
Abstract
Tuberculosis remains the leading cause of death from an infectious disease1,2. Here we report the discovery of a first-in-class small-molecule inhibitor targeting PurF, the first enzyme in the mycobacterial de novo purine biosynthesis pathway. The lead candidate, JNJ-6640, exhibited nanomolar bactericidal activity in vitro. Comprehensive genetic and biochemical approaches confirmed that JNJ-6640 was highly selective for mycobacterial PurF. Single-cell-level microscopy demonstrated a downstream effect on DNA replication. We determined the physiologically relevant concentrations of nucleobases in human and mouse lung tissue, showing that these levels were insufficient to salvage PurF inhibition. Indeed, proof-of-concept studies using a long-acting injectable formulation demonstrated the in vivo efficacy of the compound. Finally, we show that inclusion of JNJ-6640 could have a crucial role in improving current treatment regimens for drug-resistant tuberculosis. Together, we demonstrate that JNJ-6640 is a promising chemical lead and that targeting de novo purine biosynthesis represents a novel strategy for tuberculosis drug development.
Similar content being viewed by others

Main
Tuberculosis (TB) remains a critical global health crisis, persisting as a leading cause of morbidity and mortality worldwide. Current estimates indicate an alarming burden, with approximately 10.8 million new active TB infections and 1.25 million deaths reported in 2023 alone1,2. Despite extensive research efforts and the implementation of newer bedaquiline–pretomanid-containing treatment regimens, the emergence of drug-resistant TB presents a substantial challenge to global TB control initiatives. In addition, the prolonged treatment duration and potential adverse effects associated with some of the drugs in the existing treatments contribute to poor patient adherence as well as higher treatment failure rates. Therefore, there is an urgent need for more effective, shorter and well-tolerated TB treatment regimens. Furthermore, owing to emerging drug resistance, new drugs with novel modes of action are urgently required that contribute to these future regimens.
In the pursuit of novel antitubercular drugs, targeting essential metabolic pathways of Mycobacterium tuberculosis, the causative agent of TB, has gained considerable attention as exemplified by the transformational success of bedaquiline in the clinic3. Among these pathways, the de novo purine biosynthetic pathway, which results in the synthesis of inosine monophosphate (IMP), stands out for its essential role in M. tuberculosis physiology4,5,6. Purines serve as essential building blocks for nucleic acids and energy-carrying molecules, as well as signalling molecules, thus, are an attractive target for antimycobacterial drug development. The first and committed step of de novo purine biosynthesis in M. tuberculosis is catalysed by an amidophosphoribosyltransferase, known as PurF. Inhibition of PurF is predicted to disrupt the synthesis of purine nucleotides, essential for various cellular processes crucial for M. tuberculosis survival and replication. Yet, until now, PurF represented a completely unexplored target for TB drug discovery.
Here we report the discovery and validation of a first-in-class small-molecule inhibitor targeting PurF. This inhibitor represents a novel chemical scaffold, identified through phenotypic whole-cell screening of a diverse set of compounds active against non-tuberculous mycobacteria. The lead compound, JNJ-6640, demonstrated exceptional bactericidal activity in vitro. A comprehensive investigation into the molecular target of this compound, using a series of genetic and biochemical approaches, successfully identified PurF. Moreover, proof-of-concept in vivo studies showed the efficacy of the compound in mouse models. Finally, we present convincing evidence that inhibitors of PurF could have a crucial role in future combination regimens used for the treatment of drug-resistant TB. This work highlights the potential of compounds targeting PurF to contribute to future treatment regimens for drug-resistant TB.
Discovery of JNJ-6640
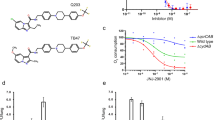
To identify new inhibitors of M. tuberculosis growth, a phenotypic whole-cell guided screen was initiated using a diversity set of 4,924 compounds previously shown to be active from a screening campaign against Mycobacterium avium. An in silico hit-triaging cascade was used to identify chemical series with limited predicted liabilities and good chemical tractability. This led to the identification of a pyrrolidinopyrimidine series, exemplified by JNJ-7310, a racemic mixture with moderate potency against M. tuberculosis (minimal inhibitory concentration of 90% (MIC90) = 328 nM; Fig. 1a). Separation of this racemic mixture into its enantiomers showed stereospecific activity against M. tuberculosis, with one enantiomer (JNJ-0999) having an MIC90 = 110 nM. As the parent compound (JNJ-7310) has the classical features present in several known kinase inhibitors, the secondary amine between the pyrimidine and pyrazole rings was changed to an ether to eliminate this property. This resulted in the preparation of compounds JNJ-6627 and JNJ-6640. Again, one enantiomer (JNJ-6640) was more active, maintaining exceptional potency against M. tuberculosis in vitro (MIC90 = 8.6 ± 3.9 nM; minimal bactericidal concentration of 99.9% (MBC99.9) = 140 ± 63 nM; Fig. 1b and Extended Data Table 1). JNJ-6640 was profiled against a panel of clinical isolates with known resistance profiles. No clear target-specific cross-resistance was observed, suggesting that this compound series had a novel mode of action and/or resistance compared with other clinical compounds (Extended Data Table 1).

a, Series evolution from the hit compound JNJ-7310 to the series lead JNJ-6640. MIC90 values against whole-cell M. tuberculosis are shown. R*, R*-enantiomer; S*, S*-enantiomer. b, Dose–response curve depicting the bactericidal activity (measured as CFU) of JNJ-6640. MBC99.9 = 140 ± 63 nM. n = 4 biological replicates. Data shown are mean ± s.d. LoD, limit of detection.
JNJ-6640 inhibits purine biosynthesis
Given the promising in vitro efficacy, we next focused on identifying and validating the molecular target of our lead molecule: JNJ-6640. M. tuberculosis was incubated with approximately 25× MIC90 of JNJ-7310, JNJ-6627 or JNJ-6640 to select drug-resistant colonies. A comparable frequency of resistance to clinical compounds was observed (Extended Data Fig. 1a). All the selected clones were confirmed to provide a 19–217-fold increase in MIC50 compared with the drug-sensitive parental strain and resistance had no effect on bacterial growth (Fig. 2a, Extended Data Table 2 and Extended Data Fig. 1b). There was clear cross-resistance against other compounds from the series; however, no cross-resistance was observed against a panel of clinical drugs, suggesting a novel mode of action from the compounds tested (Extended Data Table 2). Genomic DNA from the resistant clones was analysed via whole-genome sequencing to identify the molecular changes responsible for drug resistance. This analysis identified four different single-nucleotide polymorphisms (I241V, F428C, F428V and S470F; Extended Data Fig. 1c), all within the gene encoding the enzyme PurF (purF; Rv0808). Using an established database of genetic variance of M. tuberculosis clinical isolates7, we found that PurF is well conserved both between clinical isolates (73% positions conserved) and Mycobacterium species (93% average homology). Furthermore, none of the resistance-conferring mutations were identified among circulating clinical M. tuberculosis isolates, suggesting that there is no existing resistance (Extended Data Fig. 1c).

a, Susceptibility of JNJ-6640-resistant clones (R1–R5). MIC90 values are shown in Extended Data Table 2. b, Induced fit docking model of JNJ-6640 (cyan) in the MtPurF-binding pocket (grey). Phe428 (pink), key interactions and resistance residues are annotated. c, Percentage 15N-stable isotope incorporation following 100 nM JNJ-6640 treatment. Control metabolite (glutamine) was the most abundant nitrogen-containing metabolite. n = 5 biological replicates, representative of two independent experiments. Significance was calculated with two-sided (Bonferroni–Dunn) Student’s t-test with Welsh correction. d, Growth kinetics of CRISPRi-mediated purF transcript knockdown strains (low, medium and high) compared with ‘empty’ vector (control) and kill (atpE high) controls after ATc induction. n = 3 independent experiments. e, CRISPRi-mediated PurF ‘low’ knockdown increases susceptibility to JNJ-6640 (MIC50 = 6.4 nM for −ATc and MIC50 = 0.8 nM for +ATc). n = 3 technical replicates, representative of two independent experiments. f, Nucleobase rescue assay with CRISPRi-mediated high, medium and no (control) purF transcript knockdown in the presence of 10 µM nucleobase or nucleoside with (+ATc) and without (−ATc) induction. Starting inoculum was approximately 1 × 105 CFU ml−1. n = 3 independent experiments. g, MtPurF (IC50 = 1 nM) and Homo sapiens PPAT (HsPPAT; IC50 = 14 µM) enzymatic assays with JNJ-6640. n = 2 biological replicates. h, Cell proliferation assay with JNJ-6640 and three known cell proliferation inhibitors using 93 different cancer cell lines derived from different tissue types. The average pIC50 shown for each tissue type: pIC50 ~ 4 (IC50 ~ 100 µM) was considered non-active. Aza, azathioprine; MMPR, 6-methyl-mercaptopurine riboside; MPA, mycophenolic acid. Representative dataset from two independent experiments. For panels c–f, data shown are mean ± s.d.
PurF catalyses the transfer of a nitrogen atom from glutamine to phosphoribosylpyrophosphate (PRPP) to form glutamate and phosphoribosylamine (PRA) in the first and committed step of de novo purine biosynthesis8. A plethora of in silico methods predicted JNJ-6640 to bind near the phosphoribosyl transferase (PRTase) active site in an AlphaFold model of M. tuberculosis PurF (MtPurF)9. Pending determination of the exact binding mode via X-ray crystallography, induced fit docking into this putative binding site suggests that the pyridyl group of JNJ-6640 interacts with P31, L283, I424 and F428 (Fig. 2b). This prediction is further supported by the resistance mutations L283V and F428V, which are expected to affect the hydrophobic interactions and disrupt π–π interactions, respectively. This binding pose provides an explanation for the higher potency of R*-enantiomers (JNJ-0999 and JNJ-6640) than the S*-enantiomers (JNJ-6627), where the pyridyl group position induces weaker interactions with these amino acids. Furthermore, the pyrazole group of JNJ-6640 is predicted to form H-bond interactions with T393 and Q294. This interaction is optimized in JNJ-6640 via the oxygen linker-induced tautomeric form of the pyrazole ring, providing an explanation for the enhanced activity over JNJ-0999 (Fig. 2b).
To assess the effect of JNJ-6640 on de novo purine biosynthesis within a whole-cell context, we measured the incorporation of stable isotope-labelled nitrogen atoms into adenine and adenosine monophosphate (AMP) following a 4-h co-treatment with 100 nM JNJ-6640 and 15N-(amide)-labelled glutamine (Fig. 2c and Extended Data Fig. 2a–g). Addition of JNJ-6640 resulted in a marked reduction in the incorporation of labelled nitrogen into these purine metabolites, indicating a clear inhibition of de novo purine biosynthesis. Conversely, unlabelled experiments demonstrated an accumulation of glutamine following PurF inhibition for 24 h (Extended Data Fig. 2h).
PurF is predicted to be essential in vitro based on previous genome-wide essentiality screens4,6 and has subsequently been shown, using a transposon mutant, to be essential for M. tuberculosis survival in an animal model of infection5. Here gene essentiality was confirmed using genetic transcript knockdown with a characterized CRISPR interference (CRISPRi) system4. Multiple CRISPRi-mediated transcript knockdown strains were generated, each with a different predicted strength of purF transcript reduction (low–high knockdown). Following validation of each strain (Extended Data Fig. 3a,b), growth assays were established to measure the effect of transcript knockdown on bacterial survival (Fig. 2d). This showed that low reduction in purF transcript had limited effect on bacterial growth, whereas high knockdown had a more than 3-log bactericidal effect after 9 days, confirming essentiality of PurF. We next hypothesized that a reduction in PurF protein levels would lead to increased susceptibility of the strain to JNJ-6640. Indeed, when using the CRISPRi-mediated low-transcript knockdown strain, we showed that the induction of purF transcript knockdown led to an eightfold increase in susceptibility to JNJ-6640, providing additional evidence of target engagement (Fig. 2e). As a control, no change in the susceptibility of bedaquiline was observed following purF transcript knockdown (Extended Data Fig. 3c).
In addition to de novo purine biosynthesis, purine nucleotides can also be produced by recycling nucleobases via the purine salvage pathway. In M. tuberculosis, this salvage pathway consists of hypoxanthine-guanine phosphoribosyltransferase (Hpt; Rv3624c) and adenine phosphoribosyltransferase (Apt; Rv2584c), which utilize nucleobases, including hypoxanthine and adenine. Hpt converts guanine, xanthine and hypoxanthine back to guanosine monophosphate (GMP), xanthosine monophosphate (XMP) and IMP, respectively, whereas Apt recycles adenine back to AMP10. Although we and others have established PurF as essential for M. tuberculosis survival both in vitro4,6 and in vivo5, we hypothesized that supplementing our CRISPRi-mediated high transcript knockdown strain with exogenous hypoxanthine or adenine nucleobases could rescue the effect of inhibition of purine biosynthesis (Extended Data Fig. 4a). Indeed, the inhibition of de novo purine biosynthesis was partially rescued by hypoxanthine at concentrations exceeding 0.1 mM, whereas the presence of adenine had limited effect. As shown for genetic inhibition of PurF, the addition of hypoxanthine (0.03 mM) also led to a rescue of JNJ-6640-treated M. tuberculosis, whereas the addition of adenine, guanine or xanthine had no substantial effect on bactericidal activity (Extended Data Fig. 4b,c). Given the potential for rescue by high levels of exogenous nucleobases, we next determined the physiological levels of nucleobases and nucleosides in human lung tissue. Lung tissue samples from 15 patients with TB and 15 patients without TB (30 total) were analysed by mass spectroscopy and compared with an equivalent experiment in mice (Table 1). These patient samples were collected in the KwaZulu-Natal province of South Africa, where the first cases of extensively drug-resistant TB (XDR-TB) were identified in 2005 (ref. 11). Critically, these analyses revealed that nucleobase and nucleoside levels in humans, ranging from 0.01 to 11.79 µM, were below the concentrations required to rescue PurF inhibition (Fig. 2f). The presence of TB infection did not affect nucleobase or nucleoside levels in lung tissue. Finally, there was no evidence that the purine salvage pathway was significantly upregulated in response to JNJ-6640 treatment (Extended Data Fig. 4d). Together, this provides convincing evidence that JNJ-6640 inhibits purine de novo biosynthesis and that the physiological levels of nucleobases and nucleosides in humans and mice are insufficient to counteract its activity.
To definitively demonstrate target engagement between PurF and JNJ-6640, recombinant MtPurF protein was expressed and purified from an Escherichia coli expression system (Extended Data Fig. 5a–d). This purified PurF enzyme demonstrated catalytic activity using a coupled–coupled assay format. In brief, the glutamate produced by PurF was converted to α-ketoglutarate with a byproduct of hydrogen peroxide, which was quantified using HyPerBlu reagent. In this assay, JNJ-6640 robustly inhibited PurF in a dose-dependent manner (50% inhibitory concentration (IC50) = 1 nM; Fig. 2g).
Given the ubiquitous and essential role of purines in all organisms, it is not surprising that the MtPurF shares 56–57% homology with the human and mouse PurF homologues (PPAT) within the PRTase domain, where JNJ-6640 is predicted to bind. Therefore, to investigate the potential for off-target toxicity, the human homologue PPAT was also expressed, purified and shown to be active in the same coupled–coupled format. In this assay, JNJ-6640 was more than 10,000-fold less active against PPAT (IC50 = 14 µM; Fig. 2g). Similar inhibitory potencies and selectivity indexes were seen for other compounds from this series (Extended Data Fig. 5e). To further investigate undesirable effects in human cells, we performed a cell proliferation assay against 93 different cancer cell lines to investigate potential liabilities in host toxicity. Three known inhibitors of cell proliferation (azathioprine, MMPR and MPA) were used as controls (Fig. 2h). This analysis showed that, compared with the three controls, JNJ-6640 had minimal activity against any of the cancer cell lines tested with an average activity of negative log of the IC50 in molar (pIC50) < 4 (IC50 > 100 µM). These results demonstrate clear target engagement with MtPurF and indicate a large safety window based on selectivity over the human orthologue for purine biosynthesis.
PurF inhibition affects DNA replication
To evaluate the activity of JNJ-6640 on mycobacterial growth kinetics and heterogeneous phenotypic response at the single-cell level, we carried out timelapse imaging of M. tuberculosis grown in microfluidic devices. M. tuberculosis constitutively expressing tdTomato fluorescent protein in the cytoplasm was used in these experiments to identify cell division and lysis events upon compound treatment12. After approximately 6–10 h of treatment with 0.6 µM JNJ-6640, most of the bacteria stopped elongating and growth was arrested for the remainder of drug exposure (up to 144 h), with a significant number of lysis events (approximately 50%) detected during the latter part of exposure (Fig. 3a,b); a similar effect was observed for bedaquiline13 and ciprofloxacin (Extended Data Fig. 6a,b) under similar conditions. This cell lysis continued even after drug removal (measured for 72 h after washout), suggesting a persistent suppression of bacterial growth in the absence of compound. None of the remaining, intact M. tuberculosis exhibited regrowth after drug washout, even upon supplementation with a physiologically relevant mixture of nucleobases determined from uninfected mouse lung tissue (Table 1). In addition, timelapse microscopy was also used to assess the effect of JNJ-6640 on bacterial DNA replication. To visualize the replisome machinery, we constructed a reporter strain of M. tuberculosis, expressing DnaN as a fusion protein with GFP. This provides a real-time readout of the DNA replisome machinery, when actively replicating the bacterial chromosome14. When treated with 0.6 µM JNJ-6640, the bacteria stopped dividing, and DNA replication was inhibited, marked by the disappearance of the replisome (GFP–DnaN) foci over time, followed by cell lysis. Again, even after removal of JNJ-6640, not only was bacterial regrowth inhibited but these lysis events also continued, suggesting a persistent bactericidal effect even in the presence of a physiologically relevant mixture of nucleobases (Fig. 3c,d).

a, M. tuberculosis expressing TdTomato, cultured in a microfluidic device and imaged over 312 h at 1-h intervals, exposed to 0.6 µM JNJ-6640 (95–238 h). NB mix, nucleobase mix (239–311 h). Snapshot images of representative microcolony are shown in magenta (TdTomato fluorescence). Scale bars, 3 µm. b, Fraction of intact cells upon exposure to 0.6 µM JNJ-6640 (grey shading) and after washout and supplementation with 1× NB mix (blue shading), from the single-cell imaging experiments. The lines represent independent xy frames imaged. n = 1,195 cells, across 15 fields. A representative dataset from three independent experiments. c, Representative snapshots of M. tuberculosis replisome reporter strain (MTB::gfp-dnaN, green channel) expressing cytoplasmic TdTomato (magenta channel), before, during and after exposure to 0.6 µM JNJ-6640. Bacteria undergoing DNA replication identified by green foci representing active replisome complex. Scale bars, 3 µm. d, Number of bacteria with a GFP–DnaN foci when exposed to 0.6 µM JNJ-6640 (grey shading) and after washout and supplementation with 1× NB mix (blue shading). The lines (n = 8) represent individual xy positions imaged over time. A representative dataset from two independent experiments.
In vivo-relevant activity of JNJ-6640
To further characterize the bactericidal effects of JNJ-6640, we assessed the activity under different in vitro and ex vivo conditions. We first showed that JNJ-6640 is active in cholesterol media (MIC90 = 29.1 ± 1.7 nM), which is considered the main carbon source for M. tuberculosis in vivo15. JNJ-6640 was also active against intracellular bacteria in THP-1 cells (IC50 = 26.1 ± 10.1 nM), with comparable activity with in vitro axenic M. tuberculosis experiments (Extended Data Table 1). We further validated the intracellular activity of JNJ-6640 by carrying out imaging of macrophages infected with fluorescent M. tuberculosis over time (Extended Data Fig. 6c,d). JNJ-6640 prevented the expansion of intracellular bacteria, and this effect was maintained even when the macrophage growth media was supplemented with a mixture of nucleobases (Extended Data Fig. 6c,d).
During infection in vivo, M. tuberculosis persists in infected macrophages and in sites of caseous necrosis within granulomas. As such, the bactericidal activity of JNJ-6640 against non-replicating persistent M. tuberculosis was evaluated using the ex vivo rabbit caseum assay previously developed16,17,18. However, JNJ-6640 lacked activity in caseum at concentrations up to 128 µM (Extended Data Fig. 7a). In line with observations of hypoxia in necrotic TB granulomas19, we also assessed the bactericidal activity of JNJ-6640 in an intracellular hypoxic environment. In brief, our ‘foamy macrophage’ assay consisted of infected THP-1 cells incubated for 4 days in hypoxic conditions to generate lipid-loaded macrophages infected with non-replicating bioluminescent M. tuberculosis20 (Extended Data Fig. 7b,c). The ratio of IC50 values between the end of the hypoxic incubation (day 4) and after 1 day of normoxic regrowth (day 4 + 1) was used to characterize compounds, with IC50 ratios of more than 2 classed as bactericidal. Control compounds established to be either bactericidal (rifampicin; ratio = 96) or bacteriostatic (isoniazid; ratio = 0.65) under low-oxygen (dormancy) conditions showed the expected results, in line with their documented relative activities in other non-replicating and hypoxic assays (Extended Data Fig. 7d–f). JNJ-6640 demonstrated bactericidal activity in this hypoxic assay (ratio = 45; Fig. 4a), which is particularly encouraging as the majority of TB drugs are not effective under intracellular hypoxic conditions (Extended Data Fig. 7f).

a, Susceptibility of JNJ-6640 in a foamy macrophage assay. IC50 comparison at day 4 (30 µM) and day 4 + 1 (0.66 µM); ratio of 45 (bactericidal). n = 2 individual experiments with 4 technical replicates. b, JNJ-6640 (1,500 mg kg−1 LAI; subcutaneous) efficacy in an acute mouse model for TB administered once fortnightly (6640 1/14, 7 days post-infection and day 1 treatment phase) or once weekly (6640 2/14, 7 and 14 days post-infection, and day 1 and day 7 treatment phase). 21 Days post-infection data are shown. n = 5 animals. Representative of two independent experiments. BDQ, 25 mg kg−1 bedaquiline oral administration (PO) once daily (qd). c, JNJ-6640 (1,500 mg kg−1 LAI; subcutaneous) efficacy in the chronic mouse model for TB. 84 Days post-infection are shown. n = 6 animals. 6640 8/56, 8× doses, once weekly of JNJ-6640. d, In vitro combination studies replacing linezolid with JNJ-6640. Day 10 data are shown. 6640, 10 µM JNJ-6640; B, 0.5 µM bedaquiline; HR, 5.8 µM isoniazid and 14.58 µM rifampicin; L, 6 µM linezolid; Pa, 7 µM pretomanid. Bedaquiline, pretomanid and linezolid concentrations reflect the mouse equivalent human dose based on efficacious exposure. The full dataset is shown in Extended Data Fig. 9. n = 3 biological replicates, representative of two independent experiments. Dashed line indicates the starting inoculum. e, In vivo combination study demonstrating that JNJ-6640 can replace linezolid in a high acute model. Two week treatment. 24 Days post-infection are shown. 6,640, 1,500 mg kg−1 JNJ-6640 LAI (subcutaneous; once weekly); B, 25 mg kg−1 bedaquiline (PO qd); L, 100 mg kg−1 linezolid (PO qd); Pa, 40 mg kg−1 pretomanid (PO qd); SoT, start of treatment (10 days post-infection). n = 5 (SoT) or 6 (treatment groups) animals. For all panels, data shown are mean ± s.d. Significance was calculated with one-way analysis of variance (ANOVA) with Dunnett’s (b), Tukey’s multiple comparisons (d,e) or two-sided unpaired t-test (c).
PurF is a high-value target in vivo
Given the promising bactericidal profile of JNJ-6640, confirmation of the molecular target and clear selectivity versus the human orthologue, we next focused on demonstrating in vivo efficacy. Early ADME (absorption, distribution, metabolism and excretion) profiling revealed that JNJ-6640 was rapidly metabolized (high intrinsic clearance) in liver microsomes and hepatocytes, as well as showed inhibition of some cytochrome P450 (CYP) enzymes (Extended Data Table 3). Although the high intrinsic clearance did not translate into a high in vivo mouse clearance, the short half-life (t1/2 = 0.76 h) led to a rapid decline of JNJ-6640 plasma concentration (Extended Data Fig. 8a,b). Given the physicochemical properties of JNJ-6640 and low aqueous solubility, we used a long-acting injectable (LAI) aqueous-based suspension as an alternative approach to achieve in vivo proof of concept. LAI delivery systems allow for prolonged and sustained drug release into the bloodstream over extended periods of time21. Pharmacokinetic studies in mice showed that subcutaneous injection of a LAI formulation (1,500 mg kg−1) led to sustained exposure of JNJ-6640 for at least 4 weeks (Extended Data Fig. 8a,c). Furthermore, the dose was well tolerated in mice and no adverse clinical signs of toxicity or effect on body weight were observed for the duration of the study. On the basis of these data, JNJ-6640 was deemed to be safe and tolerable for an in vivo proof-of-concept study.
In a short acute model of infection, mice were inoculated with 200 colony-forming units (CFU) for 7 days, then treated with either one or two doses (1 dose per week) of 1,500 mg kg−1 JNJ-6640 LAI. Two doses led to a 1.8 log CFU reduction compared with vehicle control (Fig. 4b). A single injection also led to a more modest but statistical reduction in CFU, indicating a dose–response effect in in vivo activity. We were also able to demonstrate a reduction in bacterial burden in a chronic model of infection (Fig. 4c). Once weekly doses of 1,500 mg kg−1 JNJ-6640 LAI for 8 weeks resulted in a significant 0.5 log CFU reduction compared with vehicle control. These efficacy data provide clear proof of concept that an inhibitor of PurF can lead to a reduction in CFU in vivo.
JNJ-6640 contributes to TB regimens
Finally, we investigated the inclusion of JNJ-6640 in drug combinations for drug-resistant TB treatment. The recommended regimen for pre-XDR-TB requires a 6-month, all-oral combination therapy involving bedaquiline–pretomanid–linezolid22. One of the key rationales of this study was to identify compounds with novel modes of action that could become alternative combination partners for bedaquiline and pretomanid. This is owing primarily to the toxicity of linezolid as well as emerging drug resistance23,24,25. We first tested the replacement of linezolid with various concentrations of JNJ-6640 in combination with bedaquiline and pretomanid in vitro (Fig. 4d and Extended Data Fig. 9a). Replacement of linezolid with 0.1–10 µM JNJ-6640 led to a dose-dependent decrease in CFU compared with just bedaquiline and pretomanid. As JNJ-6640 led to a statistically significant reduction in CFU in vivo (Fig. 4b,c), we next replicated our combination studies in an animal model of infection (Fig. 4e). Here mice were inoculated with 10,000 CFU to effectively assess the contribution of JNJ-6640 activity to the bedaquiline–pretomanid backbone regimen. Human equivalent doses of bedaquiline and pretomanid were supplemented with either linezolid or JNJ-6640 over 2 weeks. This showed that combination with JNJ-6640 led to a similar decrease in CFU as seen for combination with linezolid, replicating our results from in vitro experiments. Finally, through DiaMOND analysis of pairwise combinations26, we have provided evidence that, as well as linezolid, JNJ-6640 also exhibits more favourable interactions with bedaquiline and pretomanid than moxifloxacin in the in vitro combinations tested (Extended Data Fig. 9b). Given the increasing prevalence of fluoroquinolone resistance, these findings highlight the potential of JNJ-6640 as a promising alternative to moxifloxacin in the standard-of-care regimen used for MDR-TB (bedaquiline, pretomanid, linezolid and moxifloxacin). Collectively, these data provide compelling evidence that inhibitors of purine biosynthesis could be an attractive addition to future treatment regimens for drug resistant-TB.
Discussion
Here we identified and validated a first-in-class small-molecule inhibitor of de novo purine biosynthesis via the inhibition of PurF, the first enzyme in the pathway. Targeting metabolic pathways in mycobacteria, such as purine biosynthesis, was previously viewed with scepticism due to possible on-target toxicity against human homologues. However, our current data, along with the previous identification of compounds targeting ATP synthase, such as bedaquiline27, or cytochrome bc1 (ref. 28) suggest that selectively targeting metabolic enzymes is an effective strategy for the development of new antibiotics to combat tuberculosis. The validation of PurF represents a much-needed new target for anti-TB drug discovery.
Starting from a phenotypic hit from a screening campaign, a medicinal chemistry programme resulted in the discovery of the lead molecule, JNJ-6640, which demonstrated remarkable potency against M. tuberculosis. A myriad of genetic and biochemical evidence showed that JNJ-6640 inhibits PurF. Correlation between enzymatic and in vitro activity, together with the selection of mutations within PurF conferring high-level resistance, provide clear evidence of target selectivity for MtPurF. Furthermore, we showed via an enzymatic assay with human PPAT, that this chemical series is highly selective for the mycobacterial PurF homologue. Finally, we demonstrated ex vivo and in vivo efficacy proof of principle for JNJ-6640 in various models, providing convincing evidence that affirm the critical role of PurF in the mycobacterial purine biosynthesis pathway and demonstrate that inhibition of PurF is a promising strategy for future TB drug discovery. The efficacy of JNJ-6640 varied across the different models used in this study and, due to the heterogeneous nature of TB disease in the clinical setting, highlights the importance of using multiple models when assessing compound efficacy.
The WHO target regimen profile outlines the need for more efficacious TB treatment regimens that allow for simpler, shorter and safer therapy of all forms of TB29. The long-term goal is the development of a pan-TB regimen consisting of three to four entirely novel drugs to overcome existing resistance mechanisms. In the near term, efforts are focused on replacing the two weakest components of the MDR or pre-XDR-TB standard-of-care regimens: moxifloxacin, due to resistance, and linezolid, due to toxicity. Our data suggest that replacing linezolid with JNJ-6640, in combination with bedaquiline and pretomanid, has comparable efficacy with bedaquiline–pretomanid–linezolid, the current standard of care for pre-XDR-TB treatment. This was supported with DiaMOND26 pairwise interaction data, which predicted that JNJ-6640 exhibits more favourable interactions with bedaquiline and pretomanid, than either linezolid or moxifloxacin. A systematic approach, supported by additional preclinical data, will be required to validate these interactions and to identify additional drug targets, thereby maximizing the potential contribution of PurF inhibitors to clinically relevant, next-generation treatment regimens.
Prolonged treatment durations in patients can contribute to non-compliance, leading to the emergence of drug-resistant strains and higher treatment failure. Shortening the duration of existing TB treatment regimens necessitates not only the discovery of new drugs but also the use of novel drug delivery approaches such as LAIs or new treatment regimens. The inclusion of LAIs in TB treatment has become an attractive option to reduce oral treatment length, overall pill burden and improve patient compliance. To this regard, an ongoing phase I study is exploring the safety and tolerability of a bedaquiline LAI in healthy volunteers30. Thus, although we utilized a JNJ-6640 LAI here to improve compound exposure and obtain in vivo efficacy, in general, assessment of compound LAI viability should be included early in TB drug discovery programmes to provide more LAI options for future TB treatment regimens.
We demonstrated that high exogenous concentrations of hypoxanthine can rescue the activity of JNJ-6640. Given the potential for rescue in patients, we quantified the physiological concentrations of nucleobases and nucleosides in human and mouse TB-infected lung tissue. Our findings clearly demonstrate that these concentrations are below those required to rescue JNJ-6640 activity. Furthermore, previous work, using a bespoke library of transposon mutants, demonstrated the essentiality of multiple components of de novo purine biosynthesis pathway (PurF, PurM and PurQ) for M. tuberculosis survival in mice, validating this pathway as a critical target for TB drug development5. Our in vivo efficacy data of JNJ-6640 also showed that the salvage pathway does not sufficiently compensate for the chemical inhibition of PurF. Together, these data suggest that de novo purine biosynthesis is critical for bacterial survival within host tissues and that host levels of nucleobases are insufficient to rescue JNJ-6640 activity. In addition, they also showed that M. tuberculosis cannot salvage purine metabolites in sufficient quantities to compensate for the loss of de novo biosynthesis.
Previous drug discovery efforts have focused on inhibitors that target GuaB2 (inosine-5′-monophosphate dehydrogenase; Rv3411c), an essential downstream member of the purine biosynthetic pathway, which converts IMP to XMP31,32,33. However, despite the promising in vitro activity of inhibitors targeting GuaB2, and demonstration that GuaB2 is essential to M. tuberculosis survival in vivo32, translating these findings into an in vivo efficacy proof of concept has proven challenging34. Nevertheless, as GuaB2 and PurF inhibitors are only rescued by high (non-physiologically relevant) concentrations of guanine and hypoxanthine, respectively, combining these inhibitors could offer a synergistic approach to enhance the efficacy of TB treatment regimens by simultaneously targeting distinct components of the same pathway.
Although JNJ-6640 represents a valuable tool compound for validating PurF as a novel drug target in M. tuberculosis, further optimization is required to progress towards a preclinical candidate. The metabolic instability of the compound is probably attributed to the central pyrrolidine core, as supported by in silico and in vitro metabolite identification studies. Although initial efforts to block this core enhanced in vitro metabolic stability, they also resulted in reduced anti-TB activity. Ongoing optimization efforts are focused on scaffold hopping of the pyrrolidine core to identify metabolically stable analogues, while maintaining the remarkable in vitro potency of JNJ-6640, for enhanced in vivo efficacy.
In conclusion, focusing on the unexplored biology and chemical space of key metabolic pathways led to the discovery and development of bedaquiline, the cornerstone component of standardized drug-resistant TB treatment. The discovery of JNJ-6640 and, more importantly the validation of PurF as a viable drug target, represents another validated metabolic target and an advancement in TB drug discovery research.
Methods
Ethics and inclusion statement
This research represents a collaborative effort between multiple research groups based in Belgium, the UK, South Africa, the USA, Canada, France and Spain. Authorship was determined solely based on substantial contributions to the conception, design, execution or interpretation of the work, and the roles and responsibilities of each author were defined according to their area of expertise. All studies involving animals or human participants received approval from the appropriate institutional ethics committees (see relevant sections) and were carried out in accordance with international and institutional ethical guidelines. All experiments involving M. tuberculosis or other pathogens were conducted in the appropriate biosafety-level facilities under strict containment protocols. No part of the research involved the use of identifiable human data or samples without consent.
Animal ethics statement
All the in vivo efficacy and lung nucleobase studies were performed at Janssen Pharmaceutica in Beerse, in a certified BSL3 facility in agreement with European Directive (2010/63/EU) and national directives for the protection of animals for experimental purposes. All procedures were approved by the Ethics Committee of Janssen Pharmaceutica (license number LA1100119), located in Beerse, Belgium, which is accredited by Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC) since 2004 under the unit number 001131 (https://www.aaalac.org/). For pharmacokinetic studies at Pharmaron, all procedures were approved by the Institutional Animal Care and Use Committee and Pharmaron holds full AAALAC accreditation.
Human ethics statement
The collection of human biological samples for this study was approved by the University of KwaZulu-Natal Biomedical Research Ethics Committee (BREC reference number BE019/13). The protocol includes the acquisition of lung tissue from anonymized patients undergoing pneumonectomy or lobectomy procedures at two public hospitals in Durban, South Africa: Inkosi Albert Luthuli Central Hospital and King Dinizulu Hospital Complex. Written, informed consent was obtained from all study participants who were routinely assessed for extent of disease and fitness for surgery according to standard imaging and clinical parameters. Of note, the patients with TB in this study cohort completed 9–24 months of anti-TB treatment before surgery, largely determined by M. tuberculosis susceptibility profiles. Lung tissue specimens from 15 patients with TB who were HIV negative (n = 15) had visible tubercles with a variable combination of associated haemorrhage, cavitation, fibrosis and bronchiectasis. Control lung specimens (n = 15) were obtained from patients with cancer who were TB and HIV negative undergoing lung surgery. Upon resection, representative tissue samples were immediately placed in conical tubes and snap frozen on dry ice before transport. Samples were then stored at −80 °C until thawed for homogenization.
Mice
Six-to-eight-week-old female Balb/cBy mice were purchased from Charles-River or Janvier. Mice were allocated in the BLS3 facility in individually ventilated cages in HEPA-filtered racks and rested for at least 5 days to allow acclimatization. Access to water and food was ad libitum.
Bacterial strains and growth conditions
The M. tuberculosis H37Rv strain was kindly provided by R. Brosch (Institut Pasteur). The strain was regularly passaged in mice to maintain virulence and retain PDIM-related genes. To prepare frozen stocks, H37Rv was grown in Middlebrook 7H9 (Becton-Dickinson) culture medium supplemented with 10% oleic acid–albumin–dextrose–catalase (OADC) complex (Becton-Dickinson), 0.2% glycerol and 0.05% Tween 80 (Sigma-Aldrich). Upon reaching stationary phase, the H37Rv culture was harvested in glycerol (15%; Becton-Dickinson) containing Middlebrook 7H9 medium and frozen at −80 °C.
Guided compound screening and MIC assay
Compounds screened in dose–response were tested in fourfold dilutions from 5 mM to 0.005 µM in black, clear bottom, 384-well microtitre plates (Greiner). Using an Echo liquid handler, a low volume dilution range in 100% DMSO was dispensed in the plates (150 nl per well). Reference plates were included as well as positive and negative control wells in each plate. M. tuberculosis H37Rv (5 × 105 CFU ml−1) diluted in Middlebrook 7H9 medium supplemented with 10% OADC, 0.2–0.5% glycerol and 0.05% Tween 80 was added to the compound plates (30 µl). Plates were incubated for 7 days at 37 °C. Before absorbance measurements (optical density at 620 nm (OD620)) using an Envision multimode plate reader (Perkin Elmer), the plates were shaken (4 min at 1,000 rpm). MIC50 values were determined as the lowest drug concentration, inhibiting at least 50% growth observed in the positive control wells using Microsoft Excel and presented as pMIC50 (Supplementary Fig. 1). For dose–response curves with JNJ-6640 or the CRISPRi-mediated transcript knockdown strain, plates were generated as above, with compounds tested in twofold dilutions, with the presence or absence of 100 ng ml−1 anhydrotetracycline (ATc). MIC90 values were determined as the lowest drug concentration inhibiting at least 90% growth observed in the positive control wells.
MBC99.9 assay
M. tuberculosis H37Rv (1 × 105 CFU ml−1) diluted in Middlebrook 7H9 medium supplemented with 10% OADC, 0.5% glycerol and 0.05% Tween 80 was added to 96-well plates containing compounds in dose–response. In each compound plate, two reference compounds in dose–response and positive control wells were included. After incubation of the plates for 17 days, 5 µl of each condition was stamped on 7H10 plates supplemented with 0.4% charcoal (Sigma-Aldrich), 0.5% glycerol and 10% OADC. In brief, a tenfold serial dilution was prepared of each concentration in PBS, plated on 7H10 six-well plates (plus 0.4% charcoal, 0.5% glycerol and 10% OADC) and incubated for 21 days before counting CFU. The MBC99.9 was defined as the concentration giving at least 3-log10 reduction compared with the initial CFU. For nucleotide rescue experiments, M. tuberculosis (5 × 105 CFU ml−1) was treated for 17 days in the presence of 1 µM JNJ-6640 supplemented with either various concentrations of hypoxanthine or adenosine.
Resistance generation
To isolate resistant colonies, agar plates containing approximately 25× MIC90 of either compound JNJ-7310, JNJ-6627 or JNJ-6640 were inoculated with WT M. tuberculosis (1–5 × 108 CFU) to select resistant colonies. Individual colonies were re-plated in the presence of compound for approximately 3 weeks to confirm resistance. Genomic DNA was isolated from resistant colonies using the Quick-DNA fungal and bacterial miniprep kit (Zymo Research). Following whole-genome sequencing, reads were aligned to the M. tuberculosis H37Rv genome (release 4, 2021-03-23; mycobrowser.epfl.ch). Comparison of the sequences of PurF homologues from different species, with the location of resistance mutations, is shown in Supplementary Fig. 2.
Molecular modelling
A model of MtPurF was generated using an AlphaFold model (AF-P9WHQ7-F1) and compared with crystal structures of other PRPP amindotransferases9,35,36,37,38. The SiteMap detection tool was used to predict putative binding pockets39,40 (D-score of 1.03 was detected at the PRTase active site; Supplementary Fig. 3), and JNJ-6640 was docked in the putative binding site using IFD-MD (Schrödinger)41,42,43. All SiteMap and IFD-MD calculations were performed using Schrödinger software (release 2024-2, Schrödinger).
Stable isotope labelling
For the 15N stable isotope labelling experiment, 10 ml M. tuberculosis H37Rv cultures (OD600 = 0.8 in 7H9 complete media) were treated with either DMSO (to 0.1% final concentration) or JNJ-6640 (100 nM final concentration) in DMSO (0.1% final concentration) and simultaneously supplemented with 15N-amide-glutamine (2 mM final concentration; Cambridge Isotope Laboratories). After 4 h incubation at 37 °C with shaking, cultures were centrifuged at 3,000g for 10 min at 4 °C. The supernatant was discarded and the pellet washed by resuspending in 150 mM ammonium acetate solution (pH 6.6) in ultra-pure water before centrifuging again as previously. Supernatant was discarded and pellet resuspended in 1 ml metabolite extraction solution (2:2:1 acetonitrile:methanol:water; all liquid chromatography–mass spectrometry (LC–MS) grade) and placed on dry ice for 10–20 min with frequent vortexing. Samples were then transferred to tubes containing 0.1-mm glass beads and the cells lysed using a Precellys 24 homogenizer (Bertin Technologies; 3 × 20 s at 6.0 ms−1). Samples were frozen at −80 °C overnight before being thawed, vortexed and centrifuged at 15,000g for 10 min at 4 °C. The supernatants were taken and filtered using prewashed Spin-X 0.22-μm centrifuge filters (Corning) by centrifuging for 15,000g for 10 min at 4 °C. Filtrates were stored at −70 °C until LC–MS analysis. For quality control purposes, additional bacterial samples were taken as above but using unlabelled glutamine, and background controls were taken by performing the same method on the media without bacteria and on the extraction solution only. Equal aliquots from each experimental sample were combined into a pooled sample. This pooled sample was used to create a dilution series and identical run control samples that were placed regularly throughout the LC–MS sample run.
LC–MS was performed using an Agilent 1290 Infinity II Bio LC system connected to an Agilent Accurate Mass 6546 QTOF mass spectrometer. Chromatography was performed using an InfinityLab Poroshell 120 HILIC-Z column (Agilent; 2.1 × 150 mm, 2.7 µm) at 15 °C. A binary gradient was used with 20 mM ammonium acetate in water (pH 9.3) containing 5 µM InfinityLab Deactivator Additive (Agilent) for mobile phase A and pure acetonitrile for mobile phase B. Flow rate was 400 μl min−1 with the solvent gradient changing linearly between the following times and holds: 0–1 min for 90% mobile phase B; 8 min for 78% phase B; 12 min for 60% phase B; and 15–18 min for 10% phase B. The solvent ratio was then returned to 90% phase B before re-equilibrating for 4.5 5 min. For MS, ionization was performed using ESI in negative mode with nebulizer pressure of 30 psig and a nitrogen drying gas flow rate of 9 l min−1 at a temperature of 225 °C. The capillary, nozzle and fragmentor voltages set to 3,000 V, 500 V and 80 V, respectively. The MS acquisition rate was 1 spectra per second. Metabolites were identified by their m/z values, MS/MS fragmentation, and comparison of fragmentation patterns and retention times to standards. Chromatogram alignment, targeted feature extraction, quantification and isotopologue analysis were performed using the Agilent MassHunter software suite. Stable isotope-labelled abundances were corrected for the natural abundance of 15N and confirmed by checking for lack of corresponding signal in the samples incubated with unlabelled glutamine. For quality control purposes, extracted metabolites with an r-squared value of less than 0.85 in the pooled sample dilution series or a relative standard deviation in the run control samples of more than 25% were removed from analysis. Metabolite abundances were normalized to the soluble protein content in each sample as measured by bichinchonic acid assay (Pierce BCA Protein assay kit, Thermo Scientific) performed as per the manufacturer’s instructions.
Measurement of glutamine levels
Ten millilitres of M. tuberculosis H37Rv cultures (OD600 = 0.8 in 7H9 complete media) was treated with either DMSO (to 0.1% final concentration) or JNJ-6640 (1,000 nM final concentration) in DMSO (0.1% final concentration). After 24 h of incubation at 37 °C with shaking, samples were processed for LC–MS analysis as described above. The LC–MS was performed using an Agilent 1290 Infinity II LC system connected to an Agilent Accurate Mass 6545 QTOF mass spectrometer. Chromatography was performed using an Cogent Diamond Hydride Type C silica HPLC column (MicroSolv) at 25 °C. A binary gradient was used with 0.2% acetic acid in water for mobile phase A and 0.2% acetic acid in acetonitrile for mobile phase B. The flow rate was 400 μl min−1 with the solvent gradient changing linearly between the following times and holds: 0–2 min for 85% mobile phase B; 3–5 min for 80% mobile phase B; 6–7 min for 75% mobile phase B; 8–9 min for 70% mobile phase B; 10–11 min for 50% mobile phase B; and 11–25 min for 20% mobile phase B. The solvent ratio was then returned to 85% phase B before re-equilibrating for 5 min. For MS, ionization was performed using ESI in positive mode with nebulizer pressure of 50 psig and a nitrogen drying gas flow rate of 5 l min−1 at 300 °C. The capillary, nozzle and fragmentor voltages were set to 1,500 V, 2,000 V and 100 V, respectively. Data were analysed as above with glutamine levels normalized to the sum of all the extracted metabolite intensities for each sample.
Generation of CRISPRi-mediated knockdown strains
DNA fragments encoding small guide RNAs (sgRNAs) that specifically target purF were cloned into the plRL2 plasmid as previously described4. In brief, complimentary single-stranded oligos, obtained from an online database of sgRNA sequences (pebble.rockefeller.edu), were annealed then cloned into plRL2 (Addgene) via BsmBI restriction sites using Golden Gate Assembly (Supplementary Table 1). Resulting colonies were Sanger sequenced to confirm the presence of the target sequence. Targeting and empty vector plRL2 plasmids (500 ng) were electroporated into WT M. tuberculosis H37Rv. Colonies were selected from 7H10 agar plates containing 50 µg ml−1 kanamycin. Inducible transcript knockdown using 100 ng ml−1 ATc was confirmed using quantitative real-time PCR (rt–qPCR). CFU counts were taken on days 7, 14 and 21 by plating serial dilutions onto 7H10 agar before incubation at 37 °C for 3 ± 1 weeks to measure the effect of transcript knockdown on growth over time.
rt–qPCR
CRISPRi-mediated knockdown strains with and without the presence of 100 ng ml−1 ATc or JNJ-6640-treated H37Rv were grown to mid-log phase and diluted to an OD600 of 0.05 in 7H9. The empty vector strain was grown in the presence of ATc. After 4 days, RNA was extracted using the RNeasy kit (Qiagen) and genomic DNA was depleted using Turbo DNAse (Invitrogen). cDNA was generated using Superscript III reverse transcriptase (Invitrogen). Relative transcript knockdown of each gene was determined using SYBR green and gene-specific primers (Supplementary Table 1). rt–qPCR was performed using the Applied Biosystems 7500 Fast Real-Time PCR System with the following conditions: 50 °C for 20 s, 95 °C for 10 min then 40 cycles of 95 °C for 10 s, 60 °C for 1 min followed by 95 °C for 15 s, 60 °C for 1 min, 95 °C for 30 s and 60 °C for 15 s. Transcripts of interest were normalized against sigA. Analysis was performed using the comparative Ct method44.
Determination of nucleobases levels in TB-infected and uninfected mouse lung tissue
Mice were infected intranasally with 200 CFU per mouse for 7 days before euthanization and lung collection. Lung tissue was snap frozen in liquid nitrogen and kept at −80 °C until further processing. Frozen lungs where homogenized in refrigerated GentleMACS M tubes containing 2.5 ml 70:30 methanol:water, using the GentleMAC Octa Dissociator on program ‘RNA_02_01’. Lung homogenate aliquots (50 µl) were transferred to screwcap Micronic tubes filled with 450 µl refrigerated acetonitrile, vortexed for 10 s, incubated on ice for 10 min and stored at −80 °C until bioanalysis.
Liquid chromatography was performed on an Acquity Premier system (Waters) and this was coupled to an Xevo TQ Absolute Triple Quadrupole Mass Spectrometer (Waters). For UHPLC analysis, a Cogent Diamond Hydride 4 u 2.1 × 150 mm column was used (MicroSolv Technology) at 30 °C. Solvent A consisted of 0.2% acetic acid in water and solvent B of 0.2% acetic acid in acetonitrile. Chromatographic separation of the nucleobases was obtained at a flow rate of 0.4 ml min−1 under the following conditions: gradient starting conditions were 95% solvent B with an isocratic hold for 2 min, then multiple linear gradient steps were applied each followed by an isocratic hold for 1 min, first to 80% solvent B in 3 min, followed by a linear gradient to 50% solvent B in 1 min and a final step to 20% solvent B in 0.5 min, afterwards the initial conditions were reached in 0.1 min. Total run time was 16 min to obtain optimal equilibration of the analytical column. The injection volume was 1 µl. Electrospray MS analysis was carried out on an Xevo TQ Absolute Triple Quadrupole Mass Spectrometer (Waters) operated in the positive ion mode. The instrument was optimized by flow injection for each nucleobase. The following internal standards were used: adenosine-13C10, 15N and xanthine-13C, 15N2. Selective reaction monitoring transitions were used as a quantifier: adenine (135.9 > 118.9), guanine (151.9 > 134.9), hypoxanthine (136.9 > 118.9), adenosine (268 > 118.9), guanosine (284.1 > 151.9), inosine (268.9 > 136.9) and xanthine (152.9 > 110); a second selective reaction monitoring transition was used as a qualifier adenine (135.9 > 92), guanine (151.9 > 110), hypoxanthine (136.9 > 109.9), adenosine (268 > 135.9), guanosine (284.1 > 134.9), inosine (268.9 > 110) and xanthine (152.9 > 135.9). For chromatographic evaluation, Waters TargetLynx software version 4.2 (Waters) was used. Calibration curves were plotted using appropriate log–log linear regression.
Human TB-infected and uninfected tissue homogenization and metabolite extraction
Wet mass of selected human lung specimens for homogenization ranged from 99 to 120 mg. Protocol for tissue homogenization was as follows: tissue sample was washed three times with 5 ml of refrigerated PBS (Gibco) and placed on adsorbent cloth to remove excess. Sample was then added to a refrigerated gentleMACs M tubes (Miltenyi Biotec) containing 1.3 ml of 70% methanol extraction solvent containing d4-alanine (Sigma) at a final concentration of 100 µg ml−1. Homogenization of tissue was performed using a gentleMACS Octo Tissue Dissociator (Miltenyi Biotec), using the ‘RNA_02_01’ protocol. Tubes were then centrifuged at 2,500g, 4 °C for 30 min and the supernatant transferred to a 70-µm nylon cell strainer (Falcon). The homogenate flow-through was then filter sterilized using 0.22-µm filter tubes (Costar Spin-X, Corning) at 13,000g at 4 °C for 10 min. Filtrates were then placed in a SpeedVac centrifugal concentrator (Thermo Scientific) and solvent removed overnight under vacuum. Dried residual solutes were reconstituted in high-purity water and placed in a Thermomixer (Eppendorf) at room temperature and shaken for 1 h (1,300 rpm). For LC–MS/MS analysis, samples were further diluted with acetonitrile (ACN) to a final solvent composition of 70% ACN.
LC–MS/MS analysis of human lung tissue
Metabolite extracts were analysed on a Thermo Q-Exactive mass spectrometer coupled to a Dionex Ultimate 3000 UPLC system. A Waters X-Bridge BEH Amide 2.1 × 100 mm HILIC column was used with mobile phase A (100% ACN) and mobile phase B (100% water), both containing 0.1% formic acid and run in gradient mode, at a flow rate of 200 μl min−1 and a temperature of 40 °C. The Q-Exactive parameters were as follows: ESI voltage of 3.5 kV was used, with a sheath gas flow rate of 45, aux gas of 10 and sweep gas of 2. Capillary temperature was kept at 250 °C. For high-resolution molecular ion scans, the range was from 50 to 750 m/z at a resolution of 70,000, in positive ion mode. For parallel reaction monitoring analysis, the basic conditions were the same, but collision energy values for each analyte were determined and were as follows: CE-40 for adenine, NCE-30 for, CE-38 for guanine, NCE-30 for guanosine, NCE-20 for inosine, CE-50 for hypoxanthine, NCE-35 for xanthine; for the internal control, d4-alanine, with a collision energy value of 10, was used, which was the minimum available and would suppress fragmentation. The gradient conditions for both methods were as follows: 0 min for 90–10% ACN to water then 15 min, 30–70% ACN to water, with a linear gradient and finally 15.1–26 min at 70% ACN. A solvent composition of 90% ACN to 10% water was used for column regeneration. Each sample was analysed twice: once using high-resolution molecular ion scan and a second time with the MS in parallel reaction monitoring mode where target molecular ions were fragmented and the spectra accumulated for further post-run analysis. Expected retention times on these methods were obtained from running high-purity, analytical standards (IROA Technologies, Thermo Scientific) for each analyte under the same conditions and used for molecule verification purposes. An external calibration series was prepared using the same high-purity, reference compounds for guanine, hypoxanthine, adenine, xanthine, guanosine, inosine and adenosine and used to calculate the concentration of analytes in the tissue samples. Peak area under curve values for analytes measured in samples were normalized to wet mass of the tissue. The RAW files obtained were processed using Skyline software (24.1.1.339) set up for this analysis and peak areas determined for both sample analytes and the external calibration standards. All solvents used for MS analysis were of highest purity.
Assembly of constructs for enzymatic assays
Gene synthesis and cloning were performed at Epoch Life Sciences. DNA encoding MtPurF (UniProt P9WHQ7) residues 35–527 was optimized for E. coli expression, synthesized and inserted into a custom-engineered pET28a vector. DNA encoding Homo sapiens PPAT (UniProt Q06203) residues 1–517 was optimized for Sf9 expression, synthesized and inserted into the baculovirus transfer vector pVL1393. The open reading frame of codon-optimized DNA sequences are documented in Supplementary Fig. 4.
Expression of MtPurF
BL21(DE3) chemically competent cells (New England Biolabs) were transformed with a pET28a vector containing a MtPurF expression cassette with a C-terminal tag for affinity purification. After transformant selection on LB agar plates supplemented with kanamycin (50 µg ml−1), a 500-ml starter culture was inoculated with a single colony and incubated overnight with shaking at 37 °C. The following day, the starter culture was diluted (1:50) into fresh medium with 50 µg ml−1 kanamycin for expression. Cultures were grown with shaking to mid-log phase (OD600 ~ 1) and induced with 1 mM isopropylthio-β-galactoside. After induction, cultures were transferred to a 16 °C shaker and expression was allowed to proceed overnight. The next day, cells were harvested by centrifugation (5,000g for 15 min at 4 °C). The pellet was transferred to a sterile bag and frozen at −80 °C. The soluble expression yield, evaluated by western blot, was estimated to be more than 25 mg l−1.
Expression of H. sapiens PPAT
Human PPAT was expressed in insect Sf9 cells using a baculoviral expression system. In brief, recombinant baculovirus was generated using the BestBac 2.0 system (Expression Systems) following the manufacturer’s instructions. Linearized Δv-cath/chiA baculovirus DNA was co-transfected with a pVL1393 transport vector containing a human PPAT expression cassette with C-terminal sortase, FLAG and 8-His tags into Sf9 cells using Cell Fectin II Reagent (Thermo Fisher) in a plate-based format. P0 virus was harvested and P1 baculovirus-infected insect cells (BIICs) were generated following protocol based on a method previously described45. Before large-scale expression, an additional round of amplification was performed to generate P2 BIICs. BIICs were stored in liquid nitrogen. Expression of human PPAT was carried out in Sf9 cells at an effective multiplicity of infection (MOI) of 0.3. Cells were expanded to reach desired volume and split back to approximately 2 × 106 cells per millilitre on the day of infection. P2 BIICs were incubated at 27 °C until almost thawed then diluted slowly in a small volume of expression medium. The diluted P2 BIICs were then added directly to the culture, and expression was allowed to proceed for approximately 72 h. After expression, cells were collected by centrifugation (1,000g for 20 min at 4 °C). The pellet was transferred to a sterile bag and frozen at −80 °C. The soluble expression yield, evaluated by western blot, was estimated to be 1–5 mg l−1.
Purification of MtPurF
All buffers were extensively sparged with N2 to minimize oxidation of the 4Fe–4S cluster. All purification steps were performed at 4 °C unless noted. The cell pellet was resuspended in 5 ml g−1 of lysis buffer containing 25 mM HEPES pH 7.5, 500 mM NaCl, 10 mM MgCl2, 10% glycerol and 1 mM dithiothreitol. The suspension was lysed by sonication and clarified by centrifugation for 30 min at 35,000g. Clarified supernatant was incubated with 1 ml l−1 CaptureSelect C-tagXL Affinity Matrix (Thermo Fisher Scientific) for 1 h with gentle rolling. Slurry was filtered over a 2.5-cm Bio-Rad Econo-Column. Bound resin was washed with 60 column volume (CV) of lysis buffer. Lysis buffer was supplemented with 3 mM C-tag peptide (Biosynth International) to generate elution buffer. Resin was eluted with 5 × 1 CV of elution buffer; each elution was incubated with the resin for 5 min before collecting. Elution pool was concentrated to 0.5 ml using an Amicon Ultra 30 kDa MWCO centrifugal filter. Concentrated sample was resolved on a Superdex 200 10/300 size-exclusion chromatography column (Cytiva) in buffer containing 25 mM HEPES pH 7.5, 150 mM NaCl, 2 mM MgCl2 and 1 mM dithiothreitol. Size-exclusion fractions containing PurF were pooled, aliquoted and frozen at −80 °C for storage.
Purification of H. sapiens PPAT
All buffers were extensively sparged with N2 in attempt to minimize oxidation of the 4Fe–4S cluster. All purification steps were performed at 4 °C unless noted. The cell pellet was resuspended in 5 ml g−1 of lysis buffer containing 25 mM HEPES pH 7.5, 500 mM NaCl, 10 mM MgCl2, 2 mM AMP, 25 mM imidazole, 10% glycerol and 10 mM dithiothreitol. The suspension was lysed by sonication and clarified by centrifugation for 30 min at 35,000g. Supernatant was applied to a 5-ml HisTrapFF column (Cytiva) and washed with a stepwise gradient of lysis buffer supplemented with either 50 mM, 75 mM or 100 mM imidazole before eluting with buffer containing 25 mM HEPES pH 7.5, 500 mM NaCl, 10 mM MgCl2, 2 mM AMP, 500 mM imidazole, 10% glycerol and 10 mM dithiothreitol. Elution fractions containing PPAT had a light brown colour. The PPAT-containing fraction was pooled and subjected to ammonium sulfate precipitation to enhance purity. The sample was brought to an ammonium sulfate saturation of 40% and incubated for 30 min. The 40% ammonium sulfate precipitated sample was centrifuged for 5 min at 16,000g. After centrifugation, brown supernatant containing PPAT was removed from the white-precipitated pellet, calibrated to an ammonium sulfate saturation of 50% and incubated for 30 min. The 50% ammonium sulfate precipitated sample was then centrifuged for 5 min at 16,000g. Clear supernatant was removed, and the brown-precipitated pellet containing PPAT was resuspended in lysis buffer, aliquoted and frozen at −80 °C for storage. The supporting information for PPAT purification can be found in Supplementary Fig. 5.
Biochemical MtPurF inhibition
A schematic of PurF enzyme function is shown in Supplementary Fig. 6. PurF (20 nM) was incubated with varying concentrations of JNJ-6640 for 30 min in 25 mM Tris pH 8, 10 mM MgCl2, 0.5 mg ml−1 BSA and 0.001% Tween-20, before the addition of 70 mM glutamine and 2.5 mM phosphoribosyl pyrophosophate in a Revvity 384w ProxiPlate Plus for a total reaction volume of 4 µl. Reactions were carried out for 3 h at room temperature before quenching with 20 mM AMP. Glutamate oxidase (0.05 U ml−1) was added and allowed to incubate for 1 h at room temperature. An equal volume of HyPerBlu reagent was added and allowed to incubate for 30 min at room temperature before reading luminescence on a BMG Pherastar FSX.
Biochemical human PPAT inhibition
PPAT (5 nM) was incubated with varying concentrations of JNJ-6640 for 30 min in 25 mM Tris pH 8, 10 mM MgCl2, 0.5 mg ml−1 BSA and 0.001% Tween-20, before the addition of 200 µM glutamine and 30 µM phosphoribosyl pyrophosophate in a Revvity 384w ProxiPlate Plus for a total reaction volume of 4 µl. Reactions were carried out for 3 h at room temperature before quenching with 25 mM EDTA. Glutamate oxidase (0.05 U ml−1) was added and allowed to incubate for 1 h at room temperature. An equal volume of HyPerBlu reagent was added and allowed to incubate for 30 min at room temperature before reading luminescence on a BMG Pherastar FSX.
Cell proliferation assay
Measurement of JNJ-6640 activity against a range of 93 cell lines was performed by Oncolead (Germany). In brief, the cell lines (see source data for Fig. 2h) were treated with varying concentrations (0.25 nM to 25 µM) of JNJ-6640, azathioprine, MMPR and MPA in vitro for 72 h. Growth inhibition was measured using sulforhodamin B, a protein staining assay, to calculate the pIC50 of the compounds in each cell line.
Timelapse microscopy experiments
WT M. tuberculosis (Erdman strain) transformed with pND257 expressing tdTomato was grown and seeded for imaging into a microfluidic device as previously described12,46. Imaging of the bacteria was carried out on an inverted wide-field fluorescent microscope (Thunder Imaging System, Leica Microsystems), equipped with an environmental chamber maintained at 37 °C (Okolab). The bacteria were imaged using a ×100/1.32 NA oil immersion objective (Leica Microsystems) on the phase and red (635 nm excitation and 642 nm emission) channels and images captured using a scientific CMOS K8 camera (Leica Microsystems). Imaging was carried out at 60-min intervals over a period of 13–16 days. The full timelapse movie can be found in Supplementary Video 1. Medium was pumped through the device with a flow rate of 10 µl min−1 using a syringe pump. When necessary, the medium was supplemented with 0.6 µM JNJ-6640 and/or 1× physiologically relevant nucleobase mix (final concentrations: 518 nM guanine, 5.2 µM hypoxanthine, 4.3 µM adenine, 1.42 µM guanosine, 15 µM inosine and 23 µM adenosine, reconstituted in DMSO; Supplementary Table 2). Experiments were carried out at least two times for each condition and about 20–30 distinct xy positions were imaged in each experiment. Images were acquired and assembled using the LAS-X software (Leica Microsystems) and analysed using FIJI software47. For imaging of M. tuberculosis-infected macrophages, mouse bone marrow-derived macrophages were isolated and differentiated as described earlier12. For infection, M. tuberculosis bacteria transformed with pND257 (expressing tdTomato) was grown to OD600 of 0.4–0.8, washed and resuspended in DMEM medium. Bacteria were filtered through a 5-μm filter to get rid of bacterial clumps and the filtrate was used to infect the macrophages at an MOI of 1:1, over a period of 4 h. Imaging of the infected macrophages was carried out on an inverted wide-field fluorescent microscope (Thunder Imaging System, Leica Microsystems), in an environmental chamber maintained at 37 °C and 5% CO2 (Okolab), using a ×20/0.8 NA dry objective (Leica Microsystems) on the brightfield and fluorescence channel 555 nm excitation and 594 nm emission) channels. Images were captured every 2 h over 7–10 days using a scientific CMOS K8 camera (Leica Microsystems). Full timelapse movies can be found in Supplementary Videos 2 and 3. At least 20 independent xy positions (3 ×1 μm z-steps) were imaged for each condition: no treatment, treatment with 3 μM JNJ-6640, treatment with 3 μM JNJ-6640 plus supplementation with 100 ng ml−1 IFNγ, treatment with 3 μM JNJ-6640 plus supplementation with 100 ng ml−1 IFNγ and 1× physiologically relevant nucleobases mix (Supplementary Table 2). Images were acquired and assembled using the LAS-X software (Leica Microsystems) and analysed using FIJI software47.
Rabbit caseum MBC assay
The rabbit caseum MBC assay was performed to assess the activity of JNJ-6640 against non-replicating bacteria in ex vivo rabbit caseum homogenate, as previously described16,17,18. In brief, a 50 mM stock solution was serially diluted in DMSO to achieve the final concentration range of 0.125–128 µM. The assay was conducted in a 96-well plate format, with a 7-day incubation at 37 °C. After incubation, caseum homogenate was sampled from each well and plated on 7H11 agar. Colony enumeration was performed after 4 weeks.
Confirming dormancy in foamy macrophage assay
The transcript levels of Tgs1 (Rv3130c), hspX (Rv2031c) and Rv3290c, known to be upregulated in stationary or low-oxygen conditions20,48, were used to confirm the induction of dormancy in our assay. Total RNA was extracted 4 days post-infection after hypoxia-infected or normoxia-infected THP-1 from at least three different batches. In brief, to protect RNA from degradation, macrophages were rinsed with PBS, then scraped with Maxwell RSC miRNA Tissue Kit homogenization solution/thioglycerol (50/1) (Promega). Followed by 10 min of incubation with Maxwell RSC miRNA Tissue Kit lysis buffer (Promega), cells and bacteria were lysed by bead beating into matrix B tubes containing silica beads (MP Biomedical) with the Super-Fast Prep-1 instrument (MP Biomedical). Finally, samples were processed into a Maxwell RSC instrument for RNA extraction. Reverse transcription was performed with 100 ng total RNA using SuperScript IV VILO Master (Applied Biosystems) or without reverse transcriptase (−RT). qPCR amplifications were run with a QuantStudio 12K Flex system (Applied Biosystems) using the oligos in Supplementary Table 1. The mRNA content was normalized to 16S expression, and relative expression was calculated following the ΔCt method (ΔCt = Ct(gene) − Ct(16S)) and expressed as 2−ΔCt. Also note that no change in CFU was observed between days 0 and 4, further confirming non-replication.
Determining bactericidality in foamy macrophages
Human THP-1 cells (American Type Culture Collection TIB-202) maintained in RPMI 1640 medium containing 10% fetal bovine serum (FBS), 1 mM pyruvate and 2 mM l-glutamine. A luminescent M. tuberculosis H37Rv reporter strain (expressing LuxABCDE) was grown in Middlebrook 7H9 broth supplemented with 10% ADC, 0.4% glycerol and 0.05% Tween 80 until the mid-log phase. THP-1 cells were infected at a MOI of 0:4 in antibiotic-free RPMI 1640 medium containing 10% FBS, 1 mM pyruvate, 2 mM l-glutamine and 40 ng ml−1 phorbol 12-myristate 13-acetate for 4 h at 37 °C with 5% CO2. Following a 4-h incubation period, infected cells were harvested, washed and plated onto 96-well plates containing compounds (up to 30 μM). Infected cells were incubated under hypoxia (using anaeroPouch, Biomerieux). After 4 days of incubation under hypoxic condition, cell luminescence was measured using an Envision plate reader. The medium was then replaced, and cells were incubated under normoxia for 1 day. The luminescence was measured a second time using an Envision plate reader. Results were calculated in percent inhibition and analysed with Genedata software using the equation shown for ‘Intramacrophage MIC90 assay with M. tuberculosis’ shown in the Supplementary Methods.
Pharmacokinetics and tolerability in mouse
The pharmacokinetics of JNJ-6640 was investigated in female BALB/c mice dosed as solution at 1 mg kg−1 intravenously. JNJ-6640 was also dosed orally at 5 and 50 mg kg−1 as a solution. For the intravenous and oral arms, three animals were used. Animals had free access to food and water through each study. Blood samples were taken at multiple timepoints up to 24 h for intravenous dosing. The test compound was also dosed at 1,500 mg kg−1 subcutaneously as an LAI aqueous suspension. Three animals were used. Animals had free access to food and water through each study. Blood samples were taken at multiple timepoints up to 672 h after subcutaneous dosing. The animals were observed for any clinical signs of toxicity and effect on body weights during the 28-day period after subcutaneous administration of 1,500 mg kg−1. At the end of 672 h, the blood samples were also collected for the clinical chemistry evaluation. Plasma samples were prepared by protein precipitation with acetonitrile, and the supernatant was analysed for concentrations of compound using a qualified LC–MS/MS method.
Animal models
All experiments used 6–8-week-old female BALB/cBy mice. Regarding sample size, various scenarios were evaluated of 4–10 animals per group. From the power analysis, it was concluded that considering 6 animals per group provided more than 80% power to detect all significant effects of 1.5 CFU (log10), assuming s.d. = 0.5, with 20 or less groups (including the reference group). No blinding or randomization was performed.
In the short acute model, experiments were performed as previously detailed49,50. Mice were infected intranasally with 200 CFU M. tuberculosis H37Rv per mouse. To verify the infection level, a subgroup of six mice were euthanized 1 day after infection. After 1 week of infection, treatment was started. Either the LAI was administered (subcutaneous) on day 1 and day 7 or daily PO administration for 12 consecutive days. Mice were then euthanized 3 days after the last dose to prevent carry over effect. To monitor the evolution of the infection, a group of mice were euthanized 7 days post-infection, when treatment started, and 21 days post-infection, when treatment had ended. Negative control mice remained untreated.
In the chronic model, mice were infected intranasally with 200 CFU M. tuberculosis H37Rv per mouse. To verify the infection level, a subgroup of six mice were euthanized 1 day after the infection. Mice were infected 1 month before treatment start and treatment lasted for 8 weeks. Six mice were euthanized at the start of treatment as a pretreatment control. Negative control mice remained untreated.
In the high acute model, mice were infected intranasally with 10,000 CFU M. tuberculosis H37Rv per mouse51. To verify the infection level, a subgroup of six mice were euthanized 1 day after the infection. Mice were infected for 10 days before treatment start and treatment lasted for 2 weeks. Six mice were euthanized at the start of treatment as a pretreatment control. As the high infection levels would lead to severe clinical signs, no ‘vehicle control’ mice were used in this study. Mice were dosed by oral gavage (100 µl, drencher with rounded end straight, 0.9 mm × 25 mm, Socorex Swiss) except for the LAI formulation that was injected subcutaneously in the upper back (100 µl) using a needle (26 G × 13 mm, BD Microlance). At euthanization, whole lungs were aseptically collected in GentleMACS tubes (M tubes with strainer, Miltenyi Biotec) containing 2.5 ml of PBS and homogenized using the RNA_01_01 settings of GentleMACS Octo Dissociator (Miltenyi Biotec). Lung homogenate was diluted in PBS and plated in 7H10 charcoal agar plates containing antibiotics (100 µg ml−1 amphotericin, 25 µg ml−1 polymyxin B, 50 µg ml−1 carbenecillin and 20 µg ml−1 trimethoprim). Plates were incubated at 37 °C during 3–5 weeks. After that, CFU counts were recorded and data were expressed in the mean log CFU per lung for each group. Statistical analysis was done by one-way analysis of variance (ANOVA) with Sidak’s test for multiple comparisons or an unpaired t-test (GraphPad Prism). An outline of the three mouse models can be found in Supplementary Fig. 7.
Measurement of pairwise drug interactions
Pairwise drug interactions with JNJ-6640 were measured using a modified checkerboard assay with DiaMOND, with interactions quantified using the fraction inhibitory concentrations at the 50% growth inhibitory levels and calculated based on Loewe additivity as the null model. Culturing conditions, experimental design and analysis for drug interaction measurements with JNJ-6640 were as previously described52 so that drug interaction profiles with JNJ-6640 (measured in this study) could be directly compared with drug interaction profiles with moxifloxacin (measured previously52). In brief, M. tuberculosis were adapted to four different in vitro conditions (with butyrate or 0.2 mM cholesterol as carbon sources, a simple dormancy model and standard growth conditions) before drug treatment for the maximum length used in these assays (10, 24 and 5 days of treatment for butyrate, cholesterol and standard, respectively, and 7 days of treatment followed by 6 days of recovery for the dormancy model). Single and combination dose–response curves to calculate fractional inhibitory concentration at 50% inhibition (FIC50) values via DiaMOND were made in these growth conditions to model aspects of the microenvironments where M. tuberculosis are resident during infection52,53. Bedaquiline and pyrazinamide were sourced from Sigma, and pretomanid was sourced from APExBIO. Single-use aliquots of stock antibiotic solutions were prepared in DMSO and stored at −30 °C.
Material availability
All unique materials used in this article are readily available from the authors.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data supporting the findings of this study are available in this published article (and its Supplementary Information). Should any raw data files be needed in another format, they are available from the corresponding authors on reasonable request. The synthesis and chemical verification of all compounds described are provided in the Supplementary Methods. Source data are provided with this paper.
Code availability
No custom codes or mathematical algorithms were used in the article. Data from the dose–response curves were measured with Perkin Elmer EnVision and modelled using Genedata. Microscopy data were processed using LAS-X and FIJI software. Mass spectroscopy data were obtained using Thermo Q-Exactive and Waters TargetLynx, then further analysed using either the Skyline software (24.1.1.339) or the Agilent MassHunter software suite. The ACD/Spectrus software (2023 v1.1) and Bruker TOPSPIN programme (v4.1) were used to support medicinal chemistry efforts. Schrödinger software was used for the computational modelling (release 2024-2). Final analysis and figure preparation were performed using Microsoft Excel and GraphPad Prism 10.
References
TB alliance. Get the facts. TB Alliance https://www.tballiance.org/why-new-tb-drugs/global-pandemic (2023).
World Health Organization. Global tuberculosis report 2023. WHO https://www.who.int/teams/global-tuberculosis-programme/tb-reports/global-tuberculosis-report-2023 (2023).
Bald, D., Villellas, C., Lu, P. & Koul, A. Targeting energy metabolism in Mycobacterium tuberculosis, a new paradigm in antimycobacterial drug discovery. mBio 8, e00272-17 (2017).
Bosch, B. et al. Genome-wide gene expression tuning reveals diverse vulnerabilities of M. tuberculosis. Cell 184, 4579–4592.e24 (2021).
Block, A. M., Wiegert, P. C., Namugenyi, S. B. & Tischler, A. D. Transposon sequencing reveals metabolic pathways essential for Mycobacterium tuberculosis infection. PLoS Pathog. 20, e1011663 (2024).
DeJesus, M. A. et al. Comprehensive essentiality analysis of the Mycobacterium tuberculosis genome via saturating transposon mutagenesis. mBio 8, e02133-16 (2017).
Phelan, J. et al. An open-access dashboard to interrogate the genetic diversity of Mycobacterium tuberculosis clinical isolates. Sci. Rep. 14, 24792 (2024).
Zhang, Y., Morar, M. & Ealick, S. E. Structural biology of the purine biosynthetic pathway. Cell. Mol. Life Sci. 65, 3699–3724 (2008).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Ducati, R. G., Breda, A., Basso, L. A. & Santos, D. S. Purine salvage pathway in Mycobacterium tuberculosis. Curr. Med. Chem. 18, 1258–1275 (2011).
Wallengren, K. et al. Drug-resistant tuberculosis, KwaZulu-Natal, South Africa, 2001–2007. Emerg. Infect. Dis. 17, 1913–1916 (2011).
Toniolo, C., Dhar, N. & McKinney, J. D. Uptake‐independent killing of macrophages by extracellular Mycobacterium tuberculosis aggregates. EMBO J. 42, e113490 (2023).
Koul, A. et al. Delayed bactericidal response of Mycobacterium tuberculosis to bedaquiline involves remodelling of bacterial metabolism. Nat. Commun. 5, 3369 (2014).
Santi, I., Dhar, N., Bousbaine, D., Wakamoto, Y. & McKinney, J. D. Single-cell dynamics of the chromosome replication and cell division cycles in mycobacteria. Nat. Commun. 4, 2470 (2013).
Pandey, A. K. & Sassetti, C. M. Mycobacterial persistence requires the utilization of host cholesterol. Proc. Natl Acad. Sci. USA 105, 4376–4380 (2008).
Ashwath, P., Osiecki, P., Weiner, D., Via, L. E. & Sarathy, J. P. Role of DNA double-strand break formation in gyrase inhibitor-mediated killing of nonreplicating persistent Mycobacterium tuberculosis in caseum. ACS Infect. Dis. 10, 3631–3639 (2024).
Sarathy, J. P. et al. Extreme drug tolerance of Mycobacterium tuberculosis in caseum. Antimicrob. Agents Chemother. 62, e02266-17 (2018).
Ramey, M. E. et al. Drug distribution and efficacy of the DprE1 inhibitor BTZ-043 in the C3HeB/FeJ mouse tuberculosis model. Antimicrob. Agents Chemother. 67, e0059723 (2023).
Sarathy, J. P. & Dartois, V. Caseum: a niche for Mycobacterium tuberculosis drug-tolerant persisters. Clin. Microbiol. Rev. 33, e00159-19 (2020).
Daniel, J., Maamar, H., Deb, C., Sirakova, T. D. & Kolattukudy, P. E. Mycobacterium tuberculosis uses host triacylglycerol to accumulate lipid droplets and acquires a dormancy-like phenotype in lipid-loaded macrophages. PLoS Pathog. 7, e1002093 (2011).
Chaudhary, K., Patel, M. M. & Mehta, P. J. Long-acting injectables: current perspectives and future promise. Crit. Rev. Ther. Drug Carrier Syst. 36, 137–181 (2019).
World Health Organization. New treatment for drug-resistant tuberculosis. WHO https://www.who.int/teams/global-tuberculosis-programme/tb-reports/global-tuberculosis-report-2023/featured-topics/new-treatment-tb (2023).
Vengurlekar, D. et al. Linezolid resistance in patients with drug-resistant TB. Int. J. Tuberc. Lung Dis. 27, 567–569 (2023).
Eimer, J. et al. Association between increased linezolid plasma concentrations and the development of severe toxicity in multidrug-resistant tuberculosis treatment. Clin. Infect. Dis. 76, e947–e956 (2022).
Wasserman, S. et al. Linezolid toxicity in patients with drug-resistant tuberculosis: a prospective cohort study. J. Antimicrob. Chemother. 77, 1146–1154 (2022).
Cokol, M., Kuru, N., Bicak, E., Larkins-Ford, J. & Aldridge, B. B. Efficient measurement and factorization of high-order drug interactions in Mycobacterium tuberculosis. Sci. Adv. 3, e1701881 (2017).
Andries, K. et al. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 307, 223–227 (2005).
Pethe, K. et al. Discovery of Q203, a potent clinical candidate for the treatment of tuberculosis. Nat. Med. 19, 1157–1160 (2013).
World Health Organization. Target regimen profiles for tuberculosis treatment, 2023 update. WHO https://www.who.int/publications/i/item/9789240081512 (2023).
Janssen Cilag International. A single ascending dose, single-centre study, to assess pharmacokinetics, safety and tolerability of a single intramuscular dose of bedaquiline long-acting injection formulation in healthy participants (EU Clinical Trial 2023-508810-41-00). EU Clinical Trials https://euclinicaltrials.eu/search-for-clinical-trials/?lang=en&EUCT=2023-508810-41-00 (2024).
Cox, J. A. et al. Novel inhibitors of Mycobacterium tuberculosis GuaB2 identified by a target based high-throughput phenotypic screen. Sci. Rep. 6, 38986 (2016).
Singh, V. et al. The inosine monophosphate dehydrogenase, GuaB2, is a vulnerable new bactericidal drug target for tuberculosis. ACS Infect. Dis. 3, 5–17 (2017).
Usha, V., Hobrath, J. V., Gurcha, S. S., Reynolds, R. C. & Besra, G. S. Identification of novel Mt-Guab2 inhibitor series active against M. tuberculosis. PLoS ONE 7, e33886 (2012).
Park, Y. et al. Essential but not vulnerable: indazole sulfonamides targeting inosine monophosphate dehydrogenase as potential leads against Mycobacterium tuberculosis. ACS Infect. Dis. 3, 18–33 (2017).
Ahmad, S. et al. An interbacterial toxin inhibits target cell growth by synthesizing (p)ppApp. Nature 575, 674–678 (2019).
Krahn, J. M. et al. Coupled formation of an amidotransferase interdomain ammonia channel and a phosphoribosyltransferase active site. Biochemistry 36, 11061–11068 (1997).
Varadi, M. et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 50, D439–D444 (2021).
Chemical Computing Group. Molecular Operating Environment (MOE), 2022.02. Chemical Computing Group https://www.chemcomp.com/en/Research.htm (2024).
Halgren, T. New method for fast and accurate binding-site identification and analysis. Chem. Biol. Drug Des. 69, 146–148 (2007).
Halgren, T. A. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 49, 377–389 (2009).
Coskun, D. et al. Using AlphaFold and experimental structures for the prediction of the structure and binding affinities of GPCR complexes via induced fit docking and free energy perturbation. J. Chem. Theory Comput. 20, 477–489 (2024).
Miller, E. B. et al. Reliable and accurate solution to the induced fit docking problem for protein-ligand binding. J. Chem. Theory Comput. 17, 2630–2639 (2021).
Xu, T. et al. Induced-fit docking enables accurate free energy perturbation calculations in homology models. J. Chem. Theory Comput. 18, 5710–5724 (2022).
Schmittgen, T. D. & Livak, K. J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 3, 1101–1108 (2008).
Wasilko, D. J. et al. The titerless infected-cells preservation and scale-up (TIPS) method for large-scale production of NO-sensitive human soluble guanylate cyclase (sGC) from insect cells infected with recombinant baculovirus. Protein Exp. Purif. 65, 122–132 (2009).
Dhar, N. & Manina, G. Single-cell analysis of mycobacteria using microfluidics and time-lapse microscopy. Methods Mol. Biol. 1285, 241–256 (2015).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Voskuil, M. I., Visconti, K. C. & Schoolnik, G. K. Mycobacterium tuberculosis gene expression during adaptation to stationary phase and low-oxygen dormancy. Tuberculosis 84, 218–227 (2004).
Ray, P. C. et al. Spirocycle MmpL3 inhibitors with improved hERG and cytotoxicity profiles as inhibitors of Mycobacterium tuberculosis growth. ACS Omega 6, 2284–2311 (2021).
Akester, J. N. et al. Synthesis, structure-activity relationship, and mechanistic studies of aminoquinazolinones displaying antimycobacterial activity. ACS Infect. Dis. 6, 1951–1964 (2020).
Lyons, M. A. et al. Use of multiple pharmacodynamic measures to deconstruct the Nix-TB regimen in a short-course murine model of tuberculosis. Antimicrob. Agents Chemother. 68, e0101023 (2024).
Larkins-Ford, J., Degefu, Y. N., Van, N., Sokolov, A. & Aldridge, B. B. Design principles to assemble drug combinations for effective tuberculosis therapy using interpretable pairwise drug response measurements. Cell Rep. Med. 3, 100737 (2022).
Larkins-Ford, J. et al. Systematic measurement of combination-drug landscapes to predict in vivo treatment outcomes for tuberculosis. Cell Syst. 12, 1046–1063.e7 (2021).
Tweed, C. D. et al. Bedaquiline, moxifloxacin, pretomanid, and pyrazinamide during the first 8 weeks of treatment of patients with drug-susceptible or drug-resistant pulmonary tuberculosis: a multicentre, open-label, partially randomised, phase 2b trial. Lancet Respir. Med. 7, 1048–1058 (2019).
Cevik, M. et al. Bedaquiline–pretomanid–moxifloxacin–pyrazinamide for drug-sensitive and drug-resistant pulmonary tuberculosis treatment: a phase 2c, open-label, multicentre, partially randomised controlled trial. Lancet Infect. Dis. 24, 1003–1014 (2024).
Acknowledgements
We thank P. Janssens for technical assistance; M. Crabbe for statistical support; A. T. Henze for medical writing support at Janssen Pharmaceutica; S. Harris and T. Alattar for technical assistance at the London School of Hygiene and Tropical Medicine; A. Upton for critical scientific review at EvoTec ID; the Imperial College London Agilent Measurement Suite; H. Florance for LC–MS support at Agilent; and S. Cloete for graphical design assistance. N.D. acknowledges support from the Canadian Institutes of Health Research (185715) and funding from Janssen Pharmaceutica. The Vaccine and Infectious Disease Organization receives operational funding from the Government of Saskatchewan through Innovation Saskatchewan and the Ministry of Agriculture and from the Canada Foundation for Innovation through the Major Science Initiatives Fund. The work at the London School of Hygiene and Tropical Medicine was supported by funding from Janssen Pharmaceutica (to A.K. and R.J.W.). Work at the Center for Discovery and Innovation was funded by Bill and Melinda Gates Foundation grant INV-040485 (to J.P.S.). Work at Tufts University was funded by Bill and Melinda Gates Foundation grant INV-027276 (to B.B.A.). This work was funded in part by NIH R61/R33AI138280 (to A.J.C.S.). We are grateful to the Global Research Resource for Human Tuberculosis supported by NIH R24AI186591 (to A.J.C.S.) for providing human TB tissue specimens. This research was funded in part by the Wellcome Trust (Africa Health Research Institute strategic core award 227167/A/23/Z). For the purpose of open access, the authors have applied a CC BY public copyright licence to any Author Accepted Manuscript version arising from this submission.
Author information
Authors and Affiliations
Contributions
A.L., D.A.L. and R.J.W. managed and coordinated the project. P.F., J.S., J.D. and J.W. generated and interpreted the in vitro data. B.T., V.P. and A.J.C.S. planned, performed and analysed the measurement of nucleobases in human lung tissue. J.E. and P.J. coordinated and guided the experimental medicinal chemistry work. M.E., P.S. and B.S. planned, coordinated and performed the drug metabolism and pharmacokinetics and toxicology analyses. C.D., E.D. and S.J.W. planned, performed and analysed experiments related to the CRISPRi strains. C.A.-P. and S.D. planned, coordinated and performed the in vivo studies. A.V. performed the molecular modelling. W.P. and G.L.-M. performed and analysed the metabolomics experiments. J.P.S. planned, performed and analysed the rabbit caseum experiments. N.D., S.L. and M.G. performed the timelapse microscopy experiments. M.D.R. and A.H. expressed and purified recombinant proteins. M.P.M., T.W., H.D. and B.B.A. performed and analysed the DiaMOND analysis. S. Saylock and K.W.C. performed and analysed the enzymatic assays. A.R., C.R. and N.C. developed, designed and analysed the foam THP-1 assay. A.D. and S. Sans developed, performed and analysed the cholesterol and intra-THP-1 assays. R.D.A., M.M., H.F., A.S.P. and T.G. provided intellectual guidance. D.A.L. and R.J.W. coordinated the assembly of the manuscript. R.J.W. wrote the manuscript with contributions from D.A.L. and A.K., on the basis of feedback from all authors. A.K. provided overall supervision of the study. All authors reviewed the manuscript for intellectual content and gave final approval to submit for publication. All authors attest that they meet the criteria for authorship.
Corresponding authors
Ethics declarations
Competing interests
D.A.L., A.L., J.S., P.F., J.W., S.D., S. Saylock, M.D.R., A.H., T.G., A.V., K.W.C., P.S., M.E., M.M., J.E., R.D.A., H.F., A.S.P., B.S., C.A.-P., P.J. and A.K. were or are all full-time employees of Janssen Pharmaceutica, a Johnson & Johnson company, and/or potential stockholders of Johnson & Johnson. J.S. and P.F. were or are employees of Charles River Laboratories, a contract research organization. A.R., C.R., N.C. and S. Sans were or are all full-time employees of Evotec. R.J.W., C.D., W.P., E.D., S.L., M.G., J.D., S.J.W., G.L.-M., N.D. and A.K. received funding from Janssen Pharmaceutica to perform contract research. The other authors declare no competing interests.
Peer review
Peer review information
Nature thanks Hyungjin Eoh, Paul Hergenrother and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 JNJ-6640 resistance profiling.
a, Mutation frequency of JNJ-6640. b, Growth of a JNJ-6640-resistant strain harbouring a I241V mutation had comparable growth to the parental WT strain based on CFU counts. n = 3 biological replicates. Data shown are mean ± SD. c, Genetic variance in 51,183 clinical TB isolates7. Protein domains predicted using InterPro domain search (ebi.ac.uk/interpro). Highlighted mutations identified from resistance generation experiments with PurF-targeting inhibitors including JNJ-6640. Sequencing of additional clones, not further profiled, also identified additional mutations in PurF. Twelve clinical isolates (from a total of 49,982) contained an amino acid change within 5 angstrom distance of JNJ-6640 binding that also maintained a genetic variation. None of these variations have been linked to JNJ-6640 resistance.
Extended Data Fig. 2 The effect of JNJ-6640 on de novo purine biosynthesis.
15N-stable isotope incorporation following co-treatment with 100 nM JNJ-6640 and 2 mM 15N- amide labelled glutamine for 4 h showing inhibition of de novo purine biosynthesis. a, Schematic of the structure of adenine, indicating the potential sources of 15N atoms, and de novo purine biosynthetic pathway showing (cyan) the amino acid donors of the purine nitrogen atoms. Note, 15N atoms may be incorporated from glutamine indirectly (grey). The total metabolite abundances, regardless of labelling, are shown for (b) adenine, (d) AMP and (f) glutamate. For the metabolites with multiple nitrogen atoms, the percentage of the total abundance made up by each labelled isotopologue are shown; (c) adenine, (e) AMP and (g) glutamine. The different isotopologue species are indicated on the x-axes by M + (x), where x= the number of 15N atoms (after accounting for the natural abundance of 15N). For (c) adenine and (e) AMP, the remaining percent is made up of the unlabelled M + 0 isotopologue species. n = 5 replicates. PRPP: Phosphoribosyl pyrophosphate; GMP: guanosine monophosphate; AMP: adenosine monophosphate. h, Measurement of total glutamine abundance (without labelling) following 24-h treatment with 1000 nM JNJ-6640. n = 5. For all panels, data shown are mean ± SD.
Extended Data Fig. 3 Validation of purF CRISPRi-mediated knockdown strains.
a, qRT-PCR demonstrating transcript knockdown for each strain (low, med and high transcript reduction) ± induction with 100 ng mL−1 anhydrotetracycline (ATc). Individual sample Ct values were normalised against the reference gene, SigA (Rv2703), followed by normalisation against the transcript levels of a control strain containing the CRISPRi machinery without specific guide RNA. n = 2 independent experiments. b, Growth defects observed from purF transcript knockdown are ATc inducible. No growth defect observed for strains grown in the absence of ATc (induction) after 9 days. Starting inoculum ~1 ×105 CFU mL−1. n = 4 biological replicates. c, Dose-response curves of bedaquiline in the presence ( + ATc; EC50 = 61 nM) and absence (- ATc; EC50 = 44 nM) of CRISPRi-mediated ‘low’ transcript knockdown of PurF. n = 3 technical replicates. Representative of two independent experiments. For all panels, data shown are mean ± SD.
Extended Data Fig. 4 Impact of the purine salvage pathway on PurF inhibition.
a, CRISPRi-mediated PurF‘high’ transcript knockdown in the presence of 0.01 - 1 mM hypoxanthine or adenine after 14 days. n = 3 independent experiments. b, Nucleobase rescue assay with 1 µM JNJ-6640 in the presence and absence of hypoxanthine and adenine. CFU count collected after 17 days treatment. n = 3 biological replicates. c, Nucleobase rescue assay with dose-response of JNJ-6640 in the presence and absence of a fixed concentration of 500 µM nucleobase. Hypoxanthine, but not other nucleobases, rescues JNJ-6640 activity. n = 3 technical replicates. For a-c, data shown are mean ± SD. d, Salvage pathway gene expression in response to treatment with 25 nM JNJ-6640 (3x MIC90) for 24 h. Transcript levels were normalised against a reference gene, sigA, then against WT relative transcript levels. Data shown are mean.
Extended Data Fig. 5 Validation of the MtPurF and HsPPAT enzymatic assays.
a, Schematic for enzymatic assays. b, SDS-PAGE gels showing MtPurF and HsPPAT protein purification. c, PurF reaction progress curves in HyPerBlu assay with 70 mM glutamine and 300 µM PRPP. n = 2 biological replicates. d, PPAT reaction progress curves in HyPerBlu assay with 200 µM glutamine and 30 µM PRPP. n = 2 biological replicates. e, IC50 values and fold selectivity from MtPurF and HsPPAT enzymatic assays. n = 2 biological replicates. Data shown are mean.
Extended Data Fig. 6 Single-cell analysis of M. tuberculosis.
a, Representative snapshots of M. tuberculosis replisome reporter strain (MTB::gfp-dnaN, green channel) expressing cytoplasmic TdTomato (magenta channel), before (67 h), during (127 h) and after (284 h) exposure to 2.5 µg mL−1 ciprofloxacin. Bacteria undergoing DNA replication are identified by the green foci which represents the active replisome complex. The bacteria cultured in microfluidic device were exposed to the compound between 73–163 h. Scale bar represents 3 µm. b, Plot depicting the number of bacteria with a GFP-DnaN foci before, during exposure to 2.5 µg mL−1 ciprofloxacin (CIP; gray area) and after washout. Cumulative number of foci observed across 13 independent xy fields. n = 242 cells. c, Mouse bone marrow-derived macrophages were infected with M. tuberculosis expressing TdTomato and imaged by timelapse microscopy at 2 h intervals for 190 h. Representative time series snapshots showing macrophages infected with fluorescent bacteria (green) over time, untreated (upper panels); treated with 3 µM JNJ-6640. Numbers on top right indicate hours elapsed. Scale bar represents 25 µm. d, Plot depicting the bacterial load in infected macrophages over time under the different treatment conditions: no treatment; 3 µM JNJ-6640; 3 µM JNJ-6640 plus 100 ng mL−1 IFNγ; 3 µM JNJ-6640 plus 100 ng mL−1 IFNγ plus 1x nucleobases mix (NB mix). Bacterial load was determined from the fluorescence area in each time frame. Data shown are median ± interquartile range. n = 11 independent xy fields for each condition. Representative dataset from two independent experiments.
Extended Data Fig. 7 Activity of JNJ-6640 under non-replicating conditions.
a, Bactericidal activity of JNJ-6640 in the ex vivo rabbit caseum assay. Bacterial burden and JNJ-6640 concentration are expressed on log scales. n = 3 biological replicates. b, Schematic outlining the foamy macrophage assay adapted from previous work20. THP-1 cells were differentiated and incubated with M. tuberculosis expressing LuxABCDE-based bioluminescence for 4 h. Infected cells were then incubated with various concentrations of compound for 4 days under hypoxic conditions. After 4 days, infected macrophages were incubated in normoxic conditions for 1 day. IC50 values were determined through measurement of bacterial bioluminescence, before (day 4) and after (day 4 + 1) regrowth in normoxic conditions. Bactericidality was measured based on ratio of these IC50 values. c, Relative expression of known dormancy genes Tgs1, HspX and Rv3290c in macrophages grown in dormancy conditions (Foam THP-1) compared to normoxic conditions (THP-1). No difference in the expression of the control gene, RpoA (Rv3457c), was observed. Relative expression compared to 16S-RSS shown. n = 3 biological replicates. Dose-response curves of control compounds d, rifampicin (bactericidal) and e, isoniazid (bacteriostatic) in a foamy macrophage assay. n = 2 individual experiments with 4 technical replicates. For a, c-e, data shown are mean ± SD. f, Foamy macrophage IC50 ratios to determine bactericidality in hypoxic conditions. IC50 values were determined based on measurement of bacterial bioluminescence before (day 4; D4) and after (day 4 + 1; D4+1) regrowth in normoxic conditions. IC50 ratios of >2 classed as bactericidal. n = 2 independent experiments containing 4 technical replicates.
Extended Data Fig. 8 Murine pharmacokinetic (PK) parameters for JNJ-6640.
a, Results are expressed as the mean ± SD. n = 3 animals were dosed for the IV and PO arms and n = 3 animals by alternating sampling (from a total of 6 dosed) for SC arms (LAI), alternating sampling per time point was performed to limit the number of times blood was collected per animal, therefore we were unable to calculate error for these treatments. CL: clearance; Cmax = maximum concentration reached; t1/2: half-life; AUC: area under the curve; MRT: Mean residence time; F: bioavailability; Vss: volume of distribution; Vz: terminal elimination phase. b, Comparison of total plasma concentration profiles of JNJ-6640 administered as IV (1 mg kg−1), PO (5 mg kg−1 and 50 mg kg−1) or SC (1500 mg kg−1; LAI). n = 3 mice ± SD (IV and PO) or n = 3 mice (from a total of 6) for LAI PK studies, alternating sampling in 3 mice per time point. c, PK profiles following one or two doses of 1500 mg kg−1 JNJ-6640 LAI over 4 weeks. n = 3 mice (from a total of 6) alternating sampling in 3 mice per time point. Data shown are mean ± SD.
Extended Data Fig. 9 Contribution of JNJ-6640 to relevant TB treatment regimens.
a, In vitro combination studies over 21 days focused on replacing linezolid with JNJ-6640. Inoculum: starting inoculum: 5 ×105 CFU; LOD: Limit of detection. R: 0.1 µM rifampicin; HR: 5.8 µM isoniazid and 14.58 µM rifampicin; B: 0.5 µM bedaquiline; Pa: 7 µM pretomanid; L: 6 µM linezolid; 6640: 0.1–10 µM JNJ-6640. n = 3 biological replicates. Representative of two independent experiments. Drug interaction profiles of partner drugs with JNJ-6640 compared to moxifloxacin. Data shown are mean ± SD. b, Drug interactions are quantified with log2 FIC50 values for JNJ-6640, linezolid and moxifloxacin in pairwise combination with bedaquiline, pretomanid, and pyrazinamide. Partner drugs were selected based on their inclusion in a Phase 2C clinical trial54,55. Lower log2 FIC50 values (towards the left) are indicative of more synergistic drug interactions. Missing bars depict pairs for which specific pairwise drug combinations did not meet quality control metrics. Drug interactions with linezolid and moxifloxacin were measured previously52 and the data replotted here for comparison. n = 3 independent experiments. Data shown is mean ± SEM.
Supplementary information
Supplementary Information (download PDF )
Supplementary Methods, Supplementary Figs. 1–8 and Supplementary Tables 1 and 2.
Supplementary Video 1 (download AVI )
Single-cell analysis of M. tuberculosis treated with JNJ-6640. M. tuberculosis expressing TdTomato was grown in a microfluidic device under a constant flow of 7H9 medium. Images were acquired every 1h on the phase (magenta) and Ex550/Em580 (green) channels and merged. Medium conditions: t = 0-94h, no compound; t = 95-239h, 0.6 µM JNJ-6640; t = 240-310h, 1x nucleobases mix.
Supplementary Video 2 (download MP4 )
Single-cell analysis of M. tuberculosis-infected macrophages without treatment. Murine bone marrow-derived macrophages were infected with M. tuberculosis expressing TdTomato and images were acquired every 2h, over a period of 190h on the phase (magenta) and Ex555/Em594 nm (green) channels and merged. Scale bar 30 µm. Numbers on top right, represents the hours elapsed.
Supplementary Video 3 (download MP4 )
Single-cell analysis of M. tuberculosis-infected macrophages treated with JNJ-6640. Murine bone marrow-derived macrophages were infected with M. tuberculosis expressing TdTomato and exposed to 3 µM JNJ-6640. Images were acquired every 2h, over a period of 190h on the phase (magenta) and Ex555/Em594 nm (green) channels and merged. Scale bar 30 µm. Numbers on top right, represents the hours elapsed.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lamprecht, D.A., Wall, R.J., Leemans, A. et al. Targeting de novo purine biosynthesis for tuberculosis treatment. Nature 644, 214–220 (2025). https://doi.org/10.1038/s41586-025-09177-7
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41586-025-09177-7
This article is cited by
-
The role of cytochrome bc1 inhibitors in future tuberculosis treatment regimens
Nature Communications (2025)
