
Abstract
Duchenne Muscular Dystrophy (DMD) is a severe genetic disorder affecting skeletal and cardiac muscles, primarily in males. While much research has focused on these systems, the role of vascular smooth muscle cells (VSMCs) remains underexplored. This study examines how dystrophin deficiency alters VSMCs plasticity using mdx mice and DMD patient-derived iPSC VSMCs. Immunohistochemistry, Western blot, electron microscopy, and transcriptomic analyses revealed significant abnormalities. In mdx mice, abnormal vascular structure and vascular degeneration were observed. DMD VSMCs showed impaired maturation, reduced contractile protein expression, and disrupted mitochondrial dynamics, including excessive fission and reduced mitochondrial area. These cells also exhibited increased apoptosis under oxidative stress. Transcriptomic profiling identified dysregulated genes related to VSMC proliferation, differentiation, and vascular development, with transcription factors such as GADD45A, SOX9, TIA1, RBBP9, and FOXM1 implicated. Under stress, apoptotic pathways were notably upregulated. These findings suggest that dystrophin deficiency drives VSMC phenotype switching and mitochondrial dysfunction, contributing to vascular pathology in DMD. These findings highlight the importance of targeting vascular abnormalities in therapeutic strategies to slow disease progression.
Similar content being viewed by others


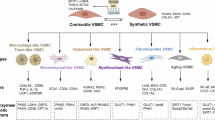
Introduction
Dystrophin is critical for maintaining the integrity of muscle cell membranes1,2. A previous clinical study demonstrated that blood loss in patients with Duchenne Muscular Dystrophy (DMD) was much higher than that in patients with spinal muscular atrophy undergoing similar surgical procedures, supporting the possibility that lack of dystrophin in vascular smooth muscle cells (VSMCs) is responsible for poor vasocontractile response leading to outcomes3. Furthermore, reduction of intramuscular blood flow has been observed in DMD patients, suggesting an existence of functional muscle ischemia4. Potential vascular dysfunction has been reported in DMD disease5,6,7. This vascular defect was considerably reduced in DMD mouse by improving angiogenesis with overexpression of growth factors8 which is consistent with the reduction of intramuscular blood flow reported in DMD patients4. Recently a study demonstrated that in dystrophin deficient dogs both vasoconstriction and vasorelaxation were comprised9. Vascular dysfunction blunts the blood supply, which impairs muscle tissue perfusion and causes muscle necrosis and degeneration, which in turn can also limit gene therapy delivery. Thus, vascular defects may be an underlying pathogenic mechanism in DMD disease.
The most common DMD mouse model is mdx mice, which carry a nonsense mutation in exon 23 of the dystrophin gene10,11. The association between muscle structure and function with age throughout the life span of the mdx mice has only recently been appreciated12. However, despite the absence of dystrophin in muscles, adult mdx mice do not exhibit the pathogenic progression characteristic of human DMD, severe muscle weakness, loss of muscle weight, accumulation of fat and fibrosis is not significant until almost two years of age11. Furthermore, an age-dependent effect on angiogenesis in mdx mice has been reported13. Significant vessel density is decreased in skeletal muscle from 12-month-old mdx mice compared with 3-month-old mdx mice and aged match wild type mice5. A previous study reported that isometric force was decreased in VSMCs from mdx mice after nitric oxide stimulation14. However, the authors did not reveal the specific age of the mdx mice they used in the study. Upon mechanical injury, increased neointima formation and smooth muscle proliferation were observed in 4–5 months old mdx mice. However, they did not report spontaneous neointima thickness in these mdx mice. As previously reported that significant vessel defects were observed in 12-month-old mdx mice, here we investigated whether any spontaneous phenotypic changes take place in VSMCs from 12 to 14 months old mdx mice. Decreased dystrophin expression was reported in synthetic smooth muscle cells compared with contractile smooth muscle cells in vitro15. Similarly, stabilization of actin filaments promoted the expression of dystrophin14 in mouse aorta smooth muscle cells. However, whether dystrophin deficiency drives phenotypic switching in VSMCs and the underlying mechanisms remain unclear. Human induced pluripotent stem cell (iPSC)-based models offer a promising platform to study rare diseases such as DMD, providing a renewable source of clinically relevant human cells.
Therefore, the objective of this study was to investigate the phenotypic changes and underlying mechanisms in VSMCs associated with dystrophin deficiency, using both aged mdx mice and DMD patient-derived iPSC VSMCs.
Results
DMD expression in VSMCs
As shown in Fig. 1A, tissue expression analysis using data from the GTEx portal revealed abundant dystrophin expression in arteries. Further analysis of single-cell RNA-seq data from normal adult mouse thoracic aorta and human aorta demonstrated that dystrophin is highly expressed in VSMCs compared to other cell types within the aorta (Fig. 1B,C). Collectively, these findings indicate that dystrophin is most abundantly expressed in VSMCs across the vascular system.

DMD gene expression across tissues and vascular cell types. (A) DMD gene expression levels in various human tissues, with notably high expression in arterial tissue. Data obtained from the GTEx Portal database. (B) Single-cell RNA sequencing (scRNA-seq) data from human aorta showing high DMD expression specifically in vascular smooth muscle cells (VSMCs), compared to other cell types including endothelial cells (EC), fibroblasts (Frb/Frbio), monocytes (Mono), and macrophages (Macro). (C) scRNA-seq data from mouse aorta confirming elevated DMD expression in VSMCs relative to other vascular cell types. Expression levels are shown as TPM (Transcripts Per Million), indicating relative transcript abundance across cell populations.
Phenotypic changes in VSMCs from Mdx mice
Gross morphology (Fig. 2A) revealed no significant dilation or overt structural abnormalities in mdx mice compared to wild type (WT) controls. Histological analysis using Trichrome Masson staining (Fig. 2B) showed disrupted vascular architecture in mdx mice, with regions of abnormally thin and thickened aortic media. In contrast, WT aortas displayed uniform medial thickness. To support the structural observations, we quantified the maximum media thickness of the aortic wall and fibrosis (P < 0.01, n = 5) (Fig. 2C,D), which were significantly increased in mdx mice (P < 0.01, n = 6) (Fig. 2C). Ultrastructural analysis by transmission electron microscopy (TEM) (Fig. 2E) revealed degenerative changes in VSMCs from mdx mice, including intranuclear and cytoplasmic vacuoles, which were absent in WT mice.

Phenotypic changes in VSMCs from mdx mice. Phenotypic changes in vascular smooth muscle cells (VSMCs) from mdx mice. (A) Gross morphology of heart and aorta from 12–14-month-old wild-type (WT) and mdx mice. No significant dilation was observed in mdx mice compared to WT controls. (B) Trichrome Masson staining of aorta sections showing loss of vascular structure and fibrosis in mdx mice. (C) Enhanced local aortic media thickness and (D) fibrosis in 12-14-month-old mdx mice. Quantification of maximum media thickness and percentage of fibrosis is shown. P < 0.01, n = 5. (E) Transmission electron microscopy (TEM) images showing degenerative changes in VSMCs from mdx mice, including intranuclear and cytoplasmic vacuoles (arrows), which were absent in WT mice. n = 5.
Characterization of vascular smooth muscle cells (VSMCs) from normal WT iPSC and DMD iPSC
Human iPSC-based disease models are promising due to unlimited supply of clinically relevant phenotypic cells of human origin, especially for rare diseases such as DMD. Accordingly, we generated VSMCs from human normal iPSC and DMD-iPSC lines (SC604A/ GM25313) by treatment with a small molecule, SB43152 followed by transforming growth factor-β (TGF-β) (Fig. 3A). With SB43152 treatment, iPSC were differentiated into mesenchymal stem cell (MSC) like cells expressing MSC markers (NG2 and CD105) with negligible expression of α-SMA and negative calponin expression (Fig. 3B). We further differentiated these cells into VSMCs using TGF-β1 as previously described16. The differentiated VSMCs expressed VSMCs markers including α-SMA and calponin (Fig. 3C). However, compared with normal VSMCs, DMD iPSC derived VSMCs displayed non-mature phenotype with low expression of contractile proteins [Calponin, smooth muscle protein 22-alpha (SM-22α), α-smooth muscle actin (α-SMA)] either by immunofluorescence (Fig. 3C,D) or Western blot (Fig. 3E,F) from two distinct normal iPSC lines and DMD iPSC lines. Mitochondrial dynamics, specifically the balance between fission and fusion, are essential for maintaining mitochondrial homeostasis and cellular function17. Interestingly, enhanced mitochondrial fission was observed in VSMCs derived from two distinct DMD iPSC with smaller area and average branch length of mitochondria (Fig. 4A–C) and in VSMCs from 12 months old mdx mice (Fig. 4D,E). Furthermore, oxidative stress was elevated in DMD VSMCs, as indicated by the increased fluorescence intensity of dihydrorhodamine 123 (DHR123) (Fig. 4F). These results suggested that dystrophin deficiency caused loss of mitochondrial homeostasis and oxidative stress in VSMCs.

Generation of VSMCs from normal and DMD iPS cells. (A) Schematic outline of vascular smooth muscle cells (VSMCs) differentiation from iPSC; (B) Differentiated MSC-like cells expressed MSC markers: CD105 and NG2, with low expression of α-SMA, calponin expression is undetectable. (C, D) VSMCs markers expression by immunostaining in VSMCs derived from two normal and DMD iPSC lines. (E, F) Representative Western blot images and semi-quantitative estimate of VSMCs markers expression. iPSC: induced pluripotent stem cells; MSC: mesenchymal stem cells. Bar = 100 μm. *P < 0.05,*P < 0.01, n = 3 correspond to independent differentiation batches.

Abnormal mitochondrial fission in dystrophin deficiency VSMCs. (A) Enhanced mitochondrial fission in VSMCs derived from DMD iPSC compared to wild type (WT) VSMCs. Mitochondria were identified by Mito-tracker. Quantification of mitochondrial area (B) and average branch length (C) in VSMCs from two WT and DMD iPSC (109.4 ± 36.02 µm2/cell for WT, 52.37 ± 25.67 µm2/cell for DMD, P < 0.001),, and shorter average branch length (1.07 ± 0.36 μm/cell for WT, 0.68 ± 0.24 μm/cell for DMD, P < 0.001). 50 cells from each group were analyzed. (D, E)TEM images showing mitochondrial fission in aortic VSMCs from 12 months old mdx mice (1.68 ± 0.53 µm2/mitochondria for WT, 0.85 ± 0.33µm2/mitochondria for DMD). Mitochondria was small and fragmented (arrows). 200 mitochondria from 3 mice of each group were measured. (F) Representative images of DHR123 staining in VSMCs derived from DMD iPSC and WT VSMCs and quantification of their intensity. ****P < 0.001, **P < 0.0, n = 3. iPSC: Induced pluripotent stem cells; VSMCs: vascular smooth muscle cells.
Transcriptome analysis of VSMCs from normal WT iPSC and DMD iPSC
Go enrichment analysis showed negative regulation of smooth muscle cell differentiation and positive regulation of osteoblast differentiation in DMD VSMCs (Fig. 5A). Vascular development was also significantly decreased in dystrophin deficient VSMCs (Fig. 5B). Using DAVID tools, we performed KEGG enrichment analysis18 for VSMCs transcriptomic profiling of normal and DMD iPSC derived VSMCs. Differentially expressed genes (DEGs) were mainly enriched in upregulation of TGF-β, P53, Rap1 and Hippo signaling pathways and downregulation of ferroptosis in DMD iPSC derived VSMCs (Fig. 5C). Heatmaps revealed enriched upregulation of genes associated with the TGF-β and P53 signaling pathways in DMD VSMCs (Fig. 5D,E ). Gene set enrichment analysis (GSEA) revelated enrichment of genes related to mitochondrial depolarization, and response to irons (Fig. 5F). Transcriptional factor, target, and motif discovery analysis of the dysregulated gene set showed enrichment of transcriptional factors (TFs) including GADD45A, SOX9, TIA1, RBBP9 and FOXM1 (Fig. 5G).

Transcriptome analysis of VSMCs derived from normal and DMD iPSC lines. GO categories of biological process (BP), molecular function (MF) and cellular components (CC) showing significant enrichment in the dataset of genes upregulated (A) and downregulated (B) in DMD-VSMCs compared to normal WT-VSMCs. Low -log10 (FDR) values are in green and high − log10 (FDR) values are in red, the size of the circle is proportional to the number of enriched genes. (C) KEGG pathway analysis of fold enrichment from up and down regulated mRNAs with top 14 in DMD-VSMCs compared to WT-VSMCs. (D) Heatmap of enriched upregulation of genes associated with the TGF-β signaling pathway in DMD VSMCs. (E) Heatmap of enriched upregulation of genes associated with the p53 signaling pathway in DMD VSMCs. (F) GSEA biological process (BP) enrichment analysis showing significant enrichment in the dataset of genes downregulated in DMD-VSMCs (P < 0.01, FDR ≤ 0.25). (G) Transcriptional factor, target, and motif discovery analysis showing enrichment of transcriptional factors GADD45A, TIA1, RBBP9, FOXM1 and SOX9 with iRegulon on the upregulated gene set (fold-change > 2, P < 0.05) in DMD-VSMCs compared to WT-VSMCs. GSEA: gene set enrichment analysis, iPSC: induced pluripotent stem cells; VSMCs: vascular smooth muscle cells; WT: wild type. n = 3 correspond to independent differentiation batches.
Transcriptome analysis of VSMCs from normal iPSC and DMD iPSC following oxidative stress
We further analyzed the response of WT-VSMCs and DMD-VSMCs to oxidative stress. After hydrogen peroxide (H2O2) treatment, Gene Ontology (GO) enrichment of biological process (BP) showed increased cell death and apoptosis, muscle contraction, reorganization of actin filament and cytoskeleton; GO enrichment of cellular components (CC) showed increased actin filament, sarcomeres, contractile fibers, vacuoles, vesicles, and lysosomes, endoplasmic reticulum; GO enrichment of molecular function (MF) showed increased binding activity to integrin, calcium, cytoskeletal protein, chemoattractant and amyloid-beta in DMD VSMCs with oxidative stress (Fig. 6A). While their response to hypoxia, oxidative stress, heparin, and fluid shear stress was decreased; cell migration and motility were also impaired. In addition, membrane components, mitochondrion and mitochondrial protein-containing complex were downregulated. Molecular function including signaling receptor, extracellular matrix binding and transmembrane receptor protein kinase activity were impaired in DMD VSMCs with oxidative stress (Fig. 6A). Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis18 showed upregulated genes related to Rap1 signaling pathways, AGE-RAGE signaling pathway in diabetic complications, fluid shear stress and atherosclerosis in DMD-VSMCs (Fig. 6B). The significantly enriched top 6 KEGG pathways in downregulated genes in DMD VSMCs included ECM-receptor interaction, arrhythmogenic right ventricular cardiomyopathy, phospholipase D signaling pathway and TGF-β signaling pathway (Fig. 6B). GSEA showed negative regulation of vascular development and calcium signaling; increased aggresomes and metabolic changes in DMD VSMCs subjected to oxidative stress (Fig. 6C,D). Muscle contraction, glycosaminoglycan biosynthesis, surfactant homeostasis was decreased in DMD VSMCs subjected to oxidative stress based on GSEA (Fig. 6E). DMD VSMCs exhibited increased vulnerability to hydrogen peroxide (H₂O₂) treatment, as evidenced by elevated oxidative stress levels indicated by enhanced DHR123 fluorescence intensity (Fig. 6F). In addition, TUNNEL analysis showed that DMD deficiency VSMCs were vulnerable to oxidative stress with increased cell apoptosis compared with normal VSMCs (Fig. 6G), which is consistent with the GO enrichment results. These data show that DMD VSMCs are vulnerable under oxidative stress conditions compared with WT-VSMCs and the pathways activated under stress conditions exacerbate the disease.

Transcriptome analysis of WT VSMCs and DMD VSMCs with oxidative stress. (A) GO categories of biological process (BP), molecular function (MF) and cellular components (CC) showing significant enrichment in the dataset of genes upregulated and downregulated in DMD-VSMCs compared to normal WT-VSMCs with oxidative stress. Low log10 (FDR) values are in blue and high − log10 (FDR) values are in purple, the size of the circle is proportional to the number of enriched genes. (B) KEGG pathway analysis of fold enrichment from up and down regulated mRNAs with top 14 in DMD-VSMCs compared to WT-VSMCs under oxidative stress. GSEA KEGG pathway enrichment analysis (C) and GO function biological process (BP), cellular components (CC), molecular function (MF) enrichment analysis (D) showing significant enrichment in the dataset of genes upregulated in DMD VSMCs with oxidative stress, and (E) BP and KEGG pathway enrichment in the dataset of genes downregulated in DMD VSMCs with oxidative stress. P < 0.01, FDR ≤ 0.25. (F) Representative images of DHR123 staining in VSMCs derived from DMD iPSC and WT VSMCs subjected to 24 h H2O2 treatment (100 µM) and quantification of their intensity. (G) VSMCs from DMD iPSC were vulnerable to oxidative stress. TUNEL positive cells in VSMCs from normal and DMD iPS cells after 24 h H2O2 (100 µM) treatment. Semi-quantitative estimate of TUNEL positive cells in VSMCs with H2O2 treatment. n = 3 correspond to independent differentiation batches. GSEA: gene set enrichment analysis, iPSC: induced pluripotent stem cells; VSMCs: vascular smooth muscle cells; WT: wild type.
Discussion
In the present study we utilized multiple models to evaluate the vascular smooth muscle from human cells as well as mdx mice which consistently show the phenotype switching directly linked to dystrophin deficiency. Here, we found loss of vascular structure and degenerative changes of VSMCs in aorta from 12 to 14 months old mdx mice. The DMD iPSC derived VSMCs (DMD VSMCs) from two patients showed maturation defects and increased vulnerability to oxidative stress compared with wild type VSMCs. Transcriptome analysis revealed dysregulation of smooth muscle proliferation and differentiation, and vascular development related biological function in DMD VSMCs. Under oxidative stress, apoptotic process was significantly enhanced in DMD VSMCs while their response to hypoxia and oxidative stress was downregulated. Transcriptional factor, target, and motif discovery analysis of the dysregulated gene set suggested potential contributions of transcriptional factors GADD45A, SOX9, TIA1, RBBP9 and FOXM to the phenotypes of DMD VSMCs. These molecular evaluation provides potential signaling pathways and medicators that are key to exacerbating DMD.
VSMCs phenotype switching plays a critical role in multiple vascular diseases. Various stimuli can modulate the plasticity of VSMCs, such as reactive oxygen species (ROS), oxidized lipids, inflammatory cytokines, and mechanical stress19. Exposure to these stimuli leads to phenotype switching of VSMCs from contraction phenotype towards synthesis phenotype, apoptosis, necrosis, degeneration, and senescence20. In fact, dystrophin is expressed in normal smooth muscle cells5. Tissue expression analysis showed an abundant expression of dystrophin in arteries from GTEx portal database. With single cell analysis in both mouse aorta and human aorta via single cell portal, we also showed higher expression of dystrophin in VSMCs compared with other cell types, indicating a potentially important role of dystrophin in VSMCs function. Although the etiological link between aortic aneurysms and DMD remains unclear, a case report of abdominal aneurysm rupture in a patient with Becker muscular dystrophy (BMD) suggests that dystrophin deficiency may contribute to vessel wall abnormalities21. However, given the differences in dystrophin expression between BMD and DMD, caution is warranted when extrapolating findings from BMD to DMD.
mdx mouse is the most widely used model to study DMD, but their phenotype is milder than that of the dystrophic patients11. The young mdx mice show only weak DMD phenotype in skeletal muscle. A greater degree of cardiac and vessel dysfunction is observed at least in 10–12 months old mdx mice not in young mdx mice13. Our study revealed that dystrophin deficiency in 12–14 months old mdx mice, resulted in spontaneous structural remodeling of the aorta. Abnormal vascular structure and degenerative changes supporting our view that VSMCs underwent degenerative phenotypes switching. These findings agree with previous studies that dystrophin expression was significantly reduced during synthetic phenotype switching upon vessel injury14.
Case reports on abdominal aneurysm rupture in patients with Becker muscular dystrophy suggests the involvement of major vessels in the pathogenesis of DMD disease. On the other hand, a lack of studies on VSMCs from DMD patients has been attributed due to nonavailability of human tissue for analysis. Therefore, in this study we differentiated VSMCs from DMD iPSC to investigate the role of dystrophin deficiency in VSMCs and its mechanism. We discovered that maturation defects are present in VSMCs differentiated from DMD iPSC compared to VSMCs differentiated from normal iPSC. It is more likely that dystrophin may be a marker of VSMCs differentiation and is critical for the maintenance of contractile phenotype. It has been reported that overexpression of myocardin and myocardin-related transcription factor A (MRTF-A) promoted expression of dystrophin14. However, it is obvious that dystrophin deficiency promoted loss of contractile phenotype of VSMCs. We performed the transcriptome analysis for VSMCs differentiated from human normal and DMD iPSC. KEGG enrichment analysis revealed upregulation of genes related to p53 and Hippo signaling pathways while genes related to ferroptosis, Notch signaling, osteoblast differentiation and inhibition of smooth muscle cell differentiation were downregulated. It is known that p53 signaling pathway is involved in the regulation of VSMCs proliferation22. p53 activation also impaired smooth muscle differentiation via inhibition of Myocd23. Hippo signaling pathway plays a critical role in the regulation of VSMCs phenotype24,25. Notch signaling pathway, mediated by basic helix–loop–helix (bHLH) transcriptional repression, controls VSMCs differentiation and modulates the transcription of endogenous contractile genes in VSMCs26,27. VSMCs mainly express Notch1, Notch2, and Notch328 receptors. Jagged 1-mediated Notch activation is required for the expression of smooth muscle contractility markers in VSMCs29,30,31,32 In addition, mitochondria are dynamic organelles and continuously undergo fission and fusion processes. VSMCs mitochondrial metabolism has been reported as one of the mechanisms involved in the complex regulation of the VSMCs phenotype33,34. GSEA analysis revealed upregulation of genes associated with mitochondrial depolarization in DMD VSMCs, consistent with our findings of disrupted mitochondrial homeostasis (excessive mitochondrial fission) in dystrophin-deficient VSMCs. However, further functional validation, such as JC-1 staining to assess mitochondrial membrane potential, is needed to confirm these observations. Mitochondrial fission is induced by membrane depolarization35. The imbalance of mitochondrial fission and fusion may contribute to the DMD VSMCs immature phenotype. Taken together, dystrophin deficiency plays an important role in VSMC homeostasis and likely promotes the loss of the contractile phenotype through dysregulation of mitochondrial and related signaling pathways.
Oxidative stress is a prominent feature of the dystrophic pathology with increased inflammation and myonecrosis in muscle tissue36. It has been reported that the lack of dystrophin renders the skeletal muscle susceptible to free radical induced injury37. However, it is unclear whether dystrophin loss also affects VSMCs vulnerability to oxidative stress. Indeed, our data support that DMD VSMCs showed enhanced oxidative stress. In addition, oxidative stress and oxidative DNA damage have a strong bearing on the DMD phenotype progression38. In this regard, we used H2O2 treatment to recapitulate phenotype changes in DMD VSMCs in comparison with wild type VSMCs. DMD VSMCs were highly vulnerable to H2O2 treatment with higher oxidative stress level and incidence of apoptosis. KEGG pathway enrichment analysis showed fluid shear stress and atherosclerosis increased in DMD VSMCs. Furthermore, the biological component of the Go analysis showed the majority of enriched categories were relevant to cell death and apoptotic process, which is consistent with increased number of apoptotic cells in DMD VSMCs exposed to oxidative stress. Go analysis also confirmed the upregulation of lytic vacuole/ vacuole and vesicle pathway in DMD-VSMCs. Per se, adult mdx mice do not exhibit the severe phenotype, but abnormalities are observed in hearts and vascular system at 10–12 months. Our results support that oxidative stress in DMD VSMCs replicated phenotype similar to 12–14 months old mdx mice where vacuole formation propensity was increased in VSMCs suggesting that DMD iPSC derived VSMCs could be used to model disease progression in vitro by applying oxidative stress. More importantly, GO enrichment analysis also supported our idea that the response of VSMCs to oxidative stress was robust. Interestingly, cellular components including actin filament, sarcomere and contractile fibers underwent increased reorganization in DMD VSMCs. MAPK signaling and regulation of contraction of smooth muscle cells were also upregulated by stress. However, GSEA and KEGG analysis showed that genes related to negative regulation of calcium were decreased in DMD VSMCs affected by oxidative stress which is known to induce actin reorganization and stress fiber formation in the vascular EC and myoblasts39,40. It is very likely that these filament/ fibers were stress fibers which needs further study. On a molecular scale, our GSEA analysis showed transcriptional changes in fatty acid metabolism and cholesterol homeostasis with increased genes expression related to fatty acid beta oxidation, sulfur lyase, lipoprotein particle receptor activity, nicotinate and nicotinamide metabolism in DMD VSMCs during disease progression. However, these bioinformatic analysis derived results need further validation. In addition to oxidative stress, nitrosative stress is a significant contributor to DMD. Studies have demonstrated that dystrophin deficiency leads to aberrant nitric oxide synthase (NOS) signaling and increased S-nitrosylation of key cardiac and muscular proteins, suggesting a role for nitrosative stress in DMD progression41,42. However, whether dystrophin deficiency directly exacerbates nitrosative stress in VSMCs derived from DMD patients remains to be elucidated.
In summary, we have established DMD iPSC derived VSMCs and mdx mouse model that the dystrophin deficiency led to VSMCs phenotype changes and disrupted mitochondrial metabolism. It is suggested that the transcriptome analysis may allow the discovery of potential signaling pathways involved in the dysregulation of transcription factors.
Methods
Human iPSC culture
The Human iPSC cell line DYS0100 (WT_1) from ATCC Company, JIPSC1000_KOLF2.1J_human iPS cell line (WT_2) from Jackson laboratory, human DMD iPS cell line SC604A (DMD_1) with a deletion of exons 3–7 in the dystrophin gene from SBI Company and human DMD iPS cell line GM25313 (DMD_2) with a deletion of exon 45 in the dystrophin gene from Coriell Institute for Medical Research were used. iPSC were cultured on vitronectin coated six-well plate in mTeSR1 medium (Stem Cell Technologies) with a daily change. iPSC were passaged using ReLeSR™ passaging reagent (Stem Cell Technologies).
Generation of vascular smooth muscle cells from iPS cells and their characterization
Human iPSC at passages 20–30 were used for VSMCs differentiation. The differentiation protocol outline is shown in Fig. 3A. Briefly, human iPSC were cultured on vitronectin using mTeSR1 medium. Upon confluency, the medium was switched to α-MEM basal medium supplemented with 20% Knock out serum (KSR), 1mM L-Glutamine, 10mM Nonessential Amino Acid and 10µM SBB-431,542 for 10 days. Next, the cells were trypsinized and seeded at a density of 4 × 104 cells/cm2 onto uncoated culture dishes in expansion medium (α-MEM basal medium + 10% FBS). After the third passage, a morphologically homogeneous population of mesenchymal stem cells (MSC)-like cells became evident and were tested for mesenchymal makers. Then the MSC-like cells were further differentiated into VSMCs using 5ng/ml TGFβ treatment for 6 days. The differentiated VSMCs were characterized with α-SMA, calponin and SM-22α staining. For immunofluorescence staining, cells were fixed with 4% PFA for 10 min and blocked with 10% FBS for 1 h at room temperature. Cells were incubated with primary antibodies including anti-CD105 (sc-18838, Santa Cruz), NG2 (MAB5384A4, Millipore Sigma), α-SMA (ab5694, Abcam) and calponin (C2687, Sigma) respectively at 4°C overnight and secondary antibody conjugated to Alexa Fluor 594 or Alexa Fluor 488 (Life Technologies) at room temperature for 1 h. DAPI was used as nuclear counterstain.
Reactive oxygen species measurement and oxidative stress induction
Reactive oxygen species (ROS) levels were assessed using dihydrorhodamine 123 (DHR123), a non-fluorescent, uncharged derivative of rhodamine 123. DHR123 passively diffuses into cells and is oxidized by ROS to form rhodamine 123 (R123), a cationic green fluorescent dye. R123 preferentially accumulates in mitochondria due to its positive charge. The increase in fluorescence intensity reflects elevated intracellular ROS levels, providing a relative measure of oxidative stress. VSMCs differentiated from either normal or DMD iPSCs were exposed to 100 µM H₂O₂ for 24 h to induce oxidative stress.
Animal and histology
Mouse aortic tissue from 12 to 14 months old wild type mice (Stock No: 000665 The Jackson Laboratory) and mdx mice (Stock No: 018018, The Jackson Laboratory) was harvested and processed for Masson’s trichrome staining as reported previously43. Maximum media wall thickness was measured as the maximum intima-media distance. Fibrosis have been shown to be normally distributed in the rodent aorta. Animal experiments were carried out according to the experimental protocol approved by the Augusta University Animal Care and Use Committee (approval number 2018 − 0940) in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. The animal is euthanized using an overdose (5%) of inhaled isoflurane, followed by pneumothorax prior to tissue collection. Animal research is reported according to the ARRIVE guidelines.
Transmission electron microscopy
Mouse aorta was processed for electron microscopy to assess ultrastructural changes in the VSMCs from mdx mice. Samples were processed and imaging was carried out by the Augusta University histology and TEM core. Briefly, aortic tissues were cut into 1mm3 pieces and fixed in 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.4). Next, tissues were post-fixed in 1% osmium tetroxide in 1% K4Fe (CN)6 buffer with 0.1 M sodium cacodylate, dehydrated through a graded series of ethanol and propylene oxide, and embedded in Epon 812. Ultrathin cryo-sections were prepared using Leica UC7 Ultramicrotomes, mounted on copper grids and stained with lead citrate and uranyl acetate. Images were captured by JEOL JEM-1230 Transmission Electron Microscope. Individual mitochondrial area measurement was used Image J.
Mito-tracker staining
Mitochondria in cultured cells were visualized by staining with MitoTracker Red CMXRos dye (Thermo Fisher Scientific). The dye was added to the live cells at a final concentration of 100 nM and incubated at 37 °C for 30 min. Cells were washed and fixed in 2% Paraformaldehyde (PFA) in phosphate-buffered saline (PBS). Images were taken by a confocal microscope (Olympus, Japan). Mitochondrial area and average branch length from VSMCs were analyzed using Image J software with mitochondrial analyzer plugin44.
TUNEL staining
Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) was performed to assess cell apoptosis using commercial apoptosis detection kit (Thermo Fisher Scientific, United States). All procedures were done according to the directions of the manufacturers. Cells were counterstained with DAPI to visualize nuclei. The number of TUNEL-positive cells was determined by randomly counting 10 fields and was expressed as a percentage of the cells with normal nuclei.
Western blot
Cell extracts were lysed with radio immunoprecipitation assay (RIPA) buffer supplemented with Protease Inhibitors Mixture (Sigma). Pierce™ BCA Protein Assay Kit (Thermo Scientific) was used to determine protein concentration. 10 µg protein was separated by SDS/PAGE and transferred to the PVDF membrane (Bio-Rad). Membranes were incubated with primary antibodies overnight at 4 °C: α-SMA (ab5694, abcam), Calponin (C2687, Sigma), SM-22α (10493-1-AP, Thermo Fisher Scientific), Tubulin (2128 S, Cell Signaling Technology) and β-actin (BDB612657, BD). On second day, membranes were incubated with an anti-mouse/rabbit peroxidase-conjugated secondary antibody at room temperature. Immunoreactive bands were visualized by the enhanced chemiluminescence method (Bio-Rad) with Fluorchem E detection system (ProteinSimple USA). The relative expression levels of target proteins were quantified by ImageJ software (National Institutes of Health, Bethesda, MD, United States).
mRNA sequencing and analysis
RNA was extracted from VSMCs derived from human iPSC lines DY0100 and SC604A, with or without oxidative stress, using the Qiagen RNeasy Kit. Cells from three independent differentiation batches were used. Sequencing was performed at Washington University’s Genome Core using the Illumina NovaSeq 6000. Libraries were prepared, indexed, pooled, and sequenced per standard protocols. Basecalling and demultiplexing were done using bcl2fastq and a custom Python script. Reads were aligned to the Ensembl release 76 genome using STAR (v2.5.1a). Gene counts were generated with featureCounts (v1.4.6-p5), and transcript quantification was done using Salmon (v0.8.2). Quality metrics were assessed with RSeQC (v2.6.4). Data were normalized using TMM in EdgeR, and low-expression and ribosomal genes were filtered out. Limma with voomWithQualityWeights was used for modeling and differential expression analysis. Genes with ≥ 2-fold change and FDR ≤ 0.05 were considered significant. Pathway enrichment for DEGs was performed using DAVID (KEGG)18 and GO tools, with p < 0.01 and FDR < 0.05 as cutoffs. Results were visualized via bioinformatics.com.cn. GSEA (v4.3.2) was used for additional KEGG and GO analysis (nominal p < 0.01, FDR < 0.25). Transcription factor analysis was done using iRegulon in Cytoscape (v3.9.1). DMD expression was examined using GTEx and Broad Institute’s Single Cell Portal datasets for human and mouse aorta.
Statistical analysis
Data were expressed as mean ± SD. Normality was tested and statistical analysis of differences among the different groups was compared by unpaired two-tailed Student’s t-tests. The differences were considered statistically significant at P < 0.05. Statistical analyses were performed by Graphpad Prism 9.5.
Data availability
The raw data of the mRNA sequence is deposited in the GEO database (GSE232219). The datasets used and/or analyzed during the current study are available from the corresponding author on request.
References
Lapidos, K. A., Kakkar, R. & McNally, E. M. The dystrophin glycoprotein complex: Signaling strength and integrity for the sarcolemma. Circ. Res. 94, 1023–1031. https://doi.org/10.1161/01.RES.0000126574.61061.25 (2004).
Houang, E. M., Sham, Y. Y., Bates, F. S. & Metzger, J. M. Muscle membrane integrity in Duchenne muscular dystrophy: recent advances in copolymer-based muscle membrane stabilizers. Skelet. Muscle. 8, 31. https://doi.org/10.1186/s13395-018-0177-7 (2018).
Noordeen, M. H. et al. Blood loss in Duchenne muscular dystrophy: Vascular smooth muscle dysfunction? J. Pediatr. Orthop. B. 8, 212–215. https://doi.org/10.1097/01202412-199907000-00015 (1999).
Dietz, A. R., Connolly, A., Dori, A. & Zaidman, C. M. Intramuscular blood flow in Duchenne and Becker muscular dystrophy: quantitative power Doppler sonography relates to disease severity. Clin. Neurophysiol. 131, 1–5. https://doi.org/10.1016/j.clinph.2019.09.023 (2020).
Latroche, C. et al. Structural and functional alterations of skeletal muscle microvasculature in dystrophin-deficient mdx mice. Am. J. Pathol. 185, 2482–2494. https://doi.org/10.1016/j.ajpath.2015.05.009 (2015).
Dabire, H. et al. Vascular endothelial dysfunction in Duchenne muscular dystrophy is restored by bradykinin through upregulation of eNOS and nNOS. Basic. Res. Cardiol. 107, 240. https://doi.org/10.1007/s00395-011-0240-6 (2012).
Timpani, C. A., Hayes, A. & Rybalka, E. Therapeutic strategies to address neuronal nitric oxide synthase deficiency and the loss of nitric oxide bioavailability in Duchenne muscular dystrophy. Orphanet J. Rare Dis. 12, 100. https://doi.org/10.1186/s13023-017-0652-y (2017).
Podkalicka, P., Mucha, O., Dulak, J. & Loboda, A. Targeting angiogenesis in Duchenne muscular dystrophy. Cell. Mol. Life Sci. 76, 1507–1528. https://doi.org/10.1007/s00018-019-03006-7 (2019).
Kodippili, K., Thorne, P. K., Laughlin, M. H. & Duan, D. Dystrophin deficiency impairs vascular structure and function in the canine model of Duchenne muscular dystrophy. J. Pathol. 254, 589–605. https://doi.org/10.1002/path.5704 (2021).
McGreevy, J. W., Hakim, C. H., McIntosh, M. A. & Duan, D. Animal models of Duchenne muscular dystrophy: From basic mechanisms to gene therapy. Dis. Model. Mech. 8, 195–213. https://doi.org/10.1242/dmm.018424 (2015).
Yucel, N., Chang, A. C., Day, J. W., Rosenthal, N. & Blau, H. M. Humanizing the mdx mouse model of DMD: The long and the short of it. NPJ Regen Med. 3:4. https://doi.org/10.1038/s41536-018-0045-4 (2018).
Chamberlain, J. S., Metzger, J., Reyes, M., Townsend, D. & Faulkner, J. A. Dystrophin-deficient mdx mice display a reduced life span and are susceptible to spontaneous rhabdomyosarcoma. FASEB J. 21, 2195–2204. https://doi.org/10.1096/fj.06-7353com (2007).
Podkalicka, P. et al. Age-dependent dysregulation of muscle vasculature and blood flow recovery after hindlimb ischemia in the mdx model of Duchenne muscular dystrophy. Biomedicines 9, 481. https://doi.org/10.3390/biomedicines9050481 (2021).
Turczynska, K. M. et al. Regulation of smooth muscle dystrophin and synaptopodin 2 expression by actin polymerization and vascular injury. Arterioscler. Thromb. Vasc Biol. 35, 1489–1497. https://doi.org/10.1161/ATVBAHA.114.305065 (2015).
Rauch, U. et al. Increased neointimal thickening in dystrophin-deficient mdx mice. PLoS One. 7, e29904. https://doi.org/10.1371/journal.pone.0029904 (2012).
Chen, Y. S. et al. Small molecule mesengenic induction of human induced pluripotent stem cells to generate mesenchymal stem/stromal cells. Stem Cells Transl Med. 1, 83–95. https://doi.org/10.5966/sctm.2011-0022 (2012).
Adebayo, M., Singh, S., Singh, A. P. & Dasgupta, S. Mitochondrial fusion and fission: The fine-tune balance for cellular homeostasis. FASEB J. 35, e21620. https://doi.org/10.1096/fj.202100067R (2021).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–462. https://doi.org/10.1093/nar/gkv1070 (2016).
Doran, A. C., Meller, N. & McNamara, C. A. Role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arterioscler. Thromb. Vasc Biol. 28, 812–819. https://doi.org/10.1161/ATVBAHA.107.159327 (2008).
Bochaton-Piallat, M. L. & Back, M. Novel concepts for the role of smooth muscle cells in vascular disease: Towards a new smooth muscle cell classification. Cardiovasc. Res. 114, 477–480. https://doi.org/10.1093/cvr/cvy031 (2018).
Yoshioka, M., Takahashi, K. H., Tanaka, T. & Onodera, H. H. Multiple aortic aneurysms in a patient with Becker muscular dystrophy. Neuromuscul Disord. 20, 664. https://doi.org/10.1016/j.nmd.2010.07.215 (2010).
Sun, X. et al. Wild-type p53-induced phosphatase 1 promotes vascular smooth muscle cell proliferation and Neointima hyperplasia after vascular injury via p-adenosine 5’-monophosphate-activated protein kinase/mammalian target of rapamycin complex 1 pathway. J. Hypertens. 37, 2256–2268. https://doi.org/10.1097/HJH.0000000000002159 (2019).
Molchadsky, A. et al. p53 plays a role in mesenchymal differentiation programs, in a cell fate dependent manner. PLoS One. 3, e3707. https://doi.org/10.1371/journal.pone.0003707 (2008).
Xie, C. et al. Yap1 protein regulates vascular smooth muscle cell phenotypic switch by interaction with myocardin. J. Biol. Chem. 287, 14598–14605. https://doi.org/10.1074/jbc.M111.329268 (2012).
Chakraborty, R. et al. Targeting smooth muscle cell phenotypic switching in vascular disease. JVS Vasc Sci. 2, 79–94. https://doi.org/10.1016/j.jvssci.2021.04.001 (2021).
Morrow, D. et al. Notch and vascular smooth muscle cell phenotype. Circ. Res. 103, 1370–1382. https://doi.org/10.1161/CIRCRESAHA.108.187534 (2008).
Karakaya, C. et al. Notch signaling regulates strain-mediated phenotypic switching of vascular smooth muscle cells. Front. Cell. Dev. Biol. 10, 910503. https://doi.org/10.3389/fcell.2022.910503 (2022).
Boucher, J., Gridley, T. & Liaw, L. Molecular pathways of notch signaling in vascular smooth muscle cells. Front. Physiol. 3, 81. https://doi.org/10.3389/fphys.2012.00081 (2012).
Boucher, J. M., Peterson, S. M., Urs, S., Zhang, C. & Liaw, L. The miR-143/145 cluster is a novel transcriptional target of Jagged-1/Notch signaling in vascular smooth muscle cells. J. Biol. Chem. 286, 28312–28321. https://doi.org/10.1074/jbc.M111.221945 (2011).
Domenga, V. et al. Notch3 is required for arterial identity and maturation of vascular smooth muscle cells. Genes Dev. 18, 2730–2735. https://doi.org/10.1101/gad.308904 (2004).
Liu, H., Kennard, S. & Lilly, B. NOTCH3 expression is induced in mural cells through an autoregulatory loop that requires endothelial-expressed JAGGED1. Circ. Res. 104, 466–475. https://doi.org/10.1161/CIRCRESAHA.108.184846 (2009).
Tang, Y., Urs, S. & Liaw, L. Hairy-related transcription factors inhibit Notch-induced smooth muscle alpha-actin expression by interfering with Notch intracellular domain/CBF-1 complex interaction with the CBF-1-binding site. Circ. Res. 102, 661–668. https://doi.org/10.1161/CIRCRESAHA.107.165134 (2008).
Chiong, M. et al. Mitochondrial metabolism and the control of vascular smooth muscle cell proliferation. Front. Cell. Dev. Biol. 2, 72. https://doi.org/10.3389/fcell.2014.00072 (2014).
Huynh, D. T. N. & Heo, K. S. Role of mitochondrial dynamics and mitophagy of vascular smooth muscle cell proliferation and migration in progression of atherosclerosis. Arch. Pharm. Res. 44, 1051–1061. https://doi.org/10.1007/s12272-021-01360-4 (2021).
Twig, G. & Shirihai, O. S. The interplay between mitochondrial dynamics and mitophagy. Antioxid. Redox Signal. 14, 1939–1951. https://doi.org/10.1089/ars.2010.3779 (2011).
Kennedy, T. L. et al. Utrophin influences mitochondrial pathology and oxidative stress in dystrophic muscle. Skelet. Muscle. 7, 22. https://doi.org/10.1186/s13395-017-0139-5 (2017).
Rando, T. A., Disatnik, M. H., Yu, Y. & Franco, A. Muscle cells from mdx mice have an increased susceptibility to oxidative stress. Neuromuscul. Disord. 8, 14–21. https://doi.org/10.1016/s0960-8966(97)00124-7 (1998).
Lazzarin, M. C., Dos Santos, J. F., Quintana, H. T., Pidone, F. A. M. & de Oliveira, F. Duchenne muscular dystrophy progression induced by downhill running is accompanied by increased endomysial fibrosis and oxidative damage DNA in muscle of mdx mice. J. Mol. Histol. https://doi.org/10.1007/s10735-022-10109-2 (2022).
Yao, Y., Lacroix, D. & Mak, A. F. Effects of oxidative stress-induced changes in the actin cytoskeletal structure on myoblast damage under compressive stress: confocal-based cell-specific finite element analysis. Biomech. Model. Mechanobiol. 15, 1495–1508. https://doi.org/10.1007/s10237-016-0779-0 (2016).
Huot, J., Houle, F., Marceau, F. & Landry, J. Oxidative stress-induced actin reorganization mediated by the p38 mitogen-activated protein kinase/heat shock protein 27 pathway in vascular endothelial cells. Circ. Res. 80, 383–392. https://doi.org/10.1161/01.res.80.3.383 (1997).
Li, D., Yue, Y., Lai, Y., Hakim, C. H. & Duan, D. Nitrosative stress elicited by nNOSmicro delocalization inhibits muscle force in dystrophin-null mice. J. Pathol. 223, 88–98. https://doi.org/10.1002/path.2799 (2011).
Lillo, M. A. et al. S-nitrosylation of connexin43 hemichannels elicits cardiac stress-induced arrhythmias in Duchenne muscular dystrophy mice. JCI Insight. 4, e130091. https://doi.org/10.1172/jci.insight.130091 (2019).
Xuan, W., Khan, M. & Ashraf, M. Pluripotent stem cell-induced skeletal muscle progenitor cells with givinostat promote myoangiogenesis and restore dystrophin in injured Duchenne dystrophic muscle. Stem Cell. Res. Ther. 12, 131. https://doi.org/10.1186/s13287-021-02174-3 (2021).
Chaudhry, A., Shi, R. & Luciani, D. S. A pipeline for multidimensional confocal analysis of mitochondrial morphology, function, and dynamics in pancreatic beta-cells. Am. J. Physiol. Endocrinol. Metab. 318, E87–E101. https://doi.org/10.1152/ajpendo.00457.2019 (2020).
Acknowledgements
We thank Syeda Roohina Ali for the technical support.
Funding
This work was supported by NIH grant AG070145 and USF startup funds (to WX) and Florida High Tech Corridor’s Undergraduate Research Initiative, and Saunders Endowment for Geriatric Pharmacotherapy (to SMT).
Author information
Authors and Affiliations
Contributions
WX conceived the study and drafted the manuscript. WX, FC, XH, SMT and MA performed the experiments, collected the data, and analyzed the data. SMT and MA reviewed and edited the manuscript. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xuan, W., Cheng, F., Han, X. et al. Impact of dystrophin deficiency on vascular smooth muscle cell. Sci Rep 15, 45254 (2025). https://doi.org/10.1038/s41598-025-29484-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-29484-3
