
Abstract
Background Ribociclib, a CDK4/6 inhibitor, is a targeted therapy for breast cancer management. However, acquired resistance to ribociclib is a common clinical challenge. This study aimed to investigate the role of LEM-Domain Protein 4 (LEM4), a nuclear envelope protein, in mediating ribociclib resistance in breast cancer cells and elucidate the underlying molecular mechanisms. Methods LEM4 expression was analyzed in breast cancer patient samples and cell lines. Ribociclib-resistant MCF-7 and T47D cell lines were established, and the effects of LEM4 knockdown on drug sensitivity, apoptosis, and β-catenin signaling were evaluated using in vitro cytotoxicity, co-immunoprecipitation, β-catenin transcription activity and in vivo tumor formation assays. Results LEM4 was found to be overexpressed in breast cancer tissues and cell lines, and its high expression correlated with worse overall survival in patients. Ribociclib-resistant breast cancer cells demonstrated significantly elevated LEM4 expression levels, and LEM4 silencing re-sensitized drug-resistant cells to ribociclib-induced cytotoxicity and apoptosis in vitro and in vivo. Mechanistically, LEM4 interacted with β-catenin, increasing its protein stability, nuclear translocation, and transcriptional activity. Inhibition of β-catenin signaling reversed ribociclib resistance. Conclusion This study identifies LEM4 as a key mediator of ribociclib resistance in breast cancer cells through its interaction with β-catenin and subsequent activation of β-catenin signaling. Targeting the LEM4/β-catenin axis represents a potential therapeutic strategy to overcome ribociclib resistance in breast cancer.
Similar content being viewed by others
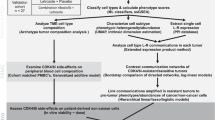


Introduction
Breast cancer represents the most prevalent malignancy and a leading cause of cancer-related mortality among women worldwide1. According to global cancer statistics, breast cancer accounted for approximately 2.3 million new cases and 666,000 deaths in 20221. While early-stage breast cancer is often treatable, metastatic or advanced disease remains a significant therapeutic challenge. Breast cancer is a heterogeneous disease with distinct molecular subtypes, including hormone receptor-positive, human epidermal growth factor receptor 2 (HER2)-positive, and triple-negative breast cancer, each requiring tailored treatment approaches2.
Hormone receptor-positive breast cancers, which express estrogen receptor (ER) and/or progesterone receptor (PR), are typically treated with endocrine therapies, such as selective estrogen receptor modulators (e.g., tamoxifen) or aromatase inhibitors (e.g., letrozole, anastrozole)3. HER2-positive breast cancers, characterized by the overexpression of the HER2 protein, are treated with HER2-targeted therapies, including monoclonal antibodies (e.g., trastuzumab, pertuzumab) and small molecule inhibitors (e.g., lapatinib)4. Triple-negative breast cancers, lacking expression of ER, PR, and HER2, present as more aggressive subtypes with limited targeted therapy options, often necessitating cytotoxic chemotherapy as the primary treatment modality5. Despite these tailored treatment strategies, a significant proportion of patients develop resistance to therapy, leading to disease progression and poor outcomes, highlighting the need for novel therapeutic approaches6,7.
Cyclin-dependent kinase (CDK) 4/6 inhibitors represent a significant advancement in the treatment of hormone receptor-positive, HER2-negative advanced or metastatic breast cancer8. These targeted therapies selectively inhibit CDK4 and CDK6, key regulators of the cell cycle, leading to cell cycle arrest and suppression of tumor growth8. Palbociclib, a first-in-class CDK4/6 inhibitor, has demonstrated remarkable efficacy in clinical trials9. The PALOMA-3 trial reported a significant improvement in progression-free survival (PFS) with the addition of palbociclib to fulvestrant in patients with hormone receptor-positive, HER2-negative metastatic breast cancer, with a median PFS of 9.5 months in the palbociclib group compared to 4.6 months in the placebo group9. CDK4/6 inhibitors are approved for the treatment of hormone receptor-positive, HER2-negative advanced or metastatic breast cancer in combination with endocrine therapy10. The estimated overall survival at 42 months among the patients who received ribociclib treatment as first-line therapy was 66.9%11. However, despite the initial clinical benefits, a significant proportion of patients eventually develop resistance to CDK4/6 inhibitors (palbociclib and ribociclib), limiting its long-term efficacy12,13.
The LAP2-Emerin-MAN1 (LEM) domain protein family is a group of inner nuclear membrane proteins involved in various cellular processes, including nuclear organization, gene regulation, and signal transduction14. While the specific functions of most LEM proteins remain incompletely characterized, compelling evidence suggests their potential involvement in cancer development and progression15,16. High levels of LEM-Domain Protein 4 (LEM4) expression are observed in tumor tissues of ER + BC, where it interacts with CDK4/6 and Rb to promote cell cycle advancement17. Overexpression of LEM4 increases resistance of ER + BC cells to endocrine therapy (ET), while depletion of LEM4 shows the opposite effect. Hence, LEM4 could serve as a predictive biomarker for ET resistance in ER-positive BC, and targeting LEM4 may enhance the efficacy of ET as a treatment strategy18. Nevertheless, the potential implication of LEM4 in CDK4/6 inhibitor drug resistance remains unexplored.
In the current study, we systematically investigated the mechanistic role of LEM4 in mediating ribociclib resistance in breast cancer cells and elucidated the underlying molecular pathways. Our findings demonstrate that LEM4 overexpression confers ribociclib resistance by enhancing the activity of the Wnt/β-catenin signaling pathway, a critical regulator of cell cycle progression and survival. Targeting the LEM4/β-catenin axis represents a potential therapeutic strategy to overcome ribociclib resistance and improve treatment outcomes in breast cancer patients.
Materials and methods
Clinical specimen collection
Tumor tissues and adjacent para-cancerous normal counterparts were collected from 60 breast cancer patients who underwent surgical resection at Yunnan Cancer Hospital, Kunming, China. The study was approved by Yunnan Cancer Hospital Ethics Committee for medical research (Approval NO.KYCS2023-014), and all patients provided written informed consent. Tissue samples were snap-frozen in liquid nitrogen and stored at −80 °C until further analysis.
Public dataset analysis
Raw RNA sequencing data from the Gene Expression Omnibus (GEO) database (accession number GSE98987) containing transcriptomic profiles of parental and palbociclib-resistant MCF7 cells were downloaded and processed using standard bioinformatics pipelines including quality control, adapter trimming, alignment to the human reference genome (GRCh38), and gene expression quantification using RSEM to generate Transcripts Per Million (TPM) normalized values. LEM4 expression levels were compared between palbociclib-sensitive and resistant cells using DESeq2 for differential expression analysis (FDR < 0.05). Gene Set Enrichment Analysis (GSEA) was performed using Molecular Signatures Database (MSigDB) Hallmark gene sets to evaluate Wnt/β-catenin pathway enrichment in resistant versus sensitive cells, with pathways considered significantly enriched at FDR q-value < 0.25 and |NES| > 1.0.
Cell culture and treatment
Human breast cancer cell lines MCF-7 (ATCC, HTB-22, Manassas, VA, USA) and T47D (ATCC, HTB-133), and normal breast epithelial cell line MCF-10 A (ATCC, CRL-10317) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM for MCF-7 and T47D, Gibco, 11965-092, Grand Island, NY, USA) or DMEM/F12 (for MCF-10 A, Gibco, 11330-032, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS, Gibco, 10099-141) and 1% penicillin-streptomycin (Gibco, 15140-122) at 37 °C in a humidified atmosphere with 5% CO2. For ribociclib treatment, cells were seeded at a density of 1 × 10^5 cells/well in 6-well plates and treated with 0.2 µM ribociclib (Selleckchem, S7440, Shanghai, China) for 48 h before cytotoxicity assay.
Transfection
Cells were transfected with siRNA targeting LEM4, siRNA targeting β-catenin or non-targeting control siRNA (GenScript, Nanjing, China) using Lipofectamine RNAiMAX transfection reagent (Invitrogen, 13778-150, Carlsbad, CA, USA) according to the manufacturer’s instructions. Briefly, cells were seeded at a density of 2 × 10^5 cells/well in 6-well plates and transfected with 50 nM siRNA for 48 h before subsequent experimental assays.
Establishment of ribociclib-resistant cell line
Ribociclib-resistant MCF-7 and T47D cell lines were established by continuously exposing parental cells to gradually increasing concentrations of ribociclib (0.1–0.8 µM) over 2–3 weeks at each concentration. Resistant cells were then maintained in the presence of 0.1 µM ribociclib.
Generation of stable knockdown in MCF-7 cells
Lentiviral particles carrying shRNA targeting LEM4 or non-targeting control shRNA (produced by Ubigene Biosciences, Guangzhou, China) were used to transduce MCF-7 and T47D cells according to the manufacturer’s protocol. Cells were seeded at a density of 2 × 10^5 cells/well in 6-well plates and transduced with lentivirus at a multiplicity of infection (MOI) of 10 for 48 h. Stable cell lines were selected with 800 ng/ml puromycin (Invitrogen, A1113803) for 2 weeks.
RT-qPCR analysis
Total RNA was extracted from cells (1 × 10^6) or tissue samples (100 mg) using TRIzol reagent (Invitrogen, 15596-026) following the manufacturer’s instructions. Reverse transcription was performed using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, 4368814, Foster City, CA, USA). Quantitative real-time PCR was conducted using PowerUp SYBR Green Master Mix (Applied Biosystems, A25742) on a StepOnePlus Real-Time PCR System (Applied Biosystems). The relative expression levels were calculated using the 2^-ΔΔCt method with GAPDH as the internal control.
Nuclear and cytoplasmic fraction collection
Nuclear and cytoplasmic fractions were isolated using the NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific, 78833, Waltham, MA, USA) according to the manufacturer’s instructions. Briefly, 1 × 10^6 cells were initially lysed using 500 µL cytoplasmic lysis buffer on ice for 15 min, followed by centrifugation at 600 g for 10 min. The collected supernatant was saved as the cytoplasmic fraction. The pellet was washed twice and then lysed using 500 µL nuclear lysis buffer on ice for 10 min. The lysate was centrifuged at 15,000 g for 10 min at 4 °C. The supernatant was saved as the nuclear fraction.
Immunoblotting
Cells (1 × 10^6) or tissue samples (50 mg) were lysed in RIPA buffer (Thermo Fisher Scientific, 89900) supplemented with Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific, 78440). Protein concentrations were determined using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, 23225). Equal amounts of protein (30 µg) were separated by SDS-PAGE and transferred to PVDF membranes (Millipore, IPVH00010, Burlington, MA, USA). After blocking with 5% non-fat dry milk in TBS-T (Tris-buffered saline with 0.1% Tween-20), membranes were incubated with primary antibodies overnight at 4 °C, followed by incubation with HRP-conjugated secondary antibodies (1: 5000, Cell Signaling Technology, 7074 S and 7076 S, Danvers, MA, USA) for 1 h at room temperature. Protein bands were visualized using the SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Fisher Scientific, 34580) and imaged using the ChemiDoc Imaging System (Bio-Rad, Hercules, CA, USA). Protein band quantification was performed using ImageJ software (NIH, Bethesda, MD, USA). The expression of target proteins was normalized against β-actin band as the loading control in each condition, and the expression levels were then presented relative to the normalized control sample. For cytoplasmic and nuclear translocation experiments, Histone H3 and GAPDH served as nuclear and cytoplasmic markers, respectively, and target protein expression was expressed as the ratio of nuclear to cytoplasmic fractions (Nuc: Cyto ratio). Primary antibodies used in this study were: LEM4 (1:1000, Abcam, ab225905, Cambridge, MA, USA), β-catenin (1:1500, Cell Signaling Technology, 8480 S), GSK3β (1:1000, Cell Signaling Technology, 9315 S), phospho-GSK3β (Ser9) (1:800, Cell Signaling Technology, 9323 S), Histone H3 (1:1000, Cell Signaling Technology, 9715 S), GAPDH (1:2000, Cell Signaling Technology, 5174 S), Ubiquitin (1;1000, Cell Signaling Technology, 3933) and β-Actin (1:1000, Cell Signaling Technology, 4967 S).
Lactate dehydrogenase (LDH) release assay
Cytotoxicity was assessed using the CytoTox 96 Non-Radioactive Cytotoxicity Assay Kit (Promega, G1780, Madison, WI, USA) according to the manufacturer’s instructions. Cells were seeded at a density of 1 × 10^4 cells/well in 96-well plates and treated under the indicated conditions. After treatment, culture supernatants were collected and mixed with the CytoTox 96 substrate solution. The absorbance was measured at 490 nm using a microplate reader (BioTek, Winooski, VT, USA).
CCK-8 cell growth analysis
Cell proliferation was evaluated using the Cell Counting Kit-8 (CCK-8, Dojindo, CK04, Kumamoto, Japan). Cells were seeded at a density of 5 × 10^3 cells/well in 96-well plates and treated under the indicated conditions. At the designated time points, 15 µL CCK-8 solution was added to each well and incubated for 2 h at 37 °C. The absorbance was measured at 450 nm using a microplate reader (BioTek, Winooski, VT, USA).
Apoptosis assay by flow cytometry
Apoptosis was detected using the Annexin V-FITC Apoptosis Detection Kit (Abcam, ab14085, Cambridge, MA, USA) according to the manufacturer’s protocol. Cells were seeded at a density of 2 × 10^5 cells/well in 6-well plates and treated under the indicated conditions. After treatment, cells were harvested, washed with cold PBS, and resuspended in 100 µL binding buffer containing 5 µL Annexin V-FITC and 5 µL propidium iodide (PI) for 15 min at room temperature in the dark. Apoptotic cells were analyzed using a BD FACSCanto II flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA).
Co-immunoprecipitation (Co-IP)
Cells (1 × 10^6) were lysed in 1mL of ice-cold IP lysis buffer (Thermo Fisher Scientific, 87788) supplemented with Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific, 78440). Lysates were pre-cleared with 100 µL of Protein A/G PLUS-Agarose Immunoprecipitation Reagent (Santa Cruz Biotechnology, sc-2003, Dallas, TX, USA) for 1 h at 4 °C. Supernatants were incubated with 6 µg of anti-LEM4 antibody (Abcam, ab225905) or anti-β-catenin antibody (Cell Signaling Technology, 8480 S) overnight at 4 °C, followed by incubation with 100 µL of Protein A/G PLUS-Agarose for 2 h at 4 °C. Immunoprecipitates were washed four times with IP lysis buffer and eluted by boiling in 2X SDS sample buffer (Bio-Rad, 1610737) for 5 min at 95 °C. Immunoblotting was performed as described above.
β-catenin transcription activity assay
The transcription-driving activity of β-catenin was measured using the TCF7 Transcription Factor Activity Assay Kit (Assay Genie, TFAB00132, Fremont, CA, USA) based on the requirement of β-catenin/TCF interaction to promote gene transcription. This assay utilizes an immobilized TCF7 consensus DNA binding sequence to capture activated β-Catenin/TCF complex, followed by the detection of captured protein using ELISA. Briefly, 100 µg nuclear protein fraction was added into each well in a 96-well plate with immobilized oligonucleotides for TCF7 binding for 2-hour incubation at room temperature. After washing three times with 200 µL washing buffer, the captured protein was detected using 100 µL of primary antibody for 1 h and 100 µL HRP-conjugated secondary antibody for 45 min. Signal development was conducted using 50 µL TMB (Tetramethylbenzidine) substrate for 15 min in the dark. The reaction was stopped with 50 µL stop solution and absorbance was measured at 450 nm using a microplate reader.
In vivo analysis of tumor formation
All animal experiments were conducted in accordance with the guidelines and protocols approved by the Ethical Review Committee for Animal Experiments of Kunming Medical University (Kmmu20240103). Female athymic nude mice (6–8 weeks old, Vital River Experimental Animal Technology, Beijing, China) were subcutaneously injected with 0.5 × 10^7 parental MCF-7 cells, ribociclib-resistant MCF-7 cells, or ribociclib-resistant MCF-7 cells expressing control shRNA (sh-NC) or shRNA targeting LEM4 (sh-LEM4) (n = 5 mice per group). After two weeks of tumor growth, mice were intraperitoneally injected with ribociclib (50 mg/kg body weight) every three days for three weeks. Tumor volumes were measured using a caliper and calculated as (length × width^2)/2. At the end of the experiment, mice were euthanized by cervical dislocation, and the xenograft tumors were excised, weighed, and processed for further analysis. TUNEL staining was performed on formalin-fixed, paraffin-embedded tumor sections cut at 5 μm thickness using a colorimetric TUNEL cell death staining kit (Baiao Leibo Technology, YT137, Beijing, China) according to the manufacturer’s instructions. TUNEL-positive cells were counted in five random fields per section at 400× magnification.
Statistics
All experiments were performed at least three times, and data are presented as mean ± standard deviation (SD). Statistical analysis was performed using GraphPad Prism version 9.0 software (GraphPad Software, San Diego, CA, USA). Differences between two groups were analyzed using two-tailed Student’s t-test, while multiple group comparisons were analyzed using one-way analysis of variance (ANOVA) followed by Tukey’s post-hoc test. Kaplan-Meier survival curves were analyzed using the log-rank (Mantel-Cox) test. A p-value < 0.05 was considered statistically significant.
Results
LEM4 overexpression is correlated with a worse overall survival in breast cancer patients
To examine LEM4 expression pattern, we collected tumor tissues and para-cancerous normal counterparts from 60 patients diagnosed with breast cancer. We detected significantly elevated LEM4 mRNA levels in the tumor specimens when compared to the paired normal samples (Fig. 1A). Immunoblotting results confirmed the overexpression of LEM4 at protein levels in breast cancer tumors (Fig. 1B). The analysis of the overall survival, after dividing the subjects into high- and low-expression groups based on the median LEM4 expression, showed a worse prognostic tendency in the high-expression group (Fig. 1C). Additionally, we observed increased protein levels of LEM4 in the cancerous cell lines (MCF-7 and T47D) in comparison to a normal breast epithelial cell line (MCF-10 A) (Fig. 1D).

Expression of LEM4 in breast cancer tissues and cell lines. (A) Comparison of LEM4 mRNA levels between matched tumor specimens and normal counterparts (n = 60 pairs). Significantly elevated LEM4 expression was observed in tumor tissues, paired Student’s t-test. (B) Immunoblotting analysis demonstrating overexpression of LEM4 protein in breast cancer tumor specimens. (C) Kaplan-Meier survival curve showing worse overall survival in breast cancer patients with high LEM4 expression (n = 60 patients, stratified by the median level of qPCR detection). (D) Increased protein levels of LEM4 in breast cancer cell lines (MCF-7, T47D) compared to a normal breast epithelial cell line (MCF-10 A). Data are summary of 3 independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
LEM4 overexpression confers ribociclib resistance
Ribociclib, a CDK4/6 inhibitor used for targeted therapy in breast cancer management19, commonly leads to resistance following chronic administration20,21. We next investigated whether LEM4 is implicated in ribociclib resistance. The resistant cell lines were established by exposing MCF-7 and T47D to increasing doses of ribociclib (0.1–0.8 µM, 2–3 weeks for each concentration). We selected 0.2 µM ribociclib for subsequent experiments as this concentration effectively inhibits parental cells (within their IC50 range of 0.2 µM) while having minimal impact on resistant cells (IC50 values above 0.8 µM), thus providing optimal conditions to distinguish between sensitive and resistant phenotypes (Figure S1). Using CCK-8 cell growth assay, we demonstrated that resistant cells continued proliferating in the presence of 0.2 µM ribociclib while the growth of parental cells was arrested (Fig. 2A). LDH cytotoxicity assay further confirmed that resistant cells showed enhanced resistance to the toxicity induced by 0.2 µM ribociclib (Fig. 2B).

LEM4 overexpression and ribociclib resistance. (A) CCK-8 cell growth assay showing the continuous proliferation of ribociclib-resistant cells (MCF-7 and T47D) in the presence of 0.2 µM ribociclib. (B) LDH cytotoxicity assay demonstrating resistance of ribociclib-resistant cells to cytotoxicity induced by 0.2 µM ribociclib. (C) Comparison of LEM4 mRNA levels between parental cells and ribociclib-resistant cells. (D) Immunoblotting analysis revealing increased protein levels of LEM4 in ribociclib-resistant cells. (E) Successful reduction of LEM4 mRNA and protein levels in ribociclib-resistant cells after siRNA interference. (F) LDH release assay showing re-sensitization of ribociclib-resistant cells to ribociclib treatment (0.2 µM ribociclib, 48 h) upon LEM4 silencing. (G) Flow cytometry analysis revealing enhanced ribociclib-induced apoptosis (0.2 µM ribociclib, 48 h) in drug-resistant cells after LEM4 knockdown. Data are summary of 3 independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
We next examined LEM4 expression levels in the parental cells and drug-resistant ones, and discovered that both the mRNA (Fig. 2C) and protein levels (Fig. 2D) of LEM4 were significantly elevated in the resistant cells. We further silenced LEM4 in the resistant cells using siRNA interference. The transfection of LEM4-targeting siRNA successfully reduced LEM4 mRNA and protein levels in drug-resistant MCF-7 and T47D cells (Fig. 2E). LDH release assays revealed that LEM4 silencing re-sensitized the resistant cells to 0.2 µM ribociclib treatment (Fig. 2F). Flow cytometry analysis further demonstrated that LEM4 knockdown in drug-resistant cells enhanced ribociclib-induced apoptosis (Fig. 2G). These data collectively indicate the requirement of LEM4 overexpression in conferring ribociclib resistance in breast cancer cells. Of note, LDH release assays revealed that ribociclib-resistant cells also showed resistance to other CDK4/6 inhibitors (palbociclib and abemaciclib), and LEM4 silencing re-sensitized the resistant cells to these inhibitors (Figure S2). Furthermore, analysis of the GSE98987 dataset revealed significantly elevated LEM4 expression in palbociclib-resistant MCF7 cells compared to parental controls, confirming our experimental findings of LEM4 upregulation in drug-resistant breast cancer cells (Figure S3A). Gene Set Enrichment Analysis demonstrated enrichment of the Wnt signaling pathway in palbociclib-resistant cells (NES = 1.669, FDR = 0.102), indicating the association between LEM4 overexpression and Wnt/β-catenin pathway activation in the context of palbociclib resistance (Figure S3B). These results collectively indicate that LEM4 overexpression not only contributes to ribociclib resistance but also applies more broadly to CDK4/6 inhibitor resistance.
Ribociclib-resistant cells exhibit upregulated β-catenin and transcriptional activity
We next sought to investigate the mechanism by which LEM4 confers resistance to ribociclib. The Wnt/β-catenin pathway is frequently deregulated, and hyperactivation of this pathway has been reported in different cancers with drug resistance22,23. Since ribociclib is an inhibitor for CDK4/6 and Wnt/β-catenin signaling controls the expression of cell-cycle genes, such as CDKs and cyclins24,25,26, we investigated whether Wnt/β-catenin signaling activation is involved in ribociclib resistance conferred by LEM4 overexpression. We examined the protein and phosphorylation levels of GSK3β (a negative regulator for β-catenin), as well as the protein level of β-catenin by immunoblotting. We observed that GSK3β phosphorylation level was reduced in drug-resistant MCF-7 and T47D cells, while there was an increased expression of β-catenin in the resistant cells (Fig. 3A). Immunoprecipitation of β-catenin followed by Western blot analysis of β-catenin and ubiquitin in parental and ribociclib-resistant cells revealed a reduction of β-catenin ubiquitination level in resistant cells despite similar total immunoprecipitated β-catenin levels (Fig. 3B). We further isolated the nuclear and cytoplasmic fractions from parental and drug-resistant cells to quantify the nuclear proportion of β-catenin. In drug-resistant cells, the relative nuclear level of β-catenin (compared to the cytoplasmic fraction) was significantly increased (Fig. 3C). The analysis of β-catenin-dependent transcription activity using the nuclear fraction also demonstrated an enhanced β-catenin activity in drug-resistant cells (Fig. 3D). Collectively, these data demonstrate augmented β-catenin activity in ribociclib-resistant cells.

Activation of β-catenin signaling pathway in ribociclib-resistant cells. (A) Immunoblotting analysis demonstrating reduced GSK3β phosphorylation and increased β-catenin expression in drug-resistant cells (MCF-7 and T47D). (B) Immunoprecipitation of β-catenin followed by Western blot analysis of β-catenin and ubiquitin in parental and ribociclib-resistant cells, showing decreased ubiquitination of β-catenin in resistant cells despite similar total immunoprecipitated β-catenin levels. (C) Quantification of nuclear β-catenin level relative to the cytoplasmic fraction, showing increased nuclear translocation of β-catenin in drug-resistant cells. (D) Measurement of β-catenin-dependent transcription activity in the nuclear fraction, revealing enhanced β-catenin activity in drug-resistant cells. Data are summary of 3 independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
LEM4 interacts with β-catenin to increase its protein stability
To investigate the possible interaction between LEM4 and β-catenin, we performed co-IP assay using both anti-LEM4 and anti-β-catenin antibodies in MCF-7 cells. The reciprocal co-IP analysis demonstrated the physical association between these two proteins (Fig. 4A). To determine whether this interaction affects the stability of β-catenin, we conducted cycloheximide (CHX, a ribosome translation inhibitor) chasing experiment by blocking protein synthesis and then analyzed the remaining β-catenin at different time points. We observed that in drug-resistant cells with high levels of LEM4 expression, β-catenin protein level decreased more slowly after CHX inhibition when compared to the parental cells. However, when LEM4 was knocked down in drug-resistant cells, β-catenin protein level decreased at a faster pace after protein translation inhibition (Fig. 4B). Consistently, LEM4 silencing reduced the total protein level of β-catenin in drug-resistant MCF-7 cells (Fig. 4C). However, the mRNA levels of β-catenin were unaffected by LEM4 interference (Fig. 4D). These findings suggest that LEM4 interacts with β-catenin to increase its protein stability.

Interaction between LEM4 and β-catenin and its impact on β-catenin stability. (A) Co-immunoprecipitation (co-IP) assay confirming the physical association between LEM4 and β-catenin in MCF-7 cells. (B) Cycloheximide (CHX) chasing experiment demonstrating slower degradation of β-catenin in drug-resistant cells with high LEM4 expression compared to parental cells, and faster degradation upon LEM4 knockdown. (C) Immunoblotting analysis showing reduced total protein level of β-catenin in drug-resistant cells after LEM4 silencing. (D) RT-qPCR analysis showing unaffected mRNA levels of β-catenin upon LEM4 interference. Data are summary of 3 independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
LEM4 knockdown suppresses the nuclear translocation of β-catenin and its transcriptional activity
We further demonstrated that LEM4 silencing in drug-resistant MCF-7 and T47D cells reduced the amount of β-catenin in the nuclear pool (Fig. 5A). This was accompanied by reduced β-catenin activity in the nuclear fraction (Fig. 5B). Moreover, we observed an increase in the expression of cell cycle-related genes (MYC, Cyclin D1, Cyclin E) and the anti-apoptotic gene (Survivin) in ribociclib-resistant cells. LEM4 knockdown significantly decreased the expression of these target genes of β-catenin (Fig. 5C).

LEM4 knockdown and its effects on β-catenin nuclear translocation and transcriptional activity. (A) Decreased amount of β-catenin in the nuclear pool of drug-resistant cells after LEM4 silencing in MCF-7 and T47D cells. (B) Reduced β-catenin activity in the nuclear fraction of drug-resistant cells upon LEM4 knockdown. (C) Expression levels of cell cycle-related genes (MYC, Cyclin D1, Cyclin E) and anti-apoptotic gene (Survivin) in ribociclib-resistant cells. LEM4 knockdown significantly decreased the expression of these β-catenin target genes. Data are summary of 3 independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
Inhibiting β-catenin activity reverses ribociclib resistance
To further establish the involvement of β-catenin activity in ribociclib resistance, we treated both parental and drug-resistant cells with an inhibitor for β-catenin activity (XAV-939). XAV-939 treatment significantly reduced β-catenin activity in the nuclear fraction in both parental and resistant cells (Fig. 6A). XAV-939 also suppressed the expression of β-catenin target genes (Fig. 6B). LDH assay revealed that inhibiting β-catenin activity not only potentiated ribociclib toxicity (0.2 µM ribociclib treatment) in parental cells, but also sensitized drug-resistant cells to ribociclib treatment (Fig. 6C). These effects were further verified by the detection of ribociclib-induced apoptosis after β-catenin inhibition (Fig. 6D). To further validate β-catenin’s role in mediating resistance, we directly knocked down β-catenin in resistant MCF-7 cells, which effectively reduced its elevated expression compared to parental cells (Figure S4A). Importantly, β-catenin knockdown significantly restored sensitivity to ribociclib in resistant cells as measured by LDH cytotoxicity assay, while control siRNA had no effect on the resistant phenotype (Figure S4B). Collectively, our findings suggest that LEM4-dependent β-catenin enhancement confers ribociclib resistance in breast cancer cells.

Reversal of ribociclib resistance by inhibiting β-catenin activity. (A) Significant reduction of β-catenin activity in the nuclear fractions of both parental and drug-resistant cells upon treatment with a β-catenin activity inhibitor (XAV-939). (B) Suppression of β-catenin target gene expression after treatment with XAV-939. (C) LDH assay showing enhanced ribociclib toxicity (0.2 µM ribociclib, 48 h) in parental cells and sensitization of drug-resistant cells to ribociclib treatment upon inhibition of β-catenin activity. (D) Flow cytometry analysis confirming increased apoptosis with ribociclib treatment (0.2 µM ribociclib, 48 h) after β-catenin inhibition. Data are summary of 3 independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
LEM4 knockdown sensitizes resistant cells to ribociclib treatment in vivo
We proceeded to verify the in vitro findings in a mouse xenograft model. Parental and resistant MCF-7 cells, or resistant cells expressing control shRNA (sh-NC) or shRNA targeting LEM4, were inoculated in nude mice subcutaneously for two weeks. Subsequently, ribociclib was administered for three weeks. At the end of the experiment, we observed that the tumor mass formed by drug-resistant cells was significantly larger than that of parental cells; while LEM4 knockdown suppressed tumor growth under ribociclib treatment conditions (Fig. 7A, tumor image; and Fig. 7B, tumor weights). TUNEL staining in the tumor tissue sections revealed that tumor samples from resistant cells showed fewer apoptotic events after ribociclib treatment. LEM4 silencing in the resistant cells promoted apoptosis induction (Fig. 7C). We also analyzed the β-catenin distribution in the cytoplasmic and nuclear fractions collected from tumor tissues. In agreement with the in vitro findings, LEM4 silencing in drug-resistant tumors reduced the amount of β-catenin in the nuclear pool (Fig. 7D). Moreover, the upregulation of β-catenin target genes in ribociclib-resistant tumor tissues was also suppressed after LEM4 knockdown (Fig. 7E). Collectively, our findings demonstrate that LEM4 knockdown sensitizes drug-resistant breast cancer cells to ribociclib treatment by dampening β-catenin activity.

LEM4 knockdown and ribociclib treatment in vivo. (A) Tumor images showing significant reduction in tumor mass formed by drug-resistant cells after LEM4 knockdown under ribociclib treatment conditions. (B) Weights of tumors from different experimental groups, demonstrating suppressed tumor growth in LEM4 knockdown group under ribociclib treatment. (C) TUNEL staining revealing increased apoptotic events in drug-resistant tumors after LEM4 silencing. (D) Immunoblotting analysis showing reduced nuclear β-catenin levels in drug-resistant tumors upon LEM4 knockdown. (E) Significant suppression of β-catenin target gene expression in ribociclib-resistant tumor tissues after LEM4 knockdown. N = 5 animals in each group. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
Discussion
The present study reveals a novel mechanism by which LEM4 overexpression confers resistance to the CDK4/6 inhibitor ribociclib in breast cancer cells. Our findings demonstrate that LEM4 interacts with and stabilizes β-catenin, leading to increased nuclear translocation and transcriptional activity of β-catenin. Consequently, the enhanced β-catenin-dependent transcription promotes cell cycle related gene expression, rendering breast cancer cells resistant to ribociclib-induced cytotoxicity and apoptosis. These results highlight the crucial role of the LEM4/β-catenin axis in mediating ribociclib resistance, suggesting that targeting this pathway could potentially overcome drug resistance and improve therapeutic outcomes.
The Wnt/β-catenin signaling pathway is a critical regulator of various cellular processes, including cell proliferation, differentiation, and survival, and its dysregulation has been implicated in the development and progression of various cancers27. In the absence of Wnt ligands, β-catenin is constantly degraded by the destruction complex, which includes AXIN, APC, and GSK-3β28. Upon Wnt activation, this complex is inhibited, leading to the cytoplasmic accumulation and nuclear translocation of β-catenin, where it interacts with TCF/LEF transcription factors to regulate the expression of target genes involved in cell cycle progression, apoptosis inhibition, and stem cell maintenance28. Aberrant activation of the Wnt/β-catenin pathway has been observed in various cancer types, including breast, colon, and lung cancers, and has been associated with tumor initiation, metastasis, and therapeutic resistance29,30,31. Notably, in breast cancer, increased β-catenin activity has been linked to endocrine therapy resistance, chemoresistance, and cancer stem cell maintenance32,33,34. Our study demonstrates that LEM4 overexpression in ribociclib-resistant cells enhanced β-catenin stability and transcriptional activity. Silencing LEM4 or pharmacologically inhibiting β-catenin activity significantly reversed drug resistance. These findings are consistent with the reported role of Wnt/β-catenin signaling in drug resistance development.
The regulation of β-catenin is a tightly controlled process involving multiple mechanisms at the protein and transcriptional levels. At the protein level, β-catenin stability and nuclear translocation are regulated by various post-translational modifications, such as phosphorylation, ubiquitination, and acetylation35. In the cytoplasm, β-catenin is targeted for proteasomal degradation by the destruction complex, which is mediated by its phosphorylation by GSK-3β36. Additionally, several nuclear pore proteins, such as RanBP3, have been shown to interact with β-catenin and facilitate its nuclear export37,38. At the transcriptional level, the activity of β-catenin is modulated by its interaction with various co-activators and co-repressors, as well as by epigenetic modifications of its target genes39,40. Our discovery that LEM4 interacts with and stabilizes β-catenin, leading to its nuclear accumulation and increased transcriptional activity, provides a novel mechanism for the dysregulation of β-catenin signaling in breast cancer and the development of ribociclib resistance.
Several LEM proteins have shown the ability to recruit and regulate transcriptional coactivators of the Wnt signaling pathway, potentially acting as oncoproteins that help drive cell-cycle progression41,42. Cancer cells, including breast cancer cells, often display abnormal changes in nuclear envelope structure, which can serve as a diagnostic marker for clinical cancer diagnosis43. The integrity of the nuclear envelope is essential for maintaining genome stability and facilitating proper nucleo-cytoplasmic exchange44,45. LEM proteins play a crucial role in interacting with the BAF complex to preserve genome organization, with LEM2 and LEM4 depletion resulting in nuclear shape defects46,47,48. Recent findings suggest that LEM proteins also contribute to monitoring nuclear envelope integrity and the nuclear transport system49,50. Our results further demonstrate that the interaction between LEM4 and β-catenin facilitates the nuclear import of β-catenin. These findings highlight a novel function of LEM4 in activating β-catenin-dependent transcription to regulate drug resistance.
While our study provides compelling evidence for the role of LEM4 in mediating ribociclib resistance through the β-catenin pathway, the findings are primarily based on breast cancer cell lines. Further validation in patient-derived drug-resistant tissues is necessary. Moreover, the precise mechanism by which LEM4 stabilizes β-catenin remains to be elucidated. Future studies, including transcriptomic and proteomic analyses, will be valuable to comprehensively decipher the interactions between LEM4 and the β-catenin pathway, providing deeper insights into this regulatory mechanism. It is also essential to investigate the interplay between LEM4 and other signaling pathways implicated in drug resistance, as cancer cells often employ multiple resistance mechanisms. From a translational perspective, the LEM4-β-catenin axis represents a promising therapeutic target that could be disrupted through small molecule inhibitors, targeted protein degraders (PROTACs), or RNA-based approaches. Recent advances in targeting protein-protein interactions suggest clinical feasibility, though optimization would be required. Future studies should focus on developing these therapeutic strategies while exploring combination approaches that address multiple resistance mechanisms simultaneously to maximize clinical benefit for patients with CDK4/6 inhibitor-resistant breast cancer.
Conclusion
In summary, our study uncovers a novel mechanism by which LEM4 overexpression contributes to ribociclib resistance in breast cancer cells through the enhancement of β-catenin signaling. By interacting with and stabilizing β-catenin, LEM4 promotes its nuclear translocation and transcriptional activity. Our findings highlight the importance of the LEM4/β-catenin axis in mediating drug resistance and provide a rationale for targeting this pathway to overcome ribociclib resistance in breast cancer management.
Data availability
The data generated in this study are available upon request to the corresponding author.
References
Filho, A. M. et al. The GLOBOCAN 2022 cancer estimates: data sources, methods, and a snapshot of the cancer burden worldwide. INT. J. CANCER. https://doi.org/10.1002/ijc.35278 (2024).
Harbeck, N. et al. Breast cancer. Nat. Rev. Dis. Primers. 5 (1), 66 (2019).
Early Breast Cancer Trialists’ Collaborative Group. Aromatase inhibitors versus Tamoxifen in early breast cancer: patient-level meta-analysis of the randomised trials. Lancet 386 (10001), 1341–1352 (2015).
Vici, P. et al. Triple positive breast cancer: a distinct subtype? Cancer Treat. Rev. 41 (2), 69–76 (2015).
Bianchini, G., Balko, J. M., Mayer, I. A., Sanders, M. E. & Gianni, L. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 13 (11), 674–690 (2016).
Saatci, O., Huynh-Dam, K. T. & Sahin, O. Endocrine resistance in breast cancer: from molecular mechanisms to therapeutic strategies. J. Mol. Med. (Berl). 99 (12), 1691–1710 (2021).
Gril, B. et al. Effect of lapatinib on the outgrowth of metastatic breast cancer cells to the brain. J. Natl. Cancer Inst. 100 (15), 1092–1103 (2008).
Gao, J. J. et al. CDK4/6 inhibitor treatment for patients with hormone receptor-positive, HER2-negative, advanced or metastatic breast cancer: a US food and drug administration pooled analysis. Lancet Oncol. 21 (2), 250–260 (2020).
Turner, N. C. et al. Overall survival with Palbociclib and fulvestrant in advanced breast cancer. N Engl. J. Med. 379 (20), 1926–1936 (2018).
Akouchekian, M., Vidhi, P. & Huang, D. CDK4/6 inhibitors in hormone receptor-positive breast cancer: current practice and future directions. Clin. Breast Cancer. 22 (5), 321–332 (2022).
Slamon, D. J. et al. Overall survival with ribociclib plus fulvestrant in advanced breast cancer. N Engl. J. Med. 382 (6), 514–524 (2020).
Portman, N. et al. Overcoming CDK4/6 inhibitor resistance in ER-positive breast cancer. Endocr. Relat. Cancer. 26 (1), R15–R30 (2019).
Mayayo-Peralta, I. et al. Ribociclib induces broad chemotherapy resistance and EGFR dependency in ESR1 wildtype and mutant breast cancer. Cancers (Basel). 13 (24), 6314 (2021).
Brachner, A. & Foisner, R. Evolvement of LEM proteins as chromatin tethers at the nuclear periphery. Biochem. Soc. Trans. 39 (6), 1735–1741 (2011).
Brachner, A. & Foisner, R. Lamina-associated polypeptide (LAP)2α and other LEM proteins in cancer biology. Adv. Exp. Med. Biol. 773, 143–163 (2014).
Denais, C. & Lammerding, J. Nuclear mechanics in cancer. Adv. Exp. Med. Biol. 773, 435–470 (2014).
Gao, A. et al. LEM4 confers Tamoxifen resistance to breast cancer cells by activating Cyclin D-CDK4/6-Rb and ERα pathway. Nat. Commun. 9 (1), 4180 (2018).
Yao, J., Deng, K., Huang, J., Zeng, R. & Zuo, J. Progress in the Understanding of the mechanism of Tamoxifen resistance in breast cancer. Front. Pharmacol. 11, 592912 (2020).
Lynce, F., Shajahan-Haq, A. N. & Swain, S. M. CDK4/6 inhibitors in breast cancer therapy: current practice and future opportunities. Pharmacol. Ther. 191, 65–73 (2018).
Park, Y. H. et al. Longitudinal multi-omics study of Palbociclib resistance in HR-positive/HER2-negative metastatic breast cancer. Genome Med. 15 (1), 55 (2023).
Huang, J., Zheng, L., Sun, Z. & Li, J. CDK4/6 inhibitor resistance mechanisms and treatment strategies (Review). Int. J. Mol. Med. 50 (4), 128 (2022).
Liu, L. et al. Inhibition of Wnt/β-catenin pathway reverses multi-drug resistance and EMT in Oct4+/Nanog + NSCLC cells. Biomed. Pharmacother. 127, 110225 (2020).
Ghandadi, M. et al. Wnt-β-catenin signaling Pathway, the achilles’ heels of cancer multidrug resistance. Curr. Pharm. Des. 25 (39), 4192–4207 (2019).
Lecarpentier, Y., Schussler, O., Hébert, J. L. & Vallée, A. Multiple targets of the canonical WNT/β-Catenin signaling in cancers. Front. Oncol. 9, 1248 (2019).
Niehrs, C. & Acebron, S. P. Mitotic and mitogenic Wnt signalling. EMBO J. 31 (12), 2705–2713 (2012).
Kafri, P. et al. Quantifying β-catenin subcellular dynamics and Cyclin D1 mRNA transcription during Wnt signaling in single living cells. Elife 5, e16748 (2016).
Nusse, R. & Clevers, H. Wnt/β-Catenin Signaling, Disease, and emerging therapeutic modalities. Cell 169 (6), 985–999 (2017).
Stamos, J. L. & Weis, W. I. The β-catenin destruction complex. Cold Spring Harb Perspect. Biol. 5 (1), a007898 (2013).
Murillo-Garzón, V. & Kypta, R. WNT signalling in prostate cancer. Nat. Rev. Urol. 14 (11), 683–696 (2017).
Krishnamurthy, N. & Kurzrock, R. Targeting the Wnt/beta-catenin pathway in cancer: update on effectors and inhibitors. Cancer Treat. Rev. 62, 50–60 (2018).
Groenewald, W., Lund, A. H. & Gay, D. M. The role of WNT pathway mutations in cancer development and an overview of therapeutic options. Cells 12 (7), 990 (2023).
Jang, G. B. et al. Blockade of Wnt/β-catenin signaling suppresses breast cancer metastasis by inhibiting CSC-associated phenotypes. Sci. Rep. 5, 12465 (2015).
Lamb, R. et al. Wnt pathway activity in breast cancer sub-types and stem-like cells. PLoS One. 8 (7), e67811 (2013).
Loh, Y. N. et al. The Wnt signalling pathway is upregulated in an in vitro model of acquired Tamoxifen resistant breast cancer. BMC Cancer. 13, 174 (2013).
Gao, C., Xiao, G. & Hu, J. Regulation of Wnt/β-catenin signaling by posttranslational modifications. Cell. Biosci. 4 (1), 13 (2014).
Park, H. B., Kim, J. W. & Baek, K. H. Regulation of Wnt signaling through ubiquitination and deubiquitination in cancers. Int. J. Mol. Sci. 21 (11), 3904 (2020).
Hendriksen, J. et al. RanBP3 enhances nuclear export of active (beta)-catenin independently of CRM1. J. Cell. Biol. 171 (5), 785–797 (2005).
Sharma, M., Johnson, M., Brocardo, M., Jamieson, C. & Henderson, B. R. Wnt signaling proteins associate with the nuclear pore complex: implications for cancer. Adv. Exp. Med. Biol. 773, 353–372 (2014).
Mosimann, C., Hausmann, G. & Basler, K. Beta-catenin hits chromatin: regulation of Wnt target gene activation. Nat. Rev. Mol. Cell. Biol. 10 (4), 276–286 (2009).
Valenta, T., Hausmann, G. & Basler, K. The many faces and functions of β-catenin. EMBO J. 31 (12), 2714–2736 (2012).
Bussolati, G., Marchiò, C., Gaetano, L., Lupo, R. & Sapino, A. Pleomorphism of the nuclear envelope in breast cancer: a new approach to an old problem. J. Cell. Mol. Med. 12 (1), 209–218 (2008).
Alvarado-Kristensson, M. & Rosselló, C. A. The biology of the nuclear envelope and its implications in cancer biology. Int. J. Mol. Sci. 20 (10), 2586 (2019).
Pan, J. et al. Altered nuclear envelopes in cancer: a common paradigm and arising opportunity. Cancers (Basel). 13 (5), 1081 (2021).
Lim, S., Quinton, R. J. & Ganem, N. J. Nuclear envelope rupture drives genome instability in cancer. Mol. Biol. Cell. 27 (21), 3210–3213 (2016).
Denais, C. M. et al. Nuclear envelope rupture and repair during cancer cell migration. Science 352 (6283), 353–358 (2016).
Lin, F. et al. MAN1, an inner nuclear membrane protein that shares the LEM domain with lamina-associated polypeptide 2 and Emerin. J. Biol. Chem. 275 (7), 4840–4847 (2000).
Asencio, C. et al. Coordination of kinase and phosphatase activities by Lem4 enables nuclear envelope reassembly during mitosis. Cell 150 (1), 122–135 (2012).
Ulbert, S., Antonin, W., Platani, M. & Mattaj, I. W. The inner nuclear membrane protein Lem2 is critical for normal nuclear envelope morphology. FEBS Lett. 580 (27), 6435–6441 (2006).
Thaller, D. J. et al. An ESCRT-LEM protein surveillance system is poised to directly monitor the nuclear envelope and nuclear transport system. Elife 8, e45284 (2019).
Borah, S., Dhanasekaran, K. & KµMar, S. The LEM-ESCRT toolkit: repair and maintenance of the nucleus. Front. Cell. Dev. Biol. 10, 989217 (2022).
Funding
This work was supported by Scientific Research Fund Project of Education Department of Yunnan Province 2023Y0652, The Joint Special Funds for the Department of Science and Technology of Yunnan Province-Kunming Medical University (NO.202301AY070001-255) and Clinical and Basic Research Project of Beijing: Kangmeng Charity Foundation TB213012.
Author information
Authors and Affiliations
Contributions
The authors confirm contribution to the paper as follows: study conception and design: Qi Tang, Xintong Zhao, Hui Li; data collection: Jiaqian Liao, Bingyu Ouyang; analysis and interpretation of results: Chunxiao Ma, Bingyu Ouyang; draft manuscript preparation: Qi Tang, Dedian Chen. All authors reviewed the results and approved the final version of the manuscript.The manuscript has been read and approved by all the authors, who confirm that the requirements for authorship as stated earlier in this document have been met, and that each author believes that the manuscript represents honest work.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
All the sample handling and data processing steps were conducted in accordance with the Declaration of Helsinki. The animal protocols received the approval of Ethical Review Committee for Animal Experiments of Kunming Medical University (Kmmu20240103). Tumor tissues and para-cancerous normal counterparts were collected from 60 breast cancer patients undergoing surgical resection at Yunnan Cancer Hospital Ethical Review Committee, Kunming, China. The study was approved by the Institutional Review Board for medical research (Approval NO.KYCS2023-014), and all patients provided written informed consent.
Arrive guidelines
The study is reported in accordance with Arrive guidelines.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tang, Q., Zhao, X., Li, H. et al. LEM4 interacts with β-catenin and promotes β-catenin-dependent transcription activity to confers ribociclib resistance in breast cancer cells. Sci Rep 15, 44435 (2025). https://doi.org/10.1038/s41598-025-28139-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-28139-7
