3 (H3K27), a mark associated with active transcription, is necessary to maintain the terminal identity of forebrain and hippocampal neurons105,106.
In the present study, we identify an additional epigenetic mechanism, i.e., ATP-dependent chromatin remodeling, as necessary for neuronal subtype identity. Our findings show that the PBAF chromatin-remodeling complex is necessary for MN diversification in C. elegans. Intriguingly, we observe a dichotomy in PBAF function. In specific nerve cord MN subtypes, PBAF represses terminal identity genes by antagonizing the terminal selector UNC-3. In caudal MN subtypes, however, PBAF and UNC-3 are both required to activate terminal identity gene expression. Our findings are consistent with previous work in non-neuronal cell types demonstrating both repressor and activator roles for PBAF107,108,109.
An important aspect of this study is the cell-type specificity of PBAF function in MNs. This finding is surprising because chromatin remodelers in general are known to be expressed broadly in various cell types and to bind DNA throughout the genome without sequence specificity110,111. By using LOF alleles for several genes encoding PBAF subunits (PBRM-1/PBRM1, SWSN-9/BRD7, and PHF-10/BAF45a), we observed remarkably specific effects in subsets of C. elegans MNs. In certain groups of nerve cord MNs (e.g., AS subclasses), PBAF tunes down gene expression, whereas in caudal MN subclasses, PBAF activates distinct terminal identity genes. What is the molecular mechanism for this dichotomy in PBAF function?
An emerging notion for the phenotypic specificity of chromatin-remodeling complexes is that they are recruited to specific gene loci by cell type-specific TFs, which are known to recognize DNA in a sequence-specific manner112. For the BAF complex, recruitment is thought to enable transcription and has been reported in various cell types, including neural stem cells and developing glia113,114,115,116,117,118,119. Inspired by these BAF studies, we propose that cell type-specific TFs operate as “recruiters” of the PBAF complex in C. elegans MNs. In caudal MNs (DA9 and VA12), a candidate recruiter is EGL-5 (Fig. 9j), a posterior Hox protein homologous to fly Abd-B and vertebrate Hox9-Hox13. We previously showed that loss of egl-5 results in failure to activate expression of caudal MN identity genes44, and the same effect is observed in PBAF-specific mutant animals lacking pbrm-1 or swsn-9 (Fig. 9). Further, PBRM-1 and SWSN-9 contain bromodomains (BrDs), which recognize acetylated histones, a hallmark of gene activation52,120, supporting a model wherein their recruitment by the cell type-specific TF EGL-5 in caudal MNs results in activation of caudal MN identity genes. Alternatively, UNC-3 itself may recruit PBAF in caudal MNs to activate gene expression. Supporting this idea, a human UNC-3 ortholog (EBF1) activates gene expression in B cells by recruiting the PBAF/BAF ATPase Brg1121.
In nerve cord MNs, however, we found that PBAF represses (tunes down) gene expression. In these cells, several lines of evidence suggest that the conserved TF MAB-9/Tbx20 acts as a PBAF recruiter to repress glr-4/GRIK4. First, MAB-9 is known to act as a transcriptional repressor in C. elegans nerve cord MNs37. In AS neurons, MAB-9 is expressed at higher levels compared to the transcriptional activator UNC-3 (Fig. 1d), possibly favoring glr-4 repression. Second, we found that animals lacking either mab-9 or PBAF activity display increased glr-4 expression in nerve cord AS MNs (Fig. 7b, c). Third, MAB-9 and PBAF-specific components (PBRM-1, SWSN-7) bind to the glr-4 locus (Fig. 7d). Fourth, mutagenesis of MAB-9 cognate binding sites indicated that MAB-9 likely acts directly by recognizing more than one of its cognate sites on the glr-4 locus. Fifth, we validated in vitro physical interactions between MAB-9/Tbx20 and SWSN-6, as well as between MAB-9/Tbx20 and SWSN-7 (Fig. 8). Interestingly, Tbx1, a Tbx20 paralog, physically interacts with a shared subunit (Baf60a) of BAF and PBAF complexes in mouse cardiac progenitors114. Last, the long PBRM-1 isoform is a signature subunit of PBAF and contains two bromo-adjacent homology (BAH) domains (Fig. 2f), which are known protein-protein interaction domains involved in transcriptional repression122,123.
Although additional mechanisms are also possible, including the documented interplay between SWI/SNF and Polycomb124,125,126, our genetic analysis suggests that the cell type-specific effects of PBAF on MN identity likely arise due to its recruitment by cell type-specific TFs, i.e., EGL-5/HOX in caudal MNs and MAB-9/Tbx20 in nerve cord MNs (Figs. 7h and 9j). If MAB-9 recruits PBAF to repress glr-4 in AS subclasses, how is glr-4 repressed by PBAF in DA and VA subclasses (Fig. 6c, d)? A possible recruiter is the transcription factor CFI-1/ARID3, which is not expressed in AS neurons, but is known to repress glr-4 in DA and VA subclasses38. In this way, PBAF essentially “borrows” the specificity of the TF that recruits it as needed, allowing for chromatin changes and targeted gene activation or repression. Altogether, our data contribute to our understanding of how broadly expressed chromatin remodelers can act with cell type-specificity.
Mutations or variants in genes encoding BAF and PBAF subunits are found in more than 20% of human cancers127,128. In addition, BAF and PBAF subunit-encoding genes are implicated in syndromic and non-syndromic neurological conditions, including autism spectrum disorder, intellectual disability, and schizophrenia45,46. In particular, mutations in the two ATPase genes (BRG1, BRM) and several other core components of BAF/PBAF complexes cause Coffin-Siris and Nicolides-Baraitser syndromes that are characterized by intellectual disability45,46. Further, variants in PBAF-specific genes ARID2/BAF200 (C. elegans swsn-7) and BAF180/PBRM1 (C. elegans pbrm-1) are also linked to Coffin-Siris syndrome and intellectual disability129,130,131,132. Because these similar neurological conditions occur when different PBAF subunits are individually mutated, it raises the possibility that the PBAF complex—as a whole—regulates chromatin dynamics and gene expression in specific neuron types. Although potential causes have been proposed (e.g., gain-of-function effects)46, how PBAF mutations lead to specific neurological phenotypes in patients remains unknown. Despite the widespread expression of PBAF-encoding genes, it is likely that such phenotypes arise due to impaired PBAF function in specific neuronal types or circuits. Therefore, the specific defects in MN diversification in C. elegans animals lacking PBAF gene activity, as well as the underlying molecular mechanism we report herein, may provide a conceptual framework to understand specific neuronal defects that arise in neurological conditions caused by mutations in broadly expressed chromatin-remodeling complexes.
Methods
Genetics
Strains were grown at 15 °C, 20 °C, or 25 °C on nematode growth media (NGM) plates with E. coli (OP50)133. Mutant alleles used: pbrm-1(ot792) (point mutation, premature stop), pbrm-1(tm415) (427 nucleotide deletion, 5 nucleotide insertion), swsn-9(ok1354) (1,010 nucleotide deletion), phf-10(rd21) (4 nucleotide deletion), unc-3(n3435) (1,008 nucleotide deletion), mab-9(e2410) (single nucleotide substitution), swsn-1(os22) (missense temperature-sensitive allele), swsn-3(tm3647) (432 nucleotide deletion), swsn-4(os13) (missense temperature-sensitive allele), swsn-5(ok622) (2,026 nucleotide deletion), and dpff-1(tm4287) (318 nucleotide deletion). All C. elegans strains used in this study are listed in Supplementary Table 2. All primers used in this study are listed in Supplementary Table 3.
Forward genetic screen
EMS mutagenesis was performed on otIs476[glr-4prom::TagRFP] fluorescent reporter animals, before screening visually at a dissecting fluorescent microscope (Zeiss SteREO Discovery V12) for changes in reporter expression in VNC MNs. To identify the causal genes of the mutant alleles obtained, Hawaiian single nucleotide polymorphism (SNP) mapping and whole-genome sequencing were performed followed by data analysis using the CloudMap pipeline51,134.
Microscopy
To prepare slides for imaging, animals were anesthetized using 100 mM sodium azide (NaN3) while being mounted on a 4% agarose pad on glass slides. Images were taken with an automated fluorescence microscope (Zeiss, Axio Imager Z2). Several z-stacks were taken with each image (0.50 μm intervals between stacks) with Zeiss Axiocam 503 mono using the ZEN software (Version 2.3.69.1000, Blue edition). Images and z-stacks were then transferred and processed in ImageJ135.
Generation of transgenic reporters
Reporter gene fusions were generated using PCR fusion136. Target gene promoter fragments were fused to the tagRFP coding sequence. Gene fusion products were then injected into wild-type young adult animals at a total DNA concentration of 100 ng/μl. The fusion product was accompanied by a myo-2::GFP or myo-2::mCherry co-injection marker at a concentration of 3-5 ng/μl. Individual lines were isolated from F1 progeny.
Generation of RNAi constructs and lines
DNA fragments spanning 2–4 exons were selected from target genes and fused in both sense and antisense orientations to enhancer elements sufficient to drive MN-specific expression, lin-39 (intron 1) and cho-1 (280 bp distal element), by PCR fusion54. Each fused construct was injected into young adult animals at a concentration of 100 ng/μl, along with myo-2::GFP or myo-2::mCherry co-injection marker plasmids at a concentration of 3–5 ng/μl. Individual lines were isolated from F1 progeny.
Motor neuron cell identification
A combination of criteria was used for MN cell ID: stereotypic cell body position of MNs2, birth order, and previously characterized fluorescent reporters for specific MN classes137. Identity was determined based on co-localization with these previously characterized fluorescent reporters.
Fluorescence intensity quantification
Images of worms were taken with identical parameters, using z-stacks that span the entire cell bodies of nerve cord MNs. These stacks were processed in ImageJ, where background was subtracted to reduce background noise from the image (rolling ball radius = 50 pixels). The MN cell bodies were manually selected (by drawing an area over MN cell bodies to be measured), and the average fluorescence intensity was measured and recorded for each visible MN in the image.
ZIF-1 mediated protein degradation
A plasmid containing GFPnanobody fused to ZIF-1 (driven by MN-specific 280 bp cho-1 promoter fragment) was injected into wild-type young adult animals along with co-injection marker myo-2::mCherry. GFPnanobody binds to GFP-tagged proteins, while ZIF-1 recruits the E3 ubiquitin ligase complex to degrade tagged proteins55. Individual lines were selected from F1 progeny. One line obtained underwent spontaneous integration into the genome and was subsequently used for our studies. This line was crossed with pbrm-1::TY1::EGFP::3xFlag to degrade PBRM-1::TY1::EGFP::3xFLAG protein.
Single-molecule RNA (smRNA) FISH
Animals were grown until plates were full of gravid worms, and were subsequently collected in an egg prep to synchronize animals. Synchronized L1 animals were then incubated in 4% paraformaldehyde in 1×PBS for 45 min at room temperature, washed two times using 1×PBS, and then permeabilized with 70% ethanol at 4 °C overnight. A glr-4 probe was designed using the Stellaris Probe Designer website (Biosearch Technologies). The probe was mixed in hybridization buffer (0.1 g/mL dextran sulfate [Sigma D8906-50G], 1 mg/mL E. coli tRNA [ROCHE 10109541001], 2 mM vanadyl ribonucleotide complex [New England Biolabs S1402s], 0.2 mg/mL RNase-free BSA [Ambion AM2618], 10% formamide) and added to the worms. The hybridization buffer was removed and worms were washed twice in wash buffer (DAPI was added during the second wash and incubated for 30 min in the dark for nuclear counterstaining). Worms were washed once in 2× SSC, incubated in GLOX buffer (0.4% glucose, 0.1 M Tris-HCl, 2× SSC) for 2 min for equilibration, and then resuspended in GLOX buffer with glucose oxidase and catalase added before imaging138.
Transcription factor motif analysis
The position weight matrix (PWM) of UNC-3 has been described49.
The MAB-9 PWM was derived from MODENCODE ChIP-seq data139 of mab-9::TY1::EGFP::3xFLAG L1 larva (experiment file identifier: ENCFF396HF). De novo motif discovery for MAB-9 was conducted with Homer140 using findMotifsGenome.pl using a sequence size of size given. The PWM of C. elegans MAB-9 is similar to the PWM of M. musculus Tbx20141. To identify putative UNC-3 and MAB-9 sites in the glr-4 locus, we used FIMO (Find Individual Motif Occurrences)142, which is one of the motif-based sequence analysis tools of MEME (Multiple Expectation maximization for Motif Elicitation) bioinformatics suite (http://meme-suite.org/). The p-value threshold for the analysis of MAB-9 and UNC-3 site in the glr-4 locus was set at p < 0.01.
Co-binding analysis of UNC-3, MAB-9, and SWSN-7 on 1000 genes
One thousand genes with expression values (transcripts per million, TPM) centered around glr-4 (TPM: 120.5) were selected from CenGEN (L4 nervous system-wide scRNA-seq) to define a list of candidates with likely similar regulatory inputs in AS MNs. The expression range for the 500 genes expressed lower than glr-4 in AS MNs was 75.9 to 120.4 TPM; the expression range for the 500 genes expressed higher than glr-4 in AS MNs was 120.5 to 282.8 TPM. A total of 5861 genes with non-zero TPM values were detected in AS MNs in this dataset, with 4414 genes expressed below (TPM range: 0.5-75.9) and 446 genes expressed above (TPM range: 283.0–216,684.1) the glr-4-centered range used for this analysis. Most known AS MN class markers were expressed well-above the glr-4-centered range, including unc-8 (351.7 TPM), elt-1 (444.1 TPM), unc-3 (499.8 TPM), mab-9 (581.2 TPM), and unc-53 (2898.7 TPM), whereas one AS-specific transcriptional repressor fell within the upper range (unc-55 = 152.4 TPM). Genes that were previously shown to not express in AS MNs typically had very low (within glr-4-centered range) or undetectable expression in this dataset (e.g., bnc-1 = 0 TPM, cfi-1 = 0 TPM, vab-7 = 0 TPM, dbl-1 = 107.8 TPM, slo-2 = 146.2 TPM). Next, we integrated ChIP-Seq data on these 1,000 genes. We found 247 genes to be co-bound by UNC-3, MAB-9 and SWSN-7, 258 genes co-bound by UNC-3 and MAB-9, and 659 genes co-bound by UNC-3 and SWSN-7 (Supplementary Data 1).
Prediction of protein–protein interactions
Protein-protein interaction was predicted using the LocalColabFold (https://github.com/YoshitakaMo/localcolabfold) implementation of Alphafold257,143, with multiple sequence alignments generated by MMseqs2144, and using 5 recycling steps. Quality of the prediction was assessed using outputted prediction confidence measures for the overall complex and for the relative subunit positions (pTM and ipTM, respectively) and PAE plots. An ipTM cut-off of 0.3 was used to identify structures warranting potential further investigation, as previously observed71. Top ranked models for structures of interest were visualized using PyMOL (https://pymol.org/).
Automated worm tracking
Worms were maintained as mixed stage populations by chunking on NGM plates with E. coli OP50 as the food source. The pbrm-1 (tm415) and pbrm-1(ot792) mutants were 6× outcrossed to the wild N2 isolate. The day before tracking, 30-40 L4 larvae were transferred to a seeded NGM plate and incubated at 20 °C for approximately 24 h. Five adults were picked from the incubated plates to each of the imaging plates (see below) and allowed to habituate for 30 min before recording for 15 min. Imaging plates are 35 mm plates with 3.5 mL of low-peptone (0.013% Difco Bacto) NGM agar (2% Bio/Agar, BioGene) to limit bacteria growth. Plates are stored at 4 °C for at least 2 days before use. Imaging plates are seeded with 50 μl of a 1:10 dilution of OP50 in M9 the day before tracking and left to dry overnight with the lid on at room temperature.
Behavioral feature extraction and analysis
All videos were analyzed using Tierpsy Tracker145 to extract each worm’s position and posture over time. These postural data were then converted into a set of behavioral features72. For each strain comparison, we performed unpaired Welch’s t-tests independently for each feature. The false discovery rate was controlled at 5% across all strain and feature comparisons using the Benjamini–Yekutieli procedure (Kim and van de Wiel, 2008).
Thrashing assay
In preparation for thrashing assay, animals were picked at the L4 stage onto an agarose plate without food. Animals were then individually picked and placed into a drop of M9 buffer on a separate agarose plate. Animals were given 30 s to acclimate and then recorded over the next 30 s via phone camera mounted onto a benchtop microscope. Video recordings of animals were analyzed at 0.5x speed to accurately count the number of thrashes (body bends) in 30 s.
Plasmid generation
Plasmids were generated by Genscript FLASH Gene DNA synthesis service. Plasmids include: (1) swsn-6 cDNA ORF clone from C. elegans (OCa343956), tagged with HA, inserted into pcDNA3.1+146, swsn-6 cDNA ORF clone from C. elegans (OCa343956), tagged with 3XFLAG, inserted into pcDNA3.1+, (3) swsn-7 cDNA ORF clone from C. elegans (OCa339589), tagged with HA, inserted into pcDNA3.1+, (4) swsn-7 cDNA ORF clone from C. elegans (OCa339589), tagged with 3XFLAG, inserted into pcDNA3.1+, (5) mab-9 cDNA ORF clone from C. elegans (OCa339423), tagged with HA, inserted into pcDNA3.1+, and (6) mab-9 cDNA ORF clone from C. elegans (OCa339423), tagged with myc, inserted into pcDNA3.1+. Control: plasmid pcDNA3.1_3xFLAG::BAP147.
Cell culture
HEK293 cells were cultured in DMEM (Gibco Dulbecco’s Modified Eagle Medium 1×11960-044) supplemented with 10% FBS (Gibco A56707-01) and 2 mM L-glutamine and grown at 37 °C with 5% CO2. Cells were cultured in 10 cm dishes.
Transfection
For each 10 cm dish, 24 μg of DNA (12 μg of plasmid 1 and 12 μg of plasmid 2) was added to 1 mL of Opti-MEM I 1× (Gibco 11058-021) and 60 μl of Lipofectamine LTX (Thermo Fisher Scientific Invitrogen 15338100). Plasmid combinations transfected into each dish include: (1) mab-9::HA and swsn-6::3XFLAG146, (2) mab-9::HA and swsn-7::3XFLAG, (3) mab-9::myc and swsn-6::HA, (4) mab-9::myc and swsn-7::HA, (5) mab-9::HA and pcDNA3.1+ containing BAP::FLAG, (6) mab-9::myc and pcDNA3.1+ containing BAP::FLAG, and (7) pcDNA3.1+ containing BAP::FLAG. BAP refers to bacteria alkaline phosphatase. After incubation for 30 min, the sample was added dropwise to each plate. Cell media was replaced 24 h after co-transfection, and protein was harvested 24 h later.
Protein harvest and quantification
Cells were collected in 3 mL of ice-cold ion-free PBS, centrifuged to a pellet at 5000 x g for 5 min at 4 °C, then lysed with 1 mL of: 50 mM Tris-HCl at pH 7.4 (Fisher Scientific BP152-500), 150 mM NaCl (Fisher Scientific S271-10), 1 mM EDTA, 1% TritonX-100, 1:100 phosphatase inhibitor cocktail 1 (Sigma Aldrich P2850-1ML), 1:100 phosphatase inhibitor cocktail 2 (Sigma Aldrich P5726-1ML), and 1:100 protease inhibitor (Sigma Aldrich P8340-1ML). Cells were incubated in lysis buffer for 20 min on ice, vortexed every 10 min. Lysed cells were centrifuged to pellet at 16,000 × g for 15 min at 4 °C. Supernatant was kept for experiments. Protein concentrations were determined using BCA Protein Assay kit (Thermo Scientific Pierce 23227).
Co-immunoprecipitation with anti-FLAG M2 magnetic beads
Anti-FLAG M2 magnetic beads (Millipore Sigma Aldrich, M8823) were washed three times with TBS (50 mM Tris-HCl Fisher Scientific BP152-500, 150 mM NaCl Fisher Scientific S271-10, 1 mM EDTA). One milligram of protein was added to 1.75 mL of TBS with 1:100 phosphatase inhibitor cocktail 1 (Sigma Aldrich P2850-1ML), 1:100 phosphatase inhibitor cocktail 2 (Sigma Aldrich P5726-1ML), and 1:100 protease inhibitor (Sigma Aldrich P8340-1ML). Each sample was incubated with 50 μl of packed beads at constant rotation overnight at 4 °C. Next day, beads were collected by magnetic stand and washed three times with 20 times the packed bead volume. Protein was eluted and denatured with Laemmli sample buffer (BioRad #1610747) with 10% 2-Mercaptoethanol (Sigma Aldrich M3148-25ML), boiled at 95 °C for 10 min.
Co-immunoprecipitation with anti-c-Myc magnetic beads
Anti-c-Myc magnetic beads (Thermo Fisher Scientific, 88842) were washed twice with 1× TBS-T buffer (25 mM Tris Fisher Scientific BP152-500, 0.15 M NaCl Fisher Scientific S271-10, 0.05% Tween-20 detergent Sigma Aldrich P1379-500ML). One milligram of protein was added to 400 μL of 1X TBS-T with 1:100 phosphatase inhibitor cocktail 1 (Sigma Aldrich P2850-1ML), 1:100 phosphatase inhibitor cocktail 2 (Sigma Aldrich P5726-1ML), and 1:100 protease inhibitor (Sigma Aldrich P8340-1ML). Each sample was incubated with 50 μL of beads at constant rotation overnight at 4 °C. Next day, beads were collected by magnetic stand and washed twice with 300 μL of 5X TBS-T buffer 125 mM Tris Fisher Scientific BP152-500, 0.75 M NaCl Fisher Scientific S271-10, 0.25% Tween-20 detergent (Sigma Aldrich P1379-500ML) and once with 300 μL of ultrapure water. Protein was eluted and denatured with Laemmli sample buffer (BioRad #1610747) with 10% 2-Mercaptoethanol (Sigma Aldrich M3148-25ML), boiled at 95 °C for 10 min.
Western blot
Lysates (20 μg of lysate, 30 μl of co-IP sample) were subjected to electrophoresis on Mini-PROTEAN TGX Stain-Free Gels (Bio-Rad), and then transferred to Amersham Hybond P 0.45 μm PVDF membranes (GE Healthcare). The membrane was blocked with 5% non-fat skim milk (Difco Skim Milk 232100) in Tris-buffered saline (Fisher Scientific BP152-500) containing 0.05% Tween-20 (Sigma Aldrich P1379-500ML) for 1 h at room temperature, and then incubated overnight at 4 °C with primary antibody against HA (1:1000, C29F4, Cell Signaling Technology). Following washing, the membrane was incubated for 1 h at room temperature with anti-rabbit IgG, horseradish peroxidase-linked whole antibody (1:5000, NA934-1ML, Cytiva). The signal was detected using SuperSignal West Dura Extended Duration Substrate (Thermo Fisher Scientific 34076) and analyzed using ChemiDoc MP Imaging System and Image Lab software (version 6.1.0, Bio-Rad). Membranes were stripped (Thermo Scientific) and reblotted with primary antibody against beta-Actin (1:1000, GeneTex GTX109639) and secondary anti-rabbit IgG, horseradish peroxidase-linked whole antibody (1:5000, NA934-1ML, Cytiva) following the same blotting, signal detection, and imaging protocol.
Statistics and reproducibility
Individual data point dot plots and box and whisker plots were used to represent the quantification of the data. All data points are presented. The horizontal line in the box represents the mean value, while the box itself represents the interquartile range. Unpaired two-sided t-test with Welch’s correction was performed and p-values were annotated. Differences with p < 0.05 were considered significant. Asterisks in figures indicate statistical significance as follows: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. For plots comparing percentage of animals, chi-square analysis was performed to determine statistical significance. Fluorescent microscopy images next to each dot plot show a representative animal. The number of animals (n) for each analysis was determined based on standards in the C. elegans field. No data were excluded from the analyses. The Investigators were not blinded to allocation during experiments and outcome assessment.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data supporting the findings of this study are included in the figures and supporting files. The ChIP-Seq data for UNC-3 are available in the NCBI Gene Expression Omnibus (GEO) database under accession code GSE143165. The ChIP-Seq data for MAB-9, PBRM-1 and SWSN-7 are available in the ENCODE database under accession codes: ENCSR032EGI, ENCSR111IAZ, ENCSR406IHG. Single-cell RNA-Sequencing data for the SAB neurons and other C. elegans nerve cord motor neurons are deposited at the NCBI GEO under the accession code GSE234962. Source data are provided with this paper.
Code availability
The code for the SAB neuron analysis can be found here: https://doi.org/10.5281/zenodo.10567647.
References
Bota, M. & Swanson, L. W. The neuron classification problem. Brain Res. Rev. 56, 79–88 (2007).
White, J. G., Southgate, E., Thomson, J. N. & Brenner, S. The structure of the nervous system of the nematode Caenorhabditis elegans. Philos. Trans. R. Soc. Lond. B Biol. Sci. 314, 1–340 (1986).
Alkaslasi, M. R. et al. Single nucleus RNA-sequencing defines unexpected diversity of cholinergic neuron types in the adult mouse spinal cord. Nat. Commun. 12, 2471 (2021).
Li, H. et al. Classifying Drosophila olfactory projection neuron subtypes by single-cell RNA sequencing. Cell 171, 1206–1220.e1222 (2017).
Taylor, S. R. et al. Molecular topography of an entire nervous system. Cell 184, 4329–4347.e4323 (2021).
Rosenberg, A. B. et al. Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science 360, 176–182 (2018).
Konstantinides, N. et al. Phenotypic convergence: distinct transcription factors regulate common terminal features. Cell 174, 622–635.e613 (2018).
Smith, J. J. et al. A molecular atlas of adult C. elegans motor neurons reveals ancient diversity delineated by conserved transcription factor codes. Cell Rep. 43, 113857 (2024).
Ozel, M. N. et al. Coordinated control of neuronal differentiation and wiring by sustained transcription factors. Science 378, eadd1884 (2022).
Ozel, M. N. et al. Neuronal diversity and convergence in a visual system developmental atlas. Nature 589, 88–95 (2021).
Bhattacherjee, A. et al. Cell type-specific transcriptional programs in mouse prefrontal cortex during adolescence and addiction. Nat. Commun. 10, 4169 (2019).
Di Bella, D. J. et al. Molecular logic of cellular diversification in the mouse cerebral cortex. Nature 595, 554–559 (2021).
Rheaume, B. A. et al. Single cell transcriptome profiling of retinal ganglion cells identifies cellular subtypes. Nat. Commun. 9, 2759 (2018).
Doe, C. Q. Temporal patterning in the Drosophila CNS. Annu. Rev. Cell Dev. Biol. 33, 219–240 (2017).
Holguera, I. & Desplan, C. Neuronal specification in space and time. Science 362, 176–180 (2018).
Briscoe, J., Pierani, A., Jessell, T. M. & Ericson, J. A homeodomain protein code specifies progenitor cell identity and neuronal fate in the ventral neural tube. Cell 101, 435–445 (2000).
Kutejova, E., Sasai, N., Shah, A., Gouti, M. & Briscoe, J. Neural progenitors adopt specific identities by directly repressing all alternative progenitor transcriptional programs. Dev. Cell 36, 639–653 (2016).
Sagner, A. & Briscoe, J. Establishing neuronal diversity in the spinal cord: a time and a place. Development 146, dev182154 (2019).
Muhr, J., Andersson, E., Persson, M., Jessell, T. M. & Ericson, J. Groucho-mediated transcriptional repression establishes progenitor cell pattern and neuronal fate in the ventral neural tube. Cell 104, 861–873 (2001).
Hobert, O. Terminal selectors of neuronal identity. Curr. Top. Dev. Biol. 116, 455–475 (2016).
Hobert, O. & Kratsios, P. Neuronal identity control by terminal selectors in worms, flies, and chordates. Curr. Opin. Neurobiol. 56, 97–105 (2019).
Liu, C. et al. Pet-1 is required across different stages of life to regulate serotonergic function. Nat. Neurosci. 13, 1190–1198 (2010).
Zhang, X. L., Spencer, W. C., Tabuchi, N., Kitt, M. M. & Deneris, E. S. Reorganization of postmitotic neuronal chromatin accessibility for maturation of serotonergic identity. Elife 11, e75970 (2022).
Kadkhodaei, B. et al. Nurr1 is required for maintenance of maturing and adult midbrain dopamine neurons. J. Neurosci. 29, 15923–15932 (2009).
Deneris, E. & Gaspar, P. Serotonin neuron development: shaping molecular and structural identities. Wiley Interdiscip. Rev. Dev. Biol. 7, e301 (2018).
Poulin, J. F., Gaertner, Z., Moreno-Ramos, O. A. & Awatramani, R. Classification of midbrain dopamine neurons using single-cell gene expression profiling approaches. Trends Neurosci. 43, 155–169 (2020).
Doan, R. N. et al. Recessive gene disruptions in autism spectrum disorder. Nat. Genet. 51, 1092–1098 (2019).
Rafati, M. et al. Identification of a novel de novo variant in OTX2 in a patient with congenital microphthalmia using targeted next-generation sequencing followed by prenatal diagnosis. Ophthalmic Genet. 43, 262–267 (2022).
Sun, C. & Chen, S. Disease-causing mutations in genes encoding transcription factors critical for photoreceptor development. Front. Mol. Neurosci. 16, 1134839 (2023).
Blackburn, P. R. et al. Novel de novo variant in EBF3 is likely to impact DNA binding in a patient with a neurodevelopmental disorder and expanded phenotypes: patient report, in silico functional assessment, and review of published cases. Cold Spring Harb. Mol. Case Stud. 3, a001743 (2017).
Chao, H. T. et al. A syndromic neurodevelopmental disorder caused by de novo variants in EBF3. Am. J. Hum. Genet. 100, 128–137 (2017).
Sleven, H. et al. De novo mutations in EBF3 cause a neurodevelopmental syndrome. Am. J. Hum. Genet. 100, 138–150 (2017).
Etchberger, J. F., Flowers, E. B., Poole, R. J., Bashllari, E. & Hobert, O. Cis-regulatory mechanisms of left/right asymmetric neuron-subtype specification in C. elegans. Development 136, 147–160 (2009).
Uchida, O., Nakano, H., Koga, M. & Ohshima, Y. The C. elegans che-1 gene encodes a zinc finger transcription factor required for specification of the ASE chemosensory neurons. Development 130, 1215–1224 (2003).
Hobert, O. Development of left/right asymmetry in the Caenorhabditis elegans nervous system: from zygote to postmitotic neuron. Genesis 52, 528–543 (2014).
Zheng, C., Jin, F. Q., Trippe, B. L., Wu, J. & Chalfie, M. Inhibition of cell fate repressors secures the differentiation of the touch receptor neurons of Caenorhabditis elegans. Development 145, dev168096 (2018).
Kerk, S. Y., Kratsios, P., Hart, M., Mourao, R. & Hobert, O. Diversification of C. elegans motor neuron identity via selective effector gene repression. Neuron 93, 80–98 (2017).
Li, Y. et al. Cell context-dependent CFI-1/ARID3 functions control neuronal terminal differentiation. Cell Rep. 42, 112220 (2023).
Hsiao, H. Y., Jukam, D., Johnston, R. & Desplan, C. The neuronal transcription factor erect wing regulates specification and maintenance of Drosophila R8 photoreceptor subtypes. Dev. Biol. 381, 482–490 (2013).
Johnston, R. J. Jr. Lessons about terminal differentiation from the specification of color-detecting photoreceptors in the Drosophila retina. Ann. N. Y. Acad. Sci. 1293, 33–44 (2013).
Kratsios, P. et al. Transcriptional coordination of synaptogenesis and neurotransmitter signaling. Curr. Biol. 25, 1282–1295 (2015).
Kratsios, P., Stolfi, A., Levine, M. & Hobert, O. Coordinated regulation of cholinergic motor neuron traits through a conserved terminal selector gene. Nat. Neurosci. 15, 205–214 (2012).
Harms, F. L. et al. Mutations in EBF3 disturb transcriptional profiles and cause intellectual disability, ataxia, and facial dysmorphism. Am. J. Hum. Genet. 100, 117–127 (2017).
Kratsios, P. et al. An intersectional gene regulatory strategy defines subclass diversity of C. elegans motor neurons. Elife 6, e25751 (2017).
Son, E. Y. & Crabtree, G. R. The role of BAF (mSWI/SNF) complexes in mammalian neural development. Am. J. Med. Genet. C Semin. Med. Genet. 166C, 333–349 (2014).
Sokpor, G., Xie, Y., Rosenbusch, J. & Tuoc, T. Chromatin remodeling BAF (SWI/SNF) complexes in neural development and disorders. Front. Mol. Neurosci. 10, 243 (2017).
Von Stetina, S. E., Treinin, M. & Miller, D. M. 3rd The motor circuit. Int. Rev. Neurobiol. 69, 125–167 (2006).
Cinar, H., Keles, S. & Jin, Y. Expression profiling of GABAergic motor neurons in Caenorhabditis elegans. Curr. Biol. 15, 340–346 (2005).
Li, Y. et al. Establishment and maintenance of motor neuron identity via temporal modularity in terminal selector function. Elife 9, e59464 (2020).
Feng, W., Destain, H., Smith, J. J. & Kratsios, P. Maintenance of neurotransmitter identity by Hox proteins through a homeostatic mechanism. Nat. Commun. 13, 6097 (2022).
Minevich, G., Park, D. S., Blankenberg, D., Poole, R. J. & Hobert, O. CloudMap: a cloud-based pipeline for analysis of mutant genome sequences. Genetics 192, 1249–1269 (2012).
Thompson, M. Polybromo-1: the chromatin targeting subunit of the PBAF complex. Biochimie 91, 309–319 (2009).
Mathies, L. D., Kim, A. C., Soukup, E. M., Thomas, A. E. & Bettinger, J. C. PBRM-1/PBAF-regulated genes in a multipotent progenitor in Caenorhabditis elegans. G3 14, jkad297 (2024).
Esposito, G., Di Schiavi, E., Bergamasco, C. & Bazzicalupo, P. Efficient and cell specific knock-down of gene function in targeted C. elegans neurons. Gene 395, 170–176 (2007).
Wang, S. et al. A toolkit for GFP-mediated tissue-specific protein degradation in C. elegans. Development 144, 2694–2701 (2017).
Hodges, C., Kirkland, J. G. & Crabtree, G. R. The many roles of BAF (mSWI/SNF) and PBAF complexes in cancer. Cold Spring Harb. Perspect. Med. 6, a026930 (2016).
Bryant, P., Pozzati, G. & Elofsson, A. Improved prediction of protein-protein interactions using AlphaFold2. Nat. Commun. 13, 1265 (2022).
Mathies, L. D. et al. SWI/SNF chromatin remodeling regulates alcohol response behaviors in Caenorhabditis elegans and is associated with alcohol dependence in humans. Proc. Natl. Acad. Sci. USA 112, 3032–3037 (2015).
Large, E. E. & Mathies, L. D. Caenorhabditis elegans SWI/SNF subunits control sequential developmental stages in the somatic gonad. G3 4, 471–483 (2014).
Katsani, K. R., Mahmoudi, T. & Verrijzer, C. P. Selective gene regulation by SWI/SNF-related chromatin remodeling factors. Curr. Top. Microbiol. Immunol. 274, 113–141 (2003).
Dykhuizen, E. C. et al. BAF complexes facilitate decatenation of DNA by topoisomerase IIalpha. Nature 497, 624–627 (2013).
Pan, J. et al. The ATPase module of mammalian SWI/SNF family complexes mediates subcomplex identity and catalytic activity-independent genomic targeting. Nat. Genet. 51, 618–626 (2019).
Richmond, E. & Peterson, C. L. Functional analysis of the DNA-stimulated ATPase domain of yeast SWI2/SNF2. Nucleic Acids Res. 24, 3685–3692 (1996).
van der Vaart, A., Godfrey, M., Portegijs, V. & van den Heuvel, S. Dose-dependent functions of SWI/SNF BAF in permitting and inhibiting cell proliferation in vivo. Sci. Adv. 6, eaay3823 (2020).
Sawa, H., Kouike, H. & Okano, H. Components of the SWI/SNF complex are required for asymmetric cell division in C. elegans. Mol. Cell 6, 617–624 (2000).
Pocock, R. et al. Neuronal function of Tbx20 conserved from nematodes to vertebrates. Dev. Biol. 317, 671–685 (2008).
Gerstein, M. B. et al. Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science 330, 1775–1787 (2010).
Humphreys, I. R. et al. Computed structures of core eukaryotic protein complexes. Science 374, eabm4805 (2021).
Rennie, M. L., Arkinson, C., Chaugule, V. K. & Walden, H. Cryo-EM reveals a mechanism of USP1 inhibition through a cryptic binding site. Sci. Adv. 8, eabq6353 (2022).
Rennie, M. L. & Oliver, M. R. Emerging frontiers in protein structure prediction following the AlphaFold revolution. J. R. Soc. Interface 22, 20240886 (2025).
Weeratunga, S. et al. Interrogation and validation of the interactome of neuronal Munc18-interacting Mint proteins with AlphaFold2. J. Biol. Chem. 300, 105541 (2024).
Javer, A., Ripoll-Sanchez, L. & Brown, A. E. X. Powerful and interpretable behavioural features for quantitative phenotyping of Caenorhabditis elegans. Philos. Trans. R. Soc. Lond. B Biol. Sci. 373, 20170375 (2018).
Yemini, E., Jucikas, T., Grundy, L. J., Brown, A. E. & Schafer, W. R. A database of Caenorhabditis elegans behavioral phenotypes. Nat. Methods 10, 877–879 (2013).
Tolstenkov, O. et al. Functionally asymmetric motor neurons contribute to coordinating locomotion of Caenorhabditis elegans. Elife 7, e34997 (2018).
Lessard, J. et al. An essential switch in subunit composition of a chromatin remodeling complex during neural development. Neuron 55, 201–215 (2007).
Yoo, A. S., Staahl, B. T., Chen, L. & Crabtree, G. R. MicroRNA-mediated switching of chromatin-remodelling complexes in neural development. Nature 460, 642–646 (2009).
Braun, S. M. G. et al. BAF subunit switching regulates chromatin accessibility to control cell cycle exit in the developing mammalian cortex. Genes Dev. 35, 335–353 (2021).
Staahl, B. T. et al. Kinetic analysis of npBAF to nBAF switching reveals exchange of SS18 with CREST and integration with neural developmental pathways. J. Neurosci. 33, 10348–10361 (2013).
Tuoc, T. C. et al. Chromatin regulation by BAF170 controls cerebral cortical size and thickness. Dev. Cell 25, 256–269 (2013).
Narayanan, R. et al. Loss of BAF (mSWI/SNF) complexes causes global transcriptional and chromatin state changes in forebrain development. Cell Rep. 13, 1842–1854 (2015).
Tuoc, T. et al. Ablation of BAF170 in developing and postnatal dentate gyrus affects neural stem cell proliferation, differentiation, and learning. Mol. Neurobiol. 54, 4618–4635 (2017).
Matsumoto, S. et al. Brg1 is required for murine neural stem cell maintenance and gliogenesis. Dev. Biol. 289, 372–383 (2006).
Vogel-Ciernia, A. et al. The neuron-specific chromatin regulatory subunit BAF53b is necessary for synaptic plasticity and memory. Nat. Neurosci. 16, 552–561 (2013).
Kim, B. et al. Neuronal activity-induced BRG1 phosphorylation regulates enhancer activation. Cell Rep. 36, 109357 (2021).
Wu, J. I. et al. Regulation of dendritic development by neuron-specific chromatin remodeling complexes. Neuron 56, 94–108 (2007).
Parrish, J. Z., Kim, M. D., Jan, L. Y. & Jan, Y. N. Genome-wide analyses identify transcription factors required for proper morphogenesis of Drosophila sensory neuron dendrites. Genes Dev. 20, 820–835 (2006).
Bachmann, C. et al. mSWI/SNF (BAF) complexes are indispensable for the neurogenesis and development of embryonic olfactory epithelium. PLoS Genet. 12, e1006274 (2016).
Weinberg, P., Flames, N., Sawa, H., Garriga, G. & Hobert, O. The SWI/SNF chromatin remodeling complex selectively affects multiple aspects of serotonergic neuron differentiation. Genetics 194, 189–198 (2013).
Waldhauser, V., Baroti, T., Frob, F. & Wegner, M. PBAF subunit Pbrm1 selectively influences the transition from progenitors to pre-myelinating cells during oligodendrocyte development. Cells 12, 1556 (2023).
Xu, Y. et al. Inactivation of BRD7 results in impaired cognitive behavior and reduced synaptic plasticity of the medial prefrontal cortex. Behav. Brain Res. 286, 1–10 (2015).
Ruijtenberg, S. & van den Heuvel, S. G1/S inhibitors and the SWI/SNF complex control cell-cycle exit during muscle differentiation. Cell 162, 300–313 (2015).
Poole, R. J., Flames, N. & Cochella, L. Neurogenesis in Caenorhabditis elegans. Genetics 228, iyae116 (2024).
Glenwinkel, L. et al. In silico analysis of the transcriptional regulatory logic of neuronal identity specification throughout the C. elegans nervous system. Elife 10, e64906 (2021).
Reilly, M. B. et al. Widespread employment of conserved C. elegans homeobox genes in neuronal identity specification. PLoS Genet. 18, e1010372 (2022).
Jafari, S. et al. Combinatorial activation and repression by seven transcription factors specify Drosophila odorant receptor expression. PLoS Biol. 10, e1001280 (2012).
Eade, K. T., Fancher, H. A., Ridyard, M. S. & Allan, D. W. Developmental transcriptional networks are required to maintain neuronal subtype identity in the mature nervous system. PLoS Genet. 8, e1002501 (2012).
Cheng, L. et al. Lmx1b, Pet-1, and Nkx2.2 coordinately specify serotonergic neurotransmitter phenotype. J. Neurosci. 23, 9961–9967 (2003).
Kitt, M. M. et al. An adult-stage transcriptional program for survival of serotonergic connectivity. Cell Rep. 39, 110711 (2022).
Ng, Y. H. et al. Efficient generation of dopaminergic induced neuronal cells with midbrain characteristics. Stem Cell Rep. 16, 1763–1776 (2021).
Stott, S. R. et al. Foxa1 and foxa2 are required for the maintenance of dopaminergic properties in ventral midbrain neurons at late embryonic stages. J. Neurosci. 33, 8022–8034 (2013).
O’Meara, M. M., Zhang, F. & Hobert, O. Maintenance of neuronal laterality in Caenorhabditis elegans through MYST histone acetyltransferase complex components LSY-12, LSY-13 and LIN-49. Genetics 186, 1497–1502 (2010).
Bordet, G., Couillault, C., Soulavie, F., Filippopoulou, K. & Bertrand, V. PRC1 chromatin factors strengthen the consistency of neuronal cell fate specification and maintenance in C. elegans. PLoS Genet. 18, e1010209 (2022).
Padeken, J., Methot, S. P. & Gasser, S. M. Establishment of H3K9-methylated heterochromatin and its functions in tissue differentiation and maintenance. Nat. Rev. Mol. Cell Biol. 23, 623–640 (2022).
Toskas, K. et al. PRC2-mediated repression is essential to maintain identity and function of differentiated dopaminergic and serotonergic neurons. Sci. Adv. 8, eabo1543 (2022).
Lipinski, M. et al. KAT3-dependent acetylation of cell type-specific genes maintains neuronal identity in the adult mouse brain. Nat. Commun. 11, 2588 (2020).
Lipinski, M. et al. CBP is required for establishing adaptive gene programs in the adult mouse brain. J. Neurosci. 42, 7984–8001 (2022).
Kaeser, M. D., Aslanian, A., Dong, M. Q., Yates, J. R. 3rd & Emerson, B. M. BRD7, a novel PBAF-specific SWI/SNF subunit, is required for target gene activation and repression in embryonic stem cells. J. Biol. Chem. 283, 32254–32263 (2008).
Kakarougkas, A. et al. Requirement for PBAF in transcriptional repression and repair at DNA breaks in actively transcribed regions of chromatin. Mol. Cell 55, 723–732 (2014).
Soshnikova, N. V. et al. PHF10 subunit of PBAF complex mediates transcriptional activation by MYC. Oncogene 40, 6071–6080 (2021).
Quinn, J., Fyrberg, A. M., Ganster, R. W., Schmidt, M. C. & Peterson, C. L. DNA-binding properties of the yeast SWI/SNF complex. Nature 379, 844–847 (1996).
Reyes, A. A., Marcum, R. D. & He, Y. Structure and function of chromatin remodelers. J. Mol. Biol. 433, 166929 (2021).
Ho, P. J., Lloyd, S. M. & Bao, X. Unwinding chromatin at the right places: how BAF is targeted to specific genomic locations during development. Development 146, dev178780 (2019).
Forcales, S. V. et al. Signal-dependent incorporation of MyoD-BAF60c into Brg1-based SWI/SNF chromatin-remodelling complex. EMBO J. 31, 301–316 (2012).
Chen, L. et al. Transcriptional control in cardiac progenitors: Tbx1 interacts with the BAF chromatin remodeling complex and regulates Wnt5a. PLoS Genet. 8, e1002571 (2012).
Vierbuchen, T. et al. AP-1 transcription factors and the BAF complex mediate signal-dependent enhancer selection. Mol. Cell 68, 1067–1082.e1012 (2017).
Ninkovic, J. et al. The BAF complex interacts with Pax6 in adult neural progenitors to establish a neurogenic cross-regulatory transcriptional network. Cell Stem Cell 13, 403–418 (2013).
Yu, Y. et al. Olig2 targets chromatin remodelers to enhancers to initiate oligodendrocyte differentiation. Cell 152, 248–261 (2013).
Limpert, A. S. et al. NF-kappaB forms a complex with the chromatin remodeler BRG1 to regulate Schwann cell differentiation. J. Neurosci. 33, 2388–2397 (2013).
Gamble, N. et al. PU.1 and BCL11B sequentially cooperate with RUNX1 to anchor mSWI/SNF to poise the T cell effector landscape. Nat. Immunol. 25, 860–872 (2024).
Zeng, L. & Zhou, M. M. Bromodomain: an acetyl-lysine binding domain. FEBS Lett. 513, 124–128 (2002).
Wang, Y. et al. A prion-like domain in transcription factor EBF1 promotes phase separation and enables B cell programming of progenitor chromatin. Immunity 53, 1151–1167.e1156 (2020).
Oliver, A. W. et al. Crystal structure of the proximal BAH domain of the polybromo protein. Biochem. J. 389, 657–664 (2005).
Callebaut, I., Courvalin, J. C. & Mornon, J. P. The BAH (bromo-adjacent homology) domain: a link between DNA methylation, replication and transcriptional regulation. FEBS Lett. 446, 189–193 (1999).
Weber, C. M. et al. mSWI/SNF promotes Polycomb repression both directly and through genome-wide redistribution. Nat. Struct. Mol. Biol. 28, 501–511 (2021).
Ho, L. et al. esBAF facilitates pluripotency by conditioning the genome for LIF/STAT3 signalling and by regulating polycomb function. Nat. Cell Biol. 13, 903–913 (2011).
Kadoch, C. et al. Dynamics of BAF-Polycomb complex opposition on heterochromatin in normal and oncogenic states. Nat. Genet. 49, 213–222 (2017).
Kadoch, C. et al. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet. 45, 592–601 (2013).
Shain, A. H. & Pollack, J. R. The spectrum of SWI/SNF mutations, ubiquitous in human cancers. PLoS ONE 8, e55119 (2013).
Wang, X., Wu, H., Sun, H., Wang, L. & Chen, L. ARID2, a rare cause of Coffin-Siris syndrome: a clinical description of two cases. Front. Pediatr. 10, 911954 (2022).
Kang, E. et al. Association between ARID2 and RAS-MAPK pathway in intellectual disability and short stature. J. Med. Genet. 58, 767–777 (2021).
Schmetz, A. et al. Delineation of the adult phenotype of Coffin-Siris syndrome in 35 individuals. Hum. Genet. 143, 71–84 (2024).
Neale, B. M. et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485, 242–245 (2012).
Brenner, S. The genetics of Caenorhabditis elegans. Genetics 77, 71–94 (1974).
Doitsidou, M., Poole, R. J., Sarin, S., Bigelow, H. & Hobert, O. C. elegans mutant identification with a one-step whole-genome-sequencing and SNP mapping strategy. PLoS ONE 5, e15435 (2010).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Hobert, O. PCR fusion-based approach to create reporter gene constructs for expression analysis in transgenic C. elegans. Biotechniques 32, 728–730 (2002).
Pereira, L. et al. A cellular and regulatory map of the cholinergic nervous system of C. elegans. Elife 4, e12432 (2015).
Ji, N. & van Oudenaarden, A. Single molecule fluorescent in situ hybridization (smFISH) of C. elegans worms and embryos. WormBook 13, 1–16 (2012).
Consortium, E. P. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
Heinz, S. et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010).
Weirauch, M. T. et al. Determination and inference of eukaryotic transcription factor sequence specificity. Cell 158, 1431–1443 (2014).
Grant, C. E., Bailey, T. L. & Noble, W. S. FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018 (2011).
Mirdita, M. et al. ColabFold: making protein folding accessible to all. Nat. Methods 19, 679–682 (2022).
Mirdita, M., Steinegger, M. & Soding, J. MMseqs2 desktop and local web server app for fast, interactive sequence searches. Bioinformatics 35, 2856–2858 (2019).
Javer, A. et al. An open-source platform for analyzing and sharing worm-behavior data. Nat. Methods 15, 645–646 (2018).
Osseward, P. J. 2nd & Pfaff, S. L. Cell type and circuit modules in the spinal cord. Curr. Opin. Neurobiol. 56, 175–184 (2019).
Hernan, R., Heuermann, K. & Brizzard, B. Multiple epitope tagging of expressed proteins for enhanced detection. Biotechniques 28, 789–793 (2000).
Boyle, A. P. et al. Comparative analysis of regulatory information and circuits across distant species. Nature 512, 453–456 (2014).
Acknowledgements
We thank the Caenorhabditis Genetics Center (CGC), which is funded by the National Institutes of Health (NIH) Office of Research Infrastructure Programs (P40 OD010440), for providing many of the strains used in this study. We thank Oliver Hobert for feedback and the ot792 allele, which was identified in his lab by Romulo Murao and Paschalis Kratsios. We thank Edwin Ferguson for invaluable advice at various stages of this project. We also thank David M. Miller III and Kratsios lab members for providing feedback on the text, and Yihan Chen and Jihad Aburas for technical assistance. This work was supported as follows: NIH 5R25GM109439 (A.O.), NSF fellowship 2140001 (H.D.), NIH T32HD055164 (M.P. and J.M.), NIH R01NS118078 (P.K.) and NIH R01NS116365 (P.K.).
Author information
Authors and Affiliations
Contributions
Conceptualization: A.O. and P.K.; Methodology: A.O., H. D., J.M., Y.L., S.R.T., M.P., I.Q.W., F.M., N.G.S., L.D.M., P.I., J.J.S. and P.K.; Investigation: A.O., H.D., J.M., Y.L., S.R.T., M.P., I.Q.W., F.M., N.G.S., L.D.M., P. I., A.E.X.B., J.J.S. and P.K.; Formal analysis: A.O., H.D., J.M., Y.L., S.R.T., M.P., I.Q.W., F.M., N.G.S., L.D.M., P.I., A.E.X.B., J.J.S. and P.K.; Visualization: A.O., H.D., J.J.S., and P.K.; Funding acquisition: P.K.; Supervision: A.E.X.B. and P.K.; Writing original draft: A.O. and P.K.; Writing—review and editing: A.O., H.D., S.R.T., M.P., L.D. M., J.J.S., and P.K.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks David Picketts and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Osuma, A., Destain, H., Mann, J. et al. Cell type-specific functions of the PBAF chromatin-remodeling complex in neuronal diversification. Nat Commun 17, 741 (2026). https://doi.org/10.1038/s41467-025-67434-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67434-9
