
Abstract
To investigate the neuroprotective mechanism of mild hypothermia (MH) in ameliorating cerebral ischemia reperfusion (IR) injury. The Pulsinelli’s four-vessel ligation method was utilized to establish a rat model of global cerebral IR injury. To investigate the role of S100A8 in MH treatment of cerebral IR injury, hippocampus-specific S100A8 loss or gain of function was achieved using an adeno-associated virus system. We examined the effect of S100A8 over-expression or knock-down on the function of the SH-SY5Y cell line subjected to oxygen-glucose deprivation reoxygenation (OGDR) injury under MH treatment and delved into the underlying mechanisms. MH significantly ameliorates IR-induced neurological injury in the brain. Similarly to MH, knock-down of S100A8 significantly reduced neuronal oxidative stress, attenuated mitochondrial damage, inhibited apoptosis, and improved cognitive function in IR rats. Conversely, over-expression of S100A8 attenuated MH’s protective effect and aggravated brain IR injury. In vitro, low expression of S100A8 significantly inhibited the decline in mitochondrial membrane potential induced by OGDR, reduced oxidative stress response, and decreased cell apoptosis, acting as a protective agent nearly equivalent to MH in SH-SY5Y cells. However, over-expression of S100A8 significantly inhibited these protective effects of MH. Mechanistically, MH down-regulated S100A8 expression, enhancing mitochondrial function via activation of the CAMKK2/AMPK signaling pathway. Moreover, with MH treatment, the administration of CAMKK2 and AMPK inhibitors STO-609 and Dorsomorphin significantly increased oxidative stress, mitochondrial damage, and cell apoptosis, thereby diminishing MH’s neuroprotective effect against cerebral IR injury. Our study identified S100A8 as a master regulator that enables MH to ameliorate neurological injury during the early stage of cerebral IR injury by enhancing mitochondrial function. By targeting the S100A8-initiated CAMKK2/AMPK signaling pathway, we may unlock a novel therapeutic intervention or develop a refined MH therapeutic strategy against cerebral IR injury.
Similar content being viewed by others


Introduction
Approximately 310 million patients worldwide undergo surgery each year, and stroke represents a common and significant complication during the perioperative period1. Perioperative stroke frequently occurs in conditions such as severe hypotensive shock, cardiac arrest, perinatal hypoxia–ischemia, asphyxia, and vascular embolism. Furthermore, reperfusion following resuscitation exacerbates brain tissue injury, commonly referred to as cerebral ischemia reperfusion (IR) injury. This type of injury poses a major challenge in perioperative medicine due to its complex pathological mechanisms, usually leading to irreversible neuronal damage and even cell death. As a neuroprotective approach, therapeutic hypothermia has demonstrated efficacy in ameliorating various brain injuries at the experimental level. Clinically, mild hypothermia (MH), maintained at 32–34 °C, is also widely utilized in the treatment of perioperative cardiac arrest and IR injury of vital organs. MH affects nearly all processes that lead to cell death, including attenuating early brain-blood barrier leakage2, enhancing glymphatic influx3, and ameliorating metabolic disturbances, calcium overload, glutamate excitotoxicity, and mitochondrial dysfunction4. However, the specific molecular mechanisms underlying MH’s effects on brain IR injury still require further investigation.
In the previous study, we examined the hippocampal proteomic characteristics of rats treated with MH following cerebral IR injury. Our findings revealed that MH reversed the elevated expression of S100A8 in the hippocampus of IR rats. Additionally, the down-regulated expression of CAMKK2 after IR was increased with MH treatment. Both of these differential proteins were found to be enriched in the metabolic and reactive oxygen species (ROS) pathways5. This enrichment may be closely related to alterations in intrahippocampal signaling pathways after brain IR injury. S100A8, known as a damage-associated molecular pattern, exists both intracellular and extracellular. Its effects have previously been reported to be primarily mediated through the advanced glycation end product receptors or toll-like receptor 4, leading to the activation of nuclear factor kappa B6,7. Clinical studies have suggested that while S100A8 is generally considered as an early alarmin or inflammatory factor, it may also play a role in brain IR disease5,8,9,10. However, the exact mechanisms remain uncertain.
Mitochondrial dysfunction plays a crucial role in the progression of brain IR injury and serves as a key mediator for neuronal survival during IR injury. Accumulating evidence indicates that neuronal survival under IR stress is influenced by a complex interplay of mechanisms that regulate mitochondrial content and quality11. Studies have demonstrated that S100A8 decreases mitochondrial membrane potential and inhibits mitochondrial fission, which promotes the production of ROS and, consequently, apoptosis12,13. Calmodulin-dependent protein kinase kinase 2 (CAMKK2), a vital secondary messenger abundantly expressed in the brain, regulates a variety of biological activities through its integrated functions14. It is recognized as a major upstream regulator of AMP-activated protein kinase (AMPK) in hippocampal pyramidal neurons, phosphorylating AMPK, which subsequently regulates mitochondrial homeostasis and influences signaling cascades related to learning and memory15. AMPK is an essential regulator of cellular energy production, maintaining mitochondrial function and cell survival, making it a pivotal molecule in IR injury16. Previous studies have shown that AMPK phosphorylation is reduced after IR injury. Its inactivation contributes to apoptosis by inducing mitochondrial dysfunction, which encompasses oxidative damage, a decrease in membrane potential, and the loss of mitochondrial crest structure17. Additionally, the knockout of S100A9 leads to the down-regulation of S100A8 expression, thereby activating AMPK, which can reduce ROS production, protect mitochondrial function, and inhibit apoptosis18,19.
Therefore, this study aims to explore the specific mechanism of neuroprotection exerted by MH and to investigate whether the S100A8-CAMKK2-AMPK axis functions in this process, potentially elucidating the underlying molecular mechanisms.
Materials and methods
Ethical statement and animals
Adult male Wistar Rats (body weight: 250–300 g, age: 6–8 weeks) were obtained from Jinan Pengyue Laboratory Animal Breeding Company (Jinan, China). All procedures were conducted in accordance with the relevant guidelines and regulations of the National Health Commission and the Ministry of Science and Technology and they adhered to the guidelines for animal research. The experimental procedures were approved by the Qingdao University Medical Ethics Committee (QDU-AEC-2024473) and every effort was made to minimize animal suffering and to reduce the number of specimens used. All the animals were maintained on a standard laboratory diet under controlled humidity (65–70%), indoor temperature (21 ± 2℃), and 12/12 h light–dark cycle conditions. Animals were grouped using a random number table method randomized for the experiments. Each experiment and the statistical calculations described below were carried out in a randomized order by the experimenter blinded to the group.
Global cerebral IR injury in rats
The rat model was established using Pulsinelli’s 4-VO method as previously described20. The rat was placed in an induction chamber and anesthesia induced by 4% isoflurane (license No. H20020267, Lunan Better Pharmaceutical Co., Ltd., China), following with endotracheal intubation. The tube was connected to an animal anesthesia ventilator (RWD Life Science, R407, China), and 0.8–1.2% isoflurane was used for maintenance anesthesia. A tidal volume of 15 ml/kg and a rate of 80–85 breaths per minute were used to maintain an appropriate peak airway pressure. Sham animals that underwent the same surgical procedure without the actual ligation served as controls. MH (33 ± 0.5℃) was induced at the beginning of reperfusion and maintained for 4 h. Core body temperature was monitored continuously using temporalis muscle and rectal temperature probes. An ice blanket was placed over the dorsum of prone rats until a body temperature of 33 ± 0.5 °C was achieved. The rats were then allowed to gradually rewarm back to their baseline temperature (37 ± 0.5 °C) during a 1-h period using heating lamps. After surgery, the animal was awakened, the tracheal tube was removed and the necessary postoperative observations were made.
Infection with S100A8 over-expressing and knock-down adeno‑associated virus and pharmacological treatments
S100A8 over-expressing adeno-associated virus type 9 (AAV9-S100A8-GFP) and adeno-associated virus type 9-negative control (AAV9-NC-GFP), and knock-down adeno-associated virus type 9 (AAV9-r-S100A8-shRNA-GFP, the shRNA promoter: U6, target sequence: 5′-GGTCACTACTGAGTGCCCTCAGTTT-3′) and adeno-associated virus type 9-negative control (AAV9-NC-GFP) were all purchased from Hanheng Biotechnology Company. The experiments were performed 4 weeks after the viral infection. The agents were microinjected bilaterally and sequentially into the lateral ventricles, with a volume of 2 μl administered on each side. Drug delivery into the lateral ventricles was carried out with a stereotaxically positioned 27-gauge stainless-steel needle connected to a 0.5–5 μl microsyringe. The stereotaxic coordinates used for the bilateral lateral ventricles were based on the rat brain atlas (Paxinos and Watson, 2007): 0.72 mm posterior to bregma, 1.5 mm from the midline, and 4.0 mm below the cortical surface. STO-609 (CAMKK2 specific inhibitor, 10 mg/kg)21, and Dorsomorphin (Dor, AMPK specific inhibitor, 10 mg/kg)22 were dissolved in DMSO, diluted to 4% DMSO in saline, and microinjected bilaterally into the lateral ventricles 30 min prior to cerebral IR injury.
Histological analysis
Histological examination was performed on rats’ brain from individuals of each group (24 h of reperfusion). After perfusion with 4% paraformaldehyde, the isolated brains were dehydrated in a series of ethanol and xylene baths and embedded in paraffin wax. Sections measuring 5–7 μm were stained with hematoxylin and eosin (HE) and Nissl staining to reflect histopathological lesions. The microscopic evaluation was performed with Axiolab light microscope at 40× and 400× magnification.
Neurological deficit score
Neuro Deficit Score (NDS) was used to evaluate the neurologic functional outcome after IR injury. The overall template of the NDS is patterned after the standard neurologic examination in humans and is applicable to rats. The NDS and its components were referenced in the study by Geocadin et al.23 and were assessed at 24 h after brain IR. The rats were removed from the cage, and a total of 7 single-blind NDS assessments were conducted, resulting in a cumulative score of 80 points. A score of 80 points indicates normal brain function, while a score of 0 points signifies brain death23. After the preparation work was completed, the rats were scored and recorded double-blind by a professionally trained experimenter.
Transmission electron microscopy (TEM)
The specimens from the CA1 region of the hippocampus were taken under a microscope and sequentially fixed in 2% paraformaldehyde, 2.5% glutaraldehyde in cacodylate buffer, and 1% osmium tetraoxide with potassium ferricyanide for 2 h. Then, the samples were dehydrated in a series of ethanol baths and embedded in resin. Sectioning is performed using a Leica UC7 ultramicrotome (Leica, Germany) at a thickness of 60–80 nm, the ultra-thin sections were then stained with uranyl acetate and lead citrate. The images were acquired in a randomized way using a JEM-1400 (Jeol, Japan) transmission electron microscope and the morphology of mitochondria in neuron cytoplasm was observed. Five sections were selected from each group, and four fields of view were randomly selected for each section under the same magnification to assess mitochondria based on the degree of mitochondrial damage by Flameng scoring method24. And a higher score indicates more severe damage.
Culture and treatment of SH-SY5Y
The SH-SY5Y cell line were purchased from Proximity Life Sciences (China). The cells were cultured in DMEM/F12 solution containing 15% fetal bovine serum (FBS) and 1% penicillin/streptomycin and placed in a constant temperature incubator at 37 °C with 5% CO₂. An in vitro cell model of cerebral IR was established using the oxygen-glucose deprivation reoxygenation (OGDR) model. Briefly, SH-SY5Y cells were cultured in glucose-free medium under hypoxic conditions (1% O₂ + 94% N₂ + 5% CO₂) at 37 °C for 2 h to establish an OGD model. The cells were then cultured in normal medium with normal oxygen and glucose content for 24 h for reperfusion. SH-SY5Y were infected with pcDNA3.1-S100A8 (Youbio, China) or S100A8 siRNA25 (RiboBio, China) for over-expressing or knock-down the expression of S100A8. STO-609 (1 μg/ml)26 or Dor (5 μM)27, dissolved in 4% DMSO, was given 30 min before OGDR injury. SH-SY5Y cells that received the same amount of DMSO served as vehicle controls.
ROS and mitochondrial membrane potential detection
ROS was detected through Dihydroethidium (DHE, Beyotime, S0063) staining. Briefly, cells or the frozen sections were incubated with 2 μM DHE staining working solution at 37 °C for 30 min and then washed gently with PBS for three times. Mitochondrial membrane potential was assessed through tetramethylrhodamine ethyl ester (TMRE, Beyotime, C2001S) staining. SH-SY5Y were treated with TMRE staining solution at 37 °C for 30 min, and then washed three times with PBS. DAPI was used to label the nuclei (Beyotime, C0003-3). Images were captured using a fluorescence microscope (Nikon, Japan).
Apoptosis assay
A terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) staining kit (TUNEL, Elabscience, E-CK-A320) was used to detect apoptotic cells according to the manufacturer’s instructions. Briefly, the frozen sections or fixed cell crawling slides were washed in PBS and then incubated in permeabilization buffer and blocked. Next, TUNEL reaction mixture was applied, and the sections were incubated for 1 h at 37 °C. Finally, sections were mounted with antifade reagent with DAPI. The images were acquired with a Nikon microscope (Nikon, Japan), and staining intensity was quantified using Image J software.
Immunofuorescence staining
Frozen sections were washed with TBS containing 0.1% Tween-20 (Beyotime, ST825), and then blocked with 5% bovine serum albumin (BSA, Sigma, A7906) for 30 min at room temperature. The sections were incubated in permeabilization solution (1% Triton X-100, Beyotime, P0096) for 15 min at room temperature and then incubated in primary antibody diluted in primary antibody dilution buffer (Beyotime, P0277) overnight at 4 °C. After rinsing with PBS, the sections were incubated with the corresponding dye-labeled secondary antibody. The sections were mounted with antifade reagent with DAPI to label the nuclei (Beyotime, C0003-3). Traced sections were examined with a Nikon microscope, and the colocalization of proteins was quantified using Image J software. The antibodies used were as follows: rabbit anti-S100A8 (1:1000; Abcam; ab180735), Cy3-labeled goat anti-rabbit IgG (H + L) (1:2000; Abcam; ab6939).
qRT‑PCR
Cells or rat hippocampus tissue were collected using Trizol reagent (Life technologies, Carlsbad, CA, USA) and lysed. The extracted RNA was reverse transcribed, and then subjected to RT-PCR using corresponding kits according to the manufacturer’s instructions (Takara, Japan). The ratio for the mRNA of interest was normalized by β-actin. Additional file Table S1 displays the qRT-PCR analysis primers used.
Western blot analysis
Total protein was extracted from rat hippocampus or SH-SY5Y and the concentration was measured by BCA kit (Elabscience, China). The primary antibodies were as follows: S100A8 (1:1000; Abcam; ab180735), CAMKK2 (1:1000; Abcam; ab135979), p-AMPK (1:1000; Abcam; ab133448), AMPK (1:1000; Abcam; ab1207442), bax (1:2000; Affinity; AF-0120), caspase3 (1:1000; Abcam; ab184787), and the corresponding secondary antibodies. The ECL luminescent solution was applied to develop the color; a fully automated chemiluminescence gel-imaging analyzing system (Beijing Sage Science and Technology Co., Ltd.) was used for exposure and photography; and Image J software was utilized for analysis. Quantitative results are expressed as gray value ratios of target proteins to β-actin, with exception of the signaling pathway proteins, such as “p-AMPK/AMPK” which are presented as a ratio of phosphorylated protein to total protein.
Morris water maze test
The Morris water maze (MWM) test is widely used to evaluate an animal’s spatial learning and memory function28. Briefly, in a circular pool of 150-cm diameter and 50-in height, which is filled with water to its half and dissolved in it ink to make it opaque. The pool is divided into four quadrants, and an invisible platform (10 cm in diameter) was placed 2 cm under the water surface in the same quadrant of training trails. The rats were placed in the room 24 h in advance to acclimatize before starting the experiment. For 4 consecutive days, each rat was trained four times per day to locate and climb on the platform. Each trail began with placing a rat in a different position in the pool and recording the rat’s escape latency. Each rat was allowed 15 s to rest on the platform. If the rat does not locate the platform within 60 s, it will be guided to and remain on the platform for 15 s. The escape latency for each rat was calculated as the mean of four trails, and the mean of each group was determined using the day’s mean. The platform was removed on the 5th day to conduct the probe test for 60 s, and the time and distance spent in the target quadrant was calculated as an indicator of memory function. Each rat’s trajectory for every training and testing in the pool was recorded using water maze software for analysis.
Statistical analysis
Statistical analysis was performed using GraphPad Prism (GraphPad Software, San Diego, CA, USA). The distribution of data was analyzed by Shapiro–Wilk normality test. Normally distributed data are presented as means ± standard deviation. Differences between three or more groups were analyzed using one-way ANOVA with Dunnett’s post-hoc test (for comparisons to controls). Student’s t-test was used to detect differences between two groups. For data with a non-normal distribution (data based on TEM microphotographs: mitochondrial injury score), the Kruskal–Wallis test followed by Dunn’s multiple comparisons test was employed and the results are presented as median. p-value < 0.05 were considered significant.
Results
MH alleviates oxidative stress and mitochondrial damage, inhibits neuronal cell damage in the hippocampal CA1 region, and improves cognitive function after cerebral IR injury in rats
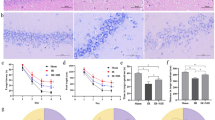
Figure 1A shows the schematic diagram of the animal experiment in this part. A Neuro Deficit Score (NDS) was determined to evaluate the neurologic functional outcome before rats were euthanized at 24 h after IR. The mean NDS value of rats in the IR group was significantly lower compared to the Sham group, while the NDS was higher in the IR + MH group compared to the IR group (Fig. 1B). This suggests that MH treatment in rats improved neurological outcomes in the early stage after cerebral IR injury compared with untreated animals.

Protective effects of MH on hippocampal neurological damage, mitochondrial oxidative stress, and cognitive dysfunction after cerebral IR injury. (A) Schematic diagram of the animal experiment. (B) Neurodeficit scores for each group (n = 10, per group). (C) HE and Nissl staining of the CA1 region in the hippocampus (Scale bar: 50 μm) (n = 5, per group). (D) The time that rats spent to find the platform during 4 training days. (E–G) Mean crossing times, and the time and distance rats spent in the target quadrant in the probe test (n = 10, per group). (H) Routes taken by rats in the probe test. (I) Representative images of DHE and TEM of neurons in the CA1 region of the hippocampus (Scale bars: DHE-50 μm, TEM-500 nm). (J) Intensity of DHE fluorescence (n = 5, per group). (K) Scores assessing mitochondrial injury (n = 5, per group). Sham: Sham-operation group; IR: Group subjected to cerebral IR injury followed by normothermia (37 °C); IR + MH: Group subjected to cerebral IR injury followed by 4 h of MH (33 ± 0.5 °C). Results are presented as the mean \(\pm\) standard deviation (SD). *p < 0.05, **p < 0.01.
Histological analysis using HE and Nissl staining revealed that brain IR in rats, compared to the Sham group, resulted in a reduction in the number of neurons in the CA1 region, a decrease in Nissler bodies in pyramidal neurons and an increase in cell death characteristics (characterized by loosely arranged neurons and fuzzy cells outlines, loss and light color staining). MH markedly ameliorated these pathological changes compared to the IR group (Fig. 1C), which is consistent with previously reported outcomes5,29.
Cognitive differences between groups were detected using the Morris water maze test. During the training period, spanning from day one to day four, the escape latency and path length for all three groups progressively decreased during the 4 training days (Fig. 1D). On the fifth day of testing, the hidden platform was removed to assess the spatial memory ability of the three groups. During the 60-s detection period, there were no statistical differences between the three groups in total swimming distance (Fig. S1). The IR group showed decreased mean crossing times and increased escape latency for the first crossing of the platform compared with the Sham group, while MH significantly improved this phenomenon (Fig. 1E–H). These observations indicate that there was memory impairment in IR rats, and MH treatment was able to improve the memory abilities in IR rats.
In order to further elucidate the cytoprotective mechanism, we examined the intracellular ROS production and the ultrastructure of mitochondria in neurons of the hippocampal CA1 region. Cerebral IR caused a large amount of ROS production in neurons in the hippocampus CA1 region (Fig. 1I), while MH treatment effectively reduced ROS production (Fig. 1J). Changes in the ultrastructure of hippocampal neurons were also detected (Fig. 1I). In Sham group, mitochondria showed a normal structure in CA1 neurons. Outer and inner mitochondrial membranes were consistent, mitochondrial cristae were preserved, and mitochondrial matrix remained electron-dense. However, in the CA1 region 24 h after IR, a great number of mitochondria showed significant edema, lacked mitochondrial cristae, and the mitochondrial matrix became lighter than the control group; the Flameng mitochondrial injury score was significantly reduced (Fig. 1K). With MH treatment, the ultrastructural damage to mitochondria, as mentioned above, was significantly improved compared to the IR group, and the mitochondrial injury score increased. Overall, these results suggest that MH could reduce the intracellular ROS production and improve the mitochondrial ultrastructural changes caused by cerebral IR, thus preventing oxidative stress and mitochondrial injury in neurons.
MH reduced S100A8 expression in the hippocampus of cerebral IR rats
In a previous study, we conducted a preliminary hippocampal proteomic study on the neuroprotective effects of MH on brain IR injury and identified several differential proteins potentially involved in its protective mechanisms, including S100A85 (Fig. 2A). This study further investigates the specific role of S100A8. We tracked the expression trend of S100A8 in the hippocampus of rats at different time points after IR. Our findings revealed a noteworthy increase in the expression of S100A8 with reperfusion time, peaking at 24 h after the IR injury, and remained elevated until day 3 (Fig. 2B and C). Consequently, the 24-h post-IR mark was selected as the time point for subsequent experiments. Next, the expression level changes of S100A8 in response to MH treatment were examined. The IR group showed significantly increased S100A8 expression in the hippocampus compared to the Sham group. However, after MH treatment, there was a significant reduction in S100A8 expression (Fig. 2D and E), particularly in neurons of the hippocampal CA1 region (Fig. 2F and G). This suggests that S100A8 may be implicated in the mechanism underlying MH’s neuroprotective action on brain IR injury.

Impact of MH on S100A8 expression after cerebral IR injury. (A) The volcano plot from proteomic analysis, proteins significantly altered are highlighted, with S100A8 indicated in red (n = 7, per group). (B) Western blot showing temporal expression of S100A8 protein at various time points after reperfusion. (C) Quantitative analysis of Western blots from (B) (n = 3, per group). (D) Western blot of S100A8 expression cross three different groups. (E) Quantitative analysis of Western blots from (D) (n = 3, per group). (F, G) Immunofluorescence staining of S100A8 in the CA1 region of the hippocampus (Scale bar: 50 μm) and its expression quantification (n = 5, per group). Sham: Sham-operation group; IR: Group subjected to cerebral IR injury followed by normothermia (37 °C); IR + MH: Group subjected to cerebral IR injury followed by 4 h of MH (33 ± 0.5 °C). Results are presented as the mean \(\pm\) SD. *p < 0.05, **p < 0.01.
In vivo, the impact of S100A8 over-expression/knock-down on MH treatment of IR neuron damage
To explore whether MH attenuates brain IR injury by modulating S100A8, we employed AVV9 vectors to either knocked-down or over-expressed S100A8 in the hippocampus (Fig. 3A). AAV9 infection achieved a strong and uniform GFP expression in the hippocampus, without a patchy distribution (Fig. S2A). AAV9.shS100A8 infection resulted in the depletion of S100A8 (Fig. S2B and C), while AAV9.S100A8 infection led to its over-expression (Fig. S2D and E).

In vivo, MH ameliorates IR-induced neuronal injury by decreasing S100A8 expression. (A) Schematic diagram of the animal experiment. (B) Western blot showing the expression of S100A8, bax, and caspase3 protein, with β-actin serving as the loading control. (C–E) Quantification of the Western blots from (B). Levels of S100A8, bax and cleaved-caspase3/pro-caspase3 normalized to the corresponding loading control across three independent trials (n = 3, per group). (F) Representative images of HE, TUNEL, DHE staining and TEM in the CA1 region of the hippocampus (Scale bars: HE-50 μm, TUNEL-50 μm, DHE-50 μm, TEM-500 nm). (G–I) Quantitative analysis of cell apoptosis ratio, DHE fluorescence intensity, and mitochondrial injury score (n = 5, per group). Sham + AAV9.NC: bilateral lateral ventricle injected with AAV9.NC 28 days before sham-operation; IR + AAV9.NC: bilateral lateral ventricle injected with AAV9.NC 28 days before cerebral IR injury; IR + AAV9.ShS100A8: bilateral lateral ventricle injected with AAV9.ShS100A8 28 days before cerebral IR injury; IR + MH + AAV9.NC: bilateral lateral ventricle injected with AAV9.NC 28 days before cerebral IR injury followed by 4 h of MH (33 ± 0.5 °C). IR + MH + AAV9.S100A8: bilateral lateral ventricle injected with AAV9.S100A8 28 days before cerebral IR injury followed by 4 h of MH (33 ± 0.5 °C). Results are presented as the mean \(\pm\) SD. *p < 0.05, **p < 0.01.
Cerebral IR significantly increased the expression of S100A8 in the hippocampus, along with an increase in apoptosis-related proteins such as bax and cleaved-caspase3; however, hypothermia effectively inhibited this increase. Echoing the effect of MH, AAV9.ShS100A8 infection significantly reduced the expression of S100A8, and levels of bax and cleaved-caspase3 were also down-regulated after IR. When compared to MH treatment alone, the S100A8 protein level was higher in the hippocampus of rats infected with AAV9.S100A8, and levels of bax and cleaved-caspase3 were similarly elevated (Fig. 3B–E). Figure 3F illustrates that, akin to MH’s effects, AAV9.ShS100A8 infection significantly inhibited apoptosis (Fig. 3G), alleviated neuronal oxidative stress (Fig. 3H), reduced mitochondrial damage (Fig. 3I), and enhanced cognitive function (Fig. S3A, B, C, D and E) in IR rats. Conversely, AAV9.S100A8 markedly weakened the protective effect of MH, resulting in worsened IR brain damage compared to MH treatment alone. This was evidenced by persistent neuronal loss, increased neuronal oxidative stress, mitochondrial damage, and neuronal apoptosis. The protective effect of MH on neurocognitive impairment was also diminished (Fig. S3A, B, C, D and E). These findings substantiate that the protective mechanism of MH involves the regulation of S100A8 level.
In vitro, the effect of S100A8 over-expression/knock-down on MH treatment of SH-SY5Y after OGDR injury
To further confirm the role of S100A8 in the neuronal protection of MH, we over-expressed/knocked-down endogenous S100A8 expression by pcDNA3.1 and RNAi strategy in SH-SY5Y (Fig. 4A). Using pcDNA3.1-S100A8 and S100A8-specific siRNA, we achieved more than 70% alteration in GFP fluorescence expression in SH-SY5Y incubated for 2 days (Fig. S4A and D). As determined by qRT-PCR (Fig. S4B and E), showing consistency with western blotting results (Fig. S4C and F).

In vitro, MH ameliorates OGDR injury by decreasing the expression of S100A8 in SH-SY5Y cells. (A) Schematic diagram of the cell experiment. (B) Western blot showing the expression of S100A8, bax, and caspase3 protein, with β-actin was serving as the loading control. (C–E) Quantification analysis of the Western blots from (B). Levels of S100A8, bax and cleaved-caspase3/pro-caspase3 normalized to the corresponding loading control across three independent trials (n = 3, per group). (F) Representative images of TUNEL, DHE and TMRE staining in SH-SY5Y cells (Scale bar: 50 μm). (G–I) Quantitative analysis of apoptosis ratio, DHE, and TMRE fluorescence intensity (n = 5, per group). Control: Control group; OGDR: oxygen-glucose deprivation 2 h and reoxygenation 24 h; OGDR + Si-S100A8: SH-SY5Y were infected S100A8 siRNA using siRNA transfection kit for knocking down the expression level of S100A8 48 h before OGDR injury; OGDR + MH: OGDR injury followed by 4 h of MH (33 ± 0.5 °C). OGDR + MH + pcDNA3.1-S100A8: SH-SY5Y were infected with pcDNA3.1-S100A8 using Lipofectamine 2000 for overexpressing the expression level of S100A8 before OGDR injury followed by 4 h of MH (33 ± 0.5 °C). Results are presented as the mean \(\pm\) SD. *p < 0.05, **p < 0.01.
As indicated in Fig. 4B, OGDR injury significantly increased the expression of S100A8 in the SH-SY5Y cells, along with increased levels of apoptosis-related proteins bax and cleaved-caspase 3, while MH inhibited the process. In a manner akin to MH, Si-S100A8 transfection significantly reduced the expression of S100A8, and correspondingly, levels of bax and cleaved-caspase3 were down-regulated post-OGDR. Compared to MH treatment alone, S100A8 protein level was elevated in SH-SY5Y cells transfected with an S100A8 plasmid, and this was accompanied by increased levels of bax and cleaved-caspase3 (Fig. 4C–E). As shown in Fig. 4F and Fig. S7, similarly to the protective effect of MH, S100A8 down-regulation significantly reduced cell apoptosis (Fig. 4G), mitigated neuronal oxidative stress (Fig. 4H) and preserved mitochondrial membrane potential (Fig. 4I). However, S100A8 over-expression largely diminished MH’s protective effect, exacerbating OGDR injury compared to MH treatment alone, which was evidenced by increased neuronal oxidative stress, decreased mitochondrial membrane potential, and enhanced neuronal apoptosis. The findings in this section underscore that, at the cellular level, MH’s neuroprotective action involves the downregulation of S100A8 expression.
Over-expression of S100A8 inhibits the activation of CAMKK2/AMPK pathway by MH in the hippocampus and SH-SY5Y cells after IR or OGDR injury.
In previous hippocampal proteomic studies on the neuroprotective effect of MH in brain IR injury, we identified several differential proteins possibly involved in its protective mechanism, including CAMKK2 and S100A85 (Fig. 5A). These findings may be closely related to changes in intrahippocampal signaling pathways following brain IR injury. S100A8 has been identified as a crucial regulatory target in the neuroprotective mechanism of MH, which mitigates neuronal apoptosis by down-regulating S100A8. Given that CAMKK2/AMPK is a key pathway for mitochondrial apoptosis, we sought to confirm whether CAMKK2/AMPK is a downstream target of S100A8. We measured the activation of CAMKK2/AMPK pathway in rat hippocampus and SH-SY5Y cells. The activity of the CAMKK2/AMPK pathway decreased after IR but increased following MH treatment (Fig. 5B–E). However, this pathway was inhibited again after S100A8 over-expression compared to MH treatment alone. Similarly, in SH-SY5Y cells, MH down-regulated S100A8 expression and enhanced CAMKK2/AMPK activation. Compared to MH treatment alone, over-expression of S100A8 again inhibited the pathway (Fig. 5F–I). These results suggest that CAMKK2/AMPK act as a downstream target of S100A8, and MH may activate the CAMKK2/AMPK pathway by downregulating the expression of S100A8.

MH activates the CAMKK2/AMPK pathway by downregulating S100A8 expression. (A) Volcano plot from proteomic analysis, highlighting potential signaling pathways, with markers for S100A8 and CAMKK2 (n = 7, per group). (B) Western blot analysis displaying in vivo expression of S100A8, CAMKK2, p-AMPK, and AMPK, with β-actin serving as the loading control. (C–E) Quantification analysis of the Western blots from (B) (n = 3, per group). (F) Western blot analysis showing in vitro expression of S100A8, CAMKK2, p-AMPK, and AMPK, with β-actin serving as the loading control. (G–I) Quantification analysis of the Western blots from (F). Levels of S100A8, CAMKK2, p-AMPK, and AMPK normalized to corresponding loading control are summarized across three independent trials (n = 3, per group). Sham + AAV9.NC: bilateral lateral ventricle injected with AAV9.NC 28 days before sham-operation; IR + AAV9.NC: bilateral lateral ventricle injected with AAV9.NC 28 days before cerebral IR injury; IR + MH + AAV9.NC: bilateral lateral ventricle injected with AAV9.NC 28 days before cerebral IR injury followed by 4 h of MH (33 ± 0.5 °C). IR + MH + AAV9.S100A8: bilateral lateral ventricle injected with AAV9.S100A8 28 days before cerebral IR injury followed by 4 h of MH (33 ± 0.5 °C); Control: Control group; OGDR: oxygen-glucose deprivation 4 h and reoxygenation 24 h; OGDR + MH: OGDR injury followed by 4 h of MH (33 ± 0.5 °C). OGDR + MH + pcDNA3.1-S100A8: SH-SY5Y were infected with pcDNA3.1-S100A8 using Lipofectamine 2000 for over-expressing S100A8 before OGDR injury followed by 4 h of MH (33 ± 0.5 °C). Results are presented as the mean \(\pm\) SD. *p < 0.05, **p < 0.01.
MH activates the CAMKK2/AMPK pathway against cerebral IR injury in rats
To elucidate the role of the CAMKK2/AMPK pathway in MH treatment of neuronal IR injury, we treated the rat hippocampus with STO-609 and Dor, inhibitors of CAMKK2 and AMPK, respectively (Fig. 6A). Following treatment with these inhibitors, the activity of the CAMKK2/AMPK pathway was suppressed, and the expression of apoptotic proteins bax and cleaved-caspase3 increased compared to the MH treatment group (Fig. 6B–G). This indicates that MH inhibits neuronal apoptosis by activating the CAMKK2/AMPK pathway. We also observed that the expression of S100A8 was up-regulated with the inhibition of the CAMKK2/AMPK pathway, suggesting a potential crosstalk between S100A8 and another perspective of the CAMKK2/AMPK pathway. MH significantly reduced neuronal oxidative stress, attenuated mitochondrial damage, and inhibited apoptosis in the hippocampus of IR rats. However, STO-609 and Dor significantly diminished the protective effects of MH, resulting in exacerbated IR brain damage in compared to MH treatment alone. This was manifested by increased neuronal oxidative stress, mitochondrial damage, and neuron apoptosis (Fig. 6H–K). These findings provide evidence that the neuroprotective mechanism of MH in IR rats involves the regulation of the CAMKK2/AMPK pathway.

Inhibition of the CAMKK2/AMPK pathway reduces the protective effect of MH on neuronal apoptosis, oxidative stress, and mitochondrial injury in IR rats. (A) Schematic diagram of the animal experiment. (B) Western blot showing in vivo expression of S100A8, CAMKK2, p-AMPK, AMPK, bax and caspase 3, with β-actin serving as the loading control. (C–G) Quantification of the Western blots of (B) (n = 3, per group). (H) Representative images of TUNEL, DHE staining and TEM in the CA1 region of the hippocampus (Scale bars: TUNEL-50 μm, DHE-50 μm, TEM-500 nm). (I–K) Quantitative analysis of cell apoptosis ratio, DHE fluorescence intensity, and mitochondrial injury score (n = 5, per group). Sham: Sham-operation group; IR: Group subjected to cerebral IR injury followed by normothermia (37 °C); IR + MH: Group subjected to cerebral IR injury followed by 4 h of MH (33 ± 0.5 °C); IR + MH + STO-609: STO-609 microinjected bilaterally into the lateral ventricles 30 min before cerebral IR injury followed by 4 h of MH (33 ± 0.5 °C); IR + MH + Dor: Dor microinjected bilaterally into the lateral ventricles 30 min before cerebral IR injury followed by 4 h of MH (33 ± 0.5 °C). Results are presented as the mean \(\pm\) SD. *p < 0.05, **p < 0.01.
MH activates the CAMKK2/AMPK pathway against OGDR injury in SH-SY5Y cells
To further elucidate the role of the CAMKK2/AMPK pathway in MH’s treatment of neuronal OGDR injury in vitro, we treated SH-SY5Y cells with STO-609 and Dor, inhibitors of CAMKK2 and AMPK, respectively (Fig. 7A). Following inhibitor administration, the activity of the CAMKK2/AMPK pathway was suppressed, and the expression of apoptotic proteins bax and cleaved-caspase3 increased, compared to the MH treatment group (Fig. 7B–G). As shown in Fig. 7H, MH significantly reduced cell oxidative stress, attenuated mitochondrial damage, and inhibited apoptosis in SH-SY5Y cells. However, STO-609 and Dor substantially reduced the protective effect of MH, leading to exacerbated neuronal oxidative stress, mitochondrial damage, and neuron apoptosis, compared to MH treatment alone (Fig. 7I–K). These findings suggest that the protective mechanism of MH in neuronal cells involves the regulation of the CAMKK2/AMPK pathway.

Inhibition of the CAMKK2/AMPK pathway reduces the protective effect of MH on apoptosis and oxidative stress in SH-SY5Y cells after OGDR injury. (A) Schematic diagram of the cell experiment; (B) Western blot analysis shows the expression of S100A8, CAMKK2, p-AMPK, AMPK, bax and caspase3 protein expression, with β-actin serving as the loading control; (C–G) Quantification of the Western blots from (B). Normalized S100A8, CAMKK2, p-AMPK/AMPK, bax and cleaved-caspase3/pro-caspase3 to corresponding loading control are summarized across three independent trials (n = 3, per group). (H) Representative images of TUNEL, DHE and TMRE staining (Scale bar: 50 μm) of SH-SY5Y cells; (I–K) Quantitative analysis of apoptosis ratio, DHE and TMRE fluorescence intensity (n = 5, per group). Control: Control group; OGDR: oxygen-glucose deprivation 2 h and reoxygenation 24 h; OGDR + MH: OGDR injury followed by 4 h of MH (33 ± 0.5 °C); OGDR + MH + STO-609: SH-SY5Y were treated with STO-609 for inhibiting the activation of CAMKK2 30 min before OGDR injury followed by 4 h of MH (33 ± 0.5 °C); OGDR + MH + Dor: SH-SY5Y were treated with Dor for inhibiting the activation of AMPK 30 min before OGDR injury followed by 4 h of MH (33 ± 0.5 °C). Results are presented as the mean \(\pm\) SD. *p < 0.05, **p < 0.01.
Discussion
In 1997, Sessler introduced the concept of perioperative MH in the New England Journal of Medicine, describing its ability to prevent cerebral ischemia30. Since then, MH has been extensively studied as one of the most robust neuroprotectants against cerebral IR injury in both laboratory and clinical settings31. MH reduces the production of free radicals, inflammation, excitotoxicity, while enhancing cerebral metabolism following cerebral IR injury, thereby protecting against neuronal damage. Although MH treatment is associated with a range of pathological and physiological changes, along with potential side effects, it remains a vital therapeutic strategy for cerebral IR injury, warranting further investigation into its underlying mechanisms32,33,34.
In previous hippocampal proteomic studies on the neuroprotective effect of MH in brain IR injury, we reported that S100A8 and CAMKK2 may be involved in its protective mechanisms5. S100A8, a member of the S100 calcium-binding family, is predominantly found as calprotectin (S100A8/A9) under physiological conditions35. Unlike S100A9, S100A8 is critical for viability as the primary active functional unit, as evidenced by the embryonic lethality observed in S100A8 knockout mice36. Moreover, variations in S100A8 expression levels, post-translational modifications, or interactions with different receptors can lead to diverse functional outcomes37,38,39,40,41. S100A8 plays a crucial role in various pathological processes during IR injury42. For instance, in myocardial IR injury, S100A8 signaling contributes to mitochondrial dysfunction and cardiomyocyte death10. Additionally, two independent multicenter cohort studies found that baseline plasma concentrations of S100A8/A9 were predictive of post-death outcomes in 4785 patients with ischemic stroke8. Exposure to hypoxia also increases the expression of the S100A8 protein in mouse neurons and microglia, which triggers neuroinflammation and neuronal apoptosis43.
Apoptosis, a form of programmed cell death, could be triggered by multiple signals, including ROS, bax or activation of the mitochondria-mediated caspase cascade, and other apoptosis-related factors44. The inhibition of CAMKK2/AMPK pathway causes mitochondrial damage, resulting in an abnormal surge in ROS, leading to cell apoptosis45. CAMKK2, abundantly expressed in hippocampus, is vital for the formation of hippocampal memory. CAMKK2-deficient mice demonstrate exacerbated brain damage, including increased infarct sizes, edema, and behavioral deficits following middle cerebral artery occlusion46. CAMKK2 sustains a certain level of AMPK activity, while activation of AMPK has been shown to protect neurons from apoptosis under ischemic conditions47,48. Thus, maintaining CAMKK2/AMPK pathway activity is essential for neuronal protection after cerebral IR injury. Calmodulin (CaM) is an indispensable protein in the CAMKK2/AMPK pathway. CAMKK2 activation is dependent on CaM, and there is evidence suggesting a potential interaction between CaM and S100 proteins49. Therefore, regulating S100A8 may influence the activity of the CAMKK2/AMPK pathway, which in turn impacts the progression of brain IR injury.
In the present study, we used the Pulsinelli’s four-vessel ligation method to establish a rat model of global cerebral IR injury. Firstly, we demonstrated the protective effects of MH in cerebral IR injury, including reducing oxidative stress, improving mitochondrial damage, reducing neuronal loss and apoptosis, and protecting neurocognitive function in rats. Secondly, we observed that S100A8 expression increased with reperfusion time, peaking at 24 h and maintaining elevated until day 3 during cerebral IR injury in the hippocampus in rats. This finding aligns with the chronological distribution of neuronal apoptosis after global cerebral IR injury reported in previous studies50. Furthermore, by manipulating S100A8 expression both in vivo and in vitro, through knock-down and over-expression, we established that MH exerts neuroprotective effects by down-regulating S100A8. The down-regulation of S100A8 mirrored the neuroprotective effect of MH treatment, whereas its over-expression attenuate these effects. Moreover, over-expression of S100A8 inhibited the activation of CAMKK2/AMPK pathway by MH, suggesting that the CAMKK2/AMPK signaling could be a downstream mediator of S100A8. Additionally, the administration of STO-609 and Dor, inhibitor of CAMKK2 and AMPK respectively, significantly reduced the protective effect of MH, leading to increased neuronal oxidative stress, mitochondrial damage, and neuron apoptosis compared to MH treatment alone. These results indicate a direct association between S100A8 and CAMKK2. MH down-regulates S100A8 expression to enhance CAMKK2-dependent AMPK activation, thereby reducing mitochondrial damage, mitigating oxidative stress, and decreasing neuronal apoptosis. Conversely, over-expression of S100A8 or inhibition of the CAMKK2/AMPK pathway can diminish the neuroprotective effect of MH on brain IR injury.
However, there are still limitations in this study. The present results only showed MH affects CAMKK2/AMPK activity by regulating the expression of S100A8-CAMKK2-AMPK axis, but the specific molecular mechanisms underlying the interaction between S100A8 and CAMKK2 have not been elucidated. Further studies are needed to deeply explore the complex regulatory mechanisms concerning the relationship between S100A8 and CAMKK2/AMPK signaling. Additionally, we observed that in vivo administration of STO-609 and Dor inhibited the activities of CAMKK2 and AMPK while concurrently promoting the expression of S100A8. In conjunction with previous studies on AMPK activation inhibiting S100A8 expression51, this observation indicating that AMPK may have a mutual feedback loop with S100A8. This suggests that no single factor can fully explain the neuroprotection provided by MH. The signal crosstalk among various cells, including microglia and neurons, merits further exploration. Understanding multifaceted effects of signal crosstalk may illuminate key neuroprotective mechanisms of MH.
Overall, this study is the first to investigate how MH ameliorates global cerebral IR injury by modulating S100A8. Our data elucidate that changes in CAMKK2/AMPK signaling, driven by S100A8 activation, lead to mitochondrial injury, which results in neuronal apoptosis after cerebral IR injury. Crucially, MH mitigates neuronal injury by inhibiting this process, highlighting its potential as a therapeutic intervention for neuroprotection (Fig. 8).

Mechanism diagram of MH regulating S100A8-CAMKK2-AMPK axis against cerebral IR injury. Cerebral IR injury leads to an increase in the expression of S100A8, which inhibits the activity of the CAMKK2/AMPK pathway. This inhibition leads to mitochondrial dysfunction, including oxidative damage, decreased membrane potential, and loss of mitochondrial criste structure, along with an increase in apoptosis-related proteins such as bax and cleaved-caspase3. Consequently, this results in neuronal apoptosis. By contrast, MH down-regulates the high expression of S100A8, thereby enhancing mitochondrial function via activation of the CAMKK2/AMPK signaling pathway. As a result, this inhibits neuronal apoptosis and ameliorates neurological injury during cerebral IR in rats.
Conclusion
In this study, we elucidated a novel neuroprotective mechanism of MH, which involves downregulating S100A8 and activating the CAMKK2/AMPK pathway, thereby mitigating mitochondrial injury. As a result, this process reduces neuronal apoptosis and alleviates cerebral IR injury. Therefore, these findings may offer new targets for enhancing MH-based treatment strategies and provide valuable insights for drug development aimed at treating cerebral IR injury.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- MH:
-
Mild hypothermia
- IR:
-
Ischemia reperfusion
- OGDR:
-
Oxygen-glucose deprivation reoxygenation
- ROS:
-
Reactive oxygen species
- CAMKK2:
-
Calmodulin-dependent protein kinase kinase 2
- AMPK:
-
AMP-activated protein kinase
- Dor:
-
Dorsomorphin
- NDS:
-
Neuro Deficit Score
- TEM:
-
Transmission electron microscopy
- DHE:
-
Dihydroethidium
- TMRE:
-
Tetramethylrhodamine ethyl ester
- TUNEL:
-
Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling
- MWM:
-
Morris water maze
References
Fanning, J. P. et al. Perioperative stroke. Nat. Rev. Dis. Primers 10, 3. https://doi.org/10.1038/s41572-023-00487-6 (2024).
Xu, Y. et al. Mild hypothermia therapy attenuates early BBB leakage in acute ischaemic stroke. J. Cerebral Blood Flow Metab. 1, 2. https://doi.org/10.1177/0271678x241275761 (2024).
Li, W. et al. Near-infrared-II imaging revealed hypothermia regulates neuroinflammation following brain injury by increasing the glymphatic influx. ACS Nano 18, 13836–13848. https://doi.org/10.1021/acsnano.4c02652 (2024).
Polderman, K. H. Mechanisms of action, physiological effects, and complications of hypothermia. Crit. Care Med. 37, S186-202. https://doi.org/10.1097/CCM.0b013e3181aa5241 (2009).
Wang, J. et al. Proteome profiling of hippocampus reveals the neuroprotective effect of mild hypothermia on global cerebral ischemia-reperfusion injury in rats. Sci. Rep. 13, 14450. https://doi.org/10.1038/s41598-023-41766-2 (2023).
Ehrchen, J. M., Sunderkötter, C., Foell, D., Vogl, T. & Roth, J. The endogenous Toll-like receptor 4 agonist S100A8/S100A9 (calprotectin) as innate amplifier of infection, autoimmunity, and cancer. J. Leukocyte Biol. 86, 557–566. https://doi.org/10.1189/jlb.1008647 (2009).
Vogl, T. et al. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat. Med. 13, 1042–1049. https://doi.org/10.1038/nm1638 (2007).
Guo, D. et al. Plasma S100A8/A9 concentrations and clinical outcomes of ischemic stroke in 2 independent multicenter cohorts. Clin. Chem. 66, 706–717. https://doi.org/10.1093/clinchem/hvaa069 (2020).
Chen, L. et al. Polymorphisms of Calgranulin genes and ischemic stroke in a Chinese population. J. Inflamm. Res. 15, 3355–3368. https://doi.org/10.2147/jir.S360775 (2022).
Li, Y. et al. S100a8/a9 signaling causes mitochondrial dysfunction and cardiomyocyte death in response to ischemic/reperfusion injury. Circulation 140, 751–764. https://doi.org/10.1161/circulationaha.118.039262 (2019).
Wu, M., Gu, X. & Ma, Z. Mitochondrial quality control in cerebral ischemia-reperfusion injury. Mol. Neurobiol. 58, 5253–5271. https://doi.org/10.1007/s12035-021-02494-8 (2021).
Ghavami, S. et al. S100A8/9 induces cell death via a novel, RAGE-independent pathway that involves selective release of Smac/DIABLO and Omi/HtrA2. Biochimica et Biophysica Acta 1783, 297–311. https://doi.org/10.1016/j.bbamcr.2007.10.015 (2008).
Ghavami, S. et al. S100A8/A9 induces autophagy and apoptosis via ROS-mediated cross-talk between mitochondria and lysosomes that involves BNIP3. Cell Res. 20, 314–331. https://doi.org/10.1038/cr.2009.129 (2010).
Marcelo, K. L., Means, A. R. & York, B. The Ca(2+)/calmodulin/CaMKK2 axis: Nature’s metabolic CaMshaft. Trends Endocrinol. Metab. TEM 27, 706–718. https://doi.org/10.1016/j.tem.2016.06.001 (2016).
Lee, A. et al. Aβ42 oligomers trigger synaptic loss through CAMKK2-AMPK-dependent effectors coordinating mitochondrial fission and mitophagy. Nat. Commun. 13, 4444. https://doi.org/10.1038/s41467-022-32130-5 (2022).
Cai, J. et al. AMPK: The key to ischemia-reperfusion injury. J. Cell. Physiol. 237, 4079–4096. https://doi.org/10.1002/jcp.30875 (2022).
Cai, Y. et al. FUNDC1-dependent mitophagy induced by tPA protects neurons against cerebral ischemia-reperfusion injury. Redox Biol. 38, 101792. https://doi.org/10.1016/j.redox.2020.101792 (2021).
Sun, J. et al. Sestrin2 overexpression attenuates osteoarthritis pain via induction of AMPK/PGC-1α-mediated mitochondrial biogenesis and suppression of neuroinflammation. Brain Behav. Immunity 102, 53–70. https://doi.org/10.1016/j.bbi.2022.02.015 (2022).
Zhang, Y., Wu, F., Teng, F., Guo, S. & Li, H. Deficiency of S100A9 alleviates sepsis-induced acute liver injury through regulating AKT-AMPK-dependent mitochondrial energy metabolism. Int. J. Mol. Sci. 24, 2112. https://doi.org/10.3390/ijms24032112 (2023).
Pulsinelli, W. A. & Buchan, A. M. The four-vessel occlusion rat model: method for complete occlusion of vertebral arteries and control of collateral circulation. Stroke 19, 913–914. https://doi.org/10.1161/01.str.19.7.913 (1988).
Cary, R. L. et al. Inhibition of Ca2⁺/calmodulin-dependent protein kinase kinase 2 stimulates osteoblast formation and inhibits osteoclast differentiation. J. Bone Mineral Res. 28, 1599–1610. https://doi.org/10.1002/jbmr.1890 (2013).
Shi, X. et al. Sestrin2, as a negative feedback regulator of mTOR, provides neuroprotection by activation AMPK phosphorylation in neonatal hypoxic-ischemic encephalopathy in rat pups. J. Cerebral Blood Flow Metab. 37, 1447–1460. https://doi.org/10.1177/0271678x16656201 (2017).
Geocadin, R. G. et al. A novel quantitative EEG injury measure of global cerebral ischemia. Clin. Neurophysiol. 111, 1779–1787. https://doi.org/10.1016/s1388-2457(00)00379-5 (2000).
Kloner, R. A., Fishbein, M. C., Braunwald, E. & Maroko, P. R. Effect of propranolol on mitochondrial morphology during acute myocardial ischemia. Am. J. Cardiol. 41, 880–886. https://doi.org/10.1016/0002-9149(78)90728-2 (1978).
Takano, A. P. C., Munhoz, C. D., Moriscot, A. S., Gupta, S. & Barreto-Chaves, M. L. M. S100A8/MYD88/NF-қB: A novel pathway involved in cardiomyocyte hypertrophy driven by thyroid hormone. J. Mol. Med. (Berlin, Germany) 95, 671–682. https://doi.org/10.1007/s00109-017-1511-y (2017).
Tokumitsu, H. et al. STO-609, a specific inhibitor of the Ca(2+)/calmodulin-dependent protein kinase kinase. J. Biol. Chem. 277, 15813–15818. https://doi.org/10.1074/jbc.M201075200 (2002).
Benito-Cuesta, I., Ordóñez-Gutiérrez, L. & Wandosell, F. AMPK activation does not enhance autophagy in neurons in contrast to MTORC1 inhibition: different impact on β-amyloid clearance. Autophagy 17, 656–671. https://doi.org/10.1080/15548627.2020.1728095 (2021).
Morris, R. Developments of a water-maze procedure for studying spatial learning in the rat. J. Neurosci. Methods 11, 47–60. https://doi.org/10.1016/0165-0270(84)90007-4 (1984).
Kawalec, M. et al. Mitochondrial dynamics, elimination and biogenesis during post-ischemic recovery in ischemia-resistant and ischemia-vulnerable gerbil hippocampal regions. Biochimica et biophysica acta Mol. Basis Dis. 1869, 166633. https://doi.org/10.1016/j.bbadis.2022.166633 (2023).
Sessler, D. I. Mild perioperative hypothermia. N. Engl. J. Med. 336, 1730–1737. https://doi.org/10.1056/nejm199706123362407 (1997).
Yenari, M. A. & Han, H. S. Neuroprotective mechanisms of hypothermia in brain ischaemia. Nat. Rev. Neurosci. 13, 267–278. https://doi.org/10.1038/nrn3174 (2012).
Choi, H. A., Badjatia, N. & Mayer, S. A. Hypothermia for acute brain injury–mechanisms and practical aspects. Nat. Rev. Neurol. 8, 214–222. https://doi.org/10.1038/nrneurol.2012.21 (2012).
Darwazeh, R. & Yan, Y. Mild hypothermia as a treatment for central nervous system injuries: Positive or negative effects. Neural Regeneration Res. 8, 2677–2686. https://doi.org/10.3969/j.issn.1673-5374.2013.28.010 (2013).
Knapp, J. et al. Mild therapeutic hypothermia after cardiac arrest - effect on survival with good neurological outcome outside of randomised controlled trials: A registry-based analysis. Eur. J. Anaesthesiol. 41, 779–786. https://doi.org/10.1097/eja.0000000000002016 (2024).
Pruenster, M., Vogl, T., Roth, J. & Sperandio, M. S100A8/A9: From basic science to clinical application. Pharmacol. Therapeut. 167, 120–131. https://doi.org/10.1016/j.pharmthera.2016.07.015 (2016).
Passey, R. J. et al. A null mutation in the inflammation-associated S100 protein S100A8 causes early resorption of the mouse embryo. J. Immunol. (Baltimore, Md.: 1950) 163, 2209–2216 (1999).
Wei, X. et al. Myocardial hypertrophic preconditioning attenuates cardiomyocyte hypertrophy and slows progression to heart failure through upregulation of S100A8/A9. Circulation 131, 1506–1517. https://doi.org/10.1161/circulationaha.114.013789 (2015) (discussion 1517).
Stephan, J. R., Yu, F., Costello, R. M., Bleier, B. S. & Nolan, E. M. Oxidative post-translational modifications accelerate proteolytic degradation of calprotectin. J. Am. Chem. Soc. 140, 17444–17455. https://doi.org/10.1021/jacs.8b06354 (2018).
Jia, J. et al. Target-selective protein S-nitrosylation by sequence motif recognition. Cell 159, 623–634. https://doi.org/10.1016/j.cell.2014.09.032 (2014).
Nolan, E. M. & Peet, J. J. Y. Post-translational modifications on the metal-sequestering protein calprotectin. Biometals Int. J. Role Metal Ions Biol. Biochem. Med. 36, 817–828. https://doi.org/10.1007/s10534-023-00493-x (2023).
Colicchia, M. et al. S100A8/A9 drives the formation of procoagulant platelets through GPIbα. Blood 140, 2626–2643. https://doi.org/10.1182/blood.2021014966 (2022).
Ma, J. et al. S100A8/A9 as a prognostic biomarker with causal effects for post-acute myocardial infarction heart failure. Nat. Commun. 15, 2701. https://doi.org/10.1038/s41467-024-46973-7 (2024).
Ha, J. S. et al. Hypoxia-induced S100A8 expression activates microglial inflammation and promotes neuronal apoptosis. Int. J. Mol. Sci. 22, 1205. https://doi.org/10.3390/ijms22031205 (2021).
Liu, W. et al. PX-478 induces apoptosis in acute myeloid leukemia under hypoxia by inhibiting the PI3K/AKT/mTOR pathway through downregulation of GBE1. Biochem. Pharmacol. 230, 116620. https://doi.org/10.1016/j.bcp.2024.116620 (2024).
Wu, Y. et al. Sodium citrate targeting Ca(2+)/CAMKK2 pathway exhibits anti-tumor activity through inducing apoptosis and ferroptosis in ovarian cancer. J. Adv. Res. 65, 89–104. https://doi.org/10.1016/j.jare.2024.04.033 (2024).
McCullough, L. D. et al. Inhibition of calcium/calmodulin-dependent protein kinase kinase β and calcium/calmodulin-dependent protein kinase IV is detrimental in cerebral ischemia. Stroke 44, 2559–2566. https://doi.org/10.1161/strokeaha.113.001030 (2013).
Guo, J. M. et al. SIRT1-dependent AMPK pathway in the protection of estrogen against ischemic brain injury. CNS Neurosci. Therapeut. 23, 360–369. https://doi.org/10.1111/cns.12686 (2017).
Xu, N. et al. Adiponectin attenuates neuronal apoptosis induced by hypoxia-ischemia via the activation of AdipoR1/APPL1/LKB1/AMPK pathway in neonatal rats. Neuropharmacology 133, 415–428. https://doi.org/10.1016/j.neuropharm.2018.02.024 (2018).
Wafer, L. N., Tzul, F. O., Pandharipande, P. P., McCallum, S. A. & Makhatadze, G. I. Structural and thermodynamic characterization of the recognition of the S100-binding peptides TRTK12 and p53 by calmodulin. Protein Sci. Publ. Protein Soc. 23, 1247–1261. https://doi.org/10.1002/pro.2506 (2014).
Li, L. X., Campbell, K., Zhao, S., Knuckey, N. W. & Meloni, B. P. The effect of blood pressure (37 vs 45 mmHg) and carotid occlusion duration (8 vs 10 min) on CA1-4 neuronal damage when using isoflurane in a global cerebral ischemia rat model. Brain Res. Bull. 86, 390–394. https://doi.org/10.1016/j.brainresbull.2011.09.005 (2011).
Grempler, R. et al. Discovery and translation of a target engagement marker for AMP-activated protein kinase (AMPK). PLoS ONE 13, e0197849. https://doi.org/10.1371/journal.pone.0197849 (2018).
Acknowledgements
Not applicable.
Funding
National Natural Science Foundation of China (82000881).
Author information
Authors and Affiliations
Contributions
W.W.Q. and M.S.W. conceived and designed the experiments, and all the experiments were performed under their guidance. D.D.Z., Y.T.D., X.Y.X., and F.G.M. performed the experiments and analyzed the data, D.D.Z. and W.W.Q. wrote the manuscript. All authors read and approve the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Male Wistar Rats (body weight: 250–300 g, age: 6–8 weeks) were obtained from Jinan Pengyue Laboratory Animal Breeding Company (Jinan, China). The animal experiments were reviewed and approved by the Qingdao University Medical Ethics Committee (QDU-AEC-2024473).
Consent for publication
Not applicable.
Statement on ARRIVE guidelines
We declared that this study was carried out in compliance with the ARRIVE guidelines.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, D., Dai, Y., Xu, X. et al. S100A8-CAMKK2-AMPK axis confers the protective effects of mild hypothermia against cerebral ischemia-reperfusion injury in rats. Sci Rep 15, 2793 (2025). https://doi.org/10.1038/s41598-025-87184-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-87184-4
