
Abstract
Isogenic bacterial populations can display probabilistic cell-to-cell variation in response to challenges. This phenotypic heterogeneity can affect virulence in animals, but its impact on plant pathogens is unknown. Previously, we showed that expression of the type III secretion system (T3SS) of the plant pathogen Pseudomonas syringae displays phenotypic variation in planta. Here we use flow cytometry and microscopy to investigate single-cell flagellar expression in relation to T3SS expression, showing that both systems undergo phenotypic heterogeneity in vitro in apoplast-mimicking medium and within apoplastic microcolonies throughout colonization of Phaseolus vulgaris. Stochastic, spatial and time factors shape the dynamics of a phenotypically diverse pathogen population that displays division of labour during colonization: effectors produced by T3SS-expressing bacteria act as ‘common goods’ to suppress immunity, allowing motile flagella-expressing bacteria to increase and leave infected tissue before necrosis. These results showcase the mechanisms of bacterial specialization during plant colonization in an environmentally and agriculturally relevant system.
Similar content being viewed by others


Main
Flagellar motility is an important trait for environmental adaptation. Members of the genus Pseudomonas can colonize soil, plant or animal tissues. The Pseudomonas syringae species complex includes most of those pathogenic to plants, collectively causing diseases in over 500 species, many agriculturally relevant1. P. syringae can live on the leaf surface, but most favour endophytic colonization, using wounds and stomata to enter the leaf. Flagellar motility confers advantages to epiphytic populations of P. syringae pv. syringae2 and facilitates leaf entry for many P. syringae pathovars3,4. Inside the leaf, P. syringae colonizes the intercellular spaces (apoplast). Once inside, flagellar motility is not required for systemic spread3, while flagellin, the main component of the pilus, triggers immunity5. Fittingly, flagellar expression is downregulated in the apoplast6,7,8.
Flagellar biosynthesis is depicted as a deterministic programme with a complex and tiered regulatory hierarchy, where promoters are sequentially activated, maintaining this state through active growth. However, single-cell studies revealed stochastic activation pulses and bimodal patterns in the flagellar system of Salmonella enterica, with clonal populations displaying phenotypic heterogeneity9,10,11,12,13. In P. syringae, regulation of flagellar synthesis remains mostly uncharacterized, but gene arrangement and promoter conservation with P. putida supports a similar hierarchy of transcriptional regulation14, with FleQ at the top15.
Apoplastic P. syringae use a type III secretion system (T3SS) to introduce effectors (T3Es) into the host-cell cytosol. T3Es suppress plant immunity including flagellin-triggered responses, allowing bacterial proliferation and disease16. T3SS gene expression requires HrpL17. We showed that a ΔhrpL mutant displays increased swimming motility in apoplast-mimicking Hrp-inducing medium (HIM), while a mutant lacking HrpV, a repressor of hrpL expression18, displays reduced motility19, suggesting a regulatory link between T3SS and flagellar expression. Later, we established that P. syringae T3SS genes display phenotypic heterogeneity during plant colonization and bistable expression with reversible ON and OFF states in HIM20.
Here we show that flagellar gene expression displays phenotypic heterogeneity in P. syringae, particularly within the plant apoplast. While average expression levels of flagellin-encoding fliC is downregulated in the apoplastic population, as previously described6,7,8, a subpopulation expresses high fliC levels during colonization. Analysing flagellar and T3SS expression simultaneously, we found all possible single-cell ON/OFF combinations, revealing the phenotypic complexity arising amid clonal populations. Nonetheless, T3SSON/FlagellaOFF and T3SSOFF/FlagellaON subpopulations were generally more abundant, suggesting counter-regulation between these loci. Expression of these systems impacts bacterial growth at the population and/or cellular level. Analysis of the spatial distribution of expression within apoplastic microcolonies shows that T3SSON bacteria are more abundant early in the infection and close to the host-cell surface, while FlagellaON bacteria become more frequent later and further away from the host cell. Following bacterial apoplastic multiplication, FlagellaON bacteria actively exit the infected tissue before the onset of necrotic symptoms. Our results support a division of labour model with T3SSON secreted effectors acting as common goods, allowing FlagellaON bacteria to increase and exit before tissue collapse.
Results
Flagellar expression in P. syringae is heterogeneous
We generated a transcriptional fusion to GFP3 (AAB18957.1) of flagellin-encoding fliC (WP_002554301.1) within its genome location, preserving context and function without affecting fitness or virulence (Extended Data Fig. 1) in model bean pathogen P. syringae pv. phaseolicola 1448A21. Using confocal laser scanning microscopy (CLSM) and flow cytometry (FC), we monitored fliC::GFP3 expression in vitro (Fig. 1a). In rich lysogeny broth (LB) medium and apoplast-mimicking HIM medium, most bacteria express fliC::GFP3 (FlagellaON) with considerable cell-to-cell variation, but a subpopulation does not (FlagellaOFF) and can be visualized only by membrane staining, overlapping with non-fluorescent bacteria in FC analyses (Fig. 1a). Expression of GFP3 from a constitutive promoter is homogeneous22, ruling out expression noise as the fliC::GFP3 source of heterogeneity.

a, Left: CLSM images and FC analysis of bacteria carrying fliC::GFP3 grown in LB or HIM medium, or apoplast extracted from leaf apoplasts at 4 dpi. FM4-64 channel (greyscale) display fluorescence from membrane staining. Scale bars, 2 µm. Right: FC data obtained from fliC::GFP3 bacteria are represented using arbitrary units in logarithmic scale. Non-GFP graphs show autofluorescence levels of wild type as reference for OFF subpopulations (vertical lines leave 99% non-GFP data to the left). Microscopy and FC panels show typical results (n = 14 independent experiments). b, Mean GFP fluorescence intensity of fliC::GFP3-carrying bacterial populations (n = 14) from FC data described in a. c, Robust coefficient of variation (RCV) obtained from FC data described in a. b and c display individual sample values, and sample sets displaying different letters are significantly different as determined using one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparisons test (P < 0.05); all comparisons showed P < 0.0001. Lines indicate the means of all individual data. Error bars represent s.e.m. d, CLSM images of fliC::GFP3 bacteria expressing a constitutive eCFP gene growing in microcolonies in planta at 3 dpi. GFP channel shows heterogeneous distribution of fliC expression. CFP channel shows homogeneous distribution of CFP (n = 3). Red in all panels corresponds to chloroplast autofluorescence. Scale bars, 5 µm. In a and d, contrast and brightness were adjusted to improve visualization and kept constant across samples and channels. e,g, Histograms showing GFP fluorescence vs cell count from fliC::GFP3 overnight LB-grown cultures (e) or 24 h HIM-grown cultures (g) before (left) and after (right) sorting. Red lines indicate gating used to separate fliC::GFP3 bacteria according to fluorescence intensity. f,h, Relative ratio between halo diameters in swimming plates seeded with cells displaying high GFP expression, divided by the diameters of halos obtained from seeding cells displaying low GFP expression, resulting from the same sorting event (identified with different shades) from cultures grown in LB (f) or HIM (h) (n = 9 as shown). Lines indicate the means of all individual data. Error bars represent s.e.m. Asterisks indicate significant differences established by an unpaired two-tailed t-test (P < 0.05). P values are indicated.
In keeping with flagellar role in facilitating leaf entry2,3,4, leaf-surface bacteria are mostly FlagellaON. Nonetheless, images of bean (Phaseolus vulgaris) leafs sprayed with fliC::GFP3 bacteria constitutively expressing eCFP show that flagellar expression is also heterogeneous on leaf surfaces (Extended Data Fig. 2). Four days post pressure inoculation (dpi), bacteria recovered from the apoplast display lower average expression than media-grown populations (Fig. 1b), agreeing with transcriptomic data6,7,8. However, most apoplast-extracted bacteria are FlagellaON (Fig. 1a). Cell-to-cell differences in fluorescence were more pronounced among apoplast-extracted bacteria than in media, with a wider distribution revealed by FC (Fig. 1a (right),c). This variability is also apparent in planta, with apoplastic microcolonies of fliC::GFP3 eCFP bacteria showing heterogeneous GFP3 fluorescence overlaid with homogeneous eCFP fluorescence distribution (Fig. 1d). Homogeneous eCFP expression was reported previously23.
LB populations were sorted by fluorescence-activated cell sorting based on GFP fluorescence (Fig. 1e), enriching populations with low or high fliC::GFP3 expression (Fig. 1e right) to use in swimming assays. Since sorting causes physical flagellar damage, it can reduce motility differences between sorted subpopulation pairs and increase variation between sorting events. Thus, we compared motility between high/low subpopulations separated in the same sorting event (Fig. 1f): we calculated the ratio between swimming halo diameters of high GFP3- and low GFP3-expressing subpopulations (per sorted event). High-to-low motility ratios for sorted LB populations were significantly higher than 1.0, indicating that fliC expression positively correlates with motility (Fig. 1f). Similar results were obtained using HIM populations (Fig. 1g,h). These results indicate that flagella are phenotypically heterogeneous in P. syringae and show that, despite flagellin being an immune elicitor, part of the apoplastic population expresses flagella.
Flagella and T3SS show counter-regulation at the cellular level
Next, we used a strain carrying transcriptional fusions fliC::tdT (to tdTomato; AAV52169 1) and hrpL::GFP3 (WP_004656240.1) to evaluate T3SS and flagella expression simultaneously. Population profiles for fliC::tdT are similar to those of fliC::GFP3 (Extended Data Fig. 3a,b). Potential interferences between fluorophores (that is, bleedthrough leading to false positives or Förster resonance energy transfer (FRET) leading to false negatives) were ruled out (Extended Data Fig. 3c,d). Both systems were expressed heterogeneously in HIM (Fig. 2a,b (left) and Extended Data Fig. 3e). FC showed two subpopulations, formed by bacteria favouring expression of T3SS (T3SSON/FlagellaOFF) or fliC (T3SSOFF/FlagellaON) (Fig. 2a,b (left)). Although these combinations were more abundant, a continuous distribution with bacteria expressing both (T3SSON/FlagellaON) or none (T3SSOFF/FlagellaOFF) was observed (Fig. 2a,b (left) and Extended Data Fig. 3e). Apoplast-extracted bacteria (4 dpi) also displayed all phenotypic combinations, although T3SSON/FlagellaOFF bacteria were more abundant (Fig. 2a,b (right)). As reported20, apoplastic populations did not display bimodal GFP distribution (Fig. 2a,b and Extended Data Fig. 3f). Analysis of T3SS effector gene hopAB1 (ref. 20)(WP_011282445.1) rendered similar results (Fig. 2c,d and Extended Data Fig. 3e,f).

a, CLSM images of dual-reporter hrpL::GFP3 fliC::tdT strain grown in HIM or extracted from leaf apoplasts at 4 dpi. Scale bars, 2 μm. Contrast and brightness were adjusted to improve visualization and used throughout. b, FC analysis of bacteria described in a. Graphs show GFP fluorescence intensity vs that of tdTomato. Results shown in the figure are representative of 3 or more independent experiments. c, CLSM images of dual-reporter hopAB1::GFP3 fliC::tdT strain grown in HIM or extracted from leaf apoplasts at 4 dpi. Scale bars, 2 μm. Contrast and brightness were adjusted to improve visualization and used throughout. d, FC analysis of bacteria described in c. Results shown in the figure are representative of at least 3 independent experiments.
Expression of T3SS or flagella carries growth penalties
Expression of Salmonella pathogenicity island 1 (SPI1) T3SS causes growth delays, which have been proposed to drive phenotypic heterogeneity24,25. We used competitive index (CI) assays (Fig. 3a) to compare growth of a ΔhrpL mutant and wild-type bacteria (Fig. 3b). CIs show that ΔhrpL mutants outgrow wild-type bacteria in HIM (CI = 1.75 ± 0.15), but not in rich medium (CI = 1.07 ± 0.08), where T3SS genes are not expressed. Growth rates of ΔhrpL bacteria are also higher in HIM (Fig. 3c). In planta, wild-type bacteria outgrow a derivative constitutively expressing HrpL (CI = 0.67 ± 0.06) (Fig. 3d). Constitutive expression of HrpL results in overexpression of T3SS genes20, supporting that T3SS expression carries a growth penalty.

a, Left: competitive index assay overview: inocula containing 1:1 mixes of wild-type and mutant (or plasmid-carrying) LB-grown cultures are used to inoculate leaves (in planta CI) or to start fresh cultures (in vitro CI). Resulting mixed cultures and apoplast-extracted bacteria (4 dpi) are serially diluted and plated with or without antibiotics for c.f.u. determination (output). Right: CI is calculated as mutant-to-wild-type c.f.u. ratio in output divided by ratio in input. CIs different from 1.0 indicate strains growing significantly differently: mutant outgrowing wild type (CI > 1.0) or vice versa (<1). b, CIs for mutants vs wild-type strain (WT) in HIM or LB (n = 3–9 as shown). c, HIM growth rates for ΔhrpL mutant and wild type. d, In planta CIs for wild type constitutively expressing hrpL (pHrpL) or ΔfliC, vs WT at 4 dpi (n = 10 as shown). Plants for ΔfliC CI pretreated with flg22. In b–d, data show mean ± s.e.m. Individual data for each biological replicate shown; different shades identify independent experiments, and results with asterisks are significantly different from 1.0 (non-parametric two-sided Student’s t-test). Significant P values indicated. e, Time-lapse images of fliC::GFP3 bacteria growing on agar pads: phase-contrast channel (top), GFP fluorescence (bottom). Contrast and brightness adjusted to improve visualization and kept constant across frames. f, Correlation between growth rates and fluorescence intensity of individual fliC::GFP3 cells. Each point represents a single cell, with fliC expression and growth rate averaged over cell lifetime. Grey-shaded area shows 95% confidence interval around linear regression model fit. Correlation tested with Pearson’s correlation coefficient using two-sided test (R = −0.17; P = 1.03 × 10−9). g, Growth rates for high-GFP- and low-GFP-signal cells (determined by splitting the population using median fluorescence intensity) show significantly slower growth in high-GFP cells (two-sided Mann–Whitney U-test, P = 3.6 × 10−8). Data presented as boxplots with first and third quartiles as bottom and top bounds, median shown as horizontal line in the box, and 1.5 interquartile range shown as whiskers. Outliers not shown. Grey violin plot shows distribution of all data points summarized in boxplot. White diamonds show mean growth rate for all cells considered.
Flagella often carries growth penalties26,27,28. However, previous CI assays did not show significant growth differences for ΔfliC bacteria in P. syringae29. Fittingly, we did not observe significant growth differences for ΔfliC or ΔfleQ (WP_011169094.1) (lacking flagellar regulator FleQ) mutants based on CI (LB: CI = 1.08 ± 0.22 and 1.00 ± 0.13, respectively; HIM: CI = 1.21 ± 0.22 and 1.31 ± 0.13, respectively), although variation in LB ΔfliC CI was large (Fig. 3b), or growth rates (Extended Data Fig. 4a). However, a ΔhrpL ΔfliC mutant remarkably outgrows wild type, with CI values (LB: CI = 4.195 ± 0.9975: HIM: CI = 25.79 ± 5.156) 4–15 fold those obtained for single ΔhrpL mutant (Fig. 3b). This supports the notion that flagellar expression carries a cost, at least when simultaneously expressed with the T3SS. Constitutive expression of fleQ from pFleQ led to severe growth delays (Extended Data Fig. 4a), although pleiotropic effects could not be ruled out given the severity of the phenotype.
In plants treated with flagellin to void differences associated with flagellin-mediated immunity, CI assays revealed growth advantages for ΔfliC mutants (CI = 1.88 ± 0.34) (Fig. 3d). However, we cannot rule out that ΔfliC mutant advantages in flagellin-treated plants may stem from other reasons.
Since T3SS and flagellar expression are both highly heterogeneous, population-level assays could be noisy and may lead to underestimating growth penalties. Thus, we analysed these impacts at the single-cell level using HIM-agar pads and time-lapse microscopy. In this setting, hopAB1::GFP3 activation takes place too late in the experiment to allow for confident quantification (Supplementary Videos 1–4). However, activation of fliC::GFP3 takes place earlier (Supplementary Videos 5 and 6, and Fig. 3e). Time-lapse analyses show that fliC expression negatively correlates with bacterial growth (Fig. 3f). Splitting the fliC::GFP3 population into two using the median fluorescence intensity value, we detected a 6.5% growth penalty for the higher-than-median-fluorescence half compared with the lower half (Fig. 3g). Constitutive expression of eGFP does not negatively correlate with growth (Extended Data Fig. 4b–f).
Phenotypic specialization during microcolony development
Apoplastic FlagellaON/T3SSOFF subpopulations are potentially more vulnerable to plant defences since they trigger but cannot suppress immunity. We used propidium iodide which binds to membrane-compromised (not viable) bacteria to determine any correlation between phenotypic variants and apoplast killing rates30,31. No significant differences were observed between dead/live ratios of Flagella or T3SS ON vs OFF subpopulations (Extended Data Fig. 5), supporting that FlagellaON/T3SSOFF cells are protected from plant responses, perhaps by T3SSON bacteria suppressing defences in trans.
We established that T3SS mutant bacteria may proliferate up to wild-type levels when growing within the same microcolony as wild-type bacteria22, while wild-type and T3SS mutant bacteria growing in separate microcolonies develop according to their respective abilities to suppress local immunity. Therefore, trans complementation between T3SSON and FlagellaON/T3SSOFF bacteria would require phenotypic variants to rise during growth of the microcolony. Fluorescence distribution of fliC:GFP3 in planta shows that for flagella, ON/OFF variants appear closely located within the microcolony (Fig. 1d). Close examination of fliC::GFP3 distribution (Supplementary Video 7) reveals areas with stronger fluorescence, suggesting a response to local stimuli (or siblings’ inheritance of an expression pattern), but also isolated bacteria displaying GFP fluorescence strikingly different from those of nearby peers, in keeping with stochastic differences.
We followed expression of T3SS and flagella genes during the development of apoplastic microcolonies, analysing leaves infiltrated with hopAB1::GFP3 fliC::tdT or hrpL::GFP3 fliC::tdT bacteria at different timepoints (Fig. 4a,b). Stochastic T3SS heterogeneity was detected throughout, while zonal distribution of flagellar and T3SS fluorescence within the microcolonies changed over time. One-dpi microcolonies displayed heterogeneous expression of T3SS, but no flagellar expression, supporting the idea that flagella is turned off in early infection stages6,7,8. Two-dpi microcolonies displayed orange and yellow tones consistent with expression of both systems. By 3–4 dpi, predominantly red (fliC::tdT) or green (hopAB1::GFP3 or hrpL::GFP3) zones with thoroughly overlaid heterogeneity becomes the norm (Fig. 4a,b). Zones with higher expression of hopAB1::GFP3 generally display lower expression of fliC::tdT and vice versa, although there are also orange and yellow spots indicative of T3SSON/FlagellaON bacteria (Fig. 4a,b). Most remarkably, this zonal distribution shows a consistent spatial pattern: bacteria closest to the lower epidermis (Fig. 4c–e), or to a spongy mesophyll host cell (Fig. 4a (right),f,g), are predominantly green, that is, T3SSON, whereas those further away from the host-cell surface, that is, the innermost or furthermost parts of the microcolonies, are predominantly red, that is, FlagellaON (Fig. 4, and Supplementary Videos 8 and 9). Although the T3SSON zone in the proximal side varies in width, it is consistently wider than a single bacterial layer, supporting the idea that the signal(s) activating T3SS expression include diffusible molecules.

a, CLSM Z-stack compilation at different dpi showing apoplastic microcolonies of hopAB1::GFP3 fliC::tdT bacteria. All images examined (n = 5 or more) per biological replicate (n = 2) and independent experiment (n = 2 for 1 and 2 dpi; n = 4 for 3 and 4 dpi). b, Images from a including orthogonal projections. The colored lines within the central images indicate the planes that generate each orthogonal section: green lines indicate the section that is displayed in the green-boxed images (images above per time point), red lines indicate the section that is displayed in the red-boxed images (images on the right per time point). In green- or red-boxed images, the blue line correspondes to the section displayed in the central image. Scale bars, 5 µm. c, Schematic leaf 3D structure with close-up showing location of apoplastic microcolonies. Position and orientation of Z-stack acquisition are indicated; 1–3 represent three planes of a bacterial microcolony growing from the lower epidermis inner surface inwards (1 closest to cell surface and 3 furthest) as shown in d and Supplementary Video 8. Plane 4 cuts through a bacterial microcolony growing wrapped around a spongy mesophyll cell (SMC) (as microcolony and frame shown in f and Z-stack shown in Supplementary Video 9). d, Frames of a Z-stack acquisition taken from an apoplastic microcolony of hrpL::GFP3 fliC::tdT bacteria at 4 dpi. Contrast and brightness were adjusted to improve visualization and kept constant throughout. Scale bars, 10 µm. Lower epidermis and mesophyll indicate relative position of frames within the leaf. e, Schematic distribution of fluorescence for fliC::tdT and T3SS::GFP3 within the type of apoplastic microcolonies indicated in c (1–3) and shown in d: planes closest to lower epidermis (abaxial side) show predominantly green bacteria and those furthest predominantly red, with intermediate plane displaying yellow. Heterogeneity is observed within each plane. f, Z-stack frame taken from an apoplastic microcolony of hopAB1::GFP3 fliC::tdT bacteria (Z-stack in Supplementary Video 9) from experiments described in a. Scale bars, 20 µm. g, Schematic fluorescence distribution for fliC::tdT T3SS::GFP3 bacteria within apoplastic microcolonies of the type indicated in c (4) and shown in f: bacteria closest to the cell are predominantly green, turning to yellow and red as they get further away. Heterogeneity is displayed throughout. In e and g, legend indicates green as T3SSON/ FlagellaOFF bacteria, red as T3SSOFF/ FlagellaON and yellow/orange as bacteria expressing both to different levels.
FlagellaON bacteria actively exit infected tissues
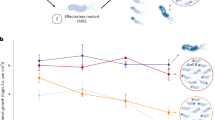
The increase of FlagellaON bacteria in older microcolonies suggests a function at later infection stages. We took leaves at 1 dpi, 2 dpi (asymptomatic), 5 dpi (onset of macroscopic symptoms) and 7 dpi (fully symptomatic) and submerged them into a sterile MgCl2 solution for 30 min. We used CLSM to evaluate hopAB1::GFP3 and fliC::tdT expression in those bacteria found in the solution. Bacteria exited even intact 1 dpi leaves. Shorter incubations also allowed bacterial recovery but not enough for reliable quantification. We compared T3SS or fliC expression between bacterial populations exiting the leaf and whole-leaf populations obtained by mechanical extraction from replicate leaves without incubation (Fig. 5 and Extended Data Fig. 6). Exiting and total apoplast-extracted populations showed similar hopAB1::GFP3 expression profiles at 1 dpi, 2 or 7 dpi (Fig. 5a,b and Extended Data Fig. 6), while significantly fewer T3SSON bacteria were detected in the exiting fraction by 5 dpi (Extended Data Fig. 6). In contrast, bacteria exiting 1 dpi leaves were significantly enriched in FlagellaON (Fig. 5a,b), even though total apoplast-extracted bacterial populations showed almost no FlagellaON bacteria (Figs. 4 and 5a, and Extended Data Fig. 6). This supports the notion of FlagellaON bacteria being more efficient in actively exiting the leaf in humid conditions. We confirmed this notion using an experimental setting mimicking rain or heavy dew, by spraying the infected leaves every 5 min for 30 min and collecting the dripping solution for microscopic examination (Fig. 5a bottom panels). The bias towards FlagellaON bacteria in the exiting populations detected at 2 and 5 dpi and progressively decreasing until they are no longer detected at 7 dpi (Fig. 5a,b and Extended Data Fig. 6), when leaf integrity has been lost, suggests that flagellar motility is not only important for bacterial entry into the leaf but also for fast and active exit of apoplast-grown bacteria before necrosis.

a, Selected images of apoplast-extracted bacteria (using negative pressure; top panels), bacteria exiting the tissue on their own (during 30 min leaf incubation within an MgCl2 solution; middle panels) or bacteria recovered from excess solution dripping from leaves sprayed every 5 min to mimic rain (bottom panels). Contrast and brightness were adjusted to improve visualization but were kept constant across the different conditions. b, Fluorescence quantification of the images obtained in a (left images) at 1 dpi (left graphs) and at 7 dpi (right graphs) of the same time-course experiment, using Fiji software. Graph shows arbitrary units of GFP fluorescence corresponding to the expression of the T3SS gene fusion hopAB1::GFP3, or tdTomato fluorescence corresponding to the expression of fliC::tdT. Each dot corresponds to an individual bacterium as analysed from the image (n = 44). Data are presented as mean ± s.e.m. Graphs show results representative of 5 independent experiments (n = 2 for strain carrying hrpL::GFP3 fliC::tdT and n = 3 for strain carrying hopAB1::GFP3 fliC::tdT). Comparisons between apoplast extraction and natural exit were carried out per sample using an unpaired t-test (****P < 0.0001) and results shown for each pair.
Discussion
Bacterial encounters with environmental changes lead to gene activation often assumed to happen in a non-fluctuating homogeneous manner. However, studies averaging population data lose cell-to-cell variation, and an increasing number of traits have been shown to manifest differently across clonal populations. Such phenotypic variation may reflect microenvironmental differences but may also be caused by stochastic cellular changes (gene expression noise) and particular regulatory circuits (enhancing noise)32.
P. syringae produce polar flagella33,34 upon activation of FleQ15 under a regulatory cascade that is mostly uncharacterized. Gene arrangement and promoter motifs conservation with P. putida14, which also produces polar flagella, support a similar organization. Nonetheless, we show that P. syringae flagellar regulation shares an important feature with that of peritrichous Salmonella: stochastic cell-to-cell variation (Fig. 1 and Extended Data Figs. 1–3)9,10,11,12,13,35,36. Bistable activation of Salmonella flagella is established by two mechanisms9,10,11,12,13: (1) a double-negative feedback loop and (2) a developmental checkpoint. A dual mechanism is also involved in turning P. syringae T3SS noisy expression into bimodality20: (1) a double-negative18,37 and (2) a positive feedback loop38. Research into the regulation of flagellar synthesis in P. syringae will help identify elements involved in phenotypic heterogeneity.
Flagellar motility is often costly26,27,28. Flagellar expression also has a cost in P. syringae (Fig. 3 and Extended Data Fig. 4), although this cost is clearer at the single-cell level (Fig. 3e–g). In Salmonella, costs linked to expression of SPI1 T3SS have been proposed as a factor favouring both counter-regulation with flagella and maintenance of heterogeneity24,25. T3SS expression also carries a growth penalty in P. syringae (Fig. 3), and the strong growth advantage of a ΔfliC ΔhrpL double mutant indicates an even greater fitness cost associated to expression of both. Phenotypic heterogeneity is considered particularly advantageous when affecting loci producing immunogenic and/or energetically costly goods, with cost to the producers and benefits to the population defining a cooperative behaviour39,40,41. The growth penalties detected for flagellar and T3SS expression in P. syringae provide selective advantages to phenotypically heterogeneous populations and may drive selection of genetic and/or epigenetic mechanism(s) underlying such heterogeneities. For bacteria engaging in host interactions, heterogeneous expression lowers the overall amount of flagellin displayed by the population, or by a microcolony at a local level, and could be also advantageous by reducing defence elicitation and facilitating immune suppression.
Cooperative virulence can also prevent the rise of non-producing mutant variants at the expense of the producers42,43,44,45. Phenotypically OFF bacteria can outcompete less frequent mutant OFF variants and safely revert to ON, thus preventing loss of function and stabilizing the cooperative behavior43. Cheater mutant variants affected in type III secretion have been described in P. syringae populations colonizing Arabidopsis, although at frequencies lower than expected considering the potential for exploitation of T3SS effectors as public goods22,46. This role of T3SS effectors as public goods has been reported using mutants32 and metapopulations, where mixed isogenic P. syringae derivatives lacking effectors, and each overexpressing a single effector, cooperatively restore virulence46.
Phenotypic heterogeneity can also allow bacterial populations to cope with rapid environmental changes, since preexisting subpopulations preadapted to incoming stresses can overcome these changes faster39,40. This is known as ‘bet-hedging’41. Phenotypic heterogeneity can also lead to ‘division of labour’ when a population diversifies, allowing the distribution of tasks among phenotypically different variants. While in bet-hedging, one subpopulation is fitter than the other in each environment, division of labour increases fitness for the entire population41. We found no evidence of any T3SS/flagellar phenotypic variant benefiting during plant colonization, as expected in a bet-hedging scenario. However, different defence elicitation contexts (for example, resistant plants) might differentially benefit certain phenotypes, as shown for genome-reorganized variants47,48. Our results support a ‘division of labour’ scenario, with T3SSON bacteria complementing T3SSOFF probably through suppressing immunity by ‘common goods’ T3SS effectors22 (Fig. 4 and Extended Data Fig. 5). The percentage of T3SSON bacteria required for effective trans complementation of T3SSOFF, since a subpopulation is expressing immunogenic flagellin, is likely to be balanced by growth trade-offs.
As eukaryotic cell types work within tissues, bacterial cooperation emerges within spatially structured populations49. However, structured cooperation has been rarely shown within the context of disease50. Indeed, although phenotypic heterogeneity is well documented in planktonic cultures, how these processes occur in spatially structured communities is less understood51. The regulatory loops involved in establishing bistable expression of the T3SS in planktonic HIM20 cultures are in place during plant colonization19,52. However, apoplastic bacteria encounter different stimuli specific to their microenvironment, which changes over time, potentially leading to cell-to-cell variation orthogonal to that generated through T3SS and flagellar stochastic heterogeneity. Time-course analysis of T3SS and flagellar expression during plant colonization shows heterogeneity throughout time and locations, but also an overlapping structured pattern (Fig. 4). On the leaf surface, most bacteria are T3SSOFF regardless of the single-cell status of flagellar expression, changing to a majority of T3SSON/ FlagellaOFF during early growth in the apoplast (Fig. 4a,b, 1 dpi). From 2 dpi onwards, heterogeneity is the norm for both loci, but predominantly T3SSON or FlagellaON areas appear. These areas show a three-dimensional (3D) pattern: those closely associated to host-cell surfaces are enriched in T3SSON bacteria, while distal areas, or the innermost parts of the microcolony, are predominantly FlagellaON (Figs. 4 and Extended Data Fig. 7, and Supplementary Videos 4 and 5). This is consistent with the notion of T3SSON bacteria predominantly located where T3SS activity is relevant to suppress cellular immunity and explains the lack of selective killing of T3SSOFF bacteria26 (Extended Data Fig. 5). Such phenotypic differentiation into spatially distributed subpopulations cooperating in a complex environment (that is, in planta vs in vitro) has rarely been investigated, particularly for more than a trait53, but it is reasonable to expect it to take place in the context of host colonization processes. Spatially structured cooperation in the context of disease has been demonstrated for cholera toxin and toxin-coregulated pilus in Vibrio cholerae in mice intestinal microcolonies, and for nitric oxide (NO)-detoxifying hmp gene in Yersinia pseudotuberculosis in spleen microcolonies49,53. Spatial heterogeneity has also been described through single-cell omics within in vitro biofilms of human pathogen P. aeruginosa, including variation in flagellar expression54. Host-cell signals, cell-to-cell interactions or lineage history55 could be involved in T3SS activation in host-proximal areas. How environmental signals combine with stochastic processes to generate phenotypic variation is a current topic of interest55, one particularly challenging to study beyond in vitro systems if positional information is to be obtained55.
As apoplastic microcolonies develop, FlagellaON bacteria increase in numbers. Motile bacteria are expected to exit the leaf more efficiently (Extended Data Fig. 7), and our results experimentally support this notion by showing that mostly FlagellaON bacteria exit colonized tissue before necrosis (Fig. 5). The final stages of the infection and host exit is an understudied domain, even for model pathogen P. syringae, so whether this active early exit is more biologically relevant than passive exit from necrotic tissue remains to be explored, but it appears a safer strategy since necrosis leads to the release of toxic compounds and to the activation of additional defence responses56,57.
Only a handful of studies have analysed expression of two bistable heterogeneous loci simultaneously49,53,58. Ours studies two loci relevant for bacterial–host interaction (Supplementary Video 10) and does it in the context of disease within a spatially structured dynamic microcolony. It provides an example of division of labour and cooperative virulence in a plant host, validating the importance of phenotypic heterogeneity in nature and shifting the emphasis from its mechanistic understanding to its ecological and biological relevance. Finally, the study establishes phenotypic heterogeneity and cooperative virulence as a conserved strategy by which bacterial pathogens cope with the fluctuating challenges encountered in plant and animal hosts.
Methods
Bacterial strains and growth conditions
Bacterial strains used and generated in this work are detailed in Supplementary Table 1. E. coli and P. syringae strains were grown with aeration in LB medium59 at 37 °C for E. coli or 28 °C for P. syringae. Antibiotics were used, when necessary, at the following concentrations: ampicillin, 100 μg ml−1 for E. coli and 500 μg ml−1 for P. syringae; kanamycin, 50 μg ml−1 for E. coli and 15 μg ml−1 for P. syringae derivative strains; gentamycin, 10 μg ml−1; nitrofurantoin, 40 μg ml−1; and cycloheximide, 2 μg ml−1.
To induce the expression of the hrp/hrc genes, bacteria were initially cultured overnight in LB at 28 °C, supplemented with the appropriate antibiotic, then washed twice in 10 mM MgCl2 before being cultured in hrp-inducing minimal medium (HIM), containing 10 mM fructose60. For this study, the pH of HIM was adjusted to 7.0 with 10 N NaOH. The initial optical density (OD) was adjusted to 0.13 and cultures were incubated at 28 °C with agitation.
Fluorescence labelling of bacterial strains
Bacterial strains carrying a chromosome-located transcriptional fusion of fliC gene to a promoterless tdTomato gene were generated using an adaptation of the method previously described in ref. 61. The plasmids used and generated for this purpose are detailed in Supplementary Table 2. The primers used are described in Supplementary Table 3. For the generation of the allelic exchange plasmid, two fragments of ~500 pb were amplified from Pph 1448A genomic DNA using Q5 High-Fidelity DNA Polymerase (New England Biolabs); one of these fragments (A) encompasses the 3′ end of the open reading frame (ORF), including the STOP codon, while the other fragment (B) covers the sequence immediately downstream of the STOP codon. All primers used are described in Supplementary Information. Each reaction was carried out at 98 °C for 1 min for the initial denaturation step, followed by 30 cycles at 98 °C for 30 s, annealing at 62 °C for 30 s and extension at 72 °C for 30 s, followed by 5 min at 72 °C for the final extension step. The reaction mixture for each PCR included 0.64 mM deoxynucleotide triphosphate (dNTP) mix, 0.4 ng of each primer, 1 ng of genomic DNA, the appropriate enzyme buffer and commercial ultrapure water (Nalgene). A volume of 2 μl of each gel-purified PCR product was employed as template for the subsequent fusion PCR, employing primers A1 and B2 in a PCR reaction conducted under the conditions described, with an extended elongation time of 1 min. The resulting bands, comprising the end of each ORF and its downstream sequence separated by an EcoRV restriction site, were A/T cloned into pGEM-T (Promega) and subjected to full sequencing to discard clones carrying mutations. This process rendered the pGT-AB-fliC plasmid necessary for generating the allelic exchange plasmid.
The sequence of the promoterless tdTomato (tdT) gene was acquired through PCR amplification from the tdTomato-pBAD plasmid (Addgene plasmid 54856) using the ProtFluorF and ProtFluorR primers. This PCR generated a tdTomato fragment with a 5′ EcoRV restriction site and a 3′ EcoRI restriction site. Similarly, the kanamycin cassette (nptII; EJQ5150444.1) was amplified using the P1 EcoRV and P2 EcoRI primers, and plasmid pKD4 served as DNA template to obtain the FRT-nptII-FRT fragment needed for the resistance to kanamycin. This fragment featured a 5′ EcoRV and 3′ EcoRI restriction site. Both amplification reactions were performed using Q5 High-Fidelity DNA Polymerase (New England Biolabs) in a 50-µl PCR reaction mixture consisting of 500 ng of plasmid as DNA template, 0.5 µM of each primer, 0.2 mM dNTPs and 0.5 µl of Q5 High-Fidelity DNA Polymerase. Each reaction followed a thermal cycling protocol that began with an initial step of 98 °C for 2 min, followed by 30 cycles at 98 °C for 10 s, 60 °C for 30 s and 72 °C for 45 s, concluding with a final extension at 72 °C for 5 min. The gel-purified fragments, both the tdTomato and the FRT-nptII-FRT, underwent digestion with EcoRI and EcoRV enzymes and were cloned into the EcoRV-digested pGT-AB-fliC plasmid, rendering the allelic exchange plasmid to be named pGT-fliC::tdT.
Strain 1448A fliC::tdT was obtained by introducing the pGT-fliC::tdT plasmid into P. syringae pv. phaseolicola 1448A through electroporation, following the method described in ref. 61. Selection was carried out on LB plates with kanamycin. Subsequently, the resulting colonies were replicated onto LB plates with ampicillin (500 μg ml−1) to discard the colonies with plasmid integration, which is indicative of a single recombination event. The colonies that exhibited kanamycin resistance but ampicillin sensitivity were then confirmed through PCR using the A1 fliC and B2 fliC primers, and through Southern blot analysis employing the nptII gene as a probe to confirm proper allelic exchange resulting from a double recombination event occurring at a unique position within the genome. To generate strains carrying two chromosomal transcriptional fusions, the pGT-fliC::tdT plasmid was transformed into the previously generated strains 1448A hrpL::GFP3, 1448A hopAB1::GFP3 and 1448A hrcU::GFP3, to generate the 1448A hrpL::GFP3 fliC::tdT, 1448A hopAB1::GFP3 fliC::tdT and 1448A hrcU::GFP3 fliC::tdT strains.
Bacterial strains carrying a chromosome-located transcriptional fusion of fliC gene to a promoterless GFP3 gene, 1448A fliC::GFP3 strains, were generated following a modified method62. The GFP3-FRT-nptII-FRT fragment was obtained by digesting the plasmid pGT-GFP+ with the EcoRI digestion enzyme. This fragment consists of the promoterless GFP3 gene, complete with its ribosomal binding site, followed by the kanamycin resistance gene (nptII) flanked by flipase recognition target (FRT) sites, with the entire construct bordered by two EcoRI restriction sites. The GFP3-FRT-nptII-FRT fragment was then blunt ended through a PCR procedure and ligated into EcoRV-digested pGT-AB-fliC through blunt-end ligation, leading to the generation of the pGT-fliC::GFP3 plasmid. Subsequently, the resulting plasmid was transformed into P. syringae pv. phaseolicola 1448A to generate the 1448A fliC::GFP3 strain.
In addition, the constitutively expressed fluorescent reporter gene eCFP was introduced into the chromosome of the 1448A fliC::GFP3 and 1448A hopAB1::GFP3 fliC::tdT strains using a Tn7 delivery system23 to generate the 1448A fliC::GFP3 eCFP and 1448A hopAB1::GFP3 fliC::tdT eCFP strains.
Generation of mutant bacterial strains
The bacterial strain carrying a deletion of the fleQ gene was generated following the method described in ref. 61, which involves the generation of gene knockouts by allelic exchange, replacing the specific ORF by a kanamycin cassette. The allelic exchange plasmid pGT-ΔfleQ was generated as previously described for the generation of the pGT-AB-fliC using primers A1 ΔfleQ, A2 ΔfleQ, B1 ΔfleQ and B2 ΔfleQ, and the same experimental settings described above. The FRT-nptII-FRT fragment was obtained by PCR amplification using P1 EcoRI and P2 EcoRI primers and pKD4 as template, and the kanamycin cassette was finally inserted by ligation in the EcoRI restriction site, generating the allelic exchange plasmid pGT-ΔfleQ. This plasmid was transformed into 1448A Pseudomonas syringae pv. phaseolicola and mutants were obtained as described in ref. 61.
The bacterial strain carrying a deletion of the hrpL gene and of the fliC gene was generated by allelic exchange as previously described, transforming the plasmid pGT-∆fliC described in ref. 29 into the ∆hrpL strain described in ref. 19 to render ∆hrpL ∆fliC double-mutant strain.
Generation of pFleQ
pFleQ was generated using the backbone of pBBRMCS-4. For its generation, the fleQ ORF was amplified using Q5 High-Fidelity DNA Polymerase (New England Biolabs) in a 50 µl PCR reaction mixture consisting of 500 ng of 1448A Pseudomonas syringae genome as DNA template, 0.5 µM of fleQF and fleQR primers, 0.2 mM dNTPs and 0.5 µl of Q5 High-Fidelity DNA Polymerase. The reaction followed a thermal cycling protocol that began with an initial step of 98 °C for 2 min, followed by 30 cycles at 98 °C for 10 s, 60 °C for 30 s and 72 °C for 45 s, concluding with a final extension at 72 °C for 5 min. The gel-purified fragment underwent digestion with KpnI and SacII enzymes, and was cloned into the KpnI/SacII-digested pBBRMCS-4 plasmid. The resulting clones were subjected to full sequencing to discard those carrying mutations, and transformed into the corresponding bacterial strains using the method described in ref. 61 for plasmid transformation of P. syringae.
Plant growth and inoculation
Phaseolus vulgaris bean cultivar Canadian Wonder plants were cultivated under controlled conditions at 23 °C and 95% humidity. Artificial light was maintained for periods of 16 h within the 24 h of the day. All experiments carried out were performed using 10-day-old plants.
For the preparation of bacterial inoculum, bacterial lawns were cultivated onto LB plates for 48 h at 28 °C. Subsequently, biomass was resuspended in 2 ml of 10 mM MgCl2. The OD600 was adjusted to 0.1, corresponding to the concentration of 5 × 107 colony-forming units per millilitre (c.f.u.s ml−1). Serial dilutions were performed to achieve the desired final inoculum concentration.
The infiltration of bean leaves for visualizing microcolonies using confocal microscopy was performed using a needleless syringe with bacterial suspension at 5 × 105 to 106 c.f.u.s ml−1.
The inoculation of bean leaves for visualizing bacteria on the surface using confocal microscopy was performed by dipping. For this, a bacterial suspension with 5 × 107 c.f.u.s ml−1 was prepared in a 10 mM MgCl2 solution, and the entire leaf was submerged in the inoculum for a few seconds. Visualization was performed at 6 h post inoculation.
For infiltrating bean leaves to extract bacteria from the apoplast for subsequent analysis by FC and CLSM, a previous report63 was followed. This involved immersing the entire leaf in a bacterial solution with a concentration of 5 × 104 c.f.u.s ml−1, containing 0.01% Silwett L-77 (Crompton Europe), and using a pressure chamber. Bacteria were recovered from the plant at 4 dpi through apoplastic fluid extraction. This extraction process63 entailed pressure infiltrating a whole leaf with 10 ml of a 10 mM MgCl2 solution inside a 20-ml syringe. After applying 5 cycles of pressure, the flow-through was collected and transferred to a fresh 50-ml tube. Three hundred microlitres of the flow-through were directly analysed by flow cytometry. Simultaneously, the 50-ml tubes were centrifuged for 30 min at low speed (900 g) at 4 °C. The resulting pellets were resuspended into 1 ml of a 10 mM MgCl2 solution and subsequently analysed by microscopy.
To compare flagellar expression in cells mechanically extracted from or naturally exiting leaves, bean leaves were infiltrated with a 5 × 107 (for 1 dpi), 5 × 106 (for 2 dpi) or 5 × 105 c.f.u.s ml−1 (for 5 and 7 dpi) bacterial suspension using a pressure chamber, as described above. For natural exit mimicking rain, leaves were sprayed with water every 5 min during a 30-min period, and drops flowing through the leaves were collected in a 50-ml cylinder tube. Bacteria were concentrated by centrifugation at 10,000 g for 5 min. For quantification at different timepoints, leaves were detached from the stem by cutting the petiole in the base of the leaf blade at the specified timepoints and incubated for 30 min in a 50-ml tube containing 30 ml of 10 mM MgCl2 to analyse natural exit. Mechanical bacterial extraction from the apoplast was carried out as described above.
Flow cytometry and cell sorting
For HIM cultures, 500 µl of an overnight P. syringae LB culture was washed twice in 10 mM MgCl2, added to 4.5 ml of HIM and incubated at 28 °C for 24 h. LB cultures were obtained from an overnight incubation in LB and apoplast-extracted bacterial suspensions were obtained as indicated in the ‘Plant growth and inoculation’ section. Of the cultures in HIM, LB or in planta, 300 µl were analysed using a BD FACS Verse cytometer (BD Biosciences), and graphs were generated with the Kaluza software (Beckman Coulter; https://www.beckman.es/flow-cytometry/software/kaluza). A minimum of 100,000 events were analysed per sample. FITC-A and PE-A filters were used to visualize GFP and tdTomato signals, respectively. To ensure bleedthrough was not taking place, strains with transcriptional single fusions to GFP3 and tdTomato were analysed with the PE-A and FITC-A filters, respectively, with the observation of fluorescence as the negative control level.
For cell sorting, stationary cultures in LB obtained after an overnight incubation were sorted using a BD FACSAria Fusion flow cytometer (BD Biosciences). Exponential cultures in HIM obtained after 24 h of incubation from 0.13 OD600 were sorted using a MoFloTM XDP cytometer (Beckman Coulter). To initiate the process of sorting, gates were drawn to distinguish cells displaying fluorescence levels overlapping with the 1448A non-GFP bacterial population, which served as the negative control, from cells expressing higher GFP levels, as indicated in the corresponding histogram. On the basis of this analysis, 1 × 105 events were sorted for cells expressing higher GFP levels and lower GFP levels. Cells from each gate were collected into separate sterile tubes. After sorting, cells were centrifuged at 12,000 g for 10 min, and the resulting pellets were resuspended into 10 mM MgCl2. An aliquot of sorted cells was run again in the cytometer to confirm the differences in expression between the separated populations. Data from cytometry experiments were analysed using Kaluza 2.1 (Beckman Coulter) for further analysis and visualization.
Confocal microscopy
For single-cell visualization of apoplast-extracted bacteria and cultured bacteria, suspensions of 2 µl were deposited over a 0.17-mm coverslip, and an agar-pad square was placed on top of the drop to create a bacterial monolayer63. To visualize all cells, bacterial suspensions were stained with FM4-64 (Life Technologies) at 20 μM, and bacterial membranes were visualized with fluorescent light; alternatively, in other cases, bright-field images were included. Images of single-cell bacteria were acquired using the Zeiss LSM880 confocal microscope (Zeiss) using ×100 objectives.
For the visualization of P. syringae microcolonies and surface cells, sections of inoculated P. vulgaris leaves (~5 mm2) were carefully excised using a razor blade and mounted on slides in double-distilled H2O, positioning the lower epidermis towards the objective. A 0.17-mm coverslip was placed over the sample. Images of the leaf mesophyll were taken using the Leica Stellaris 8 confocal microscope (Leica Microsystems) with ×40 objectives.
Filters for wavelength selection were used for the visualization of the following fluorophores (excitation/ emission): eCFP (405 nm/450–500), GFP (488 nm/500–533 nm), FM4-64 (488 nm/604–674 nm), tdTomato (514 nm/570–600) and plant autofluorescence (514/605–670 nm). Image processing was performed using Leica LAS AF (Leica Microsystems) software. To ensure bleedthrough was not taking place, strains with transcriptional single fusions to GFP3, tdTomato and strains constitutively labelled with eCFP were observed under the microscope in the visualization conditions mentioned above, with the observation of no fluorescence. Z-series imaging was performed at 1 μm using ×40 objectives.
Time-lapse microscopy
Heterogeneous flagellum expression was measured during microcolony formation in HIM + 1.25% agarose pads as follows: 2× HIM medium was mixed with a melted 2.5% agarose solution and immediately placed in the wells of a custom 3D-printed mould (template available at https://github.com/JLuneau/Pseudomonas_AgarPads_fliC/tree/main/3D_printed_AgarPad_mold) disposed on a 50-mm round coverslip (Epredia, CB005005A140MNZ0). To ensure the flatness of the pads, another coverslip was immediately placed on top of the mould. The pads were solidified for 15 min at room temperature. The bottom coverslip was removed and 4 µl of bacterial suspensions adjusted to OD = 0.005 in HIM were dropped on the pad surface. Immediately after the droplets dried, a new coverslip was placed on the mould and the assembled device was mounted on the microscope. For time-lapse experiments, images were taken every 15 min, starting at 4 h after cells were placed on the pads and for 24 h at 25 °C. Images were acquired using the NIS-Elements software on a Nikon Eclipse Ti2 inverted microscope equipped with a Hamamatsu ORCA-Flash4.0LT digital camera and a Nikon Plan Apo Lambda ×100/1.45 oil objective. The 1.5× manual knob was engaged to enhance magnification. Illumination settings: phase contrast, 100 ms, 50% intensity; GFP (470 nm excitation and 519 nm emission filters), 300 ms, 50% intensity. All imaging data are available at https://www.ebi.ac.uk/biostudies/bioimages/studies/S-BIAD1413.
Time-lapse image analysis
Time-lapse movies were visually inspected using Fiji 2.14.0 to crop the region of interest around microcolonies and to remove later frames when cells overlapped. Cells were segmented and tracked using the DeLTA 2.0 deep learning-based pipeline64 with the default pretrained models for segmentation and tracking. Time-lapse data analysis was performed using custom Python scripts adapted from ref. 65 (available at https://github.com/JLuneau/Pseudomonas_AgarPads_fliC). Visual inspection of DeLTA 2.0 output movies showed that while segmentation errors were rare, tracking errors were frequent at late timepoints. Thus, similarly to ref. 65, we filtered out erroneous cell tracks. Upon division, (1) we kept cells for which two sister cells were tracked for at least four frames after division, (2) we excluded sister cells for which the cumulated length at birth differed strongly from the length of the mother cell before division (increase or decrease of more than 20%) and (3) we excluded sister cells that showed unexpectedly large jumps in cell length between two frames (increase or decrease of more than 20%). For all retained cells, the fliC expression level was estimated as the mean fluorescence intensity in the GFP channel for all pixels belonging to a single cell, averaged over the lifetime of each individual cell. The growth rate was obtained by performing a linear regression on the log-transformed cell length over the lifetime of each cell. To estimate the cost of flagellum expression, we grouped cells into two classes: the GFP-high cells showing a mean fluorescence intensity above the median fluorescence intensity of all cells, and the GFP-low cells showing a mean fluorescence intensity below the population’s median.
Live/dead staining
One drop of the propidium iodide solution Ready Probes (Thermo Fisher) was added to 300 μl of the suspension with apoplast-extracted bacteria, and live/dead bacteria were identified by flow cytometry. For live/dead staining, bacteria were syringe infiltrated into bean leaves at 5 × 104 c.f.u.s ml−1 and extracted from the apoplast at 4 dpi.
Competitive index assay
CI was calculated by determining the ratio between the mutant strain and the wild type in the output sample divided by that in the input (which should be 1.0)66. Assays were performed after the mixed strains had been growing in either bean leaves or LB and HIM cultures.
CI assays66 were performed in bean plants (Phaseolus vulgaris cv. Canadian wonder). Bean plants were inoculated with 200 µl of a mixed bacterial suspension containing 5 × 104 c.f.u.s ml−1 of each strain, consisting of an equal proportion of wild type and mutant strains. For the assay performed in Fig. 3d, the peptide flg22 (GeneScript) was added to the bacterial suspension to a 100 nM concentration. Inoculation was performed using a 1-ml syringe without needle. Samples were extracted for quantification at 4 dpi. Bacterial recovery was carried out by taking 5 discs of 1-cm diameter from the infected leaf with a cork borer and homogenizing them by mechanical disruption into 1 ml of 10 mM MgCl2. After homogenization, serial dilutions of the bacterial suspensions were prepared and plated onto agar plates supplemented with 2 μg ml−1 cycloheximide. Bacterial enumeration was performed and CIs were calculated after 2 days of growth at 28 °C. To distinguish wild-type from mutant bacteria within the mixed infection, an aliquot from the same dilution was plated onto LB agar and LB agar plates supplemented with kanamycin.
For CI assays performed in LB cultures, 500 μl of a mixed bacterial suspension with 5 × 105 c.f.u.s ml−1 was inoculated into 4,500 μl of liquid LB in culture tubes. For CI assays performed in HIM cultures, 500 μl of a mixed bacterial suspension with 5 × 107 c.f.u.s ml−1 was inoculated into 4,500 μl of liquid LB in culture tubes. After 24 h of incubation with continuous agitation, in both LB or HIM cultures, serial dilutions were prepared and plated onto LB agar and LB agar plates supplemented with kanamycin.
To confirm dosage and relative proportion of the strains, serial dilutions of the inocula were plated onto LB agar and LB agar plates supplemented with the appropriate antibiotic. After bacterial counting, the ratio of the wild-type vs the mutant strain should be close to 1. The competitive indices represent the mean of at least three independent experiments, each with at least three replicates each. Error bars indicate standard error. Statistical analysis was performed using two-tailed Student’s t-test with a significance threshold of P < 0.05 to assess deviations from a ratio of 1.
In vitro growth curves
Growth curves to analyse growth differences between mutant and overexpressing strains were generated by culturing strains in 96-well plates (Biofil), adjusting the bacterial inoculum to an optical density (Abs600) of 0.13 in HIM in 150 µl of final volume. The inoculum was obtained from an overnight LB culture and cells were washed twice with MgCl2 before adjusting the optical density. Plates were incubated for 50 h at 28 °C with agitation in an EONC plate reader (Bio Tek Instruments).
Growth curves to compare ΔhrpL growth vs that of the wild-type strain were generated by culturing strains in culture tubes in HIM at an initial optical density of 0.13 (Abs600). The inoculum was obtained from an overnight culture in LB, washed twice with MgCl2. Samples were taken at 20, 24, 26, 28, 30, 34, 38, 44, 48 and 50 h.
To calculate bacterial growth rate, the log10 of absorbance data were calculated and represented versus time. The regression curve was calculated over the zone of exponential growth and the graph slope obtained was used as the growth rate.
Flagellar motility assay
Flagellar motility assays were conducted after sorting bacterial cultures carrying fliC::GFP3 by inoculating 2 µl of the aliquots obtained after sorting (adjusted to 10,000 sorting events) in HIM plates containing 2.5 g l−1 agar or in tryptone plates containing 3% tryptone, 5% MgCl2 and 2.5 g l−1 agar. Plates were subsequently incubated at 28 °C, and digital photographs were captured to measure the diameter of the swimming halo. Images were then taken at 1 and 3 dpi (for LB-grown sorted samples) or 3 dpi (for HIM-grown sorted samples). Halos obtained were measured and the ratio between the diameters of high versus low expression within each sorted pair was calculated.
Quantification, statistical analysis and reproducibility
All quantification and statistical analysis described in this study was performed using GraphPad Prism 9.0 (Prism; https://www.graphpad.com/). Details of the analysis used and specific P values are indicated in the figure legends for each experiment, for all or at least significantly different values. Reproducibility was tested in independent experiments and the total number of biological replicates indicated in figure legends. Software used for data quantification and analysis are further detailed in Supplementary Table 4. In all scatterplot graphs, mean ± s.e.m. are indicated. No statistical method was used to predetermine sample size. No data were excluded from the analyses, except for Figs. 1a and 5b, and Extended Data Fig. 6b,c in which, to adjust the sample size (n value) between conditions, some data were excluded following blind elections. The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Code availability
Time-lapse data analysis was performed using custom Python scripts adapted from ref. 65 (https://github.com/JLuneau/Pseudomonas_AgarPads_fliC). This paper does not report original code. Any additional information required to reanalyse the data reported in this work is available from the lead contact upon request.
References
Morris, C. E., Monteil, C. L. & Berge, O. The life history of Pseudomonas syringae: linking agriculture to earth system processes. Annu. Rev. Phytopathol. https://doi.org/10.1146/annurev-phyto-082712-102402 (2013).
Haefele, D. M. & Lindow, S. E. Flagellar motility confers epiphytic fitness advantages upon Pseudomonas syringae. Appl. Environ. Microbiol. 53, 2528–2533 (1987).
Panopoulos, N. J. Role of flagellar motility in the invasion of bean leaves by Pseudomonas phaseolicola. Phytopathol. https://doi.org/10.1094/Phyto-64-1389 (1974).
Ichinose, Y., Taguchi, F. & Mukaihara, T. Pathogenicity and virulence factors of Pseudomonas syringae. J. Gen. Plant Pathol. 79, 285–296 (2013).
DeFalco, T. A. & Zipfel, C. Molecular mechanisms of early plant pattern-triggered immune signaling. Mol. Cell 81, 3449–3467 (2021).
Yu, X. et al. Transcriptional responses of Pseudomonas syringae to growth in epiphytic versus apoplastic leaf sites. Proc. Natl Acad. Sci. USA https://doi.org/10.1073/pnas.1221892110 (2013).
Nobori, T. et al. Transcriptome landscape of a bacterial pathogen under plant immunity. Proc. Natl Acad. Sci. USA https://doi.org/10.1073/pnas.1800529115 (2018).
Nobori, T. et al. Multidimensional gene regulatory landscape of a bacterial pathogen in plants. Nat. Plants 6, 883–896 (2020).
Cummings, L. A., Wilkerson, W. D., Bergsbaken, T. & Cookson, B. T. In vivo, fliC expression by Salmonella enterica serovar Typhimurium is heterogeneous, regulated by ClpX, and anatomically restricted. Mol. Microbiol. 61, 795–809 (2006).
Saini, S., Ellermeier, J. R., Slauch, J. M. & Rao, C. V. The role of coupled positive feedback in the expression of the SPI1 type three secretion system in Salmonella. PLoS Pathog. 6, e1001025 (2010).
Koirala, S. et al. A nutrient-tunable bistable switch controls motility in Salmonella enterica serovar Typhimurium. mBio https://doi.org/10.1128/mBio.01611-14 (2014).
Zarkani, A. A. et al. Salmonella heterogeneously expresses flagellin during colonization of plants. Microorganisms 8, 815 (2020).
Wang, X., Koirala, S., Aldridge, P. D. & Rao, C. V. Two tandem mechanisms control bimodal expression of the flagellar genes in Salmonella enterica. J. Bacteriol. https://doi.org/10.1128/JB.00787-19 (2020).
Leal‐Morales, A., Pulido‐Sánchez, M., López‐Sánchez, A. & Govantes, F. Transcriptional organization and regulation of the Pseudomonas putida flagellar system. Environ. Microbiol. 24, 137–157 (2022).
Nogales, J. et al. FleQ coordinates flagellum-dependent and -independent motilities in Pseudomonas syringae pv. tomato DC3000. Appl. Environ. Microbiol. 81, 7533–7545 (2015).
Schreiber, K. J., Chau-Ly, I. J. & Lewis, J. D. What the wild things do: mechanisms of plant host manipulation by bacterial type III-secreted effector proteins. Microorganisms 9, 1029 (2021).
Fouts, D. E. et al. Genomewide identification of Pseudomonas syringae pv. tomato DC3000 promoters controlled by the HrpL alternative sigma factor. Proc. Natl Acad. Sci. USA 99, 2275–2280 (2002).
Preston, G., Deng, W.-L., Huang, H.-C. & Collmer, A. Negative regulation of hrp genes in Pseudomonas syringae by HrpV. J. Bacteriol. 180, 4532–4537 (1998).
Ortiz-Martín, I., Thwaites, R., Macho, A. P., Mansfield, J. W. & Beuzón, C. R. Positive regulation of the hrp type III secretion system in Pseudomonas syringae pv. phaseolicola. Mol. Plant Microbe Interact. 23, 665–681 (2010).
Rufián, J. S. et al. Pseudomonas syringae differentiates into phenotypically distinct subpopulations during colonization of a plant host. Environ. Microbiol. 18, 3593–3605 (2016).
Mansfield, J. et al. Top 10 plant pathogenic bacteria in molecular plant pathology. Mol. Plant Pathol. 13, 614–629 (2012).
Rufián, J. S. et al. Confocal microscopy reveals in planta dynamic interactions between pathogenic, avirulent and non-pathogenic Pseudomonas syringae strains. Mol. Plant Pathol. 19, 537–551 (2018).
López-Pagán, N., Rufián, J. S., Ruiz-Albert, J. & Beuzón, C. R. Dual-fluorescence chromosome-located labeling system for accurate in vivo single-cell gene expression analysis in Pseudomonas syringae. Methods Mol. Biol. 2751, 95–114 (2024).
Sturm, A. et al. The cost of virulence: retarded growth of Salmonella typhimurium cells expressing type III secretion system 1. PLoS Pathog. 7, e1002143 (2011).
Sánchez-Romero, M. A. & Casadesús, J. Contribution of SPI-1 bistability to Salmonella enterica cooperative virulence: insights from single cell analysis. Sci. Rep. 8, 14875 (2018).
Macnab, R. in Escherichia Coli and Salmonella: Cellular and Molecular Biology 2nd edn (eds Neidhardt, F. C. & Curtiss, R.) 119–130 (ASM Press, 1996).
Martínez‐García, E., Nikel, P. I., Chavarría, M. & de Lorenzo, V. The metabolic cost of flagellar motion Pseudomonas putida KT2440. Environ. Microbiol. 16, 291–303 (2014).
Schavemaker, P. E. & Lynch, M. Flagellar energy costs across the tree of life. eLife 11, e77266 (2022).
Leba, L. et al. CML9, an Arabidopsis calmodulin‐like protein, contributes to plant innate immunity through a flagellin‐dependent signalling pathway. Plant J. 71, 976–989 (2012).
Lehtinen, J., Nuutila, J. & Lilius, E. Green fluorescent protein–propidium iodide (GFP–PI) based assay for flow cytometric measurement of bacterial viability. Cytometry A 60A, 165–172 (2004).
Patel, R. R., Kandel, P. P., Traverso, E., Hockett, K. L. & Triplett, L. R. Pseudomonas syringae pv. phaseolicola uses distinct modes of stationary-phase persistence to survive bacteriocin and streptomycin treatments. mBio https://doi.org/10.1128/mBio.00161-21 (2021).
Ackermann, M. et al. Self-destructive cooperation mediated by phenotypic noise. Nature 454, 987–990 (2008).
Roine, E. et al. Hrp pilus: an hrp-dependent bacterial surface appendage produced by Pseudomonas syringae pv. tomato DC3000. Proc. Natl Acad. Sci. USA 94, 3459–3464 (1997).
Vargas, P. et al. Plant flavonoids target Pseudomonas syringae pv. tomato DC3000 flagella and type III secretion system. Environ. Microbiol. Rep. 5, 841–850 (2013).
Freed, N. E. et al. A simple screen to identify promoters conferring high levels of phenotypic noise. PLoS Genet. 4, e1000307 (2008).
Sánchez-Romero, M. A. & Casadesús, J. Single cell analysis of bistable expression of pathogenicity island 1 and the flagellar regulon in Salmonella enterica. Microorganisms 9, 210 (2021).
Wei, C.-F., Deng, W.-L. & Huang, H.-C. A chaperone-like HrpG protein acts as a suppressor of HrpV in regulation of the Pseudomonas syringae pv. syringae type III secretion system. Mol. Microbiol. 57, 520–536 (2005).
Wei, W. et al. The gene coding for the Hrp pilus structural protein is required for type III secretion of Hrp and Avr proteins in Pseudomonas syringae pv. tomato. Proc. Natl Acad. Sci. USA 97, 2247–2252 (2000).
Fraser, D. & Kærn, M. A chance at survival: gene expression noise and phenotypic diversification strategies. Mol. Microbiol. 71, 1333–1340 (2009).
Kamino, K., Keegstra, J. M., Long, J., Emonet, T. & Shimizu, T. S. Adaptive tuning of cell sensory diversity without changes in gene expression. Sci. Adv. 6, eabc1087 (2020).
Ackermann, M. A functional perspective on phenotypic heterogeneity in microorganisms. Nat. Rev. Microbiol. 13, 497–508 (2015).
Griffin, A. S., West, S. A. & Buckling, A. Cooperation and competition in pathogenic bacteria. Nature 430, 1024–1027 (2004).
Diard, M. et al. Stabilization of cooperative virulence by the expression of an avirulent phenotype. Nature 494, 353–356 (2013).
Davis, K. M. For the greater (bacterial) good: heterogeneous expression of energetically costly virulence factors. Infect. Immun. https://doi.org/10.1128/IAI.00911-19 (2020).
Barrett, L. G., Bell, T., Dwyer, G. & Bergelson, J. Cheating, trade-offs and the evolution of aggressiveness in a natural pathogen population. Ecol. Lett. 14, 1149–1157 (2011).
Ruiz-Bedoya, T., Wang, P. W., Desveaux, D. & Guttman, D. S. Cooperative virulence via the collective action of secreted pathogen effectors. Nat. Microbiol. 8, 640–650 (2023).
Lovell, H. C. et al. In planta conditions induce genomic changes in Pseudomonas syringae pv. phaseolicola. Mol. Plant Pathol. 12, 167–176 (2011).
Neale, H. C. et al. A low frequency persistent reservoir of a genomic island in a pathogen population ensures island survival and improves pathogen fitness in a susceptible host. Environ. Microbiol. 18, 4144–4152 (2016).
Davis, K. M., Mohammadi, S. & Isberg, R. R. Community behavior and spatial regulation within a bacterial microcolony in deep tissue sites serves to protect against host attack. Cell Host Microbe 17, 21–31 (2015).
Spratt, M. R. & Lane, K. Navigating environmental transitions: the role of phenotypic variation in bacterial responses. mBio 13, e0221222 (2022).
Schreiber, F. & Ackermann, M. Environmental drivers of metabolic heterogeneity in clonal microbial populations. Curr. Opin. Biotechnol. 62, 202–211 (2020).
Ortiz-Martín, I., Thwaites, R., Mansfield, J. W. & Beuzón, C. R. Negative regulation of the Hrp type III secretion system in Pseudomonas syringae pv. phaseolicola. Mol. Plant Microbe Interact. 23, 682–701 (2010).
Nielsen, A. T. et al. A bistable switch and anatomical site control Vibrio cholerae virulence gene expression in the intestine. PLoS Pathog. 6, e1001102 (2010).
Dar, D., Dar, N., Cai, L. & Newman, D. K. Spatial transcriptomics of planktonic and sessile bacterial populations at single-cell resolution. Science 373, eabi4882 (2021).
van Vliet, S. et al. Spatially correlated gene expression in bacterial groups: the role of lineage history, spatial gradients, and cell–cell interactions. Cell Syst. 6, 496–507.e6 (2018).
Zhou, F. et al. Co-incidence of damage and microbial patterns controls localized immune responses in roots. Cell 180, 440–453.e18 (2020).
Jacob, P., Hige, J. & Dangl, J. L. Is localized acquired resistance the mechanism for effector triggered disease resistance in plants? Nat. Plants 9, 1184–1190 (2023).
Sánchez-Romero, M. A., Olivenza, D. R., Gutiérrez, G. & Casadesús, J. Contribution of DNA adenine methylation to gene expression heterogeneity in Salmonella enterica. Nucleic Acids Res. 48, 11857–11867 (2020).
Bertani, G. Studies on lysogenesis I. J. Bacteriol. 62, 293–300 (1951).
Huynh, T. V., Dahlbeck, D. & Staskawicz, B. J. Bacterial blight of soybean: regulation of a pathogen gene determining host cultivar specificity. Science 245, 1374–1377 (1989).
Zumaquero, A., Macho, A. P., Rufián, J. S. & Beuzón, C. R. Analysis of the role of the type III effector inventory of Pseudomonas syringae pv. phaseolicola 1448a in interaction with the plant. J. Bacteriol. 192, 4474–4488 (2010).
Rufián, J. S. et al. in Methods in Molecular Biology (eds Medina, C. & López-Baena, F.) 183–199 (Humana Press2018).
Rufián, J. S., López-Pagán, N., Ruiz-Albert, J. & Beuzón, C. R. Single-cell analysis of the expression of Pseudomonas syringae genes within the plant tissue. J. Vis. Exp. https://doi.org/10.3791/64614 (2022).
O’Connor, O. M., Alnahhas, R. N., Lugagne, J.-B. & Dunlop, M. J. DeLTA 2.0: a deep learning pipeline for quantifying single-cell spatial and temporal dynamics. PLoS Comput. Biol. 18, e1009797 (2022).
Kaczmarczyk, A. et al. A genetically encoded biosensor to monitor dynamic changes of c-di-GMP with high temporal resolution. Nat. Commun. 15, 3920 (2024).
Macho, A. P., Zumaquero, A., Ortiz‐Martín, I. & Beuzón, C. R. Competitive index in mixed infections: a sensitive and accurate assay for the genetic analysis of Pseudomonas syringae–plant interactions. Mol. Plant Pathol. 8, 437–450 (2007).
López-Pagán, N., Ruiz-Albert, J. & Beuzón, C. R. Lopez_Pagan_et_al_Nature_Micro_fig_1. Zenodo https://doi.org/10.5281/zenodo.14870947 (2025).
López-Pagán, N., Ruiz-Albert, J. & Beuzón, C. R. Lopez_Pagan_et_al_Nature_Micro_fig_2. Zenodo https://doi.org/10.5281/zenodo.14865081 (2025).
López-Pagán, N., Luneau, J., Ruiz-Albert, J. & Beuzón, C. R. Lopez_Pagan_et_al_Nature_Micro_fig_3. Zenodo https://doi.org/10.5281/zenodo.14865104 (2025).
Rufián, J. S., Ruiz-Albert, J.& Beuzón, C. R. Lopez_Pagan_et_al_Nature_Micro_fig_4. Zenodo https://doi.org/10.5281/zenodo.14849825 (2025).
Rufián, J. S., Ruiz-Albert, J.& Beuzón, C. R. Lopez_Pagan_et_al_Nature_Micro_fig_5. Zenodo https://doi.org/10.5281/zenodo.14849873 (2025).
López-Pagán, N., Ruiz-Albert, J. & Beuzón, C. R. Lopez_Pagan_et_al_Nature_Micro_Extended_data_fig_1_V2. Zenodo https://doi.org/10.5281/zenodo.14844995 (2025).
López-Pagán, N., Ruiz-Albert, J. & Beuzón, C. R. Lopez_Pagan_et_al_Nature_Micro_Extended_data_fig_2. Zenodo https://doi.org/10.5281/zenodo.14849770 (2025).
López-Pagán, N., Rufián, J. S., Ruiz-Albert, J. & Beuzón, C. R. Lopez_Pagan_et_al_Nature_Micro_Extended_data_fig_3_V2. Zenodo https://doi.org/10.5281/zenodo.14865026 (2025).
López-Pagán, N., Luneau, J., Ruiz-Albert, J. & Beuzón, C. R. Lopez_Pagan_et_al_Nature_Micro_Extended_data_fig_4_V2. Zenodo https://doi.org/10.5281/zenodo.14865041 (2025).
López-Pagán, N., Ruiz-Albert, J. & Beuzón, C. R. Lopez_Pagan_et_al_Nature_Micro_Extended_data_fig_5. Zenodo https://doi.org/10.5281/zenodo.14865061 (2025).
Rufián, J. S., Ruiz-Albert, J. & Beuzón, C. R. Lopez_Pagan_et_al_Nature_Micro_Extended_data_fig_6. Zenodo https://doi.org/10.5281/zenodo.14849810 (2025).
Acknowledgements
We thank A. Zumaquero for technical assistance, I. Ortiz-Martín for contribution to preliminary experiments, E. Ruiz-Beuzón for assistance in the final stages of manuscript preparation, S. Ruiz-Beuzón for help with drawing Fig. 4a and A. García, scientific illustrator from Bio-Graphics, for help with Fig. 4c,e,g and Extended Data Fig. 7. This work was supported by project grant PID2021-127245OB-I00 awarded to C.R.B. and J.R.-A., financed by MCIN/AEI/10.13039/501100011033/ and ‘ERDP A way of making Europe’. N.L.-P. received funding for a Short-Term Training Mission from COST ACTION SUSTAIN FA1208, supported by COST (European Cooperation in Science and Technology), FEMS research and training grant, and EMBO Scientific Exchange Grant. J.S.R. was supported by Plan Andaluz de Investigación, Desarrollo e Innovación (PAIDI 2020). J.S.L. and S.v.V. were supported by the Swiss National Science Foundation (SNSF) through the Swiss National Centre of Competence in Research (NCCR) Microbiomes (grant number 51NF40_ 225148) and through an Ambizione Fellowship to S.v.V. (grant number: PZ00P3_202186). L.A. was funded by Aix-Marseille University (AMU) and the Centre National de la Recherche Scientifique (CNRS). M.-A.S.-R. was supported by PID2020-116995RB-I00 funded by MCIN/AEI/10.13039/5011100011033, and CNS2022-135641 funded by MICIU/AEI/10.13039/5011100011033, awarded to M.-A.S.-R., and by the European Union NextGenerationEU/PRTR. We also acknowledge the financial support of the Proyecto QUAL21 012 IHSM, Consejería de Universidad, Investigación e Innovación, Junta de Andalucía, and Universidad de Málaga’.
Author information
Authors and Affiliations
Contributions
N.L.-P., J.S.R., J.R.-A. and C.R.B. conceptualized the project. N.L.-P., J.S.R., L.A., M.-A.S.-R., J.L. and S.v.V. developed methodology and software and performed analysis. N.L.-P., J.S.R., J.L., M.-A.S.-R., J.R.-A. and C.R.B. procured resources. C.R.B. wrote the original draft. C.R.B., N.L.-P., J.S.R., J.L., M.-A.S.-R., L.A., S.v.V. and J.R.A. reviewed and edited the manuscript. C.R.B., J.R.-A., J.S.R., N.L.-P., M.-A.S.-R., L.A. and S.v.V. acquired funding. All authors discussed the results and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Microbiology thanks Andreas Diepold, Bradley Laflamme and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Transcriptional fusions to tdTomato do not affect gene function.
(a) A strain carrying a fliC::tdT transcriptional fusion displays wild type motility in the photographs (a) and quantifications (b) of swimming plates (n = 4). No statistical differences were observed as indicated by an unpaired two-tailed t test (P < 0.05). Scatter dot graph shown mean +/− SEM. The fliC::tdT construct is integrated in the native chromosome location of the fliC gene as established by Southern blot analysis (using the nptII gene conferring resistance to Km as a probe) of a Alw21I (Thermo Fisher Scientific, USA) digestion of genomic DNA from the wild type (Pph) and fliC::tdT derivative strains (c) showing the expected band sizes (d). (e) Representative images of the symptoms caused by the wild type strains and their relative hopAB1::GFP3 fliC::tdT, hrpL::GFP3 fliC::tdT and hrcU::GFP3 fliC::tdT strains at 5 and 7 dpi. (f) Representative images of the colonies obtained after plating serial dilutions of the indicated strains previously adjusted to 0.1 optical density (Abs600). (g) Quantification of the data obtained in f. All dilutions reach similar values indicating no differences in CFU/ml occasioned by expression of fluorescence reporter genes. f and g illustrate one experiment (>3 technical replicates), data from 3 additional independent experiments carried out in the same manner are showed in h. (h) Mean number of cfu/ml obtained after plating a dilution of the culture previously adjusted to 0.1 optical density (Abs600). Data obtained for the different strains (n = 3 biological replicates; technical replicates not used for statistical analysis) did not show significative differences according to a one-way ANOVA followed by Tukey’s multiple comparison test (P < 0.05). Graph shows individual values (scatter plot) and mean +/− SEM is also indicated.
Extended Data Fig. 2 Flagella expression is heterogeneous on plant surface.
Selected images of the hopAB1::GFP3 fliC::tdT eCFP strain on the plant surface at 6 h post inoculation (hpi) by dipping the leaf into a 5 × 107 CFU/ml bacterial suspension. tdTomato panel shows the fluorescence of tdTomato associated to fliC expression, and eCFP panel shows the fluorescence of eCFP as constitutive expression reporter, using grey scale in both cases to improve contrast. The GFP panel, corresponding to hopAB1 expression, is not shown since no fluorescence was detected. Scale bars correspond to 20 μm. Contrast and brightness were adjusted to improve visualization but were kept constant across panels. Circles highlight bacteria detected in the eCFP panel without expression in the tdTomato channel (FlagellaOFF). Right panels correspond to the magnification of the area selected in rectangles. Images are representative of 3 independent experiments.
Extended Data Fig. 3 Flagella display heterogenous expression in Pseudomonas syringae.
(a) CSLM of a strain carrying fliC::tdT grown O/N in LB (upper), or 24 h in HIM (central), or extracted from bean leaf apoplasts 4 dpi (bottom). tdTomato panels show tdTomato fluorescence as reporter of fliC expression and BF panel corresponds to bright field. Scale bars correspond to 2 µm. Images are representative of 3 independent experiments (b) FC analysis of fliC::tdT strain in conditions described in a. Dot plots show cell size versus tdTomato fluorescence intensity, represented as arbitrary units in logarithmic scale. Data collected for 100,000 events per sample. The non-tdT graph show autofluorescence levels displayed by wild type not carrying any fluorescent gene. Vertical lines leave 99% of data of non-tdT strain to the left and is used as reference to differentiate for OFF cells. a and b show representative results of at least three independent replicates. (c) FC analysis shows GFP3 and TdTomato fluorescence detection is not bias by bleed through. Left graphs correspond to fliC::GFP3 strain and right to the fliC::tdT strain. Upper graphs show tdTomato fluorescence versus cell count and bottom graphs show GFP fluorescence versus cell count. In all cases the non-fluorescent strain is included as negative control and data were treated as in b. (d) Quantification of GFP lifetime (τ) in the presence or absence of tdTomato. Bacteria expressing similar levels of hrpL::GFP3 (determined as GFP fluorescence intensity) and differing in the expressing of fliC::tdT expression (as shown by different tdTomato fluorescence intensity) were chosen for the analysis (upper panel). GFP lifetime was obtained for each pixel of the image (lower-left) and mean was calculated for each single bacterium. Average of four bacteria in each condition is represented (lower-right) and statistical significance determined with a t-test. (e and f) Dot plot graphs display fluorescence intensity of GFP or tdTomato versus cell size in non-fluorescent bacteria (wild type), or strains carrying hrpL::GFP3 fliC::tdT or hopAB1::GFP3 fliC::tdT corresponding to data shown in Fig. 4 as GFP fluorescence versus that of tdTomato. Figure show representative results of at least three independent experiments. In (e), bacteria grown 24 h in HIM after diluting an O/N LB culture, display reproducible bistable expression of both hrpL::GFP3 and hopAB1::GFP3 and heterogeneous (occasionally bistable) expression of fliC::tdT. In the case of hopAB1::GFP3 fliC::tdT strain in HIM, an additional vertical line (dashed) was added to separate the subpopulations differing in hopAB1 expression, which is higher due to the high basal levels of this gene. In (f), bacteria extracted from bean leaf apoplasts 4 dpi display typical heterogeneous (never bistable) expression of hrpL::GFP3, hopAB1::GFP3 and fliC::tdT.
Extended Data Fig. 4 T3SS and flagella expression impact on bacterial growth and growth cost associated with single cell levels of flagella production is not due to GFP.
(a) Growth rates of 1448 A wild type, a derivative carrying a plasmid that determines constitutive expression of hrpL (pHrpL), or of fleQ (pFleQ), or a fleQ deletion (ΔfleQ), in HIM. Data correspond to 4 biological replicates. Data are presented as mean values +/− SEM. Significative differences are observed between WT and pFleQ strains according to a one-way ANOVA followed by Tukey’s multiple comparison test (P < 0.05). P value is indicated in the graph. (b) Selected timelapse images of wild type constitutively expressing GFP (eGFP) during the microcolony development on agar pads in phase contrast (top) and GFP fluorescence (bottom) channels. Contrast and brightness were adjusted and maintained throughout. (c) Comparisons of mean growth rates for eGFP strain and fliC::GFP3 strain. Note that GFP in the eGFP strain has a much higher fluorescence intensity compared to fliC::GFP3. eGFP shows a significantly lower average growth rate compared to fliC::GFP (Mann-Whitney U test, P = 7.7 × 10−15). (d) Comparison of growth rate of cells displaying high or low GFP signal (determined by splitting the population into two groups using the median fluorescence intensity value) for eGFP strain shows no significant differences between growth rates of cells expressing high or low GFP levels (Mann-Whitney U test, P < 0.05). (e) Correlation between growth rates and fluorescence intensity of individual fliC::GFP3 cells indicates that higher fliC expression is associated with slower growth. The shaded area shows the 95% confidence interval. (f) Correlation between growth rates and fluorescence intensity of individual eGFP cells indicates that higher GFP expression is not associated with slower growth. The shaded area shows the 95% confidence interval.
Extended Data Fig. 5 Dead-live staining shows neither bias towards FlagellaON cells nor for T3SSOFF cells during plant growth.
Graph shows the dead/live ratio for bacteria either expressing or not fliC::GFP3, hrpL::GFP3, hopAB1:GFP3 or hrcU::GFP3 during plant growth. Apoplast-extracted bacteria at 4 dpi in bean leaves were stained with a solution of Propidium iodide (PI). Data were obtained by flow cytometry analysis (100,000 events/ per replicate) and GFP levels were used to differentiate between ON and OFF cells using the non-fluorescent wild type strain as reference, as indicated before. Fluorescence of PI was used to differentiate dead and alive bacteria comparing to the non-fluorescent wild type. Each dot represents biological replicate in two different experiments (Rep 1 and Rep 2).
Extended Data Fig. 6 Active exit from infected tissues is preferentially carried out by FlagellaON bacteria.
(a) Selected images of apoplast-extracted bacteria (using negative pressure; upper panels) or bacteria exiting the tissue on their own (during leaf incubation within a MgCl2 solution; natural exit, lower panels) at 1 dpi and 2 dpi (asymptomatic tissue), or 5 dpi (just before visible symptoms appear) and 7 dpi (fully symptomatic necrotic tissue). Contrast and brightness were adjusted to improve visualization but were kept constant across the different conditions. (b and c) Fluorescence quantification of the images obtained in a for 2 dpi and 5 dpi using Fiji software. Graph shows arbitrary units of GFP fluorescence, corresponding to the expression of the T3SS gene fusion hopAB1::GFP3 in the samples, or tdTomato fluorescence corresponding to the expression of fliC::tdT. Each dot corresponds to an individual bacterium (n = 44) as analyzed from the image. Comparisons between apoplast extraction and natural exit were carried out per each sample using an unpaired two-tailed t-test. Data are presented as mean values +/− SEM. Graphs show results representative of 3 independent experiments.
Extended Data Fig. 7 T3SS and flagella phenotypic heterogeneity overlays with dynamic and spatial expression patterns during interaction with the plant host.
Model summarizes the dynamic of expression of these two loci during in planta growth. Starts at the beginning of the infection (top left). A leaf section with different structural elements indicated, illustrates entry of swimming bacteria into the apoplast through a stoma, where bacteria expression profiles switch from FlagellaON/T3SSOFF to start the interaction with host-cell needed to suppress defences and to allow bacterial multiplication. During the initial multiplication (1 dpi) bacteria maintain a predominant FlagellaOFF/T3SSON expression profile with orthogonal stochastic switching to T3SSON, regardless of flagellar expression which is reduced to a very limited proportion of the population. However, FlagellaON bacteria are particularly capable of exiting the leaf within a humid environment and the exiting population is enriched in FlagellaON bacteria. As the microcolony grows those bacteria closest to the host cell remain T3SSON with the occasional stochastic switch to T3SSOFF, but the growing side of the microcolony, father away from the host cell switch to FlagellaON/T3SSOFF. The latter also show overlaid stochastic heterogeneity. When the infection reaches the last stages and the plant tissue becomes necrotic, bacteria of all phenotypic combinations exit the compromised tissue in wet environments. Bacteria exiting the tissues at all the stages of the infection can potentially move onto a new niche or host. Red bacteria indicate FlagellaON/T3SSOFF bacteria, and green bacteria FlagellaOFF/T3SSON, although bacteria expressing both to different levels can also be found. Grey bacteria indicate stochastically OFF bacteria for either locus.
Supplementary information
Supplementary Information (download PDF )
Supplementary Tables 1–4, references for tables, gating strategy and legend.
Supplementary Video 1 (download AVI )
Time-lapse movie of phase contrast of a strain carrying hopAB1::GFP3 multiplying in HIM.
Supplementary Video 2 (download AVI )
Time-lapse movie of GFP fluorescence of a strain carrying hopAB1::GFP3 multiplying in HIM.
Supplementary Video 3 (download AVI )
Time-lapse movie of phase contrast of a strain carrying hrpL::GFP3 multiplying in HIM.
Supplementary Video 4 (download AVI )
Time-lapse movie of GFP fluorescence of a strain carrying hrpL::GFP3 multiplying in HIM.
Supplementary Video 5 (download AVI )
Time-lapse movie of phase contrast of a strain carrying fliC::GFP3 multiplying in HIM.
Supplementary Video 6 (download AVI )
Time-lapse movie of GFP fluorescence of a strain carrying fliC::GFP3 multiplying in HIM.
Supplementary Video 7 (download AVI )
Z-stack compilation of a microcolony of strain carrying fliC::GFP3 in the apoplast at 3 dpi.
Supplementary Video 8 (download AVI )
Z-stack compilation of a microcolony of strain carrying hopAB1::GFP3 fliC::tdT in the apoplast at 4 dpi. Individual planes are displayed in Fig. 4b.
Supplementary Video 9 (download AVI )
Z-stack compilation of a microcolony of strain carrying hrpL::GFP3 fliC::tdT in the apoplast at 4 dpi. An individual plane is displayed in Fig. 4d.
Supplementary Video 10 (download MP4 )
Time-lapse movie of GFP and tdTomato fluorescence of a strain carrying hrpL::GFP3 fliC::tdT multiplying in HIM.
Source data
Source Data Fig. 1 (download XLSX )
Statistical source data.
Source Data Fig. 3 (download XLSX )
Statistical source data.
Source Data Fig. 5 and Source Data Extended Data Fig. 6 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 1 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 1 (download JPG )
Unprocessed western blot.
Source Data Extended Data Fig. 3d (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 4 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 5 (download XLSX )
Statistical source data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
López-Pagán, N., Rufián, J.S., Luneau, J. et al. Pseudomonas syringae subpopulations cooperate by coordinating flagellar and type III secretion spatiotemporal dynamics to facilitate plant infection. Nat Microbiol 10, 958–972 (2025). https://doi.org/10.1038/s41564-025-01966-0
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41564-025-01966-0
This article is cited by
-
Transcriptome-guided discovery of novel plant-associated genes in a rhizosphere Pseudomonas
Microbiome (2025)
-
Coordinated action by individuals orchestrates infection through the division of labour
Nature Reviews Microbiology (2025)

{kind=link}