
Abstract
Therapeutic targeting of mutant KRAS pathways driving cancers is being actively investigated to identify feedback mechanisms responsible for the development of adaptive resistance to mutant KRAS inhibitors undergoing clinical trials. Here we report RASH3D19 as a mediator of RAS pathway activation through a positive feedback loop involving the KRAS–microRNA signalling axis. KRAS-induced miR-222 represses ETS1 expression and downstream transactivation of miR-301a leading to elevation of its target RASH3D19. RASH3D19 facilitates activation of RAS pathways by promoting dimerization and interaction of EGFR with the SOS2, GRB2, SHP2 and GAB1 complex. Genetic deletion of RASH3D19 in mutant KRAS-expressing cancer cells exhibits growth retardation in vitro, in vivo and sensitized pancreatic ductal adenocarcinoma and colorectal cancer cells, organoids and xenografts to mutant KRAS inhibitors, suppressing feedback reactivation of RAS pathways. Therapeutic targeting of RASH3D19 is expected to lead to tumour debulking and alleviating resistance to KRAS inhibitors in mutant KRAS-expressing cancers.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$32.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others
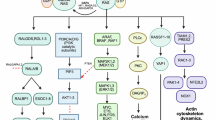


Data availability
Previously published RNA–seq data that were reanalysed here are available under accession codes GSE41368, GSE62165 and GSE101448 in the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo). The human PDAC data were derived from the TCGA Research Network (https://www.cbioportal.org) and TCGA match with GTEx (http://gepia.cancer-pku.cn/). All other data supporting the findings of this study are available from the corresponding authors on reasonable request. Source data are provided with this paper.
References
Cox, A. D., Fesik, S. W., Kimmelman, A. C., Luo, J. & Der, C. J. Drugging the undruggable RAS: mission possible?. Nat. Rev. Drug Discov. 13, 828–851 (2014).
Ostrem, J. M., Peters, U., Sos, M. L., Wells, J. A. & Shokat, K. M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503, 548–551 (2013).
Skoulidis, F. et al. Sotorasib for lung cancers with KRAS p.G12C mutation. N. Engl. J. Med. 384, 2371–2381 (2021).
Fakih, M. G. et al. Sotorasib for previously treated colorectal cancers with KRASG12C mutation (CodeBreaK100): a prespecified analysis of a single-arm, phase 2 trial. Lancet Oncol. 23, 115–124 (2022).
Strickler, J. H. et al. Sotorasib in KRAS p.G12C-mutated advanced pancreatic cancer. N. Engl. J. Med. 388, 33–43 (2023).
Hallin, J. et al. Anti-tumor efficacy of a potent and selective non-covalent KRASG12D inhibitor. Nat. Med. 28, 2171–2182 (2022).
Canon, J. et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 575, 217–223 (2019).
Awad, M. M. et al. Acquired resistance to KRASG12C inhibition in cancer. N. Engl. J. Med. 384, 2382–2393 (2021).
Ryan, M. B. et al. KRASG12C-independent feedback activation of wild-type RAS constrains KRASG12C inhibitor efficacy. Cell Rep. 39, 110993 (2022).
Yaeger, R. et al. Molecular characterization of acquired resistance to KRASG12C-EGFR inhibition in colorectal cancer. Cancer Discov. 13, 41–55 (2023).
Negrao, M. V. et al. Comutations and KRASG12C inhibitor efficacy in advanced NSCLC. Cancer Discov. 13, 1556–1571 (2023).
Singhal, A., Li, B. T. & O’Reilly, E. M. Targeting KRAS in cancer. Nat. Med. 30, 969–983 (2024).
Johnson, S. M. et al. RAS is regulated by the let-7 microRNA family. Cell 120, 635–647 (2005).
Tsunoda, T. et al. Oncogenic KRAS regulates miR-200c and miR-221/222 in a 3D-specific manner in colorectal cancer cells. Anticancer Res. 31, 2453–2459 (2011).
Galardi, S., Mercatelli, N., Farace, M. G. & Ciafre, S. A. NF-kB and c-Jun induce the expression of the oncogenic miR-221 and miR-222 in prostate carcinoma and glioblastoma cells. Nucleic Acids Res. 39, 3892–3902 (2011).
Zhang, W. & Li, P. The suppression of nuclear factor kappa B/microRNA 222 axis alleviates lipopolysaccharide-induced acute lung injury through increasing the alkylglyceronephosphate synthase expression. J. Infect. Chemother. 31, 102500 (2024).
Dittmar, R. L. et al. Plasma miRNA biomarkers in limited volume samples for detection of early-stage pancreatic cancer. Cancer Prev. Res. 14, 729–740 (2021).
Treekitkarnmongkol, W. et al. Blood-based microRNA biomarker signature of early-stage pancreatic ductal adenocarcinoma with lead-time trajectory in prediagnostic samples. Gastro Hep. Adv. 3, 1098–1115 (2024).
Xu, Q. et al. miR-221/222 induces pancreatic cancer progression through the regulation of matrix metalloproteinases. Oncotarget 6, 14153–14164 (2015).
Tanaka, M. et al. ADAM binding protein Eve-1 is required for ectodomain shedding of epidermal growth factor receptor ligands. J. Biol. Chem. 279, 41950–41959 (2004).
Lu, Z. et al. miR-301a as an NF-kappaB activator in pancreatic cancer cells. EMBO J. 30, 57–67 (2011).
du Rieu, M. C. et al. MicroRNA-21 is induced early in pancreatic ductal adenocarcinoma precursor lesions. Clin. Chem. 56, 603–612 (2010).
Yam, J. W., Jin, D. Y., So, C. W. & Chan, L. C. Identification and characterization of EBP, a novel EEN binding protein that inhibits Ras signaling and is recruited into the nucleus by the MLL-EEN fusion protein. Blood 103, 1445–1453 (2004).
Sheffels, E. et al. Oncogenic RAS isoforms show a hierarchical requirement for the guanine nucleotide exchange factor SOS2 to mediate cell transformation. Sci. Signal 11, eaar8371 (2018).
Nam, S. et al. Cell cycle progression in confining microenvironments is regulated by a growth-responsive TRPV4-PI3K/Akt-p27(Kip1) signaling axis. Sci. Adv. 5, eaaw6171 (2019).
Wang, Z. Regulation of cell cycle progression by growth factor-induced cell signaling. Cells 10, 3327 (2021).
Sheffels, E., Sealover, N. E., Theard, P. L. & Kortum, R. L. Anchorage-independent growth conditions reveal a differential SOS2 dependence for transformation and survival in RAS-mutant cancer cells. Small GTPases 12, 67–78 (2021).
Ryan, M. B. et al. Vertical pathway inhibition overcomes adaptive feedback resistance to KRASG12C inhibition. Clin. Cancer Res. 26, 1633–1643 (2020).
Batzer, A. G., Rotin, D., Urena, J. M., Skolnik, E. Y. & Schlessinger, J. Hierarchy of binding sites for Grb2 and Shc on the epidermal growth factor receptor. Mol. Cell. Biol. 14, 5192–5201 (1994).
Hagenbeek, T. J. et al. An allosteric pan-TEAD inhibitor blocks oncogenic YAP/TAZ signaling and overcomes KRAS G12C inhibitor resistance. Nat. Cancer 4, 812–828 (2023).
Kapoor, A. et al. Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell 179, 1239 (2019).
Kozer, N. et al. Recruitment of the adaptor protein Grb2 to EGFR tetramers. Biochemistry 53, 2594–2604 (2014).
Lin, C. W. et al. A two-component protein condensate of the EGFR cytoplasmic tail and Grb2 regulates Ras activation by SOS at the membrane. Proc. Natl Acad. Sci. USA 119, e2122531119 (2022).
Fedele, C. et al. SHP2 inhibition prevents adaptive resistance to MEK inhibitors in multiple cancer models. Cancer Discov. 8, 1237–1249 (2018).
Nichols, R. J. et al. RAS nucleotide cycling underlies the SHP2 phosphatase dependence of mutant BRAF-, NF1- and RAS-driven cancers. Nat. Cell Biol. 20, 1064–1073 (2018).
Radulovich, N., Qian, J. Y. & Tsao, M. S. Human pancreatic duct epithelial cell model for KRAS transformation. Methods Enzymol. 439, 1–13 (2008).
Yao, W. et al. Syndecan 1 is a critical mediator of macropinocytosis in pancreatic cancer. Nature 568, 410–414 (2019).
Katayama, H. et al. Aurora kinase-A inactivates DNA damage-induced apoptosis and spindle assembly checkpoint response functions of p73. Cancer Cell 21, 196–211 (2012).
Parte, S. et al. Cancer-associated fibroblast induces acinar-to-ductal cell transdifferentiation and pancreatic cancer initiation via LAMA5/ITGA4 axis. Gastroenterology 166, 842–858 e845 (2024).
Acknowledgements
We thank M.-S. Tsao for sharing the HPDE cells generated in his laboratory and H. Clevers and J. van Es for getting us in touch with laboratory colleagues to perform the experiments with the CRC organoid. The services received from the MD Anderson Core Facilities for animal studies (DVMS) and research histology core facility (RHCL) for immunohistochemistry assay development supported by the NCI Cancer Center Support Grant (grant no. P30 CA016672) are gratefully acknowledged. This work was supported by NCI grants (grant no. U01 CA214263 and U01 CA252965) to S.S. The Sheikh Khalifa bin Zayed Foundation supports to A.M. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
W.T., H.K., D.S. and S.S. designed the study. W.T., H.K. and D.S. performed biochemical characterization. W.T., D.S. and T.N. performed cell-based characterization. W.T., D.S., M.-C.T. and T.N. performed miRNA and RNA expression analysis. S.R. and H.C. performed organoid experiments. W.T. and T.N. performed animal experiments. D.S., W.T., M.-C.T. and H.K. performed database analysis. K.H. and H.W. performed and analysed histology. W.Y., S.K.B., J.V.H. and S.K. provided cell lines and inhibitors. F.I.T., M.P.P., I.I.W., T.D.S., T.H. and A.M. analysed data and participated in discussions on content. W.T., H.K. and S.S. wrote the paper, with input from all authors.
Corresponding authors
Ethics declarations
Competing interests
A.M. is a consultant for Tezcat Bioscience. A.M. is listed on a patent that had been licensed by Thrive Earlier Detection (an Exact Sciences Company). K.H. is funded by Nanostring Technology. The other authors declare no competing interests. A provisional patent on the clinically relevant findings of this study has been filed.
Peer review
Peer review information
Nature Cell Biology thanks Hema Adhikari, Dieter Saur and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Mutant KRAS-induction of miR-222 downregulates miR-301a by targeting ETS1.
a,b, Expression levels of miR-222 (a) in Dox inducible KRAS G12D mutant AK196 and AK12282 mouse PDAC cells untreated and treated with Dox and (b) in AsPC1 and PANC1 cells transfected with control or pre-miR-222. c, Expression levels of ETS1 protein in Dox inducible KRAS G12D mutant AK192 and AK12282 mouse PDAC cells untreated and treated with Dox. Relative expression of ETS1 normalized to β-actin is shown. d, Expression levels of miR-222 (left) and indicated proteins (right) in AsPC1 and PANC1 cells transfected with siRNA targeting KRAS G12D and/or pre-miR-222. Relative expression of ETS1 normalized to β-actin is shown. e, ChIP assay of ETS1 enrichment at 5 different predicted regions (R1-R5) on miR-301 promoter in PANC1 cells. f, Expression levels of ETS1 protein in AsPC1 and PANC1 cells transfected with control siRNA or siRNAs targeting ETS1. g,h, miR-301a promoter activity in ETS1 silenced KRAS G12D mutant stably expressed HPNE cells compared to control siRNA (n=5) (g) and in Dox inducible KRAS G12D mutant HPNE cells untreated or treated with Dox (n=5) (h). i, miR-301a expression levels in KRAS G12D mutant inducible AK192 and AK196 mouse PDAC cells untreated or treated with Dox. All blots and qRT-PCR are representative of three independent experiments. (a-b,d-e,g-i); Bar graphs are represented as mean ± SD. P values were calculated using two-sided unpaired Student’s t-tests. ns, not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Extended Data Fig. 2 RASH3D19 overexpressed in PDAC downstream of mutant KRAS is a target of miR-301a.
a, Venn diagram identifying the targets of miR-301a. b, Pearson’s correlation of miR-301a vs RASH3D19 in PDAC patient tissues in TCGA (n=177) and PDAC cell lines with high miR-301a expression in CCLE database (n=32). R = Pearson’s correlation coefficient; P values were calculated for the statistical significance of the Pearson’s correlation coefficient. c, miR-301a expression levels in AsPC1 and PANC1 cells transfected with control or pre-miR-301a. Bar graphs are represented as mean ± SD. P values were calculated using two-sided unpaired Student’s t-tests. d, Conserved region of miR-301a binding site in RASH3D19 3′UTR. e, RASH3D19 expression levels in Dox-inducible KRAS G12D mutant AK196 and AK12282 cells untreated and treated with Dox. Relative expression of RASH3D19 normalized to β-actin is shown. f, Expression levels of miR-222 and miR-301a (left) and indicated proteins (right) in BxPC3 cells untreated with treated with KRAS inhibitor BI2865. Relative protein expressions of ETS1 and RASH3D19 normalized to β-actin are shown. Bar graphs are represented as mean ± SD. P values were calculated using two-sided unpaired Student’s t-tests. g,h, RASH3D19 expression levels in normal and PDAC patients in additional GSE (g) and TCGA match with GTEx (h) datasets. Box plots are represented as mean with minima to maxima values. P values were calculated using two-sided Wilcoxon rank-sum test. All in vitro assays are representative of three independent experiments. (c,f, g-h); ns, not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Extended Data Fig. 3 RASH3D19 interacts with RAS regulatory proteins.
a,b,c, Immunoprecipitation (IP)/western blot analysis of RASH3D19 with EGFR, SHP2, SOS2 and SOS1 in PANC1 cells. d, Schematic representation of RASH3D19 full length and deletion mutants. Constructs were GST-tagged. Proline-rich region (PRR) and 5 SH3 domains (Domains A to E) are represented by orange and blue boxes, respectively. e, GST pull-down assay. Extract of 293T cells expressing EGFR, SOS2 or GRB2 was incubated with GST or GST-RASH3D19 full length immobilized on glutathione beads. Binding protein was detected with respective antibody. GST proteins were visualized by Coomassie brilliant blue (CBB) stain. f, GST pull-down assay. EGFR prepared in (e) was incubated with GST or GST-RASH3D19 full length in the presence or absence of GRB2. Binding protein was detected by western blot.
Extended Data Fig. 4 Mapping of RASH3D19 regions binding to SOS2 and GRB2, and the effect of RASH3D19 deletion in cell viability and cell cycle.
a,b, GST pull-down assay. SOS2 (a) or GRB2 (b) prepared in (Extended Data Fig. 3e) was incubated with GST, GST-RASH3D19 full length or deletion mutants. Binding proteins were detected by western blot. c, Schematic representation of GRB2 full length and deletion mutants. SH2 and SH3 domains are represented by light orange and blue boxes, respectively. d. GST pull-down assay. Extract of 293T cells expressing GFP-RASH3D19 was incubated with GST, GST-GRB2 full length or deletion mutants immobilized on glutathione beads. Binding proteins were detected by western blot. The dotted line indicates the boundary between the cropped images. e, Cell proliferation rate of sgControl and sgRASH3D19 AsPC1 and PANC1 cultured in 2D and 3D. Bar graphs are represented as mean ± SD. P values were calculated using two-sided unpaired Student’s t-tests. ns, not significant; *, P < 0.05; **, P < 0.01. f, Cell cycle distribution analysis of AsPC1 and PANC1 cell lines by quantitation of DNA content. Cells with either sgControl or sgRASH3D19 were cultured in 3D condition followed by staining with propidium iodide (PI) and DNA content was quantitated by flow cytometry. All the data are representative of three independent experiments.
Extended Data Fig. 5 RASH3D19 deletion in combination with KRAS mutant inhibition inhibits spheroid growth.
a,b, Time-lapse spheroid morphology images of sgControl and sgRASH3D19 AsPC1 (a) and PANC1 (b) cells untreated or treated with 10 nM MRTX1133. Pictures in red frame (Day 0 and Day 7 of both cell lines) were also represented in Fig. 3d.
Extended Data Fig. 6 RASH3D19 promotes adaptive response to RAS inhibitors through RAS activation.
a, RAS pull-down assay of sgControl and sgRASH3D19 MIAPaCa2 and BxPC3 cells. Cells were cultured in 3D for 24 h followed by serum starvation for additional 24 h. Then cells were treated with EGF for 10 min. Relative levels of RAS-GTP normalized to input RAS and β-actin are shown. b, EGF treated sgControl and sgRASH3D19 PANC1 cells as indicated in Fig. 4a were immunoprecipitated for GRB2 followed by western blotted with indicated antibodies. c, RASH3D19 expression levels in AsPC1, MIAPaCa2, HPAF-II cells untreated or treated with mutant KRAS inhibitor MRTX1133 or AMG510 for indicated time. d, Western blot analysis of cells prepared as in (c). Relative expression of RASH3D19 normalized to β-actin is shown. e, RASH3D19 expression levels in AsPC1, PANC1, MIAPaCa2, BxPC3 cells untreated or treated with pan-RAS inhibitor RMC7977 for indicated time. f, Western blot analysis of cells prepared as in (e). Relative expression of RASH3D19 normalized to β-actin is shown. All blots and in vitro assays are representative of three independent experiments. (c,e) Bar graphs are represented as mean ± SD. P values were calculated using two-sided unpaired Student’s t-tests. ns, not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Extended Data Fig. 7 Deletion of RASH3D19 sensitizes mutant KRAS expressing PDAC, CRC, and NSCLC cells to RAS inhibitors.
a, Cell viability assays of AsPC1 and MIAPaCa2 cells which were transfected with control sgRNA or sgRNAs targeting RASH3D19 cultured in 3D for 24 h and then treated with different concentrations of MRTX1133 or AMG510 for 3 days. b, RASH3D19 expression levels in SW837, H1972 and MIAPaCa2 cells prepared as in (a). Bar graphs are represented as mean ± SD. P values were calculated using two-sided unpaired Student’s t-tests. ***, P < 0.001. c, Cell viability assay of sgControl and sgRASH3D19 AsPC1 and PANC1 cells which were cultured in 3D for 24 h followed by treatment with different concentrations of RMC4550 for 3 days. All in vitro assays are representative of three independent experiments.
Extended Data Fig. 8 Deletion of RASH3D19 sensitizes mutant KRAS expressing PDAC and CRC organoids and xenografts to RAS inhibitors.
a,b, B40 and B106 PDAC-derived organoid cultures harboring KRAS G12D mutant, assayed for total area in percentage to control counted by Incucyte live cell imaging. Two PDAC organoids were pre-transfected with control siRNA or siRNAs targeting RASH3D19 for 6 h followed by treatment with MRTX1133 at 25 nM for 6 days. (a-b); Bar graphs are represented as percentage of organoid total area in RASH3D19 siRNA and/or MRTX1133 treated compared to untreated control siRNA from technical replicates (n=4) of two independent experiments. c,d, RASH3D19 expression levels (c) and relative survival (d) of sgControl and sgRASH3D19 CRC organoid, harboring KRAS G13D mutant, treated with DMSO or pan-RAS inhibitor RMC7997 25nM or 100 nM for 6 days compared to normal colon organoid (n=4). (c-d); Bar graphs are represented as mean ± SD. P values were calculated using unpaired two-sided Student’s t-tests. **, P < 0.01; ***, P < 0.001. e, In vivo tumor volume scatter plots on day 35 of sgControl and sgRASH3D19 PANC1 cells which were subcutaneously injected in nude mice (n=6 and 7 in the two groups) and treated daily with vehicle control or MRTX1133 at 3 mg/kg. f, In vivo tumor volume scatter plots on day 28 of sgControl and sgRASH3D19 MIAPaCa2 cells subcutaneously injected in nude mice (n=7 mice in each group) and treated daily with vehicle control or AMG510 at 3 mg/kg (e-f); Box plots are represented as mean with all data points from minima to maxima. P values were calculated using unpaired two-sided Student’s t-tests. ***, P < 0.001.
Supplementary information
Supplementary Information (download PDF )
Supplementary Fig. 1.
Supplementary Tables (download XLSX )
Supplementary Table 1: List of oligonucleotides used in this study. Supplementary Table 2: List of antibodies used in this study. Supplementary Table 3: List of purchased plasmids used in this study.
Source data
Source Data Fig. 1 (download PDF )
Unprocessed western blots and/or gels of Fig. 1.
Source Data Fig. 2 (download PDF )
Unprocessed western blots and/or gels of Fig. 2.
Source Data Fig. 3 (download PDF )
Unprocessed western blots and/or gels of Fig. 3.
Source Data Fig. 4 (download PDF )
Unprocessed western blots and/or gels of Fig. 4.
Source Data Extended Data Fig. 1 (download PDF )
Unprocessed western blots and/or gels of Extended Data Fig. 1.
Source Data Extended Data Fig. 2 (download PDF )
Unprocessed western blots and/or gels of Extended Data Fig. 2.
Source Data Extended Data Fig. 3 (download PDF )
Unprocessed western blots and/or gels of Extended Data Fig. 3.
Source Data Extended Data Fig. 4 (download PDF )
Unprocessed western blots and/or gels of Extended Data Fig. 4.
Source Data Extended Data Fig. 6 (download PDF )
Unprocessed western blots and/or gels of Extended Data Fig. 6.
Source Data Figs. 1–5 and Extended Data Figs. 1, 2, 4, 6–8 (download XLSX )
Statistical source data of Figs. 1–5 and Extended Data Figs. 1, 2, 4, 6–8.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Treekitkarnmongkol, W., Katayama, H., Sankaran, D. et al. RASH3D19 mediates RAS activation through a positive feedback loop in KRAS-mutant cancer. Nat Cell Biol 28, 197–206 (2026). https://doi.org/10.1038/s41556-025-01816-5
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41556-025-01816-5
