
Abstract
The dysfunction of HDACs is closely related to tumorigenesis and development, which has emerged as an attractive target for cancer therapy. In this study, a series of thiazole-containing hydroxamate derivatives were designed and synthesized as novel HDAC inhibitors. Among these inhibitors, compounds 15a and 15d showed excellent inhibitory activities against HDAC1 and HepG2 cancer cell line, these two compounds increased the levels of acetylated histone H3 and H4. Moreover, 15a and 15d significantly arrested HepG2 cells at the G0/G1 phase. Additionally, these two compounds could induce apoptosis and pyroptosis. Moreover, 15a exhibited significant antitumor activity in the HepG2 xenograft model. Molecular docking and molecular dynamics simulation studies revealed the possible interaction mode of compound 15a with HDAC1. Besides, the preliminary pharmacokinetics study of compound 15a in vivo was evaluated. These results suggested that these novel thiazole-based HDAC inhibitors might become a promising scaffold for further structural optimization.
Similar content being viewed by others

Introduction
Cancer is a complex life-threatening disease characterized by high fatality and morbidity rates1. Diverse epigenetic abnormalities contributed to the initiation and progression of cancers2. Histone acetylation is a critical epigenetic modification that regulates chromatin structure and gene expression. This dynamic modification is precisely balanced by the opposing activities of histone acetyltransferases (HATs) and histone deacetylases (HDACs)3,4. HATs facilitate the accumulation of acetylated histones, inducing chromatin relaxation and transcriptional activation. In contrast, HDACs facilitate the removal of acetyl groups from lysine residues, causing core histones to tightly associate with DNA through electrostatic interactions. This binding obstructs protein complexes from accessing promoter regions, thereby mediating gene repression5,6. The overexpression of HDACs has been observed in numerous cancers and is linked to the down-regulation of tumor suppressor genes7,8.
Based on structural features and catalytic mechanisms, the 18 known human HDAC isoforms were classified into four classes: class I (HDACs 1,2,3 and 8), class II (HDACs 4,5,6,7,9 and 10), class IV (HDACs 11), and class III (sirtuins 1–7). Class I, class II, and class IV are Zn2+-dependent HDACs, while class III are NAD+ -dependent sirtuins9,10. Aberrant overexpression of HDACs is frequently observed in diverse human malignancies and is mechanistically linked to tumorigenesis and cancer progression. Pharmacological inhibition of HDACs induces multifaceted antitumor effects, including cell cycle arrest, differentiation, apoptosis, autophagy, and immunomodulation.11. Recent studies further revealed that inhibition of HDAC can trigger pyroptosis-a lytic and inflammatory form of programmed cell death mediated by gasdermin protein pore formation, which bridges epigenetic regulation with antitumor12,13. Consequently, HDACs have become important therapeutic targets for cancer treatment14.
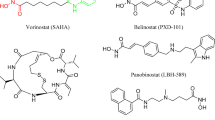
HDAC inhibitors (HDACIs) are categorized into five classes according to their chemical structure in clinical studies: hydroxamic acid, short-chain fatty acids, benzamides, epoxy ketones, and macrocyclic peptides15. To date, four HDAC-targeting drugs have been approved by FDA for treatment of hematological malignancies. Vorinostat (SAHA) and Romidepsin (FK228) have been approved for treatment of cutaneous T-cell lymphoma (CTCL), Belinostat (PDX-101) has been approved for treatment of peripheral T-cell lymphoma (PTCL), and Panobinostat (LBH-589) has been approved for multiple myeloma (MM). In 2015, Chinese Food and Drug Administration has approved Chidamide (CS055) for the treatment of PTCL16,17. Moreover, more than 20 HDAC inhibitors are now being developed as drug candidates against various cancers under different stages of clinical trials, such as Quisinostat (JNJ-26481585), Nanatinostat (CHR-3996) and Mocetinostat (MGCD-0103) (Fig. 1)18,19,20. Although these HDACIs demonstrate high efficacy in hematological malignancy, their therapeutic effects in solid tumors remain limited with reports of multiple side effects such as gastrointestinal effects, myelosuppression, QT interval prolonged, and cardiotoxicity21,22. Then there is still a need to develop novel and effective HDACIs for treating solid tumors while alleviating toxicity and expanding the therapeutic window.

FDA approved and clinical HDAC inhibitors.
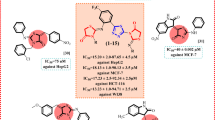
Despite the huge structural diversity, the HDACIs have a general pharmacophore model: zinc binding group (ZBG) such as hydroxamic acid and 2-amino benzamide which interacting with Zn2+ at the bottom of active site; a hydrophobic linker occupies the narrow tunnel of HDACs, connecting the ZBG to the cap group; a surface recognition group (CAP), which is essential for recognizing and interacting with residues on the rim of active site of HDACs23. In our continuing efforts to develop new HDACIs, we explored series of hydroxamate-based bis-substituted aromatic amide HDACIs, mainly contains different five-membered heterocyclic rings in the CAP region, such as thiophene or oxadiazole (1a–c), and these compounds displayed potent antiproliferative and antimetastatic activities, however with poor pharmacokinetic properties (oral bioavailability of 1b is 2.6%)24,25,26,27. To develop novel bis-substituted aromatic amide HDACIs with enhanced pharmacokinetic properties and therapeutic efficacy, we incorporated a thiazole moiety into the CAP region. Thiazole has many medicinal values and its derivatives have a wide range of biological activities, such as antioxidant, analgesic, antibacterial, anticancer, antiallergic, antihypertensive, anti-inflammatory, antimalarial, antifungal, and antipsychotic agents28,29,30,31. Moreover, several HDACIs have been shown to contain thiazole ring32,33,34. Then we introduced substituted phenylthiazole structure to the CAP region, and a series of compounds 8a–l and 15a–h were designed and synthesized, further modification on methoxyl group of 8a was made to obtain compounds 18a–m. (Fig. 2) In this study, we designed and synthesized a series of N-disubstituted hydroxamic acid derivatives incorporating a phenyl-thiazole scaffold in the CAP region, and systematically evaluated their biological activities.

Design strategy and general structures of the target compounds.
Experimental section
All methods were performed in accordance with the relevant guidelines and regulations.
Chemistry-general procedure for the synthesis of target compounds
N1-hydroxy-N7-(4-methoxyphenyl)-N7-((4-phenyl-2-thiazolyl)methyl) heptanediamide(8a).
Step-a A solution of p-anisidine (4.32 g, 35 mmol) was dissolved in 8 mL of DMF, K2CO3 (4.92 g, 35 mmol) and bromoacetonitrile (1.68 mL, 23.3 mmol) were added, and the reaction mixture was allowed to stir at room temperature for 6 h. The resulting mixture was adjusted to a pH = 6–7 with a 4.0 M aqueous solution of HCl. Then the reaction was extracted, dried over sodium sulfate, filtered and concentrated in vacuo to afford 2 (3.12 g, 82.5%).
Step-b Intermediate 2 (3.12 g, 19.23 mmol) and pimelic acid anhydride (3.28 g, 23.08 mmol) were combined in 1,4-dioxane (15.00 mL) and heated at reflux for 4 h. The reaction was cooled to room temperature, and then concentrated in vacuo to get intermediate 3 (4.34 g, 74.1%).
Step-c 3–4 drops of thionyl chloride were added to a mixture of intermediate 3 in MeOH (40 mL) at room temperature, and the reaction mixture was allowed to be heated at reflux for 5 h. Then the reaction mixture was concentrated in vacuo to obtain crude product 4, which was directly used in the next reaction step.
Step-d Ammonium sulfide (1.22 mL, 17.48 mmol) and triethylamine (1.25 mL, 8.92 mmol) were added to a mixture of compound 4 (1.42 g, 4.46 mmol) in pyridine (15.00 mL), and the reaction was heated at 50 °C for 5 h. The reaction was concentrated under reduced pressure, and then purified by chromatographic column (eluting with EA followed by 20:1 CH2Cl2/MeOH followed by 10:1 CH2Cl3/MeOH) to give the target intermediate 5 (1.15 g. 73.4%).
Step-e A solution of compound 5 (1.15 g, 3.27 mmol) and 2-chloroacetophenone (3.93 mmol) were dissolved in EtOH and heated to reflux at 80 °C with stirring for 4 h. Then the mixture was concentrated under reduced pressure and purified by chromatographic column to give intermediate 7 (0.83 g, 56%).
Step-f KOH (5.13 g, 91.5 mmol) was added to a mixture of hydroxylamine hydrochloride (6.35 g, 91.5 mmol) in methanol (20.00 mL), and the reaction was stirred at 40 °C for 30 min. The reaction mixture was cooled to room temperature and filtered. Intermediate 7 was combined into the filtrate, followed by the addition of KOH (0.51 g, 9.15 mmol), and the reaction was stirred at room temperature for 3 h. The reaction system was concentrated under reduced pressure and extracted with saturated NH4Cl and EtOAc. The organic layer was then washed with brine, dried over Na2SO4, and concentrated, and then the residue was purified by column chromatography (eluting with EA followed by 20:1 CH2Cl2/MeOH followed by 10:1 CH2Cl2/MeOH) to give compound 8a27 (0.64 g, 76.8%). 1H NMR (600 MHz, DMSO-d6) δ 10.32 (br s, 1H), 8.66 (br s, 1H), 8.04 (s, 1H), 7.89 (d, J = 7.2 Hz, 2H), 7.41 (dd, J = 7.2, 7.2 Hz, 2H), 7.32 (dd, J = 7.2, 7.2 Hz, 1H), 7.23 (d, J = 8.4 Hz, 2H), 6.96 (d, J = 7.8 Hz, 2H), 5.10 (s, 2H), 3.74 (s, 3H), 2.06 (t, J = 7.2 Hz, 2H), 1.89 (t, J = 7.8 Hz, 2H), 1.50 – 1.45 (m, 2H), 1.42 – 1.37 (m, 2H), 1.17 – 1.12 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ 172.34, 169.00, 167.21, 158.60, 153.45, 134.75, 133.99, 129.21, 128.79, 127.99, 125.93, 114.93, 114.76, 55.32, 50.71, 33.10, 32.13, 28.16, 24.93, 24.58. HRMS (ESI): calcd for [C24H27N3O4S + H]+ 454.1801, found 454.1792. HPLC purity: 99.57%, tR = 14.4 min.
The structure of our target compound has been unequivocally confirmed through its synthetic route and comprehensive analysis of NMR and mass spectrometry data—the presence of all characteristic signals verifies its structural integrity. For 1H NMR, the two broad singlets at the low field (δ ~ 10–11 ppm) correspond to the two protons of the hydroxamic acid group (–CONHOH). The singlet at δ ≈ 5.0 ppm is assigned to the methylene protons (–CH2–) adjacent to the thiazole ring, while the high-field signals (δ ≈ 1.0–2.5 ppm) originate from the five methylene protons (–CH2–) in the linker region. If the compound exhibits the aforementioned characteristic proton signals (1H NMR), combined with concordant carbon spectrum (13C NMR) and mass spectrometry (MS) data, its structure can be confidently confirmed.
Compounds 8b–l were prepared according to the procedure described for the preparation of compound 8a.
N1-hydroxy-N7-(4-methoxyphenyl)-N7-((4-(2-tolyl)-2-thiazolyl)methyl) heptanediamide(8b) (25 mg, 81.2% yield). 1H NMR (600 MHz, DMSO-d6) δ 11.96 (br s, 1H), 7.69 (br s, 1H), 7.51 (d, J = 7.2 Hz, 1H), 7.26 – 7.22 (m, 3H), 7.21 (d, J = 8.4 Hz, 2H), 6.96 (d, J = 9.0 Hz, 2H), 5.08 (s, 2H), 3.75 (s, 3H), 2.33 (s, 3H), 2.13 (t, J = 7.2 Hz, 2H), 2.06 (t, J = 7.2 Hz, 2H), 1.50 – 1.45 (m, 2H), 1.41 – 1.36 (m, 2H), 1.19 – 1.14 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ 174.39, 172.24, 165.67, 158.60, 153.75, 135.53, 134.74, 134.15, 130.79, 129.48, 129.21, 127.94, 125.86, 117.81, 114.73, 55.34, 50.48, 33.52, 33.08, 28.10, 24.57, 24.27, 20.90. HRMS (ESI) calcd for [C25 H29N3O4S + H]+ 468.1957, found 468.1951. HPLC purity: 99.87%, tR = 14.5 min.
N1-hydroxy-N7-(4-methoxyphenyl)-N7-((4-(3-tolyl)-2-thiazolyl)methyl)heptanediamide(8c) (43 mg, 73.7% yield). 1H NMR (600 MHz, DMSO-d6) δ 10.30 (br s, 1H), 8.64 (br s, 1H), 8.01 (s, 1H), 7.71 (s, 1H), 7.67 (d, J = 7.8 Hz, 1H), 7.29 (dd, J = 7.8, 7.8 Hz, 1H), 7.23 (d, J = 9.0 Hz, 2H), 7.13 (d, J = 7.2 Hz, 1H), 6.97 (d, J = 9.0 Hz, 2H), 5.09 (s, 2H), 3.75 (s, 3H), 2.34 (s, 3H), 2.06 (t, J = 7.8 Hz, 2H), 1.88 (t, J = 7.2 Hz, 2H), 1.50 – 1.45 (m, 2H), 1.41 – 1.36 (m, 2H), 1.16 – 1.12 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ 172.34, 168.97, 167.09, 158.60, 153.55, 137.90, 134.72, 133.92, 129.22, 128.68, 128.63, 126.51, 123.10, 114.76, 55.32, 50.69, 33.10, 32.12, 28.15, 24.91, 24.58, 21.08. HRMS (ESI) calcd for [C25H29N3O4S + H]+ 468.1957, found 468.1953. HPLC purity: 99.54%, tR = 15.4 min.
N1-hydroxy-N7-(4-methoxyphenyl)-N7-((4-(4-tolyl)-2-thiazolyl)methyl)heptanediamide(8d) (23 mg, 83.5% yield). 1H NMR (600 MHz, DMSO-d6) δ 10.31 (br s, 1H), 8.65 (br s, 1H), 7.96 (s, 1H), 7.77 (d, J = 8.4 Hz, 2H), 7.24 – 7.21 (m, 4H), 6.96 (d, J = 8.4 Hz, 2H), 5.08 (s, 2H), 3.75 (s, 3H), 2.31 (s, 3H), 2.06 (t, J = 7.2 Hz, 2H), 1.88 (t, J = 7.2 Hz, 2H), 1.49 – 1.44 (m, 2H), 1.41 – 1.36 (m, 2H), 1.16 – 1.11 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ 172.33, 168.99, 167.06, 158.60, 153.54, 137.33, 134.76, 131.35, 129.35, 129.22, 125.86, 114.76, 114.02, 55.33, 50.71, 33.10, 32.12, 28.15, 24.92, 24.58, 20.82. HRMS (ESI) calcd for [C25H29N3O4S + H]+ 468.1957, found 468.1954. HPLC purity: 99.76%, tR = 15.4 min.
N1-hydroxy-N7-(4-methoxyphenyl)-N7-((4-(4-methoxyphenyl)-2-thiazolyl)methyl) heptanediamide(8e) (50 mg, 74.3% yield) 1H NMR (600 MHz, DMSO-d6) δ 10.31 (br s, 1H), 8.65 (br s, 1H), 7.87 (s, 1H), 7.81 (d, J = 8.4 Hz, 2H), 7.23 (d, J = 9.0 Hz, 2H),6.97 (d, J = 2.4 Hz, 2H), 6.96 (d, J = 2.4 Hz, 2H), 5.08 (s, 2H), 3.78 (s, 3H), 3.75 (s, 3H), 2.06 (t, J = 7.2 Hz, 2H), 1.88 (t, J = 7.2 Hz, 2H), 1.49 – 1.44 (m, 2H), 1.41 – 1.36 (m, 2H), 1.16 – 1.11 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ 172.32, 168.99, 166.97, 159.10, 158.60, 153.37, 134.78, 129.23, 127.29, 126.86, 114.76, 114.14, 112.82, 55.33, 55.18, 50.69, 33.10, 32.13, 28.15, 24.92, 24.59. HRMS (ESI) calcd for [C25H29N3O5S + H]+ 484.1906, found 484.1897. HPLC purity: 99.87%, tR = 14.5 min.
N1-((4-(2-fluorophenyl)-2-thiazolyl)methyl)-N7-hydroxy-N1-(4-methoxyphenyl) heptanediamide(8f.) (36 mg, 68.9% yield) 1H NMR (600 MHz, DMSO-d6) δ 10.31 (br s, 1H), 8.66 (br s, 1H), 8.02 (dd, J = 7.2,7.2 Hz, 1H), 7.92 (s, 1H), 7.40—7.36 (m, 1H), 7.33 – 7.26 (m, 2H), 7.23 (d, J = 9.0 Hz, 2H), 6.97 (d, J = 9.0 Hz, 2H), 5.10 (s, 2H), 3.75 (s, 3H), 2.06 (t, J = 7.2 Hz, 2H), 1.88 (t, J = 7.2 Hz, 2H), 1.50 – 1.45 (m, 2H), 1.41 – 1.36 (m, 2H), 1.16 – 1.11 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ 172.41, 169.02, 166.75, 160.20, 158.64, 158.55, 146.97 (d, J = 2.3 Hz), 134.76, 129.76 (d, J = 8.6 Hz), 129.59 (d, J = 3.0 Hz), 129.21, 124.87, 124.85, 121.63 (d, J = 11.6 Hz), 119.25 (d, J = 13.5 Hz), 116.19 (d, J = 22.1 Hz), 114.80, 55.34, 50.68, 33.11, 32.14, 28.16, 24.94, 24.59. HRMS (ESI): calcd for [C24H26FN3O4S + H]+ 472.1706, found 472.1698. HPLC purity: 99.45%, tR = 14.6 min.
N1-((4-(3-fluorophenyl)-2-thiazolyl)methyl)-N7-hydroxy-N1-(4-methoxyphenyl) heptanediamide(8 g) (61 mg, 76.1% yield) 1H NMR (600 MHz, DMSO-d6) δ 10.34 (br s, 1H), 8.69 (br s, 1H), 8.14 (s, 1H), 7.73 (d, J = 7.8 Hz, 1H), 7.66 (d, J = 10.2 Hz, 1H), 7.48 – 7.43 (m, 1H), 7.22 (d, J = 9.0 Hz, 2H), 7.16 – 7.13 (m, 1H), 6.96 (d, J = 9.0 Hz, 2H), 5.08 (s, 2H), 3.74 (s, 3H), 2.05 (t, J = 7.2 Hz, 2H), 1.87 (t, J = 7.2 Hz, 2H), 1.48 – 1.43 (m, 2H), 1.40 – 1.35 (m, 2H), 1.16 – 1.10 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ 172.65, 169.29, 167.68, 162.71 (d, J = 241.65 Hz), 158.77, 152.24 (d, J = 3.0 Hz), 136.43 (d, J = 8.6 Hz), 134.84, 131.07 (d, J = 8.9 Hz), 129.35, 122.14, 116.53, 114.87 (d, J = 14.0 Hz), 112.59 (d, J = 23.0 Hz), 55.49, 50.84, 33.23, 32.25, 28.26, 25.04, 24.71. HRMS (ESI): calcd for [C24H26FN3O4S + H]+ 472.1706, found 472.1705. HPLC purity: 99.59%, tR = 14.8 min.
N1-((4-(4-fluorophenyl)-2-thiazolyl)methyl)-N7-hydroxy-N1-(4-methoxyphenyl) heptanediamide(8 h) (45 mg, 72.5% yield) 1H NMR (600 MHz, DMSO-d6) δ 10.30 (br s, 1H), 8.64 (br s, 1H), 8.03 (s, 1H), 7.93 (dd, J = 7.2, 7.2 Hz, 2H), 7.26 – 7.22 (m, 4H), 6.97 (d, J = 8.4 Hz, 2H), 5.09 (s, 2H), 3.75 (s, 3H), 2.06 (t, J = 7.2 Hz, 2H), 1.88 (t, J = 7.2 Hz, 2H), 1.49 – 1.44 (m, 2H), 1.41 – 1.36 (m, 2H), 1.16 – 1.11 (m, 2H).13C NMR (150 MHz, DMSO-d6) δ 172.34, 168.98, 167.36, 161.05, 158.60, 152.40, 134.74, 130.61, 129.21, 127.98 (d, J = 8.2 Hz), 115.73, 115.59, 114.77, 55.33, 50.67, 33.09, 32.12, 28.14, 24.91, 24.57. HRMS (ESI): calcd for [C24H26FN3O4S + H]+ 472.1706, found 472.1704. HPLC purity: 99.36%, tR = 14.6 min.
N1-((4-(2-chlorophenyl)-2-thiazolyl)methyl)-N7-hydroxy-N1-(4-methoxyphenyl) heptanediamide(8i) (56 mg, 73.4% yield) 1H NMR (600 MHz, DMSO-d6) δ 10.30 (br s, 1H), 8.64 (br s, 1H), 8.03 (s, 1H), 7.78 (dd, J = 4.8, 4.8 Hz, 1H), 7.54 (dd, J = 4.8, 4.8 Hz, 1H), 7.42 – 7.36 (m, 2H), 7.23 (d, J = 9.0 Hz, 2H), 6.97 (d, J = 9.0 Hz, 2H), 5.09 (s, 2H), 3.76 (s, 3H), 2.06 (t, J = 7.2 Hz, 2H), 1.88 (t, J = 7.2 Hz, 2H), 1.49 – 1.44 (m, 2H), 1.41 – 1.36 (m, 2H), 1.16 – 1.11 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ 172.32, 168.97, 166.11, 158.61, 149.98, 134.72, 132.76, 131.39, 130.93, 130.34, 129.55, 129.22, 127.38, 119.78, 114.76, 55.34, 50.50, 33.10, 32.12, 28.15, 24.91, 24.57. HRMS (ESI): calcd for [C24H26ClN3O4S + H]+ 488.1411, found 488.1406. HPLC purity: 97.26%, tR = 15.5 min.
N1-((4-(3-chlorophenyl)-2-thiazolyl)methyl)-N7-hydroxy-N1-(4-methoxyphenyl) heptanediamide(8j) (54 mg, 65.6% yield) 1H NMR (600 MHz, DMSO-d6) δ 10.31 (br s, 1H), 8.65 (br s, 1H), 8.19 (s, 1H), 7.93 (s, 1H), 7.86 (d, J = 7.8 Hz, 1H), 7.44 (dd, J = 7.8, 7.8 Hz, 1H), 7.38 (d, J = 8.4 Hz, 1H), 7.22 (d, J = 9.0 Hz, 2H), 6.96 (d, J = 8.4 Hz, 2H), 5.09 (s, 2H), 3.75 (s, 3H), 2.06 (t, J = 7.2 Hz, 2H), 1.88 (t, J = 7.2 Hz, 2H), 1.49 – 1.44 (m, 2H), 1.41 – 1.36 (m, 2H), 1.16 – 1.11 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ 172.44, 169.05, 167.61, 158.65, 151.83, 136.00, 134.73, 133.67, 130.78, 129.23, 127.79, 125.56, 124.52, 116.53, 114.81, 55.36, 50.70, 33.12, 32.15, 28.18, 24.94, 24.60. HRMS (ESI): calcd for [C24H26ClN3O4S + H]+ 488.1411, found 488.1404. HPLC purity: 98.64%, tR = 15.8 min.
N1-((4-(4-chlorophenyl)-2-thiazolyl)methyl)-N7-hydroxy-N1-(4-methoxyphenyl) heptanediamide(8 k) (75 mg, 69.3% yield) 1H NMR (600 MHz, DMSO-d6) δ 10.30 (br s, 1H), 8.64 (br s, 1H), 8.12 (s, 1H), 7.85 (d, J = 8.4 Hz, 2H), 7.61 (d, J = 8.4 Hz, 2H), 7.23 (d, J = 8.4 Hz, 2H), 6.96 (d, J = 8.4 Hz, 2H), 5.09 (s, 2H), 3.75 (s, 3H), 2.06 (t, J = 7.2 Hz, 2H), 1.88 (t, J = 7.2 Hz, 2H), 1.49 – 1.44 (m, 2H), 1.41 – 1.36 (m, 2H), 1.16 – 1.11 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ 172.35, 168.96, 167.53, 158.60, 152.21, 134.71, 133.18, 131.73, 129.20, 127.94, 121.09, 115.80, 114.77, 55.32, 50.67, 33.08, 32.11, 28.13, 24.90, 24.55. HRMS (ESI): calcd for [C24H26ClN3O4S + H]+ 488.1411, found 488.1409. HPLC purity: 99.47%, tR = 16.0 min.
N1-((4-(4-bromophenyl)-2-thiazolyl)methyl)-N7-hydroxy-N1-(4-methoxyphenyl) heptanediamide(8l) (46 mg, 71.3% yield) 1H NMR (600 MHz, DMSO-d6) δ 10.31 (br s, 1H), 8.65 (br s, 1H), 8.11 (s, 1H), 7.91 (d, J = 8.4 Hz, 2H), 7.47 (d, J = 9.0 Hz, 2H), 7.23 (d, J = 9.0 Hz, 2H), 6.97 (d, J = 9.0 Hz, 2H), 5.09 (s, 2H), 3.75 (s, 3H), 2.06 (t, J = 7.2 Hz, 2H), 1.88 (t, J = 7.2 Hz, 2H), 1.49 – 1.44 (m, 2H), 1.41 – 1.36 (m, 2H), 1.16 – 1.11 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ 172.36, 168.99, 167.52, 158.61, 152.17, 134.72, 132.85, 132.48, 129.21, 128.83, 127.65, 115.72, 114.78, 55.33, 50.69, 33.09, 32.13, 28.14, 24.91, 24.57. HRMS (ESI): calcd for [C24H26BrN3O4S + H]+ 534.0885, found 534.0885. HPLC purity: 98.70%, tR = 15.7 min.
General Procedure for the synthesis of 15a–15n
N1-hydroxy-N7-(4-methoxyphenyl)-N7-((2-phenyl-4-thiazolyl)methyl)heptanediamide(15a)Step-g: A solution of the benzonitrile (2.06 g, 20 mmol) was dissolved in pyridine (10.00 mL), then ammonium sulfide (1.5 mL, 22 mmol) and triethylamine (3.10 mL, 22 mmol) were added, which was heated and stirred at 50 °C for 5 h. The reaction was concentrated in vacuo to give a residue that was purified by silica gel chromatography (eluent: CH2Cl2/MeOH 20:1 to 10:1) to give 10 (2.26 g, 82.3%).
Step-h: Compound 10 was added to a mixture of 1,3-dichloroacetone (3.13 g, 24.69 mmol) in EtOH and the mixture was allowed to be stirred at reflux for 4 h. The reaction was concentrated in vacuo, and then purified by chromatographic column to give the target intermediate 11 (2.56 g, 74.77%).
Step-i: K2CO3 (1.51 g, 10.95 mmol) was slowly added to a mixture of p-anisidine (2.27 g, 10.95 mmol) in N, N-dimethylformamide (20.00 mL), the mixture was stirred at room temperature for 6 h. Then the compound 11 (2.56 g, 74.77%) was added to the mixture for 6 h at room temperature. The resulting mixture was adjusted to a pH = 6–7 with a 4.0 M aqueous solution of HCl. Then the reaction was extracted, dried over sodium sulfate, filtered and concentrated in vacuo to intermediate 12 (3.02 g, 82.8%).
Step-j: Intermediate 12 (10.18 mmol) and pimelic acid anhydride (2.17 g, 15.27 mmol) were dissolved in 20 mL of 1,4-dioxane, heated to reflux at 110 °C for 4 h, then extracted and dried to give intermediate 13 (3.12 g, 70.1%).
Step-k: To a mixture of intermediate 13 was dissolved in MeOH (30.00 mL), then 3 ~ 4 drops of thionyl chloride were added dropwise. The reaction system was heated at 80 °C to reflux for 5 h, and then concentrated in vacuo to obtain crude product 14, which was directly used in the next reaction step.
Step-l: KOH (7.01 g, 125 mmol) was slowly added to a mixture of hydroxylamine hydrochloride (0.68 g, 125 mmol) in methanol (20 mL), and the mixture was stirred at 40 °C for 30 min, then the reaction product was cooled under an ice bath and filtered. Intermediate 14 was dissolved with the filtrate, followed by the addition of KOH (0.70 g, 912.5 mmol), and the reaction was stirred for 3 h at room temperature. The reaction system was concentrated under reduced pressure and extracted with saturated NH4Cl and EtOAc. The organic layer was then washed with brine, dried over Na2SO4, and concentrated, and then the residue was purified by column chromatography (eluting with EA followed by 20:1 CH2Cl2/MeOH followed by 10:1 CH2Cl2/MeOH) to give compound 15a(0.87 mg, 76.9%).1H NMR (600 MHz, DMSO-d6) δ 10.31 (br s, 1H), 8.65 (br s, 1H), 7.88 (d, J = 7.8 Hz, 2H), 7.50 – 7.46 (m, 4H), 7.23 (d, J = 9.0 Hz, 2H), 6.93 (d, J = 9.0 Hz, 2H), 4.92 (s, 2H), 3.74 (s, 3H), 2.04 (t, J = 7.2 Hz, 2H), 1.88 (t, J = 7.2 Hz, 2H), 1.49 – 1.44 (m, 2H), 1.41 – 1.36 (m, 2H), 1.15 – 1.10 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ 171.94, 169.06, 166.71, 158.36, 153.76, 135.15, 133.04, 130.23, 129.43, 129.27, 126.04, 116.66, 114.55, 55.30, 48.89, 33.32, 32.16, 28.22, 24.96, 24.65. HRMS (ESI): calcd for [C24H27N3O4S + H]+ 454.1801, found 454.1800. HPLC purity: 97.77%, tR = 14.7 min.
Compounds 15b–n were prepared according to the procedure described for the preparation of compound15a.
N1-hydroxy-N7-(4-methoxyphenyl)-N7-((2-(2-tolyl)-4-thiazolyl)methyl)heptanediamide(15b) (59 mg, 71.7% yield). 1H NMR (600 MHz, DMSO-d6) δ 10.30 (br s, 1H), 8.65 (br s, 1H), 7.67 (d, J = 7.8 Hz, 1H), 7.51 (s, 1H), 7.37 – 7.34 (m, 2H), 7.30 (dd, J = 7.2,7.2 Hz, 1H), 7.21 (d, J = 8.4 Hz, 2H), 6.93 (d, J = 8.4 Hz, 2H), 4.93 (s, 2H), 3.74 (s, 3H), 2.48 (s, 3H), 2.03 (t, J = 7.2 Hz, 2H), 1.87 (t, J = 7.2 Hz, 2H), 1.48 – 1.43 (m, 2H), 1.40 – 1.35 (m, 2H), 1.14 – 1.09 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ 171.83, 169.03, 166.43, 158.35, 153.07, 135.78, 135.15, 132.35, 131.57, 129.51, 129.45, 129.39, 126.38, 117.20, 114.54, 55.29, 48.80, 33.33, 32.14, 28.23, 24.95, 24.65, 21.18. HRMS (ESI): calcd for [C25H29N3O4S + H]+ 468.1957, found 468.1949. HPLC purity: 94.18%, tR = 15.3 min.
N1-hydroxy-N7-(4-methoxyphenyl)-N7-((2-(3-tolyl)-4-thiazolyl)methyl)heptanediamide(15c) (101 mg, 73.6% yield). 1H NMR (600 MHz, DMSO-d6) δ 10.31 (br s, 1H), 8.65 (br s, 1H), 7.69 (s, 1H), 7.67 (d, J = 7.8 Hz, 1H), 7.44 (s, 1H), 7.36 (dd, J = 7.8,7.8 Hz, 1H), 7.28 (d, J = 7.8 Hz, 1H), 7.23 (d, J = 9.0 Hz, 2H), 6.93 (d, J = 9.0 Hz, 2H), 4.92 (s, 2H), 3.74 (s, 3H), 2.36 (s, 3H), 2.04 (t, J = 7.2 Hz, 2H), 1.88 (t, J = 7.2 Hz, 2H), 1.49 – 1.44 (m, 2H), 1.41 – 1.36 (m, 2H), 1.15 – 1.10 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ 171.92, 169.03, 166.83, 158.35, 153.66, 138.60, 135.12, 133.00, 130.87, 129.42, 129.15, 126.43, 123.26, 116.50, 114.53, 55.28, 48.86, 33.31, 32.15, 28.22, 24.95, 24.64, 20.90. HRMS (ESI): calcd for [C25H29N3O4S + H]+ 468.1957, found 468.1949. HPLC purity: 98.45%, tR = 15.7 min.
N1-hydroxy-N7-(4-methoxyphenyl)-N7-((2-(4-tolyl)-4-thiazolyl)methyl)heptanediamide(15d) (268 mg, 69.4% yield) 1H NMR (600 MHz, DMSO-d6) δ 10.30 (br s, 1H), 8.65 (br s, 1H), 7.76 (d, J = 7.8 Hz, 2H), 7.41 (s, 1H), 7.29 (d, J = 7.8 Hz, 2H), 7.23 (d, J = 9.0 Hz, 2H), 6.93 (d, J = 9.0 Hz, 2H), 4.91 (s, 2H), 3.74 (s, 3H), 2.34 (s, 3H), 2.04 (t, J = 7.4 Hz, 2H), 1.88 (t, J = 7.4 Hz, 2H), 1.49 – 1.44 (m, 2H), 1.41 – 1.36 (m, 2H), 1.15 – 1.10 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ 171.91, 169.04, 166.82, 158.34, 153.57, 140.02, 135.15, 130.48, 129.77, 129.42, 125.97, 116.04, 114.53, 55.29, 48.90, 33.31, 32.15, 28.21, 24.95, 24.63, 20.95. HRMS (ESI): calcd for [C25H29N3O4S + H]+ 468.1957, found 468.1956. HPLC purity: 98.83%, tR = 15.7 min.
N1-hydroxy-N7-(4-methoxyphenyl)-N7-((2-(4-methoxyphenyl)-4-thiazolyl)methyl) heptanediamide(15e) (65 mg, 75.1% yield) 1H NMR (600 MHz, DMSO-d6) δ 11.96 (br s, 1H), 7.81 (d, J = 8.4 Hz, 2H), 7.35 (br s, 1H), 7.23 (d, J = 9.0 Hz, 2H), 7.03 (d, J = 9.0 Hz, 2H), 6.93 (d, J = 9.0 Hz, 2H), 4.90 (s, 2H), 3.81 (s, 3H), 3.74 (s, 3H), 2.13 (t, J = 7.2 Hz, 2H), 2.04 (t, J = 7.2 Hz, 2H), 1.49 – 1.44 (m, 2H), 1.41 – 1.36 (m, 2H), 1.16 – 1.11 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ 174.41, 171.86, 166.60, 160.80, 158.33, 153.42, 135.19, 129.39, 127.57, 125.91, 115.40, 114.54, 114.49, 55.37, 55.26, 48.89, 33.50, 33.25, 28.14, 24.62, 24.26. HRMS (ESI): calcd for [C25H29N3O5S + H]+ 484.1906, found 484.1900. HPLC purity: 98.02%, tR = 15.4 min.
N1-((2-(2-fluorophenyl)-4-thiazolyl)methyl)-N7-hydroxy-N1-(4-methoxyphenyl) heptanediamide(15f.) (73 mg, 73.2% yield) 1H NMR (600 MHz, DMSO-d6) δ 10.35 (br s, 1H), 8.70 (br s, 1H), 8.12 (dd, J = 7.9,7.9 Hz, 1H), 7.58 (s, 1H), 7.53 – 7.50 (m, 1H), 7.41 – 7.34 (m, 2H), 7.22 (d, J = 8.4 Hz, 2H), 6.92 (d, J = 8.4 Hz, 2H), 4.95 (s, 2H), 3.72 (s, 3H), 2.03 (t, J = 7.2 Hz, 2H), 1.87 (t, J = 7.2 Hz, 2H), 1.48 – 1.43 (m, 2H), 1.40 – 1.34 (m, 2H), 1.14 – 1.09 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ 172.33, 169.42, 159.36 (d, J = 248.7 Hz), 159.14 (d, J = 20.9 Hz), 158.53, 152.98, 135.27, 131.99, 129.59, 128.46, 125.45 (d, J = 3.3 Hz), 120.70 (d, J = 11.4 Hz), 118.28 (d, J = 8.3 Hz), 116.64 (d, J = 21.3 Hz), 114.74, 55.48, 49.00, 33.47, 32.31, 28.35, 25.10, 24.81. HRMS (ESI): calcd for [C24H26FN3O4S + H]+ 472.1706, found 472.1698. HPLC purity: 96.36%, tR = 15.2 min.
N1-((2-(3-fluorophenyl)-4-thiazolyl)methyl)-N7-hydroxy-N1-(4-methoxyphenyl) heptanediamide(15 g). (68 mg, 77.9% yield) 1H NMR (600 MHz, DMSO-d6) δ 10.34 (br s, 1H), 8.69 (br s, 1H), 7.71 (d, J = 8.4 Hz, 1H), 7.64 (d, J = 9.6 Hz, 1H), 7.54 – 7.51 (m, 1H), 7.51 (s, 1H), 7.32 – 7.28 (m, 1H), 7.22 (d, J = 9.0 Hz, 2H), 6.93 (d, J = 9.0 Hz, 2H), 4.92 (s, 2H), 3.73 (s, 3H), 2.03 (t, J = 7.2 Hz, 2H), 1.88 (t, J = 7.2 Hz, 2H), 1.48 – 1.43 (m, 2H), 1.40 – 1.35 (m, 2H), 1.14 – 1.09 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ 172.17, 169.27, 165.29 (d, J = 3.2 Hz), 162.57 (d, J = 232.1 Hz), 158.48, 153.99, 135.27 (d, J = 8.3 Hz), 135.19, 131.59 (d, J = 8.3 Hz), 129.52, 122.41, 117.63, 117.07 (d, J = 21.2 Hz), 114.66, 112.53 (d, J = 23.3 Hz), 55.40, 48.93, 33.41, 32.25, 28.31, 25.05, 24.74. HRMS (ESI): calcd for [C24H26FN3O4S + H]+ 472.1706, found 472.1701. HPLC purity: 99.24%, tR = 15.1 min.
N1-((2-(4-fluorophenyl)-4-thiazolyl)methyl)-N7-hydroxy-N1-(4-methoxyphenyl) heptanediamide(15 h) (84 mg, 74.5% yield) 1H NMR (600 MHz, DMSO-d6) δ 10.30 (br s, 1H), 8.65 (br s, 1H), 7.93 (dd, J = 7.2, 7.2 Hz, 2H), 7.46 (s, 1H), 7.32 (dd, J = 8.4, 8.4 Hz, 2H), 7.23 (d, J = 9.0 Hz, 2H), 6.93 (d, J = 9.0 Hz, 2H), 4.92 (s, 2H), 3.74 (s, 3H), 2.04 (t, J = 7.2 Hz, 2H), 1.88 (t, J = 7.2 Hz, 2H), 1.49 – 1.44 (m, 2H), 1.41 – 1.36 (m, 2H), 1.15 – 1.10 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ 171.89, 169.02, 165.52, 163.13 (d, J = 246.5 Hz), 158.34, 153.74, 135.12, 129.71 (d, J = 3.2 Hz), 129.41, 128.3 (d, J = 8.6 Hz), 128.28, 116.75, 116.26 (d, J = 21.9 Hz), 114.53, 55.27, 48.82, 33.29, 32.14, 28.20, 24.94, 24.62. HRMS (ESI): calcd for [C24H26FN3O4S + H]+ 472.1706, found 472.1697. HPLC purity: 99.30%, tR = 14.8 min.
N1-((2-(2-chlorophenyl)-4-thiazolyl)methyl)-N7-hydroxy-N1-(4-methoxyphenyl) heptanediamide(15i) (75 mg, 72.9% yield) 1H NMR (600 MHz, DMSO-d6) δ 10.32 (br s, 1H), 8.66 (br s, 1H), 8.11 – 8.08 (m, 1H), 7.64 (s, 1H), 7.63 – 7.61 (m, 1H), 7.49 – 7.46 (m, 2H), 7.23 (d, J = 9.0 Hz, 2H), 6.93 (d, J = 9.0 Hz, 2H), 4.96 (s, 2H), 3.73 (s, 3H), 2.04 (t, J = 7.2 Hz, 2H), 1.88 (t, J = 7.2 Hz, 2H), 1.49 – 1.44 (m, 2H), 1.41 – 1.36 (m, 2H), 1.15 – 1.10 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ 171.95, 169.07, 161.91, 158.38, 152.67, 135.13, 131.20, 131.09, 130.77, 130.67, 130.63, 129.45, 127.76, 118.66, 114.56, 55.30, 48.79, 33.33, 32.16, 28.24, 24.97, 24.65. HRMS (ESI): calcd for [C24H26ClN3O4S + H]+ 488.1411, found 488.1407. HPLC purity: 99.77%, tR = 15.4 min.
N1-((2-(3-chlorophenyl)-4-thiazolyl)methyl)-N7-hydroxy-N1-(4-methoxyphenyl) heptanediamide(15j) (52 mg, 73.5% yield) 1H NMR (600 MHz, DMSO-d6) δ 10.31 (br s, 1H), 8.66 (br s, 1H), 7.88 (s, 1H), 7.82 (d, J = 7.2 Hz, 1H), 7.55 – 7.50 (m, 3H), 7.22 (d, J = 9.0 Hz, 2H), 6.93 (d, J = 9.0 Hz, 2H), 4.93 (s, 2H), 3.73 (s, 3H), 2.04 (t, J = 7.2 Hz, 2H), 1.88 (t, J = 7.2 Hz, 2H), 1.49 – 1.44 (m, 2H), 1.41 – 1.36 (m, 2H), 1.15 – 1.10 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ 171.99, 169.08, 164.91, 158.39, 153.97, 135.10, 134.90, 133.99, 131.26, 129.92, 129.45, 125.34, 124.82, 117.67, 114.56, 55.31, 48.79, 33.32, 32.17, 28.24, 24.97, 24.66. HRMS (ESI): calcd for [C24H26ClN3O4S + H]+ 488.1411, found 488.1403. HPLC purity: 99.83%, tR = 16.1 min.
N1-((2-(4-chlorophenyl)-4-thiazolyl)methyl)-N7-hydroxy-N1-(4-methoxyphenyl) heptanediamide(15 k) (36 mg, 76.8% yield) 1H NMR (600 MHz, DMSO-d6) δ 10.31 (br s, 1H), 8.65 (br s, 1H), 7.89 (d, J = 8.4 Hz, 2H), 7.54 (d, J = 9.0 Hz, 2H), 7.50 (s, 1H), 7.23 (d, J = 9.0 Hz, 2H), 6.93 (d, J = 9.0 Hz, 2H), 4.92 (s, 2H), 3.74 (s, 3H), 2.04 (t, J = 7.2 Hz, 2H), 1.88 (t, J = 7.2 Hz, 2H), 1.49 – 1.44 (m, 2H), 1.41 – 1.36 (m, 2H), 1.15 – 1.10 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ 171.90, 169.02, 165.33, 158.35, 153.92, 135.10, 134.70, 131.85, 129.40, 129.29, 127.70, 117.18, 114.54, 55.28, 48.81, 33.29, 32.15, 28.20, 24.94, 24.62. HRMS (ESI): calcd for [C24H26ClN3O4S + H]+ 488.1411, found 488.1409. HPLC purity: 95.92%, tR = 16.0 min.
N1-((2-(2-bromophenyl)-4-thiazolyl)methyl)-N7-hydroxy-N1-(4-methoxyphenyl) heptanediamide(15 l) (62 mg, 77.3% yield) 1H NMR (600 MHz, DMSO-d6) δ 10.35 (br s, 1H), 8.70 (br s, 1H), 7.92 – 7.90 (m, 1H), 7.78 (d, J = 7.8 Hz, 1H), 7.60 (s, 1H), 7.50 (dd, J = 7.2,7.2 Hz, 1H), 7.40 (dd, J = 7.8 Hz, 1H), 7.21 (d, J = 8.4 Hz, 2H), 6.93 (d, J = 8.4 Hz, 2H), 4.94 (s, 2H), 3.73 (s, 3H), 2.02 (t, J = 7.2 Hz, 2H), 1.87 (t, J = 7.2 Hz, 2H), 1.47 – 1.42 (m, 2H), 1.39 – 1.34 (m, 2H), 1.13 – 1.08 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ 172.19, 169.33, 163.79, 158.52, 152.78, 135.17, 134.16, 133.55, 131.55, 131.47, 129.60, 128.30, 120.85, 118.79, 114.70, 55.44, 48.85, 33.46, 32.27, 28.33, 25.07, 24.77. HRMS (ESI): calcd for [C24H26BrN3O4S + H]+ 534.0885, found 534.0884. HPLC purity: 99.41%, tR = 15.4 min.
N1-((2-(3-bromophenyl)-4-thiazolyl)methyl)-N7-hydroxy-N1-(4-methoxyphenyl) heptanediamide(15 m) (71 mg, 74.6% yield) 1H NMR (600 MHz, DMSO-d6) δ 10.34 (br s, 1H), 8.69 (br s, 1H), 8.00 (dd, J = 1.8, 1.8 Hz, 1H), 7.85 (d, J = 8.4 Hz, 1H), 7.66 (d, J = 7.8 Hz, 1H), 7.51 (s, 1H), 7.44 (dd, J = 7.8,7.8 Hz, 1H), 7.20 (d, J = 9.0 Hz, 2H), 6.92 (d, J = 8.4 Hz, 2H), 4.92 (s, 2H), 3.73 (s, 3H), 2.03 (t, J = 7.2 Hz, 2H), 1.87 (t, J = 7.2 Hz, 2H), 1.48 – 1.43 (m, 2H), 1.40 – 1.35 (m, 2H), 1.15 – 1.09 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ 172.24, 169.34, 164.97, 158.52, 154.04, 135.19, 135.17, 132.96, 131.67, 129.57, 128.28, 125.33, 122.58, 117.81, 114.68, 55.44, 48.89, 33.44, 32.28, 28.33, 25.07, 24.77. HRMS (ESI): calcd for [C24H26BrN3O4S + H]+ 534.0885, found 534.0868. HPLC purity: 97.23%, tR = 16.4 min.
N1-((2-(4-bromophenyl)-4-thiazolyl)methyl)-N7-hydroxy-N1-(4-methoxyphenyl) heptanediamide(15n) (45 mg, 72.7% yield) 1H NMR (600 MHz, DMSO-d6) δ 10.31 (br s, 1H), 8.65 (br s, 1H), 7.82 (d, J = 7.8 Hz, 2H), 7.68 (d, J = 8.4 Hz, 2H), 7.50 (s, 1H), 7.22 (d, J = 8.4 Hz, 2H), 6.93 (d, J = 8.4 Hz, 2H), 4.92 (s, 2H), 3.74 (s, 3H), 2.03 (t, J = 7.2 Hz, 2H), 1.88 (t, J = 7.2 Hz, 2H), 1.48 – 1.43 (m, 2H), 1.40 – 1.35 (m, 2H), 1.15 – 1.10 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ 171.92, 169.04, 165.44, 158.35, 153.95, 135.10, 132.20 (d, J = 4.9 Hz), 129.41, 127.92, 123.46, 117.21, 114.55, 55.29, 48.82, 33.30, 32.15, 28.21, 24.95, 24.62. HRMS (ESI): calcd for [C24H26BrN3O4S + H]+ 534.0885, found 534.0887. HPLC purity: 96.14%, tR = 16.3 min.
General Procedure for the synthesis of 18a-18 m.
N1-(4-ethoxyphenyl)-N7-hydroxy-N1-((4-phenyl-2-thiazolyl)methyl)heptanediamide(18a)Step-m: Boron tribromide (2.00 mL, 4 mmol) was slowly added to a mixture of compound 7 (0.45 g, 1 mmol) in CH2Cl2 (10 mL) with ice bath at 0 °C for 30 min. Then the mixture was stirring at room temperature for 6 h. Methanol was slowly added to the reaction mixture at 0 °C, and concentrated in vacuo. The mixture was purified by chromatography to give the target intermediate 16 (0.40 g, 92.3%).
Step-n: Intermediate 16 was dissolved in 8 mL of DMF, then K2CO3 (0.19 g, 1.36 mmol) was added and stirred at room temperature for 30 min. The bromoethane (0.15 g, 1.36 mmol) was added to the reaction system, which was stirred to 45 °C for 8 h. The mixture was extracted with EtOAc and purified by chromatography to give intermediate 17 (0.25 g, 60.3%).
Step-o: KOH (3.03 g, 54.1 mmol) was slowly added to a mixture of hydroxylamine hydrochloride (3.76 g, 54.1 mmol) in methanol (20 mL), and the mixture was stirred at 40 °C for 30 min, then the reaction product was cooled under an ice bath and filtered. Intermediate 17 (0.25 g, 0.541 mmol)was dissolved with the filtrate, followed by the addition of KOH (0.30 g, 5.41 mmol), and the reaction was stirred for 3 h at room temperature. The reaction system was concentrated under reduced pressure and extracted with saturated NH4Cl and EtOAc. The organic layer was then washed with brine, dried over Na2SO4, and concentrated, and then the residue was purified by column chromatography (eluting with EA followed by 20:1 CH2Cl2/MeOH followed by 10:1 CH2Cl2/MeOH) to give compound 18a(0.15 g, 59.8%). 1H NMR (600 MHz, DMSO-d6) δ 10.33 (br s, 1H), 8.67 (br s, 1H), 8.03 (s, 1H), 7.88 (d, J = 7.2 Hz, 2H), 7.41 (dd, J = 7.2,7.2 Hz, 2H), 7.32 (dd, J = 7.2,7.2 Hz, 1H), 7.21 (d, J = 9.0 Hz, 2H), 6.94 (d, J = 9.0 Hz, 2H), 5.08 (s, 2H), 4.00 (q, J = 7.2 Hz, 2H), 2.05 (t, J = 7.2 Hz, 2H), 1.88 (t, J = 7.2 Hz, 2H), 1.49 – 1.44 (m, 2H), 1.40 – 1.35 (m, 2H), 1.30 (t, J = 7.2 Hz, 3H), 1.16 – 1.10 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ 172.50, 169.16, 167.32, 158.01, 153.53, 134.67, 134.05, 129.29, 128.90, 128.11, 126.01, 115.25, 115.02, 63.40, 50.78, 33.17, 32.20, 28.22, 24.99, 24.66, 14.69. HRMS (ESI): calcd for [C25H29N3O4S + H]+ 468.1957, found 468.1951. HPLC purity: 94.89%, tR = 15.3 min.
Compounds 18b–m were prepared according to the procedure described for the preparation of compound 18a.
N1-hydroxy-N7-((4-phenyl-2-thiazolyl)methyl)-N7-(4-propoxyphenyl)heptanediamide(18b) (86 mg, 61.2% yield) 1H NMR (600 MHz, DMSO-d6) δ 10.35 (s, 1H), 8.69 (s, 1H), 8.01 (s, 1H), 7.87 (d, J = 7.8 Hz, 2H), 7.41 (dd, J = 7.8,7.8 Hz, 2H), 7.31 (dd, J = 7.2,7.2 Hz, 1H), 7.20 (d, J = 8.4 Hz, 2H), 6.94 (d, J = 9.0 Hz, 2H), 5.08 (s, 2H), 3.89 (t, J = 6.6 Hz, 2H), 2.05 (t, J = 7.2 Hz, 2H), 1.88 (t, J = 7.2 Hz, 2H), 1.72 – 1.66 (m, 2H), 1.49 – 1.43 (m, 2H), 1.40 – 1.35 (m, 2H), 1.16 – 1.10 (m, 2H), 0.94 (t, J = 7.2 Hz, 3H). 13C NMR (150 MHz, DMSO-d6) δ 172.63, 169.30, 167.40, 158.24, 153.60, 134.72, 134.10, 129.36, 128.98, 128.20, 126.07, 115.35, 115.07, 114.93, 69.33, 55.48, 50.86, 33.24, 32.26, 28.27, 25.05, 24.73, 22.14, 10.55. HRMS (ESI): calcd for [C26H31N3O4S + H]+ 482.2114, found 482.2107. HPLC purity: 99.51%, tR = 16.9 min.
N1-(4-(allyloxy)phenyl)-N7-hydroxy-N1-((4-phenyl-2-thiazolyl)methyl)heptanediamide(18c) (65 mg, 54.8% yield) 1H NMR (600 MHz, DMSO-d6) δ 10.31 (br s, 1H), 8.67 (br s, 1H), 8.04 (s, 1H), 7.89 (d, J = 7.2 Hz, 2H), 7.41 (dd, J = 7.8,7.8 Hz, 2H), 7.32 (dd, J = 7.2,7.2 Hz, 1H), 7.23 (d, J = 9.0 Hz, 2H), 6.98 (d, J = 9.0 Hz, 2H), 6.05 – 5.99 (m, 1H), 5.39 (d, J = 17.4 Hz, 1H), 5.25 (d, J = 10.2 Hz, 1H), 5.09 (s, 2H), 4.56 (d, J = 5.4 Hz, 2H), 2.06 (t, J = 7.2 Hz, 2H), 1.88 (t, J = 7.2 Hz, 2H), 1.50 – 1.44 (m, 2H), 1.41 – 1.36 (m, 2H), 1.17 – 1.11 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ 172.50, 169.05, 167.31, 157.64, 153.54, 134.93, 134.06, 133.54, 129.30, 128.91, 128.12, 126.01, 117.85, 115.55, 115.03, 68.48, 50.77, 33.19, 32.27, 28.26, 25.06, 24.68. HRMS (ESI): calcd for [C26H29N3O4S + H]+ 480.1957, found 480.1960. HPLC purity: 94.58%, tR = 15.5 min.
N1-(4-butoxyphenyl)-N7-hydroxy-N1-((4-phenyl-2-thiazolyl)methyl)heptanediamide(18d) (43 mg, 55.7% yield) 1H NMR (600 MHz, DMSO-d6) δ 10.35 (br s, 1H), 8.69 (br s, 1H), 8.01 (s, 1H), 7.87 (d, J = 7.0 Hz, 2H), 7.41 (dd, J = 7.8, 7.8 Hz, 2H), 7.31 (dd, J = 7.2, 7.2 Hz, 1H), 7.20 (d, J = 9.0 Hz, 2H), 6.94 (d, J = 9.0 Hz, 2H), 5.08 (s, 2H), 3.93 (t, J = 6.6 Hz, 2H), 2.05 (t, J = 7.2 Hz, 2H), 1.88 (t, J = 7.2 Hz, 2H), 1.68 – 1.63 (m, 2H), 1.48 – 1.43 (m, 2H), 1.41 – 1.35 (m, 2H), 1.15 – 1.10 (m, 2H), 0.90 (t, J = 7.2 Hz, 3H). 13C NMR (150 MHz, DMSO-d6) δ 172.64, 169.32, 167.41, 158.26, 153.62, 134.73, 134.11, 129.35, 128.99, 128.21, 126.08, 115.36, 115.08, 67.55, 50.87, 33.25, 32.27, 30.85, 28.28, 25.06, 24.74, 18.88, 13.85. HRMS (ESI): calcd for [C27H33N3O4S + H]+ 496.2270, found 496.2269. HPLC purity: 96.14%, tR = 16.9 min.
N1-hydroxy-N7-(4-(2-methoxyethoxy)phenyl)-N7-((4-phenyl-2-thiazolyl)methyl) heptanediamide(18e) (56 mg, 59.1% yield) 1H NMR (600 MHz, DMSO-d6) δ 10.33 (br s, 1H), 8.68 (br s, 1H), 8.02 (s, 1H), 7.88 (d, J = 7.2 Hz, 2H), 7.41 (dd, J = 7.8, 7.8 Hz, 2H), 7.32 (dd, J = 7.2, 7.2 Hz, 1H), 7.21 (d, J = 9.0 Hz, 2H), 6.97 (d, J = 9.0 Hz, 2H), 5.09 (s, 2H), 4.07 (t, J = 9.0 Hz, 2H), 3.63 (t, J = 9.0 Hz, 2H), 3.28 (s, 3H), 2.05 (t, J = 7.2 Hz, 2H), 1.88 (t, J = 7.2 Hz, 2H), 1.49 – 1.44 (m, 2H), 1.40 – 1.35 (m, 2H), 1.15 – 1.10 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ 172.54, 169.22, 167.33, 157.98, 153.57, 134.89, 134.07, 129.35, 128.94, 128.15, 126.03, 115.35, 115.04, 70.40, 67.22, 58.27, 50.79, 33.21, 32.23, 28.24, 25.01, 24.69. HRMS (ESI): calcd for [C26H31N3O5S + H]+ 498.2063, found 498.2055. HPLC purity: 99.92%, tR = 14.8 min.
N1-(4-(2-ethoxyethoxy)phenyl)-N7-hydroxy-N1-((4-phenyl-2-thiazolyl)methyl) heptanediamide(18f.) (58 mg, 54.4% yield) 1H NMR (600 MHz, DMSO-d6) δ 10.32 (br s, 1H), 8.66 (br s, 1H), 8.03 (s, 1H), 7.88 (d, J = 7.8 Hz, 2H), 7.41 (dd, J = 7.8, 7.8 Hz, 2H), 7.32 (d, J = 7.2, 7.2 Hz, 1H), 7.22 (d, J = 8.4 Hz, 2H), 6.97 (d, J = 8.4 Hz, 2H), 5.09 (s, 2H), 4.07 (t, J = 4.8 Hz, 2H), 3.67 (t, J = 4.8 Hz, 2H), 3.49 – 3.45 (m, 2H), 2.06 (t, J = 7.2 Hz, 2H), 1.88 (t, J = 7.2 Hz, 2H), 1.49 – 1.44 (m, 2H), 1.41 – 1.36 (m, 2H), 1.16 – 1.11 (m, 2H), 1.10 (t, J = 7.2 Hz, 3H). 13C NMR (150 MHz, DMSO-d6) δ 172.43, 169.09, 167.25, 157.94, 153.51, 134.83, 134.03, 129.28, 128.86, 128.07, 125.98, 115.31, 114.99, 68.32, 67.43, 65.73, 50.73, 33.15, 32.18, 28.20, 24.97, 24.64, 15.14. HRMS (ESI): calcd for [C27H33N3O5S + H] + 512.2219, found 512.2213. HPLC purity: 95.80%, tR = 14.8 min.
N1-hydroxy-N7-(4-(3-methoxypropoxy)phenyl)-N7-((4-phenyl-2-thiazolyl)methyl)) methyl)heptanediamide(18 g) (64 mg, 57.3% yield) 1H NMR (600 MHz, DMSO-d6) δ 10.32 (br s, 1H), 8.66 (br s, 1H), 8.03 (s, 1H), 7.88 (d, J = 7.2 Hz, 2H), 7.41 (dd, J = 7.8, 7.8 Hz, 2H), 7.32 (dd, J = 7.2, 7.2 Hz, 1H), 7.21 (d, J = 9.0 Hz, 2H), 6.95 (d, J = 9.0 Hz, 2H), 5.09 (s, 2H), 4.00 (t, J = 6.0 Hz, 2H), 3.44 (s, 2H), 3.22 (s, 3H), 2.06 (t, J = 7.2 Hz, 2H),1.92 (t, J = 6.0 Hz, 2H), 1.88 (t, J = 7.2 Hz, 2H), 1.49 – 1.44 (m, 2H), 1.41 – 1.36 (m, 2H), 1.16 – 1.11 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ 172.34, 169.00, 167.21, 157.97, 153.45, 134.72, 134.00, 129.23, 128.79, 128.00, 125.93, 115.22, 114.93, 68.43, 64.80, 57.95, 50.70, 48.62, 33.10, 32.13, 28.90, 28.16, 24.93, 24.59. HRMS (ESI): calcd for C27H33N3O5S [M + H]+ 512.2219, found 512.2210. HPLC purity: 93.18%, tR = 15.5 min.
N1-hydroxy-N7-(4-isobutoxyphenyl)-N7-((4-phenyl-2-thiazolyl)methyl)heptanediamide(18 h) (52 mg, 56.3% yield) 1H NMR (600 MHz, DMSO-d6) δ 10.31 (br s, 1H), 8.65 (br s, 1H), 8.04 (s, 1H), 7.89 (d, J = 7.8 Hz, 2H), 7.41 (dd, J = 7.2, 7.2 Hz, 2H),7.31 (dd, J = 7.2, 7.2 Hz, 2H), 7.21 (d, J = 8.4 Hz, 2H), 6.95 (d, J = 8.4 Hz, 2H), 5.09 (s, 2H), 3.72 (d, J = 6.6 Hz, 2H), 2.06 (t, J = 7.2 Hz, 2H), 2.01 – 1.96 (m, 1H), 1.88 (t, J = 7.2 Hz, 2H), 1.50 – 1.45 (m, 2H), 1.42 – 1.37 (m, 2H), 1.18 – 1.11 (m, 2H). 0.96–0.95 (m, 6H). 13C NMR (150 MHz, DMSO-d6) δ 172.34, 169.00, 167.22, 158.16, 153.45, 134.63, 134.00, 129.20, 128.79, 128.00, 125.93, 115.24, 114.93, 73.91, 50.71, 33.10, 32.13, 28.16, 27.69, 24.93, 24.59, 19.03. HRMS (ESI): calcd for [C27H33N3O4S + H] + 496.2270, found 496.2264. HPLC purity: 98.84%, tR = 16.9 min.
N1-(4-(cyclopropylmethoxy)phenyl)-N7-hydroxy-N1-((4-phenyl-2-thiazolyl)methyl) heptanediamide(18i) (46 mg, 52.9% yield) 1H NMR (600 MHz, DMSO-d6) δ 10.34 (br s, 1H), 8.68 (br s, 1H), 8.01 (s, 1H), 7.89 – 7.85 (m, 2H), 7.41 (dd, J = 6.6, 6.6 Hz, 2H), 7.31 (dd, J = 6.6,6.6 Hz, 1H), 7.19 (d, J = 3.0 Hz, 2H), 6.93 (d, J = 3.0 Hz, 2H), 5.07 (s, 2H), 3.78 (d, J = 6.0 Hz, 2H), 2.05 (t, J = 7.2 Hz, 2H), 1.88 (t, J = 7.2 Hz, 2H), 1.48 – 1.43 (m, 2H), 1.39 – 1.34 (m, 2H), 1.22 – 1.16 (m, 2H), 1.14 – 1.09 (m, 2H), 0.56 – 0.52 (m, 2H), 0.30 – 0.27 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ 172.63, 169.30, 167.40, 158.17, 153.60, 134.70, 134.10, 129.34, 128.99, 128.20, 126.07, 115.38, 115.09, 72.43, 50.86, 33.23, 32.26, 28.27, 25.05, 24.74, 10.23, 3.27. HRMS (ESI): calcd for [C27H31N3O4S + H]+ 494.2114, found 494.2103. HPLC purity: 97.01%, tR = 16.9 min.
N1-(4-(cyclohexylmethoxy)phenyl)-N7-hydroxy-N1-((4-phenyl-2-thiazolyl)methyl) heptanediamide(18j) (39 mg, 62.6% yield) 1H NMR (600 MHz, DMSO-d6) δ 10.31 (br s, 1H), 8.65 (br s, 1H), 8.03 (s, 1H), 7.88 (d, J = 6.6 Hz, 2H), 7.41 (dd, J = 7.2, 7.2 Hz, 2H), 7.32 (dd, J = 7.2, 7.2 Hz, 1H), 7.20 (d, J = 9.0 Hz, 2H), 6.94 (d, J = 9.0 Hz, 2H), 5.09 (s, 2H), 3.75 (d, J = 6.6 Hz, 2H), 2.06 (t, J = 7.2 Hz, 2H), 1.88 (t, J = 7.2 Hz, 2H), 1.78 – 1.76 (m, 2H), 1.71 – 1.67 (m, 3H), 1.64 – 1.61 (m, 1H), 1.49 – 1.44 (m, 2H), 1.41 – 1.36 (m, 2H), 1.26 – 1.17 (m, 3H), 1.16 – 1.11 (m, 2H), 1.04 – 0.98 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ 172.38, 169.03, 167.25, 158.23, 153.47, 134.61, 134.01, 129.20, 128.81, 128.02, 125.95, 115.22, 114.94, 72.94, 50.74, 37.08, 33.11, 32.15, 29.24, 28.17, 26.05, 25.28, 24.95, 24.61. HRMS (ESI) calcd for [C30 H37N3O4S + H]+ 536.2583, found 536.2575. HPLC purity: 97.38%, tR = 18.4 min.
N1-(4-(benzyloxy)phenyl)-N7-hydroxy-N1-((4-phenyl-2-thiazolyl)methyl) heptanediamide(18 k) (57 mg, 56.3% yield) 1H NMR (600 MHz, DMSO-d6) δ 10.32 (br s, 1H), 8.66 (br s, 1H), 8.04 (s, 1H), 7.89 (d, J = 7.2 Hz, 2H), 7.44 – 7.37 (m, 6H), 7.32 (dd, J = 7.2, 7.2 Hz, 2H), 7.24 (d, J = 10.2 Hz, 2H), 7.05 (d, J = 9.0 Hz, 2H), 5.09 (d, J = 8.4 Hz, 4H), 2.06 (t, J = 7.2 Hz, 2H), 1.88 (t, J = 7.2 Hz, 2H), 1.50 – 1.45 (m, 2H), 1.41 – 1.36 (m, 2H), 1.16 – 1.11 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ 172.38, 169.06, 167.24, 157.80, 153.48, 136.77, 134.96, 134.01, 129.28, 128.83, 128.50, 128.04, 128.00, 127.91, 125.96, 115.59, 114.98, 69.54, 50.72, 33.15, 32.16, 28.18, 24.95, 24.61. HRMS (ESI): calcd for [C30H31N3O4S + H]+ 530.2035, found 530.2114. HPLC purity: 91.14%, tR = 16.7 min.
N1-hydroxy-N7-(4-((4-methylbenzyl)oxy)phenyl)-N7-((4-phenyl-2-thiazolyl)methyl) heptanediamide(18 l) (60 mg, 58.7% yield) 1H NMR (600 MHz, DMSO-d6) δ 10.31 (br s, 1H), 8.65 (br s, 1H), 8.04 (s, 1H), 7.89 (d, J = 8.4 Hz, 2H), 7.41 (dd, J = 6.6,6.6 Hz, 2H), 7.32 (d, J = 13.2 Hz, 3H), 7.23 (d, J = 6.6 Hz, 2H), 7.18 (d, J = 6.0 Hz, 2H), 7.03 (d, J = 6.6 Hz, 2H), 5.09 (s, 2H), 5.03 (s, 2H), 2.29 (s, 3H), 2.06 (t, J = 6.6 Hz, 2H), 1.88 (t, J = 6.6 Hz, 2H), 1.50 – 1.44 (m, 2H), 1.41 – 1.36 (m, 2H), 1.17 – 1.12 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ 172.34, 169.02, 167.22, 157.81, 153.46, 137.22, 134.88, 134.00, 133.72, 129.23, 129.01, 128.80, 128.01, 127.96, 125.94, 115.56, 114.94, 69.41, 50.70, 33.12, 32.14, 28.16, 24.93, 24.59, 20.80. HRMS (ESI): calcd for [C31H33N3O4S + H]+ 544.2270, found 544.2266. HPLC purity: 98.24%, tR = 17.4 min.
N1-(4-((4-fluorobenzyl)oxy)phenyl)-N7-hydroxy-N1-((4-phenyl-2-thiazolyl)methyl) heptanediamide(18 m) (54 mg, 54.6% yield) 1H NMR (600 MHz, DMSO-d6) δ 10.33 (br s, 1H), 8.67 (br s, 1H), 8.03 (s, 1H), 7.88 (d, J = 7.2 Hz, 2H), 7.49 (d, J = 3.0 Hz, 2H), 7.41 (dd, J = 7.8 Hz, 2H), 7.32 (dd, J = 7.2,7.2 Hz, 1H), 7.24 (d, J = 8.4 Hz, 2H), 7.20 (dd, J = 9.0,9.0 Hz, 2H), 7.04 (d, J = 9.0 Hz, 2H), 5.09 (s, 2H), 5.06 (s, 2H), 2.06 (t, J = 7.2 Hz, 2H), 1.88 (t, J = 7.2 Hz, 2H), 1.49 – 1.44 (m, 2H), 1.41 – 1.36 (m, 2H), 1.16 – 1.11 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ 172.43, 169.13, 167.26, 161.89 (d, J = 242.3 Hz), 157.74, 153.51, 135.04, 134.04, 133.04 (d, J = 3.0 Hz), 130.24 (d, J = 8.4 Hz), 129.33, 128.88, 128.09, 125.99, 115.63, 115.35 (d, J = 21.2 Hz), 115.02, 68.84, 50.75, 33.18, 32.18, 28.20, 24.97, 24.64. HRMS (ESI): calcd for [C30H30FN3O4S + H]+ 548.2019, found 548.2012. HPLC purity: 98.25%, tR = 17.4 min.
Biology
HDAC inhibitory assay
The enzyme inhibitory assay was executed by Shanghai ChemPartner Corporation. HDAC enzyme (HDAC1 Active motif 31504, HDAC2 BPS Bioscience 50002, HDAC6 BPS Bioscience 50006, HDAC8 Active motif 31566) solution was preincubated with varying concentrations of SAHA or the target compounds in conjunction with the HDAC-specific substrate (Boc-Lys(Ac)-AMC) at a temperature of 37 °C for a duration of 60 min. Subsequently, lysine developer was introduced to terminate the enzymatic reaction, followed by a further 30-min incubation at 37 °C. The absorbance measurements were captured using an ELISA plate reader at a wavelength of 405 nm.
In vitro anti-proliferation assay
Cells were seeded in 96-well plates at a seeding density of 5 × 103 per well. After cell attachment, drugs were added, with control and blank groups set up accordingly. After 72 h, the culture medium was aspirated and discarded, and 100 uL of MTT solution (1 mg/mL) was added to each well, followed by incubation in the dark for 2–4 h. After incubation, the supernatant was removed, and 150 uL of DMSO was added to each well to dissolve the formazan crystals. The optical density (OD450) was determined using a microplate reader (BIO-TEK, Inc., Winooski, VT, United States). The IC50 value was computationally derived using GraphPad Prism version 8.0 software (La Jolla, CA, United States).
Antibodies and reagents
The following primary antibodies were utilized for immunoblotting, with their respective sources and dilution factors indicated: Acetylated histone H3 (Ac-H3, Beyotime, catalog #AF5620, diluted 1:1000); Acetylated histone H4 (Ac-H4, Beyotime, catalog #AF5629, diluted 1:1000); Cyclin D1 (CST, catalog #2978, diluted 1:1000); Cyclin-dependent kinase 4 (CDK4, CST, catalog #12790, diluted 1:1000); Cleaved poly (ADP-ribose) polymerase (cl-PARP, CST, catalog #5625, diluted 1:1000); Caspase 3 (CST, catalog #9662, diluted 1:1000); Cleaved caspase 3 (cl-caspase 3, CST, catalog #9661, diluted 1:1000); Anti-DFNA5/GSDME (Abcam, catalog #ab215191, diluted 1:1000); γ-H2AX (CST, catalog #9718, diluted 1:1000); β-actin (CST, catalog #3700, diluted 1:10,000); Anti-rabbit IgG Fab2 (Sigma, catalog #A0545, diluted 1:10,000); Anti-mouse IgG Fab2 (Sigma, catalog #A4416, diluted 1:10,000); Goat anti-Rabbit IgG (Invitrogen, Waltham, MA, USA, diluted 1:500). Phosphate-buffered saline (PBS) for washing, Fetal bovine serum (FBS), Trypsin–EDTA solution, and Penicillin–Streptomycin solution (PS, 100×) were procured from Gibco (Carlsbad, CA, United States). The Bicinchoninic acid (BCA) Protein assay kit was sourced from TIANGEN (Shanghai, China). The protease inhibitor cocktail was obtained from Roche (Basel, Switzerland). Cell apoptosis was assessed using the Annexin V-FITC Apoptosis Detection Kit (Beyotime, Shanghai, China). Analysis of the cell cycle was performed using a PI Cell Cycle and Apoptosis Analysis Kit (Beyotime, Shanghai, China).
Western blot analysis
Cells were exposed to varying concentrations of compounds 15a, 15d, and SAHA for a period of 24 h prior to lysis in RIPA buffer supplemented with a comprehensive protease inhibitor cocktail, followed by a 10-min incubation on ice and subsequent centrifugation at 4 °C. Protein concentrations were quantified using the BCA Protein Assay Kit (TIANGEN, Shanghai, China). Equivalent amounts of total protein were mixed with loading buffer, subjected to denaturation at 100 °C for 5 min, and resolved via 10–15% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). The resolved proteins were electrophoretically transferred onto PVDF membranes (Merck Millipore, catalog #IPFL00010, Germany). The membranes were then blocked with a 5% non-fat dry milk solution before being incubated with the specified primary antibodies in a BSA buffer overnight at 4 °C. After three washes in TBST, the membranes were incubated for 1 h at room temperature with horseradish peroxidase (HRP)-conjugated secondary antibodies. The membranes were washed again prior to enhanced chemiluminescence detection. The immunoblots were developed using the Bio-Rad ChemiDoc™ Imaging System, and band intensities were quantified using the Image Lab software, directly from the blot.
Colony formation assay
The long-term impact of compounds 15a, 15d, and SAHA on cellular proliferation was assessed using a colony formation assay. Cells were inoculated into 6-well plates at a density of 1000 cells per well. Following a 24-h incubation period, the cells were subjected to treatment with varying concentrations of 15a, 15d and SAHA. The culture medium was refreshed every other day. Thereafter, the cells were maintained in fresh medium for a continuous period of 10–14 days. The resultant colonies were fixed with methanol for 15 min, stained with a 0.5% crystal violet solution for 15 min, and then photographed. The colonies were enumerated manually using ImageJ software.
Cell cycle analysis
Cells were inoculated into 6-well plates at a seeding density of 2 × 105 cells per well and exposed to various concentrations of compounds 15a, 15d, and SAHA for a duration of 48 h, with dimethyl sulfoxide (DMSO) serving as a solvent control. Following the treatment period, the cells were harvested, washed with phosphate-buffered saline (PBS), and fixed with a 70% solution of ice-cold ethanol at 4 °C for an extended period. Post-fixation, the cells were again washed with cold PBS and stained with a mixture of 500 μL propidium iodide (PI) and RNase staining buffer, then incubated on ice for 30 min. Analysis of the cell cycle distribution was conducted using a FACSCalibur flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA). Each experimental condition was replicated three times, yielding similar results. The collected data were processed for analysis using Modfit LT 4.1 software (Verify Software House, Topsham, ME, USA).
Cell apoptosis analysis
Cell apoptosis was quantified using flow cytometry using an Annexin V-FITC apoptosis detection kit. Cells were plated at a density of 1 × 105 cells per well in 6-well plates and allowed to adhere overnight before being exposed to varying concentrations of compounds 15a, 15d and SAHA for a period of 48 h. Following treatment, the cells undergoing apoptosis were harvested and stained with Annexin V and propidium iodide (PI) in 1× binding buffer at ambient temperature for a duration of 15 min. The stained cells were subsequently subjected to analysis on a FACSCelesta flow cytometer (BD Biosciences, San Jose, CA, USA), utilizing FlowJo V10 software (FlowJo, Ashland, OR, USA) for data processing.
Reactive oxygen species (ROS) measurement
The intracellular generation of reactive oxygen species (ROS) was assessed using 2,7-dichlorodihydrofluorescein diacetate (DCFH-DA) as a fluorometric probe. Cells were plated into 6-well plates at a seeding density of 1 × 105 cells per well and exposed to various concentrations of compounds 15a, 15d and SAHA for 24 h, with dimethyl sulfoxide (DMSO) serving as a negative control. Post-treatment, cells were collected by centrifugation and incubated with 10 μM DCFH-DA at a temperature of 37 °C for 30 min. Following the incubation, cells were washed twice with PBS to remove excess probe. The mean fluorescence intensity, indicative of ROS levels, was measured using a FACSCelesta flow cytometer (BD Biosciences, San Jose, CA, USA), and the acquired data were analyzed using FlowJo V10 software (FlowJo, Ashland, OR, USA).
Immunofluorescence assay
Cells were cultured on glass coverslips placed within 6-well plates at a density of 1 × 105 cells per well and subsequently treated with compounds 15a, 15d and SAHA at a range of concentrations for a period of 24 h. Following treatment, cells were washed with PBS and fixed using a 4% formaldehyde solution. The cells were then blocked and permeabilized with a PBS-BSA solution containing 0.3% Triton X-100 for 15 min at ambient temperature. Subsequently, cells were incubated with a primary antibody specific for γH2AX overnight at 4 °C. After the primary antibody incubation, the coverslips were washed three times with PBS and incubated with a secondary antibody, Goat anti-Rabbit IgG (diluted 1:500, Invitrogen, Waltham, MA, USA), for 30 min at room temperature. Following the secondary antibody staining, cell nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI, Invitrogen, Waltham, MA, USA). Fluorescent imaging was performed using a confocal laser scanning microscope (Leica SP8, Germany).
Pharmacokinetic studies
In a pharmacokinetic study, 8-week-old female Sprague–Dawley (SD) rats were stratified based on the method of drug administration into two cohorts: one for intravenous (IV) administration and another for oral (PO) administration. The respective dosages for the IV and PO groups were established at 5 mg/kg and 30 mg/kg. Each dosage group comprised nine animals, which were further randomly allocated into three subsets, with each subset containing three animals. A crossover sampling technique was implemented to ensure that triplicate blood samples were obtained at each designated time point within each dosage group. Heparinized tubes were utilized to collect blood samples from the cheek vein at intervals of 5, 15, and 30 min, and 1, 2, 4, 8, 24, and 48 h post-administration. The collected blood samples were then centrifuged at 6000×g for 10 min at a temperature of 4 °C to isolate plasma. The harvested plasma was stored at − 80 °C for subsequent liquid chromatography-tandem mass spectrometry (LC–MS/MS) analysis. The pharmacokinetic parameters of compound 15a were derived from non-compartmental analysis, employing the plasma concentration–time data.
Molecular docking and molecular dynamics simulation studies
The molecular docking investigation was conducted utilizing AutoDock 4.2.635. The crystallographic structure of human histone deacetylase 1 (HDAC1), with the Protein Data Bank (PDB) identifier 4BKX, was sourced from the Protein Data Bank. The protein structure’s missing terminal residues were restored using the Swiss-PdbViewer software version 4.10, followed by preparation for docking, which included the removal of crystallographic water molecules and the addition of polar hydrogen atoms and Kollman charges36. The three-dimensional molecular structures of SAHA and compound 15a were constructed using ChemOffice 2019. The grid box was centered at the coordinates x, y, z = − 49.617, 18.378, − 4.783, with dimensions of 28.5 Å × 24.75 Å × 24.75 Å. Default parameters were applied for the remaining settings. The binding interactions within the protein–ligand complexes were visualized with UCSF Chimera version 1.16 and BIOVIA Discovery Studio Visualizer version 21.1.0.20298. The conformation with the lowest energy was chosen for molecular dynamics (MD) simulation analysis. All MD simulations were executed with GROMACS version 2020.6. The topological parameters for the HDAC1-15a complex were prepared using the Amber99 and General Amber Force Field (GAFF) force fields37. All molecules have been solvated into the water environment and added ions. The HDAC1-15a complex underwent energy minimization to eliminate any potential steric hindrances. Prior to the 10 ns MD simulation, NVT and NPT equilibration were perpormed. The MD simulation for the HDAC1-15a complex was carried out for a duration of 10 ns with a time step of 2 femtoseconds. Ultimately, the results of the MD simulation were analyzed.
Lactate dehydrogenase (LDH) release assay
Cells were seeded onto glass coverslips placed within 96-well plates at a density of 1 × 10^4 cells per well and subsequently exposed to various concentrations of compounds 15a, 15d and SAHA for a period of 24 h. Following treatment, cell culture supernatants were collected, and lactate dehydrogenase (LDH) activity was assessed using the LDH Assay Kit (Beyotime). A volume of 120 μL of supernatant from each well was transferred to a fresh 96-well plate, to which 60 μL of LDH detection reagent was added per well. The plates were incubated in the dark for 30 min at ambient temperature. The optical density (OD) at 490 nm was measured using a microplate reader (BioTek, CA, USA). Each sample was assayed in triplicate.
Animal experiments
Male BALB/c nude mice, aged 5–6 weeks, were procured from Nanmo Experimental Animal Co. Ltd (Shanghai, China). All in vivo experimental procedures adhered to the ARRIVE guidelines version 2.038. The mice were housed under specific pathogen-free (SPF) conditions, with a controlled environment maintaining a temperature of 25 °C and a relative humidity of 65%, on a 12-h light/dark cycle. HepG2 tumor cells, at a concentration of 1 × 106 cells, were subcutaneously implanted into 6–7 weeks-old mice. Tumor dimensions were measured using calipers, and the tumor volume was calculated employing the formula: volume = (length × width2) × 0.5. When the tumors reached a volume of approximately 100 mm3, the mice were randomly assigned to three treatment groups, each comprising five mice, and were administered intraperitoneal injections of either 15a, SAHA, or a DMSO control every 48 h for a period of three weeks. Tumor volume measurements were taken at 48-h intervals. The animal welfare and experimental protocols were reviewed and approved by the Institutional Animal Care and Use Committee at Shanghai Ocean University (Shanghai, China). (Approval No. SHOU-DW-2024-018, dated 4/3/2024).
Results and discussion
Chemistry
Compounds 8a–l were prepared by following the procedures described in Scheme 1. The direct coupling of commercially available p-methoxyaniline 2 with bromoacetonitrile gave intermediate 3, which was further condensed with pimelic acid anhydride in 1,4-dioxane to give the corresponding acid 4. Esterification of the acid in methanol at the presence of catalytic amounts of thionyl chloride generated the corresponding ester 5, and compound 6 was produced by the reaction of the ester with ammonium sulfide and triethylamine in pyridine. Compound 6 was then converted to give compounds 7a–l, which was finally stirred with the freshly prepared hydroxylamine in methanol for 1 h to give the final target compounds 8a–l.

Synthesis of Compounds 8a-l. Reagents and conditions: (a) Bromoacetonitrile, K2CO3, DMF (b): Pimelic acid anhydride, 1,4-dioxane (c): MeOH, Cat. SOCl2, reflux (d): Pyridine, (C2H5)3N, (NH4)2S5 (e): R1-Ph-CO-CH2-Cl, EtOH, reflux (f): NH2OH·HCl, KOH, MeOH.
Compounds 15a–n were prepared by following the procedures described in Scheme 2, and the key intermediates were synthesized using similar protocols as outlined in Scheme 1, followed by the reaction of a series of substituted anilines with triethylamine and ammonium sulfide under the corresponding conditions (step d) to finally give the desired compounds 15a–n.

Synthesis of Compounds 15a − n. Reagents and conditions: (g): Pyridine, (C2H5)3N, (NH4)2S5 (h): 1,3-Dichloroacetone, EtOH, reflux (i): K2CO3, DMF (j): Pimelic acid anhydride, 1,4-dioxane, reflux (k): MeOH, Cat. SOCl2, reflux (l): NH2OH·HCl, KOH, MeOH.
Compounds 18a–m were prepared by following the procedures described in Scheme 3. On the basis of compound 7a in Scheme 1, compound 16 was obtained by hydrolysis with boron tribromide. Then the intermediate 16 was coupled with different straight-chain hydrocarbons or 2-chloroacetophenone with various substituents on the benzene ring to give compound 17a–m, finally compounds 17a–m were treated with NH4OH.HCl to give the target compounds 18a–m.

Synthesis of Compounds 18a–m. Reagents and conditions: (m): DCM, BBr3 (n): K2CO3, DMF (o): NH2OH·HCl, KOH, MeOH.
Biological evaluation
In vitro HDAC1 inhibitory activity
First, the HDAC inhibitory activities were assayed using HDAC1, and SAHA was used as the control drug. In order to investigate whether the introduction of thiazole structure into the Cap region would produce effective HDAC inhibitors, and how the substituents on the phenyl affected the activities of the compounds, we first detected the activities of compounds 8a–l. The results in Table 1 revealed that the majority of the compounds displayed good inhibitory activity toward HDAC1 (IC50 range: 0.9–4.9 nM), except compounds 8b (IC50 > 10 nM). The inhibitory activity of compound 8a (IC50 = 1.6 nM) was significantly better than that of SAHA, indicating that the introduction of thiazole structure could effectively improve the binding force of the compound and HDAC1. When the electron-donating group methyl and methoxy were introduced into the benzene ring (8b–e), the activities didn’t change much. Next, the electron-withdrawing groups were introduced to the different positions of phenyl. The HDAC1 inhibition potencies of fluorine-substituted compounds 8f and 8g were identical, the para-substituted compound 8h showed the highest potency (IC50 = 0.9 nM). For chlorine-substituted compounds 8i, 8j, and 8k, the para substituted compound (8k) was more active than the ortho and meta substituted derivatives. And the bromine substituted compound 8l has no influence on the improvement of activity. In summary, there is no significant difference between electron-withdrawing and electron-donating groups substituents on compound activities.
In the second-round of optimization, we changed the orientation of the thiazole ring and synthesized compounds 15a–n. As shown in Table 2, there is no significant difference of the activities between the compounds in Table 2 and Table 1, indicating that the thiazole structure has good spatial flexibility. And similarly to 8a–l, compounds with methyl groups in the phenyl ring (15b–d) demonstrated comparable activities to those with a halogen substituent (15f–m). In general, the para- substituted compounds (15d, 15h, 15k and 15n) exhibited better activities than the other position-substituted compounds.
According to above results, the insertion of thiazole ring is beneficial for improving potency. we selected 8a for further structural modification, compounds 18a–m with different ether substitution at another phenyl were designed and synthesized. As depicted in Table 3, the HDAC1 inhibitory activity was decreased with the elongation of the alky (18a > 18b = 18c > 18d). Whereas the activity of compounds with oxygen containing alky linker decreased slightly (18e–g). And the activities of these straight alkane substituents compounds (18a–g) were higher than that of branched (18h) and cyclic substituents derivatives (18i, 18j). However, when the straight chain alkane was changed to aromatic substituents (18k–m), the potency decreased dramatically. In general, replacement of the methoxy with the more hydrophilic groups led to decreased activities, indicating that the methoxy group is optimal for HDAC1-inhibitory activity.
Selectivity profiles of representative compounds
To further study the selectivity profiles of the inhibitors toward other HDAC subtypes, the most potent inhibitors (8h, 15a, 15d and 15n) were also tested against class IIa (HDAC4, 5,7,9), class IIb (HDAC6), and class IV (HDAC11) enzymes (Table 4). The four compounds showed good inhibitory activities against HDAC1 and HDAC6. Specifically, the HDAC1 inhibitory activity of compounds 15a and 15d was tenfold better than that of SAHA (IC50 = 10.7 nM). The HDAC6 inhibitory activity of these two compounds was eightfold better than that of SAHA (IC50 = 20.4 nM). Although HDAC1 (Class I) and HDAC6 (Class IIb) belong to distinct HDAC subfamilies, both isoforms adopt the conserved arginase-deacetylase fold-a structural motif featuring a central β-sheet core (7–8 parallel β-strands) flanked by ~ 13 α-helices. Crucially, their catalytic zinc-binding sites exhibit identical coordination geometry (His-Asp-His triad), which structurally rationalizes the dual inhibitory activity of our compounds39,40.
In vitro antiproliferative activity
Based on the previous results of efficient inhibition of HDAC1, we further assessed the in vitro antiproliferative potency of five compounds (8h, 15a, 15d and 15n) against four different tumor cells (HepG2, PC9, HCT116, MCF7) by the MTT assay. As summarized in Table 5, the results showed that most of the compounds had strong anti-proliferative activities with IC50 values ranging from 0.12 to 10.25 μM, which was consistent with their high inhibitory activities. Notably, these compounds exhibited stronger cytotoxicities against HepG2 cells, especially compounds 15a and 15d displayed the best antiproliferative activities with IC50 values of 0.12 and 0.19 μM, nearly 25 and 15 times higher than SAHA, respectively. In general, 15a and 15d showed weaker cell growth inhibitory activities in the PC9 cell line than in the HepG2 line. Besides, they also exhibited lower inhibitory activities compared SAHA. In HCT116 cell line, the activities of 15a and 15d dropped almost tenfold. In addition, the anti-proliferative activities of compounds 15a and 15d decreased significantly in MCF7 cancer cell line. Based on these observations, compounds 15a and 15d showed the highest potency in inhibition of both HDAC and tumor cell growth, they were selected for further evaluation in HepG2 cancer cell line.
Efficacy of compounds 15a and 15d on tumor cell acetylated histone levels
Based on the above results, we further investigated the effects of different concentrations of 15a and 15d on histone acetylation in HepG2 cells. HepG2 cells were treated with SAHA, 15a and 15d at the indicated concentrations (0.2, 0.5 µM) for 24 h. Western blot was performed to detect the expression of acetylated H3/H4, and β-actin was used as an endogenous loading control. The results in Fig. 3 showed that compounds 15a and 15d can increased the level of acetyl-H3 and H4 in a dose-dependent manner, which was consistent with their inhibitory activities against HDAC1.

Western blot analysis of the effects of compounds 15a and 15d on the acetylated histone levels in HepG2 cells. (A) HepG2 cells were treated with 15a, 15d, and SAHA for 24 h at the indicated concentrations. The acetylated H3/H4 protein was determined by Western blot analysis, and β-actin was detected as the endogenous loading control, accordingly. (B) Quantitative analysis of (A). The data are expressed as means ± SD of three separate experiments. *p < 0.05, **p < 0.01, ***p < 0.001 versus control.
Colony formation assay
Since the tested compounds showed the best inhibitory activity against HepG2 cells, then we explored the effects of compounds 15a and 15d on colony formation of HepG2 cells with SAHA as a positive control group. As shown in Fig. 4, compounds 15a and 15d were able to suppress the formation of colonies in a dose-dependent manner. In addition, the suppressing effects of compounds 15a and 15d were stronger than that of SAHA.

Effects on the colony formation. (A) Colony-forming abilities of HepG2 cells after treating cells with the indicated concentrations of 15a, 15d, and SAHA for 14 days. (B) The statistical result of (A). The data are expressed as means ± SD of three separate experiments. *p < 0.05, **p < 0.01, ***p < 0.001 versus control.
15a and 15d arrested the cell cycle at the G0/ G1 phase
We investigated whether the anti-proliferative activity of 15a and 15d originated from cell cycle arrest. As shown in Fig. 5A,B, 15a and 15d significantly inhibited HepG2 cells in G0/G1 phase in a dose-dependent manner compared to control cells. The G0/G1 ratio of compound 15a-treated samples increased from 41.67 to 61.42% in the control group. the G2/M ratio decreased from 25.34 to 22.85% and the S-phase value decreased from 32.99 to 13.72%. The G0/G1 ratio of compound 15d treated samples increased from 41.67 to 57.36% in the control. the G2/M ratio decreased from 25.34 to 24.34% and the S-phase value decreased from 32.99 to 18.3%. These data suggested that compounds 15a and 15d induce G0/G1 phase block in HepG2 cancer cells.

15a and 15d induced HepG2 cell cycle arrest. (A) The cell cycle of HepG2 cells treated with 15a, 15d, and SAHA for 24 h at the indicated concentrations was examined by flow cytometry analyses. (B) The statistical result of (A). (C) HepG2 cells treated with 15a, 15d, and SAHA for 24 h at the indicated concentrations, the expression of CDK4 and Cyclin D1 protein levels were examined by Western blot analysis, and β-actin was detected as the endogenous loading control, accordingly. (D) The statistical result of (C). The data are expressed as means ± SD of three separate experiments. *p < 0.05, **p < 0.01, ***p < 0.001 vs. control.
In early G1, extracellular signals lead to the release of d-type cell cycle proteins (D1, D2, D3), which bind to CDK4 and CDK6 and initiate further phosphorylation of the retinoblastoma protein (Rb protein) family to accelerate the cell cycle process. To further validate the mechanism of action of compounds 15a and 15d in inducing G0/G1 phase block in HepG2 cells. We determined the expression levels of cell cycle regulators CDK4 and Cyclin D1 by Western blotting assay. The results are shown in Fig. 5C,D. Compounds 15a and 15d significantly reduced the levels of CDK4 and Cyclin D1 in a dose-dependent manner. These data suggested that compounds 15a and 15d induce G0/G1 arrest in HepG2 cells may be correlated with these regulatory proteins.
15a and 15d enhanced ROS generation and DNA damage accumulation to induce apoptosis in HepG2 cells
We further employed an Annexin V-FITC/ PI assay to observe the apoptotic induction of compounds 15a and 15d. The results are shown in Fig. 6. The morphological changes of HepG2 cells after treatment with compounds 15a and 15d was significant. 15a and 15d induced apoptosis in 28.2% and 30% of HepG2 cells at 0.2 µM and 46.5% and 41.9% of HepG2 cancer cells at 0.5 µM, respectively, which was significantly higher than that of 21% for SAHA (0.5 µM).

15a and 15d mediated HepG2 cell apoptosis. (A) Apoptosis of HepG2 cells induced by 15a, 15d, and SAHA treatment for 48 h at the indicated concentrations were examined by using Annexin V-FITC/PI double-staining analysis. (B) The statistical result of (A). (C) HepG2 cells were treated with 15a, 15d, and SAHA for 24 h at the indicated concentrations. The Cl-PARP and Cl-caspase 3 protein levels were examined by Western blot analysis, and β-actin was detected as the endogenous loading control, accordingly. (D) The statistical result of (C). The data are expressed as means ± SD of three separate experiments. *p < 0.05, **p < 0.01, ***p < 0.001 versus control.
Caspase-3 is the predominant terminal shear enzyme in apoptosis, which can promote apoptosis by activating protein hydrolysis. PARP is a cleavage substrate for cystatin protease (caspase), a core member of apoptosis. To identify the potential mechanisms underlying the induction of apoptosis induced by compounds 15a and 15d, we further monitored the expression of apoptotic proteins (Cl-PARP, Cl-caspase 3) by western blot analysis. The results in Fig. 6C,D demonstrated that 15a and 15d could upregulate the expression of Cl-PARP, Cl-caspase 3 in a dose-dependent manner. These data suggested that compounds 15a and 15d have significant ability to induce apoptosis in HepG2 cancer cells.
In order to explore the mechanism of cell apoptosis induced by 15a and 15d, we assayed the effect of these two compounds on ROS generation41. As shown in Fig. 7A,B, the ROS levels were dose-dependently increased with the treatments of 15a and 15d, which proved that 15a and 15d triggered the apoptosis of HepG2 cells by enhancing ROS levels. In addition, ROS have been reported to induce DNA damage and the formation of γH2AX foci is a well-known marker of DNA damage. Then we further assessed the nuclear γH2AX levels in 15a and 15d treated HepG2 cells. In comparison to SAHA, 15a and 15d were able to induce γH2AX expression at a much lower concentration range (Fig. 7C). Moreover, HepG2 cells treated with 15a and 15d also showed dose-dependent γH2AX foci formation. Taken together, these data indicated that 15a and 15d induced HepG2 cells apoptosis was caused by excessive ROS accumulation and DNA damage.

15a and 15d mediated HepG2 cell apoptosis via accumulation of ROS, and DNA damage. (A) The HepG2 cells treated with 15a, 15d and SAHA for 24 h followed by ROS measurement. Representative flow cytometry histograms displaying levels of fluorescent DCFH-DA in cells were presented. B The statistical result of (A). C The HepG2 cells were treated with 15a, 15d and SAHA for 24 h, and the expression of γ-H2AX was analyzed by immunofluorescence. Green represents the cells stained with anti-γ-H2AX antibody, and blue represents the nuclei stained with DAPI (Scale bar, 30 μm). *p < 0.05, **p < 0.01, ***p < 0.001 vs. control.
15a and 15d induced GSDME-mediated pyroptosis
Morphological observation of HepG2 cells induced by 15a and 15d revealed cell swelling with large bubbles, a characteristic sign of pyroptosis42 (Fig. 8A). The release of LDH is closely associated with plasma membrane rupture (PMR) during pyroptosis. The results in Fig. 8B displayed that LDH release from HepG2 cells was markedly increased, indicating damaged cell membrane integrity. The cleavage of gasdermin family proteins (GSDMs) is a key feature of pyroptosis43. Western blotting results indicated that treatment with 15a and 15d resulted in elevated levels of the N-terminal fragment of gasdermin E (GSDME) in HepG2 cells in a dose-dependent manner. Furthermore, the levels of active caspase-3 were elevated in response to 15a and 15d treatment (Fig. 8C,D), indicating that GSDME may be cleaved by the active caspase-3. Collectively, these findings demonstrate that 15a and 15d trigger pyroptosis in HepG2 cells through caspase-3 cleavage of GSDME.

15a and 15d mediated HepG2 cells pyroptosis. (A) Representative light microscopy images of HepG2 cells. (B) LDH assay of LDH level in supernatants from cell cultures. (C) HepG2 cells were treated with 15a, 15d, and SAHA for 24 h at the indicated concentrations. The Cl-caspase3, GSDME-N protein levels were examined by Western blot analysis, and β-actin was detected as the endogenous loading control, accordingly. (D) The statistical result of (C). The data are expressed as means ± SD of three separate experiments. *p < 0.05, **p < 0.01, ***p < 0.001 versus control.
Pharmacokinetic studies of 15a
Due to the excellent activity against HDAC1, significant anti-proliferative and pro-apoptotic properties, the preliminary PK study of compound 15a in vivo was further evaluated in female Sprague–Dawley (SD) rats. Compound 15a was administrated 5.0 mg/kg intravenously (IV) and 30.0 mg/kg orally (PO), as in Table 6. 15a was rapidly cleared with the clearance (CL) of 408.8 L/min/kg and the half-life (T1/2) of 0.63 h after intravenous administration. The t1/2 value of oral administration of 30 mg/kg 15a was 1.00 h. The oral bioavailability of 15a was 10.3%, suggested that oral administration was not a suitable dosing route for compound 15a. Although a certain degree of improvement was achieved compared with 1b (2.6%), there is still much room for melioration.
Antitumor effect of 15a on HepG2 xenografts
To evaluate the inhibitory effect of 15a on solid tumors in vivo, a xenograft tumor model was established by subcutaneously implanting HepG2 cells into male BALB/c nude mice. Once the tumors reached a volume of 100 mm3, the mice were stratified into three groups: control, 15a (10 mg/kg) and SAHA (10 mg/kg). The mice in all groups were administered intraperitoneally every two days, with continuous monitoring of body weight and tumor dimensions. After a three-week treatment period, the mice were euthanized, and the tumors were excised, weighed, and photographed according to their respective treatment groups. As depicted in Fig. 9A–D, the growth of tumors was dose-dependently inhibited by 15a, with a superior inhibition observed at 10 mg/kg compared to that of SAHA. Furthermore, no significant adverse effects, such as obvious weight loss, were observed in the experimental groups.

15a suppressed HepG2 xenograft growth in immunodeficient mice. HepG2 cells (1 × 106) were implanted into BALB/c nude mice, which were then divided into three groups after a week of of tumor-cell implantation. (A) The weight of the mice was measured in each group. (B) Tumor volume (length × width2 × 0.5) was assessed every 2 days, and treatments included vehicle (5% DMSO in PBS, ip, n = 5), SAHA (10 mg/kg, ip, n = 5) and 15a (10 mg/kg, ip, n = 5). (C) The scatter plot depicted the tumor weights. (D) Representative images of the tumor specimens are shown after treatment with control, SAHA, and 15a for 2 weeks. (E) Hematoxylin and eosin (HE) staining results of tumor samples. Additionally, immunocytochemical staining for Ki67, Ac-H3, Ac-H4, cleaved caspase-3, and GSDME-N was performed on tumor tissues obtained from nude mice (Magnification, 400×. Scale bar, 40 μm). (F) The IHC results were evaluated using Image-Pro Plus 6.0 (n = 5 fields of view).
To further substantiate that 15a inhibited the activity of HDAC in vivo, immunohistochemical (IHC) analysis was conducted on tumor tissues collected at the end of the treatment phase in the xenograft model. As shown in Fig. 9E,F, a dose-dependent reduction in Ki67 immunostaining was observed, which is a reliable marker of diminished tumor cell proliferation. Concurrently, 15a induced the increase of Ac- H3/H4、Cl-caspase3 and GSDME-N in a dose-dependent manner. Collectively, these findings underscore the significant antitumor efficacy of compound 15a in HepG2 xenograft model, coupled with an acceptable safety profile, thereby highlighting its potential therapeutic value.
Molecular docking and molecular dynamics simulation
To elucidate the effective inhibition of HDAC1 by compound 15a, molecular docking studies were conducted to investigate how 15a interacts with HDAC1 (PDB ID: 4BKX). As shown in (Fig. 10A,C), 15a and SAHA exhibited similar binding patterns with HDAC1. Notably, two π-π stacking interactions were identified between the phenyl group of 15a and the residues TYR204 and PHE150, both located in the surface area of HDAC1. Additionally, another π-π stacking interaction was noted between the thiazolyl group of 15a and PHE205, which further enhanced the stability of the binding interaction between 15a and HDAC1 (Fig. 10B). Furthermore, in comparison to the positive control drug SAHA, compound 15a demonstrated a higher binding affinity to HDAC1, reflected by a lower docking score of -7.32, while SAHA displayed a moderate binding affinity with a docking score of -4.82. As opposed to SAHA, three conventional hydrogen bonds were formed between 15a and amino acid residues HIS178, TYR303, and CYS151, all situated near the zinc ion-binding region, which contributed to the improved binding capacity of 15a (Fig. 10B,D). To further confirm the stability of the aforementioned binding pose, we acquired the initial complex system of 15a-HDAC1 and SAHA-HDAC1 from docking results. Subsequently, we performed 10 ns molecular dynamics (MD) simulations of the HDAC1-15a and SAHA-HDAC1 complexes in an aqueous environment using GROMACS (2020.6) with the Amber99 force field. Root-mean-square deviation (RMSD) analysis was conducted on the backbone atoms of HDAC1 and the heavy atoms of both 15a and SAHA to monitor the dynamic stability of the complexes. The results revealed that after 5 ns, the SAHA-HDAC1 complex achieved an equilibrium state, with an average RMSD of 0.8 nm during the 100 ns period. The complex system of 15a -HDAC1 demonstrated improved conformational stability, reaching equilibrium after 50 ns, with an average RMSD of 0.80 nm over the 50–100 ns timeframe, which contributed to the enhanced stabilization of the protein–ligand complex (Fig. 11A). In order to examine the fluctuation and flexibility of the residues throughout the 10 ns simulation, we calculated the Root Mean Square Fluctuation (RMSF) for both the 15a -HDAC1 and the SAHA-HDAC1 complex systems. A negative correlation was observed between the RMSF values and the side chain atoms. During the simulation, we noted similar RMSF values for both complex systems (Fig. 11B). Notably, the 15a -HDAC1 system exhibited lower RMSF values in the HIS178, TYR303, and CYS151 amino acid regions, located in the active site and stabilized by 15a (Fig. 11B). Furthermore, an analysis of the hydrogen bond interactions between the two ligands and HDAC1 indicated that 15a forms a significantly greater number of hydrogen bonds within the binding pocket than SAHA during the 50–100 ns simulation (Fig. 11C). These findings suggest that 15a possesses a strong affinity towards the active site pocket of HDAC1, thereby inhibiting its catalytic activity.

Molecular binding model involving 15a and SAHA with HDAC1. (A) The three-dimensional binding configuration of 15a (orange) in the active site of HDAC1. The representations of the protein and the ligands, HDAC1 and 15a, are depicted using cartoon and stick formats respectively, with important residues highlighted as green sticks, while hydrogen bonds are indicated by red dashed lines. (B) A diagrammatic representation illustrating the interactions between the residue sites of HDAC1 and 15a, created with the BIOVIA Discovery Studio Visualizer software. The ligand is represented by a gray line; green dashed lines signify conventional hydrogen bonds, light green dashed lines denote Van der Waals interactions, deep pink dashed lines depict Pi–Pi stacking, and light pink dashed lines illustrate Alkyl interactions. (C) The three-dimensional binding configuration of SAHA (illustrated in red) in the active site of HDAC1. The representations of the protein and ligands, HDAC1 and SAHA, follow the same format as above, with key residues marked as green sticks and hydrogen bonds indicated by red dashed lines. (D) The three-dimensional binding configuration of SAHA (illustrated in red) in the active site of HDAC1. The representations of the protein and ligands, HDAC1 and SAHA, follow the same format as above, with key residues marked as green sticks and hydrogen bonds indicated by red dashed lines.

Molecular dynamic simulation involving 15a and SAHA with HDAC1. (A) Backbone root-mean-square deviations (RMSD) of HDAC1 throughout the 100 ns molecular dynamics simulation, with the 15a-HDAC1 complex depicted by a blue line and the SAHA-HDAC1 complex shown by a red line. (B) The Root mean square fluctuation (RMSF) values of HDAC1 when complexed with 15a (red) versus those complexed with SAHA (green) over the span of 100 ns. (C) The count of hydrogen bonds formed between the protein and the ligands during their structural rearrangement.
Conclusions
In summary, a series of phenyl-thiazole containing hydroxamic acid HDACIs were designed, synthesized, and evaluated for their antitumor activity both in vitro and in vivo. Most of the compounds exhibited enhanced HDAC1 inhibitory activity and excellent antiproliferative effects against various tumor cell lines compared to SAHA. The results indicated that the introduction of the phenyl-thiazole structure improved the in vitro inhibitory activities. Through SAR studies, representative compounds 15a and 15d were selected for further biological evaluation. Mechanistically, compounds 15a and 15d could upregulate the acetylation of Ac-H3 and Ac-H4 due to their inhibition of HDAC1. Colony formation assay clearly demonstrated that compounds 15a and 15d suppressed the colony-forming ability of HepG2 cells in a dose-dependent manner. Additionally, these two compounds induced G0/G1-phase cell cycle arrest. More importantly, compounds 15a and 15d induced apoptosis and pyroptosis through the caspase-3-mediated cleavage of gasdermin E (GSDME). Furthermore, the preliminary pharmacokinetic properties of 15a were also investigated. Moreover, 15a exhibited significant antitumor activity in the HepG2 xenograft model, which were superior to the positive control SAHA. Finally, the potential binding mode of 15a with HDAC1 was further evaluated by molecular docking and molecular dynamics simulation. Collectively, our current study suggested that these hydroxamate-based phenyl-thiazole containing bis-substituted aromatic amide HDACIs could be further investigated as potential anti-tumor agents in the future.
Data availability
All data generated or analysed during this study are included in this published article [and its supplementary information files].
References
Dizon, D. S. & Kamal, A. H. Cancer statistics 2024: All hands on deck. Cancer J. Clin. 74(1), 8–9 (2024).
Yu, X. et al. Cancer epigenetics: From laboratory studies and clinical trials to precision medicine. Cell Death Discov. 10(1), 28 (2024).
Wu, K.-J. et al. Recent discoveries of the role of histone modifications and related inhibitors in pathological cardiac hypertrophy. Drug Discov. Today 29(2), 103878–103878 (2024).
Badie, A., Gaiddon, C. & Mellitzer, G. Histone deacetylase functions in gastric cancer: Therapeutic target?. Cancers 14(21), 5472 (2022).
Hontecillas-Prieto, L. et al. Synergistic enhancement of cancer therapy using HDAC inhibitors: Opportunity for clinical trials. Front. Genet. 11, 578011 (2020).
Ramaiah, M. J., Tangutur, A. D. & Manyam, R. R. Epigenetic modulation and understanding of HDAC inhibitors in cancer therapy. Life Sci. 277, 119504 (2021).
Singh, T., Kaur, P., Singh, P., Singh, S. & Munshi, A. Differential molecular mechanistic behavior of HDACs in cancer progression. Med. Oncol. 39(11), 171 (2022).
Hai, R., He, L., Shu, G. & Yin, G. Characterization of histone deacetylase mechanisms in cancer development. Front. Oncol. 11, 700947 (2021).
Shirbhate, E. et al. Dual inhibitors of HDAC and other epigenetic regulators: A novel strategy for cancer treatment. Eur. J. Med. Chem. 263, 115957 (2024).
Zhao, C. et al. Discovery of novel fedratinib-based HDAC/JAK/BRD4 triple inhibitors with remarkable antitumor activity against triple negative breast cancer. J. Med. Chem. 66(20), 14150–14174 (2023).
Psilopatis, I. et al. The emerging role of histone deacetylase inhibitors in cervical cancer therapy. Cancers 15(8), 2222–2242 (2023).
Li, X. et al. Chidamide triggers pyroptosis in T-cell lymphoblastic lymphoma/leukemia via the FOXO1/GSDME axis. Chin. Med. J. (Engl.) 137(20), 2273–2284 (2024).
Ren, H. et al. Co-delivery nano system of MS-275 and V-9302 induces pyroptosis and enhances anti-tumor immunity against uveal melanoma. Adv. Sci. (Weinheim, Ger.) 11(31), 2404375 (2024).
Roy, R., Ria, T., RoyMahaPatra, D. & Sk, U. H. Single inhibitors versus dual inhibitors: Role of HDAC in cancer. ACS Omega 8(19), 16532–16544 (2023).
Zhang, W.-X. et al. A review of progress in o-aminobenzamide-based HDAC inhibitors with dual targeting capabilities for cancer therapy. Eur. J. Med. Chem. 259, 115673 (2023).
Geurs, S., Clarisse, D., De Bosscher, K. & D’Hooghe, M. The zinc-binding group effect: Lessons from non-hydroxamic acid vorinostat analogs. J. Med. Chem. 66(12), 7698–7729 (2023).
Ru, J. et al. Technologies of targeting histone deacetylase in drug discovery: Current progress and emerging prospects. Eur. J. Med. Chem. 261, 115834 (2023).
Kell, J. Drug evaluation: MGCD-0103, a histone deacetylase inhibitor for the treatment of cancer. Curr. Opin. Investig. Drugs 8(6), 485–492 (2007).
Venugopal, B. et al. A phase I study of Quisinostat (JNJ-26481585), an oral hydroxamate histone deacetylase inhibitor with evidence of target modulation and antitumor activity, in patients with advanced solid tumors. Clin. Cancer Res. 19(15), 4262–4272 (2013).
Popat, R. et al. A phase I dose-escalation study of the class 1 selective histone deacetylase inhibitor CHR-3996 in combination with tosedostat for patients with relapsed, refractory multiple myeloma: Results of the Muk three trial. Blood 128(22), 2597–2606 (2016).
Shan, P. et al. Alteration of MDM2 by the small molecule YF438 exerts antitumor effects in triple-negative breast cancer. Can. Res. 81(15), 4027–4040 (2021).
Shan, P. et al. A novel histone deacetylase inhibitor exerts promising anti-breast cancer activity via triggering AIFM1-dependent programmed necrosis. Cancer Commun. 42(11), 1207–1211 (2022).
Liu, M. et al. Discovery of novel Src homology-2 domain-containing phosphatase 2 and histone deacetylase dual inhibitors with potent antitumor efficacy and enhanced antitumor immunity. J. Med. Chem. 65(18), 12200–12218 (2022).
Yang, F. et al. Design and optimization of novel hydroxamate-based histone deacetylase inhibitors of bis-substituted aromatic amides bearing potent activities against tumor growth and metastasis. J. Med. Chem. 57(22), 9357–9369 (2014).
Yang, F. et al. Development of hydroxamate-based histone deacetylase inhibitors containing 1,2,4-oxadiazole moiety core with antitumor activities. Bioorg. Med. Chem. Lett. 29(1), 15–21 (2019).
Yang, F. et al. Synthesis and biological evaluation of thiophene-based hydroxamate derivatives as HDACis with antitumor activities. Future Med. Chem. 12(8), 655–672 (2020).
Duan, N. et al. Targeting the E2F1/Rb/HDAC1 axis with the small molecule HR488B effectively inhibits colorectal cancer growth. Cell Death Dis. 14(12), 479 (2023).
Nayak, S. & Gaonkar, S. L. A review on recent synthetic strategies and pharmacological importance of 1,3-thiazole derivatives. Mini-Rev. Med. Chem. 19(3), 215–238 (2019).
Farghaly, T. A., Alfaifi, G. H. & Gomha, S. M. Recent literature on the synthesis of thiazole derivatives and their biological activities. Mini-Rev. Med. Chem. 24(2), 196–251 (2024).
Liu, H. et al. Current development of thiazole-containing compounds as potential antibacterials against methicillin-resistant Staphylococcus aureus. ACS Infect. Dis. 10(2), 350–370 (2024).
Pallavi, H. M., Zabiulla, & Khanum, S. A. An overview of the synthetic routes and pharmacological aspects of pyridine, isoxazole, thiazole, and indole derivatives. Polycycl. Aromat. Compd. 44(10), 7168–7196 (2023).
Liang, T. et al. Targeting histone deacetylases for cancer therapy: Trends and challenges. Acta Pharmaceut. Sin. B 13(6), 2425–2463 (2023).
Chen, W. et al. Dual NAMPT/HDAC inhibitors as a new strategy for multitargeting antitumor drug discovery. ACS Med. Chem. Lett. 9(1), 34–38 (2018).
Liang, T. et al. HDAC-bax multiple ligands enhance bax-dependent apoptosis in HeLa cells. J. Med. Chem. 63(20), 12083–12099 (2020).
Morris, G. M. et al. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 30(16), 2785–2791 (2009).
Forli, S. et al. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 11(5), 905–919 (2016).
Wang, J., Wolf, R. M., Caldwell, J. W., Kollman, P. A. & Case, D. A. Development and testing of a general amber force field. J. Comput. Chem. 25(9), 1157–1174 (2004).
Sprague, W. Arrive guidelines 2.0. Vet. Clin. Pathol. 49(3), 378–379 (2020).
Miyake, Y. et al. Structural insights into HDAC6 tubulin deacetylation and its selective inhibition. Nat. Chem. Biol. 12(9), 748 (2016).
Wagner, F. F. et al. Kinetic and structural insights into the binding of histone deacetylase 1 and 2 (HDAC1, 2) inhibitors. Bioorg. Med. Chem. 24(18), 4008–4015 (2016).
Sang, J. et al. Jolkinolide B targets thioredoxin and glutathione systems to induce ROS-mediated paraptosis and apoptosis in bladder cancer cells. Cancer Lett. 509, 13–25 (2021).
Wang, Y. et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature (London) 547(7661), 99–103 (2017).
Yu, J. et al. Cleavage of GSDME by caspase-3 determines lobaplatin-induced pyroptosis in colon cancer cells. Cell Death Dis. 10(3), 193 (2019).
Acknowledgements
This project was financially supported by the Natural Science Foundation of Shandong Province (ZR2022MB140); the Natural Science Foundation of Shanghai (Grant No. 21ZR1427300), Shanghai Frontiers Research Center of the Hadal Biosphere.
Author information
Authors and Affiliations
Contributions
Conceptualization, F.Y. and N.L.; methodology, Z.L. and H.Q.; software, R.Z.; validation, W.L., and N.D.; formal analysis, S.F.; investigation, X.L.; writing—original draft preparation, H.Q. and Z.L.; writing—review and editing, H.Z.; supervision, F.Y.; funding acquisition, F.Y. and N.L.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, Z., Qiu, H., Lu, W. et al. Design and synthesis of thiazole-based hydroxamate histone deacetylase inhibitors with potent antitumor efficacy by inducing apoptosis, pyroptosis and cell cycle arrest. Sci Rep 15, 24589 (2025). https://doi.org/10.1038/s41598-025-08474-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-08474-5
