
Abstract
Cisplatin (CDDP) serves as a vital component in the chemotherapeutic approach to treat esophageal squamous cell carcinoma (ESCC). However, prolonged CDDP application frequently culminates in resistance, compromising therapeutic outcomes. Through genome-wide CRISPR library screening, our study elucidates the mechanisms underlying this resistance, pinpointing CYFIP2 as a pivotal mediator. Notably, the involvement CYFIP2 is characterized by pronounced autophagic activity and the modulation of multiple cellular pathways. Empirical validation was achieved by treating ESCC cell lines with CDDP, which resulted in an upsurge of CYFIP2 expression. The functional impact of CYFIP2 was further delineated through knockdown experiments, where a marked suppression in cell proliferation was observed, alongside a discernible decline in reactive oxygen species levels. This was complemented by a suite of assays and microscopic techniques, including GFP-LC3, mRFP-GFP-LC3, electron microscopy and western blot, which collectively affirmed the inhibitory effect of CYFIP2 knockdown on autophagic processes, particularly impeding autophagosome formation and their subsequent fusion with lysosomes. In vivo studies have also confirmed that CYFIP2 knockdown limits tumor progression and increases CDDP efficacy. Conclusively, our findings introduce CYFIP2 as a novel contributor to CDDP resistance in ESCC, underscoring its potential as a therapeutic target. This revelation not only deepens our understanding of resistance mechanisms but also paves the way for novel oncotherapeutic strategies, promising enhanced treatment efficacy against ESCC.
Similar content being viewed by others


Introduction
Esophageal cancer (EC) is a prevalent malignancy worldwide, annually contributing to over half a million new cases and ranking as the sixth most common tumor type1. The predominant pathological forms, esophageal squamous cell carcinoma (ESCC) and esophageal adenocarcinoma, present distinct etiologies and geographical prevalences2,3. Notably, esophageal adenocarcinoma primarily emerges from Barrett’s esophagus, while ESCC is frequently associated with risk factors including smoking, alcohol intake, and the consumption of excessively hot foods4. In China, ESCC represents over 90% of EC incidences, contrasting with western regions4,5. Surgical resection stands as the principal curative approach for ESCC, yet approximately 40% of patients are deemed inoperable due to advanced tumor progression or metastasis6. For these patients, chemotherapy remains a critical component of both induction and adjuvant therapy. Recent advancements have also seen the introduction of immune checkpoint inhibitors targeting PD-1/PD-L1, like perberizumab and tislelizumab, offering promising results by impeding immune evasion mechanisms7. Ultimately, these pharmacological interventions provide patients in advanced stages with the possibility of surgical intervention or life prolongation.
Cisplatin (CDDP) is a platinum-based chemotherapeutic agent with a wide spectrum of antitumor activities, integral to ESCC treatment protocols7. Initially, ESCC are often sensitive to CDDP, but resistance frequently develops with extended treatment. The cytotoxicity of CDDP primarily stems from its ability to bind purine bases in DNA, leading to significant DNA damage, replication inhibition, and consequent cell proliferation arrest. Additionally, CDDP is known to induce reactive oxygen species (ROS) accumulation, exacerbating cellular damage8. Despite its efficacy, the emergence of CDDP resistance poses a significant challenge, with some cancer cells developing enhanced DNA repair mechanisms or altering drug uptake and efflux through membrane transporters like copper transporters and multidrug resistance protein9. Furthermore, mitochondrial adaptations, including metabolic downregulation, contribute to reduced ROS production under CDDP stress. The specific driver genes and resistance mechanisms in various cancers remain elusive, complicating the identification of universal therapeutic targets to augment CDDP’s effectiveness10.
Prior to the development of genome-wide library screening utilizing clustered regularly interspaced short palindromic repeats (CRISPR) technology, genetic screening was constrained by limited throughput and accuracy11. Small interaction RNA (siRNA) targets mRNA to induce loss of gene function and, with comprehensive libraries, can downregulate a broad spectrum of genes for driver gene identification12,13. However, siRNA-based approaches often result in partial and variable gene downregulation, leading to potential false negatives and biases in screening results14. In contrast, the CRISPR/Cas9 system, which includes small guide RNA (sgRNA, gRNA) and the Cas9 protein, enables precise genomic DNA cleavage at designated sites, facilitating nonhomologous end joining and consequent gene knockout. CRISPR/Cas9’s programmability and capacity for multiplex editing confer a significant advantage over previous gene editing technologies. With a comprehensive gRNA library, it becomes a powerful tool for genetic screening. Illustrating the utility of this approach, Li et al. employed CRISPR screening to pinpoint Kelch-like ECH-associated protein 1 as vital for the survival of breast and lung cancer cells when ataxia-telangiectasia-mutated kinase is inhibited15. Additionally, CRISPR screening revealed cyclin L1 as a significant player in gemcitabine resistance in pancreatic cancer, furthering our understanding of complex drug resistance mechanisms in oncology16.
Cytoplasmic FMR1-interacting proteins (CYFIPs), encompassing CYFIP1 and CYFIP2, were initially identified as interactors of the fragile X mental retardation protein17. CYFIP2, encoded by a gene on human chromosome 5q33.3, is implicated in a variety of vital biological processes. As a constituent of the WAVE regulatory complex, it is instrumental in modulating actin cytoskeleton dynamics. Genetic variations in CYFIP2 have been correlated with a range of neurological conditions, including intellectual disability, early-onset epileptic encephalopathy, and other developmental anomalies18,19,20. Beyond its neurological impact, CYFIP2 is posited to modulate autophagy and adipogenesis, with its deficiency linked to the upregulation of mTORC1 and inhibition of the GABA-BR signaling pathway21. In the oncological sphere, CYFIP2 has been recognized as a downstream target of insulin-like growth factor 2 mRNA-binding protein-1 IMP-1, where it exhibits a proapoptotic influence in colon cancer cells22. Its potential roles extend to the modulation of inflammation and immune evasion, factors that are critical in determining tumor behavior and patient prognosis23,24. Despite these multifaceted implications, the specific contributions of CYFIP2 to tumor resistance mechanisms remain underexplored and warrant further investigation.
Using genome-scale CRISPR knock-out (GeCKO) screening25, this study, for the first time, demonstrates that CYFIP2 promotes resistance to CDDP in ESCC. We further explored the impact of CYFIP2 inactivation in ESCC, which resulted in CDDP reactivation and reductions in ROS levels and autophagy. These findings establish CYFIP2 as a novel potential contributor that promotes CDDP resistance in ESCC, and inhibiting CYFIP2 presents a new strategy for enhancing the efficacy of CDDP therapy.
Results
Screening for CDDP-associated genes in ESCC cells
In this study, the half-maximal inhibitory concentration (IC50) of CDDP was used to screen and maintain cell resistance to the drug. Cell viability under varying concentrations of CDDP was assessed using the MTT assay. By plotting TE-1 cells viability across different logarithmic concentrations, the nonlinear best fit value determined log10IC50 (lgIC50) as 0.1892, corresponding to an IC50 of 1.546 µg/mL. And the same method determined the IC50 of EC109 cells for CDDP as 1.259 µg/mL (Supplementary Fig. S1A). Utilizing an sgRNA library, we conducted genome-wide knockouts in TE-1 cells to identify genes responsible for CDDP resistance in ESCC (Fig. 1A). Post-treatment, gRNA sequencing of survivors provided comprehensive sgRNA library coverage. The CDDP group yielded 17.1 G of data at 99.7% efficiency, outperforming the control group’s 12.73 G at 99.57% efficiency (Supplementary Material 2, 3).

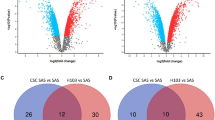
Screening for CDDP-associated genes in ESCC cells. (A) Schematic illustration of sgRNA library screening procedure to identify genes responsible for CDDP-resistance in TE-1 cells. (B) The raw data obtained from genome sequencing (gRNA sequencing) go through the bioinformatics analysis steps. (C) Volcano plot based on MAGeCK algorithms showing negatively (blue) and positively (red) selected genes in library screens, and the specific location of gRNA was determined by the fold change and p-value. (D) GSEA (KEGG gene sets) was performed based on negative screening and positive screening genes, with blue and red marked, respectively. (E) GSEA for autophagy in negative screening.
The target sequences obtained from raw data underwent quality control (QC) and were then mapped and quantified using MAGeCK software (version 0.5.7). Genes corresponding to sgRNAs that were retained or even amplified in the CDDP group were defined as positively screened, while genes corresponding to sgRNAs that were lost in the CDDP group were negative screening genes. Robust rank aggregation (RRA) scores were used to reflect the robustness of gene loss or enrichment. 483 genes were determined to be statistically significant in positive selection, and 729 genes were determined to be statistically significant in negative selection (p < 0.05 and absolute value of log2(fold change) greater than 1.5). Further enrichment analysis was conducted to elucidate potential mechanisms between molecular expression and phenotypes. The above analysis process is illustrated in Fig. 1B. Significant genes from both positive and negative screenings were visualized on a volcano plot, annotated with names and positions for those with notable fold changes and p value (Fig. 1C). Gene set enrichment analysis (GSEA) highlighted diverse pathway involvements, with discernible differences between negative and positive screening outcomes. In the positive screening, pathways related to apoptosis, Hippo signaling, and nicotine addiction were activated, while the negative screening showed enrichment in pathways related to RNA polymerase, cholesterol metabolism, and autophagy (Fig. 1D, E). Additionally, Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO) analyses were conducted to decipher auxiliary gene profile enrichments (Supplementary Fig. S2).
Identification of CYFIP2 as a novel resistant gene for CDDP
When genes conferring CDDP resistance were disrupted, cells exhibited increased vulnerability to CDDP-induced cytotoxicity, leading to a diminution of sgRNAs targeting these genes in the surviving cell population26. Consequently, genes implicated by negative screening, absent in the genomic DNA of deceased cells, were implicated in CDDP resistance. The top 20 genes identified through negative screening, ordered by RRA scores, were shortlisted as primary candidates and depicted in a colorful dot plot (Fig. 2A); alterations in sgRNA abundance are detailed in Supplementary Fig. S1B. Further refinement involved correlating these candidate genes with EC using Gene Expression Profiling Interactive Analysis (GEPIA)27 and Kaplan–Meier Plotter (KM Plotter) database28. The GEPIA, dedicated to the analysis of expression differences, revealed a predominant up-regulation trend in EC for most of the 20 candidate genes. However, only CYFIP2 and TMCO1 exhibited statistically significant differences (Fig. 2B; Table 1; Supplementary Material 4). Furthermore, the KM Plotter was utilized to investigate the correlation between candidate oncogenes and patient prognosis. The results demonstrated that higher expression levels of CYFIP2 and PRDM16 were indicative of poor prognosis in ESCC patients, with statistical significance observed in terms of overall survival (OS) and disease-free survival (DFS) (Fig. 2C, D; Table 1; Supplementary file 4). Hence, TMCO1, CYFIP2 and PRDM16, ranked according to the p value obtained in the negative screening (Fig. 2E) were selected for next validation.

Identification of CYFIP2 as a novel resistant gene for CDDP. (A) Top 20 genes in the negative screening are displayed in different colors and ranked based on RRA scores. (B) Boxplot showed the comparation of CYFIP2 expression in esophageal cancer (T, red) and normal tissues (N, black) according to GEPIA. (C,D) Using KM Plotter, we compared OS and DFS by categorizing ESCC individuals according to CYFIP2 expression levels. (E) The 3 genes with the greatest potential are displayed and sorted based on their p-values. (F) Schematic chart of CDDP resistance induced in ESCC cell lines. (G) The relative expression levels of CYFIP2 in TE-1, EC109 cells and their resistant cells measured by RT-qPCR. (H) Analysis of the GSE169337 dataset revealed a notable increase of CYFIP2 expression in CDDP-resistant ESCC cell lines. (I) The GSE116439 revealed that CDDP can induce CYFIP2 expression in multiple cancer cell lines, including human lung cancer (A549, NCI-H460), breast cancer (BT-549, MCF7), ovarian cancer (LOX) and Melanoma (SK-MEL-5). (J,K) Proteomic data from a ESCC cohort indicated a positive correlation between CYFIP2 protein levels and ESCC tumor onset and progression. NAT: normal adjacent tissues; IEN: intraepithelial neoplasia; A-ESCC: advanced ESCC; Stage 1–9 include normal tissue (Stage 1), hyperplasia (Stage 2), carcinoma in situ (Tis, Stage 3), lamina propria invasion (Stage 4), muscularis mucosa invasion (Stage 5), early submucosal invasion (Stage 6), advanced submucosal invasion (Stage 7), T2 invasion (Stage 8), and T3 invasion (Stage 9). (L,M) Proteomic data showed that genes positively correlated with CYFIP2 were significantly enriched in autophagy, mitophagy and other pathways. Error bars represent the mean ± SD for independent experiments, and “*” and “**” represent p-value < 0.05 and p-value < 0.01, respectively.
To corroborate the outcomes of the CRISPR screening, we induced CDDP resistance in ESCC cell lines including TE-1 and EC109 through a regimen of low-dose gradient escalation followed by a high-dose shock (Fig. 2F). We then measured the mRNA levels of the candidate genes in both wild-type (TE-1 and EC109) and CDDP-resistant (TE-1-cis and EC109-cis) cells using RT-qPCR. Notably, CYFIP2 expression significantly escalated in both resistant cell lines (Fig. 2G), while other candidates demonstrated unexpected or inconclusive results (Supplementary Fig. S2A). The mRNA expression profiles GSE169337 and GSE116439 from the Gene Expression Omnibus (GEO) databases revealed a close correlation between continuous CDDP treatment and resistance, along with the upregulation of CYFIP2 expression in ESCC cell lines and pan-cancer cell lines (Fig. 2H, I). In an ESCC clinical cohort with proteomic data comprising 786 trace samples from 154 patients, CYFIP2 was found to be gradually upregulated among 9 histopathological stages of ESCC carcinogenesis (Fig. 2J, K)29. Further analysis highlighted proteins positively correlated with CYFIP2 expression, which were significantly enriched in pathways such as autophagy and mitophagy (Fig. 2L, M)30. Consequently, CYFIP2 has been pinpointed as a pivotal CDDP-resistant gene, meriting deeper exploration.
Knockdown of CYFIP2 enhances CDDP sensitivity in ESCC cells
To investigate CYFIP2’s role in ESCC cells under CDDP treatment, RNA interference (RNAi) was employed. Specifically, shRNA sequences sh01 and sh04 were selected for their superior knockdown efficiency as evidenced by RT-qPCR results (Supplementary Fig. S2B). Both CDDP-resistant cells (TE-1/EC109-cis) and wild-type cells were utilized in subsequent analyses to discern changes in resistant cells and assess CYFIP2’s impact. For brevity, we denote the wild-type cells transfected with the control plasmid (NC) and not treated with CDDP as the negative control (NC) group. The TE-1/EC109-cis cells transfected with the shNC and cultured with IC50 concentration of CDDP are labelled as the IC50 group. The CDDP-resistant cells transfected with knockdown plasmids (sh01/04) and cultured with the IC50 concentration of CDDP are denoted as the IC50 + sh01/04 group.
The colony formation assays demonstrated more monoclonal formation foci in the IC50 group than in NC group, with shRNA treatment (IC50 + sh01/sh04) markedly reducing these foci (Fig. 3A, B). Correspondingly, the EdU assay revealed that resistant cell groups displayed increased fluorescence (indicative of proliferation) at the IC50 concentration compared to wild-type cells. However, this fluorescence significantly diminished in cells following CYFIP2 knockdown (Fig. 3C, D). In addition, we employed a fluorescent probe to quantify ROS levels in cells, observing elevated ROS in CDDP-resistant cells compared to naïve ESCC cells. Notably, CYFIP2 inactivation led to a reduction in ROS levels within the resistant group (Fig. 3E, F), hinting at CYFIP2’s potential role in ROS generation or mitigation pathways. Moreover, we utilized comet assays and immunofluorescence (γ-H2AX) to assess hereditary material damage. However, despite multiple attempts, the comet tails and DNA damage foci were either atypical or insufficiently prevalent for reliable statistical analysis (Supplementary Fig. 4, 5). In conclusion, our findings indicate that ESCC cells, once resistant, not only survive but proliferate under CDDP treatment, accompanied by increased ROS levels. Inhibition of CYFIP2 appears to interrupt these adaptations.

Knockdown of CYFIP2 enhances CDDP sensitivity in ESCC cells. (A,B) Colony formation assays confirmed that CDDP-resistant cells exhibited faster cloning rates (IC50 vs NC). However, when CYFIP2 was knockdown, the cloning of ESCC cells was significantly inhibited (IC50 + sh01/04 vs IC50). (C,D) EdU assays, with DAPI serving as a staining control, revealed enhanced proliferative rates in resistant TE-1 and EC109 cell lines (IC50). In addition, CYFIP2 silencing (IC50 + sh01/04) resulted in decreased proliferation. The EdU images are presented with a scale bar of 100 μm. (E,F) The level of ROS in cells was measured by fluorescent probe detection. Each photo has a scale 200 μm in the lower right corner. Error bars represent the mean ± SD for independent repeated data, and “*”, “**”, “***”, and “****” represent p-value < 0.05, p-value < 0.01, p-value < 0.001, and p-value < 0.0001, respectively.
Inhibition of CYFIP2 downregulates autophagy in ESCC cells
Building on the enrichment analysis (Fig. 1D, E; Fig. 2L, M) and the purported role of CYFIP2 in autophagy regulation, we delved into CYFIP2’s influence on autophagic processes under CDDP treatment21. GFP-LC3 assays revealed that both CDDP and rapamycin (a known autophagy inducer) escalated LC3 accumulation in phagophores, indicative of heightened autophagy. Remarkably, CYFIP2 downregulation substantially hindered the formation of these fluorescent foci (Fig. 4A, B). The mRFP-GFP-LC3 assay, assessing autophagic flux, echoed these findings, showing disrupted autophagosome-lysosome fusion in CYFIP2-suppressed (sh01 and sh04) groups compared to the CDDP and rapamycin groups (Fig. 4C, D). Scanning electron microscopy further illustrated a decrease in autophagosomes in the shRNA-treated cells (red arrow, Fig. 4E). Lastly, Western blot analysis was performed to assess the levels of Beclin 1 and LC3 proteins, with quantification achieved using ImageJ software for statistical evaluation. Notably, the LC3 II/LC3 I ratio, an indicator of autophagic activity akin to Beclin 1 levels, exhibited a significant increase upon induction with CDDP or rapamycin. Conversely, this ratio markedly decreased following CYFIP2 knockdown (Fig. 4F, G; Supplementary Material 5).

Inhibition of CYFIP2 downregulates autophagy in ESCC cells. (A,B) The accumulation of LC3 on autophagosomes was observed in cells transfected with GFP-LC3 under fluorescence microscopy (scale bar: 20 μm). (C,D) Cells transfected with mRGFP-GFP-LC3 were detected under fluorescence microscopy to dynamically analyze the fusion of autophagosomes and lysosomes (scale bar: 20 μm). (E) Electron microscopy was used to compare and analyze autophagosome formation in cells. Scale bar (up): 5 μm; Scale bar (down): 2 μm. (F,G) Beclin1 and LC3I/II were detected by western blot to evaluate the autophagy level in cells. Error bars represent the mean ± SD for independent experiments, and “*”, “**”, and “***” represent p-value < 0.05, p-value < 0.01, and p-value < 0.001, respectively.
In vivo experiments revealed the role of CYFIP2 in ESCC
RNA and proteins were extracted from stable transfection cell lines for RT-qPCR and Western blot experiments. The RT-qPCR results indicated a reduction of CYFIP2 expression in the shCYFIP2 group compared to the control group (shCtrl). Similarly, Western blot analysis revealed a downregulation of CYFIP2 protein levels in shCYFIP2 cells (Fig. 5A, B). In an in vivo study, all four groups of nude mice completed the experiment successfully. Growth curves, plotted from extradermal tumor measurements, demonstrated continuous growth of xenografts in the two untreated groups. In contrast, the xenograft volume in the CDDP-treated group peaked on Day 25 before declining, suggesting that CDDP effectively inhibited malignant tumor proliferation. Notably, tumors in the shCYFIP2 group grew more slowly than those in the shCtrl group, regardless of CDDP treatment (Fig. 5C). At the end of the experiment, tumor tissues were photographed (Fig. 5D), and tumor weight analysis confirmed that CYFIP2 knockdown significantly inhibited ESCC growth in both CDDP-treated and control groups (Fig. 5E). Subsequent HE staining confirmed accurate tumor sampling, and IHC staining showed reduced LC3 protein following CYFIP2 knockdown (Fig. 5F). Additionally, TUNEL staining was enhanced in the CDDP-treated group with CYFIP2 knockdown, suggesting that CYFIP2 may play a role in apoptosis process (Fig. 5G).

In vivo experiments revealed the role of CYFIP2 in ESCC. (A,B) RT-qPCR and western blot were used to verify the successful establishment of stably transfected cells. (C) The growth curve of subcutaneous xenograft tumor in mice during the experiment (Unit: mm3; shCtrl + PBS vs shCYFIP2 + PBS: 1264.509 ± 176.871 vs 985.390 ± 263.207, p-value = 0.0247; shCtrl + CDDP vs shCYFIP2 + CDDP: 417.525 ± 194.566 vs 206.827 ± 49.873, p-value = 0.0464; n = 5). (D) Photos of xenograft tumor and size post-euthanasia of mice. (E) Measurement and analysis of xenograft tumor weight (Unit: g; shCtrl + PBS vs shCYFIP2 + PBS: 1.13 ± 0.435 vs 0.484 ± 0.190, p-value = 0.0160; shCtrl + CDDP vs shCYFIP2 + CDDP: 0.81 ± 0.4078 vs 0.322 ± 0.182, p-value = 0.0403; n = 5). (F) The tumor tissues were prepared by paraffin wax and stained by HE and IHC (CYFIP2 and LC3I/II). (G) The paraffin sections were stained by TUNEL.
Discussion
CRISPR library screening, exemplified by the GeCKO library from Feng Zhang’s lab, represents a transformative advance in genome-wide analysis, enabling precise identification of genes linked to specific phenotypes. This technology, initially utilized to pinpoint resistance drivers in melanoma to vemurafenib, has been broadly adopted due to its comprehensive applicability25. In our research, we harnessed the GeCKO library to create stably transfected cell lines, aiming to isolate CDDP-resistant variants. Through meticulous sequencing and bioinformatic analyses post-screening, we identified a cohort of candidate genes potentially contributing to CDDP resistance. Further refinement via GEPIA analysis honed in on these candidates, leading to the construction of CDDP-resistant cells for further validation. Subsequent RT-qPCR and western blot assays underscored CYFIP2 as a prominent gene associated with CDDP resistance. We embarked on functional assays to elucidate the role of CYFIP2 in this resistance mechanism.
CYFIP2’s critical role in the nervous system is well-documented, with early research highlighting its enrichment in neurons and influence on neuronal development, synaptic structure, and function. Dysregulation of CYFIP2 is associated with a spectrum of neurological disorders, including schizophrenia, epilepsy, and cognitive impairments25,31,32,33. Intriguingly, CYFIP2 displays dichotomous oncogenic properties; it acts as a direct target of p53 and FOXD3 in gastric cancer to regulate apoptosis and exhibits variable expressions and mutations in kidney renal clear cell carcinoma, pancreatic neuroendocrine tumors, and other malignancies34,35,36,37. Its differential expression in diverse cancers, as documented in public databases, hints at its prognostic relevance23. Yet, prior to our study, CYFIP2’s role in ESCC and CDDP resistance was unexplored. Our study integrates gRNA sequencing data, the GEPIA database, GEO database, and proteomics data from the ProteomeXchange Consortium, revealing that CYFIP2 is upregulated in the occurrence and progression of ESCC and is closely associated with the acquisition of CDDP resistance in ESCC cells. Public databases suggest that CYFIP2 positively regulates autophagy, which has been validated by multiple experiments. Additionally, CYFIP2 is also associated with upregulating ROS levels. In vivo and in vitro assays displayed that targeting CYFIP2 emerges as a promising strategy to counteract CDDP resistance.
Autophagy, a complex cellular mechanism, serves the critical function of removing suboptimal products and nonessential substances, thereby recycling components to sustain cell survival under stress38. Yet, its overactivation can lead to cell death, a process beneficial for development and damage response39. In the context of cancer, autophagy paradoxically promotes tumor growth and serves as a therapeutic target. For instance, Drp1 upregulation in ESCC enhances malignant proliferation by facilitating autophagy, optimizing resource allocation for tumor growth40. Conversely, sulforaphane’s inhibitory effects on ESCC are mitigated by NRF2-induced protective autophagy, a process reversible by the autophagy inhibitor chloroquine40,41. Autophagy involves a series of molecular events, beginning with autophagosome formation and culminating in their fusion with lysosomes38. The LC3 I/II protein is a hallmark of autophagy, accumulating on forming autophagosomes and undergoing degradation by lysosomal enzymes42. Our investigations using GFP-LC3 and electron microscopy confirmed CYFIP2’s influence on autophagosome formation. Additionally, the mRFP-GFP-LC3 assay validated that CYFIP2 enhances the fusion efficiency of autophagosomes with lysosomes, highlighting its integral role in autophagic processes.
In our western blot assay, Beclin1’s expression corroborated our findings unequivocally, underscoring its pivotal role in autophagy, particularly as a part of the complex with vacuolar protein sorting 34 that generates phosphatidylinositol 3-phosphate (PI3P) on early autophagosome membranes. PI3P-rich domains are crucial for recruiting autophagy-related proteins, thus promoting autophagosome formation43. The GFP-GFP and mGFP-RFP-LC3 assays illustrated the distribution and quantity of LC3 I/II, while the western blot provided a snapshot of its total cellular content. Unlike Beclin1, LC3 I/II exhibits more dynamic behavior in actively autophagic cells, being rapidly synthesized and degraded. This dynamic turnover could result in a steady-state level when synthesis and degradation rates are balanced. Therefore, we infer that autophagy, particularly modulated by CYFIP2, plays a vital role in the development of CDDP resistance.
While ROS were previously considered mere byproducts of cellular metabolism and excessively high levels of ROS could eliminate tumor cells, subsequent studies have firmly established them as essential and active components involved in regulating various cellular pathways44. Living cells tightly regulate ROS levels through mechanisms that involve molecules such as glutathione (GSH) and other ROS elimination systems. This regulation ensures a delicate balance that adapts to the intricate requirements of life45. However, it is crucial to acknowledge that elevated ROS levels can perpetuate the activation of molecular pathways that promote tumor proliferation, spread, immune suppression among others within cancer cells, and they bear a close association with varied types of tumor drug resistance46,47. Autophagy, a critical process in tumorigenesis and resistance, is influenced by excessive ROS levels, which are considered a direct cause of aberrant autophagy48. Recent studies suggest a close interrelationship between ROS and autophagy, which together influence various aspects of life processes, including tumor development and treatment49. Synthetic drugs such as ciclopirox olamine (CPX) have been developed to promote ROS elimination through various pathways, subsequently inhibiting autophagy and ultimately decreasing proliferation48. The present study also suggests a correlation between elevated ROS levels and enhanced cell proliferation, at least within an appropriate concentration. Our findings have illuminated that CYFIP2 serves as a positive factor in increasing ROS levels. Furthermore, we infer that the elevated ROS levels induced by CYFIP2 could activate autophagy, ultimately leading to resistance against CDDP in ESCC.
Conclusion
Our study has conclusively pinpointed CYFIP2 as a critical gene instigating CDDP resistance in ESCC, unveiled through a comprehensive CRISPR library screen. The knockdown of CYFIP2 in ESCC cells curtailed cell proliferation, decreased ROS levels, and inhibited autophagy. These findings posit CYFIP2 as a promising target to enhance ESCC sensitivity to CDDP, potentially elevating the efficacy of treatments. Future endeavors should dissect the complex molecular pathways through which CYFIP2 orchestrates CDDP resistance, with a keen focus on its interplay with autophagy and ROS modulation. Additionally, more experimental validations are needed to confirm the therapeutic potential of CYFIP2 inhibition in ameliorating CDDP resistance in ESCC.
Materials and methods
Cell lines and cell culture
Human ESCC cell lines, including TE-1 and EC109, were used in this study to explore the role of CYFIP2 in ESCC. The cell lines were purchased from Procell Life (Wuhan, China) and Cellcook (Guangzhou, China), respectively, with authentication reports as an attachment (Related File 1, 2). Cells were cultured in Roswell Park Memorial Institute (RPMI)-1640 (Sigma, USA) supplemented with 10% (v/v) fetal bovine serum (FBS, NEWZERUM, New Zealand). The culture conditions involved maintaining cells at 37 °C in an atmosphere containing 5% carbon dioxide. HEK293T cells, obtained from the Key Laboratory of the Ministry of Education for Gastrointestinal Cancer (Fuzhou, China), were cultured in Dulbecco’s modified Eagle’s medium (DMEM) under similar conditions. In addition, cells were tested monthly for mycoplasma contamination, and contaminated cells were replaced or subjected to mycoplasma killing treatment.
Plasmids, lentivirus packaging, and transfection
The human CRISPR GeCKO version 2.0 sgRNA library (BioGene, Shanghai, China) was used for genomic-scale screening. This library contains library A (65,383 sgRNAs) and library B (58,028 sgRNAs). To achieve targeted gene suppression through RNAi, we integrated shRNA sequences into the pLKO.1 vector. Four shRNA sequences targeting CYFIP2 (sh01-04) were designed as detailed in Table 2. An empty pLKO.1 vector served as the control. Puromycin (Thermo Fisher Scientific, USA) was used to select the successfully transfected cells, which could be diluted by medium to the final concentration (2 μg/mL).
Cells at approximately 70% confluence were used for transfection. The transfection mixture was prepared by combining Liquid A (200 μL of Optimized Minimum Essential Medium (Opti-MEM) with 4 μg of the plasmid) and Liquid B (200 μL of Opti-MEM with 10 μL of Lipofectamine 2000) (Thermo, USA). The two liquids were mixed and allowed to sit for 20 min. The mixture, along with serum-free medium, was added to the cells. After 6 h, the old medium was replaced with fresh medium, and the cells were cultured for an additional 1–2 days. Transfection efficiency was assessed through fluorescence observation. Lentiviral shRNA was produced in HEK293T cells following a similar protocol. Additionally, we employed helper plasmids, psPAX2, and pMD2. G, and the ratio of their dosage to the core plasmids (shRNA or sgRNA libraries) was 3:1:10. After 48 h of transfection, cell medium was collected, centrifuged at 15,000 × g for 5 min at 4 °C, filtered through a 0.45 μm filter membrane, subpackaged, and stored at -80 °C for future use.
CDDP and dose‒response curve
CDDP from Hansoh Pharma (Shanghai, China) was used in this study, and dissolved in PBS solution to 1 mg/mL, stored at -20℃, eventually diluted to the specified working concentration (such as IC50) using a complete medium for use. Concentrations were measured in micrograms per (μg/mL). We performed the MTT assay to generate a dose‒response curve, determining the IC50 of CDDP. Cells were seeded at a density of 1 × 104/mL in 96-well plates and cultured for 24 h. We exposed cells to fresh culture medium containing varying concentrations of CDDP (0, 0.78125, 1.5625, 3.125, 6.25, 12.5, 25, 50, 100 μg/mL), replacing the old medium, and allowed them to incubate for 48 h before analysis. To assess cell viability, an MTT solution (Beyotime, Shanghai, China) was added following the provided guidelines. After incubation at 37 °C, the supernatant was carefully removed, and the formazan crystals were fully dissolved in dimethyl sulfoxide (DMSO, MedChemexpress, USA). The absorbance was measured at a wavelength of 490 nm. The logarithm of CDDP concentration was plotted on the horizontal axis, and the percentage of cell inhibition was plotted on the vertical axis to construct the inhibition curve.
Genome-wide CRISPR library screen
TE-1 cells were transfected with the CRISPR knockout library lentivirus at a low multiplicity of infection (MOI) of approximately 0.3. Mutant cells underwent a 7-day puromycin (2 μg/mL) selection and were subsequently divided into two groups. One group was cultured with medium containing CDDP (IC50) for 10 days, while the other group was cultured with PBS as the control. Genomic DNA was extracted from both cell groups for library construction and subsequent gRNA sequencing. The gRNA sequences were amplified and underwent adapter and index addition. Finally, double-ended sequencing (PE) with a 2 × 150 bp read length was performed using Illumina sequencing technology (Illumina, San Diego, CA, USA). The raw data underwent quality control processes and was subsequently mapped and quantified against the sgRNA library. Enrichment analyses using GO, KEGG, and GSEA (utilizing the KEGG database)50 were performed to further characterize the gene profile and better elucidate the potential mechanisms underlying the phenotype of CDDP resistance. Data processing was carried out using the R package “MAGeCKFlute” that is publicly available on GitHub (https://github.com/WubingZhang/)51. Metadata acquired from sequencing have been uploaded to the Sequence Read Archive (SRA) database and assigned the accession number PRJNA1010500.
Public databases analysis
GEPIA (http://gepia.cancer-pku.cn/) was used to analyze the expression differences of candidate genes between tumor and control samples through Boxplots27. A p value of less than 0.05 and fold change greater than 2 was deemed statistically significant and denoted with an asterisk, while those that do not meet these criteria were marked with "ns," indicating no significance. KM plotter (https://kmplot.com/analysis/) was utilized for analyzing OS and DFS of patients with ESCC28. The hazard ratios (HR) calculated by the Cox proportional hazards model indicates the risk probability of death in the high-expression group of candidate genes compared to the low-expression group. And KM plot illustrated the survival rates of different groups, with the p value displayed in the upper right corner of panel.
Transcriptomic datasets GSE169337 and GSE116439 from the GEO database were employed to explore the association between CYFIP2 and CDDP resistance. GSE169337 includes data from ESCC cell lines before and after acquiring CDDP resistance, whereas GSE116439 encompasses pan-cancer cell lines (lung cancer, breast cancer, and melanoma) at various time points following CDDP treatment.
Proteomics data (Project ID IPX0002178000) from the ProteomeXchange Consortium platform were used to examine the relationship between CYFIP2 expression and tumorigenesis, as well as the genes regulated by CYFIP2. This dataset was sourced from an ESCC-specific cohort established by Fudan University (Shanghai, China)29 to further validate the clinical significance of CYFIP2 in ESCC.
Induction of CDDP resistance in ESCC cell lines
Cells were initially revived and adjusted to a density of 1 × 105 cells/mL, followed by culturing until reaching the logarithmic growth stage. The culture medium was then replaced with CDDP at a final mass concentration of 1/9 × IC50 for 48 h. Afterward, fresh CDDP-free medium was added, and the cells were cultured until they returned to normal growth. This process was repeated, gradually increasing the CDDP concentration (according to the order: 1/9 × IC50, 1/5 × IC50, 1/2 × IC50, 3/4 × IC50, 1 × IC50) over 3–5 shocks at each concentration. Once cells could stably grow in 1 × IC50 μg/mL CDDP, intermittent induction was performed using high doses of CDDP at 2–4 × IC50 μg/mL. Ultimately, we obtained CDDP-resistant TE-1 and EC109 cells, designated TE-1-cis and EC109-cis, respectively.
Real-time fluorescence quantitative PCR (RT-qPCR)
RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, USA) to measure the levels of mRNA. RNA concentration was measured using a Nanodrop 2000 spectrophotometer (Thermo Fisher Scientific, USA). Subsequently, 1 μg of RNA was used for reverse transcription (Promega A2800, USA). The reaction was set up in triplicate, with 2 μL of cDNA from each well used for RT-qPCR reactions. The reaction protocol was as follows: Initial denaturation (95 °C, 2 min), 40 cycles of denaturation (95 °C, 15 s), annealing (60℃, 1 min) and extension (72℃). Oligo (dT) primers were used to generalize complementary DNA (cDNA) from total RNA. The Reverse Transcription Kit (Promega, Madison, USA) was used to synthesize cDNA. The PrimeScriptTM RT Master Mix Kit (Takara Bio, Kusatsu, Japan) and SYBR Green were used to perform RT-qPCR on an ABI7900 System (Thermo Fisher Scientific, USA). The sequences of primers used are presented in Table 3. β-actin was assigned the housekeeping gene. Each RT-qPCR experiment was independently performed three times, and the relative expression value was calculated using the 2−ΔΔCt method.
Western blot analysis
Cells were collected by scraping and subsequently lysed using ice-cold radioimmunoprecipitation assay (RIPA) lysis buffer, which was enriched with both protease and phosphatase inhibitors. After lysis, the samples were subjected to centrifugation at 1.2 × 104 g for 15 min at 4 °C. The supernatant containing solubilized proteins was collected for subsequent analysis. Protein concentration was determined, and Saint-Bio’s loading buffer was added to the samples, which were then boiled to prepare the final protein sample. The proteins were separated using sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and subsequently transferred to polyvinylidene fluoride membranes. And samples run on different gels for individual protein signal detection, facilitating comparison and minimizing signal interference. The membranes were then incubated with primary antibodies at 4 °C for 12 h, followed by incubation with secondary antibodies at room temperature for 60 min. The expression level of target proteins labeled by the secondary antibodies was measured using the Odyssey imaging systems (Odyssey CLx) from LI-COR Biosciences (Lincoln, USA). The primary antibodies used in this study included anti-LC3I/II (Abcam, ab84936), anti-Beclin1 (Abcam, ab207612), and anti-Beta-actin (Abbkine, ABL1010). The secondary antibodies used were DyLight 680 (KPL, 072–06-18–06) and DyLight 800 (KPL, 072–07-18–06).
Cell viability and proliferation
In this study, colony formation and EdU assays were employed to assess the viability and proliferation of ESCC cells. The EdU kit used was sourced from Abbkine (Wuhan, China). To prepare the cells, a single-cell suspension was generated and counted. The cells were then seeded into 12-well plates at a density of 2,000 cells per well. After seeding, the plates were incubated for 10 days. Following incubation, the culture medium was carefully removed, and the cells underwent a series of steps, including washing, fixation with paraformaldehyde (100 µL, 30 min, 37 °C), staining with crystal violet (30 min, 37 °C), and air-drying. The resulting foci-forming clones were analyzed and counted. Five fields were randomly selected to determine the number of positive cells. The clone formation rate was calculated using the formula: Clone formation rate = (number of clones/number of inoculated cells) × 100%.
For the EdU incorporation assay, 1000 cells were seeded into confocal dishes and allowed to grow until they reached 70–80% confluence. Next, the culture medium was replaced with fresh medium containing EdU at a final concentration of 10 µM. The reaction mixture, EdU reagent diluted by DMEM at a ratio 1:000, was then added to each dish (100 µl per dish) within 15 min. The dishes were incubated at 37℃ for 30 min in darkness. Subsequently, the reaction mixture was removed, and nuclear staining was performed using 4’,6-diamidino-2-phenylindole (DAPI, 1ug/mL) under strict light protection after thorough washing. EdU (Ex/Em = 546/565 nm) and nuclei (Ex/Em = 360/460 nm) were detected under a fluorescence microscope.
ROS detection assay
The CheKine™ ROS Kit (Abbkine, Wuhan, China) was employed to quantify ROS levels in cells. Cells were seeded into 6-well plates at 1 × 105 cells/mL cell suspension. After 24 h, DCFH-DA was prepared in serum-free medium to a final concentration of 10 μM. The cell culture medium was removed, and the DCFH-DA working medium was added to the cells. The cells were incubated at 37 °C for 30 min in darkness. Next, the cells were washed three times to remove excess DCFH-DA that did not enter the cells. Finally, the fluorescence was observed under a fluorescence microscope.
Autophagy level detection
GFP-LC3, RGFP-LC3 and electron microscopy detection were used to analyze the role of CYFIP2 in autophagy. Cells were screened, cultured and then fixed with 4% paraformaldehyde. After X-Trton treatment, the cells were stained with DAPI and cleaned. They were then observed under a confocal microscope (Leica SP8, Wetzlar, Germany) with an oil lens. The average number of green puncta per cell within each field of view was evaluated to quantify the number of autophagosomes driven by LC3. Rapamycin is known to act as an agonist of autophagy and was used as a positive control in autophagy-related assays. The mother liquor of rapamycin was prepared using DMSO as the solvent. The working concentration of rapamycin was set at 100 nM. The cells were transfected with MRFP-GFP-LC3 lentivirus vectors, generously provided by Hanbio in Shanghai, China. Subsequently, the cells underwent puromycin selection, fixation, X-Trton treatment, and DAPI staining. Imaging and observation were carried out using a Leica SP8 confocal microscope located in Wetzlar, Germany. In the merged model, the presence of yellow spots indicates autophagosomes that have not yet fused with lysosomes, while red fluorescent spots represent autophagosomes that have already merged with lysosomes and undergone acid degradation. We quantitatively assessed the average number of fluorescent dots per cell.
Cells were collected and immersed in a 2.5% glutaraldehyde solution and fixed overnight at 4 °C. After fixation, the cells were centrifuged, and the resulting pellet was washed three times with phosphoric acid buffer (pH 7.4). The cell precipitates were then suspended in a 1% agarose solution and further fixed with a 1% osmic acid solution shielded from light for 2 h. Subsequently, the samples were dehydrated through a series of alcohol gradients and were treated with varying proportions of acetone and an embedding agent. This process allowed the samples to be encased within a resin block. Finally, the resin-embedded samples were sectioned into slices with a thickness of 60–80 nm. These thin slices were supported on 150-mesh copper grids and stained with a 2% uranium acetate saturated alcohol solution. After staining, the copper mesh was cleaned by rinsing it with 70% alcohol and ultra-pure water. It was then stained using a 2.6% lead citrate solution and washed with ultra-pure water. The samples were dried at room temperature overnight and examined using an electron microscope (JEOL, Tokyo, Japan) operated by a special person.
In vivo studies
A lentiviral vector system was utilized to establish stable knockdown of CYFIP2 (shCYFIP2) and corresponding control (shCtrl) cell lines. The process involved mixing the shRNA plasmid with pMD2.G and psPAX2 helper plasmids, followed by transfection into HEK293T cells. After an 8-h incubation period, the medium was replaced, and the cells were incubated for an additional 48 h. The supernatant was then collected, centrifuged, and filtered to isolate the lentivirus, which was subsequently introduced into logarithmically-growing TE-1 cells. After an 8-h incubation, CYFIP2 gene expression was confirmed via RT-qPCR and western blot before proceeding with animal model studies.
To investigate the role of CYFIP2 in CDDP resistance in ESCC, we utilized a subcutaneous xenograft model. Balb/c nude mice, a kind of congenital athymic mouse model was acquired from Slac company (Shanghai, China) and officially commenced the experiment after a 7-day quarantine. Twenty male mice (8 weeks, 20 ± 2 g) were randomly assigned to four groups: two groups inoculated with shCtrl cells and two with shCYFIP2 cells. Among these, one group from each cell line was treated with CDDP. The cells were prepared as single-cell suspensions, and 1 × 10⁷ cells were subcutaneously inoculated into each mouse, with the inoculation day designated as Day 01. Tumor volumes were measured every 5 days during the study, calculated using the formula: volume = (width2 × length)/2, and tumor growth curves were generated.
Upon significant tumor formation (Day 20), CDDP (2 mg/kg) was administered every three days via intraperitoneal injection for 15 days, totaling six injections. After completing the drug treatment, the mice were euthanized using an intraperitoneal injection of 2% pentobarbital sodium at a dose of 35 mg/kg for anesthesia. Ten minutes later, their vital signs were re-examined, and euthanasia was confirmed once no vital signs were detected. Finally, tumor tissues were collected for weighing, followed by HE staining, immunohistochemical (IHC) staining, and TUNEL assays. Mice were also euthanized early if tumor volumes reached 2000 mm3 to prevent unnecessary suffering.
The mice were housed in a strictly controlled, pathogen-free environment (50 ± 10% relative humidity, 25 ± 1 °C), with five mice per cage, constituting a single experimental unit. To maintain consistency, all procedures were performed by the same individual following a standardized sequence. The study’s designers and data analysts were informed of the group assignments, while those involved in animal care and experimental procedures remained blinded. All animal experiments were conducted in accordance with the guidelines approved by the Institutional Animal Care and Use Committee of Fujian Medical University (Approval No. IACUC FJMU 2024-NSFC-Y-0339). And this work was presented according to the ARRIVE guidelines (2.0).
Statistical analysis
Statistical analysis was conducted and visualized using GraphPad Prism 9.5.1 (Dotmatics, USA). The mean and standard deviation (SD) were used to describe the characteristics of data conforming to a normal distribution. Statistical significance was determined using the unpaired Student’s t test or one-way analysis of variance (ANOVA), followed by Bonferroni’s multiple comparisons test, where appropriate. Three independent replicates were used for each set of experiments. A p value less than 0.05 (p value < 0.05) was considered statistically significant.
Data availability
The supplementary materials are contained in the online version. The sequencing metadata were uploaded to the SRA database (Accession NO. PRJNA1010500), and requests for other information should be addressed to M.K.
References
H. Sung, J. Ferlay, R.L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal, F. Bray, Global Cancer Statistics. GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 71(2021), 209–249 (2020).
Salem, M. E. et al. Comparative molecular analyses of esophageal squamous cell carcinoma, esophageal adenocarcinoma, and gastric adenocarcinoma. Oncologist 23, 1319–1327 (2018).
Integrated genomic characterization of oesophageal carcinoma. Nature 541, 169–175 (2017).
Smyth, E. C. et al. Oesophageal cancer. Nat Rev Dis Primers 3, 17048 (2017).
Mao, Y. S., Gao, S. G., Wang, Q., Shi, X. T., Li, Y., Gao, W. J., Guan, FD. S., Li, X. F., Han, Y. T., Liu, Y. Y., Liu, J. F., Zhang, K., Liu, S. Y., Fu, X. N., Fang, W. T., Chen, L. Q., Wu, Q. C. Xiao, G. M., Chen, K. N., Jiao, G. G., Luo, J. H., Mao, W. M., Rong, T. H., Fu, J. H., Tang, L. J., Chen, C., Xu, S. D., Guo, S. P., Yu, Z. T., Hu, J., Hu, Z. D., Sihoe, A., Yang, Y.K., Ding, N. N., Yang, D., Gao, Y. B. & He, J. Analysis of a registry database for esophageal cancer from high-volume centers in China. Dis. Esophagus, 33, 91 (2020).
Thrift, A. P. Global burden and epidemiology of Barrett oesophagus and oesophageal cancer. Nat Rev Gastroenterol Hepatol 18, 432–443 (2021).
He, S., Xu, J., Liu, X. & Zhen, Y. Advances and challenges in the treatment of esophageal cancer. Acta Pharm Sin B 11, 3379–3392 (2021).
Dasari, S. & Tchounwou, P. B. Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol 740, 364–378 (2014).
Tchounwou, P. B., Dasari, S., Noubissi, F. K., Ray, P. & Kumar, S. Advances in our understanding of the molecular mechanisms of action of cisplatin in cancer therapy. J Exp Pharmacol 13, 303–328 (2021).
Ying, W. Phenomic studies on diseases: Potential and challenges. Phenomics 3, 285–299 (2023).
Zhou, Y. et al. High-throughput screening of a CRISPR/Cas9 library for functional genomics in human cells. Nature 509, 487–491 (2014).
Li, W. et al. Deubiquitinase USP20 promotes breast cancer metastasis by stabilizing SNAI2. Genes Dev 34, 1310–1315 (2020).
Du, Y., Zhang, P., Liu, W. & Tian, J. Optical imaging of epigenetic modifications in cancer: A systematic review. Phenomics 2, 88–101 (2022).
Jiang, F. & Doudna, J. A. CRISPR-Cas9 structures and mechanisms. Annu Rev Biophys 46, 505–529 (2017).
Li, H. et al. CRISPR metabolic screen identifies ATM and KEAP1 as targetable genetic vulnerabilities in solid tumors. Proc Natl Acad Sci U S A 120, e2212072120 (2023).
Yang, H., Liu, B., Liu, D., Yang, Z., Zhang, S., Xu, P., Xing, Y., Kutschick, I., Pfeffer, S., Britzen-Laurent, N., Grützmann, R. & Pilarsky, C. Genome-wide CRISPR screening identifies DCK and CCNL1 as genes that contribute to gemcitabine resistance in pancreatic cancer. Cancers (Basel), 14, 3152 (2022).
Schenck, A., Bardoni, B., Moro, A., Bagni, C. & Mandel, J. L. A highly conserved protein family interacting with the fragile X mental retardation protein (FMRP) and displaying selective interactions with FMRP-related proteins FXR1P and FXR2P. Proc Natl Acad Sci U S A 98, 8844–8849 (2001).
Chaya, T. et al. Deficiency of the neurodevelopmental disorder-associated gene Cyfip2 alters the retinal ganglion cell properties and visual acuity. Hum Mol Genet 31, 535–547 (2022).
Nakashima, M. et al. De novo hotspot variants in CYFIP2 cause early-onset epileptic encephalopathy. Ann Neurol 83, 794–806 (2018).
Begemann, A. et al. New insights into the clinical and molecular spectrum of the novel CYFIP2-related neurodevelopmental disorder and impairment of the WRC-mediated actin dynamics. Genet Med 23, 543–554 (2021).
Manigandan, S. & Yun, J. W. Loss of cytoplasmic FMR1-interacting protein 2 (CYFIP2) induces browning in 3T3-L1 adipocytes via repression of GABA-BR and activation of mTORC1. J Cell Biochem 123, 863–877 (2022).
Mongroo, P. S. et al. IMP-1 displays cross-talk with K-Ras and modulates colon cancer cell survival through the novel proapoptotic protein CYFIP2. Cancer Res 71, 2172–2182 (2011).
Zhao, Z. et al. Analysis and experimental validation of rheumatoid arthritis innate immunity gene CYFIP2 and pan-cancer. Front Immunol 13, 954848 (2022).
Tong, J. et al. The downregulation of prognosis- and immune infiltration-related gene CYFIP2 serves as a novel target in ccRCC. Int J Gen Med 14, 6587–6599 (2021).
Shalem, O. et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87 (2014).
Yu, C. et al. Genome-wide CRISPR-cas9 knockout screening identifies GRB7 as a driver for MEK inhibitor resistance in KRAS mutant colon cancer. Oncogene 41, 191–203 (2022).
Tang, Z. et al. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res 45, W98-w102 (2017).
Győrffy, B. Integrated analysis of public datasets for the discovery and validation of survival-associated genes in solid tumors. Innovation (Camb) 5, 100625 (2024).
Li, L. et al. Integrative proteogenomic characterization of early esophageal cancer. Nature Communications 14, 1666 (2023).
Zhang, P. et al. BCLAF1 drives esophageal squamous cell carcinoma progression through regulation of YTHDF2-dependent SIX1 mRNA degradation. Cancer Lett 591, 216874 (2024).
Davenport, E. C. et al. Autism and schizophrenia-associated CYFIP1 regulates the balance of synaptic excitation and inhibition. Cell Rep 26, 2037-2051.e2036 (2019).
Tiwari, S. S. et al. Alzheimer-related decrease in CYFIP2 links amyloid production to tau hyperphosphorylation and memory loss. Brain 139, 2751–2765 (2016).
Jackson, R. S. 2nd., Cho, Y. J., Stein, S. & Liang, P. CYFIP2, a direct p53 target, is leptomycin-B sensitive. Cell Cycle 6, 95–103 (2007).
Cheng, A. S. et al. Helicobacter pylori causes epigenetic dysregulation of FOXD3 to promote gastric carcinogenesis. Gastroenterology 144, 122-133.e129 (2013).
Fu, S. et al. Identification of a novel Myc-regulated gene signature for patients with kidney renal clear cell carcinoma. J Oncol 2022, 3487859 (2022).
Vandamme, T. et al. Hotspot DAXX, PTCH2 and CYFIP2 mutations in pancreatic neuroendocrine neoplasms. Endocr Relat Cancer 26, 1–12 (2019).
Debnath, J., Gammoh, N. & Ryan, K. M. Autophagy and autophagy-related pathways in cancer. Nat Rev Mol Cell Biol 24, 560–575 (2023).
Wang, G. et al. Role of FLCN phosphorylation in insulin-mediated mTORC1 activation and tumorigenesis. Adv Sci (Weinh) 10, e2206826 (2023).
Li, Y. et al. Increased Drp1 promotes autophagy and ESCC progression by mtDNA stress mediated cGAS-STING pathway. J Exp Clin Cancer Res 41, 76 (2022).
Lu, Z. et al. Inhibiting autophagy enhances sulforaphane-induced apoptosis via targeting NRF2 in esophageal squamous cell carcinoma. Acta Pharm Sin B 11, 1246–1260 (2021).
Peña-Martinez, C., Rickman, A. D. & Heckmann, B. L. Beyond autophagy: LC3-associated phagocytosis and endocytosis. Sci Adv. 8, eabn1702 (2022).
Tremel, S. et al. Structural basis for VPS34 kinase activation by Rab1 and Rab5 on membranes. Nat Commun 12, 1564 (2021).
Winterbourn, C. C. Hydrogen peroxide reactivity and specificity in thiol-based cell signalling. Biochem Soc Trans 48, 745–754 (2020).
Glorieux, C., Liu, S., Trachootham, D. & Huang, P. Targeting ROS in cancer: Rationale and strategies. Nat Rev Drug Discov (2024).
Nakamura, H. & Takada, K. Reactive oxygen species in cancer: Current findings and future directions. Cancer Sci 112, 3945–3952 (2021).
Cui, Q. et al. Modulating ROS to overcome multidrug resistance in cancer. Drug Resist Updat 41, 1–25 (2018).
Mirzaei, S. et al. Elucidating role of reactive oxygen species (ROS) in cisplatin chemotherapy: A focus on molecular pathways and possible therapeutic strategies. Molecules 26, 2382 (2021).
Fan, H. et al. ROS generation attenuates the anti-cancer effect of CPX on cervical cancer cells by inducing autophagy and inhibiting glycophagy. Redox Biol 53, 102339 (2022).
Zhang, W., Tang, Y., Yang, P., Chen, Y., Xu, Z., Qi, C., Huang, H., Liu, R., Qin, H., Ke, H., Huang, C., Xu, F., Pang, P., Zhao, Z., Shan, H. & Xiao, F. TMX2 potentiates cell viability of hepatocellular carcinoma by promoting autophagy and mitophagy. Autophagy 1–18 (2024).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res 51, D587-d592 (2023).
Wang, B. et al. Integrative analysis of pooled CRISPR genetic screens using MAGeCKFlute. Nat Protoc 14, 756–780 (2019).
Acknowledgements
The authors would like to thank all the reviewers who participated in the review, as well as MJEditor (www.mjeditor.com), for providing English editing services during the preparation of this manuscript. We would like to extend our appreciation to SD Science Lab for their assistance in sample preparation, reagent preparation, animal feeding and analysis parameter adjustment.
Funding
This study was sponsored by the National Natural Science Foundation of China (Grant No. 82070499 & 82372680), Joint Funds for the Innovation of Science and Technology (Grant No. 2020Y9073) and Natural Science Foundation of Fujian Province (Grant No. 2023J01698).
Author information
Authors and Affiliations
Contributions
P.Z. wrote the first draft and collated most of the data. P.Z., W.Z. and J.J. finished the in vitro experiments together. P.Z. and Z.H. completed animal experiments. N.W. was involved in some of the charts and statistics. J.L. supported some experimental materials and instruction. M.K. designed and authorized this research. All authors reviewed and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, P., Zhang, W., Hong, Z. et al. Elucidating the role of CYFIP2 in conferring cisplatin resistance in esophageal squamous cell carcinoma. Sci Rep 14, 27130 (2024). https://doi.org/10.1038/s41598-024-77420-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-77420-8
