
Abstract
Whole-genome doubling (WGD) is a common feature of human cancers and is linked to tumour progression, drug resistance, and metastasis1,2,3,4,5,6. Here we examine the impact of WGD on somatic evolution and immune evasion at single-cell resolution in patient tumours. Using single-cell whole-genome sequencing, we analysed 70 high-grade serous ovarian cancer samples from 41 patients (30,260 tumour genomes) and observed near-ubiquitous evidence that WGD is an ongoing mutational process. WGD was associated with increased cell–cell diversity and higher rates of chromosomal missegregation and consequent micronucleation. We developed a mutation-based WGD timing method called doubleTime to delineate specific modes by which WGD can drive tumour evolution, including early fixation followed by considerable diversification, multiple parallel WGD events on a pre-existing background of copy-number diversity, and evolutionarily late WGD in small clones and individual cells. Furthermore, using matched single-cell RNA sequencing and high-resolution immunofluorescence microscopy, we found that inflammatory signalling and cGAS-STING pathway activation result from ongoing chromosomal instability, but this is restricted to predominantly diploid tumours (WGD-low). By contrast, predominantly WGD tumours (WGD-high), despite increased missegregation, exhibited cell-cycle dysregulation, STING1 repression, and immunosuppressive phenotypic states. Together, these findings establish WGD as an ongoing mutational process that promotes evolvability and dysregulated immunity in high-grade serous ovarian cancer.
Similar content being viewed by others

Main
Whole-genome doubling (WGD) is found in more than 30% of solid cancers and leads to increased rates of metastasis, drug resistance and poor therapeutic outcomes1,2,3,4,5. Often observed on a background of TP53 mutation, WGD leads to increased chromosomal instability (CIN) and karyotypic diversification4,7,8. Errors in chromosome segregation often lead to cytokinesis failure and the generation of polyploid cells9, indicating that WGD may be an active process during tumour evolution7. Furthermore, phenotypic consequences, such as chromatin and epigenetic compensatory changes10, replication stress11, and cell-cycle dysregulation10,11, enable cell persistence despite the expected deleterious effects of WGD. In patient tumours, the impact of WGD on tumour evolution, cancer-cell phenotypes, and the tumour microenvironment remains poorly understood, being limited in part by bulk whole-genome sequencing (WGS) approaches that do not allow the identification of WGD subpopulations. Crucially, reports from in vitro and patient-derived xenograft models have demonstrated that the temporal and evolutionary dynamics of WGD can be captured at single-cell resolution12,13. We therefore sought to use single-cell approaches to study WGD in individuals with high-grade serous ovarian cancer (HGSOC), an archetypal tumour of genomic and chromosomal instability. Our results establish WGD as both an ongoing evolutionary process and an important covariate of inflammatory signalling and immunosuppression in HGSOC.
Cohort and single-cell WGS
We generated a multimodal mapping of aneuploidy, genomic instability, and cell-intrinsic and tumour microenvironment phenotypic read-outs (Extended Data Fig. 1a). We studied a cohort of 41 treatment-naive HGSOC patients14 (Fig. 1a, Extended Data Fig. 1b, Methods and Supplementary Tables 1 and 2) using single-cell whole-genome sequencing (scWGS), multiplexed immunofluorescence and single-cell RNA sequencing (scRNA-seq), applied to 70 multi-site samples. The cohort included 18 homologous recombination-deficient (HRD)-Dup (enriched in duplications; BRCA1 mutant-like) and 8 HRD-Del (enriched in deletions; BRCA2 mutant-like) cases, as well as 14 HR-proficient foldback inversion (FBI)-bearing tumours and one tandem duplicator tumour, as inferred by integrating point mutations and structural variants13,14,15.

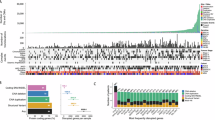
a, Overview of the MSK SPECTRUM cohort and specimen collection workflow, including numbers of patients, sites and samples processed by various means. H&E, haematoxylin and eosin; IF, immunofluorescence. b, Study design for analysing cellular ploidy and WGD in single cells using scWGS with the DLP+ protocol. The plot shows the classification of WGD multiplicity in cancer cells (0, 1 or 2 WGDs) using the fraction of the genome with major copy number (CN) ≥ 2 versus the mean allele CN difference; n = 30,260 cells. BAF, B-allele frequency; TCN, total copy number. c, Top, age at diagnosis, mutation signature, BRCA1/BRCA2 mutation status, and WGD class. Middle, distribution of cell ploidy of individual cells for each tumour, coloured by the number of WGDs. Bottom, percentage of WGDs, number of cells per patient, and fraction of cells in the minority WGD multiplicity state. Bottom right, illustrations of cell classifications. d, Heatmaps of total copy number (left) and allelic imbalance (right) for patient OV-045, with predicted WGD multiplicity and site of resection for each cell annotated. The 1×WGD population was downsampled from 1,857 to 200 cells for visualization, and the full 0×WGD and 2×WGD populations, numbering 18 and 44 cells, respectively, are shown. A-Hom, homozygous for haplotype A; A-gained, allelic imbalance with more copies of haplotype A (analogous for haplotype B); Balanced, equal copies of the two haplotypes.
To generate scWGS data, we flow-sorted tumour-derived single-cell suspensions to remove CD45+ immune cells and prepared libraries following the direct library preparation (DLP+) protocol16 (Methods and Supplementary Table 2). Sequencing yielded 100,054 single-cell whole genomes (median, 1,720 per patient) with a median coverage depth of 0.060 and a median coverage breadth of 0.057 per cell (Extended Data Fig. 2a,b and Supplementary Table 3). After extensive quality control, including filtering out non-malignant cells and doublets using the optical components of DLP+, we retained 30,260 high-quality tumour-cell genomes for downstream analysis (Extended Data Fig. 2c,d, Methods, Supplementary Note and Supplementary Table 4). The aggregated copy-number landscape was as expected for HGSOC (Extended Data Fig. 2e) and correlated with clinical panel-based bulk sequencing (Extended Data Fig. 2f) and matched bulk WGS (Extended Data Fig. 2g). From the scWGS data, we inferred the number of WGD events in the evolutionary history of each tumour cell (WGD multiplicity), based on allele-specific copy-number profiles3,17 (Fig. 1b and Extended Data Fig. 2h,i). Per-cell WGD multiplicity correlated with mitochondrial DNA copy number (Extended Data Fig. 2j), fraction of overlapping reads (Extended Data Fig. 2k), and cell size, as measured by the optical components of DLP+ (Extended Data Fig. 2l), providing orthogonal validation based on known correlates of nuclear genome scaling16,18.
Ongoing WGD
Intra-patient cellular WGD heterogeneity was pervasive across the cohort, with 40 of 41 patients exhibiting coexisting WGD multiplicities (Fig. 1c and Supplementary Note). For example, patient OV-045 (Fig. 1d) simultaneously had 0×WGD cells (1%; Extended Data Fig. 2m), a majority of 1×WGD cells (97%; Extended Data Fig. 2n) and a small fraction of 2×WGD cells (2%; Extended Data Fig. 2o). In total, 4% of all tumour cells across the cohort (n = 1,213 cells) represented non-majority WGD multiplicities (median of 2.5% of cells per patient; Extended Data Fig. 2p). Mixed WGD multiplicities were observed across sites for 16 out of 21 patients with multi-site sequencing, consistent with WGD as an ongoing process (Supplementary Note). As 39 out of 41 patients’ tumours were dominated by a single WGD multiplicity (more than 85% of cells), we divided tumours into two categories: WGD-high (over 85% of cells had at least 1×WGD; 27 out of 41 patients); or WGD-low (fewer than 15% of cells had at least 1×WGD; 14 out of 41 patients). The two tumours with intermediate (50–85%) proportions of cells having at least 1×WGD were grouped with WGD-high because they had large WGD clones. WGD-high tumours constituted 66% of the cohort, were enriched for FBI and HRD-Del mutation signatures, and occurred in patients who were significantly older at diagnosis, concordant with previous bulk genome sequencing studies14,19 (Extended Data Fig. 2q–t). Thus, the WGD-high fraction is consistent with previous bulk estimates of WGD prevalence across patients17. However, single-cell analysis established that WGD is ubiquitous across patients and exists as a distribution over coexisting 0×WGD, 1×WGD and 2×WGD cells in tumours, congruent with WGD as an ongoing mutational process.
Evolutionary histories of WGD clones
We next inferred evolutionary histories and WGD timing for each tumour to characterize the role of WGD in HGSOC clonal evolution. We developed doubleTime, a multi-step computational approach that uses somatic single nucleotide variants (SNVs) to estimate the timing of clonal divergence and WGD expansion(s) in each tumour (Fig. 2a, Methods and Supplementary Data 1–2). After excluding two patients because of technical limitations (OV-024 and OV-125; Methods), we observed four classes of WGD evolution: truncal WGD, parallel WGD, subclonal WGD, and unexpanded WGD. Truncal WGD, defined as a single WGD event ancestral to all cells and an absence of residual 0×WGD cells, was observed in 21 patients (Fig. 2b and Extended Data Fig. 4a; see Fig. 1c, Extended Data Fig. 3 and Supplementary Note for a diagram and analysis of residual 0×WGD cells). Parallel WGD, defined by multiple clones with different ancestral WGD events, was observed in two patients, OV-025 and OV-045 (Fig. 2b,c). Remarkably, for both of these patients, multiple WGD clones coexisted in different anatomical sites. In patient OV-025, all clones were present in both the right adnexa and omentum, and in patient OV-045, the left adnexa harboured one of the three WGD clones, whereas the right adnexa, omentum and peritoneal tumours were mixtures of all three WGD clones. Subclonal WGD, defined by a WGD clone coexisting with 0×WGD cells, was seen in five patients (Fig. 2b; further details below). Unexpanded WGD, defined as the absence of a discernible WGD clone, nevertheless included small populations of 1×WGD cells in all but one of the remaining 11 patients (Fig. 2b and Extended Data Fig. 4a).

a, Schematic of the approach for timing WGDs in SNV clones (Methods). cnLOH, copy-neutral loss of heterozygosity. b, Clone phylogenies and WGD timing for 18 patients (see Extended Data Fig. 4a for another 21 patients). Branch length shows the number of age-associated SNVs (C-to-T at CpG sites) assigned to each branch, adjusted for coverage-depth-related reduction in SNV sensitivity. Expanded WGD events are shown as triangles at the predicted location along WGD branches, coloured by relative timing. Branches are coloured by WGD multiplicity. Bar plots show, for each leaf, the fraction of cells in each WGD multiplicity and the fraction of cells from each anatomical site. OV-045 and OV-075 (starred) each harboured 0×WGD cells not captured in the doubleTime clone tree. The x axis is labelled with the SBMClone clone indices for each leaf. c, Variant allele frequency (VAF) of SNVs in two-copy LOH regions showing support for parallel versus shared WGD for patients OV-045 (left) and OV-025 (right). Each axis shows a different pair of clones, and each SNV is coloured according to its most likely variant copy numbers in the respective clones. SNVs that are assigned variant copy numbers 0/0 (absent from both clones) or 2/1 or 1/2 (inconsistent with the simple CNLOH WGD model) have been omitted. d, Histogram and rug plot showing the sensitivity-adjusted age-associated SNV count for WGD and diagnosis events for WGD-low (top, n = 14 patients) and WGD-high (bottom, n = 25 patients) tumours. Left, diagram showing the two time periods being measured by SNV counts. MRCA, most recent common ancestor. e, Fraction of additional-WGD cells in each clone plotted against the log binomial P value for the test that a clone has a greater fraction of additional-WGD cells than the average additional-WGD fraction across the cohort. Patients with P < 0.01 (dotted line) are annotated.
To refine our understanding of WGD heterogeneity, we timed key evolutionary events in each patient using age-associated C>T CpG mutations20. WGD-high tumours exhibited increased mutation time from conception to surgical resection compared with WGD-low tumours, similar to WGD versus non-WGD patients in previous bulk WGS analyses19. Although WGD events generally occurred early in tumour evolution19, a long tail of late events was also observed (Fig. 2d). In 8 out of 25 WGD-high tumours, the WGD event occurred more than 50% of the way through the tumour’s ancestral branch or after the most recent common ancestor (Fig. 2b,d and Extended Data Fig. 4a). Three of these late-WGD patients harboured residual populations of 0×WGD cells, consistent with a pre-WGD ancestral population coexisting with late-emerging WGD clones: OV-045 had 16 0×WGD cells (0.8%; Extended Data Figs. 3a and 4b), OV-075 had 30 0×WGD cells (3.3%; Extended Data Figs. 3c and 4c) and OV-081 had 216 0×WGD cells (35%; Fig. 2b). We speculate that the lack of residual 0×WGD populations observed in patients with earlier timing may indicate 1×WGD clonal sweeps, and therefore increased fitness associated with WGD in these patients.
Additional-WGD cells, those with one more WGD than the majority population (1×WGD in 0×WGD clones and 2×WGD in 1×WGD clones; Fig. 1c), were detected in 37 out of 41 patients, further exemplifying that WGD is ongoing. We investigated whether these additional-WGD cells shared common mutations indicative of clonal expansions (Fig. 2b,e). In patient OV-025, a small clone containing 40 2×WGD cells (and 4 1×WGD cells) harboured 296 clone-specific SNVs (Extended Data Fig. 4d). Subclonal WGD expansions in patients OV-006 (27 cells), OV-031 (7 cells) and OV-139 (17 cells) were too small to be detected by SNV analysis but nevertheless exhibited shared copy-number events across multiple WGD cells (Extended Data Fig. 4e–g). For other patients, unexpanded WGD cells were distributed across multiple clones and anatomical sites: 25 out of 31 patients had additional-WGD cells in multiple clones, and 14 out of 21 patients with multisite scWGS had additional-WGD cells in multiple sites (Extended Data Fig. 4h), indicative of ongoing WGD across clonal populations as a background mutational process.
Post-WGD genomic diversification
We then asked how WGD promotes genomic diversification and evolvability. First, we quantified cell-to-cell genomic heterogeneity using pairwise nearest-neighbour copy-number distance (NND) (Methods and Extended Data Fig. 5a). Mean NND increased with WGD multiplicity and was highest for additional-WGD cells (Fig. 3a). Some WGD-high tumours exhibited surprising levels of cellular diversity: in eight patients, the average difference between each cell and its most similar neighbour was more than 10% of the genome. The empirical distribution of NND values had a heavy tail (Extended Data Fig. 5b) consisting of cells with very distinct copy-number profiles. We therefore defined cells with NND above the 99th percentile of a beta distribution fit as divergent (Fig. 3b and Methods). These divergent cells exhibited substantial chromosome- and arm-level alterations relative to pseudobulk profiles (Fig. 3c and Extended Data Fig. 5c,d), with higher nullisomy rates across all tumours (Extended Data Fig. 5e). Increased nullisomy and lack of clonal expansion, as indicated by each cell’s unique copy-number profile, indicate that these cells have reduced proliferative capacity and decreased fitness, reminiscent of the ‘hopeful monsters’ identified in colorectal cancer organoids21. Divergent cells were present in 38 out of 41 patients (mean, 2.6% of cells), with higher rates in WGD-high tumours (Fig. 3d), and were more frequently additional-WGD cells (Extended Data Fig. 5f). Furthermore, the fraction of divergent cells was highest in late-WGD tumours and decreased with the age of the WGD event(s) (Extended Data Fig. 5g). Overall, these results suggest that expansion of WGD clones coincides with increased rates of catastrophic cell division.

a, Nearest-neighbour distance in each WGD population, where distance is calculated as the fraction of the genome with a different CN. The centre line shows the median, box boundaries show quartiles, and whiskers indicate 1.5 × the interquartile range (IQR). b, QQ plot of the beta distribution fit versus empirical quantiles of NND values for all cells, including divergent cells (greater than the 99th percentile of the beta distribution). c, CN profile of an example divergent cell from OV-004 (top) compared with the pseudobulk CN of all cells for OV-004 (bottom). Each point is a 500-kb bin coloured by assigned CN state, and y axes show normalized read counts. Shaded regions indicate CN differences. d, Fraction of divergent cells. Boxplots are defined as in a. e, Method for inferring cell-specific CN events in non-divergent cells. Chrom., chromosome. f, Ploidy-normalized event counts per cell split by WGD multiplicity and WGD-high versus WGD-low tumour status. Mann–Whitney one-sided U-test significance (FDR corrected) is annotated: *1.0 × 10−2 < P ≤ 5.0 × 10−2, **1.0 × 10−3 < P ≤ 1.0 × 10−2, ***1.0 × 10−4 < P ≤ 1.0 × 10−3, ****P ≤ 1.0 × 10−4. Only significant comparisons are shown. Boxplots are defined as in a. g, High-resolution whole-slide immunofluorescence imaging to detect micronuclei (MN) and primary nuclei (PN), and quantify micronuclei rates. Scale bars, 10 μm. h, Mean primary nuclei area. Significance was calculated using a GEE model with patients as groups, annotated as in f. Boxplots are defined as in a. i, Micronuclei rates per slide. Each point is a tumour region of interest (ROI). Bar plots show total number of cGAS+ micronuclei across tumour ROIs (top) and total number of primary nuclei across tumour ROIs (bottom). Small tumour ROIs (fewer than than 103 primary nuclei) have been excluded. Shaded boxplots indicate patients highlighted in Extended Data Fig. 5j. Boxplots are defined as in a. j, Micronuclei rate per slide. Significance was calculated using a GEE model with patients as groups, annotated as in f.
To study post-WGD diversification in non-divergent cells, we computed cell-specific copy-number aberrations (CNAs) accrued since each cell’s immediate ancestor in a phylogenetic tree (Fig. 3e, Extended Data Fig. 5h and Methods). Per-cell rates of gains and losses affecting whole chromosomes, chromosome arms, and segments (>15 Mb) increased with WGD multiplicity for all event types. Rates normalized to account for genome size yielded the same trend, indicating that rate differences were not entirely attributable to increased chromosome number, but rather were indicative of increased systemic instability after the WGD (Fig. 3f and Methods). For instance, ploidy-adjusted chromosome (2.6-fold) and arm (2.4-fold) losses were more abundant in WGD-high 1×WGD cells than in WGD-low 0×WGD cells (P = 1.2 × 10−2 and P = 2.3 × 10−3, Mann-Whitney U-test, FDR adjusted). Chromosome and arm gains both exhibited 2.3-fold increases (P = 2.1 × 10−2 and P = 6.8 × 10−3, Mann-Whitney U-test, FDR adjusted). In a multivariate generalized estimating equations (GEE) model accounting for covariates (patient age, mutation signature and anatomical site), chromosome, arm, and segmental alterations remained significantly associated with WGD (Extended Data Fig. 5i).
We next sought to validate increased CNA rates in WGD populations through immunofluorescence quantification of cGAS+ ruptured micronuclei. Missegregated chromosomes can become encapsulated in micronuclei, which are structures that have aberrant, rupture-prone nuclear envelopes. Ruptured micronuclei release genomic double-stranded DNA (dsDNA) into the cytoplasm22,23,24, resulting in activation of innate immune signalling driven by the cytosolic dsDNA-sensing pathway cGAS-STING25. Thus, we reasoned that cGAS expression can act as an orthogonal in situ marker of missegregation. We performed multiplexed immunofluorescence on formalin-fixed and paraffin-embedded (FFPE) sections (measuring DAPI, cGAS, panCK, CD8, p53 and STING), using high-resolution whole-slide microscopy imaging. We used a deep-learning approach to perform whole-slide quantification of primary nuclei and cGAS+ ruptured micronuclei. From 102 quality-filtered slides spanning 37 patients, we detected 20,988,413 primary nuclei and 896,042 ruptured micronuclei (Fig. 3g and Methods). Tumour cell nuclear area was significantly higher for WGD-high than WGD-low tumours (P = 3.5 × 10−7; Fig. 3h), further supporting biophysical correlates of WGD. The micronuclei rate, computed as the number of ruptured cGAS+ micronuclei per primary nuclei in tumour regions, ranged from 0.001 to 0.543 across regions of interest (Fig. 3i and Supplementary Tables 5 and 6). Within-patient variation was also observed, reflective of spatially heterogeneous micronuclei rates across tissues. Importantly, the micronuclei rate was 3.3-fold higher in WGD-high tumours (P = 1.8 × 10−6; Fig. 3j and exemplar regions in Extended Data Fig. 5j and Methods), providing further evidence, orthogonal to scWGS, that WGD significantly impacts CIN.
Taken together, multiple forms of CIN, including chromosomal missegregations, catastrophic mitoses, and ruptured micronuclei, exhibited elevated rates in WGD cells, firmly linking WGD to increased CIN and cellular genomic diversification in HGSOC.
Evolvability of WGD clones
Given the increased CIN associated with WGD, we next used scWGS-based phylogenies to investigate the impact of this instability on tumour evolution (Fig. 4a and Methods). We categorized CNA events on ancestral (root) branches into those inferred to occur after WGD in the ancestral branches of WGD-high tumours (post-WGD), before WGD in ancestral branches of WGD-high tumours (pre-WGD) or on the ancestral branches of WGD-low tumours (non-WGD). Ancestral gains of chromosomes and arms were rare in general, although chromosome gains were significantly more numerous post-WGD than pre-WGD, similar to previous results26 (Fig. 4b). By contrast, losses of chromosomes and arms were an order of magnitude more frequent than gains in all contexts. The ratio of losses to gains on ancestral branches was also significantly higher than the same ratio computed for cell-specific event rates (Extended Data Fig. 5k). These results, together with simulation experiments27 (Supplementary Note), indicate that the commonly observed pseudo-triploid karyotypes in HGSOC are unlikely to arise through incremental gains on a diploid background, and instead arise from WGD and both pre-WGD and post-WGD losses.

a, Pre- and post-WGD events illustrated for the ancestral branch of patient OV-044. Top, CN profile of the inferred ancestral non-WGD clone. Bottom, CN profile of the WGD clone. The plots in between show the CN changes (positive indicating gains, negative indicating losses) inferred to be pre-WGD and post-WGD, as illustrated on the left. b, Counts of ancestral arm and chromosome events detected across the cohort for non-WGD ancestral branches of WGD-low tumours, and pre- and post-WGD branches for WGD-high tumours. Bars and 95% confidence intervals show the distribution of counts on the given type of branch. Mann–Whitney U-test significance (FDR corrected) is annotated as: *1.0 × 10−2 < P ≤ 5.0 × 10−2, **1.0 × 10−3 < P ≤ 1.0 × 10−2, ***1.0 × 10−4 < P ≤ 1.0 × 10−3, ****P ≤ 1.0 × 10−4. Only significant comparisons are shown. c, Bar plots show counts of arm and chromosome events occurring post-WGD for all high-confidence clonal and subclonal WGD events detected across the cohort, split by clonality of the WGD (cell fraction threshold, 0.99). Bars and 95% confidence intervals show the distribution of counts on the root branch of the given type of WGD. Each bar indicates a clone that is labelled below and annotated above with the number of WGD events ancestral to the clone, as well as its clonality. The bottom bar plots show the fraction of cells from each patient that the clone represents. d, Boxplots summarizing c, annotated with FDR-corrected significance (Mann–Whitney U-test) as in b. NS, not significant.
To determine whether post-WGD losses were the result of immediate post-WGD instability (for example, divergent cells) or the accumulation of gradual losses, we analysed chromosome and arm CNAs in truncal and subclonal WGD clones (Fig. 4c). Truncal WGD clones harboured significantly more alterations than subclonal WGD clones (Fig. 4d), including three times as many whole chromosome and arm losses. The number of post-WGD events for some subclonal WGD clones was surprisingly low, and rarely (only one clone in OV-025) exceeded the average number of post-WGD events calculated for divergent cells (Fig. 4c). For example, the WGD clone in patient OV-081 (64% of cells) exhibited only two arm losses post-WGD compared with an average of 8.6 chromosome or arm events for divergent cells. For truncal WGD clones, the number of chromosome and arm losses was significantly correlated with the age of the WGD as measured by the number of C>T CpG mutations occurring from the WGD to the time of sample collection (Methods and Extended Data Fig. 5l). These results support a fitness model in which WGD cells are more likely to expand if they gradually accumulate post-WGD losses, rather than experience the large-scale alterations observed in divergent cells.
WGD and cellular phenotypes
Finally, we studied the phenotypic impact of WGD on cancer-cell-intrinsic, stromal, and immune cell transcriptional states using previously published patient- and site-matched scRNA-seq data14. We sought to determine whether WGD-specific phenotypic associations were independent of previously discovered links between mutation signatures, cellular states, and immune evasion in HGSOC14. We first focused on how WGD and CIN affect the cell cycle in cancer cells. WGD-high tumours exhibited a lower proportion of S-phase cells and a higher proportion of G1-phase cells, both cohort-wide and within the HRD-Dup subset (Extended Data Fig. 6a,b and Methods). Similarly, pseudotime inference of cell-cycle trajectories revealed distinct disruptions to cell-cycle progression in WGD-high versus WGD-low tumours (Extended Data Fig. 6c and Methods). In particular, MCM-complex genes involved in licensing of DNA replication origins at the G1/S transition (MCM2 and MCM6) were expressed earlier in the cell cycle in WGD-high tumours, together with factors involved in MCM-complex loading, such as CDC6 (Extended Data Fig. 6c), likely facilitating the replication of larger genomes. Mitotic cyclins (CCNE1) and genes involved in DNA repair (BRCA2 and MSH2) also had altered temporal order. Investigating differential responses to CIN, we found that the expression of E2F target genes showed strong negative correlation with chromosome losses in WGD-low tumours (Spearman’s ρ = −0.64, P = 0.015; Extended Data Fig. 6d) and an absence of correlation in WGD-high. Furthermore, the fraction of cells in G1 was correlated with rates of chromosome losses in WGD-low tumours, but not in WGD-high tumours (Spearman’s ρ = 0.64, P = 0.016; Extended Data Fig. 6e). Thus, both WGD and CIN were associated with altered cell-cycle dynamics, including delayed progression through G1, that increased with both CIN and WGD28,29.
Next we investigated CIN-dependent activation of innate immunity in cancer cells. CIN transcriptional phenotypes30 were significantly higher in WGD-high tumours (Fig. 5a), as expected given the CIN increases observed by means of scWGS and immunofluorescence. Nevertheless, WGD-high tumours showed a significant decrease in type I (IFNα and IFNβ) and type II (IFNγ) interferon, inflammatory pathways, and TNF via NF-κB signalling, relative to WGD-low tumours. The decrease was statistically significant for the cohort as a whole (Fig. 5a) and for the HRD-Dup subset (Extended Data Fig. 7a), with similar trends for the FBI subset (Extended Data Fig. 7b), indicating that the effect of WGD on cell-intrinsic immuno-phenotypic signalling may be independent of mutation signature. Interestingly, scWGS-derived rates of chromosome, arm, and segmental losses were positively correlated with immune-related expression programs in WGD-low tumours but not in WGD-high tumours (Extended Data Fig. 7c). This indicates that the innate immune response to CIN may be preserved in WGD-low tumours and abrogated in WGD-high tumours. Repression of STING1, an innate immune response gene activated by the presence of cytosolic DNA, is a well-established mechanism for evasion of the immunostimulatory effects of CIN31,32,33,34,35,36. STING1 was expressed at significantly lower levels in WGD-high tumours (Fig. 5b), whereas in WGD-low tumours, STING1 expression was positively correlated with rates of missegregation, especially chromosome losses (Spearman’s ρ = 0.75, P = 0.003; Fig. 5c). This finding was confirmed by the immunofluorescence measurements, which also showed a decrease in STING1 protein in WGD-high tumours (Fig. 5d,e). Similarly, STING1 protein was weakly correlated with micronuclei rate in WGD-low tumours, whereas in WGD-high tumours, STING1 exhibited a negative correlation with micronuclei rate (Fig. 5f). These results support a model in which WGD-high tumours adapt to increased rates of genomic and chromosomal instability by transcriptional remodelling of interferon signalling response pathways, including repression of STING1 (ref. 37).

a, Scatter plot depicting GEE regression coefficients versus Benjamini–Hochberg-adjusted P values for selected genes and pathways in WGD-high and WGD-low tumour cells. MHC, major histocompatibility complex. b, Per-sample mean gene expression of STING1 in WGD-high (n = 63) and WGD-low (n = 34) samples. Centre line shows the median, box boundaries show quartiles and whiskers indicate 1.5 × IQR. Significance calculated using two-sided Wilcoxon rank sum test is included. c, Scatter plot of STING1 gene expression versus rate (counts per cell) of chromosomal losses, split by WGD-low and WGD-high (colours). Lines indicate the result of a linear regression in either WGD-high or WGD-low tumours. Regression coefficients and significance results are shown separately for WGD-low and WGD-high tumours. d, Example immunofluorescence images of WGD-high and WGD-low tumour samples with varying STING1 expression. Top, multichannel overlay images of STING1, panCK, DAPI and cGAS intensity at high magnification (scale bars, 125 μm). Bottom, zoomed insets (locations indicated by white boxes in the top panels; scale bars, 15 μm). e, Boxplots showing distribution of per-sample mean STING1 immunofluorescence intensity over tumour cells for WGD-high and WGD-low samples. Box plots are defined as in b. Significance calculated using a GEE model is included. f, Scatter plot and density estimation of STING1 versus micronuclei rate for 1 mm × 1 mm tiles in tumour ROIs. Points, density contours and coefficients, and P values of a generalized linear model are coloured by WGD-high and WGD-low tumour status. g, Differential cell-type abundance testing results from Milo with permutation testing (Methods) for cell types in WGD-high versus WGD-low samples. h, Normalized enrichment scores (NES) in the interferon pathway for cell types in the tumour microenvironment. CAF, cancer-associated fibroblasts; cDC1, conventional type 1 DCs; DCs, dendritic cells; EC, endothelial cells; NK, natural killer; pDC, plasmacytoid DCs. i, NES in the cell-cycle pathway for cell types in the tumour microenvironment.
To validate the cell-intrinsic impacts of WGD in vitro, we used TP53 mutant hTERT-immortalized retinal pigment epithelial (RPE-1) and TP53 mutant fallopian tube epithelial (FNE1) cell lines. In each cell line, a distinct, spontaneously arising WGD clone was observed by scWGS and could be identified in scATAC-seq and scRNA-seq using clone-specific chromosome- and arm-level copy-number events (Methods and Extended Data Fig. 8a–c). We first studied how non-WGD cells from early, predominantly diploid passages of each cell line responded to the CIN-inducing drugs nocodazole and reversine. Treatment was associated with increased chromosome and arm losses and gains (Extended Data Fig. 8d) and a concomitant rise in G1 cell fraction and mean STING1 expression (Extended Data Fig. 8e,f). Untreated later passages of each cell line harboured an almost-equal mixture of WGD and non-WGD cells, allowing robust identification of WGD-specific transcriptional programs. In these mixed-WGD samples, WGD cells did not exhibit an increased G1 cell fraction (Extended Data Fig. 8g), despite increased rates of copy-number events (Extended Data Fig. 8d). However, STING1 expression was lower in WGD cells than in non-WGD cells in mixed-WGD samples and across treatment conditions in early-passage RPE-1 samples (Extended Data Fig. 8f). Together, these in vitro data indicate that WGD-induced STING1 downregulation can occur independently of the tumour immune microenvironment.
Finally, we profiled the composition of cell states in the tumour immune microenvironments of the patient tumours. We found enrichment of CXCL10+CD274+ macrophages (M2.CXCL10), and IFN-producing plasmacytoid and activated dendritic cells in WGD-low tumours in cohort-wide (Fig. 5g and Extended Data Fig. 9a) and HRD-Dup-specific analyses (Extended Data Fig. 9b). All the main cell types had significant enrichment of ISGs in WGD-low tumours, indicating a pro-inflammatory immune response (Fig. 5h). By contrast, WGD-high tumours showed enrichment for endothelial cells, pericytes, and cancer-associated fibroblasts (Fig. 5g), along with ISG suppression. WGD-high tumours also showed slight enrichment of cytotoxic CD8+ T cells, possibly because of mutual exclusivity between cytotoxic CD8+ T cells and CXCL10+CD274+ macrophages across the cohort (Extended Data Fig. 9c). Notably, all the main cell types in WGD-high tumours (except for endothelial cells) exhibited marked depletion in cell-cycle-related gene expression, consistent with a pro-angiogenic yet immunosuppressive microenvironment in WGD tumours (Fig. 5i).
Discussion
We used scWGS matched with scRNA-seq and tissue-based immunofluorescence quantification of ruptured micronuclei to reveal the impact of WGD on tumour evolvability and phenotypic states in HGSOC. Using doubleTime to infer the evolutionary histories and timing of WGD revealed a complex role for WGD in HGSOC and context-dependent selection of WGD clones. More than half of the tumours in our cohort harboured a truncal WGD event, with the timing ranging from very early to late, indicating that WGD cells can expand across the evolutionary continuum. In a subset of patients, we observed partial expansion of recently emerged late-WGD clones coexisting with populations of residual 0×WGD cells, indicating that there was active selection at the time of tumour resection. The absence of residual 0×WGD cells in early WGD cases is consistent with 1×WGD clonal sweeps, underscoring the positive selective advantage that WGD confers in ovarian cancer. Intriguingly, in tumours in which we observed parallel WGD events, these events occurred at approximately the same time in the tumour’s evolutionary history. This could indicate that cell-extrinsic promotion factors led to a WGD-permissive state in these patients, enabling the simultaneous expansion of distinct WGD subclones. In WGD-low tumours, the small fractions of cells generated by ongoing WGD indicate that fixation of WGD is not limited by the event rate, but rather by tumour contexts that are permissive of WGD expansion, raising the crucial question of which cell-intrinsic and microenvironmental factors modulate the selection of WGD in HGSOC.
The relationship between WGD and genomic diversification is evident: we found ubiquitous minor populations that have undergone additional doublings, an increased rate of cell-specific aneuploidies post-WGD, and profoundly divergent cells38. Analysis of tumour-derived single-cell data allowed measurement of CNA rates much closer to the true underlying rate of CNA in patient tumours than is possible with bulk sequencing methods. Although DLP+ sequences live cells and may miss deleterious CNAs in non-viable cells, we observed cells with large regions of homozygous deletions, indicating that we nevertheless did capture part of the non-viable population. The existence of cells with highly divergent genomes is indicative of punctuated copy-number evolution39,40,41,42,43 as a mechanism for generating the extensive losses seen in some WGD clones. However, analysis of both truncal and subclonal WGD indicates that gradual losses, rather than punctuated evolution, shape the post-WGD evolution of many WGD clones, which simultaneously requires adaptation and tolerance for the high CIN levels associated with WGD. Despite elevated CIN, WGD-high tumours showed decreased cell-intrinsic and cell-extrinsic interferon signalling and a pro-angiogenic, immunosuppressive tumour microenvironment, consistent with previous findings on chronic CIN-induced immune suppression37,44. The disrupted correlation between CIN and STING1 in WGD-high tumours implicates STING1 transcriptional repression as a prerequisite for the clonal expansion of WGD. Given the very early timing of WGD in some patients, our results also prompt further investigation of STING1 repression as an early event that may precede WGD in the evolutionary history of some HGSOC tumours. Studying WGD and cGAS-STING in the context of serous tubal epithelial carcinoma (STIC) precursor lesions45,46 could yield important insights into how WGD and cGAS-STING modulation contributes to tumorigenesis in HGSOC.
Our data introduce a critical covariate for therapeutic stratification of patients: nearly every tumour harbours WGD cells with co-existing multiplicities. Even with the modest cohort size presented here, we anticipate that studying how WGD clones affect responsiveness to HRD-stratified PARP inhibitors, or to anti-angiogenic therapies such as bevacizumab, will advance the rational administration of therapeutic strategies for HGSOC47,48. Intriguingly, the genomic and phenotypic consequences of WGD were evident even within HRD subtypes, indicating the potential for composite biomarkers involving mutational process and WGD to stratify patients. Moreover, given that emerging approaches targeting the WGD process itself and/or the downstream consequences of CIN49,50,51 are in early phase clinical trials, we anticipate that further insight into WGD evolutionary dynamics will be required to interpret the efficacy and durability of response. The relevance of our findings to other tumour types remains unclear, although in vitro12, breast patient-derived xenograft models13 and pancreatic cancer mouse7 studies indicate that ongoing WGD dynamics may be pervasive across TP53 mutant cancers. Thus, future studies should prioritize investigating how the evolutionary dynamics of ongoing WGD affect therapeutic responses52 across tumour types.
Methods
Experimental methods
Sample collection
All the enrolled patients were consented to an institutional biospecimen banking protocol and MSK-IMPACT testing53, and all analyses were performed per a biospecimen research protocol. All protocols were approved by the Institutional Review Board (IRB) of Memorial Sloan Kettering Cancer Center. Patients were consented following the IRB-approved standard operating procedures for informed consent. Written informed consent was obtained from all patients before conducting any study-related procedures. The study was conducted in accordance with the Declaration of Helsinki and the Good Clinical Practice guidelines (GCP).
We collected fresh tumour tissues from 41 HGSOC patients at the time of up-front diagnostic laparoscopic or debulking surgery. Ascites and tumour tissue from multiple metastatic sites, including bilateral adnexa, omentum, pelvic peritoneum, bilateral upper quadrants and bowel, were procured in a predetermined, systemic fashion (a median of four primary and metastatic tissues per patient) and were placed in cold RPMI for immediate processing. Blood samples were collected before surgery for the isolation of peripheral blood mononucleated cells (PBMCs) for normal whole-genome sequencing (WGS). The isolated cells were frozen and stored at –80 °C. Tissue was also snap-frozen for bulk DNA extraction and tumour WGS. Tissue was also subjected to FFPE for histological, immunohistochemical and multiplex immunophenotypic characterization.
Sample processing
We profiled patient samples using five different experimental assays:
-
1.
Viably frozen single-cell suspensions were derived from fresh tissue samples and processed for scWGS of 70 sites from 41 patients (mean of 1,429 cells per site; Supplementary Table 3). CD45− cells were flow-sorted in samples with low tumour purity.
-
2.
CD45+ and CD45− flow-sorted cells were previously reported fresh tissue samples and were processed for scRNA-seq of 123 sites from 32 patients (about 6,000 cells per site).
-
3.
For each specimen with scWGS and/or scRNA-seq, site-matched FFPE tissue sections were stained by multiplexed immunofluorescence for micronuclei and DNA-sensing mechanisms, together with adjacent sections used for whole-slide haematoxylin and eosin (H&E) staining (102 tissue samples from 37 patients).
-
4.
FDA-approved clinical sequencing of 468 cancer genes (MSK-IMPACT) was obtained on DNA extracted from FFPE tumour and matched normal blood specimens for each patient (Extended Data Fig. 1b).
-
5.
Snap-frozen tissues were processed to obtain matched tumour-normal bulk WGS on a single representative site from 33 of 41 patients with scWGS, scRNA-seq and immunofluorescence, to derive mutational processes from genome-wide single-nucleotide and structural variants.
Single-cell DNA sequencing
Tissue dissociation
Tumour tissue was immediately processed for tissue dissociation. Fresh tissue was cut into 1-mm pieces and dissociated at 37 °C using a human tumour dissociation kit (Miltenyi Biotec) on a gentleMACS Octo Dissociator. After dissociation, single-cell suspensions were filtered and washed with ammonium-chloride-potassium (ACK) lysing buffer. Cells were stained with Trypan blue, and cell counts and viability were assessed using a Countess II automated cell counter (ThermoFisher). For a detailed protocol, see ref. 54. Freshly dissociated cells were processed for scRNA-seq as described previously14. Viably frozen dissociated cells were stored for scWGS.
Cell sorting
Viably frozen dissociated cells used for scWGS were thawed and then stained with a mixture of GhostRed780 live/dead marker (TonBo Biosciences) and Human TruStain FcX Fc receptor blocking solution (BioLegend). For samples with low tumour purity, the stained samples were then optionally incubated and stained with Alexa Fluor 700 anti-human CD45 antibody (BioLegend). After staining, they were washed and resuspended in RPMI plus 2% FCS and submitted for cell sorting. The cells were sorted into CD45-positive and CD45-negative fractions by fluorescence assisted cell sorting on a BD FACSAria III flow cytometer (BD Biosciences). Positive and negative controls were prepared and used to set up compensations on the flow cytometer. Cells were sorted into tubes containing RPMI plus 2% FCS for sequencing.
Library preparation and sequencing
Single-cell whole-genome library preparation was done as described previously16. In brief, single cells were dispensed into nanowells with protease (Qiagen) and DirectPCR cell lysis reagent (Viagen). After overnight incubation, cells were subjected to heat lysis and protease inactivation followed by tagmentation in a tagmentation mix (14.335 nl TD buffer, 3.5 nl TDE1 and 0.165 nl 10% Tween-20) at 55 °C for 10 min. When the tagmentation reaction was neutralized, eight cycles of PCR followed. The indexed single-cell libraries were recovered from the nanowells by centrifugation into a pool and sequenced at the MSKCC Integrated Genomics Core on an Illumina NovaSeq 6000 (paired-end 150-base pair reads).
Immunofluorescence
Overview
We profiled matched FFPE tissues by immunofluorescence to quantify the rate of micronuclei formation in tumours using a six-colour assay (DAPI, cGAS, STING, p53, panCK and CD8). Immunofluorescence detection was done at the Molecular Cytology Core Facility of Memorial Sloan Kettering Cancer Center using a Discovery XT processor (Ventana Medical Systems, Roche-AZ). Antigen retrieval was done using ULTRA Cell Conditioning (Ventana Medical Systems, 950-224). The tissue sections were blocked first for 30 min in background blocking reagent (Innovex, NB306). Multiplex assay antibodies and conditions are described in Supplementary Table 6.
Tissue staining
Automated multiplex immunofluorescence was done using a Leica Bond BX staining system. Paraffin-embedded tissues were sectioned at 5 μm and baked at 58 °C for 1 h. Slides were loaded in Leica Bond and immunofluorescence staining was done as follows. Samples were dewaxed at 72 °C before being pretreated with EDTA-based epitope retrieval ER2 solution (Leica, AR9640) for 20 min at 100 °C. The 5-plex antibody staining and detection was done sequentially. The primary antibody against cGas (1.25 μg ml−1, rb, CST, 7997), Sting (0.075 μg ml−1, rb, CST, 13647), p53 (0.005 μg ml−1, rb, Abcam, ab32389), panCK (ms, 1:500, DAKO, M3515) or CD8 (rb, ventana, 1/40) was incubated for 1 h at room temperature followed by application of Leica Bond polymer anti-rabbit HRP secondary antibody (included in the Polymer Refine detection kit (Leica, DS9800)) for 8 min at room temperature. For the mouse primary antibody, the rabbit anti-mouse linker (Leica Bond post-primary reagent included in Polymer Refine detection kit (Leica, DS9800)) was incubated for 8 min before the application of Leica Bond polymer anti-rabbit HRP. After that, Alexa Fluor tyramide signal amplification reagents (Life Technologies, B40953, B40958) or CF dye tyramide conjugates (Biotium, 92172, 96053, 92174) were used for detection. After each round of immunofluorescence staining, epitope retrieval was done for denaturation of primary and secondary antibodies before another primary antibody was applied. When the run was finished, slides were washed in PBS and incubated in 5 μg ml−1 4′,6-diamidino-2-phenylindole (DAPI) (Sigma Aldrich) in PBS for 5 min, rinsed in PBS and mounted in Mowiol 4–88 (Calbiochem). Slides were kept overnight at −20 °C before imaging.
RPE-1 cell-line experiments
We explored the phenotypic effects of chromosomal instability and WGD in TP53-knockout RPE-1 cells. TP53-knockout RPE-1 was a gift from the Maciejowski laboratory at the Memorial Sloan Kettering Cancer Center (MSKCC). RPE-1 cells were cultured in DMEM (Corning) supplemented with 10% fetal bovine serum (Sigma-Aldrich), 1% penicillin-streptomycin (Thermo Fisher) at 37 °C and 5% CO2. All cells were periodically tested for mycoplasma contamination.
TP53−/− RPE-1 cells were treated with nocodazole, reversine and DMSO control to induce varying levels of chromosomal instability, then subjected to both 10× multiome sequencing and scWGS using DLP+ (Supplementary Table 7). For nocodazole treatment, RPE-1 cells were seeded at 20% confluence at the time of nocadazole addition. Cells were treated with 100 ng ml−1 nocodazole (Sigma-Aldrich) or DMSO for 8 h. After 8 h, cells were washed three times with PBS to remove the drug. After 48 h, the cells were collected. For reversine (Cayman Chemical Company) treatment, cells were treated at a concentration of 0.5 µM reversine for 48 h. After 48 h, cells were washed three times with PBS to remove the drug. Cells were collected after 12 h. We collected 10,000 cells per condition for 10x Genomics Chromium Single Cell Multiome ATAC+ gene expression according to the manufacturer’s protocol. Library preparation and sequencing were done in the MSKCC Integrated Genomics Core. We subjected 1 million matched cells per condition to scWGS DLP+ as described above.
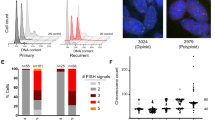
A spontaneously arising WGD subclone was observed as a minor population of TP53-knockout RPE-1 cells (Extended Data Fig. 8a). The relative fraction of this WGD population was monitored by DNA FISH every 5 passages. After 30 further passages (sample RPE-1 mixed), the WGD subclone, as measured by DLP+, comprised 37% of the population. Sample RPE-1 mixed was subjected to scWGS DLP+ and 10× scRNA-seq.
FNE1 cell-line experiments
FNE1 cells were a gift from Tan Ince. Cells were cultured in FOMI (US Biological Life Science, 506388.500) at 5% O2 and 5% CO2 at 37 °C, as described previously55. All cells were periodically tested for mycoplasma contamination. TP53 knockout was performed by electroporation (Lonza 4D nucleofector) of a ribonucleoprotein complex of Alt-R Cas9 (IDT 1081058) and the guide sequence mC*mC*mA* rUrUrG rUrUrC rArArU rArUrC rGrUrC rCrGrG rUrUrU rUrArG rArGrC rUrArG rArArA rUrArG rCrArA rGrUrU rArArA rArUrA rArGrG rCrUrA rGrUrC rCrGrU rUrArU rCrArA rCrUrU rGrArA rArArA rGrUrG rGrCrA rCrCrG rArGrU rCrGrG rUrGrC mU*mU*mU* rU. Cells were treated with 10 µM nutlin-3a to select for TP53-deficient cells for one week; at that time point, cells treated with a control guide were no longer proliferating. Loss of p53 was also confirmed by sequencing. For reversine (Cayman Chemical Company) treatment, early passage cells were treated at a concentration of 0.25 µM reversine for 48 h. After 48 h, cells were washed three times with PBS to remove the drug. Cells were collected after 12 h. We collected 20,000 cells for each condition. Cells were passaged and monitored for the emergence of a WGD population by DNA FISH as described above. By passage 15 after TP53 loss, nearly 50% of the cells were polyploid, as quantified by DNA FISH.
Monitoring for WGD using DNA FISH
After every five passages, cells were frozen and assessed for WGD using DNA FISH. In brief, cells were pelleted, incubated in 5 ml 75 mM KCl for 15–30 min. Cells were subsequently washed two times in ice-cold 3:1 methanol:glacial acetic acid solution. Cells were then spotted on a slide and dried overnight at 37 °C. Slides were washed twice in 2× SSC for 2 min each, then dehydrated sequentially in 70%, 85% and 100% ethanol, and air-dried for 2 min. FISH probes (MetaSystems, D-6008-100-OG) were applied to cells on glass slides, sealed with a coverslip using rubber cement and co-denatured with the samples at 72 °C for 5 min. After denaturation, hybridization was performed overnight at 37 °C in a humidified chamber. After hybridization, slides were washed in 2× SSC three times for 2 min each, rinsed in PBS, counterstained with DAPI and dehydrated in 70%, 85% and 100% ethanol before being mounted in ProLong Gold antifade solution. Quantification of tetraploid cells was performed on a Zeiss LSM880 (Carl Zeiss Microscopy) using a Plan-Apochromat 63×/1.4 NA oil objective lens.
Computational methods
Computational analyses of multimodal datasets were enabled by the Isabl platform56.
Single-cell DNA sequencing
Overview
The single-cell DNA analysis pipeline is a suite of workflows for analysing the single-cell data generated by the DLP+ platform16. The workflow takes dual-indexed reads from Illumina paired-end sequencing data as the input and performs various alignment and postprocessing tasks. The pipeline is publicly available on GitHub (https://github.com/mondrian-scwgs/mondrian), which we run within the Isabl framework56.
Alignment
We used Trim Galore to remove adapters and FastQC to generate QC reports before running alignment. The reads were then aligned with bwa-mem v0.7.17 (ref. 57) (with support for bwa-aln). PCR duplicates were marked using Picard v.2.27.4 with the MarkDuplicates tool, and alignment metrics were computed for each cell with the Picard tools CollectWgsMetrics and CollectInsertSizeMetrics. The pipeline also generated plots for each alignment metric for a quick overview.
Copy-number segmentation
Reads were tabulated for non-overlapping 500-kilobase regions. A modal regression normalization16 was performed to reduce GC bias. The pipeline then ran HMMcopy with six different ploidy settings and the best fit was chosen automatically58. The pipeline also generated heatmaps with cell clustering, per-cell copy-number profiles and the modal regression curve for visualization.
Quality control
The scWGS data were first subjected to quality control and filtering to remove non-cancer cells, S-phase replicating cells, low-quality cells, and doublets, resulting in 30,260 high-quality cancer-cell genomes (Extended Data Fig. 2c,d and Supplementary Note). The quality-control pipeline compiled the results from the total copy-number analysis and alignment, and we then used a random forest classifier to predict the quality of each cell based on the alignment and HMMcopy metrics16. We then inferred allele-specific copy-number profiles for each of these cells using SIGNALS13. Patient-level average ploidy ranged from 1.6 to 4.4, and the average fraction of LOH ranged from 0.12 to 0.57. Ploidy and LOH estimates were concordant with matching bulk WGS and clinical panel sequencing by MSK-IMPACT, and losses and gains from scWGS coincided with known drivers of HGSOC (Extended Data Fig. 2e–g). Thus, at a pseudobulk level, the genomic characteristics of our scWGS cohort matched those of both whole-genome and targeted bulk data.
Haplotype-specific copy number
In a bulk WGS matched normal sample for each patient, we measured reference and alternate allele counts for SNPs from the 1000 Genomes Phase 2 reference panel. We used a binomial exact test to identify SNPs that were heterozygous in the normal sample. Using SHAPEIT59 and the 1000 Genomes phase 2 reference panel, we computed haplotype blocks. Next we measured per-cell reference and alternate allele counts for heterozygous SNPs in the tumour scWGS data.
Mitochondrial DNA copy number
To infer the mitochondrial DNA copy number, we first computed the average read depth of the mitochondrial genome in each cell, restricting it to reads with a mapping quality of at least 30. Then we converted the mitochondrial genome coverage for each cell to an approximate copy number by dividing by the nuclear genome coverage and multiplying by the cell’s average (nuclear) ploidy.
Cell filtering
We established stringent filters to maximize the removal of problematic cells without losing sensitivity to rare, interesting populations, including those representing cell-specific WGD.
Removal of low-quality cells
We removed cells with a quality score lower than 0.75. The quality score was computed using the classifier presented in ref. 16.
Removal of normal cells
After copy-number calling, we identified normal cells as those with an average copy-number state between 1.95 and 2.05 with a standard deviation of less than 0.5. We removed these normal cells from further analysis. We also manually inspected cells with aneuploidy slightly outside this range but much less than tumour cells in the same sample, and manually selected ‘aberrant normal’ cells for removal (see Supplementary Note for examples). These cells typically did not share SNVs with the tumour cells and may correspond to other epithelial cells affected by field cancerization60 or immune/stromal cells with rare chromosomal aberrations.
Removal of S-phase cells
It is necessary to remove S-phase cells before downstream analysis because the observed HMMcopy profiles of these cells reflect a mixture of both somatic (heritable) copy number and transient doubling of replicated genomic loci. We nominated S-phase cells through a combination of features known to correlate with S-phase cells. We aimed to isolate the high-quality G1/2-phase cells for downstream analysis, so we did not need to distinguish between S-phase cells and low-quality cells (noisy HMMcopy profiles resulting from other factors, such as under-tagmentation before sequencing or incomplete cell lysis).
We first computed the following three features for each cell:
-
1.
The Spearman correlation between the HMMcopy state profile for a cell of interest and the RepliSeq replication timing profile from MCF-7 cells. S-phase cells have higher correlations than G1/2-phase cells.
-
2.
The number of HMMcopy breakpoints per cell, that is, the number of pairs of adjacent bins with different integer copy-number states. S-phase cells have more breakpoints than G1/2-phase cells.
-
3.
The median breakpoint prevalence across all HMMcopy breakpoints. This statistic was calculated by first computing the mean prevalence of each breakpoint across all cells belonging to a particular patient. Then, for each cell of interest, we subset to only the genomic loci with detected breakpoints in that cell and calculated the median of the mean breakpoint prevalences for those loci. S-phase cells have low median breakpoint frequency scores, because they have lots of rare breakpoints.
All three features varied widely across patients because of each patient’s unique number, positioning and heterogeneity of somatic copy-number alteration. We therefore used a strategy of examining each feature’s distribution across all cells in a patient, manually inspecting outlier cells and selecting custom thresholds for each patient. We used a filtering approach whereby cells are called as S-phase if any two of the three features are beyond the threshold. This conservative strategy ensured that all remaining cells were truly in the G1/2 phase and therefore had HMMcopy profiles that accurately reflected the somatic copy number. The thresholds used for each patient are included as Supplementary Table 4.
Removal of doublets
We applied several orthogonal approaches to remove doublets from the DLP data. First, under the assumption that the chromosome 17 LOH should be clonal in ovarian cancer, we removed tumour cells that lacked LOH of chromosome 17. Then we used a combination of mutation-based features to manually identify tumour-normal doublets, including LOH (much lower than typical tumour cells), the proportion of SNVs with alternate reads (higher than typical normal cells) and copy-number profiles that were similar to tumour cells with the addition of two copies across the genome. Finally, two raters separately reviewed the brightfield image of each cell in the clear microfluidic nozzle before deposition in the microwell array for sequencing and flagged any images that appeared to contain more than one cell. Any cell with an image that was flagged by at least one reviewer was removed from analysis. Example doublet copy-number profiles and spotter images are included in the Supplementary Note.
Removal of suspect high-ploidy cells
We restricted analysis to cells with high-confidence ploidy calls. Absolute ploidy is unidentifiable from the copy-number data of an individual cell, so we took a parsimony approach and assumed the true ploidy to be the lowest ploidy value that provided a reasonable fit to the data. One failure mode in the automatic determination of ploidy by HMMCopy occurred when HMMCopy converged on a solution with double the true ploidy, driven by the overfitting of isolated outlier bins. Such cells were characterized by mostly even copy-number states, except for isolated bins with odd copy numbers. To remove such potential artefacts, we required there to be at least one segment longer than 10 megabases in length with a copy number of 1, 3 or 5. Cells with no segments longer than 10 megabases with copy number 1, 3, or 5 were removed from further analysis. Note that as a result of this conservative approach, G2-phase cells and cells that had sustained perfect doublings would be detected as half their true ploidy or omitted from this study.
In conclusion, we performed several filtering steps including both automatic classification and manual review to remove low-quality cells, normal cells, S-phase cells, doublets and dubious high-ploidy cells (see also Supplementary Note). The requirement that predicted copy-number profiles include at least one 10-megabase or larger segment with a copy-number state of 1, 3 or 5 ruled out a non-WGD solution with half of the inferred copy number. However, it should be noted that individual cells that had sustained perfect doublings and non-aberrant G2 phase cells would be detected as half of their true ploidy in this study.
Comparison with bulk copy number
We used the WGS copy number inferred by ReMixT61 to validate the average ploidy in the MSK SPECTRUM cohort. Similarly, we used the IMPACT copy number inferred by FACETS62 for further orthogonal validation.
Detecting WGD in single cells using allele-specific copy number
WGD events were identified in single cells based on the allele-specific copy number state, as previously described for bulk WGS3. We computed two metrics from SIGNALS results: the fraction of the genome with two or more copies for the main allele (FM2) and the fraction of the genome with three or more copies for the main allele (FM3). Similar to the results in bulk WGS, a clear separation could be seen between subpopulations using each metric (Extended Data Fig. 2h,i). We classified any cell with FM2 > 0.5 as having undergone at least one WGD, and any cell with FM3 > 0.5 as having undergone at least two WGDs.
Patient-level WGD classifications
Tumours were classified as WGD-high at the patient level if the fraction of cells with at least one WGD exceeded 50% of the cells sequenced for that patient. The remaining tumours were classified as WGD-low.
Subclonal WGD classification
We classified cells for each patient as comprising a subclonal WGD subpopulation if they were predicted to have one more WGD than the ‘background’ WGD multiplicity, which we define as the lowest WGD multiplicity representing at least 25% of cells. For all WGD-low tumours, this was 0×WGD. For most WGD-high tumours, this was 1×WGD, with the exception of cells from patients OV-081 and OV-125, which had a background WGD multiplicity of 0×WGD as they had more than 25% 0×WGD cells.
Variant calling
SNV calling
Because the low per-cell coverage in scWGS was insufficient to resolve variants at nucleotide resolution, we merged all the single cells together to create a pseudo-bulk genome for each library. We ran the Mutect2 variant caller63 on the merged data across all the libraries from each patient. We computed the reference and alternate counts for each cell at all variant loci detected across all samples from a given patient.
SV calling
We used a similar approach for breakpoint calling by creating pseudo-bulk libraries, then running deStruct64 and Lumpy65 on each library. Only consensus SVs detected by both methods were retained; SVs from both methods were considered consensus if their coordinates were within 200 base pairs and their orientations matched. The SV calls were further post-processed as described in a previous study66.
Filtering somatic variant calls using ArtiCull
We applied ArtiCull67 to remove artefactual SNVs resulting from the short insert sizes in the scWGS data. ArtiCull was trained on high-confidence correct and artefactual calls based on manually labelled clones from seven patients (OV-004, OV-022, OV-045, OV-046, OV-052, OV-081 and OV-083), then applied it to all variants from all patients.
SBMClone
We applied SBMClone68 to the filtered somatic variants for each patient. SBMClone was run ten times for each patient with different random initializations, and the solution with the highest likelihood was kept (for patient OV-024, two of the initializations exceeded the runtime limit of seven days so the best solution of eight initializations was used).
Evolutionary histories of SNV clones using doubleTime
We developed doubleTime, which is a method for computing the evolutionary histories of the SNV clones in each patient, including accurate placement of WGD events in the clonal phylogeny of each patient. We have made doubleTime publicly available on GitHub (https://github.com/shahcompbio/doubleTime). It involves three main steps. First, we constructed a clonal phylogeny relating the clones identified by SBMClone. Second, we assigned WGD events to branches in the clonal phylogeny. For each pair of WGD clones, we assessed whether those clones arose from a single shared WGD or two parallel WGD events. Given this information, we were able to unambiguously assign WGD events to branches of each patient’s clonal phylogeny. Third, we used a probabilistic model to assign SNVs to branches of the clonal phylogeny, including assignment before and after WGD events on WGD branches. To control for the effect of small clones on sensitivity to detect mutations, terminal branch lengths were corrected for the total haploid coverage of the corresponding clone (Supplementary Note). We describe each of the three steps in detail below. Patient OV-024 was excluded because the clones were predominantly 2×WGD, which is not supported. Patient OV-125 was excluded owing to low cell counts (no SBMClone clone with at least 20 cells).
SBMClone SNV-based clonal phylogenies
We reconstructed phylogenetic trees with SBMClone clones as leaves using a binarized version of the implicit block structure inferred by SBMClone. We first computed a density matrix D, in which each row corresponded to a clone (cell block), each column corresponded to an SNV cluster (SNV block) and each entry Di,j contained the number of pairs (a,b), in which cell a in clone i had at least one alternative read covering SNV b in cluster j, divided by the total number of possible pairs (the size of clone i times the size of cluster j). We then computed a binary matrix B by rounding up those entries of D that exceeded a density of 0.01, removing empty columns, and collapsing identical rows (combining clones that contained the same blocks of mutations). We then attempted to infer a phylogenetic tree by applying the perfect phylogeny algorithm. Matrices B that did not permit a perfect phylogeny were manually modified with the minimum number of changes required to permit a perfect phylogeny; this typically occurred when mutations shared between two or more clones had been lost owing to a deletion in a subset of the clones.
Discerning parallel from shared WGD
To identify cases in which sequenced WGD cells arose from distinct WGD events, we analysed SNVs from the single-cell DNA sequencing data. Specifically, for each patient, we focused exclusively on those regions that exhibited copy-neutral loss of heterozygosity (cnLOH; major copy number 2 and minor copy number 0) among nearly all (90% or more) tumour cells with a single WGD. Given a candidate bipartition of the 1×WGD cells, under the infinite-sites assumption, each cnLOH SNV can be assigned to one of the following categories:
-
two mutant copies in both clones (shared pre-WGD and pre-divergence);
-
one mutant copy in one clone (private post-divergence);
-
no mutant copies (false-positive variant);
-
one mutant copy in both clones (shared post-WGD and pre-divergence);
-
two mutant copies in one clone (private pre-WGD and post-divergence).
The last two categories of SNVs present evidence for or against multiple parallel WGD events. SNVs that are shared at one variant copy (VAF ~ 0.5) would indicate that the two sets of cells underwent the same ancestral WGD event, because they share mutations that must have followed the WGD. Conversely, SNVs that are private at two variant copies (VAF ~ 1) would indicate that the two sets of cells underwent distinct WGD events, because they have private mutations that preceded the WGD. Specifically, we considered the following hypotheses:
-
1.
single-WGD: shared one-copy SNVs are allowed but private two-copy SNVs are not allowed;
-
2.
multiple-WGD: shared one-copy SNVs are not allowed, but private two-copy SNVs are allowed.
To evaluate the relative strength of these hypotheses, we developed a likelihood ratio test that compared the probability of observing the given variant counts for cnLOH SNVs under these two hypotheses: for each patient, we evaluated P(multiple-WGD)/P(single-WGD) using a simple binomial model of read counts. We then tested the significance of this likelihood ratio by generating an empirical null distribution: we fixed the total SNV read counts and their best-fitting variant copy numbers under the single-WGD hypothesis and resampled alternate read counts.
Assigning SNVs to branches and estimating branch lengths
From the previous steps, we have a tree relating the clones detected by SBMclone. We place WGD events on branches such that all WGD-high tumours had a WGD event placed on the root of the tree, except those in which parallel WGD events had been identified (patients OV-025 and OV-045) or WGD only affected a subset of clones (patient OV-081), in which case those specific events were placed further down the tree. We used a probabilistic model to assign SNVs to branches and estimate branch lengths based on read-count evidence for SNVs in each clone (for each leaf, we collected read counts only from those cells in the majority WGD multiplicity). For WGD branches, the model assigns SNVs as occurring before or after the WGD and estimates the length of the branch before and after the WGD. This strategy effectively splits each branch with a WGD event into two unique positions in the tree, meaning that the total number of positions in the tree to which an SNV can be assigned is equal to the number of branches plus the number of branches with WGD events.
For this analysis, we considered only those SNVs in regions where, for each SBMClone clone, more than 80% of cells shared the same copy-number state. We further restricted analysis to SNVs in regions with allele-specific copy-number states whose multiplicity (the variant copy number, or the number of copies of the genome containing the SNV), and thus the expected VAF, could be uniquely determined by the combination of tree placement and WGD status (that is, whether or not the clone was affected by an ancestral WGD event). Specifically, we analysed regions with the following copy-number states across all clones:
-
1:0 in both WGD and non-WGD clones;
-
1:1 in both WGD and non-WGD clones;
-
2:0 in WGD clones, 1:0 in non-WGD clones;
-
2:1 in WGD clones, 1:1 in non-WGD clones;
-
2:2 in WGD clones, 1:1 in non-WGD clones.
In each of these scenarios, we assumed that the WGD and copy-number events immediately following the WGD accounted for the differences in copy number between WGD and non-WGD clones. Note that the only patient in the cohort with different WGD status for different leaves was patient OV-081, so for nearly all patients, we analysed only those SNVs with clonal copy-number states (matching the above listed states depending on WGD status). The multiplicity for an SNV on a particular allele placed on a particular branch of the tree was as follows:
-
0, if the corresponding allele had 0 copies;
-
equal to the allele-specific copy number of the allele in the clone, if the SNV occurred pre-WGD and the leaf was affected by WGD;
-
equal to 1 otherwise.
Each SNV was assigned to a tree position by fitting the observed total and alternative counts of said SNV to the expected VAFs for all clones. SNVs were assigned to positions in the tree using a Dirichlet categorical distribution, and a beta-binomial emission model was used to relate observed SNV counts to expected VAFs. The model was implemented in Pyro and fitted using black-box variational inference69. Note that when computing branch lengths, we only used C>T SNVs at CpG sites because these SNVs have been reported to correspond most closely to chronological age20.
To account for the differences in genome size and copy-number heterogeneity between different patients with varying amounts of aneuploidy, we normalized the number of C>T CpG SNVs on each branch by the number of bases being considered. First, we computed the effective genome length of each clone as the total size of the bins considered to be clonal for a valid copy-number state as defined above, with each bin weighted by its total copy number. Then, for the internal nodes of the tree, we assumed that the only copy-number changes to these bins were directly coupled to WGD events. Thus, for post-WGD branches, the genome length was identical to that of the leaves; and for pre-WGD branches, the genome length was computed using the correspondence described above between pre- and post-WGD copy numbers.
Estimating pre- and post-WGD changes in WGD subpopulations
We used a maximum parsimony-based method to estimate pre- and post-WGD changes from estimated ancestral and descendent copy-number profiles. We proceeded independently for each bin. Let x be the ancestral copy-number state and y be the descendent copy-number state, and assume that y is produced by a combination of pre-WGD copy-number change followed by WGD followed by post-WGD copy-number change. We can relate x and y using
where b represents the pre-WGD copy-number change and a represents the post-WGD copy-number change. Let the cost of any given a and b be |a| + |b|. Conveniently, every combination of x and y results in a unique a and b that minimize this cost. Thus, for each x and y, we computed the associated b and a as the pre- and post-WGD changes, respectively, and |a| + |b| as the cost of those changes.
Computing the percentage genome different
We computed the percentage genome different for a pair of cells as follows. First, we computed the bin-level difference in total copy number and identified consecutive segments of changed and unchanged bins. We then removed segments less than or equal to 2 megabases in size (that is, affecting fewer than four consecutive 500-kb bins). Finally, we counted the number of bins for which the two genomes have different total copy numbers and divided by the total number of bins considered.
Classification of divergent cells
We defined divergent cells as outliers of the NND, using the percentage genome different as the distance metric. For each index cell, we identified its nearest neighbour as the other cell in the population with the minimal percentage genome different. The NND for each cell is thus the percentage genome different with respect to this neighbour cell. We then fitted a beta distribution to the NND values of all cells in the cohort and called divergent cells as those cells that have NND values in the 99th percentile of this beta distribution.
Cell phylogenies using MEDICC2
We derived estimates of chromosome missegregation rates per cell for each patient from copy-number phylogenies inferred using MEDICC2 (ref. 70). In addition to the cell filtering applied for all analyses, we removed divergent cells before running MEDICC2. First, we refined the single-cell haplotype-specific copy-number profiles for each patient by applying the dynamic programming formulation from asmultipcf71 to GC-corrected read counts and phased B-allele frequencies for each bin across all cells from the patient. Using this method, we identified segment boundaries for each patient and then summarized the number of copies of each segment and haplotype in each cell by rounding. Next, we ran MEDICC270 on these refined haplotype-specific single-cell copy numbers, which infers a tree with single cells corresponding to leaves. We used the –wgd-x2 flag for MEDICC2 which represents WGD as an actual doubling of all copy-number segments in the genome, rather than the default behaviour of adding 1 to all segments.
Reconstruction of ancestral copy number
To infer the ancestral haplotype-specific copy-number profiles associated with internal nodes of the cell phylogeny inferred by MEDICC2, we used a maximum-parsimony approach that treats each bin independently and aims to minimize the total number of changes on the tree. For each branch, the parsimony score is the absolute difference between the haplotype-specific copy-number profiles of the parent and the child. Transitions from 0 to any other copy number are given a score of infinity to prevent gain from 0 copies. The score for a WGD branch (assuming WGD placement from MEDICC2 is correct) is the sum of two parsimony scores: the parsimony score for copy-number changes between the parent and an intermediate genome, and the parsimony score for copy-number changes between a doubled version of the intermediate genome and the child (this is described above in the Estimating pre- and post-WGD changes in WGD subpopulations section). The state of each bin at each branch in the tree was chosen to minimize this parsimony score using the Sankoff algorithm72,73. We assumed that the MEDICC2 placement of WGD on branches of the phylogeny is correct in most cases, with the following exceptions.
-
1.
For patients OV-025 and OV-045, we adjusted the WGD placement to be concordant with SNV evidence indicating a distinct clonal origin of multiple parallel WGD clones.
-
2.
For 10 patients (OV-002, OV-003, OV-014, OV-024, OV-036, OV-044, OV-051, OV-052, OV-071 and OV-083), MEDICC2 failed to identify an ancestral WGD affecting a large proportion (97–100%) of cells that were indicated as WGD by the cell-specific CNA-based classifier. To correct this, we added a WGD event for each of these patients such that the number of WGD events ancestral to each cell in the MEDICC2 tree was identical to the number of ancestral WGD events indicated by the CNA-based classification.
-
3.
For a further 5 patients (OV-004, OV-022, OV-050, OV-087 and OV-139), MEDICC2 disagreed with the cell-specific CNA-based classifier on the WGD classification of a small number (at most five) of cells. These cells were removed from the tree before ancestral reconstruction.
Classifying events from copy-number differences
Given a phylogenetic tree in which both leaves and internal nodes are labelled by haplotype-specific copy-number profiles, we identified the copy-number events on each branch using a greedy approach. First, we identified the differences between the parent haplotype-specific copy-number profile and the child copy-number profile. Then, for each chromosome and haplotype, we explained the copy-number differences between parent and child using events that are as large as possible:
-
1.
if more than 90% of bins in the chromosome were altered in the same direction, we called a chromosome gain or loss that accounted for a change of one copy for all bins in the chromosome;
-
2.
if no chromosome gain or loss was found, but 90% of the bins in one of the two arms is altered in the same direction, we called an arm-level gain or loss that accounted for a change of one copy for all bins in the chromosome arm;
-
3.
if no chromosome- or arm-level gain or loss was found, we called a gain or loss of the largest contiguous segment that had a change in the same direction.
We then adjusted the copy-number difference by the selected event and repeated until all copy-number changes between parent and child have been accounted for. Note that if nearly all of the bins of a chromosome are gained (or lost), our method will first predict a chromosome gain (or loss), then another small segment loss (or gain) to account for the few bins that were predicted as unchanged. We selected this approach because we consider a whole chromosome (or arm) change to be more parsimonious if most of a chromosome’s (or arm’s) bins are altered. Our approach is also more robust to bin-level noise than a strategy that requires 100% of the bins to be altered.
For branches with WGD, we computed the intermediate pre-doubling profile that would result in the fewest copy-number changes (see Estimating pre- and post-WGD changes in WGD subpopulations above). Using our bin-independent parsimony model, we can compute this optimal intermediate profile analytically. We then performed the event-calling procedure described above twice: once on the differences between the parent and the intermediate pre-WGD profile, and once between the doubled intermediate profile and the child.
Normalizing missegregation rates to account for cell ploidy
We controlled for the opportunity for each cell to missegregate by dividing the number of copy-number events for each cell by the number of chromosomes (for chromosome-level missegregations) or arms (for arm-level missegregations) in the inferred parent node of each cell in the tree (the source of the terminal branch). This yields a rate of missegregation events per cell and per parental copy. For shorter segmental copy-number events, we divided the number of events in each cell by its parent’s genome length to control for opportunity. Although the resulting rate is not comparable to segment- and arm-level rates, it makes the cell-specific segmental rates more comparable between cells and across patients.
Enumerating events on ancestral branches
We classified copy-number events on the root branch of each patient’s cell phylogeny into three classes of event timing. Events were classified as non-WGD if they were predicted to occur on the root branch of a WGD-low tumour; pre-WGD if they were predicted to occur before the WGD event on the root branch of a WGD-high tumour; and post-WGD if they were predicted to occur after the WGD event on the root branch of a WGD-high tumour. Patients OV-025, OV-045, and OV-081 were omitted from this analysis because their WGD history precludes this categorization of copy-number events.
Calculating post-WGD changes in WGD clones
We catalogued all the high-confidence WGD clones detected in our cohort. This included all predicted WGD clades with at least 20 cells in the MEDICC2 phylogenies. We also included three small WGD clones from patients OV-006, OV-031 and OV-139 (Extended Data Fig. 4e–g). Counts of shared post-WGD events were calculated from the ancestral reconstruction on MEDICC2 trees as described above (see the Reconstruction of ancestral copy number section).
Single-cell RNA sequencing
Cell type assignment
Using scRNA-seq of CD45+/− sorted cells, we assigned the main cell types by supervised clustering using CellAssign74, as described in ref. 14.
InferCNV and scRNA-seq-derived copy-number clonal decomposition
InferCNV (v.1.3.5) was used to identify large-scale copy-number alterations in ovarian cancer cells identified by CellAssign75,76. For each patient, 3,200 non-cancer cells annotated by CellAssign were randomly sampled from the cohort and used as the set of reference ‘normal’ cells. After subtracting the reference expressions in non-cancer cells, chromosome-level smoothing and de-noising, we derived a processed expression matrix that represents copy-number signals. Cancer-cell subclusters are identified by ward.D2 hierarchical clustering and random_trees partition method using P < 0.05.
WGD classification
Identification of WGD cells from scRNA-seq data is technically challenging, because inferred copy number from expression data is typically noisy, allele-specific markers are sparse, and, as shown in our scWGS analysis, the prevalence of non-WGD cells in WGD-high tumours and WGD cells in WGD-low tumours is generally low, confounding identification of non-clonal ploidy populations within samples. Leveraging the high concordance between scWGS- and scRNA-derived copy number, even between non-site-matched patient samples (see Supplementary Note), we propagated scWGS-derived WGD status labels to all available patient-matched scRNA-seq samples for the purposes of transcriptional phenotyping analysis. Within-sample absolute normalization of unique molecular identifier (UMI) counts between tumour and non-tumour cells showed a significant increase in overall transcript counts per cell in WGD-high versus WGD-low tumours (see Supplementary Note), which was highly concordant with established estimates of transcriptional changes in WGD versus non-WGD samples in bulk RNA77. Thus, we concluded that site-matched scRNA-seq data effectively capture WGD transcriptional phenotypes. Any analyses correlating scWGS-derived missegregation rates to transcriptional phenotypes were restricted to site-matched samples with at least 20 cells in both scWGS and scRNA-seq.
Cell-cycle analysis
Discrete cell-cycle phase information was computed using Seurat’s CellCycleScoring function, excluding samples with fewer than 20 malignant cells. To estimate the association between WGD and cell-cycle phase, we used binomial GEE models cohort wide. We included tumour site and added interaction terms for WGD and age, and for WGD and mutation signature subtype. We repeated this analysis within the HRD-Dup signature subset.
We identified circular trajectories linked to cell-cycle progression in cancer cells using Cyclum78. Across the cohort, 10,000 cancer cells annotated by CellAssign were randomly sampled across tumours and used for cell-cycle trajectory inference. Pseudotime inference was performed on the scaled cell-by-gene matrix, limiting genes to cell-cycle markers included in cell-cycle GO terms (GO:0007049). Discretization of the continuous pseudotime trajectories was accomplished using a three-component Gaussian mixture model. Smoothed pseudotime trajectories of cell-cycle-related genes previously reported in the literature79 were then evaluated to interpret phase-specific gene activity and phase transitions as a function of pseudotime (Extended Data Fig. 6e).
Differential gene and pathway activity
Pathways were curated from single-cell hallmark metaprograms80, 50 hallmark pathways81 or CIN-associated gene signatures manually curated from the literature, including inflammatory signalling and ER stress30,37, and scored in single cells using Seurat’s AddModuleScore function. Owing to the hierarchical nature of the data, with multiple samples from patients, we used GEE on sample mean gene or pathway expression levels, adding tumour site (adnexa or non-adnexa) as a covariate in the model and restricting analysis to samples with at least 20 cells to compare WGD multiplicities. We repeated this procedure subsetting for HRD-Dup samples and adding an interaction term for age and WGD status as well as tumour site (HRD-Dup-only model). P values were adjusted for multiple testing using FDR. In parallel, we also performed differential expression analysis using a pseudobulked generalized linear mixed model (DREAMLET82), accounting for random patient and fixed tumour-site effects, and performed gene-set enrichment analysis (GSEA) with the same set of pathways.
Differential cell-type abundance
To determine cell populations that were differentially abundant between WGD-low and WGD-high samples, we used miloR v.1.8.1 (ref. 83), setting prop to 0.2 and using tumour_megasite (adnexa or non-adnexa) as a contrast in the differential abundance testing. To obtain significance values for each cell population, we ran permutation tests by swapping the sample WGD status labels 1,000 times and computing the proportion of tests in which the resulting non-permuted median log2(fold change) was more extreme than the permuted median values for each cell type.
Immunofluorescence
ROIs
We defined ROIs containing tumours on immunofluorescence images by delineating regions with tumour foci based on panCK, p53 and DAPI signal, and contrasting these with images of the immunofluorescence-adjacent H&E section. ROI annotations were drawn in QuPath. To ensure that complex tissue regions within ROIs used for analysis only included tumour, we classified regions of tumour, stroma, vasculature and glass within each ROI. We trained a pixel classifier with examples of tumour, stroma, vasculature and glass from each of the ROIs and slides using the panCK, p53 and DAPI signal in immunofluorescence, and verifying the region classification against the immunofluorescence-adjacent H&E section. ROIs with high cGAS background were excluded from analysis to minimize false-positive segmentations of cGAS+ micronuclei.
Segmentation of primary nuclei and micronuclei
Whole-slide immunofluorescence images stained with DAPI, cGAS, STING, p53, panCK and CD8 were analysed to characterize primary nuclei and micronuclei in ROIs. Segmentation of primary nuclei was done in QuPath v.0.5.1 using the StarDist algorithm on the DAPI channel84. We used a segmentation model pretrained on single-channel DAPI images (dsb2018_heavy_augment.pb). Applying the primary nuclei segmentation model across all ROIs yielded 20,988,413 primary nuclei in tumour regions. Segmented primary nuclei ranged between 5 μm2 and 100 μm2 in size, with a minimum fluorescence intensity of 1 a.u. The cell membrane for each primary nuclei was approximated using a cell expansion of 3 μm of the nuclear boundary.
MN were detected by StarDist segmentation of cGAS spots. We trained a new segmentation model on single-channel cGAS images using a U-Net architecture. We manually annotated cGAS+ micronuclei in a set of 256-pixel x 256-pixel tiles encompassing tumour regions across all slides. We created training and test sets using a 70:30 split, resulting in a training set of 70 tiles and a test set of 30 tiles. To ensure that the model generalized across patients and samples, we applied augmentation to the training set by applying random rotations, flips and intensity changes. We monitored the loss function during model training and saved the trained model with frozen weights.
This allows for whole-slide quantification and cell-level annotation of primary nuclei and micronuclei. Nuclear segmentation was also done using StarDist on the DAPI channel. Each micronucleus was assigned to the closest primary nucleus. Micronuclei were included for analysis if they were 10 μm or less from the centroid of the closest nucleus, had an area of 20 μm2 or less, a circularity of more than 0.65 and a minimum object probability of more than 0.75.
Validation of micronuclei segmentation
We evaluated our method on a test dataset with held-out micronuclei labels, showing good performance of predicted micronuclei segmentations with high average precision and F1 scores (intersection-over-union (IoU) < 0.5). We quantitatively evaluated the segmentation performance on the test data by considering cGAS+ micronuclei objects in the ground truth to be correctly matched if there were predicted objects with overlap. We used IoU as an overlap criterion, demonstrating good performance with a chosen IoU threshold of more than 0.5.
Micronuclei rates
Micronuclei rupture rates were estimated on the basis of the number of cGAS+ micronuclei and primary nuclei segmented in tumour ROIs. The rate of micronuclei rupture was estimated by localization of cGAS+ micronuclei neighbouring primary nuclei. The micronuclei rate was calculated as the fraction of primary nuclei with one or more micronuclei. Applying the micronuclei segmentation model across all ROIs yielded 896,042 cGAS+ micronuclei in tumour ROIs, with a mean micronuclei area of 0.76 μm2, ranging between 0.1 μm2 and 6.8 μm2. Slide-level and ROI-level micronuclei rates were calculated and are summarized in Supp Tab. 5, excluding small ROIs with 1,000 primary nuclei or fewer in downstream analyses.
Statistical comparisons of micronuclei rates
To compare the micronuclei rate between WGD-high and WGD-low, we used GEE. We used binary WGD-high versus WGD-low as the dependent variable with gaussian distribution and log(micronuclei rate) as the independent variable, adding patient as a group variable in the model. Reported effect size of WGD was calculated from the coefficient of log(micronuclei rate) in the learned model.
Analysing the relationship between micronuclei rate and STING1
We used a linear mixed effects model to evaluate the relationship between STING1 protein intensity and micronuclei rate separately for WGD-high and WGD-low tumours. We first divided each image into a regular grid of 1 mm × 1 mm tiles. For each primary nucleus, we computed the mean STING1 protein intensity in the combined nuclear and cytoplasmic region. For each tile we then computed the micronuclei rate in the tile and the mean STING1 protein intensity for tumour cells detected in the tile. We log-transformed this micronuclei rate and mean STING1 intensity and used a linear mixed effects model with the formula log(STING1) ~ log(micronuclei rate) with images as the group variable. We then report the coefficient and P -value of the coefficient of log(micronuclei rate) in the model.
Mutational signatures
We analysed mutational signatures by integrating SNVs and structural variations detected by either bulk WGS or scWGS in a unified probabilistic approach called multimodal correlated topic models (MMCTM)15.
For bulk WGS samples, we obtained signature labels in the MSK SPECTRUM cohort (n = 41) using MMCTM, as presented in ref. 14. Mutational signatures for cases without bulk WGS data were assigned on the basis of mutational signatures inferred from scWGS. For scWGS samples, we obtained signature labels in the MSK SPECTRUM cohort (n = 41) using a ridge classifier with default regularization strength (α = 1.0). This classifier was trained on the integrated SNV and SV signature probabilities, which were obtained using MMCTM13 from HGSOC bulk whole genomes13 (n = 170).
Consensus mutational signatures were preferentially derived based on MMCTM signatures derived from bulk WGS and MMCTM signatures from scWGS. Mutational signatures for cases without bulk WGS data (OV-006, OV-044, OV-046, and OV-071) or inconclusive bulk WGS assignments (OV-004, OV-045, OV-080, and OV-081) were resolved on the basis of scWGS.
Analysis of RPE-1 and FNE1 cell-line experiments
10x scRNA-seq preprocessing
Raw 10x Genomics sequencing data for RPE-1-mixed and FNE1-mixed were aligned using CellRanger (v.7.0.0), which also performed barcode filtering and UMI gene counting using the 10× GRCh38 reference transcriptome.
10x Multiome preprocessing
Raw 10x Genomics sequencing data for RPE-D, RPE-Noco and RPE-Rev were aligned to the 10x Genomics GRCh38 reference transcriptome using CellRanger ARC (v.2.0.2). CellRanger ARC also performed barcode filtering and UMI gene counting to generate feature-barcode matrices for both RNA and ATAC modalities.
scATAC-seq copy-number analysis
Copy number was inferred from the scATAC-seq component of the 10x Genomics multiome data for the RPE-D, RPE-Noco and RPE-Rev samples. Blacklist-filtered fragments were first counted in 10-megabase genome bins. Bins with a GC content of less than 30% were removed before GC correction using modal regression16. Cells with more than 5% of their bins containing NA values after GC modal correction were removed from subsequent analysis. GC-corrected counts were smoothed using the DNACopy R package (v.1.73.0) smooth.CNA function, setting smooth.region = 4. Smoothed counts were mean-normalized per cell before clustering using Seurat (v.5)85. For visualization, mean-normalized and smoothed counts were scaled bin-wise to emphasize copy differences between clusters.
scRNA-seq copy-number analysis
Copy number was inferred from 10× scRNA-seq for the RPE-1-Mixed and FNE1-Mixed samples using Numbat (v.1.4.0)86 to preprocess and smooth expression counts. Smoothed counts were then rebinned to 500-kilobase bins, reduced to 50 dimensions by PCA and then clustered using Leiden clustering at 1.0 resolution on a SNN graph.
Identification of WGD subclones
A spontaneously arising WGD copy-number clone was observed in all DLP+ samples for RPE-1, characterized by gain of chromosome 1p and loss of chromosomes 1q, 2q, 4q and 21 (Extended Data Fig. 8a). The same WGD clone was evident in copy number inferred from scATAC-seq for RPE-1-D, RPE-1-Noco, and RPE-1-Rev, and from scRNA-seq for RPE-1-Mixed (Extended Data Fig. 8a). For event rate analyses in RPE-1-D, RPE-1-Noco and RPE-1-Rev we excluded scRNA-seq cells in the scATAC-seq-inferred WGD clone from further analysis to characterize the phenotypic impact of CIN in non-WGD cells. For RPE-1-Mixed, we aimed to characterize the phenotypic differences between WGD and non-WGD cells. We therefore used the scRNA-based copy-number clusters to label cells in that sample as either WGD or non-WGD.
In the FNE1-Mixed cell line, from the DLP+ data we identified a WGD clone characterized by loss of chromosomes 4, 18 and 21, and gain of chromosomes 5 and 20 (Extended Data Fig. 8b). The same WGD clone was evident in the copy number inferred from scRNA-seq (Extended Data Fig. 8b).
Estimating rates of cell-specific events from DLP+
We inferred cell-specific rates of copy-number change from the RPE-1 and FNE1 DLP+ data using a clustering-based method. We first removed low-quality and cycling cells as described above. We then clustered all cells from each cell line to identify a stable non-WGD copy-number profile. Next, for each cell, we computed the number of copy-number events between the stable non-WGD profile and the cell profile under two scenarios: including a WGD along the path from stable profile to cell profile, and not including a WGD. For each scenario, we classified events using the same greedy approach as in the patient data to identify chromosome, arm and segment events. We kept the smaller set of events for each cell; if this corresponded to the scenario with a WGD, then the cell was called WGD. For the FNE1 data, we ignored the small number of WGD cells in preceding samples (9.2–13.2% of cells) because only those in the FNE1-Mixed sample represented the clone identifiable in scRNA-seq (Extended Data Fig. 8b).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Publicly accessible and controlled-access data generated and analysed in this study are documented at Synapse (accession number: syn66366718). Raw scWGS data are available by requesting authorization to the Data Access Committee through dbGaP (accession number: phs002857.v3.p1). Processed scWGS data are available on Synapse (accession number: syn66366960). Raw 10 × 3′ scRNA-seq data are available from the NCBI Gene Expression Omnibus (accession number: GSE180661). Processed scRNA-seq data are available at Synapse (accession numbers: syn33521743 and syn66477498). The raw microscopy data have not been uploaded owing to their large size (1.5 Tb) but are available from the corresponding authors upon reasonable request.
Code availability
The pipeline to process DLP+ scWGS is available at https://github.com/mondrian-scwgs. SIGNALS13 was used for most scWGS analysis and is available at https://github.com/shahcompbio/signals. doubleTime is available at https://github.com/shahcompbio/doubleTime. Code used for further analysis and to generate figures is available at https://github.com/shahcompbio/spectrum_wgd_paper.
References
Bielski, C. M. et al. Genome doubling shapes the evolution and prognosis of advanced cancers. Nat. Genet. 50, 1189–1195 (2018).
Zack, T. I. et al. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 45, 1134–1140 (2013).
Frankell, A. M. et al. The evolution of lung cancer and impact of subclonal selection in TRACERx. Nature 616, 525–533 (2023).
Dewhurst, S. M. et al. Tolerance of whole-genome doubling propagates chromosomal instability and accelerates cancer genome evolution. Cancer Discov. 4, 175–185 (2014).
Kuznetsova, A. Y. et al. Chromosomal instability, tolerance of mitotic errors and multidrug resistance are promoted by tetraploidization in human cells. Cell Cycle 14, 2810–2820 (2015).
Newcomb, R., Dean, E., McKinney, B. J. & Alvarez, J. V. Context-dependent effects of whole-genome duplication during mammary tumor recurrence. Sci. Rep. 11, 14932 (2021).
Baslan, T. et al. Ordered and deterministic cancer genome evolution after p53 loss. Nature 608, 795–802 (2022).
Zeng, J., Hills, S. A., Ozono, E. & Diffley, J. F. X. Cyclin E-induced replicative stress drives p53-dependent whole-genome duplication. Cell 186, 528–542 (2023).
Shi, Q. & King, R. W. Chromosome nondisjunction yields tetraploid rather than aneuploid cells in human cell lines. Nature 437, 1038–1042 (2005).
Lambuta, R. A. et al. Whole-genome doubling drives oncogenic loss of chromatin segregation. Nature 615, 925–933 (2023).
Gemble, S. et al. Genetic instability from a single S phase after whole-genome duplication. Nature 604, 146–151 (2022).
Salehi, S. et al. Clonal fitness inferred from time-series modelling of single-cell cancer genomes. Nature 595, 585–590 (2021).
Funnell, T. et al. Single-cell genomic variation induced by mutational processes in cancer. Nature 612, 106–115 (2022).
Vázquez-García, I. et al. Ovarian cancer mutational processes drive site-specific immune evasion. Nature 612, 778–786 (2022).
Funnell, T. et al. Integrated structural variation and point mutation signatures in cancer genomes using correlated topic models. PLoS Comput. Biol. 15, e1006799 (2019).
Laks, E. et al. Clonal decomposition and DNA replication states defined by scaled single-cell genome sequencing. Cell 179, 1207–1221 (2019).
Dentro, S. C. et al. Characterizing genetic intra-tumor heterogeneity across 2,658 human cancer genomes. Cell 184, 2239–2254 (2021).
Kim, M. et al. Single-cell mtDNA dynamics in tumors is driven by coregulation of nuclear and mitochondrial genomes. Nat. Genet. 56, 889–899 (2024).
Gerstung, M. et al. The evolutionary history of 2,658 cancers. Nature 578, 122–128 (2020).
Alexandrov, L. B. et al. Clock-like mutational processes in human somatic cells. Nat. Genet. 47, 1402–1407 (2015).
Bollen, Y. et al. Reconstructing single-cell karyotype alterations in colorectal cancer identifies punctuated and gradual diversification patterns. Nat. Genet. 53, 1187–1195 (2021).
Zhang, C.-Z. et al. Chromothripsis from DNA damage in micronuclei. Nature 522, 179–184 (2015).
Crasta, K. et al. DNA breaks and chromosome pulverization from errors in mitosis. Nature 482, 53–58 (2012).
Umbreit, N. T. et al. Mechanisms generating cancer genome complexity from a single cell division error. Science 368, eaba0712 (2020).
Mackenzie, K. J. et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548, 461–465 (2017).
Baker, T. M. et al. The history of chromosomal instability in genome-doubled tumors. Cancer Discov. 14, 1810–1822 (2024).
Dinh, K. N. et al. CINner: Modeling and simulation of chromosomal instability in cancer at single-cell resolution. PLoS Comput. Biol. 21, e1012902 (2025).
Santaguida, S. et al. Chromosome mis-segregation generates cell-cycle-arrested cells with complex karyotypes that are eliminated by the immune system. Dev. Cell 41, 638–651 (2017).
Thompson, S. L. & Compton, D. A. Proliferation of aneuploid human cells is limited by a p53-dependent mechanism. J. Cell Biol. 188, 369–381 (2010).
Bakhoum, S. F. et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 553, 467–472 (2018).
Xia, T., Konno, H., Ahn, J. & Barber, G. N. Deregulation of STING signaling in colorectal carcinoma constrains DNA damage responses and correlates with tumorigenesis. Cell Rep. 14, 282–297 (2016).
Konno, H. et al. Suppression of STING signaling through epigenetic silencing and missense mutation impedes DNA damage mediated cytokine production. Oncogene 37, 2037–2051 (2018).
Xia, T., Konno, H. & Barber, G. N. Recurrent loss of STING signaling in melanoma correlates with susceptibility to viral oncolysis. Cancer Res. 76, 6747–6759 (2016).
Kitajima, S. et al. Suppression of STING associated with LKB1 loss in KRAS-driven lung cancer. Cancer Discov. 9, 34–45 (2019).
Amiji, M. M. & Milane, L. S. (eds) Cancer Immunology and Immunotherapy. Vol. 1. Delivery Strategies and Engineering Technologies in Cancer Immunotherapy (Academic, 2021).
de Queiroz, N. M. G. P., Xia, T., Konno, H. & Barber, G. N. Ovarian cancer cells commonly exhibit defective STING signaling which affects sensitivity to viral oncolysis. Mol. Cancer Res. 17, 974–986 (2019).
Li, J. et al. Non-cell-autonomous cancer progression from chromosomal instability. Nature 620, 1080–1088 (2023).
Sauer, C. M. et al. Molecular landscape and functional characterization of centrosome amplification in ovarian cancer. Nat. Commun. 14, 6505 (2023).
Gao, R. et al. Punctuated copy number evolution and clonal stasis in triple-negative breast cancer. Nat. Genet. 48, 1119–1130 (2016).
Navin, N. et al. Tumour evolution inferred by single-cell sequencing. Nature 472, 90–94 (2011).
Baca, S. C. et al. Punctuated evolution of prostate cancer genomes. Cell 153, 666–677 (2013).
Cross, W. et al. The evolutionary landscape of colorectal tumorigenesis. Nat. Ecol. Evol. 2, 1661–1672 (2018).
Minussi, D. C. et al. Breast tumours maintain a reservoir of subclonal diversity during expansion. Nature 592, 302–308 (2021).
Burdett, N. L. et al. Timing of whole genome duplication is associated with tumor-specific MHC-II depletion in serous ovarian cancer. Nat. Commun. 15, 6069 (2024).
Cheng, Z. et al. The genomic trajectory of ovarian high-grade serous carcinoma can be observed in STIC lesions. J. Pathol. 264, 42–54 (2024).
Kader, T. et al. Multimodal spatial profiling reveals immune suppression and microenvironment remodeling in fallopian tube precursors to high-grade serous ovarian carcinoma. Cancer Discov. https://doi.org/10.1158/2159-8290.CD-24-1366 (2025).
Tewari, K. S. et al. Final overall survival of a randomized trial of bevacizumab for primary treatment of ovarian cancer. J. Clin. Oncol. 37, 2317–2328 (2019).
Lorusso, D. et al. Updated progression-free survival and final overall survival with maintenance olaparib plus bevacizumab according to clinical risk in patients with newly diagnosed advanced ovarian cancer in the phase III PAOLA-1/ENGOT-ov25 trial. Int. J. Gynecol. Cancer 34, 550–558 (2024).
Phillips, A. F. et al. Targeting chromosomally unstable tumors with a selective KIF18A inhibitor. Nat. Commun. 16, 307 (2025).
Payton, M. et al. Small-molecule inhibition of kinesin KIF18A reveals a mitotic vulnerability enriched in chromosomally unstable cancers. Nat Cancer 5, 66–84 (2024).
Quinton, R. J. et al. Whole-genome doubling confers unique genetic vulnerabilities on tumour cells. Nature 590, 492–497 (2021).
Hobor, S. et al. Mixed responses to targeted therapy driven by chromosomal instability through p53 dysfunction and genome doubling. Nat. Commun. 15, 4871 (2024).
Zehir, A. et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 23, 703–713 (2017).
Bykov, Y., Kim, S. H. & Zamarin, D. Preparation of single cells from tumors for single-cell RNA sequencing. Methods Enzymol. 632, 295–308 (2020).
Merritt, M. A. et al. Gene expression signature of normal cell-of-origin predicts ovarian tumor outcomes. PLoS ONE 8, e80314 (2013).
Medina-Martínez, J. S. et al. Isabl Platform, a digital biobank for processing multimodal patient data. BMC Bioinformatics 21, 549 (2020).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Ha, G. et al. Integrative analysis of genome-wide loss of heterozygosity and monoallelic expression at nucleotide resolution reveals disrupted pathways in triple-negative breast cancer. Genome Res. 22, 1995–2007 (2012).
Delaneau, O., Marchini, J. & Zagury, J.-F. A linear complexity phasing method for thousands of genomes. Nat. Methods 9, 179–181 (2012).
Curtius, K., Wright, N. A. & Graham, T. A. An evolutionary perspective on field cancerization. Nat. Rev. Cancer 18, 19–32 (2018).
McPherson, A. W. et al. ReMixT: clone-specific genomic structure estimation in cancer. Genome Biol. 18, 140 (2017).
Shen, R. & Seshan, V. E. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 44, e131 (2016).
Benjamin, D. et al. Calling somatic SNVs and indels with Mutect2. Preprint at bioRxiv https://doi.org/10.1101/861054 (2019).
McPherson, A., Shah, S. & Cenk Sahinalp, S. deStruct: accurate rearrangement detection using breakpoint specific realignment. Preprint at bioRxiv https://doi.org/10.1101/117523 (2017).
Layer, R. M., Chiang, C., Quinlan, A. R. & Hall, I. M. LUMPY: a probabilistic framework for structural variant discovery. Genome Biol. 15, R84 (2014).
Wang, Y. K. et al. Genomic consequences of aberrant DNA repair mechanisms stratify ovarian cancer histotypes. Nat. Genet. 49, 856–865 (2017).
Satas, G., Myers, M. A., McPherson, A. & Shah, S. P. Inferring active mutational processes in cancer using single cell sequencing and evolutionary constraints. Preprint at bioRxiv https://doi.org/10.1101/2025.02.24.639589 (2025).
Myers, M. A., Zaccaria, S. & Raphael, B. J. Identifying tumor clones in sparse single-cell mutation data. Bioinformatics 36, i186–i193 (2020).
Ranganath, R., Gerrish, S. & Blei, D. Black box variational inference. In Proc. Seventeenth International Conference on Artificial Intelligence and Statistics (eds Kaski, S. & Corander, J.) vol. 33, 814–822 (PMLR, 2014).
Kaufmann, T. L. et al. MEDICC2: whole-genome doubling aware copy-number phylogenies for cancer evolution. Genome Biol. 23, 241 (2022).
Ross, E. M., Haase, K., Van Loo, P. & Markowetz, F. Allele-specific multi-sample copy number segmentation in ASCAT. Bioinformatics 37, 1909–1911 (2021).
Fitch, W. M. Toward defining the course of evolution: minimum change for a specific tree topology. Syst. Zool. 20, 406–416 (1971).
Sankoff, D. Minimal mutation trees of sequences. SIAM J. Appl. Math. 28, 35–42 (1975).
Zhang, A. W. et al. Probabilistic cell-type assignment of single-cell RNA-seq for tumor microenvironment profiling. Nat. Methods 16, 1007–1015 (2019).
Tirosh, I. et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature 539, 309–313 (2016).
Tirosh, I. et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016).
Zatzman, M. et al. Widespread hypertranscription in aggressive human cancers. Sci. Adv. 8, eabn0238 (2022).
Liang, S., Wang, F., Han, J. & Chen, K. Latent periodic process inference from single-cell RNA-seq data. Nat. Commun. 11, 1441 (2020).
Whitfield, M. L. et al. Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Mol. Biol. Cell 13, 1977–2000 (2002).
Gavish, A. et al. Hallmarks of transcriptional intratumour heterogeneity across a thousand tumours. Nature 618, 598–606 (2023).
Liberzon, A. et al. The Molecular Signatures Database hallmark gene set collection. Cell Syst. 1, 417–425 (2015).
Hoffman, G. E. et al. Efficient differential expression analysis of large-scale single cell transcriptomics data using dreamlet. Preprint at bioRxiv https://doi.org/10.1101/2023.03.17.533005 (2024).
Dann, E., Henderson, N. C., Teichmann, S. A., Morgan, M. D. & Marioni, J. C. Differential abundance testing on single-cell data using k-nearest neighbor graphs. Nat. Biotechnol. 40, 245–253 (2022).
Bankhead, P. et al. QuPath: open source software for digital pathology image analysis. Sci. Rep. 7, 16878 (2017).
Butler, A., Hoffman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018).
Gao, T. et al. Haplotype-aware analysis of somatic copy number variations from single-cell transcriptomes. Nat. Biotechnol. 41, 417–426 (2023).
Acknowledgements
This project was primarily supported through a Department of Defense Congressionally Directed Medical Research Program award (W81XWH-20-1-0565), an Ovarian Cancer Research Alliance (OCRA) Collaborative Research Development Grant (648007), an NIH R01 CA281928-01 and by the Seidenberg Family Foundation. Further support was provided by the Halvorsen Center for Computational Oncology and Cycle for Survival supporting Memorial Sloan Kettering Cancer Center. S.P.S. holds the Nicholls Biondi Chair in Computational Oncology and is a Susan G. Komen scholar. The OCRA Ann Schreiber Mentored Investigator Award to I.V.-G. (650687), OCRA Liz Tilberis Award to D.Z. (657721), the Marie-Josée and Henry R. Kravis Center for Molecular Oncology and the National Cancer Institute (NCI) Cancer Center Core Grant (P30-CA008748) provided additional support. D.H.A.-R. is supported by the Mary Jane Milton Endowed Fellowship in Gynecologic Oncology, NIH T32-CA009207, an OCRA Mentored Research Award (MIG-2023-1-1012), Conquer Cancer Research YIA and Gerstner Physician Scientist Award. S.F.B. is funded by NIH/NCI grants (P50CA247749, DP5OD026395, R01CA256188 and P30-CA008748), the Department of Defense Congressionally Directed Medical Research Program (BC201053), the Burroughs Wellcome Fund, the Josie Robertson Foundation, the Pershing Square Sohn Alliance for Cancer Research and the Mary Kay Ash Foundation. B.W. is funded in part by the Breast Cancer Research Foundation and NIH/NCI P50 CA247749 01 grants. D.Z. is funded by NIH grant R01 CA269382. A.C.W. is supported by an NCI Ruth L. Kirschstein National Research Service Award for Predoctoral Fellows F31-CA271673. R.F.S. is a professor at the Cancer Research Center Cologne Essen funded by the Ministry of Culture and Science of the State of North Rhine-Westphalia, and was partly funded by the German Ministry for Education and Research as BIFOLD − Berlin Institute for the Foundations of Learning and Data (01IS18025A and 01IS18037A). This work used the resources of the High-Performance Computing Group at Memorial Sloan Kettering Cancer Center.
Author information
Authors and Affiliations
Contributions
A.M. and S.P.S.: project conception and oversight. A.M., D.H.A.-R., I.V.-G., M.A.M., M.Z., N.R., S.F.B. and S.P.S.: manuscript writing and editing. A.M., F.U., I.V.-G., S.F.B. and S.P.S.: study design. B.W., C.A., J.L.P.L., M.W. and N.R.A.-R.: clinical research coordination. B.W., E.A., H.G., J.L.P.L., M.W. and N.V.: tissue procurement and data generation. D.Z. and S.H.K.: dissociation protocols. A.V., D.H.A.-R., D.K., J.C., N.M., R.P., S.A., S.R. and Y.W.: DLP+ sequencing. A.V. and N.M.: scRNA-seq and genome sequencing. D.H.A.-R., D.N., J.L. and M.D.: immunofluorescence imaging. D.H.A.-R. and D.N.: in vitro experiments. A.M., A.C.W., I.V.-G., M.A.M., R.F.S. and T.K.: computational method development. A.M., A.C.W., A.W.Z., F.U., I.V.-G., K.N.D., M.A.M., M.J.W., M.Z., N.C., R.F.S., S.T. and T.K.: computational biology and data analysis. F.U. and I.V.-G.: data curation. A.M., D.G., E.H., F.U., G.S., H.S., I.V.-G., J.T., M.A.M., M.J.W., M.K., M.Z., R.K., S.C. and S.F.: data processing and visualization. D.S.C., G.J.G., K.L.R., N.R.A.-R., O.Z., V.B. and Y.S.: surgery. C.A., C.F.F., D.Z., R.N.G. and Y.L.L.: clinical data review. A.W.Z. and L.H.E.: pathology review. Y.L.: radiology review. A.M., S.F.B. and S.P.S.: discussion.
Corresponding authors
Ethics declarations
Competing interests
B.W. reports grant funding by Repare Therapeutics paid to the institution, outside the scope of this paper, and employment of a direct family member at AstraZeneca. C.A. reports grants from Clovis, Genentech, AbbVie and AstraZeneca, and personal fees from Tesaro, Eisai/Merck, Mersana Therapeutics, Roche/Genentech, Abbvie, AstraZeneca/Merck and Repare Therapeutics, outside the scope of this paper. C.A. also reports clinical-trial funding to the institution from Abbvie, AstraZeneca and Genentech/Roche; participation on a data safety monitoring board or advisory board at AstraZeneca and Merck; unpaid membership of the GOG Foundation board of directors and the NRG Oncology board of directors. C.F.F. reports research funding to the institution from Merck, AstraZeneca, Genentech/Roche, Bristol Myers Squibb and Daiichi; uncompensated membership of a scientific advisory board for Merck and Genentech; and is a consultant for OncLive, Aptitude Health, Bristol Myers Squibb and Seagen, all outside the scope of this paper. D.S.C. reports membership of the medical advisory board of Verthermia Acquio and Biom’up, is a paid speaker for AstraZeneca and holds stock of Doximity, Moderna and BioNTech. D.Z. reports institutional grants from Merck, Genentech, AstraZeneca, Plexxikon and Synthekine, and personal fees from AstraZeneca, Xencor, Memgen, Takeda, Astellas, Immunos, Tessa Therapeutics, Miltenyi and Calidi Biotherapeutics. D.Z. holds a patent on the use of oncolytic Newcastle disease virus for cancer therapy. N.R.A.-R. reports grants to the institution from Stryker/Novadaq and GRAIL, outside the scope of this paper. R.N.G. reports funding from GSK, Novartis, Mateon Therapeutics, Corcept, Regeneron, Clovis, Context Therapeutics, EMD Serono, MCM Education, OncLive, Aptitude Health and Prime Oncology, outside the scope of this paper. S.F.B. owns equity in, receives compensation from and serves as a consultant and on the scientific advisory board and board of directors of Volastra Therapeutics. He also serves on the scientific advisory board of Meliora Therapeutics. S.P.S. reports research funding from AstraZeneca and Bristol Myers Squibb, outside the scope of this paper. Y.L.L. reports research funding from AstraZeneca, GSK/Tesaro, Artios Pharma and Tesaro Therapeutics, outside the scope of this paper. Y.L. reports serving as a consultant for Calyx Clinical Trial Solutions, outside the scope of this paper. All the remaining authors declare no competing interests.
Peer review
Peer review information
Nature thanks Seishi Ogawa and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Study and cohort overview.
a. Schematic of the MSK SPECTRUM specimen collection workflow including primary debulking surgery or laparoscopic biopsy, single-cell suspensions for scWGS and scRNA-seq, and biobanking of snap-frozen and FFPE tissue samples. b. Cohort overview. Top panel: Oncoprint of selected somatic and germline mutations per patient and cohort-wide prevalence. Single nucleotide variants (SNVs), indels, and fusions shown are detected by targeted panel sequencing (MSK-IMPACT). Focal amplifications and deletions are detected by single-cell whole genome sequencing (scWGS). Patient data include WGD class, mutational signature subtype, patient age, staging following FIGO Ovarian Cancer Staging guidelines, and type of surgical procedure. Bottom panel: Sample and data inventory indicating number of co-registered multi-site datasets: single-cell whole genome sequencing, single-cell RNA sequencing, H&E whole-slide images, immunofluorescence, bulk WGS and bulk MSK-IMPACT.
Extended Data Fig. 2 Quality control of scWGS data and WGD inference.
a. Number of high-quality cells generated per patient, divided into and colored by anatomical site. b. Box plots of per-cell coverage depth per patient (n = 41 patients). Center line shows the median, box boundaries show quartiles, and whiskers indicate 1.5 × IQR. c. Fraction of cells called as tumor, non-tumor, doublet, and S-phase for each patient. d. Example doublet identified from an image taken during DLP+ sequencing (see Supplementary Note for additional examples). e. Frequency of gains (red, above the horizontal) and losses (blue, below the horizontal) among all single-cell genomes in the cohort, with known drivers genes annotated. f. Tumor ploidy (mean tumor copy number) inferred by FACETS in MSK IMPACT data (x-axis) compared to average ploidy (mean copy number per cell, averaged across cells) for each patient in the SPECTRUM cohort (y-axis). The dashed line denotes the linear regression fit, grey regions indicate 95% confidence intervals, and two-sided Spearman’s rank correlation coefficient and p-value are shown in the upper left. g. Tumor ploidy (mean tumor copy number) inferred by ReMixT in bulk WGS data (x-axis) compared to average ploidy (mean copy number per cell, averaged across cells) for each patient in the SPECTRUM cohort (y-axis). The dashed line denotes the linear regression fit, grey regions indicate 95% confidence intervals, and two-sided Spearman’s rank correlation coefficient and p-value are shown in the upper left. Two patients (OV-052 and OV-068) were omitted due to poor quality bulk WGS copy number. h. Shown for all quality-filtered cells in the cohort is the mean difference between major and minor copy number (y-axis) versus the fraction of the genome with major copy number ≥ 2 (x-axis), with cells colored by WGD multiplicity. The dashed line at 0.5 denotes the decision boundary for 0 vs 1 WGDs. i. Shown for all quality-filtered cells in the cohort is the mean difference between major and minor copy number (y-axis) versus the fraction of the genome with major copy number ≥ 3 (x-axis), with cells colored by WGD multiplicity. The dashed line at 0.5 denotes the decision boundary for 1 vs 2 WGDs. j. Mitochondrial DNA copy number (log10) for each scWGS cell grouped by WGD multiplicity for 0 × (n = 13,069), 1 × (n = 16,782), and 2×WGD (n = 409) cells. Each datapoint is a cell. Box plots are defined as per b. Mann-Whitney two-sided U test significance is annotated as ‘ns’: 5.0 × 10−2 < p <= 1.0, ‘*’: 1.0 × 10−2 < p <= 5.0 × 10−2, ‘**’: 1.0 × 10−3 < p <= 1.0 × 10−2, ‘***’: 1.0 × 10−4 < p <= 1.0 × 10−3, ‘****’: p <= 1.0 × 10−4. Both p-values < 10−22. k. Average fraction of overlapping reads for each scWGS cell, grouped by WGD multiplicity (same n cells as j). Box plots are defined as per b. Significance was calculated and annotated as per j. Both p-values < 10−51. l. Cell diameter measured from DLP+ images for each scWGS cell, split by WGD multiplicity (same n cells as j). Boxplots are defined as per b. Significance was calculated and annotated as per j. Both p-values < 10−26. m-o. Example 0×WGD, 1×WGD, and 2×WGD cells from patient OV-045. Each point is a 500 kb bin. Top track shows GC corrected read count scaled by the inferred ploidy and colored by total copy number state, and bottom track shows B-allele frequency colored by allelic imbalance. p. Distribution of the fraction of additional-WGD cells per patient. q. Age at diagnosis for patients in the SPECTRUM cohort split by WGD-high (n = 27) vs WGD-low (n = 14). Box plots are defined as per b. p-value was calculated using a Mann-Whitney U one-sided test. r. Age at diagnosis for patients in the PCAWG ovarian cohort split by WGD (n = 67) vs non-WGD (n = 42). Box plots are defined as per b. p-value was calculated using a Mann-Whitney U one-sided test. s. Fraction of WGD-high and WGD-low tumors in the SPECTRUM cohort for each mutational signature. t. Fraction non-WGD and WGD patients in the Ovarian Metacohort15 for each mutation signature.
Extended Data Fig. 3 Residual 0xWGD cells in WGD-high patients.
a-f. Total copy-number profiles for patient OV-045 pseudobulk and all non-divergent 0xWGD cells. Each point is a 500 kb bin colored by its assigned copy-number state, the y-axis shows scaled GC-corrected read depth, and the x-axis shows genomic position. The top track of each panel shows the pseudobulk profile for all filter-passing cells (note that average total copy number, i.e., ploidy, is close to 3N-4N indicating WGD), and each lower track shows a single cell. b. Total copy-number profiles for OV-051 pseudobulk and all non-divergent 0xWGD cells as defined in a. c. Total copy-number profiles for OV-075 pseudobulk and all non-divergent 0xWGD cells as defined in a. d. Total copy-number profiles for OV-087 pseudobulk and all non-divergent 0xWGD cells as defined in a. e. Total copy-number profiles for OV-107 pseudobulk and all non-divergent 0xWGD cells as defined in a. f. Total copy-number profiles for OV-110 pseudobulk and all non-divergent 0xWGD cells as defined in a.
Extended Data Fig. 4 WGD evolution, non-WGD subclones, and subclonal WGD.
a. Clone phylogenies and WGD timing for 21 additional patients in our cohort (18 patients are shown in Fig. 2b). Branch length shows the number of age-associated SNVs (C-to-T at CpG) assigned to each branch, adjusted for coverage-depth-related reduction in SNV sensitivity. Clone size as a fraction of the patient’s total sequenced cells is shown by the size of the triangle for each leaf. Expanded WGD events are represented as triangles at the predicted location along WGD branches, with the color of the triangle indicating relative timing (early vs late). Branches are colored according to the number of WGD at that point in each evolutionary history. Bar plots below each clone tree show, for each SBMClone-derived leaf, the fraction of cells in each WGD multiplicity and the fraction of cells from each anatomical site. Patients are grouped by WGD evolution class. The x-axis is labeled with the SBMClone clone indices for each leaf. b-c. SBMClone clones and 0×WGD subpopulations in patients OV-045 (A) and OV-075 (B). Shown for each patient is the total copy number (left) and allelic imbalance (middle) for each clone (y-axis). Barplots on the right show the fraction of cells from that clone found in each anatomic site (left) and the number of cells for each clone (right). d. SBMClone block density matrix for patient OV-025 showing the proportion of SNVs detected for each clone (y-axis) and SNV block (x-axis). The SBMClone cluster and WGD status of each cell are shown on the right. The mostly-2×WGD clone in patient OV-025 is distinguished by clone-specific SNVs (arrow). e. Copy number for chromosomes 7, 8, and 9 for cells in patient OV-006, separated into non-WGD cells (top), WGD cells (middle), and inferred post-WGD changes in WGD cells (bottom; gains are indicated in red and losses are indicated in blue). The cell order is the same for the middle and bottom plots. Arrows indicate shared post-WGD changes that represent a WGD subclone. f. Copy number for chromosomes 2 and 8 for cells in patient OV-031, shown as per e. g. Copy number for chromosomes 1, 4, 15 and X for cells in patient OV-139, shown as per e. h. Absolute (upper bar plot) and relative (lower bar plot) number of malignant scWGS cells by WGD multiplicity (color) and sample (x-axis). Samples are separated by patient and ordered by the proportion of cancer cells with at least 1 WGD. Bottom tracks indicate the anatomical site for each sample and the WGD class for each patient.
Extended Data Fig. 5 Single cell measurement of chromosomal instability.
a. Schematic of nearest neighbor difference (NND) using fraction of the genome different as a distance measure (left). Shown are two pairs of example nearest nearest neighbor cells and regions of the genome that are different for a 0×WGD cell (middle) and a 1×WGD cell (right). Each point is a 500 kb bin colored by the assigned copy-number state, and the y-axis shows ploidy-scaled GC-normalized read counts. b. Empirical distribution of NND for all cells, and beta distribution fit (red). c. NND (y-axis) by ploidy (x-axis) for cells from patient OV-081. Color indicates divergent status, and WGD multiplicity for non-divergent cells. d. Copy-number profiles for example 0×WGD (top), 1×WGD (middle) and divergent (bottom) cells from patient OV-081. Arrows highlight homozygously deleted regions. e. Arm nullisomy rates (counts per cell) for divergent and non-divergent cells in WGD-low and WGD-high tumors. Shown is the distribution of mean rates per population in each patient (only those populations with at least 10 cells are included): WGD-low non-divergent n = 14, WGD-low divergent n = 6, WGD-high non-divergent n = 20, WGD-high divergent n = 12 populations. Mann-Whitney U one-sided test significance is annotated as ‘ns’: 5.0 × 10−2 < p <= 1.0, ‘*’: 1.0 × 10−2 < p <= 5.0 × 10−2, ‘**’: 1.0 × 10−3 < p <= 1.0 × 10−2, ‘***’: 1.0 × 10−4 < p <= 1.0 × 10−3, ‘****’: p <= 1.0 × 10−4. Center line shows the median, box boundaries show quartiles, and whiskers indicate 1.5×IQR. WGD-low p = 2.6 × 10−5, WGD-high p = 2.0 × 10−6. f. Boxplots comparing fraction of divergent cells between WGD multiplicity populations for WGD-low and WGD-high tumors (only those populations with over 20 cells are included): WGD-low 0xWGD n = 14, WGD-low 1xWGD n = 8, WGD-high 0xWGD n = 2, WGD-high 1xWGD n = 25, WGD-high 2xWGD n = 4 populations. Mann-Whitney U one-sided test significance is annotated as per e. Boxplots are defined as per e. WGD-low (0xWGD vs 1xWGD) p = 8.2 × 10−4, WGD-high (1xWGD vs 2xWGD) p = 8.4 × 10−5. g. Fraction of divergent cells (y-axis) by age of the WGD as measured by C > T CpG mutations gained since WGD (x-axis). Shown is the p-value of a two-sided Spearman correlation after removing the three patients with the oldest WGDs. Two-sided Spearman correlation retaining these outliers is ρ = −0.47 p = 0.019. Shaded region indicates 95% confidence interval. h. MEDICC2 phylogeny (left) total copy number (center) and inferred cell-specific copy number changes (right) for patient OV-110. i. Coefficients, 95% confidence intervals, and p-values for the WGD term of a GEE model of chromosome, arm and segment loss and gain rates (counts per cell, normalized for genome size) for n = 54 adnexa vs non-adnexa subpopulations from the 37 patients with event rate estimates. The GEE model includes patient age, WGD status (high vs low), mutation signature (FBI vs non-FBI) and site (Adnexa vs non-Adnexa). j. Example immunofluorescence images of WGD-high and WGD-low tumor samples with varying MN rates. Images are annotated with the slide-level MN rates, calculated as the median MN rate across all tumor ROI regions within the slide. Top panels: Multi-channel overlay images of DAPI, cGAS and panCK intensity at high magnification. Bottom panels: Segmentation masks for cGAS+ MN and PN, including examples of micronuclei with annotated area size in μm2. k. Ratio of losses to gains for chromosomes (left) and chromosome arms (right). Cell specific refers to changes on leaf branches of the MEDICC2 phylogeny which are split by WGD-low and WGD-high tumor type. Ancestral changes are split into non-WGD, pre-WGD, and post-WGD as defined for Fig. 4B. Each datapoint for the cell specific distributions is a ratio of losses to gains for a single patient. Each datapoint for non-WGD, pre-WGD, and post-WGD distributions is a ratio computed from the root branch of the MEDICC2 phylogeny for a patient, distinguishing pre- and post-WGD changes for WGD-high tumors and including all changes for WGD-low tumors. Error bars indicate 95% confidence interval. Patients OV-045 and OV-025 with multiple parallel WGD events were excluded from this analysis. Mann-Whitney one-sided multiple-hypothesis-corrected U test p < 1.3 × 10−3 for chromosome event ratios, p = 0.016 for WGD-low vs. non-WGD arm event ratios, and p < 7.1* × 10−3 for remaining arm comparisons. WGD-low n=non-WGD n = 14, WGD-high n = 27, and pre-WGD n=post-WGD n = 21. l. Number of post-WGD chromosome and arm gains and losses (x-axis) compared to the mutation time in C > T CpG counts (y-axis) measured since the WGD event. Spearman correlation coefficients and p-values are shown.
Extended Data Fig. 6 Cell cycle progression in the context of WGD.
a. Absolute and relative compositions of cell cycle fractions in CD45− sorted samples based on scRNA-seq. Samples are separated by patient, and ordered within each patient by proportion of S-phase cells out of all cancer cells. b. Coefficients (x-axis) of a Generalized Estimation Equation (GEE) fit to the difference in cancer cell cycle fractions between WGD-low and WGD-high samples, corrected for patient effects, age and tumor site and mutational signature subtype. Bars indicate 95% confidence intervals. * indicates p < 0.05. c. Scaled expression of phase-specific genes in WGD-high (left panel) vs WGD-low (middle panel) tumors as a function of cell cycle pseudotime. Right panel: Differences in scaled gene expression of phase-specific genes in WGD-high vs WGD-low tumors as a function of cell cycle pseudotime. d. Scatter plot of hallmark E2F module score (y-axis) by rate (counts per cell) of chromosomal losses (x-axis) split by WGD-low and WGD-high (color). Lines indicate the result of a linear regression within either WGD-high or WGD-low tumors. Regression coefficients and significance are shown separately for WGD-low and WGD-high tumors. Each point is a tumor sample. e. Scatter plot of the fraction of cancer cells in G1 (y-axis) by rate (counts per cell) of chromosomal losses (x-axis) split by WGD-low and WGD-high (color). Lines indicate the result of a linear regression within either WGD-high or WGD-low tumors. Regression coefficients and significance are shown separately for WGD-low and WGD-high tumors. Each point is a tumor sample.
Extended Data Fig. 7 Tumor cell phenotypes in the context of WGD and mutation signatures.
a. Scatter plot depicting regression coefficients (x-axis) and significance (y-axis) for selected genes and pathways in WGD-high vs WGD-low tumor cells in the HRD-Dup mutation signature subset. b. Violin plots of per-sample mean expression for select cancer-cell-intrinsic signaling pathways faceted by mutation signature subset and WGD status: FBI (WGD-low n = 3 samples, WGD-high n = 27 samples), HRD-Del (WGD-high n = 17 samples), and HRD-Dup (WGD-low n = 31 samples, WGD-high n = 16 samples). Dot indicates median and bars indicate quartiles. c. Dotplot of correlations between missegregation rates derived from scWGS (column) and cancer-cell-intrinsic pathways from scRNA-seq in site-matched samples (row). Spearman’s rho significance is annotated as ‘ns’: 5.0 × 10-2 <p <= 1, ‘*’: 1.0 × 10−2 < p <= 5.0 × 10−2, ‘**’: 1.0 × 10−3 < p <= 1.0 × 10−2, ‘***’: 1.0 × 10−4 < p <= 1.0 × 10−3, ‘****’: p <= 1.0 × 10−4.
Extended Data Fig. 8 Transcriptional consequences of WGD in RPE-1 and FNE1 cell lines.
a. Clone copy number inferred from scWGS DLP+ (top), scATAC-seq (middle), and scRNA-seq (bottom) for RPE-1 cells across treatment conditions. Two clones were identified in all modalities: one WGD and one non-WGD. b. Clone copy number inferred from scWGS DLP+ (top) and scRNA-seq (bottom) for FNE1 cells. Two clones were identified in both modalities: one WGD and one non-WGD. c. Expression UMAP from scRNA-seq of FNE1 and RPE-1 mixed-WGD samples with cells colored by assignment to the WGD and non-WGD clones. d. Chromosome and arm loss and gain events per cell for RPE-1 and FNE1 cells split by WGD status (upper panels), and number of cells in each condition (bottom row) from scWGS DLP+. e. Cell-cycle phase fractions inferred from scRNA-seq for RPE-1 and FNE1 samples treated with DMSO (RPE-1-D and FNE1-D), Nocodazole (RPE-1-Noco and FNE1-Noco) and Reversine (RPE-1-Rev and FNE1-Rev). Cell-cycle phase fractions for RPE-1 samples were computed after excluding the spontaneously arising WGD present in these samples. f. STING1 expression in STING1 positive cells (top), mean STING1 expression (middle), and proportion of WGD cells (bottom) for non-WGD and WGD RPE-1 (left) and FNE1 (right) cells by treatment condition. Note that proportion of WGD cells was estimated from scRNA whereas in d, number of cells was computed from scWGS DLP+. Center line shows the median, box boundaries show quartiles, and whiskers indicate 1.58×IQR/sqrt(n). Each point is a cell: RPE-1-D non-WGD n = 744, WGD n = 63; RPE-1-Noco non-WGD n = 1013, WGD n = 77; RPE-1-Rev non-WGD n = 1050, WGD n = 29; RPE-1-Mixed non-WGD n = 79, WGD n = 117; FNE1-D non-WGD n = 486; FNE1-REF non-WGD n = 454; FNE1-Mixed non-WGD n = 66, WGD n = 203; cells. Wilcoxon two-sided test with Bonferroni-Hochberg correction, all comparisons p < 5.04 × 10−4. g. Cell-cycle phase fractions inferred from scRNA-seq for WGD and nonWGD populations in untreated mixed-WGD RPE-1 and FNE1 samples.
Extended Data Fig. 9 Microenvironment remodeling in the context of WGD and mutation signatures.
a. UMAPs showing differential cell state enrichment in WGD-high vs WGD-low samples in different TME cell types. b. Dotplot of log2 fold-changes and significance for TME cell type differential abundance testing between WGD-high and WGD-low in the whole cohort and in the HRD-Dup subset. Significance was calculated from Milo results using a permutation test (Methods). c. Cytotoxic CD8+ T cells (y-axis) and CXCL10+CD274+ Macrophages (x-axis) as fractions of CD45+ cells across CD45+ samples. Points are colored by the WGD class of the patient from which the sample originated. Spearman’s ρ and p-value are annotated.
Supplementary information
Supplementary Information (download DOCX )
The Supplementary Information file contains Supplementary Notes 1–10, Supplementary Figs. 1–32 and legends for Supplementary Data files 1 and 2.
Supplementary Table 1 (download XLSX )
Clinical overview of the MSK SPECTRUM patient cohort. Data include patient age at diagnosis, staging following FIGO Ovarian Cancer Staging guidelines, type of surgical procedure, WGD class, VAF and mutational signature subtype.
Supplementary Table 2 (download XLSX )
Sample inventory. Metadata associated with scWGS, scRNA-seq, H&E, immunofluorescence, bulk tumour and normal WGS, and tumour and normal MSK-IMPACT datasets.
Supplementary Table 3 (download CSV )
Sample-level sequencing statistics and cell counts for scWGS datasets.
Supplementary Table 4 (download CSV )
Thresholds used for each patient for filtering out S-phase cells in scWGS datasets.
Supplementary Table 5 (download XLSX )
Sample-level and ROI-level statistics for immunofluorescence datasets.
Supplementary Table 6 (download XLSX )
Antibodies and staining conditions for immunofluorescence datasets.
Supplementary Table 7 (download XLSX )
Description of cell line datasets.
Supplementary Data 1 (download PDF )
Pairwise VAF plots for the 13 WGD-high tumours with sufficient data showing clonal cnLOH SNVs plotted for each pair of SBMClone clones.
Supplementary Data 2 (download PDF )
Plots showing doubleTime-assigned SNVs for each branch for all patients.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
McPherson, A., Vázquez-García, I., Myers, M.A. et al. Ongoing genome doubling shapes evolvability and immunity in ovarian cancer. Nature 644, 1078–1087 (2025). https://doi.org/10.1038/s41586-025-09240-3
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41586-025-09240-3
This article is cited by
-
Genome doubling as a dynamic driver of ovarian cancer evolution: insights from single-cell sequencing
Journal of Ovarian Research (2025)
-
Large scale quantification of natural killer cell-induced apoptosis in patient-derived organoids reveals intratumoral response heterogeneity
npj Precision Oncology (2025)
-
Evolution of drug-resistant ovarian-cancer clones tracked in patients
Nature (2025)
-
Genome doubling fuels ovarian cancer evolution and immune dysregulation
Nature (2025)
-
Tracking clonal evolution during treatment in ovarian cancer using cell-free DNA
Nature (2025)
