
Abstract
Induced pluripotent stem cell (iPSC)-derived motor neurons (MNs) offer a promising model system for understanding motor neuron diseases (MNDs) and advancing drug discovery. However, variability in differentiation outcomes presents a major barrier to reproducibility and model reliability. This study evaluates a widely adopted small molecule protocol for MN differentiation to quantify variability and identify its sources within an industrial setting. Analysing data from 15 differentiation sets across 8 cell lines, we found that non-genetic factors – particularly induction set and operator – were the predominant sources of variability, outweighing the contribution from cell line genetics. We further demonstrated that iPSC genomic instability, as assessed by a targeted RT-qPCR assay for common karyotypic abnormalities, significantly affected differentiation efficiency and purity. Cultures derived from genomically stable iPSCs exhibited reduced variance and improved MN marker expression profiles. These findings support routine genomic assessment of iPSCs as a practical and effective strategy to enhance the reliability of iPSC-derived MN models, thereby improving their utility in preclinical MND research and therapeutic development.
Similar content being viewed by others


Introduction
Motor neuron disease (MND), the third most prevalent neurodegenerative condition after Alzheimer’s and Parkinson’s, is a highly fatal, rapidly progressive, and complex disease. Typical patient survival is 2–3 years post symptom onset, and given the greatest incidence is observed between 50 and 85 year olds, there is a rapid expected increase in global burden as populations age1. Current therapeutic options yield minimal benefit in broad patient populations2,3,4, and as such there is a strong demand for a new pipeline of developed therapeutics.
Unfortunately, modelling MND for drug discovery purposes remains challenging, partly due to the range of pathological mechanisms implicated, many of which are backed by genetic associations5,6,7,8,9,10. Given the cellular complexity and mechanistic heterogeneity of MND, strong support exists for use of patient derived iPSC derived motor neurons as a model to enable disease interrogation and preclinical drug discovery.
However, these iPSC models encounter significant reproducibility challenges. When in a pluripotent state, they are highly sensitive to external environmental cues, modifying internal genomic architecture, becoming unsuitable for differentiations11. Whilst protocols using transcription factor driven differentiation can overcome some of these challenges, demonstrating greater reproducibility, purity, and decreased cost/generation time12,13,14, these genetic methods fail to mimic neurodevelopment.
Given the importance of cellular identity for hypothesised subtle phenotypes within MND, mimicking these developmental pathways are key. As such, preference exists for small molecule driven protocols that can provide highly guided developmental routes via promotion of key signalling cascades. Such protocols have been developed, and provide motor neurons reproducibly from iPSCs15,16, however inconsistencies remain17.
Here, we examine suitability of the widely used Hall et al. small molecule driven protocol for motor neuron differentiation18 to support early drug discovery in an industrial environment. We assess variability in the purity of differentiated cells and using statistical analysis, attempt to identify which factors (namely operator, induction set and cell line) are the main sources of variability. We conclude that assessing iPSC genomic stability is an accessible monitoring factor for reducing model variability.
Results
Variability is high and more greatly influenced by non-genetic factors
Using data generated across 15 separate rounds of differentiation (where a round can include more than one cell line), herein termed “induction sets”, and 8 cell lines (with a minimum of 3 induction sets per cell line, and a minimum of 3 induction sets per operator), motor neuronal characteristic, herein referred to as QC (Quality Control) metrics (detailed in Table 1), were analysed to describe variability and assess robustness of the differentiation protocol described by Hall et al.18. Statistical testing focused on exploring the magnitude of variance generated, and how much variation was attributable to key factors (namely cell line, induction set, and operator). Selection of factors was based upon importance described from literature in similar settings19, and those pertaining to handling of cells, namely “induction set” given each differentiation attempt will be subject to expected undefinable variations in environmental and reagent conditions.
Coefficient of variance analysis suggested that most quantifiable factors displayed high levels of variation (> 30% – see Table 2), with over half of these metrics exceeding 40%, far beyond acceptable levels for an industrial environment (ideally < 20%)20. Further linear modelling suggested that “Operator”, followed by “Induction Set” explained large amounts of variation within each of the QC metrics as demonstrated by high R2 values (i.e. the amount of variation that can be attributed to this factor) and low p-values associated with each (p < 0.05 in all cases). Whilst “Cell Line” was a significant explanatory factor for variation (p < 0.05 in all cases), generally this remained low in magnitude, with R2 values < 12% in the majority of cases.
Testing to assess independence between key factors and metrics with a binary outcome (i.e. “Yes” or “No”) highlighted that a significant association existed between “Induction Set” and whether non-neuronal morphology was present or cultures possessed cellular clusters > 5000µm2 (Non-neuronal morphology: χ2 = 20.79, df = 12, p < 0.05; Cellular clusters > 5000µm2: χ2 = 23.04, df = 12, p < 0.05). This relationship was however lacking when examined against “Cell Line” and “Operator”.
Overall statistical data highlights that variability in differentiation quality can be linked strongly to “Induction Set” and “Operator”, and less so to “Cell Line”. Whilst some variability continues to remain attributable to cell line (see Table 2), it remains low and provides remit to investigate factors more amenable to being controlled (such as handling procedures) to improve differentiation quality.
iPSC genomic stability is a key determinant of differentiation protocol performance and variability
Following previous studies in the literature, it is known that iPSC genomic architecture can change over time whilst these cells are in culture11 and this can have a noticeable impact on differentiation protocols21. We assessed iPSC karyotypic abnormalities on motor neuron differentiations using the Hall et al. protocol18, to understand if this knowledge can be used to reduce variability of QC metrics in produced material.
Using a bulk RT-qPCR based assay, with primers designed to target the nine most common karyotypic abnormalities in human iPSCs (as designed by STEMCELL Technologies), five rounds of differentiations were carried out across two cell lines to assess the impact of genomic stability. Cell lines with chromosomal copy numbers < 1.5 or > 2.5 (< 0.7 or > 1.3 for chromosome X for lines from male donors) were considered “abnormal”. One-way ANOVAs comparing chromosomal copy number across all loci with post hoc Tukey test p-value adjustment were then used to confirm these chromosomal deletions.
Differentiations from cell lines with no detectable abnormalities commonly displayed decreased coefficient of variance values for QC metrics when compared against all differentiation sets, consequently meaning these were less variable (see Table 3). Differentiations from these cells also showed significantly greater purities at the neural precursor and terminal differentiation day 3 stage, as determined by quantitative immunocytochemistry co-expression analysis for relevant neural precursor and motor neuronal markers (see Table 3 for details).
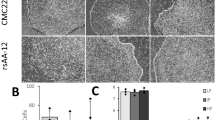
Morphological abnormalities were clearly present in cell lines harbouring confirmed or suspected chromosomal deletions, with confirmed cases (i.e. copy number < 1.5, adjusted p-value < 0.05) displaying complete lack of motor neuronal organisation (see Fig. 1A) and suspected cases (i.e. copy number < 1.5, adjusted p-value > 0.05) retaining some neuronal networks but with extremely dense clusters and rapidly proliferating cells with non-neuronal morphology (see Fig. 1B). In cell lines with no chromosomal deletions, typical neuronal structuring with no clear morphological abnormalities were observed (see Fig. 1C). Detected chromosomal abnormalities are detailed in Table 4.

Representative images of motor neurons terminally differentiated for 10 days subject to karyotypic abnormality analysis with states as follows: (A) detected chromosomal copy numbers of Chr8q, Chr17q, ChrXp < 1.5, lowest adjusted p-value < 0.034; (B) detected chromosomal copy numbers of Chr1q, Chr18q < 1.5, lowest adjusted p-value < 0.93; (C) detected chromosomal copy numbers normal. Targeted karyotyping for common abnormalities performed via bulk qPCR assay. Merge image colourings: Blue = Hoechst; Red = MAP2; Orange = ISL1; scale bar = 100 μm.
Discussion
“Induction set” shown to be greatest contributor to variability of culture purity.
Variance and statistical analysis show that factors affecting variability in order of largest to smallest contributor were: induction set > operator > cell line.
Large scale studies into reproducibility of differentiation success from iPSCs have described similar contributors22, largely grouped into genetic and non-genetic factors, with each contributing 20% and 40–60% to overall variation respectively19. These numbers align with calculated values from variability studies carried out in this study for genetic (i.e. cell line) and non-genetic factors (i.e. induction set and operator) (~ 2–30% and 30–70% respectively).
Interestingly, variability attributable to genetic factors (i.e. cell line) remained low yet persistent across most QC metrics. Given this is an inherent biological property that cannot be readily controlled or changed, it is encouraging that the magnitude of this response is low across metrics and multiple studies19,22. However, proposed approaches to account for this genetic variation, including use of “Rosetta lines” to normalise data between multiple cell lines and remove technical variation19, warrant further investigation. Cell lines used in this study are widely available from the HipSci (Human Induced Pluripotent Stem Cell Initiative) project23 and National Institute of Neurological Disorders and Stroke (NINDS) repository, and as such, provide datasets to normalise studies against if required.
Variability attributable to non-genetic factors (i.e. induction set and operator) by far outweigh contribution to variance from genetic factors (i.e. cell lines). This was explored further in the data with linear modelling of self-reported operator years of cell culture experience (general, complex model, and iPSC, ranging 6–20 years), however no results of significance were observed (see supplementary table S1). This most likely suggests handling techniques that cannot be accurately recorded, unique to each operator, play a key role and should be considered in future similar studies. It has been previously demonstrated variation due to cellular heterogeneity (i.e. cell line) is amplified during technical handling of iPSCs22. As handling time increases, this variation continues to grow and amplify, aligning with statistical findings here that non-genetic factors/overall handling was the biggest contributor to variability. Furthermore, additional studies examining gene expression profiles show a higher variability in iPSC derived models when compared against the primary cell culture counterparts they are intended to model24 confirming that the differentiation process itself (analogous to “induction set” in our analysis) is introducing a significant amount of variation, supporting our statistical analysis. The use of large scale automated systems have been shown to reduce this source of variation25, effectively controlling for “induction set” and “operator” given the repeatable environment and handling cells are subject to. However, such systems are not widely available, and other handling techniques beyond operator training to reduce this variability are required.
iPSC genomic instability dictates differentiation culture purity and variability
Assessment of iPSC karyotypic abnormality status via bulk qPCR was shown to provide a quantifiable approach to reducing differentiation failure rates and improving variability. It has been well characterised that iPSCs with abnormalities possess reduced differentiation capacity, for example loss of neuroectoderm differentiation capacity following a 20q11.21 gain, or a failure to complete hepatic progenitor generation following a 12p13.31 gain26,27.
Interestingly, instability of chromosomes 12,17,18q and 20q all pertained to increased oncogenic activity in cells28,29,30,31 – this is a major issue within a clinical transplantation setting but also not desirable within a drug discovery setting. Within differentiations that were impure, a high proliferation rate was seen of either motor neuronal cells, or those of unknown origin. In the case of a 17q deletion, an extremely large increase in cells was observed in cultures, akin to previous studies29. Whilst they may appear motor neuronal in origin, these are effectively cancerous cells that are not physiologically representative or usable within a drug discovery setting.
Given low level chromosomal mosaicism has been shown in multiple routine iPSC cultures32,33, continued monitoring of cells for genomic stability is key to prevent oncogenesis and adverse differentiations. Ultimately this will reduce overall variability of produced cellular material.
Conclusion
In summary, this study highlights that whilst variation in motor neuronal purity remains high using the Hall et al. protocol (with coefficient of variance values exceeding 20%), this can partially be controlled for via regular iPSC chromosomal assessment. This assessment can be performed through simple, accessible and cost efficient methods such as RT-qPCR. Current variability defined here (both genetic and non) remains largely in line with values described elsewhere in literature19, and whether this can be reduced further remains a current hurdle within the field. A significant portion of this variability is attributable to non-genetic factors (~ 30–70%), and as such further in-depth technical analysis is required and encouraged for groups using this differentiation protocol regularly. At the very least both induction set and operator need to be included as factors in experimental design and as potential confounders in statistical analysis.
However in lieu of this knowledge, we show focussing on defining chromosomal stability is an accessible strategy for providing reproducible cellular models, especially within the motor neuronal space. Subsequently, this facilitates improved model interrogation and therapeutic discovery strategies within the MND field.
Methods
Cell line details
Cell lines sourced from HipSci (line ID): WTSli010-A, WTSli022-A, WTSli076-A; cell lines sourced from National Institute of Neurological Disorders and Stroke (line ID): NH500217, NH50305, NH50306, NH50218, NH50219. Cells thawed from minimum of passage 10 and maximum of passage 20. Cell line authenticity, sterility and mycoplasma free status confirmed internally.
iPSC maintenance
6 well tissue culture plates (Corning) for iPSC growth were coated with Geltrex (Gibco) at a 1:100 dilution in DMEM/F12 (Gibco) – plates were incubated at 37 °C, 5% CO2 1 h minimum/1 week maximum prior to cell seeding.
iPSC vials frozen in liquid nitrogen were thawed using E8 media with supplement (Gibco) and 10µM Y-27,632 (ROCK inhibitor; STEMCELL Technologies) (original Y-27632 stock dissolved in water). Media was changed next day for E8 with no Y-27,632 – cells were incubated at 37 °C, 5% CO2.
Cells were passaged with 0.5mM EDTA (Invitrogen) in PBS (Thermo Fisher), receiving daily media changes of 2mL/well E8 media with supplement warmed to room temperature.
Motor neuron differentiation
Protocol originally described by Hall et al.18. Briefly, iPSCs were allowed to reach 90–100% confluency during routine maintenance before starting a differentiation. Basal differentiation media was made as 50:50 aliquots of Neurobasal plus and DMEM/F12 with 1X B27 plus supplement, 1X Glutamax, 1X non essential amino acids, 1X N2 supplement, 10µM 2-mercaptoethanol, and 0.5 µg/mL insulin. Relevant small molecules were added for each differentiation stage, and possessed a final DMSO concentration of < 0.01%.
Daily differentiation media changes (warmed to room temperature) were performed (minimum of 2mL/well) and incubated at 37 °C, 5% CO2. Cells were treated with differentiation media across an 18 day period as follows: Day 0–6 = 1µM Dorsomorphin (Sigma), 2µM SB431542 (STEMCELL Technologies), 3µM CHIR99021 (STEMCELL Technologies); Day 7–13 = 0.5µM Retinoic acid (Tocris), 1µM Purmophamine (Sigma); Day 14–17 = 0.1µM Purmorphamine (Sigma); Day 18 (19–38 optional for cycling at neural precursor stage) = 10ng/mL FGF (Peprotech); Day 19 earliest for motor maturation = 0.1µM Compound E (Enzo Life Sciences).
Cells were split using dispase (Gibco) dissolved in PBS (10 mg/mL), and reseeded in appropriate differentiation media with 10µM Y-27,632. Splits were performed no earlier than day 4 of the differentiation (1:2 splitting ratio), and no more than 2 additional times (1:3 splitting ratios) over the whole 18 day period. Once cells had reached day 18 and achieved a neural precursor identity, cells were passaged with accutase (Invitrogen) and reseeded in differentiation media containing 10ng/mL FGF and 10µM Y-27,632.
Maturation was performed in 96 well imaging plates (Revvity) coated with Geltrex. Neural precursor cells were seeded in differentiation media containing 10ng/mL FGF and 10µM Y-27,632, and after a 24 h incubation at 37 °C, 5% CO2 for 24 h, media was aspirated and replaced with differentiation media containing 0.1µM Compound E.
Cell culture purity assessment – Immunocytochemistry
Motor neurons were matured in 96 well plates and fixed with 4% formaldehyde for 20 min at room temperature. Cells were then washed with PBS, permeabilised with 0.5% triton in PBS (Sigma) for 20 min, blocked with 10% bovine serum albumin (Sigma) and 20% fetal bovine serum in PBS (Gibco), and treated with primary antibody in 0.25% triton and 5% bovine serum albumin overnight at 4 °C. Wells were then washed with PBS and treated with secondary antibody in 0.25% triton and 5% bovine serum albumin for 1 h in the dark at room temperature. Cells were then washed with PBS and stained with Hoechst 33,342 (Thermo Fisher) in PBS for 10 min in the dark at room temperature. Cells were then washed in PBS, and imaged on the INCell Analyzer 6500 (GE Healthcare).
Images were analysed in Signals Image Artist (Revvity) using an automated pipeline and custom built script for each plate, with intensity thresholds for positive expression defined by iPSCs seeded on the plate (known to not express targets of interest28 and Hoechst intensity thresholds defining cell viability where appropriate.
Primary antibodies: NANOG (MABD24 – Merck), OCT4 (ab19857 – abcam), PAX6 (ab195054 – abcam), OLIG2 (157H00010215-M02 – Tebu), SMI32 (801701 – Biolegend), ISL1 (ab109517 – abcam), MAP2 (ab92434 – abcam).
Secondary anitbodies: Alexa Fluor 488 (A32723 – Invitrogen, Alexa Fluor 594 (A32740 – Invitrogen), Alexa Fluor 647 (A32933 – Invitrogen).
iPSC karyotypic abnormality assessment
iPSCs were cultured as previously described and prior to starting a differentiation, DNA was extracted from iPSCs grown in parallel from the same starting cellular material and purified using a DNA isolation kit (STEMCELL technologies) following the manufacturers’ protocol. Samples were frozen at -20 °C until ready to process.
Upon thawing on ice, DNA concentration and quality was assessed on a nanodrop (Thermo Fisher). Material with an A260/280 reading of 1.8–2.0 and an A260/230 reading of 1.9–2.2 was considered acceptable for further processing. Material was then prepared for qPCR assessment via a genetic analysis kit as per the manufacturers’ protocol (STEMCELL technologies) – primers were designed to detect 70% of the most recurrent karyotypic abnormalities reported in human stem cell cultures found in Chr 1q, 8q, 10p, 12p, 17q, 18q, 20q, Xp (primers available from STEMCELL technologies - #07550).
The ViiA 7 Real-Time PCR System (Thermo Fisher) was used to take readings in a 384 well format, ΔCT values per loci were calculated via normalisation against Chr 4p (following manufacturer recommendations), and data was analysed using one way ANOVAs across all loci and post-hoc tukey tests to confirm p-values.
Linear model variability analysis
The following linear model was used to calculate variation attributable to key factors:
Where:
Analysis was performed in R, with linear models being created for each quantitative QC (quality control) metric as defined in the QC pipeline. Diagnostic plots (residuals vs. fitted, normal Q-Q, scale-location, and residuals vs. leverage) were examined in each case to determine suitability of linear modelling – in cases where models were not acceptable, data was log transformed if appropriate.
Statistics
Significance testing via t-tests (for quantitative data) and chi-squared tests (for qualitative data) were performed in in Prism (version 9.4.1; GraphPad) and Excel (Microsoft). Variance testing (F testing) was carried out prior to significance testing to determine whether heteroscedastic or homoscedastic testing should be applied.
Data availability
The datasets used and analysed during the current study are available from the corresponding author on reasonable request.
References
Logroscino, G. et al. Global, regional, and National burden of motor neuron diseases 1990–2016: a systematic analysis for the global burden of disease study 2016. Lancet Neurol. 17 (12), 1083–1097 (2018).
Hawrot, J., Imhof, S. & Wainger, B. J. Modeling cell-autonomous motor neuron phenotypes in ALS using iPSCs. Neurobiol. Dis. 134, 104680 (2020).
Fang, T. et al. Stage at which riluzole treatment prolongs survival in patients with amyotrophic lateral sclerosis: a retrospective analysis of data from a dose-ranging study. Lancet Neurol. 17 (5), 416–422 (2018).
Okada, M. et al. Long-term effects of Edaravone on survival of patients with amyotrophic lateral sclerosis. ENeurologicalSci 11, 11–14 (2018).
Mead, R. J., Shan, N., Reiser, H. J., Marshall, F. & Shaw, P. J. Amyotrophic lateral sclerosis: a neurodegenerative disorder poised for successful therapeutic translation. Nat. Rev. Drug Discovery. 22 (3), 185–212 (2023).
Hardiman, O. et al. Amyotrophic lateral sclerosis. Nat. Reviews Disease Primers. 3 (1), 1–19 (2017).
Gregory, J. M., Fagegaltier, D., Phatnani, H. & Harms, M. B. Genetics of amyotrophic lateral sclerosis. Curr. Genetic Med. Rep. 8, 121–131 (2020).
Shepheard, S. R. et al. Value of systematic genetic screening of patients with amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry. 92 (5), 510–518 (2021).
Chiò, A. et al. The multistep hypothesis of ALS revisited: the role of genetic mutations. Neurology 91 (7), e635–e642 (2018).
Bajc Česnik, A., Motaln, H. & Rogelj, B. The impact of ALS-associated genes hnRNPA1, MATR3, VCP and UBQLN2 on the severity of TDP-43 aggregation. Cells 9 (8), 1791 (2020).
Yoshihara, M., Hayashizaki, Y. & Murakawa, Y. Genomic instability of iPSCs: challenges towards their clinical applications. Stem cell. Reviews Rep. 13, 7–16 (2017).
Fernandopulle, M. S. et al. Transcription factor–mediated differentiation of human iPSCs into neurons. Curr. Protocols cell. Biology. 79 (1), e51 (2018).
Hester, M. E. et al. Rapid and efficient generation of functional motor neurons from human pluripotent stem cells using gene delivered transcription factor codes. Mol. Ther. 19 (10), 1905–1912 (2011).
Canals, I., Quist, E. & Ahlenius, H. Transcription factor-based strategies to generate neural cell types from human pluripotent stem cells. Cell. Reprogramming. 23 (4), 206–220 (2021).
Bianchi, F. et al. Rapid and efficient differentiation of functional motor neurons from human iPSC for neural injury modelling. Stem cell. Res. 32, 126–134 (2018).
Shimojo, D. et al. Rapid, efficient, and simple motor neuron differentiation from human pluripotent stem cells. Mol. Brain. 8, 1–15 (2015).
Sances, S. et al. Modeling ALS with motor neurons derived from human induced pluripotent stem cells. Nat. Neurosci. 19 (4), 542–553 (2016).
Hall, C. E. et al. Progressive motor neuron pathology and the role of astrocytes in a human stem cell model of VCP-related ALS. Cell. Rep. 19 (9), 1739–1749 (2017).
Volpato, V. & Webber, C. Addressing variability in iPSC-derived models of human disease: guidelines to promote reproducibility. Dis. Models Mech. 13 (1), dmm042317 (2020).
Jelliffe, R. W., Schumitzky, A., Bayard, D., Fu, X. & Neely, M. Describing assay precision—reciprocal of variance is correct, not CV percent: its use should significantly improve laboratory performance. Ther. Drug Monit. 37 (3), 389–394 (2015).
Puigdevall, P., Jerber, J., Danecek, P., Castellano, S. & Kilpinen, H. Somatic mutations alter the differentiation outcomes of iPSC-derived neurons. Cell. Genomics 3 (4), 100280 (2023).
Volpato, V. et al. Reproducibility of molecular phenotypes after long-term differentiation to human iPSC-derived neurons: a multi-site omics study. Stem cell. Rep. 11 (4), 897–911 (2018).
Streeter, I. et al. The human-induced pluripotent stem cell initiative—data resources for cellular genetics. Nucleic Acids Res. 45 (D1), D691–D697 (2017).
Schwartzentruber, J. et al. Molecular and functional variation in iPSC-derived sensory neurons. Nat. Genet. 50 (1), 54–61 (2018).
Crombie, D. E. et al. Development of a modular automated system for maintenance and differentiation of adherent human pluripotent stem cells. Slas Discovery: Adv. Life Sci. R&D. 22 (8), 1016–1025 (2017).
Markouli, C. et al. Gain of 20q11. 21 in human pluripotent stem cells impairs TGF-β-dependent neuroectodermal commitment. Stem cell. Rep. 13 (1), 163–176 (2019).
Keller, A. et al. Gains of 12p13. 31 delay WNT-mediated initiation of hPSC differentiation and promote residual pluripotency in a cell cycle dependent manner. bioRxiv, 2021.05.22.445238 (2021).
Kraggerud, S. M. et al. Genome profiles of familial/bilateral and sporadic testicular germ cell tumors. Genes Chromosom. Cancer. 34 (2), 168–174 (2002).
Bown, N. et al. 17q gain in neuroblastoma predicts adverse clinical outcome. Med. Pediatr. Oncology: Official J. SIOP—International Soc. Pediatr. Oncol. (Societé Int. D’Oncologie Pédiatrique). 36 (1), 14–19 (2001).
Popat, S. & Houlston, R. S. A systematic review and meta-analysis of the relationship between chromosome 18q genotype, DCC status and highectal cancer prognosis. Eur. J. Cancer. 41 (14), 2060–2070 (2005).
Nguyen, H. T. & Duong, H. The molecular characteristics of colorectal cancer: implications for diagnosis and therapy. Oncol. Lett. 16 (1), 9–18 (2018).
Keller, A. et al. Uncovering low-level mosaicism in human embryonic stem cells using high throughput single cell shallow sequencing. Sci. Rep. 9 (1), 14844 (2019).
Jacobs, K. et al. Low-grade chromosomal mosaicism in human somatic and embryonic stem cell populations. Nat. Commun. 5 (1), 4227 (2014).
Author information
Authors and Affiliations
Contributions
FG, DM, EV, ZY contributed to data generation. FG, EH, CL, RM, WS contributed to analysis. FG, RM, WS contributed to manuscript preparation. All authors contributed equally to manuscript review.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Gaffey, F., McCoull, D., Vaitone, E. et al. Examining iPSC derived motor neuron variability and genome stability monitoring as a solution. Sci Rep 15, 39670 (2025). https://doi.org/10.1038/s41598-025-23378-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-23378-0
This article is cited by
-
When the Scaffold Becomes the Signal: Making MSC Secretome Therapies Decision-Grade Through “Context-of-Manufacture” Potency Standards
Stem Cell Reviews and Reports (2026)
