
Abstract
Cancer cells reprogram their energy metabolism pathways, but the mechanisms that enable them to meet their energy demands remain poorly understood. This study investigates the anticancer effects of ethyl p-methoxycinnamate (EMC) in Ehrlich ascites tumor cells (EATCs) and reveals that de novo fatty acid synthesis, rather than glycolysis, plays a pivotal role in sustaining energy homeostasis in cancer cells. EMC significantly reduced ATP levels despite enhancing glycolytic activity. It suppressed the expression of key enzymes involved in de novo fatty acid synthesis, including Acly, Acc1, and Fasn, resulting in decreased intracellular triglyceride (TG) levels. The addition of exogenous palmitic acid reversed EMC-induced ATP depletion and mitigated its anti-proliferative effects. Mechanistically, the ATP reduction caused by EMC was associated with inhibition of the c-Myc/SREBP1 pathway and arrest of the G1/S cell cycle transition. These findings demonstrate that EMC inhibits EATC proliferation by reducing ATP levels via suppression of de novo fatty acid synthesis. This study highlights the critical role of de novo fatty acid synthesis, rather than glycolysis, in maintaining energy homeostasis in cancer cells and provides novel insights into targeting cancer metabolism.
Similar content being viewed by others


Introduction
Cancer cells require substantial amounts of ATP to sustain their rapid proliferation and survival. When intracellular energy deficits are sensed at cell cycle checkpoints, cells may arrest the cell cycle or exit it altogether to prevent uncontrolled division and potential cell damage1. To meet the increased energy demands, cancer cells reprogram their metabolic pathways for glucose, fatty acids, and amino acids2. A fundamental feature of cancer metabolism is the Warburg effect, a phenomenon where cancer cells predominantly generate ATP through glycolysis rather than oxidative phosphorylation, even in oxygen-rich conditions. Initially observed by Warburg in Ehrlich ascites tumor cells (EATCs), this effect has since been demonstrated across various cancer cell types3,4,5. Based on the Warburg effect, it has been suggested that cancer cells rely on glycolysis for ATP production due to impaired mitochondrial function6,7.
However, recent studies have questioned this assumption, showing that mitochondria in cancer cells are not necessarily impaired and retain various functional capacities, including oxidative phosphorylation, even in highly glycolytic cancer cells8. Furthermore, recent research has reported that fatty acid metabolism plays an important role as the primary ATP source rather than glycolysis in some cancer cells9. Both de novo fatty acid synthesis and fatty acid oxidation are increasingly recognized as prominent features of cancer cell metabolism10. To meet fatty acid demand, dietary fatty acids are utilized in normal cells, but de novo fatty acid synthesis is preferred in cancer cells11,12. These findings underscore the complexity of cancer cell energy metabolism, as it involves coordinated reprogramming of multiple metabolic pathways, making a comprehensive understanding of cancer metabolism challenging and subject to ongoing research.
Natural compounds have garnered significant interest in cancer prevention for several decades due to their potent anticancer effects and low toxicity13. Compounds such as curcumin, resveratrol, and epigallocatechin gallate have been shown to inhibit the proliferation of cancers, including colorectal, liver, nasopharyngeal, breast, and pheochromocytoma, by modulating energy metabolism14,15. However, most studies have focused on the effects of these compounds on specific metabolic pathways, often without examining the broader interactions within energy metabolism. Kaempferia galanga L., a medicinal plant from the Zingiberaceae family, contains ethyl p-methoxycinnamate (EMC) as a primary active compound16,17. Our recent studies have demonstrated that Kaempferia galanga L. extract and EMC inhibit the proliferation of EATCs by suppressing the G1 to S phase transition through a reduction in the transcriptional activity of the oncogene c-Myc18. However, the effects of EMC on energy metabolism in EATCs remain unclear. Therefore, this study aimed to elucidate the anticancer mechanisms of EMC by investigating its influence on energy metabolism, focusing on glycolysis and fatty acid metabolism. This comprehensive investigation seeks to provide a deeper understanding of the interplay between metabolic pathways in cancer cells, using EATCs as a model to evaluate the impact of EMC on energy metabolism.
Results
Effect of EMC on Glycolysis and ATP levels in EATCs
In a previous study, we demonstrated that 100 µM EMC inhibits the proliferation of EATCs without cytotoxic effects on normal cells18. Based on these findings, a concentration of 100 µM EMC was employed in the present study. Considering the Warburg effect, EMC’s anticancer effect in EATCs may involve ATP depletion through glycolysis inhibition. To evaluate this hypothesis, we examined glucose consumption and lactate production in the culture medium of EMC-treated EATCs. The same medium samples were analyzed for both glucose consumption and lactate release. EATCs treated with EMC significantly increased glucose consumption in the culture medium (Fig. 1A). Lactate release levels were also significantly increased in EMC-treated EATCs (Fig. 1B). Conversely, intracellular ATP levels were significantly decreased following EMC treatment (Fig. 1C).
Effect of EMC on fatty acid synthesis and oxidation in EATCs
To investigate the mechanism by which EMC reduces ATP levels, we examined its effects on fatty acid synthesis and oxidation. Key genes involved in de novo fatty acid synthesis, including ATP citrate lyase (Acly), acetyl-CoA carboxylase alpha (Acc1), and fatty acid synthase (Fasn), which collectively convert citrate to PA, were analyzed. The mRNA expression levels of Acly, Acc1, and Fasn were significantly downregulated in EMC-treated EATCs 12 h after treatment (Fig. 2A, B and C). However, the mRNA levels of these fatty acid synthesis-related genes did not significantly change at 1–6 h after EMC treatment (Fig. 2D, E and F).
De novo synthesized fatty acids are typically incorporated into triglycerides (TGs), a primary component of lipid droplets used for energy storage in cancer cells19. We therefore measured intracellular TG levels and found a significant decrease in TG content in EMC-treated EATCs (Fig. 2G).
To assess the effect of EMC on fatty acid oxidation, we measured mRNA expression levels of carnitine palmitoyltransferase 1a (Cpt1a) and carnitine palmitoyltransferase 1b (Cpt1b), enzymes essential for fatty acid oxidation. No significant changes were observed in Cpt1a and Cpt1b mRNA expression levels following EMC treatment (Fig. 2H and I).
Effect of co-addition of EMC and palmitic acid (PA) on ATP levels in EATCs
To determine if the reduction in ATP levels caused by EMC is related to its inhibition of de novo fatty acid synthesis, exogenous PA was added to the culture medium to restore the intracellular fatty acid levels decreased by the inhibition of de novo synthesis, and ATP levels were subsequently measured. EMC treatment significantly decreased ATP levels; however, this reduction was effectively reversed by the addition of exogenous PA (Fig. 3). There was no significant difference in ATP levels between the control group and the palmitic acid alone group.
Effect of EMC on ATP levels and DNA content in EATCs
Cellular energy shortage is detected by cell cycle checkpoints, which can result in cell cycle arrest1. We previously demonstrated that EMC inhibits the G1 to S phase transition in EATCs18. Based on this, we examined the relationship between EMC-induced ATP reduction and the inhibition of G1/S phase transition. An increase in DNA content over time indicates that EATCs have progressed from the G1 to S phase. EATCs were treated with or without EMC and analyzed at 1, 6, 12, and 24 h. Up to 6 h after EMC treatment, there was no significant difference in DNA content between the groups (Fig. 4A). However, at time points beyond 12 h, the control group exhibited an increase in DNA content, whereas this increase was suppressed in the EMC-treated EATCs. Similarly, ATP levels showed no significant difference up to 6 h after EMC treatment but were significantly reduced in the EMC-treated group after 12 h (Fig. 4B). These results indicate that EMC-induced ATP depletion coincides with the inhibition of the G1 to S phase transition.
Effect of co-addition of EMC and PA on cell number and viability of EATCs
To investigate whether the anti-proliferative effect of EMC on cancer cells is related to intracellular fatty acid depletion, we supplemented the culture medium with exogenous PA and evaluated its effect on cell number and viability in EATCs. EMC treatment significantly reduced cell number; however, this reduction was reversed by the addition of exogenous PA (Fig. 5A). This result indicate that fatty acid depletion is a contributing factor in EMC-induced suppression of EATC proliferation. In contrast, no significant differences in cell viability were observed across the treatment groups, indicating that apoptosis was not induced in any of the groups (Fig. 5B).
Effect of EMC on sterol regulatory element binding transcription factor 1 (SREBP1) expression levels in EATCs
To further elucidate the mechanism behind EMC-induced inhibition of de novo fatty acid synthesis, we examined the expression of SREBP1, a key regulator of genes involved in fatty acid and triglyceride biosynthesis20. SREBP1 directly regulates the expression of target genes including Acly, Acc1, and Fasn21. Our results showed that EMC treatment significantly reduced SREBP1 mRNA levels in EATCs (Fig. 6A).
SREBP1 protein is synthesized as a precursor (pSREBP1) and initially resides in the endoplasmic reticulum. Upon transport to the Golgi apparatus, pSREBP1 undergoes cleavage by Site-1 and Site-2 proteases to generate the mature, active form (mSREBP1), which then translocates to the nucleus to function as a transcription factor22. As shown in Fig. 6B, both pSREBP1 and mSREBP1 protein levels were significantly decreased in EMC-treated EATCs.
Effect of EMC on the levels of phosphorylated c-Myc at Serine 62 in EATCs
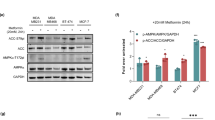
The oncogene c-Myc directly binds to the promoter region of SREBP1, thereby activating its transcriptional expression23. Phosphorylation of Ser62 on c-Myc enhances its transcriptional activity24. In a previous study, we showed that EMC treatment suppresses phosphorylation at serine 62 of c-Myc in EATCs after 12 h18. In this study, we examined the effect of EMC on c-Myc phosphorylation at serine 62 at an earlier time point to gain further insights into the mechanisms underlying EMC’s anticancer effects via c-Myc regulation. As shown in Fig. 7A and B, EMC did not affect the total expression levels of c-Myc but significantly reduced the levels of serine 62-phosphorylated c-Myc. These results indicated that EMC significantly decreased the levels of serine 62-phosphorylated c-Myc compared to the total c-Myc expression levels (Fig. 7C).
Discussion
Cancer cells are characterized by their ability to reprogram energy metabolism to sustain uncontrolled proliferation, leading to distinct metabolic traits compared to normal cells25. Despite significant advances in understanding ATP-generating pathways in cancer biology, the mechanisms underlying ATP reduction in cancer cells remain poorly understood. In this study, we provide a novel perspective on cancer metabolism by demonstrating that EMC suppresses ATP production via the inhibition of de novo fatty acid synthesis, which is accompanied by enhanced glycolytic activity. Notably, the ATP reduction was significantly reversed by exogenous supplementation with PA, highlighting the critical role of de novo fatty acid synthesis in maintaining energy homeostasis in cancer cells. These findings suggest that targeting de novo fatty acid synthesis could be a promising therapeutic strategy for cancer treatment.
ATP is produced from carbon substrates via two distinct metabolic pathways: glycolysis and mitochondrial oxidative phosphorylation. Glycolysis has long been regarded as the primary ATP-generating pathway in cancer cells7. However, our results revealed a paradoxical increase in glycolytic activity, evidenced by enhanced glucose uptake and lactate production, despite EMC-induced ATP depletion. This finding is particularly significant, as it indicates that ATP reduction is not caused by glycolytic inhibition. While mitochondrial dysfunction has been implicated in various metabolic shifts in cancer cells, emerging evidence, including our study, challenges this assumption. Specifically, synthetic phosphoethanolamine has been shown to induce apoptosis via mitochondrial membrane potential loss in EATCs26, reinforcing the importance of mitochondria as a therapeutic target. Taken together, our findings emphasize the complementary roles of glycolysis and mitochondrial oxidative phosphorylation in sustaining the high ATP demands of cancer cells.
A key finding of this study is that EMC-induced ATP depletion primarily results from the inhibition of de novo fatty acid synthesis. Our previous study has demonstrated that EMC did not affect the mitochondrial membrane potential and mitochondrial DNA copy number in EATCs18, suggesting that there is no inhibition of mitochondrial ATP synthesis activity. This finding further supports the idea that EMC-induced ATP depletion is unrelated to mitochondrial dysfunction, suggesting the involvement of other energy production processes. Fatty acids serve as one of the energy substrates for ATP production in mitochondria. There are distinct differences in fatty acid metabolism between normal and cancer cells. While normal cells utilize exogenous fatty acids, cancer cells prefer endogenous fatty acids to sustain their uncontrolled proliferation12,27. In human cancer tissues, Acly, Acc1, and Fasn are overexpressed compared to normal tissues28, and de novo synthesized fatty acids are efficiently oxidized and utilized as substrates for ATP production in cancer cells29,30. In this study, EMC did not significantly affect the expression levels of Cpt1a and Cpt1b, the rate-limiting enzymes of fatty acid oxidation. However, EMC significantly reduced the expression of key enzymes involved in de novo fatty acid synthesis, including Acly, Acc1, and Fasn. Furthermore, intracellular triglyceride levels were also reduced. The restoration of ATP levels by PA supplementation further supports the notion that de novo fatty acid synthesis is essential for ATP production in cancer cells. Additionally, the expression levels of Acly, Acc1, and Fasn remained unchanged at 1 and 6 h after EMC treatment but were significantly downregulated at 12 h. There was no temporal relationship between the expression of these fatty acid synthesis-related genes and glucose consumption. Therefore, it was suggested that fluctuations in glucose consumption are not involved in the downregulation of fatty acid synthesis-related gene expression. This mechanistic insight highlights the unique vulnerability of cancer cells to disruptions in fatty acid metabolism.
Cancer cells are known for their ability to adapt to energy stress by reprogramming metabolic pathways. For example, inhibition of mitochondrial oxidative phosphorylation often leads to enhanced glycolysis31. Similarly, our study demonstrates for the first time that EMC-induced inhibition of de novo fatty acid synthesis triggers a compensatory upregulation of glycolysis, potentially as a survival mechanism. This adaptive response likely contributes to cancer cell survival under metabolic stress and is recognized as a key mechanism underlying resistance to metabolism-targeting therapies32. While EMC inhibits EATC proliferation, its failure to induce cell death may be attributed to this metabolic plasticity. These findings underscore the potential of combining de novo fatty acid synthesis and glycolysis inhibitors as a more effective strategy for suppressing cancer cell growth.
This study also provides important evidence linking ATP depletion to cell cycle arrest at the G1/S phase transition. Previous reports indicate that ATP levels peak during the transition from G1 to S phase and that compounds such as gramicidin A inhibit this transition by depleting ATP33,34. Our results show that EMC-induced ATP reduction coincides with G1/S phase arrest and that the suppression of EATC proliferation by EMC is reversed by PA supplementation. These findings suggest that ATP depletion caused by the inhibition of de novo fatty acid synthesis disrupts energy-dependent cell cycle progression, thereby suppressing EATC proliferation. This mechanistic link between fatty acid metabolism, ATP production, and cell cycle regulation highlights a critical vulnerability that can be exploited for therapeutic purposes.
Schcolnik-Cabrera et al. have shown that orlistat is an appropriate positive control for FASN inhibition and demonstrated that orlistat induces apoptosis in colon cancer cells by regulating fatty acid metabolism35,36. In contrast, our results showed that EMC suppressed cancer cell proliferation without inducing apoptosis. Given this difference and the absence of a suitable positive control that aligns with the mechanism of action of EMC, we were unable to include a FASN inhibitor as a positive control in this study. In future studies, establishing an appropriate positive control will be crucial for further elucidating the specific effects of EMC.
Another novel finding of this study is the identification of SREBP1 and c-Myc as key regulators of EMC-induced inhibition of de novo fatty acid synthesis. SREBP1 serves as a master regulator of de novo fatty acid biosynthesis, while c-Myc functions as its transcriptional regulator20,23. SREBP1 was significantly downregulated at both the mRNA and protein levels following EMC treatment. Additionally, EMC inhibited c-Myc phosphorylation at Ser62. Inhibition of Ser62 phosphorylation reduces c-Myc’s binding affinity for the promoter regions of target genes, leading to a decrease in transcriptional activity37,38. These findings reveal a mechanism by which EMC disrupts fatty acid metabolism through the inactivation of the c-Myc/SREBP1 axis. Further studies are required to elucidate how EMC inhibits c-Myc phosphorylation and to explore its broader implications in cancer biology.
In conclusion, this study demonstrates that EMC suppresses EATC proliferation by reducing ATP levels through the inhibition of de novo fatty acid synthesis, rather than glycolysis. Furthermore, this ATP depletion is linked to the inactivation of the c-Myc/SREBP1 pathway and disruption of the G1/S phase transition. These findings provide critical new insights into the metabolic vulnerabilities of cancer cells and underscore the therapeutic potential of targeting de novo fatty acid synthesis in combination with glycolysis inhibition. To fully understand the impact of EMC on cancer cell metabolism, future studies should explore its effects on other pathways, such as glutamine metabolism, the pentose phosphate pathway, and the TCA cycle.
Materials and methods
Cell culture
EATCs (JCRB9090), spontaneously derived cancer cells collected from mouse ascites fluid, were obtained from the Japanese Cancer Research Resource Bank (Tokyo, Japan). EATCs were grown in Dulbecco’s modified Eagle’s medium (DMEM; 05915, Nissui Pharmaceutical, Tokyo, Japan) at 37 °C under humidified conditions with 5% CO2. DMEM contained 10% fetal bovine serum (FBS; S1810, Biowest, Nuaille, France) and 0.1% streptomycin/penicillin (02008/00906, Meiji Seika Kaisha, Tokyo, Japan). In all experiments, EATCs were pre-incubated at a density of 400 cells/φ 91-mm glass petri dish (FS-90 A, FLAT Co.,Ltd, Chiba, Japan) in 10 ml of culture medium for 48 h. Subsequently, EATCs were adjusted to a concentration of 1.0 × 10⁶ cells/mL and treated with or without EMC (M1204, Tokyo Kasei Kogyo Co., Tokyo, Japan) and palmitic acid (PA; 165 − 00102, FUJIFILM Wako Pure Chemical, Osaka, Japan). EMC was dissolved in dimethyl sulfoxide (048-21985, FUJIFILM Wako Pure Chemical). PA was dissolved in a 10% bovine serum albumin solution free of fatty acids in phosphate-buffered saline (PBS), heated at 37 °C for 30 min, and subsequently underwent sterilizing filtration (0.22 μm).
Extraction of protein and measurement of protein levels
After the pre-incubation described above, EATCs at a concentration of 1 × 10⁶ cells/ml were seeded at 2 × 10⁶ cells/φ 48-mm glass petri dish (P-2, FLAT Co.,Ltd) or 3.2 × 10⁶ cells /φ 64-mm glass petri dish (FS-60, FLAT Co.,Ltd). EATCs were rinsed twice with PBS and lysed in radioimmunoprecipitation (RIPA) buffer containing 20 µM leupeptin, 1 mM phenylmethylsulfonyl fluoride, and 15 µM pepstatin as protease inhibitors, as well as 5 mM NaF and 1 mM Na₃VO₄ as phosphatase inhibitors. The cell lysates were incubated on ice for 30 min, and intracellular proteins were extracted by freeze-thawing with liquid nitrogen. The total protein concentration in the extract was determined using a Pierce BCA Protein Assay Kit (23227, Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s protocol. Briefly, 25 µl of the sample was mixed with 200 µl of working reagent and incubated at 37 °C for 30 min. After incubation, the absorbance was measured at 550 nm using a microplate reader (Wallac ARVO SX 1420 multilabel counter; PerkinElmer, Waltham, MA, USA).
Measurement of glucose consumption
Following the pre-incubation described above, EATCs at a concentration of 1 × 10⁶ cells/ml were seeded at 2 × 10⁶ cells/φ 48-mm glass petri dish. After cultivating EATCs with or without EMC, the culture media were collected. Glucose consumption in the medium was measured using the Glucose Assay Kit-WST (G264, Dojindo, Kumamoto, Japan) following the manufacturer’s protocol. Absorbance at 450 nm, reflecting glucose consumption, was determined using a Wallac ARVO SX 1420 multilabel counter. Glucose consumption was calculated by subtracting the post-culturing glucose concentration from the pre-culturing glucose concentration. Glucose consumption between groups was normalized to intracellular total protein levels.
Measurement of lactate release levels
EATCs at a concentration of 1 × 10⁶ cells/ml were seeded at 2 × 10⁶ cells/φ 48-mm glass petri dish subsequent to the pre-incubation described above. After cultivating EATCs with or without EMC, the culture media were collected. Lactate release levels in the medium were measured using the Lactate Assay Kit-WST (L256, Dojindo) following the manufacturer’s protocol. Absorbance at 450 nm, reflecting lactate release levels, was determined using a Wallac ARVO SX 1420 multilabel counter. Lactate release levels between groups were normalized to intracellular total protein levels.
Measurement of ATP levels
After the pre-incubation described above, EATCs at a concentration of 1 × 10⁶ cells/ml were seeded at 2 × 10⁶ cells/φ 48-mm glass petri dish. ATP levels were measured using the CellTiter-Glo 2.0 Assay (Promega, Madison, WI, USA) following the manufacturer’s protocol. Briefly, after culturing EATCs with or without EMC and PA, the cells were collected. The cells suspension was mixed with an equal volume of CellTiter-Glo 2.0 Reagent in a 96-well white plate (204012, Porvair Sciences, Norfolk, UK). After incubating at room temperature for 10 min, luminescence was measured using the Infinite F200 PRO microplate reader (Tecan Group Ltd., Männedorf, Switzerland). ATP levels were normalized to intracellular total protein levels between groups.
RNA extraction, synthesis of cDNA and quantitative reverse transcription PCR (qRT-PCR)
Following the pre-incubation described above, EATCs at a concentration of 1 × 10⁶ cells/ml were seeded at 2 × 10⁶ cells/φ 48-mm glass petri dish. EATCs were processed for total RNA extraction using the FastGene RNA Basic Kit (FG-80050, Nippon Genetics, Tokyo, Japan) following the manufacturer’s protocol. The quality and quantity of the extracted RNA were assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). cDNA was synthesized by reverse transcription of RNA using the ReverTra Ace qPCR RT Master Mix with gDNA Remover (FSQ-301, Toyobo, Osaka, Japan). qRT-PCR analysis was carried out using the GeneAce SYBR qPCR Mix α Low ROX (316–07693, Nippon gene, Tokyo, Japan) and a Stratagene Mx3005P instrument (Agilent Technologies). The β-actin gene was used as a reference gene to normalize mRNA expression levels. The mRNA levels were determined using relative quantification (2−ΔΔCt). The Primer-BLAST tool (http://www.ncbi.nlm.nih.gov/tools/primer-blast/) was utilized to design all primers (Table 1). The accuracy of the qRT-PCR products was verified through melting curve analysis.
Measurement of triglyceride (TG) levels
EATCs at a concentration of 1 × 10⁶ cells/ml were seeded at 2 × 10⁶ cells/φ 48-mm glass petri dish subsequent to the pre-incubation described above. After EATCs were rinsed twice with PBS, they were lysed in RIPA buffer and kept on ice for 30 min. Cell lysates were subjected to freeze-thaw cycles in liquid nitrogen, followed by centrifugation to extract intracellular TG. Intracellular TG levels were measured using the LabAssay Triglyceride (632-50991, FUJIFILM Wako Pure Chemical) following the manufacturer’s protocol. Absorbance at 600 nm, reflecting TG levels, was determined using a Wallac ARVO SX 1420 multilabel counter. Intracellular TG levels in each group were normalized to intracellular protein.
Measurement of DNA content
After the pre-incubation described above, EATCs at a concentration of 1 × 10⁶ cells/ml were seeded at 2 × 10⁶ cells/φ 48-mm glass petri dish. The DNA content of EATCs was quantified using diphenylamine, as previously described39. EATCs were rinsed twice with PBS and lysed in 300 µl of 0.4 N perchloric acid. After centrifugation, the precipitate was resuspended in 700 µl of 0.4 N perchloric acid and heated at 70 °C for 20 min. The supernatant was diluted 4-fold with 0.4 N perchloric acid, and 0.4 ml of the diluted sample was mixed with 0.1 ml of ultrapure water and 1 ml of the DNA reaction solution. The DNA reaction solution consisted of 980.3 µl of 88.6 mM diphenylamine in acetic acid, 14.7 µl of sulfuric acid, and 5 µl of 405.3 mM acetaldehyde in ultrapure water. The mixture was incubated overnight at 36 °C, and absorbance was measured at 600 nm using a microplate reader.
Trypan blue assay
Following the pre-incubation described above, EATCs at a concentration of 1 × 10⁶ cells/mL were seeded at 1 × 10⁵ cells per well in a 96-well microplate and incubated for 24 h with or without EMC and PA. After cultivation, an equal volume of 0.4% trypan blue solution was added to the cell suspension. The numbers of viable and non-viable cells were then counted using a hemocytometer and a microscope. Cell viability was calculated using the following formula: cell viability (%) = [(total cells – non-viable cells) / total cells] × 100.
Western blot analysis
After the pre-incubation, EATCs at a concentration of 1 × 10⁶ cells/ml were seeded at 3.2 × 10⁶ cells /φ 58-mm glass petri dish. Protein extraction and quantification were performed as described above. SDS-PAGE was employed to separate proteins using an AE-8155 myPowerII 500 (ATTO, Tokyo, Japan), and the proteins were subsequently transferred to a PVDF membrane (IPFL00010; Merck Millipore, Billerica, MA, USA). The membranes were blocked at room temperature for 1 h using skim milk dissolved in Tris-buffered saline containing Tween 20. Subsequently, the primary antibody incubation was performed at room temperature for 2 h. The primary antibodies used were anti-SREBP1 (sc-13551, 1:600, Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-c-Myc (sc-40, 1:1000, Santa Cruz Biotechnology), anti-phospho-c-Myc (Ser62) (#13748, 1:1000, Cell Signaling Technology, Danvers, MA, USA), and anti-GAPDH (#5174, 1:15000, Cell Signaling Technology). Membranes were incubated with Goat anti-Mouse IgG (H + L) Secondary Antibody, Biotin (#31800, 1:2000, Thermo Fisher Scientific) or Anti-rabbit IgG (H + L), Biotinylated Antibody (#14708, 1:2000, Cell Signaling Technology) at room temperature for 1 h, followed by exposure to horseradish peroxidase-conjugated streptavidin (P0397, Agilent Technologies) for an additional hour. Chemiluminescent bands of the proteins were detected using EzWestLumi Plus (2332638, ATTO) and quantified with ImageJ.
Statistical analysis
All numerical data are expressed as mean ± standard deviation (SD). Comparisons among the groups were performed by one-way ANOVA, followed by the Tukey-Kramer test in Statcel-4 (OMS Inc., Tokorozawa, Japan). Comparisons between two groups were evaluated for statistical significance by Student’s t-test. Differences were considered statistically significant at p < 0.05.

Effect of EMC on glycolysis and ATP levels in EATCs. EATCs were treated with or without 100 µM EMC for various time points. (A) The glucose consumption levels in the medium were determined by the Glucose Assay Kit-WST. Results are expressed as mean ± SD (n = 4). (B) The lactate release levels in the medium were determined by the Lactate Assay Kit-WST. Results are expressed as mean ± SD (n = 4). (C) ATP levels were determined by the CellTiter-Glo 2.0 Assay. Results are expressed as mean ± SD (n = 5). Statistical significance between the two groups was evaluated using Student’s t-test. The data were found to be significantly different compared to the control: ** p < 0.01.



Effect of EMC on fatty acid synthesis and oxidation in EATCs. (A-C, H, I) EATCs were treated with or without 100 µM EMC for 12 h. (D-F) EATCs were treated with or without 100 µM EMC for various time points. mRNA levels of genes related to de novo fatty acid synthesis (Acly, Acc1, and Fasn) and oxidation (Cpt1a and Cpt1b) determined by using qRT-PCR. Results are expressed as mean ± SD (n = 3). (G) Intracellular TG levels were determined by LabAssay Triglyceride. Results are expressed as mean ± SD (n = 4). Statistical significance between the two groups was evaluated using Student’s t-test. The data were found to be significantly different compared to the control: ** p < 0.01.

Effect of co-addition of EMC and PA on ATP levels in EATCs. EATCs were treated with 100 µM EMC, 40 µM palmitic acid (PA), a combination of both, or left untreated for 12 h. ATP levels were determined by the CellTiter-Glo 2.0 Assay. Results are expressed as mean ± SD (n = 10). Statistical significance between the four groups was evaluated using one-way ANOVA, followed by the Tukey-Kramer test. The data were found to be significantly different: * p < 0.05 and ** p < 0.01.

Effect of EMC on ATP levels and DNA content in EATCs. EATCs were treated with or without 100 µM EMC for various times. (A) DNA content was determined using diphenylamine. Results are expressed as mean ± SD (n = 4). (B) ATP levels were determined by the CellTiter-Glo 2.0 Assay. Results are expressed as mean ± SD (n = 3). Statistical significance between the two groups was evaluated using Student’s t-test. The data were found to be significantly different comparison to the control: ** p < 0.01.

Effect of co-addition of EMC and PA on cell number and viability of EATCs. EATCs were treated with 100 µM EMC, 40 µM palmitic acid (PA), a combination of both, or left untreated for 24 h. (A) Cell number and (B) cell viability were determined using trypan blue. Results are expressed as mean ± SD (n = 5). Statistical significance between the four groups was evaluated using one-way ANOVA, followed by the Tukey-Kramer test. The data were found to be significantly different comparison to the control: ** p < 0.01.

Effect of EMC on sterol regulatory element binding transcription factor 1 (SREBP1) expression levels in EATCs. EATCs were treated with or without 100 µM EMC for 12 h. (A) SREBP1 mRNA expression levels were determined by qRT-PCR. Results are expressed as mean ± SD (n = 3). (B) SREBP1 protein expression levels were determined by Western blot analysis. The images of pSREBP1 and mSREBP1 were obtained from the same blot with different exposure times. After detecting SREBP1, the membrane was stripped of the SREBP1 antibody and reprobed with GAPDH antibody to obtain the GAPDH bands. Results are expressed as mean ± SD (n = 4). Statistical significance between the two groups was evaluated using Student’s t-test. The data were found to be significantly different comparison to the control: ** p < 0.01.

Effect of EMC on the levels of phosphorylated c-Myc at Ser-62 in EATCs. EATCs were treated with or without 100 µM EMC for 6 h. The levels of (A) c-Myc and (B) phosphorylated c-Myc at Ser-62 were determined by Western blot analysis. The images of c-Myc and GAPDH were obtained from the same blot with different exposure times. After detecting c-Myc, the membrane was stripped of the c-Myc antibody and reprobed with GAPDH antibody to obtain the GAPDH bands. The images of the phosphorylated c-Myc at Ser-62 and GAPDH were obtained from the same blot but with different exposure times. (C) Results show the ratio of phosphorylated c-Myc at Ser-62 to c-Myc. Results are expressed as mean ± SD (n = 4). Statistical significance between the two groups was evaluated using Student’s t-test. The data were found to be significantly different comparison to the control: ** p < 0.01.
Data availability
The datasets used and/or analyzed during the current study available from the corresponding author on reasonable request.
References
Salazar-Roa, M. & Malumbres, M. Fueling the cell division cycle. Trends Cell. Biol. 27, 69–81. https://doi.org/10.1016/j.tcb.2016.08.009 (2017).
Xu, X. et al. Metabolic reprogramming and epigenetic modifications in cancer: from the impacts and mechanisms to the treatment potential. Exp. Mol. Med. 55, 1357–1370. https://doi.org/10.1038/s12276-023-01020-1 (2023).
Warburg, O. On the origin of cancer cells. Science 123, 309–314. https://doi.org/10.1126/science.123.3191.309 (1956).
Warburg, O., Wind, F. & Negelein, E. Ueber Den Stoffwechsel von tumoren Im Körper. Klin. Wochenschr. 5, 829–832. https://doi.org/10.1007/BF01726240 (1926).
Gogvadze, V., Zhivotovsky, B. & Orrenius, S. The Warburg effect and mitochondrial stability in cancer cells. Mol. Aspects Med. 31, 60–74. https://doi.org/10.1016/j.mam.2009.12.004 (2010).
Pelicano, H., Martin, D., Xu, R., Huang, P. & & Glycolysis Inhibition for anticancer treatment. Oncogene 25, 4633–4646. https://doi.org/10.1038/sj.onc.1209597 (2006).
Ganapathy-Kanniappan, S. & Geschwind, J. F. H. Tumor Glycolysis as a target for cancer therapy: progress and prospects. Mol. Cancer. 12, 1–11. https://doi.org/10.1186/1476-4598-12-152 (2013).
Zu, X. L. & Guppy, M. Cancer metabolism: facts, fantasy, and fiction. Biochem. Biophys. Res. Commun. 313, 459–465. https://doi.org/10.1016/j.bbrc.2003.11.136 (2004).
Lee, H., Woo, S. M., Jang, H., Kang, M. & Kim, S. Y. Cancer depends on fatty acids for ATP production: A possible link between cancer and obesity. Semin Cancer Biol. 86 (Pt2), 347–357. https://doi.org/10.1016/j.semcancer.2022.07.005 (2022).
Butler, L. M. et al. Lipids and cancer: emerging roles in pathogenesis, diagnosis and therapeutic intervention. Adv. Drug Deliv Rev. 159, 245–293. https://doi.org/10.1016/j.addr.2020.07.013 (2020).
Medes, G., Thomas, A. & Weinhouse, S. Metabolism of neoplastic tissue. IV. A study of lipid synthesis in neoplastic tissue slices in vitro. Cancer Res. 13, 27–29 (1953).
Menendez, J. A. & Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer. 7, 763–777. https://doi.org/10.1038/nrc2222 (2007).
Ma, X. & Wang, Z. Anticancer drug discovery in the future: an evolutionary perspective. Drug Discov. Today, 14, 1136–1142. https://doi.org/10.1016/j.drudis.2009.09.006 (2009).
Dias, A. S., Helguero, L., Almeida, C. R. & Duarte, I. F. Natural compounds as metabolic modulators of the tumor microenvironment. Molecules 26, 3494. https://doi.org/10.3390/molecules26123494 (2021).
Wang, T., Ma, F. & Qian, H.-l. Defueling the cancer: ATP synthase as an emerging target in cancer therapy. Mol. Ther. -Oncolytics. 23, 82–95. https://doi.org/10.1016/j.omto.2021.08.015 (2021).
Hikmawanti, N. P. E. & Dwita, L. P. Chemical component of Kencur (kaemferia Galanga L.) ethanolic extract using gas chromatography-mass spectrometry. IOP Conf. Ser. Earth Environ. Sci. 819, 012057. https://doi.org/10.1088/1755-1315/819/1/012057 (2021).
He, Z. H., Yue, G. G. L., Lau, C. B. S., Ge, W. & But, P. P. H. Antiangiogenic effects and mechanisms of trans-ethyl p-methoxycinnamate from Kaempferia galanga L. J. Agric. Food Chem. 60, 11309–11317. https://doi.org/10.1021/jf304169j (2012).
Sasaki, Y. et al. Kaempferia galanga L. extract and its main component, Ethyl p-methoxycinnamate, inhibit the proliferation of Ehrlich Ascites tumor cells by suppressing TFAM expression. Heliyon 9, e17588. https://doi.org/10.1016/j.heliyon.2023.e17588 (2023).
Munir, R., Lisec, J., Swinnen, J. V. & Zaidi, N. Lipid metabolism in cancer cells under metabolic stress. Br. J. Cancer. 120, 1090–1098. https://doi.org/10.1038/s41416-019-0451-4 (2019).
Su, F. & Koeberle, A. Regulation and targeting of SREBP-1 in hepatocellular carcinoma. Cancer Metastasis Rev. 43, 673–708. https://doi.org/10.1007/s10555-023-10156-5 (2024).
Snaebjornsson, M. T., Janaki-Raman, S. & Schulze, A. Greasing the wheels of the cancer machine: the role of lipid metabolism in cancer. Cell. Metab. 31, 62–76. https://doi.org/10.1016/j.cmet.2019.11.010 (2020).
Edwards, P. A., Tabor, D., Kast, H. R. & Venkateswaran, A. Regulation of gene expression by SREBP and SCAP. Biochim. Biophys. Acta Mol. Cell. Biol. Lipids. 1529, 103–113. https://doi.org/10.1016/s1388-1981(00)00140-2 (2000).
Chen, J. et al. ACSL4 reprograms fatty acid metabolism in hepatocellular carcinoma via c-Myc/SREBP1 pathway. Cancer Lett. 502, 154–165. https://doi.org/10.1016/j.canlet.2020.12.019 (2021).
Seth, A., Alvarez, E., Gupta, S. & Davis, R. J. A phosphorylation site located in the NH2-terminal domain of c-Myc increases transactivation of gene expression. J. Biol. Chem. 266, 23521–23524. https://doi.org/10.1016/S0021-9258(18)54312-X (1991).
Schiliro, C. & Firestein, B. L. Mechanisms of metabolic reprogramming in Cancer cells supporting enhanced growth and proliferation. Cells 10 (5), 1056. https://doi.org/10.3390/cells10051056 (2021).
Ferreira, A. K. et al. Anticancer effects of synthetic phosphoethanolamine on Ehrlich Ascites tumor: an experimental study. Anticancer Res. 32, 95–104 (2012).
Swinnen, J. V., Brusselmans, K. & Verhoeven, G. Increased lipogenesis in cancer cells: new players, novel targets. Curr. Opin. Clin. Nutr. Metab. Care. 9, 358–365. https://doi.org/10.1097/01.mco.0000232894.28674.30 (2006).
Furuta, E., Okuda, H., Kobayashi, A. & Watabe, K. Metabolic genes in cancer: their roles in tumor progression and clinical implications. Biochim. Biophys. Acta. 1805, 141–152. https://doi.org/10.1016/j.bbcan.2010.01.005 (2010).
Zaytseva, Y. Y. et al. Increased expression of fatty acid synthase provides a survival advantage to colorectal cancer cells via upregulation of cellular respiration. Oncotarget 6, 18891–18904. https://doi.org/10.18632/oncotarget.3783 (2015).
Pike, L. S., Smift, A. L., Croteau, N. J., Ferrick, D. A. & Wu, M. Inhibition of fatty acid oxidation by etomoxir impairs NADPH production and increases reactive oxygen species resulting in ATP depletion and cell death in human glioblastoma cells. Biochim. Biophys. Acta. 1807, 726–734. https://doi.org/10.1016/j.bbabio.2010.10.022 (2011).
Hao, W., Chang, C. P., Tsao, C. C. & Xu, J. Oligomycin-induced bioenergetic adaptation in cancer cells with heterogeneous bioenergetic organization. J. Biol. Chem. 285, 12647–12654. https://doi.org/10.1074/jbc.M109.084194 (2010).
Navarro, C. et al. Metabolic Reprogramming in Cancer Cells: Emerging Molecular Mechanisms and Novel Therapeutic Approaches. Pharmaceutics 14, 1303. (2022). https://doi.org/10.3390/pharmaceutics14061303
Mitra, K., Wunder, C., Roysam, B., Lin, G. & Lippincott-Schwartz, J. A hyperfused mitochondrial state achieved at G1-S regulates Cyclin E buildup and entry into S phase. Proc. Natl. Acad. Sci. U S A. 106, 11960–11965. https://doi.org/10.1073/pnas.0904875106 (2009).
Xue, Y. W., Itoh, H., Dan, S., Inoue, M. & Gramicidin A accumulates in mitochondria, reduces ATP levels, induces mitophagy, and inhibits cancer cell growth. Chem. Sci. 13, 7482–7491. https://doi.org/10.1039/d2sc02024f (2022).
Schcolnik-Cabrera, A. et al. Pharmacological Inhibition of tumor anabolism and host catabolism as a cancer therapy. Sci. Rep. 11 (1), 5222. https://doi.org/10.1038/s41598-021-84538-6 (2021).
Schcolnik-Cabrera, A. et al. A combination of inhibitors of glycolysis, Glutaminolysis and de Novo fatty acid synthesis decrease the expression of chemokines in human colon cancer cells. Oncol. Lett. 18, 6909–6916. https://doi.org/10.3892/ol.2019.11008 (2019).
Janghorban, M. et al. Targeting c-MYC by antagonizing PP2A inhibitors in breast cancer. Proc. Natl. Acad. Sci. U S A. 111, 9157–9162. https://doi.org/10.1073/pnas.1317630111 (2014).
Benassi, B. et al. c-Myc phosphorylation is required for cellular response to oxidative stress. Mol. Cell. 21, 509–519. https://doi.org/10.1016/j.molcel.2006.01.009 (2006).
Burton, K. [105] determination of DNA concentration with diphenylamine. Methods Enzymol. 12, 163–166. https://doi.org/10.1016/0076-6879(67)12127-7 (1968).
Funding
This work was supported by JSPS KAKENHI (Grant Number 20K11626).
Author information
Authors and Affiliations
Contributions
Y. S., T. N., I .M-Y. and A. K-Y. designed the research; A. K-Y. conducted the research; R. F. and L. L. provided essential materials; Y. S., T. N., I. M-Y. and A. K-Y analyzed data; Y. S., I. M-Y. and A. K-Y wrote the manuscript; A. K-Y had primary responsibility for final content; and all authors have provided important intellectual input and have read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sasaki, Y., Mizushima, N., Norikura, T. et al. Ethyl p-methoxycinnamate inhibits tumor growth by suppressing of fatty acid synthesis and depleting ATP. Sci Rep 15, 15317 (2025). https://doi.org/10.1038/s41598-025-00131-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-00131-1
