
Abstract
Pediatric brain tumors (PBTs) are the leading cause of cancer-related mortality in children. Despite advances in next-generation sequencing (NGS) deepening our understanding of the molecular features of PBT, the lack of preclinical models that capture their complexity and diversity remains a significant barrier to developing less toxic and more effective treatments. We have established 20 ex-vivo patient-derived tumoroid (PDTs) cultures from fresh surgical material of a wide range of PBTs, including low- and high-grade gliomas, medulloblastomas, and even rarer tumor entities such as a CNS tumor with BCOR alteration. Immunofluorescence, NGS analysis, and DNA methylation profiling revealed that the PDTs faithfully mirrored the cellular nature, the genetic and epigenetic landscape of their matched primary tumors. Our study shows the feasibility of generating PDTs, even from rarer entities, that recapitulate the genetic and epigenetic features of primary tumors, highlighting their potential as models for tumor biology studies and precision medicine.
Similar content being viewed by others


Introduction
Pediatric brain tumors (PBT) are the most common solid tumors in children and represent the leading cause of cancer-related mortality in this age group1. Despite advances in treatment, survival rates for these tumors have remained unsatisfactory and are accompanied by significant morbidity2. PBTs are a heterogeneous group in terms of their histopathology, molecular characteristics, and prognosis3. Gliomas, particularly low-grade gliomas (pLGGs), are the most frequent, followed by medulloblastomas (MBs)4. The current standard of care for PBTs is a multimodal approach that comprises resection surgery, chemotherapy, and radiotherapy, which carries toxicity and has a significant impact on the quality of life of patients and their families5. Consequently, there is a pressing need for the development of new, less toxic therapeutic strategies.
Recent advances in next-generation sequencing (NGS) technologies have greatly enhanced our understanding of the genetic and epigenetic features of these tumors6,7,8,9,10,11, leading to the integration of molecular characteristics into their classification12,13. However, these insights have not yet been effectively translated into new, less harmful treatments, largely due to the lack of adequate preclinical models that accurately reflect the diversity and complexity of PBTs.
In this context, patient-derived tumoroids (PDTs) have emerged as promising tools for preclinical research14,15. These three-dimensional models, initially developed from adult brain tumors16,17,18,19, have demonstrated the ability to recapitulate the genomic and epigenomic landscapes of their original tumors, as well as patient-specific intratumoral heterogeneity and drug responses16,20. Though already applied in pediatric oncology, the progress of PDT technology has been marginal compared to its application in adult tumors21,22. Our work aims to address this gap by demonstrating the effectiveness of PDT generation from a variety of PBTs, ranging from pLGGs to high-grade gliomas (pHGGs), MBs, and rarer entities such as atypical teratoid rhabdoid tumors (ATRTs), a central nervous system (CNS) tumor with BCOR internal tandem duplication (CNSt-BCOR), and meningiomas (MNGs). These PDT cultures were successfully established and mimicked the genomic and epigenomic landscapes of their primary tumors, offering a promising platform for advancing PBT research and therapeutic development.
Results
Establishment of a pipeline for pediatric brain tumor sample collection
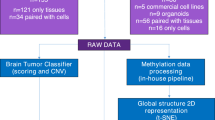
Our PBT collection pipeline effectively addressed all the logistical demands associated with tissue collection and PDTs establishment. The tight coordination between Ipatimup/i3S, and the departments of Pathology, Neurosurgery, and Pediatric Oncology, at Centro Hospitalar Universitário São João (CHUSJ), was crucial for efficiently implementing sample collection, PDTs generation, and molecular characterization (Fig. 1a). PBT samples for PDTs generation were successfully obtained in 90% of cases (30/33 surgeries). All tumor specimens were collected and processed for PDTs within 1 h (Fig. 1a). The diagnostic assessment by the neuropathologist assured that the specimen accurately represented the original primary tumor and guaranteed the representativeness and quality of the collected PBT specimens. The preliminary diagnosis provided by the neuropathologist enabled us to adjust the PBT culture protocol tailored to the tumor type, for instance, including or not growth factors in the culture media. Between November 2022 and September 2024, we collected 30 PBT samples from 33 surgeries performed at CHUSJ. In three cases, the resected material was not sufficient for PDT generation and was therefore prioritized for pathological analysis. Four specimens were lost from the initial 30 PBT collection set due to transportation or sample processing issues. In the resulting 26 PBT samples, we successfully generated 20 PDT cultures, i.e. a success rate of 77% (Fig. 1b).

a Processing steps for PDT generation and timelines for sample collection, processing, and tumoroid generation. b Summary of the PBT cohort from sample collection to processing as PDT culture. c Comparison of PDT culture conditions applied to different PBT entities. PDT Patient-derived tumoroids, PBT Pediatric brain tumors, LGG low-grade gliomas, HGG high-grade gliomas, MB medulloblastomas, EPM ependymomas, MNG meningiomas, ATRT atypical teratoid rhabdoid tumor.
Pediatric brain tumor cohort
Our PBT cohort consisted of 30 cases, categorized into pLGGs (n = 16), pHGGs (n = 5), MBs (n = 3), and other PBTs, including an ependymoma (EPM) (n = 1), MNG (n = 1), pineoblastoma (PNB) (n = 1), ATRT (n = 1), MNG (n = 1) and a CNSt-BCOR (n = 1), in this last case it was possible to collect material from both primary and recurrent tumor (Table 1). Most tumors were located in the posterior fossa (16 out of 30) and brain hemispheres (7 out of 30), followed by the basal ganglia (2 out of 30). The remaining tumors were located in different brain regions (Table 1). The mean age at onset was nine years, ranging from seven months to 17 years (Table 1). The most common tumors in our cohort were pilocytic astrocytomas (PA), which accounted for 11 out of the 30 cases, followed by pHGG and MB. Many pLGGs exhibited alterations in the BRAF gene (10 out of 16, 63%), specifically the KIAA1549::BRAF fusion in six cases and the BRAFV600E mutation in four cases (Table 1). Two pLGG (LGG_02, LGG_15) were associated with neurofibromatosis type 1, harboring NF1 germline mutations (Table 1 and Table S2). The pHGG group, the second most frequent tumor, comprised five patients with different tumor entities including two diffuse high-grade gliomas, a high-grade astrocytoma with piloid features, a diffuse midline glioma H3K27-altered (H3K27DMG) and an infant-type hemispheric glioma (IHG). These tumors presented typical molecular alterations of pHGG, such as mutations and amplifications in TP53, H3F3A, ATRX, PDGFRA, and PIK3CA genes (Table 1). The IHG infant-type hemispheric glioma (IHG) carried a GFAP::ALK fusion (Table 1). The MB group (n = 3) included patients aged between four and 17 years old and all were diagnosed with Group 4 MB, located in the posterior fossa (Table 1). The cohort included six additional PBTs, namely a PNB with a DROSHA mutation, a supratentorial hemispheric EPM with a ZFTA::RELA fusion, a CNSt-BCOR (primary and recurrent tumors) with a BCOR mutation an ATRT, and a MNG (Table 1 and Table S2). Three collected specimens were from recurrent tumors, two PA, and a CNSt-BCOR (Table 1).
Generation of patient-derived tumoroid cultures from different PBT types
Among the 26 PBTs that were placed under culture, we successfully generated PDTs from 10/13 (77%) pLGG, 3/5 (60%) pHGG, and 3/3 (100%) MB samples (Table 1). Additionally, we established PDT cultures from an EPM, an ATRT, a CNSt-BCOR, and a MNG (Table 1). Details regarding culture conditions for different PBT entities are summarized in Fig. 1c. In all cultures, PDT formation was detectable 7−14 days after the initial seeding with a diameter that ranged between 200 and 800μm (Fig. 2). Moreover, the PDT cultures were capable of being passaged and maintained in culture for a minimum of two months. All the PDT cultures fulfilled the elected criteria for an effective PDT generation: remained viable for > two weeks in culture, acquired a spherical morphology, and showed continuous growth in culture (Fig. 2).

Brightfield microscopy images illustrating the morphological evolution of PDTs derived from pediatric low-grade gliomas, a meningioma, an ependymoma, pediatric high-grade gliomas, a CNS tumor with BCOR alteration, medulloblastomas, and an atypical teratoid/rhabdoid tumor (ATRT). Scale bar: 100 μm. Images were acquired using a 20 ×/ 0.4 objective with a digital imaging system (ZOE, Bio-Rad). PDT Patient-derived tumoroids.
Certain protocol parameters needed to be adjusted according to the PBT diagnosis (Fig. 1c), namely, the addition of growth factors that were not included in the initial medium composition16. Indeed, while pLGG-derived and MNG-derived PDT cultures thrived in the initial medium, yielding 3–4 PDT per well, pHGG- and EPM-derived PDT cultures exhibited only one PDT per well. To address this issue, we supplemented the medium with growth factors (EGF, bFGF, PDGF-AA), which resulted in a rapid PDT expansion in both HGG_02 and EPM PDT cultures (Fig. 2). The same strategy was employed for the following pHGG, MB-, CNSt-BCOR- and ATRT-derived PDT, given their high-grade nature and previous studies supporting the use of growth factors in these PDTs23,24.
To propagate PDT cultures, we tested two different approaches: an enzymatic strategy using TripleX (Gibco, 12605010) or Accutase (Merck, A6964) and a mechanical one, by cutting PDT into small fragments, using scalpels and scissors, and reseeding in the same culture conditions, in a 1:2 to 1:4 ratio16. pLGG-derived PDT did not recover after enzymatic passage with either TripleX or Accutase. However, they recovered after mechanical passage, as depicted in Fig. 2 for LGG_01, which was mechanically passaged on day 22 and showed 660μm-diameter PDTs on day 55 (Fig. 2). Only the ATRT- and MNG-PDT managed to recover from both enzymatic and mechanical approaches. The PDT cultures were propagated and maintained in culture for at least two months.
Among the six PBT samples that were cultured but did not yield PDTs, three were derived from pLGG, two from pHGG, and one from a PNB (Table 1). The LGG_05 sample had no viable cells 1 month post-seeding, while LGG_08 developed round structures that failed to sustain continuous growth, and the LGG_10 sample, being sparse and with low cellularity, resulted in cell debris after 25 days in culture. The HGG_01 sample was notably deficient in tumor cells, as indicated by the neuropathologist at the time of the biopsy. The HGG_03 sample generated round structures that did not show continuous growth in culture, failing the criteria of successful PDT generation. The PNB, although having viable cells for four months, failed to generate round structures.
Patient-derived tumoroids retain the expression of key diagnostic markers of their respective primary tumors
To assess the tissue architecture and cell composition of each PDT, we performed immunofluorescence and immunohistochemistry (IHC) analysis, employing the same markers used in PBT diagnosis (Fig. 3). When sufficient material was available, with priority given to genomic assays, PDTs were harvested three to six weeks after initial seeding, depending on the development of each culture. A total of 11 PDT cultures were analyzed using immunofluorescence, while an additional four PDTs were collected for IHC. LGG_12, a ganglioglioma with a BRAFV600E mutation, was characterized by a population of astrocytic cells with fibrillary cytoplasm and scattered ganglion cells, and diffuse GFAP expression was observed in the astrocytic population (Fig. 3a). Similarly, the matched tumoroid exhibited a predominance of astrocytic cells with abundant cytoplasm and moderate nuclear pleomorphism (Fig. 3a). IHC revealed variable GFAP expression, and cytoplasmic expression of the BRAFV600E mutation was also detected in the tumoroid cells (Fig. 3a). Likewise, HGG_05, a H3K27DMG, presented astrocytic differentiation. The neoplastic cells demonstrated diffuse GFAP expression along with nuclear expression of the H3 K27M mutant protein (Fig. 3a). Correspondingly, the matched tumoroid displayed a population of astrocytic cells with more prominent nuclear pleomorphism (Fig. 3a). Consistent with the primary tumor, diffuse cytoplasmic expression of GFAP and nuclear expression of the mutant H3 K27M protein were also observed in the matched PDTs (Fig. 3a). In the case of BCOR_02, the primary tumor exhibited a neoplasm with a fibrillary matrix and a population of neoplastic cells characterized by monotonous oval nuclei, vesicular chromatin, and scattered mitotic figures (Fig. 3a). Similarly, the matched PDTs retained these histological features, displaying a highly cellular neoplasm with oval nuclei and vesicular chromatin (Fig. 3a). Finally, MNG was identified as a meningothelial neoplasm with a syncytial and whorled growth pattern (Fig. 3a). Importantly, the matched PDTs closely recapitulated this histological architecture, maintaining a similar structural organization (Fig. 3a). The confocal imaging revealed that PDTs from glial/neuroglial tumors (LGG_04, EPM, HGG_02, and HGG_04) were positive for the expression of the glial fibrillary acidic protein (GFAP) marker, showing that these structures kept the glial differentiation of the matched primary tumors (Fig. 2b). In addition, PDTs from MB_02 and an MNG were shown to express synaptophysin, a neuronal cell marker25, and mucin 1 (MUC-1), a glycoprotein typically expressed in MNG, respectively, resembling their primary tumors (Fig. 2b). Altogether, these immunohistology analysis showed that the PDTs can replicate the histological features, cell lineage, and cell markers of their corresponding primary tumors.

a Comparison of morphological features and expression markers between primary PBT samples and their respective PDT cultures. Hematoxylin and Eosin (H&E) staining and immunohistochemistry analysis are shown. PDTs were harvested for analysis - LGG_12 at day 15, HGG_05 at day 11, and BCOR_02 at day 18 of culture. Markers: GFAP (green): glial fibrillary acidic protein; BRAFV600E: marker for the hotspot mutation; H3K27M: marker for the mutated form of H3 protein. Scale bar: 100 μm (b). Confocal microscopy images of PDTs derived from pediatric low-grade gliomas (LGG_04), an ependymoma (EPM), a medulloblastoma (MB_01), pediatric high-grade gliomas (HGG_02, HGG_04) and a meningioma (MNG), highlighting the expression of lineage/ tumor-specific markers: GFAP (green, glial fibrillary acidic protein), SYP (red, synaptophysin), and MUC-1 (brown, mucin 1). Nuclei are stained in blue (HOESCHT). All images were acquired using the OperaPhenix Plus system. Scale bar: 100 μm. PBT pediatric brain tumor, PDT patient-derived tumoroid.
Patient-derived tumoroids retain the genomic profile of their corresponding primary tumor
To analyze the genomic profile of each PDT culture and assess whether they retain the genomic characteristics of their corresponding primary tumors, PDTs were harvested after one month in culture, and their matched DNA underwent NGS analysis or PCR followed by Sanger sequencing analysis (HGG_04 and BCOR_02). LGG_06 is not represented in Fig. 4 due to culture contamination that hampered NGS analysis. As shown in Fig. 4, PDTs successfully preserved key driver alterations and closely mirrored the genomic landscape of their respective primary tumors. This was consistent across all types of molecular alterations, including the KIAA1549::BRAF gene fusion observed in three PAs, a ZFTA::RELA fusion in the EPM case, as well as point mutations affecting BRAF, PIK3CA, FGFR1, NF1, H3F3A and ATRX, homozygous CDKN2A deletions, and copy number amplifications in genes such as KRAS, CDK4, MDM2, GLI1, PTPN11, and PIK3CA. Moreover, these alterations were maintained with similar allelic fractions (for single-nucleotide variants, SNVs), read counts (for fusions), or copy numbers (for amplifications) (Table S2). The heatmap depicted in Fig. 4 highlights additional alterations, some of which were classified as benign and likely constitutional, such as the SMARCA4 (p.Tyr372His) mutation, and other variants, likely to be subclonal events that were either lost or gained during PDT generation (Fig. 4). Overall, the NGS results demonstrated that PDTs effectively preserved the driver alterations of the primary tumors.

Heatmap depicting somatic alterations and cancer cell fractions in primary tumors of pediatric low-grade gliomas (LGGs), pediatric high-grade gliomas (HGGs), an ependymoma (EPM), and a medulloblastoma (MB_02) and their matched PDTs. HGG_04 and BCOR_02 molecular profile was not determined using the OCCRA panel but through PCR and Sanger sequencing analysis (Fig. S1, S2 and Table S3). Cases are shown in columns, and genes are in rows. Mutations and cancer cell fractions are color-coded according to the legend.
Patient-derived tumoroids recapitulate DNA methylation and copy number variation profiles of primary tumors
DNA methylation array profiling was performed to evaluate global methylation and copy number variations (CNVs) in MBs, HGGs, and PBTs without detectable driver alterations or with characteristic methylation-defined subtypes (n = 7), when sufficient material was available. According to the 2021 WHO classification of central nervous system tumors, the diagnosis of PBTs, including entities such as MBs and ATRT, should follow an integrated approach that combines histopathological assessment with molecular features, including DNA methylation profiling13. DNA methylation-based classification uses a CNS tumor classifier, which generates a calibrated score indicating the degree of similarity between the sample’s methylation profile and predefined methylation classes26. Additionally, it also determines CNV profiles, providing an overview regarding structural alterations such as amplifications, deletions, and numerical chromosome aberrations. The DNA methylation profiles of each PDTs matched those from the corresponding primary tumors in all samples (Table 2), retaining the methylation score level in all but on case: the DNA methylation profile of the MB_03 PDT partially matched the primary tumor: it showed a high score (0.98) in defining a non-WNT/non-SHH MB, but a lower score (0.58 vs. 0.82) in the Group 4 subgrouping. t-SNE analysis of DNA methylation profiles demonstrated that patient-derived tumoroids clustered closely with their corresponding primary tumors (Fig. 5a) and reference cohorts (Fig. 5b). MBs, ATRTs, and HGGs each formed distinct clusters consistent with their established methylation classes, while MNG samples grouped separately (Fig. 5b). Moreover, the CNV profiles of PDTs were similar to their matched primary tumors (Fig. 5c). For instance, the PDT from MB_01 kept the imbalanced CNV profile of its primary tumor, including the loss of chromosome 7 and gain of chromosome 8, a typical cytogenetic alteration of group 4 subclass Vii MB (Fig. 5c)27. This indicates that the tumoroids faithfully recapitulate the epigenetic landscape of the originating tumors.

a Visualization of DNA methylation patterns of primary tumors and corresponding PDT cultures using t-distributed stochastic neighbor embedding (t-SNE). Each case is represented by a dot. PDTs are represented in purple, and primary tumors in red. b t-SNE analysis of DNA methylation data from our study cohort (primary tumors and respective PDT) in comparison with 41 published DNA methylation classes (Table S4). c Copy number variation profiles of primary PBTs (left panel) and their matched PDT (right panel) from HGG_04, MB_01, an ATRT, and an MNG. PBT pediatric brain tumor, PDT patient-derived tumoroid, HGG high-grade glioma, IHG infant-type hemispheric glioma, ATRT atypical teratoid rhabdoid tumor, EPM ependymoma, MB medulloblastoma, MNG meningioma.
Discussion
Despite significant advances in understanding PBTs through evolving molecular technologies, translating these insights into improved patient outcomes remains challenging. Effective development of new, personalized therapeutic strategies requires robust preclinical models for these tumors. Our work represents a significant step forward in demonstrating the feasibility of a streamlined pipeline for generating PDT cultures, achieving a 77% success rate across a diverse range of PBT types, including glial, embryonal, and very rare PBT. This collection encompasses difficult-to-model tumors such as pLGG28, pHGG, EPM, and MB, as well as rarer types like ATRT, CNSt-BCOR, and MNG.
Regarding the success rate of our pipeline, two critical factors must be carefully considered when generating PDT cultures: the quantity of tissue available and its quality, particularly the tumor cell representation within the sample16,23,29. In PBTs, the tissue samples are usually obtained from biopsies, meaning the sample size is often limited. This constraint can impact PDT generation, as demonstrated by the LGG_11 and LGG_13 cases, in which sample scarcity resulted in tissue loss during processing. The fact that PDT generation success heavily relies on high tumor cell content29, pinpoints the importance of having an experienced neuropathologist for extemporaneous examination. The importance of tumor cell content was highlighted in the case of HGG_01, a difficult PBT sample to obtain, which was reported to have a low percentage of tumor cells. Despite the aggressiveness of the tumor and its unique value, our attempt to generate PDTs ultimately failed, emphasizing the critical role of sufficient tumor cell content for successful PDT generation. Another important consideration is the challenge of culturing recurrent tumors, as they often exhibit increased necrosis compared to primary tumors30. In our cohort, only two recurrent tumors were successfully cultured, limiting the sample size and preventing definitive conclusions from being drawn.
We defined a simple tissue processing protocol that could be applied to different types of PBT. The samples were only mechanically minced and processed as small tumor fragments, as described by Jacob et al. for glioblastoma samples16. This mechanical processing preserves original tumor cell-cell interactions and minimizes clonal selection among tumor cells16. Our results align with those of Lago et al., who showed that mechanical dissociation of PBT-derived tumoroids yields better results than the enzymatic method31.
Previous work on protocols for solid tumor-derived tumoroids used Matrigel™ or basement membrane extract as an extracellular matrix (ECM) substitute, while protocols for brain tumor PDTs from both pediatric23,24,31 and adult tumors16,32 usually employ a distinct approach, in which tumor fragments are placed onto ultra-low adherence plates, without the need for ECM substitutes. Notably, in rarer PBTs such as ATRT, the matched PDTs were only generated under suspension conditions, proving to be unviable when embedded in basement membrane extract (BME)23. Our study further demonstrates that this approach can be effectively applied to generate PDTs from different PBT subtypes, offering a simplified, ECM-independent alternative with broader potential for clinical and research applications.
Unlike pHGG and MB, PDTs from pLGGs and MNG PDT cultures did not require exogenous growth factors (EGF, bFGF, and PDFG-AA) in the culture medium. This may be because most of our pLGG samples displayed molecular alterations affecting BRAF or NF1, leading to constitutive activation of the MAPK signaling pathway. Since this pathway functions downstream of EGF, FGF, and PDGF receptors, it is plausible that these tumors can still grow without the need for supplemental growth factors. The remaining PBTs (non-pLGG or MNG) either showed (i) alterations in receptor tyrosine kinases (PDGFR or EGFR), (ii) lacked alterations in the MAPK signaling pathway (histone H3-mutated glioma), or (iii) had no mutations identified, which could explain why these tumors seem more dependent on exogenous growth factors. These observations emphasize the diversity within PBT and the impact of the oncogenic nature in their etiologies.
We demonstrated that PDT cultures preserve the expression of key protein markers, routinely used in the PBT diagnosis and management, found in primary tumors and the genomic landscape of their original tumors, particularly their driver genetic alterations. Notably, this genomic stability between primary tumor and PDTs is seen across different molecular alterations, from qualitative – like point mutations – to quantitative alterations like CNVs. Some alterations could be present only in the primary tumor and not in PDTs, and vice versa. The former might be related to an initial underrepresentation of these subclones in the original PBT specimen or a limited proliferation under the culture conditions, while the latter might suggest alterations that emerged during PDT culture.
DNA methylation profiling is being increasingly used as a classification method to complement molecular and histopathologic classification, especially in PBT26. Both DNA-methylation scores and CNV profiles are important tools in the clinical setting, and they have already been shown to be reliable methods for assessing the similarity between PBTs and respective in vitro models33, as highlighted by our work as well as other studies23,31,34. The finding that PDTs keep the DNA methylation profile of the corresponding primary tumor, with a match in the methylation classes, indicates that PDTs retain not only structural DNA alterations (point mutations, fusions, gains, and losses) but also epigenetic DNA modifications, as the whole genome DNA methylation patterns.
Our work present some limitations. Although we analyzed the genomic profile of all PDT cultures, this analysis was performed at a single time point, a month post-seeding. PDTs analysis at additional timepoints could be important to assess potential genetic drift over time. Nevertheless, other studies using similar PBT-derived PDT methodologies showed minimal genetic drift over extended periods24,31, suggesting that PDTs are genomically stable. We also did not assess whether PDTs retain the transcriptomic features of the primary tumors, although previous work indicates that PDTs can successfully preserve these transcriptomic characteristics overtime16,24,31.
Compared to other studies on PBT-derived tumoroids, our work presents a streamlined and efficient protocol, tested in a diverse set of PBTs, including pLGG and rare PBTs with non-glial origin, achieving a high success rate. To the best of our knowledge, this is the first study to report the generation of PDTs from particularly aggressive PBT types, such as a CNSt-BCOR, and the fusion gene-enriched IHG. These are characterized by their aggressive nature and poor prognosis35,36, underscoring the significance of our PDT protocol for enabling further research into these challenging tumors. Additionally, pLGG are notably difficult to model in vitro28, as existing preclinical models, such as primary cell lines, often fail to capture the supportive tumor microenvironment and cellular heterogeneity inherent to these tumors28. In this context, PDT cultures offer a more advanced and representative model, overcoming some limitations of the traditional 2D in vitro models. Our protocol enables the rapid generation of PDTs, and we observed no difference in the generation of PDT derived from pLGG compared to those from higher-grade tumors, underlining the robustness and applicability across various tumor grades. These models and respective protocol details and support are available to the scientific community upon a previous contact with our team.
Future work should focus on assessing the ability of PDTs to preserve tumor heterogeneity and tumor microenvironment elements from their original tumors. This feature is a major advantage and innovation of PDT models, potentially significantly impacting the study of these rare tumors by more accurately representing their complex biology. Additionally, implementing drug screening tests will be essential to evaluate PDTs as predictive tools for patient-specific drug responses. Integrating drug sensitivity profiles into clinical workflows will offer healthcare providers valuable insights for devising tailored and effective therapeutic strategies.
In summary, we successfully established a robust pipeline for collecting PBT specimens and generating PDT cultures from a wide range of PBT, including pLGG and rare types of PBTs. This achievement not only paves the way for further studies but also supports the establishment of a biobank through cryopreservation of PDT cultures. Together, these advancements will contribute to the evolution of precision medicine, ultimately improving outcomes for children with PBTs.
Methods
Human tissue collection
Fresh surgical material of PBTs was collected in collaboration with CHUSJ, particularly with the Departments of Neurosurgery, Pediatric Oncology, and Pathology. Patients’ samples and clinical data were obtained according the Declaration of Helsinki, with the approval of the CHUSJ ethical committee (reference 50/22; CHUSJ). We included all PBTs treated in the CHUSJ, regardless of their diagnosis, to cover all possible PBT subtypes. A PBT was included in the study if all of the following criteria were met: (i) age 18 years or younger; (ii) newly diagnosed PBT, or PBT on relapse, or PBT with progressive disease; (iii) PBT scheduled for surgical intervention, tumor removal or biopsy and (iv) signed informed consent. Freshly resected PBT tissue (biopsies or surgical resection specimens) was transported from the surgical theatre to the Pathology Department of CHUSJ, where it underwent extemporaneous examination by a neuropathologist to confirm the neoplastic nature of the lesion and provide, whenever possible, a preliminary PBT diagnosis. The freshly resected PBT tissue was then divided into two portions: one was used for detailed pathology evaluation, and the second was placed in Hibernate A (Gibco, A1247501), 1% Penicillin/streptomycin (Biowest, L0022-100), to maintain tissue viability and prevent microbial contamination, and transported on ice to the laboratory for PDT culture generation. All PBT samples were processed within 30 min to an hour following surgical resection to maximize tissue integrity and cell viability. In total, we collected and processed 30 PBT samples (Fig. 1) and were able to assess their clinical features as described in Table 1.
Tissue processing and patient-derived tumoroid generation
The PDT cultures were generated following a previously described protocol for PDT generation from glioblastomas18. Tumor tissue was transferred to a sterile 10 cm cell culture glass dish, with 5 mL of H + GPSA medium, composed of Hibernate A (Gibco, A1247501), Glutamax (ThermoFisher, 35050061), Penicillin/streptomycin (Biowest, L0022-100) and Amphotericin B (Corning, 1397893). The tumor tissue was mechanically dissociated using scalpels and scissors in a laminar flow biosafety cabinet. The resulting tumor pieces were washed twice with H + GPSA, centrifuged at 1600 rpm for 5 min to remove cell debris, and further incubated in a red blood cell (RBC) lysis buffer (ThermoFisher, 00-4333-57), at room temperature for 10 min. After removing the RBC lysis buffer, the tumor pieces were washed again with H + GPSA and were resuspended in cerebral tumoroid medium (CTM) for distribution in ultra-low attachment 24-well culture plates (Sarstedt, 83.3922.500) (around 2–3 fragments per well). CTM was composed of 1:1 DMEM F12 (ThermoFisher, 11320033) and Neurobasal (ThermoFisher, 21103049) medium, Glutamax, Non-Essential Amino Acids Solution (ThermoFisher, 11140050), Penicillin/streptomycin (Biowest, L0022-100), N2 supplement (ThermoFisher, 17502048), B27 without Vitamin A (ThermoFisher, 12587010), 2-βmercaptoethanol (Gibco, 31350-010), Human insulin (Sigma, I9278) and Primocin (InvivoGen, ant-pm-05). In tumor samples from pHGGs and EPMs, we supplemented the CTM with growth factors, namely human epidermal growth factor (EGF, 78006, StemCell Technologies), human basic fibroblast growth factor (bFGF, StemCell Technologies, 78003), human platelet-derived growth factor (PDGF-AA, StemCell Technologies, 78095), and Heparin solution 0.2% (StemCell Technologies, 7980). This combination of growth factors has been previously documented for the generation of PDTs from high-risk PBT and ATRT-derived PDT23,24. The plates were placed on a sterile incubator with orbital shaking (80 rpm, 37°C, 5% CO2, and 90% humidity).
The culture medium was changed every 48h. The morphological aspect of the PDTs was monitored every 48h by optical microscopy. When PDTs reached a diameter of over 500 µm, they were cut into smaller pieces, using scalpels and scissors, and propagated into new wells. To store the PDTs through cryopreservation, they were collected at least three weeks after being cultured and underwent a 1h incubation in CTM supplemented with 10 µM Y-27632 (StemCell Technologies, 72304), which served to enhance cell viability, as previously described18. The PDTs were resuspended in a cryopreservation medium composed of CTM, 10% DMSO, and 10 µM Y-27632 for the freezing process.
Histology and immunohistochemistry analysis
Serial tissue sections (3 µm thick) of representative tumor samples were prepared on the slide for immunohistochemistry (IHC) analysis, as previously described37. Immunostainings were performed using the Ventana Benchmark Ultra automated immunostainer (Ventana Medical System, Tucson, AZ, USA)37. Antibodies information can be found in Table S1. PDT samples were embedded in HistoGel™ (Epredia™, HG-4000-012) to facilitate histological processing. HistoGel™ was liquefied by heating to 60 °C and maintained at 50 °C to prevent premature solidification. Tumoroid suspensions were centrifuged at 2000 rpm for 2 min in 1.5 mL tubes, and the supernatant was carefully removed. Once liquefied, 300–350 µL of HistoGel™ was added to each sample, gently mixed, and centrifuged again at 2000 rpm for 2 min. Tubes were placed at 4 °C for 20 min to solidify the gel, then cut open to retrieve the gel plug. Samples were transferred to labeled histocassettes and fixed in 10% formaldehyde for at least 2 h before histological processing38.
Immunofluorescence
Immunofluorescence (IF) analysis was conducted in each PDT culture to assess their cellular composition. Each PDT culture was stained with appropriate markers, based on the diagnosis and features of the corresponding primary tumor, using a protocol adapted from various works describing immunostaining in tumoroids39,40,41. The list and dilution of each antibody are presented in Table S1. For fixation, PDTs were incubated with 4% paraformaldehyde for 20 min at room temperature, washed once with PBS, and stored for up to two weeks before immunostaining. PDT blocking and permeabilization were performed by incubating the samples in a solution composed of PBS, 0,3% Triton X100, 1% DMSO, and 1% bovine serum albumin (BSA) for 1 h at room temperature, followed by washing twice with PBS 1% BSA. Primary antibodies were diluted in a blocking solution, and PDTs were incubated overnight at 4°C, with rocking. On the next day, the PDTs were washed twice with PBS 1% BSA and incubated, with rocking, in the dark for 3h at room temperature with diluted secondary antibodies (Table S1). PDTs were then incubated for 30 min at room temperature, protected from the light, with diluted PhenoVue HOECHST 33342 (1:1500 in PBS) for nuclei staining.
High-content confocal microscopy and image processing
PDTs were imaged using an Opera Phenix Plus (Revvity) in confocal mode, acquiring several z-planes using the following objectives: 10x/0.3 Air, 20x/1.0 water, and 40x/1.1 water. Images were analyzed and cropped using Harmony software (Revvity) and Fiji/ImageJ software42.
Targeted next-generation sequencing
DNA and RNA of 29 collected PBT samples (Table S2) and 13 PDTs were sequenced using the Oncomine™ Childhood Cancer Research Assay (OCCRA; ThermoFisher) NGS panel. DNA and RNA were extracted from PDTs using the Maxwell® RSC DNA formalin-fixed paraffin-embedded (FFPE) kit and the Maxwell® RSC RNA FFPE kit, respectively, with minor modifications. Briefly, PDTs were collected in PBS, centrifuged for 5 min at 1600 rpm, and resuspended in lysis buffer supplemented with proteinase K. A proportion of 60:40 of PDT resuspension was prepared to proceed with DNA and RNA extractions, respectively. Samples were incubated for 30 min at 56 °C and then heat-inactivated for 10 min at 80 °C. PDT-derived DNA and RNA samples were further extracted with the Maxwell robot, according to the manufacturer’s instructions. DNA and RNA quantitation were assessed on a Qubit®2.0 fluorometer with Qubit™ dsDNA HS Assay and Qubit™ RNA HS Assay kits, respectively. DNA and RNA libraries were prepared manually, and high-depth sequencing was performed with Ion 540™ chips with Ion S5™ XL sequencer. Data was assembled with Torrent Suite Software (v.5.16.1 or higher). Sequencing data were mapped to the Human Genome Build 37 (GRCh37/hg19) in the Ion Reporter™ software (v. 5.18.4.0 or higher) and analyzed with “Oncomine Childhood Cancer Research (OCCRA) - w2.8 - DNA and Fusions - Single Sample” workflow. Coverage analysis reports from the Ion Reporter™ Software providing measurements of mapped reads, mean depth, uniformity, and alignment over a target region were used as quality assessments of the sequencing reactions. For the genomic comparison between the PDTs and matched primary tumors (n = 26, 13 pairs), diploid variants with a variant allele frequency (VAF) ranging from 45-55% (heterozygous variants), or with a VAF > 90% (homozygous variants) were considered non-somatic variants and were hence excluded from the analysis. To characterize the clonal states of all somatic mutations identified in the PDTs and matched primary tumors, we used the VAFs and copy number data to estimate the fraction of cancer cells harboring a particular mutation43. Somatic variants deemed “likely subclonal” had a VAF ranging from 2−16%, and those deemed “likely clonal” had a VAF ranging from 15-88% (Table S2). For two cases, HGG_04 and BCOR_02 (Table 1), the primary tumors exhibited a GFAP::ALK fusion and a BCOR intern tandem duplication (ITD), respectively, which could not be detected using the OCCRA panel. In the HGG_04 case, the fusion was first detected using RNA sequencing and subsequently validated through PCR and Sanger sequencing for diagnosis purposes. The GFAP::ALK fusion was also confirmed in the matched PDTs using the same PCR and Sanger sequencing techniques. In the BCOR_02 case, the BCOR tandem duplication was verified by PCR (primer list provided in Table S3) and Sanger sequencing (Figs. S1 and S2), following the methodology described by Roy et al.44.
DNA methylation arrays
DNA samples of five additional PDTs were extracted and quantified as described in the previous section. DNA samples were bisulfite-treated using the EZ DNA Methylation™ Kit (Zymo Research, USA), following the alternative incubation conditions, according to the manufacturer’s recommendations, and then restored with Infinium HD FFPE protocol (Illumina, San Diego, USA). Samples were prepared with Illumina Infinium HD Methylation manual protocol (Illumina, San Diego, USA), according to the manufacturer’s instructions, processed with Infinium MethylationEPIC v2.0 BeadChip, and analyzed on Illumina NextSeq 550 platform (Illumina, San Diego, USA). Data produced for each sample (unprocessed IDAT files) were uploaded into the Heidelberg brain tumor methylation classifier (version 12.8)26. This online platform (https://app.epignostix.com) enables the classification of brain tumors (or derived material) based on the genome-wide methylation profiles obtained from ~930 K methylation sites targeting the human methylome in the Infinium MethylationEPIC v2.0 BeadChip, along with the samples´ copy number profile26. The classifier’s calibrated score ranges from 0 to 1, where 1 indicates the highest confidence level. Scores exceeding 0,9 are considered a match for a tumor type or subtype and are deemed valid for diagnostic purposes26. Scores below 0,9 may still be informative, particularly in cases involving suboptimal DNA quality or limited tumor cell representation.
Raw DNA methylation data were processed from IDAT files using minfi package (v1.52.0) (PMID: 24478339; PMID: 28035024) in the R statistical environment (v4.4.3). Quality control was assessed using the built-in minfi functions, and HGG_05 was excluded due to failed experiment. To correct for technical biases between arrays, including probe-type and color-channel biases, the raw data were normalized using Functional Normalization (preprocessFunnorm function). For statistical analysis and visualization, normalized methylation Beta-values were converted to M-values, as M-values have statistical properties more appropriate for differential analysis. To visualize the relationships between samples, we performed t-distributed Stochastic Neighbor Embedding (t-SNE). The analysis was conducted on the 5000 most variable CpG sites across all samples, as determined by the highest variance in M-values.
Reference data from the Heidelberg cohort (PMID: 37875569, GSE90496), generated using 450 k methylation assay, were used for comparison. To ensure compatibility, we intersected probes between the EPIC and 450 k platforms and repeated the t-SNE analysis on the shared 5000 most variable CpG sites.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Data is provided in the manuscript or supplementary information files. Sensitive data is not openly available, in accordance with the approval from the local ethical committee, and can be available from the corresponding author upon reasonable request.
Code availability
The code for analysing the DNA methylation data is available in the supplementary data.
References
Siegel, R. L., Miller, K. D., Wagle, N. S. & Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 73, 17–48 (2023).
Erdmann, F. et al. Childhood cancer: survival, treatment modalities, late effects and improvements over time. Cancer Epidemiol. 71, 101733 (2021).
Pollack, I. F., Agnihotri, S. & Broniscer, A. Childhood brain tumors: current management, biological insights, and future directions. J. Neurosurg. Pediatr. 23, 261–273 (2019).
Adel Fahmideh, M. & Scheurer, M. E. Pediatric brain tumors: descriptive epidemiology, risk factors, and future directions. Cancer Epidemiol. Biomark. Prev. 30, 813–821 (2021).
Suh, E. et al. Late mortality and chronic health conditions in long-term survivors of early-adolescent and young adult cancers: a retrospective cohort analysis from the childhood cancer survivor study. lancet Oncol. 21, 421–435 (2020).
Ryall, S. et al. Integrated molecular and clinical analysis of 1,000 pediatric low-grade gliomas. Cancer Cell 37, 569–583.e565 (2020).
Ma, X. et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature 555, 371–376 (2018).
Clarke, M. et al. Infant high-grade gliomas comprise multiple subgroups characterized by novel targetable gene fusions and favorable outcomes. Cancer Discov. 10, 942–963 (2020).
Guerreiro Stucklin, A. S. et al. Alterations in ALK/ROS1/NTRK/MET drive a group of infantile hemispheric gliomas. Nat. Commun. 10, 4343 (2019).
Reinhardt, A. et al. Anaplastic astrocytoma with piloid features, a novel molecular class of IDH wildtype glioma with recurrent MAPK pathway, CDKN2A/B and ATRX alterations. Acta Neuropathol. 136, 273–291 (2018).
Johnson, A. et al. Comprehensive genomic profiling of 282 pediatric low- and high-grade gliomas reveals genomic drivers, tumor mutational burden, and hypermutation signatures. Oncologist 22, 1478–1490 (2017).
Louis, D. N. et al. The 2016 world health organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 131, 803–820 (2016).
Louis, D. N. et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol. 23, 1231–1251 (2021).
Drost, J. & Clevers, H. Organoids in cancer research. Nat. Rev. Cancer 18, 407–418 (2018).
Clevers, H. Modeling development and disease with organoids. Cell 165, 1586–1597 (2016).
Jacob, F. et al. A patient-derived glioblastoma organoid model and biobank recapitulates inter- and intra-tumoral heterogeneity. Cell 180, 188–204.e122 (2020).
Gamboa, C. M. et al. Generation of glioblastoma patient-derived organoids and mouse brain orthotopic xenografts for drug screening. STAR Protoc. 2, 100345 (2021).
Jacob, F., Ming, G. L. & Song, H. Generation and biobanking of patient-derived glioblastoma organoids and their application in CAR T cell testing. Nat. Protoc. 15, 4000–4033 (2020).
Golebiewska, A. et al. Patient-derived organoids and orthotopic xenografts of primary and recurrent gliomas represent relevant patient avatars for precision oncology. Acta Neuropathol. 140, 919–949 (2020).
Verduin, M. et al. Patient-derived glioblastoma organoids reflect tumor heterogeneity and treatment sensitivity. Neurooncol. Adv. 5, vdad152 (2023).
Ferreira, B., Peixoto, J. & Lima, J. Reference Module in Biomedical Sciences (Elsevier, 2023).
Blattner-Johnson, M., Jones, D. T. W. & Pfaff, E. Precision medicine in pediatric solid cancers. Semin Cancer Biol. 84, 214–227 (2022).
Paassen, I. et al. Atypical teratoid/rhabdoid tumoroids reveal subgroup-specific drug vulnerabilities. Oncogene 42, 1661–1671 (2023).
Peterziel, H. et al. Drug sensitivity profiling of 3D tumor tissue cultures in the pediatric precision oncology program INFORM. NPJ Precis Oncol. 6, 94 (2022).
Coffin, C. M., Braun, J. T., Wick, M. R. & Dehner, L. P. A clinicopathologic and immunohistochemical analysis of 53 cases of medulloblastoma with emphasis on synaptophysin expression. Mod. Pathol. 3, 164–170 (1990).
Capper, D. et al. DNA methylation-based classification of central nervous system tumours. Nature 555, 469–474 (2018).
Hovestadt, V. et al. Medulloblastomics revisited: biological and clinical insights from thousands of patients. Nat. Rev. Cancer 20, 42–56 (2020).
Milde, T. et al. Optimizing preclinical pediatric low-grade glioma models for meaningful clinical translation. Neuro Oncol. 25, 1920–1931 (2023).
Bose, S., Clevers, H. & Shen, X. Promises and challenges of organoid-guided precision medicine. Med. 2, 1011–1026 (2021).
Yoo, J. et al. The extent of necrosis in brain metastases may predict subtypes of primary cancer and overall survival in patients receiving craniotomy. Cancers (Basel) 14, 1694 (2022).
Lago, C. et al. Patient- and xenograft-derived organoids recapitulate pediatric brain tumor features and patient treatments. EMBO Mol. Med. 15, e18199 (2023).
Abdullah, K. G. et al. Establishment of patient-derived organoid models of lower-grade glioma. Neuro Oncol. 24, 612–623 (2022).
Pedace, L. et al. Evaluating cell culture reliability in pediatric brain tumor primary cells through DNA methylation profiling. NPJ Precis. Oncol. 8, 92 (2024).
Calandrini, C. et al. An organoid biobank for childhood kidney cancers that captures disease and tissue heterogeneity. Nat. Commun. 11, 1310 (2020).
Weller, M. et al. Glioma. Nat. Rev. Dis. Prim. 10, 33 (2024).
Sturm, D. et al. New brain tumor entities emerge from molecular classification of CNS-PNETs. Cell 164, 1060–1072 (2016).
Carvalho, B. et al. The role of c-Met and VEGFR2 in glioblastoma resistance to bevacizumab. Sci. Rep. 11, 6067 (2021).
Zhang, S. W. et al. An efficient and user-friendly method for cytohistological analysis of organoids. J. Tissue Eng. Regen. Med. 15, 1012–1022 (2021).
Eiraku, M. et al. Self-organized formation of polarized cortical tissues from ESCs and its active manipulation by extrinsic signals. Cell Stem Cell 3, 519–532 (2008).
Sato, T. et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 141, 1762–1772 (2011).
Sato, T. et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459, 262–265 (2009).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Pereira, B. et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat. Commun. 7, 11479 (2016).
Roy, A. et al. Recurrent internal tandem duplications of BCOR in clear cell sarcoma of the kidney. Nat. Commun. 6, 8891 (2015).
Acknowledgements
We thank all the patients and families. This work was funded by the Fundação Rui Osório de Castro and Liga Portuguesa Contra o Cancro (LPCC). BSF was supported by an Oncology fellowship from LPCC and a La Caixa PhD fellowship (LCF/BQ/DR24/12080023). J.P. is financed through a junior researcher contract from Fundação para a Ciência e a Tecnologia (2022.02137.CEECIND). Instituto de Investigação e Inovação em Saúde da Universidade do Porto (i3S) is an Associated Laboratory of the Portuguese Ministry of Science, Technology, and Higher Education that is partially supported by Fundação para a Ciência e a Tecnologia.
Author information
Authors and Affiliations
Contributions
Conceptualization: J.L., B.S.F., and MJ. GdC.; Methodology: B.S.F., J.Peixoto, A.Ferro., B.E, J.Pinheiro, R.S., A.M., M.A, L.F and L.C.; Data curation: B.S.F., J.L., A.P., Z.Y. and A.Ferro; Resources: J.Pereira, J.O., S.N., A.Fernandes and MJ. GdC.; Software: J.L., A.P., and A.M; Writing -original draft: B.S.F.; Writing: review and editing: J.L., J. Peixoto, A.Ferro. and A.P.; Supervision: J.L and P.S.; Funding acquisition: B.S.F., J.Peixoto and J.L. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Soares-Ferreira, B., Peixoto, J., Ferro, A. et al. Patient-derived tumoroids recapitulate the morphologic and molecular features of pediatric brain tumors. npj Precis. Onc. 9, 371 (2025). https://doi.org/10.1038/s41698-025-01151-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41698-025-01151-w
This article is cited by
-
Modelling brain tumours with organoids: towards precision medicine in neuro-oncology
Nature Reviews Neurology (2026)
-
Human thyroid organoids: tools for understanding thyroid diseases and development
Nature Reviews Endocrinology (2026)
