
Abstract
Herpes simplex virus 1 (HSV-1) and influenza A viruses (IAV) induce Z-form-nucleic-acid-binding protein 1 (ZBP1)-initiated cell death1,2,3,4,5,6,7,8. ZBP1 is activated by Z-RNA1,7,9, and the Z-RNAs that trigger ZBP1 during HSV-1 and IAV infections were assumed to be of viral origin1. Here, however, we show that host cell-encoded Z-RNAs are major and sufficient ZBP1-activating ligands after infection by these two human pathogens. The majority of cellular Z-RNAs mapped to intergenic endogenous retroelements embedded within abnormally long 3′ extensions of host cell mRNAs. These aberrant host cell transcripts arose as a consequence of disruption of transcription termination (DoTT)—a virus-driven phenomenon that disables cleavage and polyadenylation specificity factor (CPSF)-mediated 3′ processing of nascent pre-mRNAs10,11,12,13,14,15. Mutant viruses lacking ICP27 or NS1—the virus-encoded proteins responsible for inhibiting CPSF and triggering DoTT13,15—did not induce host cell Z-RNA accrual and were attenuated in their ability to stimulate ZBP1. Ectopic expression of HSV-1 ICP27 or IAV NS1 or pharmacological blockade of CPSF activity induced accumulation of host cell Z-RNAs and activated ZBP1. These results demonstrate that DoTT-generated cellular Z-RNAs are bona fide ZBP1 ligands, and position ZBP1-activated cell death as a host response to counter viral disruption of the cellular transcriptional machinery.
Similar content being viewed by others

Main
ZBP1 activates cell death after infection by HSV-1 and IAV1,2,3,4,5,6,7,8. ZBP1 senses left-handed (Z-form) double-helical nucleic acids (Z-DNA and Z-RNA, hereafter Z-NA)16,17,18,19,20 and associates with receptor-interacting protein kinase 3 (RIPK3), initiating apoptosis and necroptosis1,21,22,23. How HSV-1 and IAV infections activate ZBP1 is currently unclear. Although the HSV-1 genome is enriched in Z-DNA-forming sequences24, and de novo RNA transcription was found to be necessary for ZBP1 activation during lytic HSV-1 infections4, HSV-1-generated ligands for ZBP1 are unknown. We previously implicated IAV-produced defective viral genomic (DVG) RNAs in the activation of ZBP1 (refs. 6,7). However, low-DVG preparations of IAV still triggered ZBP1 in cells7, suggesting the existence of additional, possibly non-viral, sources of Z-RNA capable of instigating ZBP1. Here we report that host cell-derived Z-RNAs arising from viral disruption of host transcription termination are abundant and bona fide ZBP1 ligands.
HSV-1 infections generate both viral- and host cell-derived Z-RNAs
To identify HSV-1-generated Z-NA ligands for ZBP1, we stably reconstituted immortalized Zbp1−/− mouse embryonic fibroblasts (MEFs) with wild-type (WT) Flag-tagged ZBP1 (Flag–ZBP1 MEFs) and infected them with WT HSV-1. We fixed and stained these cells with a monoclonal antibody (clone Z22) that recognizes both Z-RNA and Z-DNA7,25. We readily detected a Z-NA signal in the infected cells, which first appeared in the nucleus, and became increasingly evident in the cytoplasm (Fig. 1a,b). DNase I and RNase A each partially diminished, and together abolished, the Z-NA signal (Fig. 1c,d). The HSV-1 Z-NA signal in primary MEFs and human cells displayed similar susceptibility to DNase I and RNase A treatment (Extended Data Fig. 1a–f). Flag–ZBP1, but not a ZBP1 mutant lacking its Zα domains (Flag–ZBP1(ΔZα)), co-localized with Z-NA primarily in the nucleus of HSV-1-infected cells (Extended Data Fig. 1g–i). We also detected an A-form dsRNA (A-RNA) signal in the infected cells (Extended Data Fig. 1j,k).

a,c,e, Z-NA accrual in HSV-1-infected (F strain; multiplicity of infection (MOI) = 5) (gB+) Flag–ZBP1 MEFs. a, Time course of Z-NA formation. c,e, MEFs were exposed to the indicated nucleases after fixation (c) or were treated with ActD 1 h post-infection (h.p.i.) (e), before fixation at 9 h.p.i. b,d,f, The fluorescence intensity (arbitrary units, a.u.) of the Z-NA signal in a, c and e, respectively. g, Cell death kinetics of empty vector (Vec) or Flag–ZBP1 MEFs (Flag–ZBP1) infected with HSV-1 ICP6 RHIMmut virus (MOI = 5) in the presence or absence of ActD. h, The genomic distribution of viral RNAs enriched in anti-Flag RIP–seq from HSV-1 infected Flag–ZBP1 MEFs. The outer and inner sectors represent genomic annotation and scaled fold-change values on the forward and reverse strands, respectively. The black caps denote the 5′ ends. i, The estimated proportion of canonical and uncharacterized viral RNA (vRNA) in anti-Flag RIP–seq data. j,k, Exemplar Z-RNA forming viral transcripts for canonical vRNA (j) and uncharacterized vRNA (k). qPCR analysis of the indicated viral transcripts after Z22 RIP analysis of WT HSV-1-infected Flag–ZBP1 MEFs (left). Putative Z-RNA-forming regions within exemplar transcripts (right). Each arc denotes a base-pairing event and red arcs show Z-prone stems (enumerated above the MFE Circos plots). Exemplar Z-RNAs (black arrowheads) and potential Zα-binding sites (pink boxes) are detailed at the bottom. l, qPCR analysis of viral RNAs after Flag RIP from either Flag–ZBP1 or Flag–ZBP1(ΔZα) mutant MEFs infected with HSV-1. Data are mean ± s.d. n = 30 cells per group (b, d and f), and n = 4 (g and i) and n = 3 (j–l) biologically independent samples. Statistical analysis was performed using one-way analysis of variance (ANOVA) with Dunnett’s multiple-comparison test (d, f and l), two-way ANOVA (g) and two-tailed unpaired t-tests with Welch’s correction (j and k). ***P < 0.0005 (except for in d, f, g and l, for which P < 0.0001). Data are representative of at least two (a, c and e) or three (g) independent experiments. Scale bars, 10 μm (a, c and e (right)) and 20 μm (e (left and middle)).
To determine the relative importance of Z-RNA versus Z-DNA to ZBP1 activation, we infected Flag–ZBP1 MEFs with HSV-1 and exposed cells to phosphonoformic acid (PFA), which inhibits viral DNA synthesis (Extended Data Fig. 1l) and therefore restricts viral gene expression primarily to the immediate-early (IE) genes4,26. RNase A, but not DNase I, almost completely abolished the Z-NA signal in PFA-treated cells (Extended Data Fig. 1m,n). The RNA polymerase II (RNA Pol II) inhibitor actinomycin D (ActD), which prevents both viral and cellular mRNA synthesis, greatly diminished the nuclear Z-NA signal and completely suppressed cytoplasmic Z-RNA accrual (Fig. 1e,f). ActD, but not PFA, prevented ZBP1-dependent cell death (Fig. 1g and Extended Data Fig. 1o), indicating that Z-RNAs generated as a consequence of RNA Pol II activity are essential ZBP1-activating ligands, whereas incoming viral genomic DNA, tegument proteins and the majority (that is, non-IE) of viral transcripts are not required for ZBP1 activation during HSV-1 infections. We used the RHIMmut virus because this virus has mutations (VQCG to AAAA) in the receptor-interacting protein homology interaction motif (RHIM) of the viral protein ICP6, which otherwise interferes with cell death signalling downstream of ZBP1 (refs. 3,4,5,27,28) (Extended Data Fig. 1p). Mutating the RHIM in ICP6 permits ZBP1-dependent cell death in both mouse and human cells3,4,5. HSV-1-induced death of Flag–ZBP1 MEFs was accompanied by MLKL phosphorylation and cleavage of procaspase-3, and was blocked combined inhibition of caspases and RIPK3 kinase activity (Extended Data Fig. 1q,r). Notably, the N-terminal Flag tag did not affect ZBP1-initiated cell death (Extended Data Fig. 1s). Activated MLKL was first seen in the nucleus, rupturing the nuclear envelope in a notable fraction of necroptotic cells (Extended Data Fig. 1t–y).
We next sequenced ZBP1-bound RNAs from HSV-1-infected MEFs. Viral RNA reads in the Flag–ZBP1 pull-downs mapped across the entire HSV-1 transcriptome (Fig. 1h). Although canonical HSV-1 transcripts constituted the majority (around 75%) of these viral Z-RNAs, a substantial proportion (around 25%) originated from RNAs expressed antisense to canonical viral mRNAs, or aligned with viral RNAs of uncharacterized biological function29 (Fig. 1i). Minimum free-energy (MFE) analyses of the predicted secondary structures of two canonical (RS1 and UL19/20) and two non-canonical (UL54.5, which is antisense to UL54, and UL46.5/UL47.5, which is antisense to UL48) exemplar RNAs indicated that each could form multiple Z-prone RNA stems (Fig. 1j,k and Supplementary Fig. 1a,b (red arcs)) in cis through ‘fold-back’ mechanisms (Fig. 1j,k and Supplementary Fig. 1a,b (bottom, pink boxes)). We confirmed the enrichment of these vRNAs in the Z22 pull-downs (Z22 RNA immunoprecipitation (RIP)) by quantitative PCR with reverse transcription (RT–qPCR), demonstrating that both types of viral RNA contribute to Z-RNA formation (Fig 1j,k and Supplementary Fig. 1a,b). When assessed using RT–qPCR, the same transcripts were readily pulled down by Flag–ZBP1, but less so by Flag–ZBP1(ΔZα) (Fig. 1l). We also noted broad and highly equal enrichment in the Flag–ZBP1 pull-downs of both sense and antisense transcripts arising from several regions across the HSV-1 genome (Fig. 1h and Supplementary Fig. 1c). A fraction of viral Z-RNAs bound by ZBP1 therefore presumably arose by hybridization of canonical transcripts and their overlapping antisense counterparts.
Notably, Flag–ZBP1 and Z22 pull-downs from HSV-1-infected mouse and human cells also recovered a substantial number of RNA fragments mapping to the host cell genome (Supplementary Fig. 1d). Indeed, host cell-derived fragments comprised a majority of all reads, indicating that these signals are neither rare nor incidental. These results raised the possibility that acute HSV-1 infections generate host cell-derived Z-RNAs capable of engaging and activating ZBP1.
Host cell Z-RNAs arise from aberrant RNA transcription
To trace the origins of host-derived Z-RNAs, we first catalogued all putative dsRNAs and recurrent, but unresolved, RNA sequences detected in Z22 or Flag–ZBP1 pull-downs (Extended Data Fig. 2a). We then applied a filtering pipeline (Extended Data Fig. 2b) to this reference set (Methods). A total of 1,646 host cell RNA species was significantly enriched by the Z22 antibody from HSV-1-infected mouse cells (Fig. 2a), on the basis of our criteria. These virus-induced host cell Z-RNAs demonstrated widespread evidence of A to I editing (68.3%), indicative of dsRNA formation in cellulo, and mostly mapped to host transcripts with extensively elongated 3′ ends (around 78%) or to RNAs transcribed de novo from cryptic intergenic transcription initiation sites frequently found in close proximity to cellular genes (around 12%, as confirmed by stranded sequencing) (Fig. 2a,b). Most (around 88%) of the 1,646 cellular Z-RNAs harboured inverted intergenic endogenous retroelements (EREs) capable of forming dsRNA, with inverted long interspersed nuclear element-1 (LINE1) and inverted B1/B2 short interspersed nuclear element (SINE) repeats being the most numerous (Fig. 2c,d). Z-RNAs enriched in the Flag–ZBP1 pull-downs almost completely overlapped with those seen in the Z22 pull-downs (approximately 87% overlap) and mapped primarily to host transcripts with extensively elongated 3′ ends (Extended Data Fig. 2c, d), and were predominantly formed by inverted EREs, with a higher proportion of inverted LINE1 elements compared with in the Z22 pull-downs (Extended Data Fig. 2e,f).

a, The enrichment (RIP/input) of virus-induced host cell Z-RNAs in Z22 RIP–seq analysis of Flag–ZBP1 MEFs infected with WT HSV-1 (MOI = 2, 8 h.p.i.). Z-RNAs with evidence of A to I editing are circled in black. Statistical significance was assessed using a one-sided Wald test in DESeq2. Only RNAs significantly induced by infection (adjusted P ≤ 0.1, fold change ≥ 1.5) are shown, as determined using separate two-sided Wald tests comparing the RIP/RIP or input/input between infected and matched mock cells (Methods). P values were adjusted for multiple testing using the Benjamini–Hochberg procedure. FDR, false-discovery rate. b, The genomic distribution of HSV-1-induced host cell Z-RNAs enriched in Z22 RIP–seq data. c, Source of inverted reverse-complement sequences within HSV-1-induced host cell Z-RNAs enriched in Z22 RIP–seq data. The ‘unresolved’ category includes Z-forming RNAs of which the secondary structures could not be readily solved. d, Detailed distribution of inverted EREs in HSV-1-induced host cell Z-RNAs enriched in Z22 RIP–seq data. e–h, Coverage tracks for exemplar Z-RNAs (Nabp1 (e), Gtpbp4 (f), H2ac18/19 (g) and U1 locus (h)) showing Z22 RIP (green), Flag RIP (purple), input total RNA (orange), A to I editing sites (red ticks, top), positions of qPCR primers (red arrows) and a schematic of the putative Z-RNA-forming structure (bottom). Coverage exceeding the indicated limits is denoted by thick black caps.
For example, the aberrant Nabp1 transcript enriched in both the Z22 and Flag–ZBP1 pull-downs contained an elongated 3′ sequence, 45 kb in length, which is significantly longer (around 16-fold) than the canonical protein-coding isoform encoded by this locus (Fig. 2e). This 3′ extension included two pairs of widely separated (around 1.8 kb and 8.8 kb apart) inverted LINE1 elements, which formed fold-back Z-RNAs enriched in both pull-downs. Similarly, the aberrant 3′ extension of the Gtpbp4 transcript, which was also enriched in both Z22 and Flag–ZBP1 pull-down datasets, had an inverted pair of long terminal repeats (LTRs) flanking fold-back sequences capable of forming a complex Z-RNA (Fig. 2f). As examples of host cell Z-RNAs arising from de novo intergenic transcription, the H2ac18/19 locus generated Z-RNAs after bidirectional transcription from cryptic intergenic start sites between the H2ac18 and H2ac19 genes (Fig. 2g), and the U1 locus gave rise to a single de novo transcript that probably forms Z-RNA by folding back on itself (Fig. 2h).
Other examples of host cell Z-RNAs enriched in the Z22 RIP followed by sequencing (RIP–seq) analysis included aberrantly elongated transcripts studded with inverted pairs of mouse SINEs (Ptbp1, Pcna, Haus2, Tlcd1), LINEs (Btbd3) and LTRs (Gnpda1, Rock1) (Extended Data Fig. 3a–g). Notably, aberrant 3′ transcription downstream of Pcna resulted in ‘read-in’ transcription of the Tmem230 gene and consequent generation of a Z-RNA-forming pair of inverted SINEs within an intron in Tmem230. Other examples of de novo intergenic transcription include transcripts upstream of Hmga1 and Anapc4 promoters extending into inverted LTR repeat regions in the opposite direction from these genes (Extended Data Fig. 3h,i). Notably, although these exemplar Z-RNAs were enriched only in the Z22 pull-down datasets, they were nonetheless robustly pulled-down by Flag–ZBP1, but not by a Zα mutant of ZBP1, when assessed using RT–qPCR (Extended Data Fig. 3j,k).
In human (HT-29) cells, the landscape of HSV-1-induced host cell Z-RNAs largely mirrored that found in MEFs. A total of 1,818 host cell Z-RNAs was induced by HSV-1 and significantly enriched in the Z22 RIP–seq dataset (Extended Data Fig. 4a), with 396 (around 49%) of these also enriched in the Flag–ZBP1 pull-downs (Extended Data Fig. 4b). Most endogenous Z-RNAs in human cells mapped to extended 3′ ends of known genes, and a subset to introns (Extended Data Fig. 4c). These Z-RNAs were formed by inverted repeat EREs, particularly Alu SINEs (Extended Data Fig. 4d,e). Other examples of Z-RNA-forming EREs within abnormally elongated 3′ extensions included inverted LINEs located within introns of a downstream gene (RABGGTB), α satellite repeats in pericentromeric regions (ROCK1) or, notably, even pairs of inverted gene paralogues (CGB2 and CGB3, downstream of RUVBL) (Extended Data Fig. 4f–h). Paralogous genes in opposite orientations to each other also produced Z-RNA after de novo transcription from a normally silent transcription start site (TSS) upstream of the ZNF132/ZNF584 genes (Extended Data Fig. 4i). Using RIP followed by qPCR (RIP–qPCR), we confirmed that each of these host cell Z-RNAs was selectively bound by Z22 (Extended Data Fig. 4j) and by Flag–ZBP1 (Extended Data Fig. 4k).
In agreement with these findings, ADAR1-mediated A to I editing of host cell RNAs was increased in intergenic regions (both proximal and distal to genic loci) after HSV-1 infection of both mouse and human cells (Extended Data Fig. 4l). Using 4-thiouridine labelling (4sU-seq)11 to follow the kinetics of nascent viral and host transcript production in infected human cells, ADAR1-mediated A to I editing of host cell transcripts also switched from intragenic to intergenic sequences downstream of annotated genes, reflecting the increasing accrual of intergenic dsRNA-forming species as the infection progressed (Extended Data Fig. 4m). Thus, inverted EREs and other repeats embedded within aberrantly long 3′ extensions of host RNAs, or RNA species generated de novo from cryptic intergenic loci, are unanticipated and abundant sources of Z-RNA in HSV-1-infected mammalian cells.
Notably, several vRNAs were significantly enriched in Flag pull-downs from infected MEFs, accounting for about one-third of all enriched hits in these pull-downs, but were not enriched to the same extent in Flag pull-downs from human cells, or in Z22 pull-downs from cells of either species (Discussion and Supplementary Fig. 2a–c).
HSV-1 ICP27 induces host cell Z-RNAs and activates ZBP1
We next compared the extent of aberrant transcription (that is, downstream and upstream of the gene, occurring in the opposite orientation of the canonical gene product, or resulting in intron accumulation) to the levels of canonical (that is, exonic) transcription of each host mRNA in HSV-1-infected cells (Extended Data Fig. 5a). In both human and mouse cells, HSV-1 infection caused a marked induction of aberrant transcription, with the most notable increase in transcription occurring downstream of genes (Extended Data Fig. 5b). In MEFs, downstream transcription was increased in over 80% of all expressed mRNAs, a large subset (around 30%) of which displayed grossly altered downstream transcription levels of 10% or higher (Fig. 3a and Extended Data Fig. 5b). In human cells, these abnormalities first manifested around 2 h after infection (Extended Data Fig. 5c), aligning well with the first observed accumulation of Z-RNAs in mouse cells (Fig. 1b), and with our earlier studies4, demonstrating that blocking Pol-II-driven transcription within the first 2–3 h of infection completely rescues HSV-1-triggered cell death. Together, these findings show that HSV-1-induced aberrant host cell mRNA transcription, particularly downstream of canonical protein-coding genes, is widespread, occurs early during infection, accounts for most host RNAs enriched in Z22 and Flag pull-downs and may therefore be responsible for the accrual of the earliest host cell Z-RNA ligands for ZBP1.

a, Downstream transcription levels in mock- or HSV-1-infected Flag–ZBP1 MEFs. Each datapoint represents a single mRNA, with transcription downstream of all annotated termination sites normalized to transcription levels of exons. Significant changes are colour coded by directionality, with changes exceeding 10% marked as strong. b, HSV-1 ICP27 binds to and interferes with the function of the CPSF complex, preventing proper termination of host cell pre-mRNAs, resulting in DoTT. c, Time course of Z-RNA formation in immortalized Zbp1−/− MEFs transfected with either empty vector (Vec) or Flag-ICP27-expressing plasmid. Scale bar, 10 μm. d, The fluorescence intensity of the Z-RNA signal in c. n = 30 cells per group (empty vector; ICP27-transfected, 6 h; and ICP27-transfected, 18 h, +RNase A) and n = 32 cells (ICP27 transfected, 18 h). e, Immunoblot analysis of phosphorylated MLKL (pMLKL) in immortalized Zbp1−/− MEFs stably reconstituted with either empty vector or with MYC-tagged ZBP1 (MYC–ZBP1), transfected with Flag–ICP27, in the presence of zVAD with or without RIPK3 kinase inhibitor (RIPK3i). f,h, Kinetics of cell death in MYC–ZBP1 or MYC–ZBP1(ΔZα) mutant MEFs transfected with control vector (ctrl vector), Flag–ICP27 or Flag–ICP27 n504 mutant constructs in the presence of zVAD (f) or in Vec and Flag–ZBP1 MEFs infected with HSV-1 RHIMmut (MOI = 5) or HSV-1 RHIMmutΔICP27 (MOI = 15) (h). g, The levels of viral proteins in MEFs infected with HSV-1 RHIMmut (MOI = 5) or HSV-1 RHIMmutΔICP27 (MOI = 15), at 8 h after infection. Data are mean ± s.d. n = 4 (f and h). Statistical analysis was performed using one-way ANOVA with Dunnett’s multiple-comparison test (d). Data are representative of at least two (c, e and g) or three (f and h) independent experiments.
We next examined what causes these aberrancies in host cell mRNA transcription. HSV-1 is known to induce the phenomenon of DoTT, a process in which pre-mRNAs fail to undergo proper 3′ cleavage and processing10,11,14. Mechanistically, the HSV-1 protein ICP27 drives DoTT by associating with the CPSF complex and preventing this complex from properly processing the 3′ ends of pre-mRNAs13 (Fig. 3b). As a result, Pol II transcription fails to terminate, producing very lengthy read-through RNAs that are not typically observed in uninfected cells. An ICP27-null HSV-1 mutant virus (ΔICP27) induces substantially less aberrant host cell mRNA transcription13 (Extended Data Fig. 5d), prompting us to hypothesize that ICP27-driven DoTT may be responsible for the generation of abnormal host cell mRNAs and, therefore, for the accrual of Z-RNAs in these abnormal transcripts.
Ectopically expressed ICP27 robustly induced Z-RNA formation within 6 h of transfection in MEFs (Fig. 3c,d). Although the Z-RNA signal was also seen in the cytoplasm (Fig. 3c), the Z-RNAs are probably nuclear in origin because the nuclear export inhibitor leptomycin B was able to trap these Z-RNAs in the nucleus (Extended Data Fig. 5e,f). ICP27 also generated A-RNA in the transfected cells (Extended Data Fig. 5g,h). An ICP27 mutant (n504), which is unable to interfere with 3′ pre-mRNA cleavage13, was significantly compromised in its ability to induce Z-RNA (Extended Data Fig. 5i–k). The same Z-RNA ligands for ZBP1 generated by HSV-1 infection were also enriched in Z22 and Flag–ZBP1 pull-downs from ICP27-transfected cells (Extended Data Fig. 5l,m). ICP27 triggered ZBP1-dependent phosphorylation of MLKL and cell death, both of which were reduced in cells transfected with the ICP27 n504 mutant (Fig. 3e,f).
We generated an ICP27-null virus on the RHIMmut background (HSV-1 RHIMmutΔICP27, Fig. 3g). This double mutant induced substantially less ZBP1-dependent cell death than the ICP27-containing RHIMmut virus in Flag–ZBP1-expressing MEFs (Fig. 3h), as well as in IFNβ-pretreated primary MEFs expressing endogenous ZBP1, in human HT-29 Flag–ZBP1 cells and in IFNβ-pretreated HS68 cells expressing endogenous ZBP1 (Extended Data Fig. 5n–r). Notably, IFNβ pretreatment was not required for HSV-1 RHIMmut induction of endogenous ZBP1 or activation of ZBP1-dependent cell death; it simply hastened the kinetics of such cell death without affecting its eventual magnitude (Extended Data Fig. 5s,t).
The RHIMmutΔICP27 virus also induced significantly less (and, in many cases, completely failed to trigger) DoTT-driven host cell Z-RNA formation and consequent sensing of these Z-RNAs by ZBP1 (Supplementary Figs. 3a–d and 4a,b). We next used another pair of HSV-1 mutants, d103 and d106 (refs. 30,31), which both have deletions of the key viral transactivator ICP4 and are therefore compromised in E and L viral gene expression (Supplementary Fig. 5a). They also lack two of the remaining four IE genes, ICP22 and ICP47. However, whereas d103 still expresses ICP27, this gene is deleted in d106. Importantly, we previously demonstrated that d103 still induces substantial DoTT, whereas d106 does not13. We found that d103, but not d106, induced ZBP1-dependent death in the majority of infected cells (Supplementary Fig. 5b,c). Critically, d106 did not induce detectable levels of Z-forming DoTT transcripts in infected cells (Supplementary Fig. 5d–g).
IAV infections generate host cell Z-RNAs
As IAV has been reported to induce DoTT10,14,15,32,33, we examined whether IAV infections generated cellular Z-RNA, and whether these cellular Z-RNAs are also ZBP1 ligands. We infected MEFs with the IAV H1N1 strain A/PuertoRico/8/1934 (PR8), isolated Z-RNAs and ZBP1-bound RNAs from these cells, and mapped the recovered sequences. As in HSV-1-infected cells, the majority of RNA fragments in both pull-downs aligned to the host genome (Supplementary Fig. 6a). Processing these reads through our analysis pipeline (Extended Data Fig. 2a,b) identified 1,445 distinct host RNAs induced by IAV and selectively enriched in Z22 pull-downs from infected cells (Fig. 4a). The vast majority of RNAs found in the Flag–ZBP1 pull-downs were also seen in the Z22 pull-downs (around 90% overlap; Supplementary Fig. 6b). As we observed in HSV-1 infections, the bulk of host cell Z-RNAs induced by IAV infection mapped to aberrantly elongated 3′ ends of host mRNA transcripts (Fig. 4b and Supplementary Fig. 6c), with the remainder arising from de novo intergenic transcription and intronic sources.

a, The enrichment (RIP/input) of virus-induced host cell Z-RNAs in Z22 RIP–seq data of Flag–ZBP1 MEFs infected with IAV (PR8, MOI = 2, 8 h.p.i.). Statistical methods, significance thresholds, symbols and legend are as described in Fig. 2a. b, The genomic distribution of IAV-induced host cell Z-RNAs enriched in Z22 RIP–seq data. c, The source of inverted reverse-complement sequences within IAV-induced host cell Z-RNAs enriched in Z22 RIP–seq. The ‘unresolved’ category includes Z-forming RNAs of which the secondary structures could not be readily solved. d, Detailed distribution of inverted EREs in IAV-induced host cell Z-RNAs enriched in underlying Z22 RIP–seq data. e–g, Coverage tracks for exemplar Z-RNAs (Gtpbp4 (e), 2310057M21Rik (f) and Vbp1 (g)), showing Z22 RIP (green), Flag RIP (purple), input total RNA (orange), A to I editing sites (red ticks), qPCR primer location (red arrows) and putative Z-RNA structures (bottom). Thick black caps indicate coverage exceeding the indicated limits. h, IAV NS1 associates with CPSF4, preventing proper termination of host cell pre-mRNAs and triggering DoTT. i, Z-RNA accrual in Flag–ZBP1 MEFs transfected with either an empty vector (Vec) or with a plasmid expressing V5-tagged NS1 derived from IAV strain PR8, at 6 h after transfection. MEFs were subjected to limited proteinase K digestion and treated with RNase A after fixation. Scale bar, 10 μm. j, Fluorescence intensity of Z-RNA signal in i. n = 32 (Vec), n = 47 (NS1-transfected) and n = 39 (NS1-transfected + RNase A) cells. k, Kinetics of cell death in Vec or Flag–ZBP1 MEFs transfected with control vector (ctrl vector) or V5-NS1 (NS1) constructs in the presence of zVAD (50 µM). l, The kinetics of cell death in empty vector or Flag–ZBP1 MEFs infected with IAV (MOI = 5) and IAV ΔNS1 (MOI = 5). Data are mean ± s.d. n = 4 (k and l). Statistical analysis was performed using one-way ANOVA with Dunnett’s multiple-comparison test (j). Data are representative of at least two (i) and three (k and l) independent experiments.
The majority of IAV-induced host cell Z-RNAs were formed by inverted repeat sequences (Fig. 4c,d and Supplementary Fig. 6d). For example, the abnormal 3′ extensions of the Gtpbp4 and 2310057M21Rik mRNA transcripts, enriched in both the Z22 RIP–seq and Flag–ZBP1 RIP–seq datasets from IAV-infected cells, generated Z-RNA through inverted LTRs and fold-back sequences (Fig. 4e), or through inverted gene paralogues (Fam24a and Fam24b; Fig. 4f), respectively. The Z22-enriched 3′ extensions of Vbp1 carried inverted intergenic LINE1 elements, which formed Z-RNA (Fig. 4g). We also found cases in which Z-RNA was formed by complex composite mouse repeats, such as satellite-like sequences in the 3′ extension of Gm14419 transcript (Supplementary Fig. 6e). Notably, Z-RNAs downstream of Gtpbp4 seen in HSV-1 infected cells (Fig. 2f) were also identified in IAV-infected cells (Fig. 4e). Each of these RNAs were readily pulled-down by the Z22 antibody and by Flag–ZBP1, but not by a Zα mutant of ZBP1 (Supplementary Fig. 6f,g).
In IAV-infected human (HT-29 Flag–ZBP1) cells, a total of 2,771 IAV-induced Z-RNAs was enriched by the Z22 antibody, with around 69% of these also bound by Flag–ZBP1 (Supplementary Fig. 6h,i). Most IAV-induced human Z-RNAs mapped to elongated 3′ ends of host RNAs and were formed by inverted ERE repeats, predominantly arising from Alu-family SINEs (Supplementary Fig. 6j–l). Exemplar Z-RNAs formed in IAV-infected human cells include inverted satellite repeats embedded in the elongated ROCK1 transcript (also seen in HSV-1-infected cells), inverted LTR repeats in the 3′ extended KBTBD2 transcript, and inverted SINE or SINE–LINE pairs embedded in the elongated 3′ ends of the NCL and C6orf62 precursor mRNAs (Extended Data Fig. 6a–d). These human Z-RNAs were readily pulled-down by the Z22 antibody and by Flag–ZBP1 (Extended Data Fig. 6e,f).
A few viral RNA species were enriched in both Flag and Z22 RIPs from mouse and human cells (Extended Data Fig. 6g). These viral RNAs resulted exclusively from positive-sense viral transcripts, with many mapping to DVG RNAs derived from the three longest viral RNA segments, PB1, PB2 and PA, consistent with our previous findings that IAV-generated DVG RNAs are the primary source of viral ligands for ZBP1 (refs. 6,7).
The overall repertoires of virus-induced host cell Z-RNAs generated in HSV-1- or IAV-infected cells were largely similar to each other (around 49% overlap in MEFs, around 76% in HT-29 cells; Extended Data Fig. 7a). Indeed, a comparison of virus-induced transcription events downstream of host genes showed that more than 80% of all expressed host cell mRNAs in MEFs and over 70% in HT-29 cells were equivalently disrupted after infection with HSV-1 or IAV (that is, differences within 10%, shaded areas), indicative of largely similar overall levels of DoTT induction by these viruses (Extended Data Fig. 7b). Notably, in cases in which the same mRNAs were subject to DoTT by both viruses, the propensity of many of these aberrant mRNAs to actually form Z-RNA varied between the two infection settings (red and green dots; Extended Data Fig. 7b). We attribute this variability in Z-RNA formation to the stochastic nature of the A to Z flip, the stability of the ensuing Z-RNA species34 and the activity of virus- or host-encoded dsRNA-binding proteins specific to each infection.
IAV infections triggered a significant repositioning of A to I editing events towards intergenic regions in MEFs and HT-29 cells (Extended Data Fig. 7c), an effect that was accurately reflected in an independent dataset from IAV-infected human monocyte-derived macrophages35 (Extended Data Fig. 7d). We found that IAV infections led to a significant global increase in aberrant transcription events, with the strongest aberrancies occurring in regions downstream of genes (Extended Data Fig. 8a).
The IAV protein NS1 induces DoTT by associating with CSPF4 (also called CPSF30)15,33 (Fig. 4h), and our analysis of an independent dataset36 showed that DoTT is significantly diminished in cells infected with an NS1-null virus (Extended Data Fig. 8b). These results suggested that NS1, by activating DoTT, was responsible for IAV-induced host cell Z-RNA formation, analogous to what we have shown with HSV-1 ICP27. We found that NS1 on its own was indeed able to induce an RNase-A-sensitive signal in transfected cells, after limited proteinase K digestion of these cells post-fixation (Fig. 4i,j) as we had previously observed in the setting of IAV infection itself7. Ectopic expression of NS1 generated the same Z-RNAs also induced by IAV infection (Extended Data Fig. 8c). These Z-RNAs were bound by Flag–ZBP1 in NS1-transfected cells (Extended Data Fig. 8d), resulting in ZBP1-dependent cell death (Fig. 4k). An IAV mutant lacking NS1 did not provoke the generation of host cell Z-RNA ligands for ZBP1 in either mouse (Extended Data Fig. 8e–i) or human (Extended Data Fig. 6e,f) cells, and induced significantly less ZBP1-dependent death in primary and immortalized MEFs, as well as in human cells, compared with the WT virus (Fig. 4l and Extended Data Fig. 8j,k).
Host cell Z-RNAs are sufficient to activate ZBP1
To directly demonstrate that HSV-1- and IAV-induced host cell Z-RNAs are sufficient ligands for ZBP1, we transfected Flag–ZBP1 MEFs with expression plasmids encoding either ICP27 or NS1, and isolated the resulting Z-RNAs and ZBP1-bound RNAs from these cells. We then transfected these RNAs into Flag–ZBP1-expressing MEFs, as well as into primary Zbp1+/+ and Zbp1−/− MEFs (Extended Data Fig. 9a). We found that both Z22- and Flag–ZBP1-bound RNAs formed Z-RNA that co-localized with Flag–ZBP1 in the challenged cells (Fig. 5a–c and Extended Data Fig. 9b–q). Both Z22- and Flag–ZBP1-bound RNAs triggered robust ZBP1-dependent cell death in these cells (Fig. 5d and Extended Data Fig. 9r–x). Next, we identified Z-prone structures from our representative exemplar host cell Z-RNAs, including inverted LINE repeats downstream of Vbp1, inverted LTRs upstream of Hmga1 and inverted SINEs downstream of Ptbp1 (Extended Data Fig. 10a (top)). We generated fluorescently (FAM) tagged hairpin RNAs from each sequence and modified the hairpins with 2′-O-methyl-8-methylguanosine (m8Gm) to stabilize them in the Z conformation37 (Extended Data Fig. 10a (bottom)). m8Gm-modified or unmodified GC-repeat hairpins served as positive (Z-RNA) and negative (A-RNA) controls7. We transfected these synthetic FAM-labelled RNA hairpins into cells, and found that each of the predicted host cell Z-forming RNA sequences did indeed form Z-RNA within cells (Fig. 5e,f and Extended Data Fig. 10b,c), co-localize with ZBP1 (Extended Data Fig. 10d,e) and potently trigger ZBP1-dependent cell death (Fig. 5g and Extended Data Fig. 10f,g).

a, Immunofluorescence staining of Z-RNA and Flag–ZBP1 in Flag–ZBP1 MEFs transfected with the indicated RNAs. These RNAs were eluted from Z22 RNA immunoprecipitates from MEFs ectopically expressing ICP27 at 9 h after transfection. b, Fluorescence intensity (a.u.) of the Z-RNA signal in a. n = 38 (no treatment), n = 41 (IgG RIP), n = 64 (Z22 RIP) and n = 54 (Z22 RIP + RNase A treatment) cells. c, Quantification of co-localized ZBP1 and Z-RNA in a. n = 30 (IgG) and n = 32 (Z22 RIP) cells. d,g,k, The kinetics of cell death of Flag–ZBP1 MEFs transfected with the RNAs eluted from Z22 RNA immunoprecipitates as indicated in a (d), transfected with the indicated hairpin RNAs (g) or treated with vehicle or JTE-607, in the presence of zVAD. For k, vehicle, DMSO + zVAD. e, Immunofluorescence staining of Z-RNA and FAM in Flag–ZBP1 MEFs transfected with the indicated hairpin RNAs. f, Quantification of co-localized Z-RNA and FAM in e. h, The structure of active JTE-607 (top). Bottom, magnified view of the interaction between active JTE-607 and human (solid) or mouse (translucent) CPSF3 (bottom). i, Z-NA accrual in primary MEFs treated with vehicle or JTE-607. j, The fluorescence intensity of the Z-RNA signal in i. l, The proposed model for the generation of cellular Z-RNAs during HSV-1 and IAV infection. Both viruses trigger DoTT, which generates 3′-extended transcripts containing otherwise-intergenic inverted-repeat EREs, which form Z-RNA and activate ZBP1-induced cell death. Data are mean ± s.d. n = 4 (d, g and k) and n = 10 (f) fields per group, and n = 30 cells per group (j). Statistical analysis was performed using one-way ANOVA with Dunnett’s multiple-comparison test (b and f) and two-tailed unpaired t-tests with Welch’s correction (c). For a and e, the small white rectangles indicate the regions magnified in the insets. Data are representative of at least two (a, e and i) or three (d, g and k) independent experiments. For a, e and i, scale bars, 10 μm.
After determining that ablating expression of CPSF3 by RNA interference (RNAi) was sufficient to generate Z-RNA and activate ZBP1-dependent cell death (Extended Data Fig. 10h–k), we took a pharmacological approach to disabling CPSF and examined whether this intervention was also sufficient to drive DoTT and generate host cell Z-RNA ligands for ZBP1. The compound JTE-607 is a CPSF3 inhibitor pro-drug that undergoes ester cleavage in cells, exposing a critical carboxylic acid to form the active inhibitor (Fig. 5h). CPSF3 is highly conserved (>98% identity) between mice and humans, with differences occurring outside of the JTE-607-binding site. Aligning the structure of human JTE-607-bound CPSF3 (Protein Data Bank (PDB): 6MQ8) to the mouse CPSF3 orthologue (modelled using AlphaFold38) shows that JTE-607 will engage and inhibit mouse CPSF3 as well (Fig. 5h and Extended Data Fig. 10l,m). Indeed, JTE-607 induced Z-RNA and triggered highly selective and rapid ZBP1-dependent necroptosis in both primary and Flag–ZBP1 MEFs (Fig. 5i–k and Extended Data Fig. 10n).
Discussion
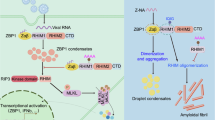
This study positions ZBP1-induced cell death as a key host cell response to limit viral pathogens that induce DoTT and target the termination of host cell transcription (Fig. 5l). DoTT disables mRNA 3′ processing and the export and translation of host mRNAs10,11,12,14,15,32,39. It therefore represents a mechanism by which viruses induce host shut-off, free up ribosomes for translation of viral RNAs and evade transcription-dependent innate-immune responses39,40. As a cellular countermeasure to DoTT, aberrant cellular RNAs generated by DoTT (or by de novo intergenic transcription) are potent cell-death-activating ZBP1 ligands. These RNAs contain numerous dsRNA-forming inverted-repeat EREs, as well as other repeats capable of forming Z-RNAs by folding back on themselves. Such ERE-derived Z-RNAs, which we call flipons, are probably under positive selection to defend against existing and emerging viral threats, which, by licensing DoTT, have compromised cellular mRNA transcription. While inverted repeat SINEs were previously shown to form Z-RNA41, this study provides examples of inverted LINEs, inverted LTRs and more complex sequences (such as exons from inverted gene pairs) as Z-RNA-forming species.
HSV-1 transcripts escape DoTT as ICP27 binds to GC-rich sequences immediately upstream of the polyadenylation signal of these transcripts and recruits the CPSF complex to facilitate efficient 3′-end processing and subsequent export of viral mRNAs13. The notably higher GC content of HSV-1 mRNAs therefore safeguards these transcripts from ICP27-mediated DoTT. This high GC content also represents a liability, as it increases the propensity of HSV-1-encoded vRNAs to adopt the Z-conformation. Our observation that vRNAs are significantly enriched in ZBP1 pull-downs from mouse but not human cells (Supplementary Fig. 2) suggests that HSV-1 may have evolved ways to mask its Z-prone vRNAs from being sensed by human ZBP1. The human cell therefore cannot rely on viral Z-RNAs to drive ZBP1 activation, making host cell Z-RNAs substantially more critical for activation of human ZBP1. We also observed that the vRNAs bound by mouse ZBP1 were not enriched in parallel Z22 pull-downs from the same cells (Supplementary Fig. 2a), suggesting that these are Z-prone vRNAs that are stabilized in the Z-conformation after association with the Zα domain.
IAV mRNAs are not transcribed by host RNA polymerase II but by the virus-encoded RNA-dependent RNA polymerase. Their 3′ processing and polyadenylation are therefore not dependent on the activity of the CPSF complex but result from reiterative copying of a stretch of 5–7 uracil bases present at the 5′ ends of each IAV gene segment42,43. As NS1 from all of the tested IAV strains associates with CPSF4 and induces DoTT, and as differences between IAV strains in the affinity of their NS1 isoforms for CPSF4 correlate well with the ability of these strains to dampen IFN- and NF-κB-driven immune responses33, NS1-induced DoTT appears to be an effective means of blunting intrinsic innate immune responses without affecting viral RNA transcription.
DoTT is a very rapid phenomenon, activated within around 2 h of infection (Extended Data Fig. 5c). Host-derived Z-RNAs produced as a consequence of DoTT therefore enable the host to activate ZBP1 and kill the infected cell before virus host shut-off, and before the cell becomes a virus factory. More broadly, cellular Z-RNAs may activate ZBP1 in other settings of nuclear stress, such as after exposure to salt, heat and oxygen free radicals, which also result in extensive transcription downstream of genes14,44,45. Small-molecule approaches to engaging DoTT and activating ZBP1 may also serve as immunogenic adjuvants for cancer immunotherapies46.
Methods
Cells and viruses
Primary MEFs were generated from E14.5 C57BL/6J embryos, and immortalized using a 3T3 protocol. Primary and immortalized MEFs were maintained in DMEM supplemented with 15% FBS, 1 mM sodium pyruvate, 1× GlutaMax and 1% penicillin–streptomycin. HT-29 cells (American Type Culture Collection (ATCC), HTB-38) were maintained in DMEM supplemented with 10% FBS, 1× GlutaMax and 1% penicillin–streptomycin. HS68 cells (ATCC, CRL-1635), were maintained in DMEM supplemented with 10% FBS, 1× GlutaMax and 1% penicillin–streptomycin. All cells were cultured at 37 °C under 5% CO2, and were routinely tested for mycoplasma. Flag–ZBP1 and Flag–ZBP1(ΔZα) MEFs were generated by stable retroviral transduction (pRetroX, Clontech) of immortalized Zbp1−/− MEFs with expression vectors encoding either Flag-tagged full-length mouse ZBP1 or ZBP1 lacking both Zα domains (ΔZα). HT-29 cells were stably reconstituted with Flag-tagged human ZBP1 expression constructs using the Retro-X retroviral transduction system. WT HSV-1 (F strain) was obtained from the ATCC (VR-733). HSV-1 RHIMmut has been described previously3. HSV-1 RHIMmut∆ICP27 was generated using a bacterial artificial chromosome (BAC) clone of HSV-1 RHIMmut (ref. 3). In brief, Escherichia coli DH10B cells containing the HSV-1 BAC clone were cultured to an optical density at 600 nm of 0.4–0.6, then incubated at 42 °C to induce recombination activity and made electrocompetent by multiple washes in ice-cold water. The DH10B cells were then electroporated with plasmid pSC101-ccdA-gbaA47 (Vector Builder) for expression of the ccdA gene. The ccdB-ampicillin (amp) cassette was amplified from the plasmid p15A-amp-ccdB48 (Vector Builder) with 50-nucleotide overhangs corresponding to sequences adjacent to the ICP27 gene of HSV-1 The primers used were as follows: forward: GGGGATGCGCCCGGGCGGGAAGCTCCTAAGGGCACTATGACACCGGCCTTGCTAGCGCTTTGTTTATTTTTCTA; reverse: CCGTGGTGGCCGGGGTGGTGCTCGTGGCGCTTCACTACGAGCAGGAGATCTCTAGATTTCAGTGCAATTTATCTCT.
The ccdB-amp cassette containing the 50-nucleotide overhangs was subsequently electroporated into the HSV-1 BAC-containing DH10B cells, after which ICP27 in the BAC clone was replaced with the ccdB-amp cassette by lambda-RED homologous recombination. Recombinant BACs lacking ICP27 were obtained by selection of the DH10B cells in ampicillin-containing (100 μg ml−1) agar at 30 °C. The ccdB-amp cassette was subsequently deleted by Red/ET homologous recombination, and pSC101-ccdA-gbaA was eliminated by culturing the DH10B cells at 37 °C. After confirmation by sequencing that ICP27 was successfully deleted and no further mutations were introduced in the BAC clone during recombination, 1 μg each of purified BAC DNA was transfected into Vero cells at 90% confluency using polyethylenimine (Polysciences). At 72 h after transfection, when visible fluorescent plaques appeared, a clone of the HSV-1 RHIMmut∆ICP27 virus from an isolated plaque was selected for propagation.
HSV-1 d103 and d106 mutants30 were propagated in Vero-derived complementing (ICP4+ICP27+) E11 cells at an MOI of 0.005 and 0.01, respectively, and collected after 3 days. Viruses were purified by Ficoll gradient ultracentrifugation and titrated by plaque assay in E11 cells30.
IAV H1N1 strain A/Puerto Rico/8/1934 (PR8) was propagated by allantoic cavity inoculation of 10-day embryonated chicken eggs, or in MDCK cells (ATCC, CCL-34). The PR8 ΔNS1 virus was propagated in NS1-expressing MDCK cells. IAV titres were determined by plaque assay or by indirect immunofluorescence microscopy with an anti-NP polyclonal antibody on MDCK cells.
Assessing cell death
MEFs and HS68 cells were seeded in 48-well plates (10,000–15,000 cells per well), in 24-well plates (30,000 cells per well) or 6-well plates (150,000 cells per well) 1 day before treatment. HT-29 cells were seeded at 30,000 cells per well in 96-well plates or 60,000 cells per well in 48-well plates. On the day of treatment, the medium was replaced with fresh medium including Cytotox Green Dye (250 nM, Sartorius) and the indicated treatment. Dead cells were stained with Cytotox Green Dye and Cytotox Green+ cells were quantified at each timepoint using custom processing definitions on the Incucyte Live-Cell Analysis System (Sartorius) at 37 °C and 5% CO2. In parallel, separate cells seeded in identical numbers were stained with Cytolight Rapid Dye (3.3 µM, Sartorius) for live-cell labelling. The percentage of cell death was calculated as Cytotox Green+/Cytolight+ at each timepoint. In some cases, cell viability was determined by Trypan Blue exclusion. The percentage of cell death was calculated as follows: (total cell count − live cell count)/total cell count. zVAD (50 µM), RIPK3 kinase inhibitor GSK843 (5 µM) and JTE-607 (100 µM) were added at the indicated final concentrations immediately before Incucyte scanning. PFA (300 µg ml−1) and ActD (200 nM) were added at final concentrations 1 h after virus inoculation. For assessment of endogenous ZBP1-driven dell death, primary MEFs and HS68 cells were pretreated with recombinant IFNβ to ensure that enough endogenous ZBP1 was available for synchronous activation by virus (HSV-1) or Z-RNA.
Producing and testing synthetic Z-RNAs
Synthetic hairpins of putative Z-prone sequences from host cell-derived inverted-repeat EREs with or without m8Gm modifications were produced by Lumi D&NA. The hairpins were transfected into cells using Lipofectamine 3000 (Invitrogen) and cells were either immediately assessed for viability or were fixed for immunofluorescence microscopy.
RNA interference
For Cpsf3 ablation, immortalized MEFs were transiently transfected with small interfering RNA (siRNA) against mouse Cpsf3 (GCATGACATACCCATTTACTA) (Thermo Fisher Scientific) or with a negative control siRNA (Thermo Fisher Scientific) using the Lipofectamine RNAiMAX transfection reagent (Thermo Fisher Scientific). After 48 h, cells were collected and examined for CPSF3 (CPSF73) protein expression by immunoblotting, immunofluorescence microscopy and cell death assay.
RIP–seq
Cells were infected with or without HSV-1 or IAV (MOI = 2) for 8 h, collected and RIP was conducted using the EZ-Magna RIP (Cross-Linked) RNA-binding protein immunoprecipitation kit (Millipore) according to the manufacturer’s instructions. In brief, cross-linked cell pellets were lysed in RIP lysis buffer, followed by incubation with RIP buffer containing magnetic beads conjugated with anti-Z-NA antibody (Z22, Absolute Antibody, 1:50), anti-Flag antibody (F1804, Sigma-Aldrich, 1:50) or their respective isotype control antibodies, at 4 °C overnight. The samples were then incubated with proteinase K and immunoprecipitated RNAs were recovered by phenol:chloroform:isoamyl alcohol purification. RNA was quantified using the Quant-iT RiboGreen RNA assay (Thermo Fisher Scientific) and assessed for quality with the 2100 Bioanalyzer RNA 6000 Nano assay (Agilent) or 4200 TapeStation High Sensitivity RNA ScreenTape assay (Agilent) before library generation. Libraries were prepared from total RNA with the TruSeq Stranded Total RNA Library Prep Kit according to the manufacturer’s instructions (Illumina). Libraries were analysed for insert size distribution using the 2100 BioAnalyzer High Sensitivity kit (Agilent), 4200 TapeStation D1000 ScreenTape assay (Agilent) or 5300 Fragment Analyzer NGS fragment kit (Agilent). Libraries were quantified using the Quant-iT PicoGreen ds DNA assay (Thermo Fisher Scientific) or by low-pass sequencing using the a MiSeq nano kit (Illumina). Paired-end 150 cycle sequencing was performed on the NovaSeq X+ (Illumina) system.
qPCR analysis
RNA from RIP samples was quantified by Qubit and equal amounts of RNA was reverse-transcribed into cDNA using the SuperScript IV VILO Master Mix (Thermo Fisher Scientific). Equal amounts of cDNA were used as template and qPCR was performed using SYBR Green (Thermo Fisher Scientific). RT–qPCR was performed for the target fragments in the RIP samples and the input samples, and the Ct values for each target in the RIP samples were normalized to the input samples by subtracting the Ct value of the Input from the Ct value of the RIP: ΔCt = Ct(RIP) − Ct(input). The ΔΔCt value was then calculated by subtracting the ΔCt value of the IgG sample from the ΔCt value of the Flag/Z22 RIP sample: ΔΔCt = ΔCt(Flag/Z22-RIP) − ΔCt(IgG). Finally, the fold enrichment of the target Flag/Z22-binding locus sequence over IgG-binding locus sequence was calculated as fold enrichment = 2\(-\mathrm{\varDelta \varDelta }{C}_{{t}}\). Primers used were as follows: RS1-fwd: TGATCACGCGGCTGCTGTACAC; RS1-rev: GGTGATGAAGGAGCTGCTGTTG; UL54-AS-fwd: CCAGGCCGAGGTCAATTAG; UL54-AS-rev: ACCAGAGGCCATATCCGAC; UL19/20-fwd: TCCTTAGCACGATCGAGGTG; UL19/20-rev: GACAGGGTGTTGCAATACGAC; UL48-AS-fwd: GTACAGGGCCGAGCAGAAGTTG; UL48-AS-rev: CGAAGCGCTCTCTCGTTTCTT; Gtpbp4-fwd: TGTAGTTCAGTAGTGATGTGTG; Gtpbp4-rev: CTTTAGTTTGTGTCAAGTTGAC; Btbd3-fwd: ATACTGATTTCCAGAGTGGTTG; Btbd3-rev: AGCAGATGCTAGTGAGGATG; H2ac18/19-fwd: CCAAGTTAAGCAATTTCTGACC; H2ac18/19-rev: AGGAAAGCATTAGGCAGTCC; Ptbp1-fwd: AGGCAGGAGGATCAGGAGTTC; Ptbp1-rev: TGTGCTTTGAACCGCTGAGC; Nabp1-fwd: TGAAATTCCTAGGCAAATGG; Nabp1-rev: CCTCAGTTTCTGGGCTAATATC; Hmga1-fwd: TCTCTGCTAGGTTGTTGCTATC; Hmga1-rev: TTTGCATCAACTCCTGTCTG; U1-fwd: CTGTGTAAATCTCCCACTTG; U1-rev: TTTCTCTTGAGTCCTATCCC; Gnpda1-fwd: TGCCTTCAGATCAAGATGTAG; Gnpda1-rev: TTATCATCATGGCAGGAAAC; Rock1-fwd: TTGGTCGTCTGTTCGTGATTC; Rock1-rev: GCCTCCAAATGAAAGAACCTAG; Anapc4-fwd: ACATACCTACTCCAACAAGG; Anapc4-rev: ACAGTAAGTGAAGTTGTGGG; Pcna-fwd: CTCAGCAGTTAAGAGCACTCAC; Pcna-rev: TAGAGAGGGTTGTGAGCCAC; Haus2-fwd: GGGTCAGATCTCATTACAGATG; Haus2-rev: ACTGACTGCTCTTCCAGAGG; Tlcd1-fwd: AACCAGGCTGACCTCAAACTC; Tlcd1-rev: ATTCATGCTGGGCAGTGGTG; 2310057M21Rik-fwd: CCTCTCTGGAAAGAGTTCATC; 2310057M21Rik-rev: GAATGAGGTGTCCCTCCTAC; Gm14419-fwd: CACACGTGAAATTGTTCTCG; Gm14419-rev: TCCATAACGAGGAGAACACC; Vbp1-fwd: GAAGGGTGTTCCTCTTTGTC; Vbp1-rev: AGATTTCACCTCACACCAGTC; RABGGTB-fwd: GAACTAGCCTTCCATCCCAG; RABGGTB-rev: AATACTGGCAAACCGAATCC; RUVBL2-fwd: GGTGAGCTCGACACTAACCC; RUVBL2-rev: TGGACCAGTGAGAGGAGAGG; ZNFs-fwd: CTGGTGTAGAACAAGTGCGTC; ZNFs-rev: CACCAGAAGGTTCACACAGG; KBTBD2-fwd: ATCACCCTTACCCTGCTCAAC; KBTBD2-rev: CCATACTGTAGCAGGCGAGTG; C6orf62-fwd: GGCAATTCCTCAAGGATCTAC; C6orf62-rev: GCATGATTTATAATCCTTTGGG; NCL-fwd: TGGTGTCACGCTCTTATAGTCC; NCL-rev: CAATCACAGCTCACTGCAGC; ROCK1-rev: TCACAGAGTTGAACATTCCC; ROCK1-fwd: TTTCACCAAAGTCGTCAAAG.
Fold enrichment values of the immunoprecipitation product were normalized to the input samples. Data presented show the average of three independent biological repeats.
Immunoblotting
Cells were lysed in RIPA lysis buffer (Thermo Fisher Scientific) supplemented with protease and phosphatase inhibitors (Thermo Fisher Scientific). Cell lysates were incubated on ice for 10 min, and briefly sonicated to shear chromatin, then cleared by high-speed centrifugation (20,000g, 10 min) at 4 °C. The supernatants were subjected to immunoblot analysis as described previously49 (Fig. 3e,g, Extended Data Figs. 1g,q, 5o,q,t, 8e and 10h and Supplementary Fig. 5a). Sources and dilutions of primary antibodies were as follows: phosphorylated mouse MLKL (Ab196436, Abcam, 1:2,000), total mouse MLKL (MABC60, EMD Millipore, 1:2,000), RIPK3 (2283, ProSci, 1:2,000), Flag (A00187, GenScript, 1:2,000), cleaved caspase-3 (9664, Cell Signaling Technology, 1:2,000), caspase-3 (9662, Cell Signaling Technology, 1:2,000), GAPDH (60004-1-Ig, Proteintech, 1:4,000), ICP27 (sc-69806, Santa Cruz Biotechnology, 1:2,000), MYC (9E10) (MA1-980, Invitrogen, 1:2,000), ICP0 (sc-53070, Santa Cruz Biotechnology, 1:2,000), ICP5 (sc-56989, Santa Cruz Biotechnology, 1:2,000), NS1 (sc-130568, Santa Cruz Biotechnology, 1:2,000), NS1 (1A7, gift from A. García-Sastre), NP (GTX125989, GeneTex, 1:5,000), human MLKL (14993, Cell Signaling Technology, 1:2,000), human RIPK3 (13526, Cell Signaling Technology, 1:2,000), mouse ZBP1 (AG-20B-0010-C100, AdipoGen Life Sciences, 1:2,000), human ZBP1 (PA5-20455, Thermo Fisher Scientific), CPSF3 (ab72295 Abcam, 1:2,000). Gel source data are provided in Supplementary Fig. 7.
Immunofluorescence microscopy
Cells were plated onto eight-well glass slides (EMD Millipore). After infection or treatment, cells were fixed for 10 min with fresh 4% (w/v) paraformaldehyde in PBS, permeabilized in 0.5% (v/v) Triton X-100 in PBS for 10 min, blocked with MAXblock blocking medium (Active Motif) for 1 h and incubated overnight with primary antibodies at 4 °C. After three washes in PBS, cells were incubated with fluorophore-conjugated secondary antibodies for 1 h at room temperature. After an additional three washes in PBS, the slides were mounted in ProLong Gold antifade reagent (Thermo Fisher Scientific) and imaged by confocal microscopy on a Leica SP8 instrument. For immunofluorescence staining of tissue fixed with 4% (w/v) paraformaldehyde in PBS and dehydrated with 30% sucrose in PBS, frozen tissue sections were cut at 10 µm thickness in a cryostat microtome. The sections were permeabilized with 0.5% (v/v) Triton X-100 in PBS for 10 min, blocked with MAXblock Blocking Medium (Active Motif) for 1 h and incubated overnight with primary antibodies at 4 °C. After three washes in PBS, the sections were incubated with fluorophore-conjugated secondary antibodies for 1 h at room temperature. After an additional three washes in PBS, the sections were mounted in ProLong Gold antifade reagent (Thermo Fisher Scientific) and imaged by confocal microscopy on the Leica SP8 instrument. Random sampling was carried out near the centre of the wells in an eight-well chamber slide, excluding the peripheral regions, to avoid any potential edge effects on fluorescence intensity. Images were acquired of serial sections from each stained cell (around 10 sections per cell, each of 0.3-μm thickness). These serial sections were then z-stacked and the signal fluorescence intensity was quantitatively analysed using the Leica LAS X software package. When required, proteinase K (0.008 U ml−1) was used for 20 min at 37 °C; RNase A (1 mg ml−1) and DNase 1 (25 U ml−1) were used for 1 h at 37 °C before primary antibody incubation. Primary antibodies were used for immunofluorescence studies: Z-DNA and Z-RNA (Z22, Absolute Antibody, 1:200), A-RNA (9D5, Millipore, 1:500), phosphorylated mouse MLKL (37333, Cell Signaling, 1:500), Flag (A00187, GenScript, 1:500), emerin (ab40688, Abcam, 1:500), HSV-1/2 gB (ab6506, Abcam, 1:500), HSV-1 ICP5 (ab6508, Abcam, 1:500), anti-NS1 antibody (1A7, gift from A. García-Sastre, 1:500), V5 (R960-25, Thermo Fisher Scientific, 1:500), FAM (A-889, Thermo Fisher Scientific, 1:500) and NP (MCA400, Bio-Rad, 1:500).
RNA-seq data processing
Public RNA-seq datasets were retrieved from the SRA/ENA databases, and their accession numbers are provided in Supplementary Table 1. The mouse GRCm39 genome assembly and annotation were obtained from GENCODE (M36)50; the human CHM13v2 assembly was sourced from the T2T consortium51,52. The GENCODE annotation (v47) for CHM13v2 was lifted from the GRCh38 assembly using Liftoff (v.1.6.3)53. Viral assemblies and supplementary host annotations were downloaded from RefSeq54. The HSV-1 genome annotation was adopted from our previous work29 and extended with several RefSeq-annotated genes (such as LAT RNA) and miRNAs from the VIRmiRNA database55.
To ensure comparability, both public and newly generated data underwent processing through an optimized fork of the community-curated Nextflow rnaseq pipeline (v.3.15.1)56,57,58, which was executed in the following order:
Read preprocessing
Adapters, low-quality base pairs, and poly(A) and poly(G) tails were trimmed using the fastp59 program (v.0.23.4). Poly(G) clipping was necessary to eliminate the known technical artifacts of Illumina’s two-colour chemistry, whereby a weak or no signal at the end of a read is detected as a high-quality G base.
Alignment
Trimmed libraries were initially mapped with relaxed parameters to a chimeric reference comprising the host genome assembly, all viral assemblies and a curated collection of eukaryotic rRNA sequences using the STAR aligner (v.2.7.10a)60. Reads uniquely mapped to rRNA species were excluded to reduce technical noise arising from variable rRNA depletion efficiency (for example, some 4sU HFF samples lacked rRNA removal). These in silico rRNA-depleted reads were then remapped to the main reference, sorted with SAMtools (v.1.2)61 and passed to Picard (https://broadinstitute.github.io/picard/, v.3.1.1) to mark duplicates.
Quality control
Quality control was conducted using FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/), DESeq2 (ref. 62) and RSeQC63, with the results visualized using MultiQC64. All of the libraries exhibited acceptable sequencing and mapping quality metrics, and the samples clustered as expected based on condition and protocol on the principal component analysis plot.
Quantification of transcript expression
RNA abundance was estimated using Salmon (v1.10.1)65 running in pseudo-alignment mode.
If not specified otherwise, the genomic annotation used for downstream analyses is based on all GENCODE basic RNAs, excluding automatically annotated loci not verified manually (annotation level 3), pseudogenes, to be experimentally confirmed (TEC) transcripts and aberrant RNA models (for example, arising due to intron retention). GENCODE-specific ncRNAs not supported by the RefSeq annotation (NR models) were also excluded. This decision is based on the observation that the vast majority of these RNAs are undetectable in mock samples and, in infected cells, are transcribed only as part of much longer aberrant RNAs, typically driven by virus-induced DoTT events occurring in upstream mRNAs.
RIP–seq analysis
All RIP experiments were subjected to pre-processing and alignment according to the same protocol as for the RNA-seq samples. Subsequent analysis of RIP data was conducted using a custom procedure outlined below.
Peak calling
First, a relaxed set of RNA sequences enriched by Z22 or Flag antibodies was identified using a peak-calling approach. Each RIP sample and its corresponding RIP input control were normalized using the median-of-ratios method62, calculated across all intergenic, exonic and intronic regions of annotated genes. To account for technical variability, the control signal for each annotated mature RNA, intron or intergenic region (collectively termed RNA models) was smoothed using a running average. The final control signal for each genomic locus was defined as the maximum value across all overlapping RNA models.
Fold-change values for each genomic position were calculated by comparing the normalized signal to the normalized and smoothed control coverage. Adjacent positions meeting enrichment criteria (fold change ≥ 2) were merged (Extended Data Fig. 2a (peak calling)) and then refined to include bases proximal to putative binding summits. The refinement was accomplished by comparing the signal of each peak to its surrounding neighbourhood (Extended Data Fig. 2a (non-maximum suppression)), defined as ± the peak length (minimum, 1 kb; maximum, 10 kb), and bounded by RNA model limits. Loci exhibiting at least twofold enrichment relative to the adjacent signal across all overlapping RNA models were retained for subsequent analysis.
DsRNA reconstruction and clustering
All detected peaks were further filtered to eliminate overlaps with a manually curated list of technically challenging regions, such as homopolymers (Extended Data Fig. 2a (global filtering)). Peaks smaller than the empirical RIP resolution limit (36 bp, around one-quarter of the read length) or detected in only one experiment were excluded unless they overlapped with newly identified or REDIportal-annotated A to I editing sites, markers of dsRNA formation.
Filtered peaks served as seeds for reconstructing dsRNA structures (Extended Data Fig. 2a (dsRNA reconstruction)) using the following steps.
Sequence extraction
Peaks within 15 kb were grouped, and RNA sequences spanning each group and its 15 kb flanking regions were retrieved. Grouping across splicing sites of Ensembl Canonical or GENCODE primary mRNAs was disabled to limit groups length.
Alignment analysis
Sequences were aligned against their reverse complements, allowing both canonical (A–U, G–C) and non-canonical (G–U) base pairing. All locally optimal alignments representing putative dsRNAs longer than 36 bp and with at least 12 bp overlap with peaks were extracted.
Scoring and optimization
Candidate dsRNA structures were scored based on RIP signal intensity. A greedy algorithm was used to identify the optimal combination of dsRNAs that explained at least 90% of the observed RIP signal. This scoring and optimization process was conducted independently for each sample. dsRNAs supported by at least two experiments were retained.
Clustering
Selected dsRNAs and peaks that either overlapped them or were strongly reproducible (that is, supported by all experiments in at least one condition) were clustered on the basis of genomic or transcriptomic proximity, as measured by overlapping RNA models, to reduce redundancy and minimize correlations between adjacent hits (Extended Data Fig. 2a (clustering)).
Statistical analysis and dsRNA annotation
The total number of reads overlapping each dsRNA cluster was calculated and normalized using the median-of-ratios method described above (Extended Data Fig. 2b (read counting)). These values were then subjected to a classical DESeq2-based analysis62,66 (Extended Data Fig. 2b (statistical analysis)). Clusters were classified as virus induced if they displayed higher concentrations in either Z22 RIP or RIP input from infected cells compared with the matched mock cells (fold change ≥ 1.5, adjusted P ≤ 0.1, two-sided test). In parallel, dsRNAs groups were defined as RIP enriched if they were overrepresented in RIP from infected cells relative to the matched input control (fold change ≥ 1.5, adjusted P ≤ 0.01, one-sided test). Virus-inducible Z-RNAs passing both criteria were subjected to downstream analyses (Extended Data Fig. 2b (downstream analyses)).
The sequence composition of identified endogenous Z-RNAs was analysed by matching them against the RepeatMasker annotation retrieved from the UCSC Table Browser67. For each Z-RNA cluster, the repeat type (or lack thereof) with the highest RIP signal was selected. Inverted repeats were naturally incorporated into this scheme by jointly annotating arms underlying inferred Z-RNA stems. Annotation was conducted independently for each RIP replicate, and the most frequent category across replicates was selected as representative. Similarly, the genomic distribution of Z-clusters was determined by mapping them against the GENCODE annotation and a curated set of regions, such as virus-induced de novo intergenic RNAs, using a RIP-weighted approach.
The statistical significance of overlaps between different pull-downs was assessed using a hypergeometric test, with the set of all identified dsRNA clusters serving as the background population.
A global comparison of RIP enrichment levels between host dsRNA clusters and viral RNAs was conducted (Supplementary Fig. 2 and Extended Data Fig. 6) by merging generated endogenous dsRNA and viral annotations. IAV genome segments were included either as full-length units or, for PA, PB1, and PB2, as intact central regions and separate 5′ and 3′ fragments to specifically capture defective viral RNAs. Overlapping HSV-1 transcripts were segmented into canonical (for example, UL18/19/20) or uncharacterized (for example, UL46.5/47.5) regions. The combined annotation was then reanalysed using methodology described in the ‘Statistical analysis and annotation’ section.
HSV-1 viral RNA enrichment profile
The pyCirclize package (github.com/moshi4/pyCirclize, v.1.7.1) was used to visualize the global enrichment profile of viral transcripts. Fold-change values were calculated based on the median-of-ratios normalization strategy, incorporating pseudocounts of one. Sites meeting the fold-change criterion (fold change ≥ 2) were scaled by RIP coverage to account for uneven read distribution (scale = coverage/mean viral coverage) so that comparisons between loci were valid and not dependent on the region-specific sequencing efficiency. The HSV-1 terminal repeats, which are prone to mapping and sequencing errors, were excluded from the visualization. Canonical viral transcripts (RL, RS, US, UL and LAT) were classified as immediate early, early, late and latency-associated based on published literature68,69,70,71,72,73. Novel HSV-1 transcripts29 were labelled as uncharacterized and partially omitted from the plot (for example, NAGNAG splicing isoforms with alternative acceptor sites 3 bp apart) for clarity of visualization.
Viral RNA secondary structure prediction
MFE structures for selected vRNAs were predicted using the ViennaRNA software package (v.2.7.0)74. To evaluate the thermodynamic propensity of predicted RNA stems to transition from A-RNA to Z-RNA, we used the Z-Hunt[rs] (ZH) program (v.0.0.4, https://github.com/biomancy/zhuntrs), a Rust-based reimplementation of the original Z-Hunt algorithm (v.3)75. Although Z-Hunt was originally developed for Z-DNA analysis, its usefulness extends to Z-RNA prediction as the ranking of the energetic cost for individual RNA and DNA sequences to adopt the Z conformation will be similar, even though the absolute energetic cost may be different in RNA due to the 2′ hydroxyl group and the variability of A–Z junction energy costs. We considered an RNA stem longer than 6-nucleotides to be Z-prone if its predicted ZH score ranked within the top 5% of all possible ZH-scores for RNA stems of the same length. For stems exceeding 12 nucleotides, Z-prone stems were defined as those containing a 12-nucleotide subsequence meeting this threshold.
A to I editing
The genomic distribution of A to I editing sites in each RNA-sequencing experiment, including RIP–seq, was analysed using a custom pipeline. In brief, candidate sites were initially selected based on the following criteria: coverage of ≥8 reads, low technical noise (the sum of frequencies of other mismatches ≤ editing frequency) and strong experimental support (editing frequency ≥ 5%). Subsequently, sites near annotated splicing sites, which are prone to alignment noise76, and putative genomic SNPs with editing frequencies above 45% were excluded. Finally, only sites reproducibly identified in ≥3 experiments or present in our data and recorded in the REDIportal database77 were retained for subsequent annotation and analysis.
Analysis of aberrant host cell transcription
To investigate the global effects of viral infection on abnormal host transcription, we first defined transcription boundaries based on annotated splicing sites and transcription start/end sites (TSS, TES) recorded in the filtered GENCODE annotation or high-quality RefSeq models (NM and NR transcripts). For each protein-coding gene, we selected the primary mRNA (marked as Ensembl Canonical), cropped its 5′ and 3′ exons to the shortest exon variant among all annotated secondary isoforms of the same gene (Extended Data Fig. 5a) and called this the reference mRNA. Next, each reference mRNA was matched to several aberrant transcription regions, defined as follows:
-
Downstream region: located downstream of the gene, extending beyond all recorded transcription end sites.
-
Upstream region: spanning from the 5′-most TSS of the gene to the upstream transcription boundary, typically the TES of the upstream gene.
-
Divergent transcription region: running in the divergent orientation from the 5′-most TSS of the gene to the next transcription boundary.
-
Intronic regions: defined as regions from the donor site to the nearest downstream transcription boundary and from the acceptor site to the nearest upstream transcription boundary. This definition excludes exons of secondary isoforms encoded within the intron, ensuring a focus on unambiguously intronic regions.
Downstream, upstream and divergent regions were trimmed to a maximum length of 2.5 kb and were excluded if they were shorter than this threshold. All constitutive exons of the primary mRNA served as a ‘normal’ transcription reference for downstream, upstream and divergent regions, while individual constitutive 5′ exons served as the reference for intronic regions.
The relative level of aberrant transcription (Extended Data Fig. 5a) in infected cells was statistically compared to control cells using the DEXSeq statistical framework78. Libraries were normalized using previously described median-of-ratios procedure and for each comparison, only references with a coverage of ≥1 fragment per kilobase per million were included. Relative changes in aberrant transcription with a fold change ≥ 1.5 (adjusted P ≤ 0.01, two-sided test) were considered significant. These changes were further classified as strong if the difference in aberrant transcription levels between conditions exceeded 10%.
AlphaFold modelling of murine CPSF3
The structure of human JTE-607 bound CPSF3 (PDB: 6MQ8) was aligned with the mouse CPSF3 orthologue AlphaFold database model (AF-Q9QXK7-F1-v4) in PyMOL.
Statistics
Statistical significance was determined using unpaired Student’s t-tests with Welch’s correction for comparisons between two groups, one-way ANOVA with Dunnett’s multiple-comparison test or two-way ANOVA with Tukey’s multiple-comparison test between multiple (>2) groups. P values of 0.05 or lower were considered to be significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data discussed in this publication have been deposited in the NCBI Gene Expression Omnibus (GEO) under GEO Series accession number GSE308489.
Code availability
All code used to analyse sequencing data in this study is publicly available at GitHub (https://github.com/alnfedorov/Z-DoTT).
References
Balachandran, S. & Mocarski, E. S. Viral Z-RNA triggers ZBP1-dependent cell death. Curr. Opin. Virol. 51, 134–140 (2021).
Mocarski, E. S., Guo, H. & Kaiser, W. J. Necroptosis: the Trojan horse in cell autonomous antiviral host defense. Virology 479–480, 160–166 (2015).
Huang, Z. et al. RIP1/RIP3 binding to HSV-1 ICP6 initiates necroptosis to restrict virus propagation in mice. Cell Host Microbe 17, 229–242 (2015).
Guo, H. et al. Species-independent contribution of ZBP1/DAI/DLM-1-triggered necroptosis in host defense against HSV1. Cell Death Dis. 9, 816 (2018).
Guo, H. et al. Herpes simplex virus suppresses necroptosis in human cells. Cell Host Microbe 17, 243–251 (2015).
Thapa, R. J. et al. DAI senses influenza A virus genomic RNA and activates RIPK3-dependent cell death. Cell Host Microbe 20, 674–681 (2016).
Zhang, T. et al. Influenza virus Z-RNAs induce ZBP1-mediated necroptosis. Cell 180, 1115–1129 (2020).
Kuriakose, T. et al. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci. Immunol. 1, aag2045 (2016).
Maelfait, J. et al. Sensing of viral and endogenous RNA by ZBP1/DAI induces necroptosis. EMBO J. 36, 2529–2543 (2017).
Proudfoot, N. J. Transcriptional termination in mammals: stopping the RNA polymerase II juggernaut. Science 352, aad9926 (2016).
Rutkowski, A. J. et al. Widespread disruption of host transcription termination in HSV-1 infection. Nat. Commun. 6, 7126 (2015).
Hennig, T. et al. HSV-1-induced disruption of transcription termination resembles a cellular stress response but selectively increases chromatin accessibility downstream of genes. PLoS Pathog. 14, e1006954 (2018).
Wang, X. et al. Herpes simplex virus blocks host transcription termination via the bimodal activities of ICP27. Nat. Commun. 11, 293 (2020).
Vilborg, A. & Steitz, J. A. Readthrough transcription: how are DoGs made and what do they do? RNA Biol. 14, 632–636 (2017).
Nemeroff, M. E., Barabino, S. M., Li, Y., Keller, W. & Krug, R. M. Influenza virus NS1 protein interacts with the cellular 30 kDa subunit of CPSF and inhibits 3′ end formation of cellular pre-mRNAs. Mol. Cell 1, 991–1000 (1998).
DeAntoneo, C., Herbert, A. & Balachandran, S. Z-Form nucleic acid-binding protein 1 (ZBP1) as a sensor of viral and cellular Z-RNAs: walking the razor’s edge. Curr. Opin. Immunol. 83, 102347 (2023).
Brown, B. A. 2nd, Lowenhaupt, K., Wilbert, C. M., Hanlon, E. B. & Rich, A. The Zα domain of the editing enzyme dsRNA adenosine deaminase binds left-handed Z-RNA as well as Z-DNA. Proc. Natl Acad. Sci. USA 97, 13532–13536 (2000).
Placido, D., Brown, B. A. 2nd, Lowenhaupt, K., Rich, A. & Athanasiadis, A. A left-handed RNA double helix bound by the Zα domain of the RNA-editing enzyme ADAR1. Structure 15, 395–404 (2007).
Kim, K. et al. Solution structure of the Zβ domain of human DNA-dependent activator of IFN-regulatory factors and its binding modes to B- and Z-DNAs. Proc. Natl Acad. Sci. USA 108, 6921–6926 (2011).
Herbert, A. The ancient Z-DNA and Z-RNA specific Zα fold has evolved modern roles in immunity and transcription through the natural selection of flipons. R. Soc. Open Sci. 11, 240080 (2024).
Thomas, P. G., Shubina, M. & Balachandran, S. in Alternate Programmed Cell Death Signaling in Antiviral Host Defense, Vol. 442 (eds Mocarski, E.S. & Mandal, P.) 41–63 (Springer, 2020).
Upton, J. W., Kaiser, W. J. & Mocarski, E. S. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 11, 290–297 (2012).
Upton, J. W., Shubina, M. & Balachandran, S. RIPK3-driven cell death during virus infections. Immunol. Rev. 277, 90–101 (2017).
Herbert, A., Fedorov, A. & Poptsova, M. Mono a mano: ZBP1’s love-hate relationship with the kissing virus. Int. J. Mol. Sci. https://doi.org/10.3390/ijms23063079 (2022).
Sanford, D. G. & Stollar, B. D. Characterization of anti-Z-DNA antibody binding sites on Z-DNA by nuclear magnetic resonance spectroscopy. J. Biol. Chem. 265, 18608–18614 (1990).
Helgstrand, E. et al. Trisodium phosphonoformate, a new antiviral compound. Science 201, 819–821 (1978).
Wang, X. et al. Direct activation of RIP3/MLKL-dependent necrosis by herpes simplex virus 1 (HSV-1) protein ICP6 triggers host antiviral defense. Proc. Natl Acad. Sci. USA 111, 15438–15443 (2014).
Yu, X. et al. Herpes simplex virus 1 (HSV-1) and HSV-2 mediate species-specific modulations of programmed necrosis through the viral ribonucleotide reductase large subunit R1. J. Virol. 90, 1088–1095 (2015).
Whisnant, A. W. et al. Integrative functional genomics decodes herpes simplex virus 1. Nat. Commun. 11, 2038 (2020).
Samaniego, L. A., Neiderhiser, L. & DeLuca, N. A. Persistence and expression of the herpes simplex virus genome in the absence of immediate-early proteins. J. Virol. 72, 3307–3320 (1998).
Hobbs, W. E. 2nd & DeLuca, N. A. Perturbation of cell cycle progression and cellular gene expression as a function of herpes simplex virus ICP0. J. Virol. 73, 8245–8255 (1999).
Bauer, D. L. V. et al. Influenza virus mounts a two-pronged attack on host RNA polymerase II transcription. Cell Rep. 23, 2119–2129 (2018).
Bergant, V. et al. mRNA 3′UTR lengthening by alternative polyadenylation attenuates inflammatory responses and correlates with virulence of influenza A virus. Nat. Commun. 14, 4906 (2023).
Guo, X. et al. ADAR1 Zα domain P195A mutation activates the MDA5-dependent RNA-sensing signaling pathway in brain without decreasing overall RNA editing. Cell Rep. 42, 112733 (2023).
Heinz, S. et al. Transcription elongation can affect genome 3D structure. Cell 174, 1522–1536 (2018).
Lork, M. et al. Regulated localization of transposable element RNA during influenza A virus infection. EMBO Rep. 26, 3506–3528 (2025).
Balasubramaniyam, T., Ishizuka, T., Xiao, C. D., Bao, H. L. & Xu, Y. 2′-O-methyl-8-methylguanosine as a Z-form RNA stabilizer for structural and functional study of Z-RNA. Molecules 23, 2572–2579 (2018).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Friedel, C. C. et al. Dissecting herpes simplex virus 1-induced host shutoff at the RNA level. J. Virol. https://doi.org/10.1128/JVI.01399-20 (2021).
Chen, L. L. & Carmichael, G. G. Altered nuclear retention of mRNAs containing inverted repeats in human embryonic stem cells: functional role of a nuclear noncoding RNA. Mol. Cell 35, 467–478 (2009).
Maelfait, J. & Rehwinkel, J. The Z-nucleic acid sensor ZBP1 in health and disease. J. Exp. Med. https://doi.org/10.1084/jem.20221156 (2023).
Poon, L. L., Pritlove, D. C., Fodor, E. & Brownlee, G. G. Direct evidence that the poly(A) tail of influenza A virus mRNA is synthesized by reiterative copying of a U track in the virion RNA template. J. Virol. 73, 3473–3476 (1999).
Robertson, J. S., Schubert, M. & Lazzarini, R. A. Polyadenylation sites for influenza virus mRNA. J. Virol. 38, 157–163 (1981).
Vilborg, A., Passarelli, M. C., Yario, T. A., Tycowski, K. T. & Steitz, J. A. Widespread inducible transcription downstream of human genes. Mol. Cell 59, 449–461 (2015).
Yang, T. et al. Triggering endogenous Z-RNA sensing for anti-tumor therapy through ZBP1-dependent necroptosis. Cell Rep. 42, 113377 (2023).
Zhang, T. et al. ADAR1 masks the cancer immunotherapeutic promise of ZBP1-driven necroptosis. Nature 606, 594–602 (2022).
Wang, H. et al. Improved seamless mutagenesis by recombineering using ccdB for counterselection. Nucleic Acids Res. 42, e37 (2014).
Yin, J. et al. Direct cloning and heterologous expression of the salinomycin biosynthetic gene cluster from Streptomyces albus DSM41398 in Streptomyces coelicolor A3(2). Sci. Rep. 5, 15081 (2015).
Chen, P. et al. Anti-CD70 Immunocytokines for exploitation of interferon-γ-induced RIP1-dependent necrosis in renal cell carcinoma. PLoS ONE 8, e61446 (2013).
Frankish, A. et al. GENCODE: reference annotation for the human and mouse genomes in 2023. Nucleic Acids Res. 51, D942–D949 (2023).
Nurk, S. et al. The complete sequence of a human genome. Science 376, 44–53 (2022).
Rhie, A. et al. The complete sequence of a human Y chromosome. Nature 621, 344–354 (2023).
Shumate, A. & Salzberg, S. L. Liftoff: accurate mapping of gene annotations. Bioinformatics 37, 1639–1643 (2021).
O’Leary, N. A. et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–D745 (2016).
Qureshi, A., Thakur, N., Monga, I., Thakur, A. & Kumar, M. VIRmiRNA: a comprehensive resource for experimentally validated viral miRNAs and their targets. Database https://doi.org/10.1093/database/bau103 (2014).
Di Tommaso, P. et al. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 35, 316–319 (2017).
Ewels, P. A. et al. The nf-core framework for community-curated bioinformatics pipelines. Nat. Biotechnol. 38, 276–278 (2020).
da Veiga Leprevost, F. et al. BioContainers: an open-source and community-driven framework for software standardization. Bioinformatics 33, 2580–2582 (2017).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience https://doi.org/10.1093/gigascience/giab008 (2021).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Wang, L., Wang, S. & Li, W. RSeQC: quality control of RNA-seq experiments. Bioinformatics 28, 2184–2185 (2012).
Ewels, P., Magnusson, M., Lundin, S. & Kaller, M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048 (2016).
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A. & Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017).
Zhu, A., Ibrahim, J. G. & Love, M. I. Heavy-tailed prior distributions for sequence count data: removing the noise and preserving large differences. Bioinformatics 35, 2084–2092 (2019).
Karolchik, D. et al. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 32, D493–D496 (2004).
Perng, G. C. et al. A novel herpes simplex virus type 1 transcript (AL-RNA) antisense to the 5′ end of the latency-associated transcript produces a protein in infected rabbits. J. Virol. 76, 8003–8010 (2002).
Eide, T. et al. Identification of the UL4 protein of herpes simplex virus type 1. J. Gen. Virol. 79, 3033–3038 (1998).
Patel, A. H. & MacLean, J. B. The product of the UL6 gene of herpes simplex virus type 1 is associated with virus capsids. Virology 206, 465–478 (1995).
Yamauchi, Y. et al. The UL14 tegument protein of herpes simplex virus type 1 is required for efficient nuclear transport of the alpha transinducing factor VP16 and viral capsids. J. Virol. 82, 1094–1106 (2008).
Reynolds, A. E., Fan, Y. & Baines, J. D. Characterization of the UL33 gene product of herpes simplex virus 1. Virology 266, 310–318 (2000).
Kulej, K. et al. Time-resolved global and chromatin proteomics during herpes simplex virus type 1 (HSV-1) infection. Mol. Cell. Proteom. 16, S92–S107 (2017).
Lorenz, R. et al. ViennaRNA Package 2.0. Algorithms Mol. Biol. 6, 26 (2011).
Ho, P. S., Ellison, M. J., Quigley, G. J. & Rich, A. A computer aided thermodynamic approach for predicting the formation of Z-DNA in naturally occurring sequences. EMBO J. 5, 2737–2744 (1986).
Pinto, Y. & Levanon, E. Y. Computational approaches for detection and quantification of A-to-I RNA-editing. Methods 156, 25–31 (2019).
Mansi, L. et al. REDIportal: millions of novel A-to-I RNA editing events from thousands of RNAseq experiments. Nucleic Acids Res. 49, D1012–D1019 (2021).
Anders, S., Reyes, A. & Huber, W. Detecting differential usage of exons from RNA-seq data. Genome Res. 22, 2008–2017 (2012).
Acknowledgements
We thank R. Cadagan and A. García-Sastre for NS1-mutant IAV, NS1 constructs and anti-NS1 antibodies; N. DeLuca for HSV-1 d103 and d106 viruses; J. Han for the HSV1 RHIMmut BAC clone; and J. Qu and Y. Zhou for review of the bioinformatics code. This work was supported by NIH grants AI135025, AI144400, AI161624 and CA168621 to S.B., NIH grant R21AI175590 and COBRE P20GM134974 to H.G., R21CA303392 to H.G. and S.B., NIH grant R01 AI121832 and the St Jude Center of Excellence for Influenza Research and Surveillance (SJCEIRS) NIAID contract HHSN272201400006C to P.G.T., funding from the Association of Lebanese and Syrian American Charities at St Jude Children’s Hospital to J.C.C. and P.G.T.; T.Z. was supported by Grant 32370157 from the National Natural Science Foundation of China, the National Science and Technology Major Project of China (2023ZD0500502) and 1.3.5 project for disciplines of excellence from West China Hospital of Sichuan University (ZYYC24008). J.R. acknowledges funding from UKRI Medical Research Council, grant number MR/Y013212/1; V.B. by the Slovenian Research and Innovation Agency (P1-0242, J4-60082); A.P. by the European Research Council (ERC-CoG- 817798-ProDAP), the German Research Foundation (CRC-TRR179/TP24, CRC-TRR237/A07, CRC-TRR353/B04), the State of Bavaria (P3M, BayVFP 2024-2027) and the Danish National Research Foundation (DNRF 164; CiViA). L.D. was supported by the European Research Council (ERC-2021-CoG-101041177-DecipherHSV), the German Research Foundation (DFG), SFB 1583/1, project number 492620490 and funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy, EXC 2155, project number 390874280; A.F. and M.P. by the Basic Research Program of the National Research University Higher School of Economics, for which A.H. is an International Supervisor. A.F. and A.R. acknowledge funding from the Clarendon Fund, the Radcliffe Department of Medicine and Keble and St Cross Oxford Colleges. Additional funds were provided by Boehringer Ingelheim Funds to A.R. and NIH Cancer Center Support Grant P30CA006927 to S.B.
Author information
Authors and Affiliations
Contributions
C.Y., A.F., H.G. and T.Z. carried out most of the biological experiments and data analyses, with assistance from J.C.C., C.R., X.Z., R.M.W., A.G., H.S.K., A.W.W., T.H., A.R., Y.Z., S.L., V.B., S.W. and S.M.; P.D. provided anti-Z-NA antibodies. M.P., J.R., A.P., P.G.T., E.S.M., L.D., T.Z., A.H. and S.B. designed experiments and analysed the data. C.Y., A.F., L.D., T.Z., A.H. and S.B. wrote the manuscript. All of the authors contributed to editing the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 (Related to Fig. 1). HSV-1 produces Z-RNAs in both murine and human cells and activates ZBP1-dependent nuclear necroptosis.
a, Primary MEFs infected with HSV-1 (MOI = 5) at 12 h after infection were fixed with fresh 4% (w/v) paraformaldehyde in PBS and exposed to the indicated nucleases for 45 min, before staining with anti-Z-NA (clone Z22) and anti-HSV-1gB antibodies. b, Fluorescence intensity of Z-NA signal in a. n = 41 cells (mock), n = 38 cells (HSV-1), n = 40 cells (HSV-1 + DNase I), n = 35 cells (HSV-1 + RNase A), n = 31 cells (HSV-1 + DNase I + RNase A). c, Z-NA accrual in HS68 human fibroblasts infected with WT HSV-1(MOI = 10, 18 h post-infection). Viral protein ICP5 staining marks infected cells. Cells were exposed to the indicated nucleases for 45 min, before staining with anti-Z-NA and anti- ICP5 antibodies. Nuclei are outlined with dashed white lines. d, Fluorescence intensity of Z-NA signal in c. e, Z-NA accrual in HT29 cells infected with HSV-1 (MOI = 10) at 9 h after infection. Viral protein ICP5 (green) staining marks infected cells. Cells were exposed to the indicated nucleases for 45 min, before staining with anti-Z-NA and anti-ICP5 antibodies. Nuclei are outlined with dashed white lines. f, Fluorescence intensity of Z-NA signal in e. n = 35 cells (mock), n = 37 cells (HSV-1), n = 38 cells (HSV-1 + DNase I), n = 39 cells (HSV-1 + RNase A), n = 49 cells (HSV-1 + DNase I + RNase A). g, Expression levels of FLAG-ZBP1 constructs in retrovirally-reconstituted Zbp1−/− MEFs, and of endogenous ZBP1 in primary WT MEFs, were assessed by immunoblotting for the FLAG tag and for murine ZBP1, following exposure of cells to mIFNβ (100 ng/ml, 24 h). h, Heat map of Z-NA and FLAG immunofluorescence signals in FLAG-ZBP1 or FLAG-ZBP1 ΔZα mutant MEFs infected with HSV-1. Nuclei are outlined with dashed white lines. i, Co-localization of FLAG and Z-NA signals in h, as assessed by evaluation of the Pearson Correlation Coefficient between the two signals. j, A-RNA accumulation in HSV-1-infected FLAG-ZBP1 MEFs (MOI = 5; 9 h post-infection). Nuclei are outlined with dashed white lines. k, Fluorescence intensity of A-RNA signal in j. n = 33 cells (mock), n = 37 cells (HSV-1 infection), n = 36 cells (HSV-1 infection + RNase A treatment). l, HSV-1 viral DNA levels in infected FLAG-ZBP1 MEFs with or without PFA (300 µg/ml) treatment was detected by qPCR using primers targeting US1 and RL1 genomic sequences. MEFs were infected with HSV-1 (MOI = 5, 9 h post-infection), exposed to PFA (300 µg/ml) 1 h post-infection, and examined by qPCR at the indicated times. m, Z-NA accrual in HSV-1-infected FLAG-ZBP1 MEFs in the presence or absence of PFA (300 µg/ml). FLAG-ZBP1 MEFs infected with HSV-1 were fixed and exposed to the indicated nucleases for 45 min, before staining with anti-Z-NA and anti-ICP5 antibodies. Viral protein ICP5 staining marks infected cells. Nuclei are outlined with dashed white lines. n, Fluorescence intensity of Z-NA signal in m. o, Kinetics of HSV-1 ICP6 RHIM mutant virus-induced cell death in Vec and FLAG-ZBP1 MEFs with or without PFA (300 µg/ml) treatment 1 h post-infection. p, Kinetics of HSV-1 ICP6 RHIM mutant virus-induced cell death in Vec and FLAG-ZBP1 MEFs. q, Immunoblots of phosphorylated MLKL (pMLKL) and cleaved caspase 3 (CC3) after infection of Vec and FLAG-ZBP1 MEFs with HSV-1 ICP6 RHIM mutant virus (MOI = 5). r, Viability of Vec or FLAG-ZBP1 MEFs infected with HSV-1 ICP6 RHIM mutant virus (MOI = 5) for 12 h in the presence or absence of the pan-caspase inhibitor zVAD (50 µM) and/or the RIPK3 kinase inhibitor (RIPK3 inh) GSK’843 (5 µM). s, Kinetics of cell death in immortalized Zbp1–/– MEFs reconstituted with either empty vector (Vec), FLAG-ZBP1, or (WT) ZBP1, following infection with HSV-1 RHIM mut (MOI = 5). t, Immunofluorescence staining for pMLKL and HSV-1 gB in primary Zbp1+/+ and Zbp1–/– MEFs infected with HSV-1 ICP6 RHIM mutant virus (MOI = 5). u, Kinetics of pMLKL positivity in cells infected with HSV-1 ICP6 RHIM mutant virus (MOI = 5) as in t. v, Immunofluorescence staining for pMLKL and HSV-1 gB in FLAG-ZBP1 MEFs infected with HSV-1 ICP6 RHIM mutant virus (MOI = 5). w, Kinetics of pMLKL positivity (black line) and localization of pMLKL signal (bars) of cells infected as in v. x, 3D reconstruction of HSV-1 ICP6 RHIM mutant virus-infected nuclei stained for the inner envelope protein Emerin (red) and HSV-1 gB (green). Arrows show areas of nuclear envelope rupture. y, Kinetics of nuclear envelope breakdown in HSV-1 ICP6 RHIM mutant virus -infected MEFs. Data are mean ± s.d. (n = 30 cells per group in d, n; n = 15 cells per group in i, n = 3 biologically independent samples in l; n = 4 in o, p, r, s; n = 5 fields per group in u, w; n = 6 per group in y). Two-tailed unpaired t-test with Welch’s correction (l). Two-way ANOVA test (p, u, y), One-way ANOVA with Dunnett’s multiple comparisons test (b, d, f, i, k, n, r). *P < 0.05, **P < 0.005, ***P < 0.0005 (P < 0.0001 in b, d, f, i, k, p, r, u, y). Data are representative of at least two independent experiments (a-k, m, n, q, t -y) or three independent experiments (o, p, r, s).
Extended Data Fig. 2 (Related to Fig. 2). Analysis of host cell Z-RNAs and ZBP1-bound RNAs.
a, Overview of peak calling and identification of unique dsRNA clusters. Arrows extending from the RIP track indicate the processing stages at which the RIP signal is utilized. Dashed boxes highlight dsRNA clusters which include one or more putative dsRNAs (e.g., the central and rightmost clusters, based on reverse-complementary sequences) and/or unresolved - but highly reproducible - RIP enrichment sites lacking a clear complementary pair (e.g., the leftmost cluster). b, Overview of the dsRNA analyses. Virus-induced host cell Z-forming dsRNAs were defined as those showing RIP enrichment in the target RIP (Z22 or FLAG) and increased levels in infected cells, either in total RNA or Z22 RIP reads, compared to mock-treated controls. These are summarized in a Rx2 table where R is the number of unique RIP-enriched Z-RNAs which are significantly overexpressed in infected cells after exclusion of the K dsRNAs unaffected by the infection. c, Global overlap of virus-induced host cell Z-RNAs (Z22-enriched) and ZBP1-bound (anti-FLAG enriched) RNAs from FLAG-ZBP1 MEFs infected with WT HSV-1 (MOI = 2, 8 h post-infection). Hypergeometric p-value, indicative of the odds that the overlap between the two pulldowns arises from chance alone, is shown above the Venn diagram. d, Genomic distribution of virus-induced host cell Z-RNAs enriched in FLAG RIP-seq from FLAG-ZBP1 MEFs infected with WT HSV-1 (MOI = 2, 8 h post-infection). e, Source of inverted reverse complement sequences within virus-induced host cell Z-RNAs enriched in FLAG RIP-seq from FLAG-ZBP1 MEFs infected with WT HSV-1 (MOI = 2, 8 h post-infection). The ‘Unresolved’ category includes Z-forming RNAs whose secondary structures could not be readily solved. f, Detailed distribution of inverted EREs in virus-induced host cell Z-RNAs enriched in FLAG RIP-seq from FLAG-ZBP1 MEFs infected with WT HSV-1 (MOI = 2, 8 h post-infection).
Extended Data Fig. 3 (Related to Fig. 2). Characterization of host cell Z-RNAs generated during HSV-1 infections in murine cells.
a-i, Coverage tracks for exemplar Z-RNAs showing Z22 RIP (green), input total RNA (orange), REDIportal-annotated or newly identified A → I editing sites (red ticks, top), positions of qPCR primers (red arrows), and a schematic of the putative Z-RNA structure (bottom). Coverage exceeding indicated limits is denoted by thick black caps. Here, Z-RNAs are produced by 3′ extension of host cell mRNA transcription (a-g), including into neighbouring genes (Tmem230 in b or Rab34 in d), or by de novo transcripts generated in the opposite direction from Hmga1 and Anapc4 genes (h, i). j, RNA eluted from Z22 or control IgG antibody pulldowns from WT HSV-1-infected cells (MOI = 2, 8 h post-infection) were examined by qPCR for exemplar Z-RNAs in panels (Fig. 2e–h, Extended Data Fig. 3a–i). Positions of qPCR primers are shown as red arrows in Fig. 2e–h, Extended Data Fig. 3a–i. Data were normalized to Input. k, RNA eluted from anti-FLAG antibody pulldowns from either FLAG-ZBP1 or FLAG-ZBP1 ΔZα mutant MEFs infected with HSV-1 (MOI = 2; 8 h post-infection) were examined by qPCR for exemplar Z-RNAs in panels (Fig. 2e–h, Extended Data Fig. 3a–i). Positions of qPCR primers are shown as red arrows in Fig. 2e–h, Extended Data Fig. 3a–i. Data were normalized to Input. Data are mean ± s.d. (n = 3 biologically independent samples in j, k). Two-tailed unpaired t-test with Welch’s correction (j). One-way ANOVA with Dunnett’s multiple comparisons test (k). *P < 0.05, **P < 0.005, ***P < 0.0005.
Extended Data Fig. 4 (Related to Fig. 2). Characterization of HSV-1 generated host cell Z-RNAs in human cells.
a, Enrichment of virus-induced host Z-RNAs in Z22 RIP-seq from FLAG-ZBP1 HT-29 cells infected with WT HSV-1 (MOI = 2, 8 h post-infection). Z-RNAs with evidence of A → I editing (i.e, overlapping REDIportal-annotated sites or identified in this study) are circled in black. Statistical significance was assessed using a one-sided Wald test in DESeq2. Only RNAs significantly induced by infection (adj. p-value ≤ 0.1, fold change ≥ 1.5) are shown, as determined by separate two-sided Wald tests comparing RIP/RIP or input/input between infected and matched mock cells (see Methods). P values were adjusted for multiple testing using the Benjamini-Hochberg procedure. b, Overlap of virus-induced host Z-RNAs enriched in Z22 or FLAG RIP-seq from FLAG-ZBP1 HT-29 cells infected with WT HSV-1 (MOI = 2, 8 h post-infection). Hypergeometric p-value, indicative of the odds that the overlap between the two pulldowns arises from chance alone, is shown above the Venn diagram. c, Genomic distribution of virus-induced host cell Z-RNAs enriched in Z22 (left) or FLAG (right) RIP-seq from FLAG-ZBP1 HT-29 cells infected with WT HSV-1 (MOI = 2, 8 h post-infection). d, Source of inverted reverse complement sequences within virus-induced host cell Z-RNAs enriched in Z22 (left) or FLAG (right) RIP-seq from FLAG-ZBP1 HT-29 cells infected with WT HSV-1 (MOI = 2, 8 h post-infection). The ‘Unresolved’ category includes Z-forming RNAs whose secondary structures could not be readily solved. e, Detailed distribution of inverted EREs in virus-induced host cell Z-RNAs enriched in Z22 or FLAG RIP-seq from FLAG-ZBP1 HT-29 cells infected with WT HSV-1 (MOI = 2, 8 h post-infection). f-i, Coverage tracks of exemplar Z-RNAs showing Z22 RIP (green), FLAG RIP (purple), input total RNA (orange), REDIportal-annotated or newly identified A → I editing sites (red ticks, top), positions of qPCR primers (red arrows), and a schematic of the Z-RNA structure (bottom). Coverage exceeding indicated limits is denoted by thick black caps. Here, Z-RNAs are produced either by 3′ extension of mRNA transcription (f-h), including into downsteam genes (e.g., MSH4 in f), or by de novo transcription from a normally silent genic or intergenic locus (i). j, RNA eluted from Z22 or control IgG antibody pulldowns from WT HSV-1-infected HT-29 cells (MOI = 2, 8 h post-infection) were examined by qPCR for Z-RNAs formed in 3′ extended transcripts arising from RABGGTB, ROCK1, and RUVBL2 loci, or generated via de novo transcription at the the ZNF132/584 locus. Positions of qPCR primers are shown as red arrows in panels f-i. Data were normalized to Input. k, RNA eluted from anti-FLAG antibody pulldowns from either FLAG-ZBP1 HT29 cells infected with HSV-1 (MOI = 2; 8 h post-infection) were examined by qPCR for Z-RNAs formed in aberrant transcripts arising from RABGGTB, ROCK1, RUVBL2 and ZNF132/584 (ZNFs) loci. Positions of qPCR primers are shown as red arrows in panels f-i. Data were normalized to Input. l, Proportion of proximal (i.e., within 10 kbp of TES) and distal (>10 kbp of TES) A → I intergenic editing sites identified in total RNA-seq from mock- or HSV-1-infected FLAG-ZBP1 MEFs (left) and FLAG-ZBP1 HT-29 cells (right). The total number of editing sites (including both genic and intergenic sites) detected in each condition is indicated above the respective bars. n = 8, 10, 4, 8 in order of conditions from left to right. m, Distribution of A → I editing sites in newly transcribed RNAs collected at the indicated time points from HSV-1-infected (WT, strain 17) human foreskin fibroblasts (HFF). The total number of editing sites (including both genic and intergenic sites) detected at each time-point is indicated on top. Sequencing data were obtained from the Sequence Read Archive (accession PRJNA256013). Data are mean ± s.d. (n = 3 biologically independent samples per group in j, k). Two-tailed unpaired t-test with Welch’s correction (j, k). *P < 0.05, **P < 0.005.
Extended Data Fig. 5 (Related to Fig. 3). Characterization of HSV-1 ICP27-triggered aberrant host cell transcription.
a, Aberrant transcription regions were defined for each mRNA as regions spanning individual introns (Intronic), regions upstream or downstream of annotated gene boundaries (Upstream, Downstream) or transcription in the divergent orientation from the same promoter (Divergent). Transcription within each region was normalized to reference constitutive exon(s), to calculate relative levels. b, Proportion of aberrant transcription regions with significant changes in normalized transcription levels in FLAG-ZBP1 MEFs and FLAG-ZBP1 HT-29 cells infected with HSV-1 (MOI = 2, 8 h post-infection). The total number of tested regions associated with expressed mRNAs is indicated on the right. Changes exceeding 10% were classified as strong. c, Temporal dynamics of aberrant transcription changes in HSV-1-infected HFF cells, estimated using 4sU-seq data. 4sU RNA labelling time windows are indicated on the left; the total number of tested regions associated with expressed mRNAs is indicated on the right. Changes exceeding 10% were classified as strong. Sequencing data were obtained from the Sequence Read Archive (accession PRJNA256013). d, Proportion of aberrant transcription regions with significant changes in normalized transcription levels in HFF cells infected with either wild-type HSV-1 (WT, strain 17) or a matched ICP27-null mutant (ΔICP27). The total number of tested regions associated with expressed mRNAs is indicated on the right. Changes exceeding 10% were classified as strong. Sequencing data were obtained from the Sequence Read Archive (accession PRJNA637636). e, Z-RNA accumulation in ICP27-transfected MEFs with or without leptomycin B (LepB, 5 nM) treatment was assessed at 6 h post-transfection. f, Fluorescence intensity of Z-RNA signal in e. n = 33 cells (no treatment), n = 39 cells (LepB treatment). g, A-RNA accumulation in MEFs transfected with FLAG-ICP27 construct was assessed 6 h post-transfection. Cells were either treated with RNase A, or were left untreated, before staining. Nuclei are outlined with dashed white lines. h, Fluorescence intensity of A-RNA signal in g. n = 61 cells (vector), n = 50 cells (ICP27-transfected), n = 53 cells (ICP27-transfected + RNase A). i, Schematic of ICP27 domain organization and location of the n504 mutation. LRR: leucine-rich putative nuclear export sequence (NES), D/E: acidic region, NLS: nuclear localization signal, RGG: arginine-/glycine-rich RNA binding motif, R2: second arginine-rich region, KH1-3: three predicted hnRNP K homology (KH) domains, CCHC: zinc-finger-like domain. The n504 mutation introduces a premature stop codon, generating a C-terminally truncated ICP27 mutant which is compromised in its ability to bind CPSF. j, Z-RNA formation in MEFs transfected with empty vector, FLAG-ICP27 or FLAG-ICP27 n504 mutant constructs assessed at 6 h post-transfection. k, Fluorescence intensity of Z-RNA signal in j. n = 34 cells (vector), n = 37 cells (ICP27-transfected), n = 35 cells (ICP27 n504-transfected). l, RNA eluted from Z22 or control IgG antibody pulldowns from ICP27 transfected cells were examined by qPCR for Z-RNAs formed within the 3′ extended transcripts downstream of Nabp1, Btbd3 or Ptbp1 loci, or in a de novo transcript generated from the H2ac18/19 locus or in the opposite orientation from the Hmga1 gene. Data were normalized to Input. m, RNA eluted from anti-FLAG antibody pulldowns of either FLAG-ZBP1 or FLAG-ZBP1 ΔZα mutant MEFs transfected with ICP27 were examined by qPCR for Z-RNAs formed in 3′ extended transcripts downstream of Nabp1, Btbd3 or Ptbp1 loci, or in a de novo transcript generated from the H2ac18/19 locus or in the opposite orientation from the Hmga1 gene. Data were normalized to Input. n, Kinetics of cell death in IFNβ pre-treated (100 ng/ml, 16 h) primary Zbp1+/+ and Zbp1–/– MEFs infected with HSV-1 RHIM mut (MOI = 5) and HSV-1 RHIM mut ΔICP27 (MOI = 15). o, Protein levels of ZBP1, RIPK3, and MLKL in HT-29 cells stably reconstituted with either empty vector (Vec) or FLAG-ZBP1 (FLAG-ZBP1), following IFNβ treatment (100 ng/ml, 48 h). p, Kinetics of cell death in Vec and FLAG-ZBP1 HT29 cells infected with either HSV-1 RHIM mut (MOI = 5) or HSV-1 RHIM mut ΔICP27 (MOI = 15). q, Protein levels of ZBP1, RIPK3 and MLKL in HS68 human fibroblasts in the presence or absence of IFNβ treatment (100 ng/ml, 48 h). r, Kinetics of cell death in hIFNβ pretreated (100 ng/mL, 16 h) HS68 cells infected with either HSV-1 RHIM mut (MOI = 5) or HSV-1 RHIM mut ΔICP27 (MOI = 15). s, Kinetics of cell death in WT and Zbp1–/– primary MEFs which were either left untreated, or were pre-treated with IFNβ (100 ng/ml, 16 h), followed by infection with HSV-1 RHIM mut virus (MOI = 5). t, Immunoblot showing induction of endogenous ZBP1 after infection of primary WT (Zbp1+/+) or Zbp1–/– MEFs with HSV-1 RHIM mut virus (MOI = 5, 8 h). Blotting for the HSV-1 protein ICP0 shows equal levels of virus replication in Zbp1+/+ and or Zbp1–/– MEFs. Data are mean ± s.d. (n = 3 biologically independent samples in l, m; n = 4 in n, p, r, s). Two-tailed unpaired t-test with Welch’s correction (f, l). One-way ANOVA with Dunnett’s multiple comparisons test (h, k, m). *P < 0.05, **P < 0.005, ***P < 0.0005. (p < 0.0001 in f, h, k). Data are representative of at least two independent experiments (e, g, j, o, q, t) or three independent experiments (n, p, r, s).
Extended Data Fig. 6 (Related to Fig. 4). Additional characterization of IAV-induced host cell Z-RNAs and ZBP1-bound RNAs.
a-d, Coverage tracks for exemplar Z-RNAs showing Z22 RIP (green), FLAG RIP (purple), input total RNA (orange), REDIportal-annotated or newly identified A → I editing sites (red ticks, top), positions of qPCR primers (red arrows), and a schematic of the Z-RNA structure (bottom). Coverage exceeding indicated limits is denoted by thick black caps. Here, Z-RNAs are found within aberrantly elongated 3′ extensions of ROCK1, KBTBD2, NCL, or C6orf62 mRNAs. e, f, RNA eluted from Z22- (e), anti-FLAG antibody (f) or control IgG antibody pulldowns from FLAG-ZBP1 HT29 cells infected with IAV (MOI = 5) or IAV ΔNS1 (MOI = 5) were examined by qPCR for Z-RNAs formed in 3′ extended transcripts downstream of ROCK1, KBTBD2, NCL, and C6orf62 loci. Data were normalized to Input. g, Enrichment of viral (green) and virus-induced endogenous (orange) RNAs in RIP-seq experiments from HSV-1-infected FLAG-ZBP1-expressing MEFs (left) or HT-29 cells (right). Host RNAs assessed by qPCR are labelled. The number of enriched viral hits in these pulldowns are indicated within each scatterplot. Data are mean ± s.d. (n = 3 biologically independent samples per group in e, f). Two-way ANOVA with Tukey’s multiple comparisons test (e, f). **P < 0.005, ***P < 0.0005. NS, no significance.
Extended Data Fig. 7 (Related to Fig. 4). Additional characterization of IAV-induced host cell Z-RNAs and ZBP1-bound RNAs.
a, Overlap between HSV-1- and IAV-induced Z-RNAs enriched in Z22 or FLAG pulldowns from FLAG-ZBP1 MEFs (left) or FLAG-ZBP1 HT-29 cells (right) infected with these viruses. b, Correlation between downstream transcription levels of individual mRNAs in FLAG-ZBP1 MEFs (left) and FLAG-ZBP1 HT-29 cells (right) infected with HSV-1 or IAV. Each mRNA is colour-coded based on the presence of virus-inducible Z-RNA downstream of the transcript. Normalized density plots of transcription levels for each mRNA class are shown on the marginal axes. The shaded area bounded by dashed lines represents mRNAs with similar downstream transcription levels (difference ≤ 10%) in both HSV-1- and IAV-infected cells. Only mRNAs meeting expression thresholds in both conditions are shown. The regression line for all mRNAs and its 95% confidence interval are shown in grey; r denotes the Pearson Correlation Coefficient. c, Proportion of proximal (within 10 kbp of TES) and distal ( > 10 kbp of TES) A → I intergenic editing sites identified in total RNA-seq from mock- or IAV-infected FLAG-ZBP1 MEFs (left) and FLAG-ZBP1 HT-29 cells (right). The total number of editing sites (both genic and intergenic) detected in each condition is indicated above the respective bars. Data are mean ± s.d. (n = 6, 6, 4, 8 biologically independent samples in order of conditions from left to right). d, Distribution of A → I editing sites in total RNA sampled at different time points from IAV- (A/Wyoming/03/03) infected human monocyte-derived macrophages. The total number of editing sites (both genic and intergenic) detected at each time-point is indicated on top. Sequencing data were obtained from the Sequence Read Archive (accession PRJNA382632).
Extended Data Fig. 8 (Related to Fig. 4). IAV NS1 mediates IAV-induced host cell Z-RNA formation and ZBP1-driven cell death.
a, Proportion of aberrant transcription regions with significant changes in normalized transcription levels in FLAG-ZBP1 MEFs and FLAG-ZBP1 HT-29 cells infected with IAV H1N1 strain A/Puerto Rico/8/1934 (MOI = 2, 8 h post-infection). The total number of tested regions associated with expressed mRNAs is indicated on the right. Changes exceeding 10% were classified as strong. b, Proportion of aberrant transcription regions with significant changes in normalized transcription levels in the nucleus of A549 cells infected with either wild-type IAV (WT, H1N1 strain A/WSN/1933) or a matched NS1-null mutant (ΔNS1) at 16 h post-infection. The total number of tested regions associated with expressed mRNAs is indicated on the right. Changes exceeding 10% were classified as strong. Sequencing data were obtained from the European Nucleotide Archive (accession PRJEB75711). c, RNA eluted from Z22- or control IgG antibody pulldowns from cells transfected with an NS1 expression construct were examined by qPCR for Z-RNAs formed within 3′ extended transcripts downstream of 2310057M21Rik and Gm14419. Data were normalized to Input. d, RNA eluted from anti-FLAG antibody pulldowns of either FLAG-ZBP1 or FLAG-ZBP1 ΔZα mutant MEFs transfected with an NS1 expression construct were examined by qPCR for Z-RNAs formed within 3′ extended transcripts downstream of 2310057M21Rik and Gm14419. Data were normalized to Input. e, Levels of IAV NS1 and NP in MEFs infected with IAV (MOI = 5) or IAV ΔNS1 (MOI = 5) at 8 h after infection. f, g, RNA eluted from Z22- (f), anti-FLAG antibody- (g) or IgG antibody pulldowns from FLAG-ZBP1 MEFs infected with IAV (MOI = 5) and IAV ΔNS1 (MOI = 5) were examined by qPCR for Z-RNAs formed within 3′ extended transcripts downstream of 2310057M21Rik, Gtpbp4, Vbp1 and Gm14419 loci. Data were normalized to Input. h, i, RNA eluted from Z22- (h), anti-ZBP1 antibody- (i) or IgG antibody pulldowns from primary MEFs infected with IAV (MOI = 5) and IAV ΔNS1 (MOI = 5) were examined by qPCR for Z-RNAs formed in 3′ extended transcripts downstream of 2310057M21Rik, Gtpbp4, Vbp1 and Gm14419 loci. Data were normalized to Input. j, Kinetics of cell death in primary Zbp1+/+ and Zbp1–/– MEFs infected with IAV (MOI = 5) and IAV ΔNS1 (MOI = 5). k, Kinetics of cell death in Vec or FLAG-ZBP1 HT29 cells infected with IAV (MOI = 5) and IAV ΔNS1 (MOI = 5). Data are mean ± s.d. (n = 3 biologically independent samples per group in c, d, f- i; n = 4 in j, k). Two-tailed unpaired t-test with Welch’s correction (c). One-way ANOVA with Dunnett’s multiple comparisons test (d). Two-way ANOVA with Tukey’s multiple comparisons test (f- i) *P < 0.05, **P < 0.005, ***P < 0.0005. Data are representative of three independent experiments (j, k).
Extended Data Fig. 9 (Related to Fig. 5.) Ectopic expression of ICP27 and NS1 induce formation of host cell Z-RNAs.
a, Schematic of experimental setup for the experiments shown in Fig. 5a–d, Extended Data Fig. 9b–x. Expression vectors encoding either HSV-1 ICP27 or IAV NS1 were transfected into FLAG-ZBP1 MEFs. Host cell Z-RNAs, as well as ZBP1-bound RNAs were isolated from these cells (by Z22 or FLAG RIP), respectively. These RNAs were eluted, purified, and subsequently re-transfected into fresh challenge cells for immunofluorescence and cell death analyses. b, Immunofluorescence staining of Z-RNA and FLAG-ZBP1 in FLAG-ZBP1 MEFs transfected with the indicated RNAs eluted from FLAG-ZBP1 or IgG pulldowns from MEFs expressing ectopic ICP27. FLAG-ZBP1 MEFs were fixed with fresh 4% (w/v) paraformaldehyde in PBS at 9 h after transfection and exposed to RNase A for 45 min, before staining with anti-Z-NA (clone Z22) and FLAG antibodies. Small white rectangles indicate regions that are magnified in bottom-left insets, showing colocalization of Z-RNA and FLAG-ZBP1. c, Fluorescence intensity in arbitrary units (a.u.) of Z-RNA signal in b. n = 65 cells (no treatment), n = 66 cells (IgG RIP), n = 59 cells (FLAG RIP), n = 59 cells (FLAG RIP+ RNase A treatment). d, Quantitation of colocalized ZBP1 and Z-RNA in c. n = 30 cells (IgG), n = 31 cells (FLAG RIP). e, Immunofluorescence staining of Z-RNA and FLAG-ZBP1 in FLAG-ZBP1 MEFs transfected with the indicated RNAs eluted from Z22 or IgG pulldowns from MEFs expressing ectopic NS1. FLAG-ZBP1 MEFs were fixed with fresh 4% (w/v) paraformaldehyde in PBS at 9 h after transfection and exposed to RNase A for 45 min, before staining with anti-Z-NA (clone Z22) and FLAG antibodies. Small white rectangles indicate regions that are magnified in bottom-right insets, showing colocalization of Z-RNA and FLAG-ZBP1. f, Fluorescence intensity in arbitrary units (a.u.) of Z-RNA signal in e. n = 40 cells (no treatment), n = 44 cells (IgG RIP), n = 45 cells (Z22 RIP), n = 45 cells (Z22 RIP + RNase A treatment). g, Immunofluorescence staining of Z-RNA and FLAG-ZBP1 in FLAG-ZBP1 MEFs transfected with the indicated RNAs eluted from FLAG-ZBP1 or IgG pulldowns from MEFs expressing ectopic NS1. FLAG-ZBP1 MEFs were fixed with fresh 4% (w/v) paraformaldehyde in PBS at 9 h after transfection and exposed to RNase A for 45 min, before staining with anti-Z-NA (clone Z22) and FLAG antibodies. Small white rectangles indicate regions that are magnified in bottom-right insets, showing colocalization of Z-RNA and FLAG-ZBP1. h, Fluorescence intensity in arbitrary units (a.u.) of Z-RNA signal in g. n = 66 cells (no treatment), n = 71 cells (IgG RIP), n = 69 cells (FLAG RIP), n = 48 cells (FLAG RIP+ RNase A treatment). i, Quantitation of colocalized ZBP1 and Z-RNA in e and g. n = 32 cells (Z22 RIP), n = 32 cells (FLAG RIP). j, Immunofluorescence staining of Z-RNA in primary MEFs transfected with indicated RNAs eluted from Z22- or control IgG antibody pulldowns from MEFs expressing ectopic ICP27. Primary MEFs were fixed with fresh 4% (w/v) paraformaldehyde in PBS at 6 h after transfection and exposed to RNase A for 45 min, before staining with anti-Z-NA (clone Z22) antibody. k, Fluorescence intensity in arbitrary units (a.u.) of Z-RNA signal in j. n = 31 cells (no treatment), n = 31 cells (IgG RIP), n = 34 cells (Z22 RIP), n = 47 cells (Z22 RIP+ RNase A treatment). l, Immunofluorescence staining of Z-RNA in primary MEFs transfected with indicated RNAs eluted from FLAG-ZBP1 pulldowns from FLAG-ZBP1 MEFs expressing ectopic ICP27. Primary MEFs were fixed with fresh 4% (w/v) paraformaldehyde in PBS at 9 h after transfection and exposed to RNase A for 45 min, before staining with anti-Z-NA (clone Z22) antibody. m, Fluorescence intensity in arbitrary units (a.u.) of Z-RNA signal in l. n = 35 cells (no treatment), n = 35 cells (IgG RIP), n = 37 cells (FLAG RIP), n = 40 cells (FLAG RIP+ RNase A treatment). n, Immunofluorescence staining of Z-RNA in primary MEFs transfected with indicated RNAs eluted from Z22 or IgG pulldowns from MEFs expressing ectopic NS1. Primary MEFs were fixed with fresh 4% (w/v) paraformaldehyde in PBS at 6 h after transfection and exposed to RNase A for 45 min, before staining with anti-Z-NA (clone Z22) antibody. o, Fluorescence intensity in arbitrary units (a.u.) of Z-RNA signal in n. n = 33 cells (no treatment), n = 31 cells (IgG RIP), n = 38 cells (Z22 RIP), n = 37 cells (Z22 RIP + RNase A treatment). p, Immunofluorescence staining of Z-RNA in primary MEFs transfected with indicated RNAs eluted from FLAG-ZBP1 or IgG pulldowns from FLAG-ZBP1 MEFs expressing ectopic NS1. Primary MEFs were fixed with fresh 4% (w/v) paraformaldehyde in PBS at 6 h after transfection and exposed to RNase A for 45 min, before staining with anti-Z-NA (clone Z22) antibody. q, Fluorescence intensity in arbitrary units (a.u.) of Z-RNA signal in p. n = 32 cells (no treatment), n = 34 cells (IgG RIP), n = 38 cells (FLAG RIP), n = 33 cells (FLAG RIP + RNase A treatment). r, Kinetics of cell death of Vec or FLAG-ZBP1 MEFs transfected with indicated RNAs eluted from FLAG or IgG pulldowns from MEFs expressing ectopic ICP27. s, t, Kinetics of cell death of Vec or FLAG-ZBP1 MEFs transfected with indicated RNAs eluted from Z22- (s), FLAG- (t) or IgG pulldowns from MEFs expressing ectopic NS1. u, v, Kinetics of cell death of IFNβ pre-treated (100 ng/ml, 16 h) primary Zbp1–/– or WT MEFs transfected with indicated RNAs eluted from Z22 (u), FLAG (v) or IgG pulldowns from MEFs expressing ectopic ICP27. w, x, Kinetics of cell death of IFNβ pre-treated (100 ng/ml, 16 h) primary Zbp1–/– or WT MEFs transfected with indicated RNAs eluted from Z22 (w), FLAG (x) or IgG pulldowns from MEFs expressing ectopic NS1. Data are mean ± s.d. (n = 4 in r-x).One-way ANOVA with Dunnett’s multiple comparisons test (c, f, h, k, m, o, q). Two-tailed unpaired t-test with Welch’s correction (d). ***P < 0.0005 (P < 0.0001 in c, d, f, h, k, m, o, q). Data are representative of at least two independent experiments (b, e, g, j, l, n, p) or three independent experiments (r-x).
Extended Data Fig. 10 (Related to Fig. 5). Synthetic host cell-derived Z-RNAs and pharmacological CPSF3 inhibition activate ZBP1.
a, Schematic of synthetic RNA hairpins generated from predicted virus-induced host Z-RNAs. The coverage track displays the Z22 RIP signal from infected cells, with red ticks indicating REDIportal-annotated or newly identified A → I editing sites. Synthetic hairpins were generated from predicted Z-RNA stems within Z22 peaks (orange highlight) formed by pairs of inverted LINE (Vbp1), LTR (Hmga1), and SINE (Ptbp1) elements. The minimum free energy structure of each hairpin is shown at the bottom, with the artificial outer UUUC loop rendered semitransparent. 2′-O-methyl-8-methylguanosine (m8Gm) modified bases are circled in red. b, Immunofluorescence staining of Z-RNA and FAM in primary MEFs transfected with RNA hairpins designed from putative Z-forming sequences withing aberrant host cell transcripts. Small white rectangles indicate regions magnified in bottom-right insets, showing colocalization of Z-RNA and FAM-labelled hairpins. c, Quantitation of colocalized Z-RNA and FAM in b. n = 11 fields (LINE, SINE), n = 10 fields (LTR, A-RNA and Z-RNA). d, Immunofluorescence staining of Z-RNA and FLAG in FLAG-ZBP1 MEFs transfected with RNA designed from aberrant Z-forming transcripts. Small white rectangles indicate regions that are magnified in bottom-right insets, showing colocalization of Z-RNA and FLAG-ZBP1. e, Quantitation of colocalized FLAG-ZBP1 and Z-RNA in c. n = 10 fields (LINE, LTR, A-RNA and Z-RNA), n = 8 fields (SINE). f, g, Kinetics of cell death in IFNβ pretreated (100 ng/mL, 16 h) primary Zbp1+/+ MEFs (f) or primary Zbp1–/– MEFs (g) transfected with the indicated RNA hairpins. h, Levels of CPSF3 protein in MEFs transfected with either control siRNA or CPSF3 siRNA. i, Z-RNA accrual in MEFs transfected with either control siRNA or CPSF3 siRNA. Cells were exposed to RNase A post-fixation, before staining with anti-Z-NA antibody. Nuclei are outlined with dashed white lines. j, Fluorescence intensity of Z-RNA signal in i. n = 35 cells (Si Ctrl), n = 34 cells (Si Cpsf3 and Si Cpsf3 + RNase A). k, Kinetics of cell death in Vec and FLAG-ZBP1 MEFs transfected with either control siRNA or CPSF3 siRNA. l, Alignment of JTE607-bound human CPSF3 crystal structure (bold) and murine CPSF3 AlphaFold model (transluscent). JTE-607 is shown as ball and stick (carbons; black, chlorine; green, oxygen; red, nitrogen; blue) with mesh (grey) representation to highlight occupied chemical space. The metallo-β-lactamase domain (green; residues 1-208 and blue; residues 396–459) and β-CASP domain (yellow; residues 209–395) display near identical folds, even in the absence of bound ligand. m, Active JTE-607 possesses the carboxylic acid moiety necessary for the bifurcated interaction with G330 and F241 backbone amides in CPSF3. Electrostatic interactions between the piperazine functional group of JTE-607 with E48 side chain and L47 backbone amide are also shown (orange dashes). n, Kinetics of cell death in IFNβ pretreated (100 ng/mL, 16 h) primary Zbp1+/+ and Zbp1–/– MEFs treated with DMSO (vehicle) or JTE-607 (100 μM), in the presence of zVAD (50 µM). Data are mean ± s.d. (n = 4 in f, g, k, n). One-way ANOVA with Dunnett’s multiple comparisons test (c, e, j). ***P < 0.0005 (P < 0.0001 in c, e, j). Data are representative of at least two independent experiments (b, d, h, i) or three independent experiments (f, g, k, n).
Supplementary information
Supplementary Figs. 1–7 (download PDF )
Supplementary Figures 1–7.
Supplementary Table 1 (download XLSX )
Public data accession. The accession numbers of public RNA-seq datasets used in this publication.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yin, C., Fedorov, A., Guo, H. et al. Host cell Z-RNAs activate ZBP1 during virus infections. Nature 648, 707–716 (2025). https://doi.org/10.1038/s41586-025-09705-5
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41586-025-09705-5
This article is cited by
-
The evolutionary arms race on the Z-form-nucleic- acid-binding protein 1 front: endogenous Z-RNA as a self-danger signal
Molecular Biomedicine (2026)
-
ZBP1 links infections to cancer immunotherapy
Cell Research (2025)
