
Abstract
Emerging infectious diseases pose a threat to pollinators. Virus transmission among pollinators via flowers may be reinforced by anthropogenic land-use change and concomitant alteration of plant–pollinator interactions. Here, we examine how species’ traits and roles in flower-visitation networks and landscape-scale factors drive key honeybee viruses—black queen cell virus (BQCV) and deformed wing virus—in 19 wild bee and hoverfly species, across 12 landscapes varying in pollinator-friendly (flower-rich) habitat. Viral loads were on average more than ten times higher in managed honeybees than in wild pollinators. Viral loads in wild pollinators were higher when floral resource use overlapped with honeybees, suggesting these as reservoir hosts, and increased with pollinator abundance and viral loads in honeybees. Viral prevalence decreased with the amount of pollinator-friendly habitat in a landscape, which was partly driven by reduced floral resource overlap with honeybees. Black queen cell virus loads decreased with a wild pollinator’s centrality in the network and the proportion of visited dish-shaped flowers. Our findings highlight the complex interplay of resource overlap with honeybees, species traits and roles in flower-visitation networks and flower-rich pollinator habitat shaping virus transmission.
Similar content being viewed by others
Main
Transmission of newly emerging diseases can profoundly impact human and animal health1, as recently exemplified by the Covid-19 pandemic. Pathogen host shifts2 leading to emerging diseases could potentially harm pollinator health and may contribute to pollinator declines3,4, jeopardizing the provision of vital pollination services5,6. RNA viruses such as the deformed wing virus complex (DWV-A and DWV-B) and black queen cell virus (BQCV) commonly detected in the western honeybee, Apis mellifera7 show active replication in other bee species8,9. Indeed, shared viral strains between honeybees and wild pollinators, including non-bee taxa such as hoverflies2,10,11, suggest ongoing transmission. This may impact the health of co-occurring pollinators12,13,14 and affects the structure and functioning of pollinator assemblages15. Flowers shared by pollinators can be primary hubs of faecal–oral virus transmission16. Thus, anthropogenic land-use changes that alter the composition and interaction structure of plant–pollinator communities17,18 may profoundly impact pathogen transmission among pollinators15.
The risk of exposure to pathogens of a potential pollinator host is probably mediated by the traits and roles of pollinator and plant species in an interaction network that, together with landscape effects, have rarely been investigated within the same framework. For example, broader pollinator diets (that is, low floral specialization) and a well-connected network (and thus pollinator species with central roles) are predicted to dilute the risk of picking up a pathogen when the pollinator is visiting many different flowers19.
Flower morphology may also shape pathogen transmission risks. For instance, open dish flowers may increase the probability of insect defecation in the flower20,21, increase viral exposure to denaturing ultraviolet (UV) light and attract multiple pollinators (potential hosts) to easily accessible nectar rewards22,23,24. Flowers providing high amounts of nectar and pollen rewards (correlated to flower volume)25 that are readily accessible are expected to be visited frequently by a broad range of pollinators. This will increase the probability of being visited by infected (or vectoring) hosts and the corresponding spread of pathogens to coforaging pollinators via faecal–oral virus transmission24. In contrast, easy accessibility of (open) flowers with short corollas could also lead to shorter flower handling times26, thus potentially reducing the temporal risk of depositing or picking up pathogens.
Community properties such as high flower and pollinator diversity may also reduce pathogen transmission through dilution processes27,28,29. Greater plant diversity might reduce overlap in floral resource use between (mostly) managed honeybees—predicted to act as a viral reservoir host9—and wild pollinators30, thereby potentially reducing pathogen transmission via shared flowers. In contrast, hotspots of attractive floral resources may concentrate foraging pollinators and the proportional abundance of competent hosts to increase transmission probabilities and pathogen prevalence in the community29,31. Consequently, landscapes poor in flower-rich pollinator habitat may lead to pollinators aggregating on the few available floral resource patches, thereby increasing the likelihood of pathogen transmission32. Thus, an holistic understanding of the factors affecting interspecific transmission of pollinator pathogens at species, interaction network, community and landscape levels will help to design conservation strategies that mitigate pathogen transmission among pollinators.
Here, we analyse plant–pollinator interaction networks, species traits and landscape composition to identify potential transmission pathways affecting prevalence and loads of RNA viruses (DWV-A, DWV-B and BQCV) in 19 wild bee (non-A. mellifera) and hoverfly pollinator species (hereafter wild pollinators for simplicity). We collected plant–pollinator interaction network data from 12 agricultural or more urbanized landscapes in northern Switzerland with varying amounts of pollinator-friendly habitat (flowering habitats playing important roles in sustaining pollinators, including urban green spaces) (Extended Data Fig. 1).
We hypothesized that viral prevalence and loads in wild pollinator species will increase with: (1) high floral resource overlap with managed honeybees (likely reservoir host); (2) high diet specialization; (3) low centrality in flower-visitation networks; (4) greater proportion of open (dish-bowl-shaped) flowers in the diet offering easy access to highly rewarding pollen/nectar; and (5) greater abundance of potential pollinator ‘hosts’ and high viral loads in honeybees in a landscape. We also hypothesized that viral prevalence and loads will decrease with (6) flowering plant diversity and (7) the amount of pollinator-friendly habitat in a landscape, (8) which together contribute to reduced honeybee–wild pollinator resource overlap (dilution of pathogen transmission) (Fig. 1).

Virus transmission in plant–pollinator communities is hypothesized to be influenced by various mechanisms operating at the species, community and landscape levels. At the species and network level, we hypothesize that specific foraging traits such as (1) a high floral resource overlap of pollinators with other species, in particular honeybees (which are expected to act as a reservoir host9), (2) a high diet specialization and (3) a low centrality in the network19, as well as flower morphology of the visited plants such as (4) open dish (dish-bowl-shaped) flowers22,24 will lead to an increased viral prevalence and load in wild pollinators. At the landscape level, (5) a high abundance of honeybees and/or high viral loads or prevalence in honeybees and high abundance of wild pollinators or a low pollinator diversity as well as (6) a low amount of pollinator-friendly habitat and (7) flower diversity are predicted to increase viral prevalence and load in wild pollinators28,29, for example, (8) through increasing the floral resource overlap among pollinators, in particular between honeybees and wild pollinators30. Figure created using Procreate (https://procreate.com/).
Results
Viral prevalence and loads across pollinator communities
All three viruses were detected in honeybees (A. mellifera), with DWV-A being the least abundant virus (5.83% of individuals tested positive), while more than ten times more individuals tested positive for DWV-B (64.17%) and BQCV (86.39%). Moreover, the maximum DWV-A viral load in honeybees (absolute number of genome copies per µg of RNA per individual sample) was lower by a factor of 105 and 109 than the maximum DWV-B and BQCV viral loads, respectively (Supplementary Table 1). This pattern was mirrored in the wild pollinator community, where 10 out of 19 (53%) screened species tested positive for DWV-A, whereas all 19 species (100%) tested positive for DWV-B and 18 out of 19 species (95%) tested positive for BQCV, although with loads and prevalence varying greatly among species (Extended Data Fig. 2 and Supplementary Table 1). For example, regarding numbers of copies per µg of RNA per individual: DWV-A ranged from 7 × 101 (Lasioglossum glabriusculum) to 9.7 × 104 (Bombus terrestris), DWV-B from 4.3 × 102 (Lasioglossum malachurum) to 9.0 × 107 in a single Syrphus vitripennis individual, while BQCV ranged from 2.3 × 102 (L. glabriusculum) to 3.5 × 108 (B. humilis). Low titres of DWV-A precluded statistical analysis of DWV-A (Methods). Thus, a total of 588 samples of 17 wild pollinator species and 240 samples of honeybees were used for statistical analyses. Median viral loads (log) of DWV-B and BQCV within a wild pollinator species (across landscapes) were positively correlated (Spearman’s rank correlation rho = 0.64, P = 0.002). Viral load (median (log)) and prevalence in the wild pollinator community in a landscape were also strongly positively correlated (Spearman’s rank correlations—BQCV for May/June rho = 0.81, P = 0.001 and for July rho = 0.81, P = 0.002; DWV-B for May/June rho = 0.79, P = 0.002 and for July rho = 0.88, P = 0.0003). Confidence intervals for apparent virus prevalence (BQCV and DWV-B) in each species can be found in Supplementary Table 3.
Floral species’ traits and pollinator network roles
In the best models (ΔAICc < 2) predicting BQCV load (Fig. 2a and Supplementary Table 4) and prevalence (Supplementary Table 5 and Extended Data Fig. 3a) in wild pollinators, floral resource overlap of a wild pollinator species with honeybees (community weighted mean (CWM) number of honeybee visits on flowers visited by a given wild pollinator species) had the strongest effect, positively affecting both BQCV prevalence (Supplementary Table 6) and load in wild pollinators (Table 1 and Fig. 3a; see Extended Data Fig. 4 for an example plant–pollinator network visualizing floral resource overlap). Weighted betweenness as a measure of a pollinator species’ centrality within a network and the proportion of visited dish-bowl flowers were negatively related to its BQCV load (Table 1 and Fig. 3b,c) but only weakly to BQCV prevalence (Supplementary Table 6). Corolla length (CWM) of visited flowers was weakly negatively related to BQCV load (Table 1 and Fig. 3d), while specialization d′ of pollinators or flower volume (CWM) of visited flowers did not affect BQCV prevalence (Supplementary Table 6) or load (Table 1).

a–d, Averaged model estimates (modelled slopes of the relationships) and 95% confidence intervals of explanatory variables in best models (LMMs, ΔAICc < 2) explaining viral load of BQCV (n = 304 virus-positive individuals) (a,c) and DWV-B (n = 288 virus-positive individuals) (b,d) in wild pollinators (the response variable was log-transformed number of genome copies per microgram RNA of virus-positive individuals). a,b, display the results from modelling the effects of plant species traits and pollinator network roles (centrality measured as weighted betweenness (Betweenness), corolla length, specialization d′, proportion of dish-bowl flowers among visited flowers (Dish flowers) and floral resource overlap with honeybees (Resource overlap)). c,d, display the results from modelling the effects of pollinator community properties (Shannon diversity of flowers (Flower diversity), honeybee density (HB density) and honeybee viral load (HB BQCV or DWV-B load), wild pollinator abundance (WP abundance) and percentage cover of pollinator habitat (Pollinator habitat)) in a landscape.

a–d, Relationships of BQCV load of virus-positive wild pollinator individuals with floral resource overlap with honeybees (CWM number of honeybee visits on visited flowers) (P < 0.0001, corrected P < 0.0001) (a), weighted betweenness as a measure of centrality in the network (P = 0.009, corrected P = 0.027) (b), proportion of dish-bowl flowers among visited flowers (P < 0.0001, corrected P = 0.001) (c) and corolla length (mm) of visited flowers (CWM) (P = 0.045, corrected P = 0.082) (d). Z-tests were used to derive P values from averaged LMMs and P values were corrected with the FDR correction. Averaged model predictions of the best models (ΔAICc < 2) are plotted with 95% Bayesian credible intervals (shaded areas). All explanatory variables present in the best models (ΔAICc < 2) except the ones for which relationships with response variables are plotted were fixed at their mean values. Points show raw data. Figure created using Procreate (https://procreate.com/).
In the best models (ΔAICc < 2) predicting DWV-B load (Fig. 2b and Supplementary Table 4) and prevalence (Supplementary Table 5 and Extended Data Fig. 3b) in wild pollinators, floral resource overlap with honeybees similarly had the strongest effect, positively affecting both DWV-B prevalence (Supplementary Table 6) and load (Table 1). Weighted betweenness, corolla length, specialization d′ and flower volume did not affect DWV-B prevalence (Supplementary Table 6) or load (Table 1), respectively. Floral resource overlap between wild pollinators and honeybees decreased with increasing proportion of pollinator-friendly habitat in a landscape (linear model; R2m = 0.021, R2c = 0.028, pollinator-friendly habitat (%)—F1,10 = 12.01, P = 0.006; sampling round—F1,228 = 0.13, P = 0.717).
Plants receiving the highest number of honeybee visits in May/June and July had open dish-bowl flowers (for example, Centaurea jacea and Knautia arvensis) as well as closed flag or gullet flowers (for example, Origanum vulgare and Trifolium repens) (Fig. 4a,b). However, non-metric multidimensional scaling (NMDS) of pollinator species composition and flower traits of their visited flowers showed that wild pollinators visiting mainly dish-bowl flowers were those with generally lowest viral loads (permutation test: BQCV—R2 = 0.39, P = 0.014; DWV-B—R2 = 0.60, P = 0.001) (Fig. 4c,d), confirming the results reported above. A similarity of percentage analysis (SIMPER)33 corroborated that flower volume, corolla length and dish-bowl shape were the traits of visited flowers most strongly associated with differences in viral loads among pollinators (Supplementary Table 7). To better understand the findings that dish-bowl flowers are highly visited by honeybees, while at the same time pollinators visiting a high proportion of dish-bowl-shaped flowers have lower viral loads, we separately tested the effect of resource overlap between honeybees and wild pollinators for dish-bowl and non-dish-bowl shaped flowers on viral loads. We found an interactive effect of resource overlap with flower shape on both BQCV and DWV-B loads: the positive relationship between viral load and resource overlap was steeper for resource overlap on non-dish-bowl flowers versus on dish-bowl flowers (Extended Data Fig. 5, LMMs; BQCV— interaction resource overlap × flower shape F1,473 = 5.8, P = 0.0167 and sampling round F1,479 = 68.0, P < 0.001; DWV-B—interaction resource overlap × flower shape F1,444 = 6.7, P = 0.01 and sampling round F1,460 = 10.5, P = 0.001).

a, Plant species that received the most visits by honeybees in May/June and July (mean values ± s.e.m., n = 123 observations from 12 landscapes). The insert is displaying the types of flowers most frequently visited by honeybees. b, The six species with the highest mean number of visits were Helianthus annuus, Phaseolus vulgaris, Origanum vulgare, Centaurea jacea, Trifolium repens and Knautia arvensis and their flower type is indicated. Additionally, examples for the flower types ‘tube’, ‘brush’, ‘bell-trumpet’ and ‘stalk-disk’ are given with images of Symphytum officinale, Phyteuma spicatum, Campanula spp. and Pulmonaria spp. as example taxa are shown. c,d, NMDS visualizing Bray–Curtis dissimilarity distances among the 20 pollinator species screened for viruses preferring plant species with different flower traits. The pollinator species are grouped by load (mean number of genome copies per microgram RNA) of BQCV (c) and DWV-B (d). Numbers in c and d indicate specific pollinator species: HB, honeybees; 1, Andrena humilis; 2, Bombus hortorum; 3, B. humilis; 4, B. hypnorum; 5, B. lapidarius; 6, B. lucorum; 7, B. pascuorum; 8, B. ruderatus; 9, B. subterraneus; 10, B. sylvarum; 11, B. terrestris; 12, Halictus scabiosae; 13, Lasioglossum glabriusculum; 14, L. malachurum; 15, L. morio; 16, L. pauxillum; 17, Melanostoma mellinum; 18, Sphaerophoria philanthus; 19, Syrphus vitripennis. Credit: Phaseolus vulgaris photo © Thomas Bresson, https://www.flickr.com/photos/computerhotline/, under a Creative Commons licence CC BY 2.0.
Influence of landscape and pollinator community properties
Testing for the influence of landscape and pollinator community properties at the landscape scale on BQCV loads (best models with ΔAICc < 2; Fig. 2c and Supplementary Table 8) and prevalence (Supplementary Table 9 and Extended Data Fig. 3c) in wild pollinators revealed that wild pollinator abundance was positively related to BQCV prevalence (Supplementary Table 6) and weakly to load (Table 1 and Fig. 5a). BQCV load in honeybees also explained a significant amount of variation of BQCV load in wild pollinators (Table 1 and Fig. 5b) but BQCV prevalence in honeybees did not explain prevalence in wild pollinators (Supplementary Table 6). The proportion of pollinator-friendly habitat was weakly negatively related to BQCV prevalence (Fig. 5c and Supplementary Table 6).

a,b, Relationships of either BQCV load of virus-positive wild pollinator individuals with wild pollinator abundance (P = 0.020, corrected P = 0.051) (a) or BQCV load in honeybees (log) (median load of ten honeybees per landscape and sampling round) (P = 0.0002, corrected P = 0.001) (b). c, The relationship between BQCV prevalence in wild pollinators and cover of pollinator-friendly habitat (P = 0.024, corrected P = 0.054). Z-tests were used to derive P values from averaged LMMs (viral load) or GLMMs (viral prevalence) and P values were corrected with the FDR correction. Averaged model predictions of the best models (ΔAICc < 2) with 95% Bayesian credible intervals (shaded areas) are plotted. All explanatory variables present in the best models (ΔAICc < 2) except the ones for which relationships with response variables are plotted were fixed at their mean values. Points show raw data. Figure created using Procreate (https://procreate.com/).
In the best models (ΔAICc < 2) predicting DWV-B load (Fig. 2d and Supplementary Table 8) and prevalence (Supplementary Table 9 and Extended Data Fig. 3d) in wild pollinators, wild pollinator abundance was positively related to DWV-B load but not to prevalence (Table 1 and Supplementary Table 6). DWV-B load or prevalence in honeybees did not significantly affect DWV-B loads or prevalence in wild pollinators (Table 1 and Supplementary Table 6).
Discussion
Our study demonstrates how pathogen transmission pathways among potential hosts are the product of complex, multiple interactions and processes operating across levels of ecological organization. The interplay of pollinator foraging roles within networks, preferences for particular plant traits, viral loads in coforaging honeybees and landscape composition together influenced viral prevalence and loads in wild pollinators. Both BQCV and DWV-B viruses were more prevalent with higher loads in wild pollinator species that had a high degree of floral resource overlap with honeybees and with a high wild pollinator abundance in the landscape. However, a more central position of a pollinator within a network, a high proportion of dish-bowl flowers in the diet and high amounts of pollinator-friendly habitat in the landscape reducing floral resource overlap, decreased the chance of pathogen transmission.
BQCV and DWV-B loads were markedly increased in many field-sampled bee and hoverfly species with high floral resource overlap with honeybees and with high viral loads in coforaging honeybees (for BQCV). Our results indicate that transmission of BQCV was more widespread, while transmission of DWV-B affected especially those wild pollinators with a particularly high floral resource overlap with honeybees. This is in line with our finding that BQCV was clearly more widespread in honeybees, wild bee and hoverfly pollinators screened in the study region than was DWV-B. Although we did not test for active replication of the virus in a pollinator (and thus infection), these findings, together with the generally substantially higher viral loads in honeybees compared to wild pollinators, provide strong evidence for virus spillover from honeybees to wild pollinators through foraging on shared floral resources. However, our data show also that a few wild bee species, especially bumblebee species such as B. lapidarius and B. terrestris which were frequently sampled in the plant–pollinator networks, had similarly high median loads or prevalence of BQCV and/or DWV-B, although clearly lower maximum viral loads, compared to honeybees. It is therefore conceivable that virus transmission could occur in both directions among honeybees and these wild bee species, potentially also including spillback21 of viruses following spillover from honeybees (but see ref. 34 finding experimental evidence that the directionality of DWV transmission is predominantly from honeybees to bumblebees). We found some support for this hypothesis based on analyses showing that BQCV prevalence in B. lapidarius significantly predict BQCV prevalence in honeybees and similar positive associations between DWV-B prevalence in B. terrestris and honeybees as well as DWV-B loads in B. lapidarius and honeybees (Supplementary Table 10). It is important to note, however, that while these findings corroborate virus transmission among honeybees and these bumblebee species, we cannot unravel the directionality or determine the dominance of transmission in one or the other direction, which would require, for example, a controlled experimental approach34.
Transmission of viruses among pollinators via flowers can either occur through direct contact while foraging on the same flower34 or more likely via visiting flowers contaminated through the transmission of contaminated pollen or oral secretions and faeces from infected pollinators16,35. In other pollinator–pathogen systems, trypanosomatid transmission can be strongly affected by flower morphology36. For example, increased defecation rates of bumblebees on large open flowers (dish-bowl flowers) may increase the spread of trypanosome pathogens21. In contrast to our hypothesis, our results indicate lower BQCV loads in pollinators visiting a high proportion of open dish-bowl flowers, suggesting that other factors associated with the dish shape of flowers played an important role in BQCV transmission24. For example, viruses in nectar and pollen of open dish-bowl flowers might degrade faster due to higher UV light exposure37,38, thereby potentially reducing transmission rates. Additionally, the open dish-bowl flowers and flowers with shorter corollas are generally easier to handle for many pollinators, leading to shorter flower handling times26, which may decrease the risk of picking up viruses. These potential mechanisms might explain the lower virus loads in species visiting proportionally more dish flowers and flowers with shorter corollas compared to species preferentially visiting flowers of other shapes, despite some pollinators having overlapping resources with honeybees. The pollinator species with lower virus loads most frequently visiting these dish flowers belonged to the families Halictidae, Andrenidae and Syrphidae, as revealed by the NMDS analysis. Thus, together with the potential mechanisms of increased UV light exposure and decreased handling time discussed above, it is conceivable that the observed usually high diversity of pollinators visiting these readily accessible dish flowers included a particular high proportion of species with very low viral loads which may not act as viral reservoir hosts. This, along with a disproportionally high densities of dish flowers (as revealed by the flower abundance estimates for the sample plant–pollinator networks), could be associated with a reduced probability that individual flowers are actually shared among hosts, possibly further contributing to a reduced risk of virus transmission among pollinators through a dilution effect mediated by flower shape24.
Moreover, several of our other findings point to a dilution effect27 at the pollinator species and the landscape level, although some of our findings also indicate processes that counter those of pathogen dilution. At the pollinator species level, our finding that pollinators with a central position in the networks (high weighted betweenness) are associated with lower viral loads points towards a dilution pathway: pollinators that visit a variety of different plant species also visited by other pollinators may experience a reduced risk of pathogen exposure compared to species focusing on few plant species19,29.
Finally, at the landscape level, our findings suggest that high amounts of pollinator-friendly habitat are related to reduced BQCV prevalence, both in bee and hoverfly pollinators. This was probably driven by the reduced floral resource overlap between honeybees and wild pollinators as a consequence of increased availability of different foraging habitat offering higher floral diversity levels39. High habitat amount and flower diversity generally reduce the probability of inter- and intra-species contact due to increasing complementarity in flowering plant species visitation30,40,41. At the same time, high amounts of suitable foraging habitat probably promote optimal nutritional conditions and thus general health of pollinators42. Therefore, our study of 17 wild bee and hoverfly species corroborates evidence for such a mitigation function of high-quality foraging habitats that have been found to reduce levels of viruses or microsporidia in four bee species in the United States39,43 and to decrease virus prevalence in pollinators on farms participating in agri-environment schemes in the United Kingdom29. Thus, pollinator-friendly habitat can reduce pathogen transmission and loads in wild pollinators via direct benefits through enhanced foraging, nesting and overwintering opportunities, and indirect benefits through reduced resource competition, in particular with honeybees30. However, such hotspots of high-quality foraging habitats attract at the same time a high abundance and diversity of pollinators. This can lead to increased pathogen transmission of density-dependent pathogens, such as BQCV and DWV-B (here) and for slow bee paralysis virus, microsporidia and acute bee paralysis virus29,31,44. This finding suggests that such high-quality foraging habitats may also trigger processes that counter those of a pathogen dilution effect, potentially confounding the beneficial effects of the latter on wild pollinator health. In fact, our findings imply that there is a potentially complex trade-off between the direct and indirect benefits of diverse floral resources enhancing pollinator nutrition and diluting pathogen transmission risks on the one hand and density-dependent elevation of pathogen transmission on the other hand.
In conclusion, by combining species traits, species interactions and landscape level factors, our study identifies several key drivers of viral prevalence and loads and potential transmission pathways of viral pathogens in pollinator communities. Our results highlight the important role of floral resource overlap with honeybees, identified as a reservoir host9, as a primary driver of viral prevalence and loads in wild pollinators. Our findings also provide insights into how virus transmission and viral loads are mediated by foraging traits and roles of pollinator species in interaction networks, floral trait composition and the amount of pollinator-friendly habitat at the landscape scale. However, the direction of transmission and the extent to which detected viral loads affect health, fitness and population dynamics of wild pollinators in the field remains an important research gap14 but see ref. 45; our results therefore serve to inform future hypothesis testing.
To follow the precautionary principle and reduce virus transmission among pollinators, our results highlight how conservation and restoration of flower-diverse pollinator-friendly habitat in a landscape may not only contribute to pollinator conservation through the provision of vital resources and a reduction in floral resource competition but also help to mitigate transmission of key viruses. Such habitats should be diverse in flowering plant species and not exclusively contain plant species that are highly visited by honeybees. Further, our study highlights the important role of high viral loads in managed honeybees as a driver of virus transmission to wild pollinators. It underpins the view that it is vital to increase efforts to implement best management practices to control Varroa mites in honeybee colonies as a key measure to reduce viral loads of honeybees, to minimize virus transmission to wild pollinators. In light of concerns about ongoing pollinator declines, our findings thus provide strong support for measures that promote pollinator-friendly habitats to maintain diverse and healthy pollinator communities and to secure the pollination functions and services they provide.
Methods
Study design
We selected 12 landscapes in the lowland of northern Switzerland (n = 12, 1 km radius, separated by >3 km; Extended Data Fig. 1). This accounts for the typical foraging ranges of most of the studied pollinator groups being considerably less than 3 km (refs. 46,47), although honeybees can forage further if foraging resources dictate it but see refs. 46,48. The region is characterized by agricultural landscapes, seminatural habitats and settlements. The study landscapes thus varied from landscapes dominated by mixed production agriculture to landscapes dominated by settlements (many areas with one-family homes with gardens and other green space), with lower or higher amounts of seminatural habitat (forest, hedgerows and extensively managed grasslands). We classified each major land-use type as either a potential pollinator-friendly habitat or not based on previous studies in the study region that assessed the value of different land-use types to sustain bee and hoverfly pollinators49,50,51,52,53,54. Accordingly, grasslands (that is, extensively managed meadows and pastures; including traditional orchards and vineyards largely composed of extensively managed permanent grassland vegetation), sown flower strips, hedgerows and urban green spaces (urban area with >25% green space, that is, houses with gardens, parks, green roadsides) were classified (ArcGIS Pro v.10.7, ESRI) as pollinator-friendly habitats. Arable crops, intensively managed grasslands, forests, urban space with <25% green space area and water bodies were classified as not pollinator-friendly habitats. Although arable crops and forests can provide floral resources for pollinators, their overall importance for pollinators in the study region was found to be inferior in the studies cited above and their floral resource provision mainly concentrates on early in the season25, a period finally excluded from our analyses due to low numbers of species with minimum sample size for virus screening (see section on ‘Pollinator sample selection criteria and viral targets’). Consequently, we did not categorize them as pollinator-friendly in the present study. On the basis of raster maps (pixel size: 2 × 2 m2), we calculated the percentage of pollinator-friendly habitat in a 500 m radius around the centre of each landscape using the R package landscapemetrics55. The resulting gradient in pollinator-friendly habitat ranged from 2.6% to 74.1% (Supplementary Table 11).
Sampling of plant–pollinator networks and flower surveys
Within each landscape, plant–pollinator visitation networks were sampled along different transect sections in habitats providing flowers at the time of the transect walks (for example, grasslands, forest edges, hedgerows, flowering crops, field edges and gardens) summing up to a total length of 1 km (2 m wide) per landscape in each sampling round (three rounds in total, April, May/June and July 2020). The transect section length per flowering habitat type was proportional to the cover of that specific flowering habitat type within the core sector (500 m radius) of a landscape52. Non-flowering habitats such as conifer plantations or cereal fields that were unlikely to contribute to flower–pollinator interactions in a landscape were not sampled. All flower-visiting bees (Hymenoptera: Anthophila), including the western honeybee, A. mellifera L. and hoverflies (Diptera: Syrphidae) were sampled as they are the most common pollinators in the study region4. The specimens were collected with a tube or a self-made ‘net’ consisting of a plastic bag fixed on a stick. After each catch, a fresh plastic bag was fixed on the stick to avoid contamination among specimens. Additionally, the species identity of all visited plants were recorded using the key ‘Flora Helvetica Exkursionsflora’56 to allow construction of plant–pollinator interaction networks. Transect walks were standardized (3 min per 25 m, pausing the clock for catching and processing pollinator specimens) and conducted between 09:00 and 18:00 during dry and warm weather (>14 °C). After each transect walk, we walked back the same transect at a steady pace to obtain an additional independent measure of honeybee densities by counting (without catching) each foraging or flying honeybee encountered within the transect. Immediately after sampling, all insects were placed on dry ice in the field and then at −80 °C in the laboratory. All wild bee and hoverfly samples were assigned to species by barcoding the cytochrome oxidase subunit I gene region57 by the company Microsynth Ecogenics GmbH. Barcoding is an objective and highly accurate method to identify species58. There are only a few cryptic species complexes that cannot be unequivocally identified using barcodes58,59, with only one present in our samples (Halictus simplex and H. eurygnathus). Abundance of wild pollinators was calculated as the number of flower-visiting wild pollinator individuals caught during the transect walks per landscape and sampling round, while wild pollinator richness was calculated as the number of different species caught per landscape and sampling round.
To obtain measures of flower abundance and flowering plant species richness independent of visitation (plant–pollinator network) data, flower abundance and richness were quantified in ten plots (2 × 0.5 m2) per 100 m along the transects (between one and three plots per 100 m for flowering crop monocultures) by a trained botanist, using the key ‘Flora Helvetica Exkursionsflora’56. The plots were either placed horizontally for herbaceous flowering vegetation or vertically along woody vegetation of hedgerows and forest edges. We estimated flower abundance as the number of single flowers (or inflorescences in the case of Asteraceae, K. arvensis and Plantago spp.) multiplied by their size (calculated as circle area as a proxy of floral resource availability, following ref. 25) for each flowering plant species. For flower/inflorescence size trait information see section on ‘Plant–pollinator network metrics and species traits’. The total number of flowering plant species per landscape and their flower abundance in each sampling round was used to calculate flower Shannon diversity.
Virus quantification
Pollinator sample selection criteria and viral targets
Wild bee and hoverfly pollinator species were considered with n ≥ 7 samples (insect specimens per species) in a particular landscape and sampling round. A maximum of n = 10 samples per wild pollinator species and ten individual honeybees (A. mellifera) were screened per landscape and sampling round—a sample size used previously60. Further, this approach meant that we covered the most abundant species most likely to affect the plant–pollinator network structure in the respective landscapes. Samples (n = 986, 20 species (A. mellifera, 16 wild bee species and three hoverfly species); Supplementary Table 2) were screened for three viruses which are common in A. mellifera: DWV-A, DWV-B and BQCV7, all associated with the presence of and probably transmitted to A. mellifera by ectoparasitic mites Varroa destructor61. Owing to low numbers of species with minimum sample size for virus screening in the first sampling round (April) (Supplementary Table 2), data from this sampling round were not considered for statistical analyses. Further, due to very low titres of DWV-A, we refrained from data analyses of this virus (Supplementary Table 1 and Extended Data Fig. 2). Since the samples from April had to be dropped (n = 120 honeybees, n = 38 wild pollinator samples), this resulted in n = 588 samples of 15 wild bee and two hoverfly species and 240 samples of honeybees which were used for statistical data analysis.
RNA extraction and reverse transcription
The pollinators were not surface sterilized before extraction because our main objective was to better understand possible virus transmission pathways rather than to detect and understand patterns of virus infections (we therefore did not test for replicating virus). This should be considered accordingly when interpreting results. Before RNA extraction, samples were randomized to avoid introducing any undesired bias (regarding species or collection site) during the sample preparation for viral quantification (RNA extraction, reverse transcription and PCR). Individual samples were crushed in PBS buffer (0.5 mg of tissue per μl) with a 5 mm glass bead in 2 ml Eppendorf tubes using a Retsch MM 300 mixer mill for 1 min at the frequency 25 s−1 (ref. 37). Then, RNA was extracted from 50 μl (25 mg of tissue) of this homogenate using a NucleoSpin RNA II kit (Macherey-Nagel) following the manufacturer’s recommendations. RNA was eluted in 60 μl of elution buffer and stored at −80 °C (ref. 37).
The RNA was reverse transcribed using a M-MLV RT Kit (Promega) following the manufacturer’s recommendations. Briefly, 0.75 μl of a random hexamer oligonucleotide (100 μM) and water were incubated for 5 min at 70 °C with 0.5 µg of template RNA using a thermocycler (Biometra). Then, for a 25 µl volume reaction, 5 μl of 5× buffer, 1.125 μl of dNTPs (10 mM) and 1 μl of reverse transcriptase (M-MLV) were added and incubated at 37 °C for 60 min to synthesize complementary DNA.
Quantitative PCR
Absolute quantification of viral loads was performed by quantitative PCR (qPCR). Reactions were prepared with 6 µl of 2× reaction buffer (SensiFAST SYBR No-ROX Kit, Meridian Bioscience), 0.24 µl of forward and reverse primer (Supplementary Table 12), 2.52 µl of water and 3 µl of tenfold diluted cDNA. Then qPCR reactions were performed in a CFX96TM Real-Time PCR Detection Systems (Bio-Rad) with the following conditions: 3 min for 95 °C, 40 cycles of 3 s at 95 °C and 30 s at 57 °C. After amplification, we analysed the melting curve profile of all PCRs (strand dissociation) to verify product specificity (a single product with the correct dissociation temperature) by reading the fluorescence at 0.5 °C increments from 55 to 95 °C. Each sample was run in duplicate for each of the targeted viruses. A third technical run was included for samples that differed by more than one cycle. Amplification of the host insect’s 28S ribosomal RNA gene using primer sequences conserved across suborder Apocrita62, which has been shown to perform well also for hoverflies63, was used as reference gene to assess the quality of the process (RNA extraction, cDNA synthesis and qPCR). Furthermore, each plate was run with seven tenfold serial dilutions (10−2 to 10−8 ng) of synthetic strands (standard curve; Supplementary Table 13) which served as positive controls for the viral sequence target, plus two no-template negative controls64. For quantification, qPCR efficiencies (E) were calculated on the basis of the slope of the standard curve, according to the following formula: E = 10(−1/slope) (DWV-A—E = 1.93, slope = −3.479, y intercept = 38.907, R2 = 0.990; DWV-B—E = 1.97, slope = −3.373, y intercept = 39.187, R2 = 0.991; BQCV—E = 2.06, slope = −3.183, y intercept = 38.195, R2 = 0.992). Plates with no-template negative controls showing a signal matching the melting curve peak of the target viral gene were re-run. The quantification threshold (Cq) was set in the Bio-Rad CFX MAESTRO 1.0 software as auto calculated for all runs per target, assuring Cq was always set in the exponential phase of the amplification curve64. A qPCR quantification cycle (Cq) threshold of Cq < 35 was used to define a positive sample. This threshold is commonly accepted as it is interpreted that in general with an input of ten template copies in the reaction and a suitable PCR efficiency (between 1.8 and 2), a Cq value of ~35 will be observed65. Moreover, melt curve analysis was also used to consider a sample positive or negative if their melting temperatures (Tm) match or not those of their respective controls (81.5–82.5 °C Tm for DWV-A, 79.0–80.0 °C Tm for DWV-B and 82.5–83.0 °C Tm for BQCV). To calculate 95% confidence intervals (Clopper–Pearson interval) around the apparent virus prevalence in each species (per site and sampling round), we used the function propCI from the R package prevalence66. Although sequencing of viral variants from different species and sites would be necessary to provide unequivocal support for cross-species viral sharing (for example, ref. 67), the limited geographic scale of our sampling in northern Switzerland is unlikely to provide sufficient resolution to separate sharing of variants across species versus across sites.
Plant–pollinator network metrics and flower species traits
We analysed flower–insect visitor networks for each landscape and sampling round with plant and pollinator species as nodes and interaction frequencies as links. The networks were constructed with the entire plant–pollinator community that was sampled during transect walks. We refer to plant–pollinator networks for simplicity, while acknowledging that not every flower visit necessarily results in a pollination event68. To test whether floral niche overlap with managed honeybees (hypothesis 1) influences virus prevalence and loads of wild pollinators, we calculated the CWM number of honeybee visits to flowering plant species visited by a given wild pollinator species. This metric of floral resource overlap correlated strongly positively with the potential for apparent competition (PAC) metric (Spearman’s rank correlation rho = 0.82) often used to quantify resource overlap69 but is simpler to interpret compared to PAC. To assess how pollinator species specialization determines viral loads (hypothesis 2), we calculated specialization d′ based on observed floral resource use of a pollinator species in each network for those pollinator species that were screened for viruses. Specialization d′ accounts for the presence and abundance of partner species available in an interaction network and denotes the degree of diet specialization of each species (0, no specialization; 1, perfect specialist)70. To assess how viral loads related to centrality of a pollinator species in a network (hypothesis 3), we calculated weighted betweenness of each screened pollinator species. It measures the proportion of shortest paths that pass through a focal species, indicating how central and closely connected a species is to other pollinators in the network. All network analyses were performed using R package bipartite71.
To assess the role of traits of flowering plants in the network hypothesized to affect pathogen transmission (flower morphology characterized through flower type and corolla length and flower volume as proxy for nectar rewards24), we linked information from the plant–pollinator networks with published information about traits of visited flowers. For hypothesis 4, we obtained data on flower (inflorescence) type from ref. 72 classified after ref. 73 as either dish-bowl type (for example, Asteraceae and Rosaceae), flag (for example, Fabaceae), gullet (for example, Lamiaceae), brush (for example, Cirsium and Plantago), bell-trumpet (for example, Campanulaceae), tube (for example, Symphytum officinale) or stalk-disk (for example, Primula acaulis and Dianthus carthusianorum), corolla length (mm) and average flower volume (cm3, approximated as cylinder volume with height = corolla length, radius = 0.5 × flower diameter)25. Flower diameter and corolla length were retrieved from a floral trait database including most plant species from the study region (D. Frey, L. Amman, M. A. & M. Moretti, manuscript in preparation) and from Info Flora (https://www.infoflora.ch/), PlantNET (https://plantnet.rbgsyd.nsw.gov.au/) and Naturegate (https://luontoportti.com/). Flower volume was calculated for individual flowers, except for Asteraceae, K. arvensis and Plantago spp., for which the volume of inflorescences (for example, entire flower heads) was calculated. We then calculated the CWM of each trait for all flowers visited by a pollinator species using R package FD74.
Statistical analyses
Floral species’ traits and pollinator network roles
To investigate how floral species’ traits and pollinator roles in the plant–pollinator network drive viral prevalence and loads in wild pollinators (hypotheses 1–4), we used generalized linear mixed effects models (GLMMs, binomial error distribution) with viral prevalence (presence or absence of BQCV or DWV-B) in wild pollinators as response variable and linear mixed effects models (LMM, Gaussian error distribution) with viral load (log BQCV or log DWV-B) of each wild pollinator individual (considering only virus-positive individuals) as the response variable. Floral resource overlap of wild pollinators with honeybees, wild pollinator specialization d′, weighted betweenness of a wild pollinator, mean flower volume of visited flowers (CWM), mean corolla length of visited flowers (CWM) and the proportion of visited plant species with dish-bowl flowers were fitted as explanatory variables in the full models, with landscape ID and species ID as random factors. Exploratory analyses revealed no interaction between predictors and sampling round, which therefore was not included as a candidate interaction term in the full model. However, we accounted for the influence of sampling round by including it as a fixed factor in all models.
To visualize groups of pollinators visiting plants with similar floral traits, we used NMDS based on Bray–Curtis dissimilarity of the entire interaction matrix (pooled across landscapes and sampling rounds) using the R package vegan75. In this matrix, the rows represented the pollinator species, the columns represented the trait of the visited plants (flower volume, corolla length and inflorescence type) and the values were the CWMs of the respective trait. We then classified the pollinator species into three groups according to the lower, intermediate and upper third of the mean virus load (number of genome copies per µg RNA per individual sample) distribution (<33%, >33%, <66% and >66% quantiles; BQCV—<44,000; >44,000 < 900,000; >900,000 genome copies; DWV-B—<1,600; >1,600 < 10,000; >10,000 genome copies) and applied a permutation test to evaluate whether these groups of distinct levels of virus loads are associated with floral trait dependent visitation by pollinators. Since these virus-load groups significantly explained the differences in flower visitation, we performed a similarity percentage (SIMPER)33 analysis in R package vegan73 to evaluate which traits of visited flowers contributed most to these differences.
Landscape and pollinator community properties
To test how the explanatory variables landscape composition (proportion of pollinator-friendly habitat), flowering plant community composition (Shannon diversity of flowers at the landscape scale) and pollinator community composition (honeybee density and viral prevalence or loads in honeybees, species richness and abundance of wild pollinators at the landscape scale) affected the response variables viral prevalence or loads (log-transformed, virus-positive individuals only) of the wild pollinator community (hypotheses 5–8), we fitted GLMMs (binomial error distribution) and LMMs, respectively. We included sampling round as covariate and landscape ID and species ID as random factors. Viral loads in honeybees were calculated as the median number of viral copies (log-transformed) of the ten screened honeybees per landscape in a given sampling round, while viral prevalence in honeybees was the proportion of honeybees with viral load >0 per landscape in a given sampling round. Because wild pollinator abundance and species richness were strongly positively correlated (Spearman’s rank correlation rho = 0.7), we only used wild pollinator abundance in the full models due to its higher predictive power and goodness of model fit (lower AICc in univariate models).
Finally, we tested whether floral resource overlap between wild pollinators and honeybees (calculated as described above) was related to the proportion of pollinator-friendly habitat in a landscape (hypothesis 8). Owing to heteroscedasticity in the residuals of the model, we used an LMM with a ‘power of the covariate’ variance structure (varPower) to ensure correct estimation of regression parameters and standard errors76, with resource overlap as the response variable, proportion of pollinator-friendly habitat as the explanatory variable and landscape ID as a random factor using the R package nlme77.
Assessing correlation among explanatory variables in all full models using variance inflation factors (VIF)78 in R package performance79 showed low multicollinearity (VIF < 2.5). For all analyses, the set of best models was selected on the basis of the Akaike information criterion corrected for small sample size (AICc) using the R package MuMIn80. We considered all models with ΔAICc < 2 (that is, the difference of the AICc of the focal model to the best model <2) and used model averaging (conditional average) to extract the final model coefficients81. To visualize the major effects (of the terms whose confidence intervals do not overlap with zero), we used a Bayesian framework to calculate averaged model predictions by drawing samples from the joint posterior distribution with the function sim of the R package arm82. Since we ran several models exploring species-specific as well as landscape and community level effects on viral load and prevalence, we did a false discovery rate (FDR) correction using the function p.adjust of R package stats83.
For all statistical analyses the software R v.4.2.1 (ref. 83) was used and models were fitted with the package lme4 (ref. 84), except where stated differently. All continuous variables were scaled and centred to improve model convergence. Model assumptions were checked by inspection of residual plots using the R packages DHARMa85 and performance79. The data used for all analyses of this study are openly available86.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data is openly available via Figshare at https://doi.org/10.6084/m9.figshare.25101977 (ref. 86).
References
Daszak, P., Cunningham, A. A. & Hyatt, A. D. Emerging infectious diseases of wildlife—threats to biodiversity and human health. Science 287, 443–449 (2000).
Manley, R. et al. Knock-on community impacts of a novel vector: spillover of emerging DWV-B from Varroa-infested honeybees to wild bumblebees. Ecol. Lett. 22, 1306–1315 (2019).
Cameron, S. A. et al. Patterns of widespread decline in North American bumble bees. Proc. Natl Acad. Sci. USA 108, 662–667 (2011).
IPBES. The assessment report of the Intergovernmental Science-Policy Platform on Biodiversity and Ecosystem Services on pollinators, pollination and food production. Zenodo https://doi.org/10.5281/zenodo.3402857 (2016).
Vanbergen, A. J. Threats to an ecosystem service: pressures on pollinators. Front. Ecol. Environ. 11, 251–259 (2013).
Wilfert, L., Brown, M. J. F. & Doublet, V. OneHealth implications of infectious diseases of wild and managed bees. J. Invertebr. Pathol. 186, 107506 (2021).
Gisder, S. & Genersch, E. Viruses of commercialized insect pollinators. J. Invertebr. Pathol. 147, 51–59 (2017).
Radzeviciute, R. et al. Replication of honey bee-associated RNA viruses across multiple bee species in apple orchards of Georgia, Germany and Kyrgyzstan. J. Invertebr. Pathol. 146, 14–23 (2017).
Tehel, A., Streicher, T., Tragust, S. & Paxton, R. J. Experimental infection of bumblebees with honeybee-associated viruses: no direct fitness costs but potential future threats to novel wild bee hosts. R. Soc. Open Sci. 7, 200480 (2020).
Bailes, E. J. et al. First detection of bee viruses in hoverfly (syrphid) pollinators. Biol. Lett. 14, 20180001 (2018).
Murray, E. A. et al. Viral transmission in honey bees and native bees, supported by a global black queen cell virus phylogeny. Environ. Microbiol. 21, 972–983 (2019).
Fürst, M. A., McMahon, D. P., Osborne, J. L., Paxton, R. J. & Brown, M. J. Disease associations between honeybees and bumblebees as a threat to wild pollinators. Nature 506, 364–366 (2014).
Manley, R., Boots, M. & Wilfert, L. Condition-dependent virulence of slow bee paralysis virus in Bombus terrestris: are the impacts of honeybee viruses in wild pollinators underestimated? Oecologia 184, 305–315 (2017).
Streicher, T., Tehel, A., Tragust, S. & Paxton, R. J. Experimental viral spillover can harm Bombus terrestris workers under field conditions. Ecol. Entomol. 48, 81–89 (2023).
Proesmans, W. et al. Pathways for novel epidemiology: plant–pollinator–pathogen networks and global change. Trends Ecol. Evol. 36, 623–636 (2021).
Burnham, P. A. et al. Flowers as dirty doorknobs: deformed wing virus transmitted between Apis mellifera and Bombus impatiens through shared flowers. J. Appl. Ecol. 58, 2065–2074 (2021).
Grass, I., Jauker, B., Steffan-Dewenter, I., Tscharntke, T. & Jauker, F. Past and potential future effects of habitat fragmentation on structure and stability of plant–pollinator and host–parasitoid networks. Nat. Ecol. Evol. 2, 1408–1417 (2018).
Wilson, C. J. & Jamieson, M. A. The effects of urbanization on bee communities depends on floral resource availability and bee functional traits. PLoS ONE 14, e0225852 (2019).
Figueroa, L. L. et al. Landscape simplification shapes pathogen prevalence in plant–pollinator networks. Ecol. Lett. 23, 1212–1222 (2020).
Figueroa, L. L. et al. Bee pathogen transmission dynamics: deposition, persistence and acquisition on flowers. Proc. Biol. Sci. 286, 20190603 (2019).
Pinilla-Gallego, M. S., Ng, W. H., Amaral, V. E. & Irwin, R. E. Floral shape predicts bee–parasite transmission potential. Ecology 103, e3730 (2022).
Adler, L. S. et al. Disease where you dine: plant species and floral traits associated with pathogen transmission in bumble bees. Ecology 99, 2535–2545 (2018).
Durrer, S. & Schmid-Hempel, P. Shared use of flowers leads to horizontal pathogen transmission. Proc. R. Soc. Lond. B 258, 299–302 (1994).
Nicholls, E., Rands, S. A., Botías, C. & Hempel de Ibarra, N. Flower sharing and pollinator health: a behavioural perspective. Philos. Trans. R. Soc. B 377, 20210157 (2022).
Ammann, L. et al. Spatio-temporal complementarity of floral resources sustains wild bee pollinators in agricultural landscapes. Agric. Ecosyst. Environ. 359, 108754 (2024).
Harder, L. D. Flower handling efficiency of bumble bees: morphological aspects of probing time. Oecologia 57, 274–280 (1983).
Civitello, D. J. et al. Biodiversity inhibits parasites: broad evidence for the dilution effect. Proc. Natl Acad. Sci. USA 112, 8667–8671 (2015).
Fearon, M. L. & Tibbetts, E. A. Pollinator community species richness dilutes prevalence of multiple viruses within multiple host species. Ecology 102, e03305 (2021).
Manley, R. et al. Conservation measures or hotspots of disease transmission? Agri-environment schemes can reduce disease prevalence in pollinator communities. Philos. Trans. R. Soc. B 378, 20220004 (2023).
Cappellari, A. et al. Functional traits of plants and pollinators explain resource overlap between honeybees and wild pollinators. Oecologia 198, 1019–1029 (2022).
Graystock, P. et al. Dominant bee species and floral abundance drive parasite temporal dynamics in plant–pollinator communities. Nat. Ecol. Evol. 4, 1358–1367 (2020).
Piot, N. et al. Establishment of wildflower fields in poor quality landscapes enhances micro-parasite prevalence in wild bumble bees. Oecologia 189, 149–158 (2019).
Clarke, K. R. Non‐parametric multivariate analyses of changes in community structure. Aust. J. Ecol. 18, 117–143 (1993).
Tehel, A., Streicher, T., Tragust, S. & Paxton, R. J. Experimental cross species transmission of a major viral pathogen in bees is predominantly from honeybees to bumblebees. Proc. R. Soc. B 289, 20212255 (2022).
Yañez, O. et al. Bee viruses: routes of infection in Hymenoptera. Front. Microbiol. 11, 537174 (2020).
Van Wyk, J. I., Lynch, A.-M. & Adler, L. S. Manipulation of multiple floral traits demonstrates role in pollinator disease transmission. Ecology 104, e3866 (2023).
de Miranda, J. R. et al. Standard methods for virus research in Apis mellifera. J. Apic. Res. 52, 1–56 (2013).
Ignoffo, C. M., Hostetter, D. L., Sikorowski, P. P., Sutter, G. & Brooks, W. M. Inactivation of representative species of entomopathogenic viruses, a bacterium, fungus, and protozoan by an ultraviolet light source. Environ. Entomol. 6, 411–415 (1977).
Fearon, M. L., Wood, C. L. & Tibbetts, E. A. Habitat quality influences pollinator pathogen prevalence through both habitat–disease and biodiversity–disease pathways. Ecology 104, e3933 (2023).
Doublet, V. et al. Increasing flower species richness in agricultural landscapes alters insect pollinator networks: Implications for bee health and competition. Ecol. Evol. 12, e9442 (2022).
Gómez-Martínez, C., González-Estévez, M. A., Cursach, J. & Lázaro, A. Pollinator richness, pollination networks, and diet adjustment along local and landscape gradients of resource diversity. Ecol. Appl. 32, e2634 (2022).
Alaux, C. et al. A ‘landscape physiology’ approach for assessing bee health highlights the benefits of floral landscape enrichment and semi-natural habitats. Sci. Rep. 7, 40568 (2017).
McNeil, D. J. et al. Bumble bees in landscapes with abundant floral resources have lower pathogen loads. Sci. Rep. 10, 22306 (2020).
Bailes, E. J. et al. Host density drives viral, but not trypanosome, transmission in a key pollinator. Proc. Biol. Sci. 287, 20191969 (2020).
Bosco, L. et al. Landscape structure affects temporal dynamics in the bumble bee virome: landscape heterogeneity supports colony resilience. Sci. Total Environ. 946, 174280 (2024).
Danner, N., Molitor, A. M., Schiele, S., Härtel, S. & Steffan‐Dewenter, I. Season and landscape composition affect pollen foraging distances and habitat use of honey bees. Ecol. Appl. 26, 1920–1929 (2016).
Greenleaf, S. S., Williams, N. M., Winfree, R. & Kremen, C. Bee foraging ranges and their relationship to body size. Oecologia 153, 589–596 (2007).
Beekman, M. & Ratnieks, F. L. W. Long‐range foraging by the honey‐bee, Apis mellifera L. Funct. Ecol. 14, 490–496 (2000).
Bartual, A. M. et al. The potential of different semi-natural habitats to sustain pollinators and natural enemies in European agricultural landscapes. Agric. Ecosyst. Environ. 279, 43–52 (2019).
Casanelles-Abella, J., Fontana, S., Fournier, B., Frey, D. & Moretti, M. Low resource availability drives feeding niche partitioning between wild bees and honeybees in a European city. Ecol. Appl. 33, e2727 (2023).
Ganser, D., Albrecht, M., Knop, E. & Peralta, G. Wildflower strips enhance wild bee reproductive success. J. Appl. Ecol. 58, 486–495 (2020).
Maurer, C., Sutter, L., Martínez‐Núñez, C., Pellissier, L. & Albrecht, M. Different types of semi‐natural habitat are required to sustain diverse wild bee communities across agricultural landscapes. J. Appl. Ecol. 59, 2604–2615 (2022).
Pfiffner, L., Ostermaier, M., Stoeckli, S. & Müller, A. Wild bees respond complementarily to ‘high-quality’ perennial and annual habitats of organic farms in a complex landscape. J. Insect Conserv. 22, 551–562 (2018).
Sutter, L., Albrecht, M. & Jeanneret, P. Landscape greening and local creation of wildflower strips and hedgerows promote multiple ecosystem services. J. Appl. Ecol. 55, 612–620 (2018).
Hesselbarth, M. H. K., Sciaini, M., With, K. A., Wiegand, K. & Nowosad, J. landscapemetrics: an open-source R tool to calculate landscape metrics. Ecography 42, 1648–1657 (2019).
Eggenberg, S. et al. Flora Helvetica Exkursionsflora (Haupt Verlag, 2018).
Hebert, P. D. N., Cywinska, A., Ball, S. L. & deWaard, J. R. Biological identifications through DNA barcodes. Proc. R. Soc. Lond. B 270, 313–321 (2003).
Schmidt, S., Schmid-Egger, C., Moriniere, J., Haszprunar, G. & Hebert, P. D. DNA barcoding largely supports 250 years of classical taxonomy: identifications for Central European bees (Hymenoptera, Apoidea partim). Mol. Ecol. Resour. 15, 985–1000 (2015).
Gueuning, M., Frey, J. E. & Praz, C. Ultraconserved yet informative for species delimitation: ultraconserved elements resolve long‐standing systematic enigma in Central European bees. Mol. Ecol. 29, 4203–4220 (2020).
Fleites-Ayil, F. A. et al. Trouble in the tropics: pathogen spillover is a threat for native stingless bees. Biol. Conserv. 284, 110150 (2023).
Doublet, V. et al. Shift in virus composition in honeybees (Apis mellifera) following worldwide invasion by the parasitic mite and virus vector Varroa destructor. R. Soc. Open Sci. 11, 231529 (2024).
Jones, L. J., Ford, R. P., Schilder, R. J. & López-Uribe, M. M. Honey bee viruses are highly prevalent but at low intensities in wild pollinators of cucurbit agroecosystems. J. Invertebr. Pathol. 185, 107667 (2021).
Slone, J. R. Integrated Management Practices for Improved Pollinator Health. DPhil thesis, North Carolina State Univ. (2019); https://repository.lib.ncsu.edu/server/api/core/bitstreams/08d86d58-4442-4461-9d53-4f57c9a4b6ec/content
Bustin, S. A. et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 55, 611–622 (2009).
Ruiz-Villalba, A., Ruijter, J. M. & van den Hoff, M. J. Use and misuse of Cq in qPCR data analysis and reporting. Life 11, 496 (2021).
Devleesschauwer, B. et al. prevalence: Tools for prevalence assessment studies. R package version 0.4.1 (2022).
Manley, R., Temperton, B., Boots, M. & Wilfert, L. Contrasting impacts of a novel specialist vector on multihost viral pathogen epidemiology in wild and managed bees. Mol. Ecol. 29, 380–393 (2020).
Popic, T. J., Wardle, G. M. & Davila, Y. C. Flower-visitor networks only partially predict the function of pollen transport by bees. Austral Ecol. 38, 76–86 (2013).
Page, M. L. & Williams, N. M. Evidence of exploitative competition between honey bees and native bees in two California landscapes. J. Anim. Ecol. 92, 1802–1814 (2023).
Blüthgen, N., Menzel, F. & Blüthgen, N. Measuring specialization in species interaction networks. BMC Ecol. 6, 9 (2006).
Dormann, C. F., Gruber, B. & Fründ, J. Introducing the bipartite package: analysing ecological networks. R News 8/2, 8–11 (2008).
Casanelles-Abella, J. et al. A dataset of the flowering plants (Angiospermae) in urban green areas in five European cities. Data Brief. 37, 107243 (2021).
Faegri, K. & van der Pijl, L. in Principles of Pollination Ecology 3rd edn (eds Faegri, K. & Van der PiJl, L.) 88–95 (Pergamon, 1979).
Laliberté, E., Legendre, P., Shipley, B. & Laliberté, M. Measuring functional diversity from multiple traits, and other tools for functional ecology. R package version 1.0.12.1 (2014).
Oksanen, J. et al. vegan: Community ecology package. R package version 2.6.2 (2016).
Zuur, A. F., Ieno, E. N., Walker, N. J., Saveliev, A. A. & Smith, G. M. Mixed Effects Models and Extensions in Ecology with R (Springer, 2009).
Pinheiro, J. C., Bates, D. M. & R Core Team. nlme: Linear and nonlinear mixed effects models. R package version 3.1.160 (2022).
Zuur, A. F., Ieno, E. N. & Elphick, C. S. A protocol for data exploration to avoid common statistical problems. Methods Ecol. Evol. 1, 3–14 (2010).
Lüdecke, D. et al. performance: an R package for assessment, comparison and testing of statistical models. J. Open Source Softw. 6, 3139 (2021).
Barton, K. MuMIn: Multi-model inference. R package version 1.47.1 (2022).
Harrison, X. A. et al. A brief introduction to mixed effects modelling and multi-model inference in ecology. PeerJ 6, e4794 (2018).
Gelman, A. & Su, Y. arm: Data analysis using regression and multilevel/hierarchical models. R package version 1.13.1 (2022).
R Core Team. R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2022).
Bates, D., Maechler, M., Bolker, B. & Walker, S. Fitting linear mixed-effects models using lme4. J. Stat. Softw. 67, 1–48 (2015).
Hartig, F. DHARMa: Residual diagnostics for hierarchical (multi-level/mixed) regression models. R package version 0.4.6 (2022).
Maurer, C. et al. Species traits, landscape quality and floral resource overlap with honeybees determine virus transmission in plant–pollinator networks. Figshare https://doi.org/10.6084/m9.figshare.25101977 (2024).
World Topographic Map. ESRI www.arcgis.com/home/item.html?id=7dc6cea0b1764a1f9af2e679f642f0f5 (2024).
Acknowledgements
We thank S. Bossart and L. Bona for their help with the field work and plant species identification and E. Rabêlo for his help with the laboratory work. Furthermore, we are grateful to C. Grünig and J. Kast from Microsynth Ecogenics GmbH for conducting the barcoding. We thank J. M. Schwarz for her support with the drawing of the icons. We are grateful to all farmers and landowners for permission to work on their properties. The VOODOO (viral eco-evolutionary dynamics of wild and domestic pollinators under global change) project (https://voodoo-project.eu/) was funded through the 2018-2019 BiodivERsA joint call for research proposals, under the BiodivERsA3 ERA-Net COFUND programme and with the funding organizations Swiss National Science Foundation (SNSF) grant no. 31BD30_186532/1 (P.N. and M.A.); France, ANR-19-EBI3-0006 (A.J.V.); Germany, German Research Foundation (DFG) PA 623/10-1, PA 623/12-1 (R.J.P.), Federal Ministry of Education and Research (BMBF) and German Aerospace Research and Technology Centre (as executing organization), grant no. 01LC1905A (O.S.); and Poland, NCN UMO-2019/32/Z/NZ8/00006 (H.S.).
Author information
Authors and Affiliations
Contributions
A.J.V., O.S., H.S., R.J.P., P.N., A.G., O.Y. and M.A. conceived the study and developed the sampling and laboratory methodological protocols. C.M. collected the data. A.S., O.Y. and C.M. carried out the molecular analyses and performed the laboratory work. C.M., M.A. and L.P. conceived the data analyses. C.M. and M.A. analysed the data. C.M. led the writing of the manuscript. All authors contributed to the writing and gave final approval for publication.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Ecology & Evolution thanks Scott McArt, Margarita Lopez-Uribe and Lena Wilfert for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Map of the study landscapes.
The maps are showing the study landscapes located in the northern lowlands of Switzerland (orange = intensive agricultural landscape; green = rural habitat mosaic landscapes; grey = urban landscapes). Raster maps (1 km radius) show the land cover classes for three example landscapes (orange = arable crops; pink = pollinator-friendly habitat; dark green = forest; light green = intensively managed grasslands; grey = urban space ( < 25 % green areas); blue = water bodies). Maps created using ArcGIS Pro software by Esri. ArcGIS Pro is the intellectual property of Esri and is used herein under licence. Copyright © Esri. All rights reserved. Additional credits for the map are listed in ref. 87.
Extended Data Fig. 2 Overview of virus loads in pollinators.
Displayed are boxplots of BQCV (A), DWV-A (B) and DWV-B (C) virus loads (log-transformed number of genome copies per individual) of all screened individuals per species in each landscape and sampling round. Wild bee and hoverfly species were screened when they were sampled with at least N = 7 individuals per landscape and sampling round (maximum N = 10 individuals per landscape and sampling round). Boxplots show the median, the box representing the first and the third quartile, and the whiskers extend from the hinge to the largest and lowest values, respectively, no further than 1.5 times the interquartile range from the hinge. Points show raw log-transformed data. Colours represent the different sampling rounds. (Green = April, yellow = May/June, blue = July; BQCV = Black Queen Cell Virus; DWV = Deformed Wing Virus; see Table S1 for full species names).
Extended Data Fig. 3 Best ranking models predicting viral prevalence in wild pollinators.
Averaged model estimates (that is the modelled slopes of the relationships) and 95% confidence intervals of explanatory variables in best models (ΔAICc < 2) explaining viral prevalence of BQCV (A, C) and DWV-B (B, D) in wild pollinators (n = 588 individuals). (A, B) display the results from modelling the effects of species traits and roles in the network (weighted betweenness (Betweenness), corolla length, specialisation d’, proportion of dish-bowl flowers among visited flowers (Dish flowers) and floral resource overlap with honeybees (Resource overlap)). (C, D) display the results from modelling the effects of landscape and pollinator community properties (Shannon diversity of flowers (H flowers), honeybee density (HB density), wild pollinator abundance (Poll. abundance) and percentage cover of pollinator habitat (Poll. hab. %)).
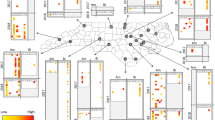
Extended Data Fig. 4 Example plant-pollinator network.
Plant-pollinator network of a landscape with 56% cover of pollinator-friendly habitat in July. Nodes and links are weighted by the abundance of the species and number of observed interactions, respectively. The colours of the pollinator species and links indicate their mean viral load (log-transformed) of black queen cell virus (BQCV copies; yellow = 0-400 BQCV copies; orange = 400-10000 BQCV copies; red = >10000 BQCV copies, grey = these species were not screened). Plant species coloured in dark green were visited by a pollinator carrying BQCV, while plant species coloured in light green were not visited by a pollinator carrying BQCV or a pollinator that was not screened for viruses.
Extended Data Fig. 5 Floral resource overlap on dish-bowl vs. non-dish-bowl flowers.
Model predictions and 95% confidence intervals of models (LMMs with landscape ID and species ID as random factors) testing the interactive effect of resource overlap between wild pollinators and honeybees on dish-bowl shaped flowers vs. on non-dish-bowl shaped flowers on virus load (A: BQCV or B: DWV-B). Orange solid line = dish-bowl flowers; blue dashed line = non-dish flowers. BQCV = black queen cell virus; DWV = deformed wing virus. Points show raw data.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Maurer, C., Schauer, A., Yañez, O. et al. Species traits, landscape quality and floral resource overlap with honeybees determine virus transmission in plant–pollinator networks. Nat Ecol Evol 8, 2239–2251 (2024). https://doi.org/10.1038/s41559-024-02555-w
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41559-024-02555-w
