
Abstract
Plasma cell-free DNA (cfDNA) is a promising biomarker for liquid biopsy, essential for diagnosing and monitoring diseases. Current methods for estimating tissue contributions primarily rely on methylation markers, which can damage cfDNA, limiting clinical use. While research shows cfDNA coverage near transcription start sites (TSS) of actively transcribed genes decreases due to open chromatin, a comprehensive cross-tissue atlas has been lacking. Here, we identify 2549 tissue-specific, highly expressed genes across 12 human tissues and develop the Tissue Contribution Index (TCI) to quantify tissue contributions to plasma cfDNA using TSS coverage. TCI is validated in cfDNA origin models, including pregnant women and transplant recipients, demonstrating high accuracy. We establish reference intervals using plasma cfDNA from 460 healthy individuals and explore TCI’s diagnostic utility in monitoring tissue damage and predicting outcomes. This study presents a simple, cost-effective method for tissue deconvolution of cfDNA, advancing liquid biopsy for disease detection and personalized medicine.
Similar content being viewed by others


Introduction
Cell-free DNA (cfDNA) in blood circulation contains DNA fragments originating from various tissues within the body1. During pregnancy, cfDNA is released from both the mother and the fetus, enabling the development of non-invasive prenatal testing (NIPT) for trisomy screening by examining the fetal-derived cfDNA2. In the field of oncology, the analysis of circulating tumor DNA (ctDNA) facilitates cancer diagnosis and monitoring. Similarly, in organ transplant recipients, monitoring the proportion of donor-derived cfDNA in plasma offers a non-invasive and time-efficient approach to detect organ rejection. Recent studies have further revealed elevated levels of plasma cfDNA originating from damaged tissues in patients with autoimmune diseases3, injuries4, and infections5. Hence, assessing the tissue origin and contributions of cfDNA in plasma has emerged as a crucial step in detecting disease through the abnormal representation of the affected tissue DNA.
Current approaches for measuring the tissue contribution of cfDNA can be broadly categorized into the use of genetic and non-genetic markers. Genetic markers, such as fetal specific single nucleotide polymorphism (SNP) in maternal plasma, tumor specific copy number variations (CNV), and somatic mutations in cancer patients, serve as direct signatures to trace the tissue origin of cfDNA and estimate the tissue contribution6,7. These markers have demonstrated high accuracy and can be considered a gold standard. However, their application is limited to single-tissue assessments, and they suffer from high heterogeneity, necessitating the development of stable, widely applicable non-genetic markers. Epigenetic markers, particularly tissue-specific methylation, offer stable alternatives for cfDNA tissue deconvolution1,8,9,10. The current widely used method to assess DNA methylation from cfDNA is bisulfite sequencing (BS-seq). However, BS-seq requires large blood volumes and complex processing, and may degrade DNA and introduce GC content bias11,12, limiting its utility for clinical practice.
Unlike genomic DNA, cfDNA is non-randomly fragmented during the release and generation. CfDNA fragmentomic characteristics, such as fragment size13, preferred ending sites14, ends motifs15, and jagged-ends16 are emerging promising non-genetic biomarkers for disease detection and have demonstrated specificity to its tissue-of-origin. Moreover, it is known that nucleosome positioning varies across tissues17. By inferring nucleosome footprints through short cfDNA coverage, Synder et al. estimated cell types contributing to cfDNA in cancer patients18. However, such methods can only provide a rank of contributing tissues rather than quantifying their contributions. To address this limitation, Sun et al. leveraged the cfDNA cleavage pattern within tissue-specific open chromatin regions to develop the orientation-aware plasma cfDNA fragmentation (OCF) method for evaluating cfDNA contributions from multiple tissues19. However, OCF was not applicable to single-end sequencing data and has accuracy limitations.
Previous studies have demonstrated that nucleosome occupancy in regions around transcription start sites (TSSs) results in read-depth coverage patterns related to gene expression levels17,20. These findings suggested the possibility of accurately inferring tissue contributions of cfDNA from its fragmentation pattern in such regions. One recent study provided a proof-of-concept approach to predict the ctDNA levels in cancer patients by measuring the nucleosome-dependent cfDNA degradation at TSSs of tumor highly expressed genes21.
Here, we present a multi-tissue traceability method named Tissue Contribution Index (TCI) to quantify the cfDNA release across multiple tissues. Our method is based on the cfDNA fragmentation pattern in TSS regions of tissue-specific genes. We validated the predictions in pregnant women and liver and bone marrow transplant recipients. We further constructed a benchmark for TCI by sequencing a healthy cohort of 460 individuals to determine reference intervals. We predicted the tissue contributions in COVID-19 patients and cancer patients and applied TCI for the prediction of tissue damage, prognosis, and cancer status.
Results
Overview of study design
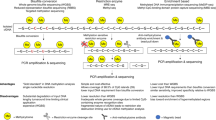
In this study, we developed an algorithm, referred to as the Tissue Contribution Index, to quantify the contribution of cfDNA from various tissues in human plasma. The workflow of our study is schematically illustrated in Fig. 1. Previous study has shown that cfDNA exhibits reduced genome coverage in the TSS region of highly expressed genes due to nucleosome depletion17. With this knowledge, we analyzed transcriptomic data from the Genotype-Tissue Expression (GTEx) database and Pregnancy Outcome Prediction Study (POPS) to identify tissue specific highly expressed genes (SH-genes) in 12 different human tissues. These SH-genes were hypothesized to reflect the relative contribution of cfDNA from their respective tissues (Fig. 1a).

a Coverage pattern of highly and lowly expressed gene around transcriptional start site (TSS), identification of tissue-specific highly expressed genes (SH-genes), and the TSS coverage pattern of SH-genes for each tissue in plasma. b TCI calculation workflow for individual tissues. c Construction of baseline of plasma TCI in 460 healthy individuals, validation in plasma of pregnant women and organ transplant recipients and applications of TCI for cancer detection and tissue injury assessment. Some elements in (a, b) were created in BioRender. Li, L. (2025) https://BioRender.com/8hdbkw8.
To quantify the cfDNA coverage profile, we interrogated the genome coverage within the 2 kb regions centered around the TSS of each SH-gene for the 12 tissues. The TCI algorithm was developed to assess the contribution of cfDNA from each tissue based on the normalized TSS coverages of their respective SH-genes (Fig. 1b) (See Methods). The performance of the TCI algorithm was validated through two key evaluations: tracking the fraction of fetal DNA in maternal plasma among pregnant women and determining the proportion of donor organ cfDNA in the plasma of transplant recipients. Additionally, we used plasma DNA data from 460 healthy individuals to establish reference intervals for TCI values. These reference intervals serve as benchmarks for detecting tissue damage and diseases. Finally, we applied the TCI algorithm along with the established reference intervals for cancer diagnosis and assessment of tissue damage (Fig. 1c).
We finally identified 2549 SH-genes from 4561 tissue samples across 12 tissues. The number of SH-genes per tissue varied, with a median of 201 and a range of 91 to 500 genes (Fig. 2a and https://github.com/lingguoli/TCI/blob/main/Ref/TPM_atlas.csv). The tissue-specificity of these SH-genes was displayed by unsupervised t-distributed Stochastic Neighbor Embedding (t-SNE) analysis on 4561 tissue samples from the GTEx and POPS database (Fig. 2b).

a SH-gene counts of each 12 tissues with corresponding z-scored Transcripts Per Million (TPM) from Genotype-Tissue Expression (GTEx) and Pregnancy Outcome Prediction Study (POPS) databases. b t-distributed Stochastic Neighbor Embedding (t-SNE) visualization of 4561 tissue samples (from GTEx and POPS database) based on expression patterns of 2549 SH-genes.
Validation of TCI in pregnant women and organ transplant recipients
To validate whether the TCI could accurately reflect tissue contribution of cfDNA in plasma, we applied TCI analysis to two well-studied scenarios: detecting fetal fraction in plasma cfDNA of pregnant women and donor fraction in plasma cfDNA of organ transplant recipients.
Placenta TCI in pregnant women
To explore the effectiveness of TCI in measuring fetal DNA fraction in maternal plasma, we analyzed cfDNA data from 101 plasma samples of pregnant women carrying a male fetus that were recruited in Shenzhen. Given that the placenta is the primary source of cell-free fetal DNA in maternal plasma22, we focused on measuring the cfDNA coverage of placental SH-genes and calculated the TCI of the placenta to estimate the fetal cfDNA fraction. We stratified these samples into four groups based on the fetal cfDNA fraction: < 10%; 10 - 20%; 20 - 30%; ≥ 30%. We adopted Base Counts Per Million mapped bases (BCPM) at base-pair resolution to show the normalized cfDNA coverage across 2 kb TSS regions. Figure 3a illustrates the average BCPM at base-pair resolution of placental SH-genes for 4 groups with varied fetal DNA fractions. CfDNA coverages progressively reduced as the fetal fraction increased, suggesting that the depletion of cfDNA coverage is a strong indicator of the placental contribution. This trend was consistent across all 101 samples (Supplementary Fig. 1a). We then calculated the placental TCI for each sample and compared it with the fetal DNA fraction measured by Y-chromosome (chrY) proportion in cfDNA, which originates from the male fetus. As shown in Fig. 3b, we observed a positive and strong linear correlation between the placental TCI and the fetal DNA fractions (r = 0.93, 95% confidence interval (CI): 0.88–0.95, p < 0.0001, Pearson’s correlation), suggesting the high accuracy of TCI in estimating the placental contribution in maternal plasma. Furthermore, we examined the correlations between 12 distinct types of TCI and fetal fractions. As illustrated in Supplementary Fig. 2a, only placental TCI positively correlated with fetal fractions, highlighting that the increase in TCI is tissue-specific.

a The average Base Counts Per Million mapped bases (BCPM) profiles at single base pair resolution across 1,000 bp upstream and downstream of transcriptional start site (TSS) of placental tissue-specific highly expressed genes (SH-genes) in pregnant women grouped by fetal DNA fraction intervals. b Correlation between placenta Tissue Contribution Index (TCI) and fetal cfDNA fractions in 101 samples from pregnant women with male fetus. c Comparison of the correlations between fetal cfDNA fraction and plasma placental contribution predicted by TCI and OCF methods in simulation samples with different cfDNA amounts. Central dot in (c) indicate Pearson correlation coefficients, with two bars showing 95% confidence intervals. The shaded area in (a, b) represents the 95% confidence interval.
Clinical practice typically employs shallow-depth or single-end (SE) sequencing to reduce costs. To assess the applicability of our TCI method for low-depth and SE data, we simulated various sequencing conditions by in silico downsampling samples into different number of cfDNA molecules. SE 35 bp data were generated by truncating the first 35 bp of cfDNA fragments from our pair-end data. We compared the performance of fetal DNA fraction estimation among three methods: TCI based on paired-end (PE) data, TCI based on SE data, and the previously published OCF method, which utilizes cfDNA end patterns in tissue-specific open chromatin areas19. Figure 3c illustrates that the performance of the 3 methods improved as the number of cfDNA molecules increased. Notably, TCI approaches using both PE and SE data exhibit comparable performance. Moreover, TCI methods demonstrate consistently higher Pearson correlation coefficients than the OCF method across different data amounts, particularly when exceeding 100 million cfDNA molecules.
TCI in organ transplantation recipients
To further validate our TCI method, we analyzed published 12 plasma cfDNA sequencing data from liver transplant recipients19 and 54 plasma cfDNA sequencing data from bone marrow transplant recipients23. The fraction of donor-derived cfDNA was obtained from the original studies19,23, which was calculated based on donor-recipient informative allele types. For liver transplant recipients, we measured the cfDNA coverage across the TSS regions of liver SH-genes and calculated the liver TCI. We observed a decrease in average BCPM of liver SH-genes as the donor fraction increased in the recipients’ plasma (Fig. 4a and Supplementary Fig. 1b). We found a positive correlation between the liver TCI and the donor cfDNA fraction in these recipients (r = 0.96, 95% CI: 0.56–0.99, p < 0.0001, Pearson’s correlation, Fig. 4b). We further examined the correlations between the TCI for 11 non-placental tissues and donor fractions in liver transplant recipients. As shown in Supplementary Fig. 2b, a strong linear positive correlation was observed specifically between liver TCI and donor fractions.

a The average Base Counts Per Million mapped bases (BCPM) profiles at single base pair resolution across 1000 bp upstream and downstream of transcriptional start site (TSS) of liver tissue-specific highly expressed genes (SH-genes) in liver transplant recipients grouped by donor cfDNA fraction. b Correlation between liver Tissue Contribution Index (TCI) and donor cfDNA fraction in 12 liver transplant recipients. c The average BCPM at single base pair resolution across 1000 bp upstream and downstream of TSS of blood cell SH-genes in bone marrow transplant recipients grouped by donor cfDNA fraction. d Correlation between blood cell TCI and donor cfDNA fraction in bone marrow transplant recipients. The shaded area in (a–d) represents the 95% confidence interval.
Similarly, for bone marrow transplant recipients, we analyzed the SH-genes coverage and TCI for blood cells, which are generated and released from the bone marrow. We observed a gradual reduction in BCPM of blood cell SH-genes as the fraction of blood cells increased (Fig. 4c and Supplementary Fig. 1c). Correlation analysis revealed a positive correlation between the blood cell TCI and donor cfDNA fraction in these recipients (r = 0.85, 95% CI: 0.78–0.90, p < 0.0001, Pearson’s correlation, Fig. 4d). Pearson correlation analysis was conducted between the TCI of 11 non-placental tissues and donor fractions in bone marrow transplant recipients. As depicted in Supplementary Fig. 2c, blood cell TCI showed a specific positive correlation with donor fractions in these recipients.
Accurate quantification of donor-derived cfDNA in plasma is crucial for monitoring post-transplant tissue damage and potential immune rejection in organ transplant recipients. Among these bone marrow transplant recipients, one patient (recipient I6) was reported to suffer liver damage due to acute graft-versus-host disease (aGVHD). To explore the potential of liver TCI in detecting such liver damage, we traced the dynamic liver TCI in the plasma DNA of this patient after the transplantation surgery. As illustrated in Fig. 5a, the blood cell TCI showed a consistent trend with the donor fraction across different timepoints. Notably, the liver TCI exhibited a dramatic increase coinciding with the onset of aGVHD in the patient’s liver. Among recipients without aGVHD (Fig. 5b–e), the blood cell TCI exhibited similar trends to the donor fraction, achieving relatively high levels during later monitoring periods. In contrast, the liver TCI maintained relatively low levels. These findings demonstrate that the TCI approach is able to accurately estimate the tissue contribution in plasma DNA and can serve as a promising biomarker for post-transplant surveillance.

a Liver and blood cell TCI dynamics in a recipient with acute graft-versus-host disease (aGVHD) affecting the liver. b–e Liver and blood cell TCI dynamics in recipients without aGVHD. Left Y-axis: donor cfDNA fraction (black dots); right Y-axis: liver TCI (red squares) and blood cell TCI (blue crosses).
Establishing TCI Reference in Healthy Subjects
A key application of cfDNA analysis is the potential to predict disease status by detecting abnormal contributions of cfDNA from affected tissues. To achieve this goal, we sought to establish reference TCI values for various tissues under healthy conditions. We collected plasma DNA data from 460 healthy individuals during their routine physical examinations (Supplementary Table 1). We analyzed TCI values for 12 different tissues across these cfDNA samples. To establish healthy reference intervals, we used the central 95% of TCI from this cohort. As shown in Fig. 6, we defined the upper and lower reference limits of TCI for each tissue using the 97.5th and 2.5th percentiles of the TCI distribution, respectively. Among the tissues analyzed, blood cells exhibited the highest TCI reference interval (23.92 to 26.51), while the artery showed the lowest TCI reference interval (13.90 to 15.67). These reference intervals provide a baseline for distinguishing abnormal tissue contributions from healthy conditions.

The blue and red dashed lines represent the 2.5th and 97.5th percentiles of TCI for each tissue, respectively. The grey shaded area represents the central 95% of TCI determined from 460 healthy individuals, which was defined as the reference interval.
Plasma DNA TCI in COVID-19 patients
Severe infections often lead to tissue damage in multiple organs. This is particularly evident in critical cases of COVID-1924. To investigate the potential clinical uses of TCI in infectious diseases, we examined 194 plasma cfDNA data from critically ill COVID-19 patients from our previous study25.
Elevated serum levels of lactate dehydrogenase (LDH) and alanine aminotransferase (ALT) are clinical indicators of liver damage. Clinically, liver injury is defined as LDH ≥ 500 U/L or ALT ≥ 80 U/L, which is twice the established clinical upper limits of 250 U/L and 40 U/L, respectively26,27. Patients with clinically defined liver injury, as determined by high LDH, exhibited significantly higher liver TCI (median: 21.58; range: 18.70 to 30.64) compared to those with normal LDH levels (median: 18.89; range: 17.48 to 23.52; p < 0.0001, Mann-Whitney U test) (Fig. 7a). We also observed similar trends in patients with elevated ALT levels. Patients with liver injury based on high ALT also demonstrated significantly higher liver TCI (median: 21.41; range: 18.94 to 30.64) than those with normal ALT levels (median: 18.85; range: 16.97 to 23.52; p < 0.0001, Mann-Whitney U test) (Fig. 7b). As depicted in Supplementary Fig. 3a, b, patients with abnormal LDH or ALT levels showed a specific increase in liver TCI compared to normal samples (p < 0.0001, Mann-Whitney U test). Conversely, the blood cell TCI exhibited a specific decrease (p < 0.01, Mann-Whitney U test).

a Boxplot of liver TCI in patients with normal serum lactate dehydrogenase (LDH) level and those with liver injury determined by high LDH (LDH ≥ 500 U/L). b Boxplot of liver TCI in patients with normal alanine aminotransferase (ALT) level (ALT < 80 U/L) and those with liver injury (ALT ≥ 80 U/L). c Receiver operator characteristic (ROC) analysis of liver TCI for discriminating between patients with liver injury (LDH ≥ 500 U/L or ALT ≥ 80 U/L) versus normal (LDH < 250 U/L and ALT < 40 U/L). d Boxplot of artery TCI for patients in the intensive care unit (ICU) and patients not in ICU (non-ICU). e Boxplot of artery TCI for patients with cured and deceased outcomes. f ROC analysis of artery TCI for distinguishing ICU/non-ICU status and cured-deceased outcomes. g Boxplot of lung TCI for patients in ICU and patients not in ICU. h Boxplot of lung TCI for patients with cured and deceased outcomes. i ROC analysis of lung TCI for distinguishing ICU/non-ICU status and cured-deceased outcomes. In ROC curves (c, f, i), square brackets indicate 95% confidence intervals for AUC values. In boxplots (a, b, d, e, g, h), red/blue dashed lines represent reference TCI limits (derived from 460 healthy controls).
To evaluate the diagnostic performance of our method, we used the upper limit of liver TCI established from our healthy cohort (i.e., 19.38) as a threshold. For detecting clinically defined liver injury, we achieved high sensitivity (86% for abnormal LDH and 94% for abnormal ALT) and moderate specificity (73% for abnormal LDH and 76% for abnormal ALT) (Supplementary Table 2). The area under the curve (AUC) values were 0.88 (95% CI: 0.78–0.98) for LDH-based assessment and 0.92 (95% CI: 0.85–0.99) for ALT-based assessment (Fig. 7c). A previous study has suggested that multi-organ vascular endothelial cell injury may contribute to poor prognosis in COVID-19 patients24. To investigate this, we evaluated artery TCI in patients with different outcomes and intensive care unit (ICU) statuses. Our analysis revealed that ICU patients had significantly higher artery TCI compared to non-ICU patients (median: 16.54 vs. 15.65, p < 0.0001, Mann-Whitney U test) (Fig. 7d, Supplementary Fig. 3c). Similarly, deceased patients exhibited higher artery TCI (median: 16.64, range: 14.93 to 22.10) than survivors (median: 15.50, range: 17.00 to 19.98, p < 0.0001, Mann-Whitney U test) (Fig. 7e, Supplementary Fig. 3d). The AUC for differentiating ICU from non-ICU patients using artery TCI was 0.71, while the AUC for distinguishing deceased from surviving patients was 0.83 (Fig. 7f). Besides, other TCI values (e.g. pancreas, colon, and heart) were also significantly increased alongside the artery TCI in the deceased group of COVID19 patients (Supplementary Fig. 3d). Using the upper limit of artery TCI (i.e. 15.67) as a threshold, we achieved sensitivities of 73% and 86%, specificities of 53% and 65% for predicting ICU admission and mortality, respectively (Supplementary Table 2). Lung TCI also showed significant differences between ICU and non-ICU patients (Fig. 7g), and between deceased and surviving patients (Fig. 7h). The AUC for differentiating ICU from non-ICU patients using lung TCI was 0.63, while the AUC for distinguishing deceased from surviving patients was 0.74 (Fig. 7i). These findings suggest that artery TCI may reflect multi-organ vascular endothelial cell injury and could serve as a potential biomarker for predicting prognosis and ICU status in COVID-19 patients.
Plasma DNA TCI in Cancer patients
The abnormally elevated levels of cell-free DNA from diseased tissues have been established as a key biomarker in cancer diagnosis10,28. To evaluate the diagnostic utility of plasma TCI in cancer diagnosis, we downloaded publicly available sequencing data of plasma DNA for 32 healthy individuals and 74 hepatocellular carcinoma (HCC) patients29 from FinaleDB30. Tumor DNA fraction (TF) for these HCC patients was obtained from Sun et al.’s study19, where it was calculated using the ichorCNA method31. Sun et al.19 stratified HCC subjects into low (TF ≤ 10%) and high (TF > 10%) tumor burden groups. We examined the distribution of 11 types of non-placental tissue TCI in both healthy samples and HCC samples. Our analysis revealed that the liver TCI in HCC samples with high tumor burden (median: 20.48, range: 19.76 to 23.02) was significantly elevated compared to healthy controls (median: 18.31, range: 16.55 to 19.45, p < 0.0001, Mann-Whitney U test, Fig. 8a and Supplementary Fig. 4a). Notably, the blood cell TCI also exhibited similar potential in distinguishing HCC patients from healthy subjects. In high tumor burden HCC samples (median: 22.38, range: 21.03 to 23.97), the blood TCI were significantly lower than in healthy controls (median: 24.47, range: 23.17 to 26.49, p < 0.0001, Mann-Whitney U test, Fig. 8a and Supplementary Fig. 4b). Furthermore, among the 32 healthy controls, 29 showed liver TCI values within the reference interval, whereas all 8 high tumor burden HCC samples exceeded the upper limit (Supplementary Fig. 4a). Additionally, we observed a positive correlation between liver TCI and tumor fraction in 74 HCC samples (r = 0.41, 95% CI: 0.20–0.59, p = 0.0002, Spearman’s correlation, Supplementary Fig. 4c).

a TCI levels of 11 non-placental tissues in HCC patients versus healthy subjects. b Receiver operator characteristic (ROC) analysis for distinguishing HCC patients with high tumor burden using liver TCI and blood cell TCI ratio, liver TCI, blood cell TCI, liver OCF, motif diversity score (MDS), and proportion of short cfDNA fragments (< 150 bp). c ROC analysis for distinguishing all HCC patients using the same six biomarkers as in (b). d TCI level of 11 non-placental tissues in cancer patients and healthy subjects from Cristiano et al.’s study. ns: p ≥ 0.05, **: p < 0.01, ****: p < 0.0001.
As reported in previous studies, the proportions of short fragments (less than 150 bp) in plasma cfDNA29, the OCF values of the cancerous sites19, and the ends motif diversity score of cfDNA fragments15 were elevated in cancer patients. In this study, we calculated these fragmentomic characteristics for comparison. Compared with the healthy samples, these biomarkers were all observed to be elevated in high tumor burden HCC samples (Supplementary Fig. 4d–f). We performed ROC analysis to compare diagnostic performance of TCI with these fragmentomic features for HCC detection. As depicted in Fig. 8b, when differentiating HCC patients with high tumor burden from healthy samples, both liver TCI (AUC = 1.00, with 100% sensitivity at 99% specificity) and blood cell TCI (AUC = 0.95, with 88% sensitivity at 99% specificity), as well as their ratio (TCI(L/B)) (AUC = 1.00, with 100% sensitivity at 99% specificity), showed consistently higher performance than OCF, MDS, and fragment size-based biomarker. Similarly, when distinguishing HCC patients with all stages from healthy samples, the TCI methods also exhibited higher performance (AUC of liver TCI, blood TCI and TCI(L/B): 0.68, 0.75 and 0.74; sensitivity at 99% specificity of liver TCI, blood TCI and TCI(L/B): 39%, 50% and 41%) than others (AUC of size, MDS and OCF: 0.49, 0.34 and 0.45; sensitivity at 99% specificity of size, MDS and OCF: 19%, 5% and 14%) (Fig. 8c).
Using the upper limit of liver TCI (17.15) as a cutoff, we distinguished high tumor load group from healthy individuals with 100% sensitivity, 94% specificity, and a positive likelihood ratio (PLR) of 16. When applying the same cutoff to differentiate all 74 HCC patients from healthy controls, the sensitivity, specificity, and PLR were 40%, 94%, and 6.27, respectively (Supplementary Table 3).
We further analyzed TCI in plasma DNA from studies of Cristiano et al. (healthy samples: 262, cancer samples: 276)32, Snyder et al. (healthy samples: 4, cancer samples: 48)18, and Liang et al. (healthy samples: 10, cancer samples: 20)33. We dissected the TCI of all 11 non-placental tissues between cancer patients and healthy subjects in Cristiano et al.‘s study32. Compared with healthy samples, the artery TCI were increased in cancer patients (median: 15.62 vs. 15.89, p = 0.0049, Mann-Whitney U test, Fig. 8d). Meanwhile, we also observed significantly decreased blood TCI in cancer patients (median: 22.95, range: 18.07 to 26.67) in comparison to healthy samples (median: 23.55, range: 21.20 to 26.54, p < 0.0001, Mann-Whitney U test, Fig. 8d). Similar decreased trends were observed in cancer samples from Snyder et al.18 (median: 24.76 vs. 23.33, p = 0.0705, Mann-Whitney U test, Supplementary Fig. 5a) and Liang et al.33 (median: 26.06 vs. 25.03, p = 0.0679, Mann-Whitney U test, Supplementary Fig. 5b).
Among the cancer patients in Cristiano et al.‘s study32, we noticed that there were 27 patients diagnosed with colorectal cancer, and 8 of them had liver metastasis. We further compared the distribution of TCI of liver, colon, and blood cells among these colorectal cancer patients with and without metastases, as well as in healthy samples. Healthy controls demonstrated significantly higher blood cell TCI values (median: 23.56, range: 21.20 to 26.54) compared to colorectal cancer patients with liver metastasis (median: 21.67, range: 18.66 to 26.24, p = 0.0060, Mann-Whitney U test, Supplementary Fig. 5c). Meanwhile, the colorectal cancer patients with liver metastasis (median: 17.66, range: 14.22 to 20.77) showed higher colon TCI than those without metastases (median: 15.18, range: 13.98 to 17.16, p = 0.08, Mann-Whitney U test) and healthy controls (median: 15.58, range: 13.40 to 19.55, p = 0.0236, Mann-Whitney U test, Supplementary Fig. 5d). Furthermore, liver TCI was significantly elevated in colorectal cancer patients with liver metastases (median: 19.10, range: 17.88 to 20.89) compared to both non-metastatic patients (median: 17.47 range: 16.79 to 22.31, p = 0.0162) and healthy controls (median: 17.88, range: 15.45 to 20.90, p = 0.0027, Mann-Whitney U test, Supplementary Fig. 5e).
We next estimated the tumor DNA fraction in cancer samples from studies of Cristiano et al.32, Snyder et al.18 and Liang et al.33 by using ichorCNA31. In samples with detectable TF (TF > 0%), blood cell TCI was negatively correlated with TF (Spearman r = −0.37, −0.72, −0.54, and −0.66 for Jiang et al.29, Snyder et al.18, Cristiano et al.32, and Liang et al.33 datasets, respectively, Supplementary Fig. 6a, c, e, and g). A similar negative correlation was observed when restricting the analysis to samples with very low TF (0% < TF ≤ 5%) (Spearman r = −0.34, −0.36, and −0.52 for Jiang et al.29, Snyder et al.18 and Cristiano et al.32 datasets, respectively, Supplementary Fig. 6b, d, and f).
Discussion
In this study, we presented the Tissue Contribution Index, an approach for inferring the relative contributions of different tissues to plasma cfDNA. This method analyzes cfDNA fragmentation patterns in TSS regions, focusing on tissue-specific highly expressed genes. We validated the TCI approach using various biological scenarios, including the detection of plasma DNA fractions from (i) the placenta in pregnant women, (ii) the liver in liver transplantations, and (iii) blood cells in bone marrow transplantation recipients. The strong linear correlations observed between the tissue contribution assessed by TCI and genetic markers demonstrate the accuracy and robustness of our method. We showed that when the contribution of a certain tissue to cfDNA increases, only its corresponding TCI shows a strong linear positive correlation, demonstrating that the increasing TCI is tissue-specific (Supplementary Fig. 2).
We used plasma cfDNA data of 460 healthy subjects to establish reference intervals for TCI of different tissues. By comparing the individual tissue TCI with the reference intervals, one can identify the abnormal contributions from the target tissues, thus aiding in detecting tissue damage and diseases. We explored the clinical applications of TCI in critically ill COVID-19 patients, showing its potential as a biomarker for monitoring disease status and predicting clinical outcomes in infectious diseases. Additionally, we demonstrated the diagnostic utility of liver TCI in detecting HCC, highlighting its potential as a cancer biomarker. Besides, our analysis revealed a significant decrease in blood TCI among cancer patients compared to non-cancer individuals. Notably, this blood TCI reduction was even more pronounced than the tumor-related tissue TCI differences between these groups (Fig. 8c). This finding suggests that tumorigenesis and progression may substantially impact the turnover of hematopoietic and immune cells. An additional potential contributor to the blood cell TCI reduction could be altered tissue contribution to plasma DNA. Specifically, the elevated release of liver-derived DNA fragments into plasma in HCC patients dilutes the relative contribution from blood cells, thereby amplifying the observed decrease in blood TCI. While the current AUC for liver cancer detection using TCI remains moderate (about 0.75), further research could focus on identifying SH-genes of tumor tissue to calculate tumor TCI for improved diagnostic performance.
Unlike previous bisulfite-sequencing-based tissue deconvolution methods, TCI analysis can be performed using standard genome sequencing of cfDNA. This approach preserves cfDNA fragmentation profiles, which have shown promise as biomarkers for disease diagnosis15,29,34,35. Thus, the TCI approach allows for the simultaneous analysis of tissue contribution and fragmentomic characteristics to enhance the liquid biopsy applications.
Our simulation data revealed that TCI is applicable to both paired-end and single-end sequencing data. Notably, we observed a strong linear correlation (Pearson’s correlation r = 0.7) between placenta TCI and fetal fraction in plasma cfDNA at sequencing amounts of 50 million reads (about 0.6-fold coverage depth for SE 35 bp), indicating the method’s suitability for cost-effective, low-depth sequencing applications. Furthermore, our approach offers notable scalability. Researchers can adapt it to specific populations or tissues of interest. For instance, establishing age-specific TCI reference ranges for elderly populations, or incorporating epithelial cell analysis in COVID-19 studies. Moreover, TCI calculations are based on relatively small genomic regions surrounding the TSSs of tissue-specific genes, comprising only 2–3% of the human genome. This feature allows for the potential use of targeted capture sequencing, further improving cost-effectiveness. We further speculate that coverage patterns at enhancers and transcription factor binding sites (TFBS) may similarly enhance tissue-specific cfDNA tracking. Future studies could integrate these regulatory regions to improve tissue-of-origin detection.
Despite these promising results, our study has several limitations. First, this study adopted bulk RNA sequencing data to identify tissue-specific genes. With the complexity of tissue heterogeneity, it would be interesting to explore further to ascertain whether the accuracy of the TCI could be improved by using SH-genes from single-cell RNA sequencing data. Second, our reference intervals were established using a cohort of 460 Chinese individuals from Shenzhen. Geographical and ethnic diversity or different laboratory protocols may impact the generalizability of reference TCI intervals in different datasets. For instance, samples from Cristiano et al. demonstrate a notable difference in the plasma cfDNA fragment size profile compared to the physical examination cohort in this study, characterized by an increase in 120 - 166 bp fragments and the absence of a dinucleosomal peak at ~330 bp (Supplementary Fig. 7). Previous research has indicated that dinucleosomal fragments are enriched around transcription factor binding sites of highly expressed genes36. Given that fragment length distribution varies with experimental or data preprocess methods, re-establishing reference intervals is recommended. Lastly, the current TCI quantifies relative tissue contributions to cfDNA but cannot compare contributions across tissue types. Future studies could leverage enzymatic methyl-seq37, which preserves both methylation and fragmentomic features, to measure absolute tissue contribution percentages. Subsequently, machine learning models could be trained to map TCI values to these absolute percentages, enabling next-generation TCI methods to directly output accurate tissue contribution estimates.
In summary, we have developed a promising tool for assessing tissue contributions in cfDNA that is compatible with both single-end and paired-end whole-genome sequencing data, even at low sequencing depths. This approach offers a fast and cost-effective method for integrating tissue contribution and fragmentation pattern analysis, potentially enhancing liquid biopsy applications in various clinical settings.
Methods
Subjects and Ethics
For the pregnant women, 101 plasma samples were collected from 58 pregnant women in Shenzhen, China. For COVID-19 patients, 194 plasma samples were collected from COVID-19 subjects in Wuhan, China, from March to April 2020. These COVID-19 patients were diagnosed through real-time fluorescent quantitative polymerase chain reaction (qPCR). For the physical examination cohort, 460 plasma samples were collected from 460 healthy individuals during their routine physical examinations in Shenzhen, China in 2022. Inclusion criteria of the physical examination cohort required adult age, no malignant tumor history, and no active infectious diseases. Institutional Review Board (IRB) approval was obtained from BGI-IRB (BGI-IRB 24073) to use sequencing data housed at BGI-Research (BGI-IRB 20020, BGI-IRB 17087, and BGI-IRB 21157 for COVID-19 patients, pregnant women, and physical examination cohort, respectively). All participants provided written informed consent. All ethical regulations relevant to human research participants were followed in this study.
Public plasma cfDNA data collection
We obtained plasma cfDNA data from FinaleDB30, including Jiang et al. (healthy samples: n = 32, cancer samples: n = 90)29, Sun et al. (liver transplant samples: n = 13)19, Cristiano et al. (healthy samples: n = 262, cancer samples: n = 276)32, and Snyder et al. (healthy samples: n = 4, cancer samples: n = 48)18. The plasma cfDNA data from Liang et al. (healthy samples: n = 10, cancer samples: n = 20)33 were obtained from China National GeneBank (CNGB) Nucleotide Sequence Archive (CNSA, CNP0000680). The 76 plasma DNA data of bone marrow transplant recipients were acquired from Sequence Read Archive (SRA, SRP093037)23. From Jiang et al.‘s 90 cancer samples29, we retained 74 cases with tumor TF > 0%, and among Sun et al.‘s 13 liver transplant recipients19, sample EE85745 was excluded due to discordance between calculated and originally reported liver OCF values. Among the 76 bone marrow transplant recipient plasma DNA samples, we excluded six samples (SRR5006888, SRR5006890, SRR5006919, SRR5006924, SRR5006868, and SRR5006915) for failing quality control. We additionally removed 16 samples collected either pre-transplantation or within 2 days post-transplantation.
Blood sample processing, cfDNA extraction, and DNA sequencing
Plasma samples were prepared within 6 hours after blood sampling by initially centrifuging from blood samples at 1600 g for 10 minutes at 4 °C. A second centrifugation at 16,000 g for 10 minutes at 4 °C was performed to remove the impurities. Genomic DNA was extracted from plasma using the MGIEasy Circulating DNA Isolation Kit (MGI, Shenzhen, China). Libraries were prepared using the MGI Easy Cell-free DNA Library Prep Kit (1000012701, MGI) as described previously38. Paired-end 100 bp sequencing data were produced from these libraries on the DNBSEQ platform.
Sequencing Data Processing
The quality of sequencing data was evaluated based on three dimensions: GC content, proportion of N bases, and sequencing quality (Supplementary Fig. 8). The raw FASTQ data were preprocessed using Fastp (v 0.23.4)39 to trim sequencing adapters and filter low-quality reads (containing >10 “N” bases or Phred scores < 5). Quality-filtered reads were then aligned to the human reference genome (GRCh38/hg38) using Minimap2 (v 2.28)40, with duplicate reads marked by Samblaster (v 0.1.26)41. Final alignments were sorted and converted to BAM format using Samtools (1.21)42. In BED files, we retained only paired-end reads that were properly aligned to the same chromosome, had the correct orientation, were non-duplicated, and featured an insertion length of less than 600 base pairs. The median effective cfDNA fragments were 398 million (range: 107 - 699 million) for pregnant women, 257 million (15 - 755 million) for COVID-19 patients, and 454 million (236 - 1095 million) for the physical examination cohort.
GC Content Bias Correction
We applied a modified Griffin43 algorithm to correct GC bias in all BED files (from FinaleDB or generated in this study). The adjusted coverage for each cfDNA fragment accounted for fragment-level GC bias. The modified code is publicly available on GitHub (https://github.com/lingguoli/TCI).
Identification of tissue-specific highly expressed genes
The gene expression data for various tissues, including whole blood (n = 755), lung (n = 578), breast (n = 459), artery (n = 432), heart (n = 429), colon (n = 406), stomach (n = 359), pancreas (n = 328), liver (n = 226), ovary (n = 180), and kidney (n = 85), were obtained from the Genotype-Tissue Expression Portal (https://storage.cloud.google.com/adult-gtex/bulk-gex/v8/rna-seq/GTEx_Analysis_2017-06-05_v8_RNASeQCv1.1.9_gene_tpm.gct.gz)44. Additionally, gene expression data for protein-coding genes in the placenta (n = 324) were obtained from the Pregnancy Outcome Prediction Study45. The mean gene expression levels were quantified by calculating the Transcripts Per Million (TPM) for each tissue. A stringent filtering process was applied to identify tissue-specific highly expressed genes. Gene expressions in 12 tissues were ranked in descending order based on their TPM values. Two ratios were calculated:
-
1)
SH_1: The ratio between (a) the difference of the maximum TPM and the upper quartile TPM, and (b) the difference between the upper and lower quartile TPM.
-
2)
SH_2: The ratio between (a) the difference of the maximum TPM and the second-highest TPM, and (b) the maximum TPM.
A lower limit of 10 was set for SH_1 to ensure the gene was expressed substantially higher in one tissue compared to the others. For SH_2, a lower limit of 0.3 was established, guaranteeing that the gene expression in one tissue was at least 42.86% higher than in any other tissue. Genes were classified as potential SH-genes only if they satisfied both SH_1 and SH_2 threshold criteria. Among these genes, the top 500 genes with high TPM in whole blood and the top 250 genes with high TPM for each of the remaining tissues were selected as blood SH-genes and SH-genes for other tissues, respectively.
Calculation of Tissue Contribution Index
We defined the TSS region as the upstream 1000 bp and downstream 1000 bp surrounding the TSS. For paired-end data analysis, we employed the intersect module of BEDTools (v 2.25.0)46 to count DNA fragments between 150 to 210 bp47 that overlapped with the TSS regions. The coverage of each DNA fragment has been adjusted based on the GC bias matrix. The total number (GC bias corrected) of base pairs located in each TSS region was considered the TSS coverage.
To standardize the TSS coverage, we calculated the total base count of sequenced cfDNA located across all 38,865 TSSs located on the autosomes. We then normalized the base count for each TSS as Base Counts Per Million mapped bases (BCPM) as follows:
Here, BCPM(i) represents the base counts per million mapped bases for the i-th TSS; Base count(i) denotes the raw base pair count for that TSS.
We identified SH-genes among the 12 tissues, noting that the BCPM of SH-gene is negatively correlated with tissue contribution. With an average BCPM of approximately 25 (i.e., 106/38,865), we calculated the TCI as the negative value of the mean BCPM of SH-genes plus 50 to ensure a positive correlation with actual cfDNA tissue contributions.
BCPM(i,t) refers to the BCPM of the i-th TSS in the t-th tissue, k is the total number of TSSs of SH-genes in each tissue, and TCI(t) represents the tissue contribution index for the t-th tissue.
For single-end data analysis, the total number of mapped reads within a TSS region was defined as the TSS reads count. Afterwards, the TSS reads count was normalized as Reads Per Million mapped reads (RPM) as follows:
Here, RPM(i) represents the reads per million mapped reads for the i-th TSS; TSS reads count (i) denotes the reads count for the i-th TSS.
Similarly, we calculated the mean RPM of the TSSs of these SH-genes in each tissue to define a tissue contribution index as follows:
TCI(t) represents the tissue contribution index for the t-th tissue. k denotes the total number of TSSs of SH-genes in each tissue. RPM(i,t) refers to the reads per million mapped reads of the i-th TSS in the t-th tissue.
Calculation of fetal fraction
Fetal DNA fractions in maternal plasma were calculated following Hudecova et al.‘s method48. First, we determined the proportion of chrY-mapped reads as Y(i) for each plasma sample i:
We then analyzed plasma samples from healthy males and females to determine chrY read proportion in males (\(\bar{Y}\)(male)) and the average background noise in females (\(\bar{Y}\)(female)). The fetal fraction (FF) for each sample s was calculated using the formula:
Statistics and reproducibility
The computation and visualization of correlation coefficients, area under the curve (AUC), Operating Characteristic (ROC) curve, t-Distributed Stochastic Neighbor Embedding (t-SNE), box plots, line plots, and scatter plots were all performed using custom Python scripts and Python packages. A p value below 0.05 was considered statistically significant. In the figures, the symbols ‘*‘, ‘**‘, ‘***‘, and ‘****‘ denote p values below 0.05, 0.01, 0.001, and 0.0001, respectively. The p values from multi-comparison were corrected by the Bonferroni approach.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The BED format data of pregnant women and COVID-19 patients have been deposited into China National GeneBank (CNGB) Nucleotide Sequence Archive (CNSA, https://db.cngb.org/cnsa/) with accession numbers CNP0005249 (DOI: 10.26036/CNP0005249) and CNP0005212 (DOI: 10.26036/CNP0005212), respectively. The following public datasets were also used: GTExv8 TPM values for bulk tissues (https://storage.cloud.google.com/adult-gtex/bulk-gex/v8/rna-seq/GTEx_Analysis_2017-06-05_v8_RNASeQCv1.1.9_gene_tpm.gct.gz), transcriptome data of placenta (https://github.com/sung/POPS-Placenta-Transcriptome-2020/tree/master/Data/placenta.T3.rZ). The numerical source data for graphs is available in Supplementary Data 1.
Code availability
Implementation codes for the TCI method have been uploaded to GitHub (https://github.com/lingguoli/TCI).
References
Sun, K. et al. Plasma DNA tissue mapping by genome-wide methylation sequencing for noninvasive prenatal, cancer, and transplantation assessments. Proc. Natl. Acad. Sci. USA 112, E5503–E5512 (2015).
Lo, Y. M. D. Non-invasive prenatal diagnosis by massively parallel sequencing of maternal plasma DNA. Open Biol. https://doi.org/10.1098/rsob.120086 (2012).
Chen, J. A. et al. Sensitive detection of plasma/serum DNA in patients with systemic lupus erythematosus. Autoimmunity https://doi.org/10.1080/08916930701356317(2007).
Lam, N. Y. L., Rainer, T. H., Chan, L. Y. S., Joynt, G. M. & Lo, Y. M. D. Time course of early and late changes in plasma DNA in trauma patients. Clin. Chem. https://doi.org/10.1373/49.8.1286 (2003).
Andargie, T. E. et al. Cell-free DNA maps COVID-19 tissue injury and risk of death and can cause tissue injury. JCI Insight https://doi.org/10.1172/jci.insight.147610 (2021).
Schütz, E. et al. Graft-derived cell-free DNA, a noninvasive early rejection and graft damage marker in liver transplantation: A prospective, observational, multicenter cohort study. PLoS Med. https://doi.org/10.1371/journal.pmed.1002286 (2017).
Jiang, P. et al. FetalQuant: Deducing fractional fetal DNA concentration from massively parallel sequencing of DNA in maternal plasma. Bioinformatics https://doi.org/10.1093/bioinformatics/bts549 (2012).
Moss, J. et al. Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nat. Commun. 9, 1–12 (2018).
Loyfer, N. et al. A DNA methylation atlas of normal human cell types. Nature 613, 355–364 (2023).
Li, S. et al. Comprehensive tissue deconvolution of cell-free DNA by deep learning for disease diagnosis and monitoring. Proc. Natl. Acad. Sci. USA https://doi.org/10.1073/pnas.2305236120 (2023).
Tanaka, K. & Okamoto, A. Degradation of DNA by bisulfite treatment. Bioorg. Med. Chem. Lett. https://doi.org/10.1016/j.bmcl.2007.01.040 (2007).
Shen, L., Guo, Y., Chen, X., Ahmed, S. & Issa, J.-P. J. Optimizing Annealing Temperature Overcomes Bias in Bisulfite PCR Methylation Analysis. BioTechniques 42, 48–58 (2007).
Yu, S. C. Y. et al. Size-based molecular diagnostics using plasma DNA for noninvasive prenatal testing. Proc. Natl. Acad. Sci. USA 111, 8583–8588 (2014).
Sun, K. et al. Size-tagged preferred ends in maternal plasma DNA shed light on the production mechanism and show utility in noninvasive prenatal testing. Proc. Natl. Acad. Sci. USA. https://doi.org/10.1073/pnas.1804134115 (2018).
Jiang, P. et al. Plasma DNA end-motif profiling as a fragmentomic marker in cancer, pregnancy, and transplantation. Cancer Discov. 10, 664–673 (2020).
Jiang, P. et al. Detection and characterization of jagged ends of double-stranded DNA in plasma. Genome Res. 30, 1144–1153 (2020).
Ulz, P. et al. Inferring expressed genes by whole-genome sequencing of plasma DNA. Nat. Genet. https://doi.org/10.1038/ng.3648 (2016).
Snyder, M. W., Kircher, M., Hill, A. J., Daza, R. M. & Shendure, J. Cell-free DNA Comprises an in Vivo Nucleosome Footprint that Informs Its Tissues-Of-Origin. Cell 164, 57–68 (2016).
Sun, K. et al. Orientation-aware plasma cell-free DNA fragmentation analysis in open chromatin regions informs tissue of origin. Genome Res. https://doi.org/10.1101/gr.242719.118 (2019).
Chen, X. et al. Transcriptional Start Site Coverage Analysis in Plasma Cell-Free DNA Reveals Disease Severity and Tissue Specificity of COVID-19 Patients. Front. Genet. https://doi.org/10.3389/fgene.2021.663098 (2021).
Zhu, G. et al. Tissue-specific cell-free DNA degradation quantifies circulating tumor DNA burden. Nat. Commun. 12, 1–11 (2021).
Chim, S. S. C. et al. Detection of the placental epigenetic signature of the maspin gene in maternal plasma. Proc. Natl. Acad. Sci. USA https://doi.org/10.1073/pnas.0503335102 (2005).
Sharon, E. et al. Quantification of transplant-derived circulating cell-free DNA in absence of a donor genotype. PLoS Comput. Biol. https://doi.org/10.1371/journal.pcbi.1005629 (2017).
Wu, T. et al. Multi-organ Dysfunction in Patients with COVID-19: A Systematic Review and Meta-analysis. Aging Dis. 11, 874 (2020).
Bai, Y. et al. Integrating plasma cell-free DNA with clinical laboratory results enhances the prediction of critically ill patients with COVID-19 at hospital admission. Clin. Transl. Med. 12, 1–6 (2022).
Liang, H. et al. Elevated levels of pre-treatment lactate dehydrogenase are an unfavorable predictor factor in patients with EML4-ALK rearrangement non-small cell lung cancer treated with crizotinib. Cancer Manag. Res. https://doi.org/10.2147/CMAR.S213572 (2019).
Gao, P. et al. The effect of hepatitis B virus on T lymphocyte and its subsets in chronic hepatitis B patients in different ALT stages: A new concept ALT in HBV infection. Int. Immunopharmacol. https://doi.org/10.1016/j.intimp.2021.108182 (2021).
Kustanovich, A., Schwartz, R., Peretz, T. & Grinshpun, A. Life and death of circulating cell-free DNA. Cancer Biol. Ther. 20, 1057–1067 (2019).
Jiang, P. et al. Lengthening and shortening of plasma DNA in hepatocellular carcinoma patients. Proc. Natl. Acad. Sci. USA 112, E1317–E1325 (2015).
Zheng, H., Zhu, M. S. & Liu, Y. FinaleDB: A browser and database of cell-free DNA fragmentation patterns. Bioinformatics https://doi.org/10.1093/bioinformatics/btaa999 (2021).
Adalsteinsson, V. A. et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat. Commun. 8, 1–13 (2017).
Cristiano, S. et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature 570, 385–389 (2019).
Liang, H. et al. Whole-genome sequencing of cell-free DNA yields genome-wide read distribution patterns to track tissue of origin in cancer patients. Clin. Transl. Med. https://doi.org/10.1002/ctm2.177 (2020).
Moldovan, N. et al. Genome-wide cell-free DNA termini in patients with cancer. medRxiv 2021 09, 21264176 (2021).
Zhou, Z. et al. Fragmentation landscape of cell-free DNA revealed by deconvolutional analysis of end motifs. Proc. Natl. Acad. Sci. USA 1, 11 (2023).
Lazzeri, I., Spiegl, B. G., Hasenleithner, S. O., Speicher, M. R. & Kircher, M. LBFextract: Unveiling transcription factor dynamics from liquid biopsy data. Comput. Struct. Biotechnol. J. 23, 3163–3174 (2024).
Bie, F. et al. Multimodal analysis of cell-free DNA whole-methylome sequencing for cancer detection and localization. Nat. Commun. 14, 1–13 (2023).
Xu, Y. et al. A new massively parallel nanoball sequencing platform for whole exome research. BMC Bioinformatics https://doi.org/10.1186/s12859-019-2751-3 (2019).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. in Bioinformatics https://doi.org/10.1093/bioinformatics/bty560 (2018).
Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics https://doi.org/10.1093/bioinformatics/bty191 (2018).
Faust, G. G. & Hall, I. M. SAMBLASTER: Fast duplicate marking and structural variant read extraction. Bioinformatics 30, 2503–2505 (2014).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics https://doi.org/10.1093/bioinformatics/btp352 (2009).
Doebley, A. L. et al. A framework for clinical cancer subtyping from nucleosome profiling of cell-free DNA. Nat. Commun. 13, 1–18 (2022).
Lonsdale, J. et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 45, 580–585 (2013).
Gong, S. et al. The RNA landscape of the human placenta in health and disease. Nat. Commun. https://doi.org/10.1038/s41467-021-22695-y (2021).
Quinlan, A. R. & Hall, I. M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics https://doi.org/10.1093/bioinformatics/btq033 (2010).
Esfahani, M. S. et al. Inferring gene expression from cell-free DNA fragmentation profiles. Nat. Biotechnol. https://doi.org/10.1038/s41587-022-01222-4 (2022).
Hudecova, I. et al. Maternal plasma fetal DNA fractions in pregnancies with low and high risks for fetal chromosomal aneuploidies. PLoS ONE https://doi.org/10.1371/journal.pone.0088484 (2014).
Acknowledgements
We gratefully acknowledge the participants for their generous contribution of blood samples to this research. We also thank the Core Facilities of BGI research and China National GeneBank (CNGB) for their technical support. This work has been supported by the National Natural Science Foundation of China (32171441 and 32000398) and the National Key R&D Program of China (2022YFC2502402). Some elements were created using BioRender.com.
Author information
Authors and Affiliations
Contributions
L.L., H.Z., and X.J. designed the research. Y.G. and F.S. collected the blood samples. F.S., Y.Z., and Z.T. performed DNA extraction and library construction. L.L. analyzed the sequencing data. L.L. and H.Z. wrote the paper. X.J., X. L., Y.L., and J.S. Revised the paper. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Benjamin Spiegel and the other anonymous reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Ana Rita Grosso and Mengtan Xing. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, L., Li, X., Li, Y. et al. Tracing the tissue origin of cell-free DNA through open chromatin footprint. Commun Biol 8, 1845 (2025). https://doi.org/10.1038/s42003-025-09232-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-09232-z
