
Abstract
Non-coding RNAs, including piwi-interacting RNAs (piRNAs), are known to regulate osteogenic differentiation in bone marrow-derived mesenchymal stem cells (BMSCs); their role in mesenchymal stem cells (MSCs) from diverse origins remains unclear. In this study, we identified piR48444 as a key regulator that is downregulated during the osteogenic differentiation of stem cells from exfoliated deciduous teeth (SHED) but is upregulated in inflamed and aged BMSCs. Functionally, piR48444 inhibited, while its knockdown enhanced osteogenic differentiation across MSCs from multiple sources. Notably, piR48444-depleted MSCs exhibited superior bone defect repair capacity. PiR48444 antagomir promoted bone regeneration in LPS-induced osteolysis mice and aging mice. Mechanistically, we demonstrated that piR48444 targets METTL7A, suppressing BMP2 mRNA m6A methylation. Furthermore, we discovered that the METTL7A/eIF4E complex binds to BMP2 mRNA, thereby enhancing its translational efficiency. Our findings establish piR48444 as a negative regulator of MSC osteogenesis through METTL7A-mediated BMP2 m6A methylation, highlighting its potential as a therapeutic target to enhance MSC-based bone regeneration strategies.

Similar content being viewed by others


Introduction
Mesenchymal stem cells (MSCs) are stromal cells capable of self-renewal and multilineage differentiation1. Isolated from various tissues, including bone marrow, adipose tissue, umbilical cord, periosteum, and dental pulp-MSCs can differentiate into osteogenic, adipogenic, chondrogenic, neurogenic, and myogenic lineages2. Due to these properties, MSCs are widely used as seed cells for regenerating tissues such as bone, cartilage, nerve, and muscle3. Different MSCs exhibit distinct advantages in bone regeneration, yet there is no consensus on the optimal source for bone tissue engineering. Additionally, the osteogenic potential of MSCs is influenced by complex in vivo conditions, such as inflammation and aging. Therefore, understanding the common regulatory mechanisms governing osteogenic differentiation across MSC types is crucial, particularly for applications such as bone defect repair, osteoporosis treatment, and addressing related bone loss.
Piwi-interacting RNAs (piRNAs) are small non-coding RNAs (24–32 nucleotides) characterized by 2ʹ-O-methyl-modified 3ʹ termini. Unlike microRNAs (miRNAs) and small interfering RNAs (siRNAs), piRNAs guide PIWI-clade Argonaute proteins and are generated through the ping-pong cycle and self-amplification mechanisms4,5,6. The PIWI-piRNA complex silences transposable elements and post-transcriptionally regulates protein-coding mRNAs7. Functionally, piRNAs modulate key processes, including spermatogenesis, wound healing, and tumorigenesis8,9,10. Emerging evidence highlights their role in MSCs biology. piRNAs are differentially expressed in exosomes from bone marrow-derived MSCs (BMSCs) and apical papilla-derived MSCs9. Specific piRNAs, such as piR36741, promote osteogenic differentiation of BMSCs by enhancing BMP2 expression via METTL3-mediated DNA methylation, thereby mitigating osteoporosis11. Conversely, piR63049 is upregulated in osteoporosis and inhibits osteogenesis by suppressing the Wnt2b/β-catenin pathway12,13,14. However, the pan-MSC regulatory role of piRNAs in osteogenesis remains unexplored. Notably, no studies have identified piRNAs that simultaneously modulate osteogenic differentiation across MSC types or their implications in bone defect repair, aging, or LPS-induced bone loss.
In this study, we sought to elucidate the regulatory role of piRNAs in the osteogenic differentiation of MSCs and their underlying mechanisms. Through piRNA sequencing, we identified piR48444 as a key regulator negatively modulating osteogenesis across MSCs derived from multiple tissue sources. Strikingly, inhibition of piR48444 enhanced bone defect repair and mitigated LPS-induced and aging-associated bone loss, demonstrating its therapeutic potential. We unraveled that piR48444 inhibits the osteogenic differentiation and bone regeneration of MSCs by targeting METTL7A/eIF4E-mediated BMP2 m6A methylation and translation. These findings establish piR48444 as a novel molecular target to augment MSC-based bone regeneration strategies.
Results
PiR48444 was related to the osteogenic differentiation of MSCs
When cultured in an osteogenic medium, SHED exhibited enhanced matrix mineralization compared to those in a proliferation medium (Fig. 1A). This was accompanied by significantly elevated expression of osteogenic markers ALP and RUNX2 (Fig. 1B). To investigate the underlying mechanisms, we performed piRNA and mRNA sequencing on SHED cultured for 7 days in an osteogenic vs. proliferation medium. The osteogenic group showed 907 upregulated and 500 downregulated piRNAs (Fig. S1A, C), along with 699 upregulated and 659 downregulated mRNAs (Fig. S1B, D). Gene Ontology (GO) analysis linked the differentially expressed genes (DEGs) to processes such as, cell adhesion and neural development (synapse organization, telencephalon development). KEGG pathway analysis further highlighted enrichment in cytokine-cytokine receptor interaction, axon guidance, platelet activation, and cell adhesion molecules (Fig. S1E, F).

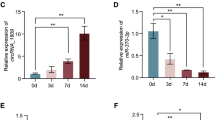
A Alizarin red staining at day 14. B Expression pattern of osteogenic differentiation markers at day 7. C The correlation analysis heatmap of differentially expressed piRNAs and mRNAs in SHED at day 7 of osteogenic differentiation. D Differentially expressed piRNAs targeting BMP2 in SHED. E The bioinformatic expression pattern of piR48444. F The RT-qPCR analysis of piR48444 in different MSCs (G), LPS-treated BMSCs (H), and aged BMSCs (I). The bioinformatics analysis of piR48444 expression in mononuclear cells from peripheral blood (n = 3) (GSE159121). J RT-qPCR analysis of piR48444 expression in plasma (n = 15) of osteoporosis patients. K The cytoplasm and nucleus separation analysis of piR48444 in the nucleus and cytoplasm. L The piR48444 probe staining. M The binding of piR48444 with PIWIL1 protein. N The expression level of piR48444 in PIWIL1-KD SHED. Data in (B, E–J, L, N) presented as mean ± SD; statistical analysis via Student’s t test for two-group comparisons; significance levels: *p < 0.05, **p < 0.01 and ***p < 0.001. PM proliferation medium; OM osteogenic medium.
Through correlation analysis of differentially expressed piRNAs and their predicted target genes (|fold change| ≥ 2), we identified strong negative correlations (r < −0.9) (Fig. 1C). Notably, piR48444 demonstrated significant downregulation during SHED osteogenic differentiation. Given the established critical role of BMP2 in osteogenic differentiation across MSC types, we specifically analyzed piRNAs correlated with BMP2 expression. Among downregulated piRNAs, piR48444 exhibited the strongest negative correlation with BMP2 mRNA levels (r = −0.98022) (Fig. 1D), suggesting its potential as a key regulator of BMP2-mediated osteogenesis. Furthermore, we confirmed that the expression of piR-1917696 and piR361914 was downregulated during osteogenic differentiation, while knocking down these piRNAs did not influence the ALP staining of SHED (Fig. S2A–C).
Bioinformatics analysis confirmed the downregulation of piR48444 during osteogenic differentiation (Fig. 1E). Subsequent RT-qPCR validation revealed consistent downregulation of piR48444 across multiple MSC types, including SHED, BMSCs, periodontal ligament stem cells (PDLSCs), and adipose stem cells (ASCs) (Figs. 1F). Notably, we observed an inverse relationship between piR48444 and osteogenic markers in pathological conditions. While osteogenic markers were suppressed in LPS-treated and aged BMSCs (Fig. S3A, B), piR48444 showed significant upregulation in these models (Fig. 1G, H). Clinical correlation studies demonstrated elevated piR48444 levels in osteoporosis patients. Bioinformatics analysis of peripheral blood mononuclear cells revealed a non-significant upward trend (Fig. 1I). Meanwhile, direct measurement in patient plasma samples showed a 2.87-fold increase compared to healthy controls (Fig. 1J), suggesting its potential as a diagnostic biomarker. Subcellular localization analysis in SHED cells revealed the predominant cytoplasmic distribution of piR48444, as determined by cytoplasmic/nuclear separation and probe staining (Fig. 1K, L), providing important context for its potential mechanism of action.
The PIWI-piRNA complex is known to play crucial regulatory roles in gene expression7. Our miRNA pulldown assays specifically identified PIWIL1, but not PIWIL2 or PIWIL4, as the direct binding partner of piR48444 (Fig. 1M). Supporting this functional relationship, PIWIL1 knockdown resulted in significantly reduced piR48444 levels (Fig. 1N). These results collectively demonstrate that piR48444, associated with PIWIL1, functions as a negative regulator of osteogenic differentiation across multiple MSC types and may contribute to bone loss mechanisms.
PiR48444 knockdown promoted osteogenic differentiation of MSCs from different sources
PiR48444 knockdown consistently enhanced osteogenic differentiation across multiple MSC types, including SHED, BMSCs, PDLSCs, and ASCs, as demonstrated by increased ALP activity and ARS staining, calcium deposition, and upregulation of osteogenic markers (ALP, RUNX2, OCN, COL1A) (Figs. 2A, C, E, F, I, J, M, N, and S4A–C). Conversely, piR48444 overexpression suppressed these osteogenic effects in all tested MSCs (Fig. 2B, D, G, H, K, L, O, and P), confirming its role as a conserved negative regulator of osteogenesis in MSCs from diverse origins.

A–D SHEDs were transfected with a piR48444 knockdown (piR48444-KD) or overexpression (piR48444-OE) construct. A, B Representative images (left) and quantification (right) of ALP staining at day 7 and Alizarin Red S (ARS) staining at day 14. C, D mRNA expression levels of the osteogenic markers ALP, RUNX2, OCN, COL1A1, and BSP at day 7, measured by RT-qPCR. E–P The effects of piR48444 modulation were validated in BMSCs (E–H), PDLSCs (I–L), and ASCs (M–P). E, I, M Representative ALP staining at day 7 following piR48444-KD. F, J, N Osteogenic gene expression at day 7 following piR48444-KD. G, K, O Representative images and quantification of ALP staining at day 7 following piR48444-OE. H, L, P Osteogenic gene expression at day 7 following piR48444-OE. All quantitative data are presented as mean ± SD (n = 3 independent experiments). Statistical significance was determined by a two-tailed Student’s t test (*p < 0.05, **p < 0.01, ***p < 0.001). KD knockdown, OE overexpression.
PiR48444-knockdown in MSCs promoted calvarial bone defect repair
PiR48444-knockdown SHED demonstrated significantly enhanced bone regeneration capacity in nude mouse calvarial defect models. Micro-CT analysis revealed substantially more new bone formation in defects treated with piR48444-knockdown SHED cell sheets compared to controls (Fig. 3A, B), with 3.34-fold higher BV/TV, 2.75-fold greater Tb.N, and 1.26-fold increased Tb.Th (Fig. 3C). Histological examination further confirmed enhanced bone formation in the knockdown group (Fig. 3D, E). When delivered via hydrogel, piR48444-knockdown SHED maintained this superior regenerative capacity (Fig. S5A–C). These findings collectively demonstrate the therapeutic potential of piR48444-modulated SHED for bone regeneration applications.

A Representative MicroCT images. B MicroCT images in vertical view. C Bone volume/total volume (BV/TV), trabecular number (Tb.N), trabecular thickness (Tb.Th), and trabecular separation (Tb.Sp) (n = 6). D H&E staining. E Masson staining. Data in (C) are presented as mean ± SD; statistical analysis via Student’s t test for two-group comparisons. Significance levels: **p < 0.01. NB new bone.
PiR48444 inhibition protected against LPS-induced and aging-related trabecular bone loss
PiR48444 inhibition demonstrated significant therapeutic potential in protecting against LPS-induced and aging-related trabecular bone loss, as evidenced by micro-CT and histopathological analyses showing that piR48444 antagomir treatment effectively rescued LPS-induced trabecular bone loss in both male and female mice (Fig. 4A, B), while significantly improving key bone parameters, including increased BMD, BV/TV, and Tb.N along with decreased Tb.Sp (Fig. 4C). Importantly, this protective effect was specific to trabecular bone, with no observed impact on cortical bone parameters (Fig. S6A, B).

A HE staining in LPS-treated male and female mice. B Representative micro-CT images for trabecular bone in LPS-treated male and female mice. C Bone mineral density (BMD), bone volume/total volume (BV/TV), trabecular thickness (Tb.Th), trabecular number (Tb.N), and trabecular separation (Tb.Sp) in LPS-treated male mice and female mice (n = 8). D HE staining in 18-month-old male and female mice. E Representative micro-CT images for trabecular bone in 18-month-old male mice and female mice (n = 6). F Bone mineral density (BMD), bone volume/total volume (BV/TV), trabecular thickness (Tb.Th), trabecular number (Tb.N), and trabecular separation (Tb.Sp) in male mice and female mice (n = 6). Data are presented as mean ± SD (C, F). Statistical significance was determined by one-way ANOVA (C) and Student’s t test (F). Significance levels: *p < 0.05 and **p < 0.01, and ***p < 0.001.
PiR48444 antagomir treatment significantly ameliorated age-related trabecular bone loss in both male and female 18-month-old mice, as demonstrated by improved trabecular bone structure (Fig. 4D, E). MicroCT analysis revealed increased trabecular BMD and BV/TV with decreased Tb.Sp in aging males, while treated females showed significant increases in Tb.N and non-significant improvements in BMD, BV/TV and Tb.Sp (Fig. 4F). Notably, cortical bone parameters remained unaffected by piR48444 antagomir treatment in aging mice (Fig. S6C, D), confirming the specific protective effect of piR48444 inhibition on trabecular bone loss during aging.
PiR48444 inhibited SHED osteogenic differentiation by targeting METTL7A mRNA
The regulatory mechanism of piR48444 on osteogenic differentiation was further revealed. Our results demonstrate that piR48444 negatively regulates the osteogenic differentiation of SHED by targeting METTL7A and BMP2, as predicted by bioinformatic analysis (Fig. 5A). During osteogenic induction, piR48444 expression was inversely correlated with METTL7A and BMP2 mRNA levels (Fig. 5B), both of which were upregulated during differentiation (Fig. 5C, D). Functional studies revealed that piR48444 overexpression suppressed, while its knockdown enhanced, METTL7A and BMP2 protein expression (Fig. 5E, F) and increased their staining intensity in vivo (Fig. 5G). The predicted binding sites of piR48444 on METTL7A and BMP2 were shown in Fig. S7. Luciferase reporter assays confirmed direct binding of piR48444 to the 3’UTR of METTL7A (Fig. 5H, left panel), but not BMP2, suggesting that piR48444 directly regulates METTL7A while indirectly modulating BMP2 expression during SHED osteogenic differentiation. These findings identify piR48444 as a novel repressor of osteogenesis through its interaction with METTL7A.

A Predicated target mRNAs of piR48444 in SHED. B PiR48444 expression negatively correlated with METTL7A, BMP2 expression (n = 3). C, D The expression pattern and RT-qPCR analysis of METTLA, BMP2. E, F METTL7A and BMP2protein expression patterns in piR48444-OE and piR48444-KD SHED (n = 3). G IHC for METTL7A and BMP2 in piR48444 knockdown cell sheets for calvarial defect repair. H Luciferase reporter assay validation of piR48444 targets METTL7A and BMP2 (n = 4). Data are presented as mean ± SD. Statistical significance was determined by Student’s t test (C, D, F) and one-way ANOVA (H). Significance levels: *p < 0.05 and **p < 0.01, and ***p < 0.001. OE overexpression, KD knockdown, Wt wildtype, Mu mutant.
Our findings demonstrate that METTL7A plays a critical anabolic role in SHED osteogenic differentiation, as its overexpression led to a 9.57-fold increase in mRNA levels (Fig. 6A) and significantly enhanced ALP and ARS staining intensities (Fig. 6B), along with upregulated expression of osteogenic markers (ALP, RUNX2, and OCN; Fig. 6C). Conversely, METTL7A knockdown reduced ALP activity, matrix mineralization, and osteogenic gene expression (Fig. 6D–F). Importantly, METTL7A overexpression rescued the inhibitory effects of piR48444 on SHED osteogenic differentiation (Fig. 6G, H), confirming that piR48444 directly targets METTL7A to suppress osteogenesis. Together, these results establish METTL7A as a key promoter of SHED differentiation and reveal piR48444 as its functional repressor.

A Expression pattern of METTL7A in METTL7A-OE SHED. B Staining and quantification of alkaline phosphatase (ALP) staining at day 7 and alizarin red staining (ARS) at day 14. C ALP, RUNX2, and OCN mRNA expression pattern in METTL7A-OE SHED. D Expression pattern of METTL7A in METTL7A-KD SHED. E Staining and quantification of ALP staining at day 7 and ARS at day 14. F ALP, RUNX2, and OCN mRNA expression pattern in METTL7A-KD SHED. G, H Alkaline phosphatase (ALP) staining and quantification at day 7, and alizarin red staining (ARS) and quantification at day 14. Data are presented as mean ± SD. Statistical significance was determined by Student’s t test (A–F) and one-way ANOVA (H). Significance levels: *p < 0.05, **p < 0.01, and ***p < 0.001, n = 3. OE overexpression, KD knockdown.
METTL7A promoted BMP2 stabilization and translation by m6A methylation
To further elucidate the regulatory mechanism of METTL7A in SHED osteogenic differentiation, we investigated its potential role in modulating BMP2 expression. While METTL7A overexpression upregulated and its knockdown downregulated BMP2 protein levels (Fig. 7A, B), no significant changes were observed in BMP2 mRNA expression (Fig. 7C), suggesting post-transcriptional regulation. Given the established role of m6A methylation in mRNA translation, we explored whether METTL7A mediates this process. Although key m6A methyltransferases (METTL3, METTL14, METTL16, and WTAP) showed no upregulation during osteogenic differentiation (Fig. S8A, B). METTL7A itself exhibited increased expression in both cytoplasmic and nuclear compartments (Fig. 7D, E). Bioinformatic analysis predicted seven m6A modification sites on BMP2 mRNA (Fig. S9A, B), and RIP assays confirmed direct binding between METTL7A and BMP2 mRNA (Fig. 7F). Importantly, meRIP assays revealed that METTL7A overexpression enhanced m6A methylation at sites 2 and 3 of BMP2 mRNA by 2.88-fold, while METTL7A knockdown reduced methylation at these sites (Fig. 7G). No enrichment was detected at other predicted sites, indicating site-specific regulation. These findings demonstrate that METTL7A promotes BMP2 translation through m6A methylation, providing a mechanistic link between METTL7A and BMP2 during SHED osteogenic differentiation, independent of transcriptional regulation.

A, B METTL7A and BMP2 protein expression in METTL7A-OE and METTL7A-KD SHED. C BMP2 mRNA expression in METTL7A-OE and METTL7A-KD SHED. D Immunostaining and quantification for METTL7A in SHED. E METTL7A expression in the cytoplasm and nucleus. F The direct binding of METTL7A with BMP2 mRNA by RIP assay. G BMP2 m6A modification level in METTL7A-OE and METTL7A-KD SHED. H BMP2 mRNA stability in METTL7A-OE and METTL7A-KD SHED. BMP2 protein synthesis in METTL7A-OE (I, J) and METTL7A-KD (K, L) SHED. BMP2 protein degradation in METTL7A-OE (M, N) and METTL7A-KD (O, P) SHED. Data are presented as mean ± SD. Data are presented as mean ± SD. Statistical significance was determined by Student’s t test (B, C, D, F, G), two-way ANOVA (H), and one-way ANOVA (J, L, N, P). Significance levels: *p < 0.05, **p < 0.01, and ***p < 0.001, n = 3. OE overexpression, KD knockdown.
Our results demonstrate that METTL7A enhances BMP2 expression through m6A-mediated post-transcriptional regulation, as evidenced by increased BMP2 mRNA stability (Fig. 7H) and translational efficiency (Fig. 7I–L) without affecting protein degradation (Fig. 7M–P). Mechanistically, METTL7A binds directly to BMP2 mRNA (Fig. 7F) and specifically methylates sites 2 and 3 (Fig. 7G), with mutation of these sites abolishing the translational enhancement (Fig. S9C–F). Importantly, exogenous BMP2 rescued the impaired osteogenic differentiation caused by METTL7A knockdown (Fig. S9G), confirming the functional significance of this regulatory axis. Furthermore, we observed consistent upregulation of both METTL7A and BMP2 proteins upon piR48444 knockdown across multiple MSC types (Fig. S10A, B), establishing the piR48444-METTL7A-BMP2 pathway as a conserved mechanism regulating osteogenic differentiation. These findings collectively reveal METTL7A as a critical epigenetic regulator that promotes osteogenesis through m6A-mediated stabilization and translation of BMP2 mRNA, while piR48444 serves as an upstream negative regulator of this process.
METTL7A-eIF4E complex regulated piR48444-induced BMP2 protein expression
Our findings reveal that the METTL7A/eIF4E complex is pivotal in regulating piR48444-induced BMP2 protein expression during SHED osteogenic differentiation. Bioinformatics and RT-qPCR analysis showed that eIF4EBP1, an inhibitor of eIF4E, was significantly downregulated during osteogenesis (Fig. 8A, B) and further suppressed by piR48444 overexpression (Fig. 8C), while eIF4E expression remained unchanged (Fig. 8A–C), suggesting increased availability of eIF4E for translational initiation. Cluspro and PyMOL analysis confirmed METTL7A-eIF4E interaction (Fig. 8D), which was further validated by co-IP assays (Fig. 8E). Functional studies demonstrated that eIF4E overexpression enhanced osteogenic markers (ALP, RUNX2, OCN) and ALP activity (Fig. 8F–H), while METTL7A knockdown attenuated eIF4E’s pro-osteogenic effects (Fig. 8G, H). And eIF4E knockdown diminished METTL7A-mediated osteogenesis (Fig. 8I–K). Importantly, eIF4E overexpression increased BMP2 protein levels, whereas its knockdown reduced it (Fig. 8L, M), and exogenous BMP2 supplementation rescued impaired osteogenesis in eIF4E-deficient SHED (Fig. 8N, O). Collectively, these results demonstrate that piR48444 suppresses osteogenesis by inhibiting METTL7A, which, in complex with eIF4E, enhances BMP2 translation via m6A-mediated mechanisms, thereby driving osteogenic differentiation.

A, B Bioinformatic and RT-qPCR analysis of eIF4EBP1 and eIF4E in SHED during osteogenic differentiation. C The expression of eIF4EBP1 and eIF4E in piR48444-KD SHED. D The binding prediction analysis of METTL7A and eIF4E by ClusPro and PyMOL. E Co-IP analysis of the binding between METTL7A and eIF4E. F Expression pattern of eIF4E in eIF4E-OE SHED. G The expression levels of osteogenic genes, including ALP, RUNX2, and OCN, at day 7. H The staining of ALP and quantification at day 7. I Expression pattern of eIF4E in eIF4E-KD SHED at day 7. J The expression levels of osteogenic genes, including ALP, RUNX2, and OCN, at day 7. K The staining of ALP and quantification at day 7. L, M BMP2 expression and quantification in eIF4E-OE and eIF4E-KD SHED. N The expression levels of osteogenic genes, including ALP, RUNX2, and OCN, at day 7. O The staining of ALP and quantification at day 7. Data are presented as mean ± SD (n = 3). Statistical significance was determined by Student’s t test (A–C, F, I, M), and one-way ANOVA (G, H, J, K, N, O). Significance levels: *p < 0.05, **p < 0.01, and ***p < 0.001. OE overexpression, KD knockdown.
Discussion
MSCs serve as crucial candidates for stem cell-based bone tissue engineering and regeneration15,16, necessitating a deeper understanding of their osteogenic differentiation mechanisms to enhance therapeutic potential. While piRNAs have been implicated in regulating osteogenesis in bone marrow-derived MSCs12,13,14, their role across MSCs from diverse origins remains unexplored. Our study demonstrates that piR48444 is consistently downregulated during osteogenic differentiation in multiple MSC types, whereas it is upregulated in both inflamed and aged bone marrow MSCs, suggesting a context-dependent regulatory role. Functionally, overexpression of piR48444 inhibited, whereas its knockdown enhanced osteogenic differentiation across MSC sources. In vivo studies confirmed that piR48444 knockdown in SHED accelerated bone defect repair, and that systemic administration of piR48444 antagomir ameliorated LPS- and aging-induced bone loss, collectively establishing piR48444 as a negative regulator of osteogenesis. Mechanistically, we identified METTL7A as a direct target of piR48444, whose suppression by piR48444 impaired METTL7A-mediated m6A methylation of BMP2 mRNA, consequently reducing BMP2 mRNA stability and translation efficiency through disrupted METTL7A/eIF4E complex formation. These findings delineate a novel piR48444/METTL7A/BMP2 axis governing osteogenic differentiation and propose piR48444 inhibition as a promising strategy to enhance the bone regenerative capacity of MSCs from various tissue sources.
PiRNAs, a class of small non-coding RNAs, play crucial roles in post-transcriptional regulation across various cell types, including cardiac cells, somatic cells, and male germ cells12,17,18,19. While recent studies have identified specific piRNAs (piR36741 and piR63049) as regulators of BMSC osteogenesis12,13,14. Our study represents the first comprehensive investigation of a piRNA (piR48444) that can modulate osteogenic differentiation across MSCs from diverse origins. Through differential expression analysis during SHED osteogenic differentiation, we identified piR48444 as the most significantly downregulated piRNA exhibiting a strong negative correlation with BMP2, a master regulator of osteogenesis. Functional validation confirmed piR48444’s inhibitory role, as its overexpression was suppressed while knockdown enhanced osteogenic differentiation in SHED, BMSCs, PDLSCs, and ASCs. Importantly, piR48444 knockdown in SHED significantly enhanced bone defect repair in vivo, and its elevated expression in inflamed and aged BMSCs suggested pathological relevance. The therapeutic potential was further demonstrated through piR48444 antagomir treatment, effectively mitigating LPS-induced and aging-related bone loss. These findings establish piR48444 as a broad-spectrum negative regulator of MSC osteogenesis, highlighting its inhibition as a promising therapeutic strategy for bone regeneration and the treatment of osteoporosis.
Emerging evidence reveals the diverse regulatory mechanisms of piRNAs in post-transcriptional gene regulation, ranging from translational activation through the formation of super-complexes with MIWI/piRNA/eIF3F/HuR in mouse spermatids10 to mRNA silencing via deadenylation and decay pathways resembling miRNA function20,21,22,23. Our study adds a new dimension to piRNA biology by demonstrating their inhibitory role in mesenchymal stem cell osteogenesis. Through comprehensive binding prediction and luciferase reporter validation, we identified METTL7A as a direct target of piR48444 in SHED. While METTL7A was previously known to support BMSC survival and osteogenic potential under metabolic stress24, we now establish its crucial pro-osteogenic function in SHED, where its overexpression was enhanced while its knockdown impaired differentiation. The complete rescue of piR48444-mediated osteogenic inhibition by METTL7A overexpression provides compelling evidence that piR48444 exerts its suppressive effects primarily through METTL7A targeting. These findings not only expand our understanding of piRNA-mediated regulation in stem cell differentiation but also reveal METTL7A as a pivotal mediator of piR48444’s function in dental stem cell osteogenesis, suggesting tissue-specific yet conserved regulatory mechanisms across MSC populations.
m6A methylation, a key epigenetic mRNA modification, regulates diverse physiological and pathological processes25,26,27. While METTL3, METTL14, METTL16, and WTAP are well-known m6A “writers,” we found they were not upregulated during SHED osteogenic differentiation. In contrast, METTL7A, linked to cancer prognosis28, emerged as a critical regulator. Although piRNAs influence m6A modification in cancers and osteogenesis8,13,29, our study reveals a novel piR48444-METTL7A-BMP2 axis. Overexpressing METTL7A increased BMP2 protein (but not mRNA), while knockdown had the opposite effect. METTL7A localized to both the nucleus and cytoplasm and directly bound BMP2 mRNA, inducing m6A modifications in its coding region (confirmed by RIP, meRIP, and luciferase assays). These modifications enhanced BMP2 mRNA stability and translation-consistent with m6A’s known roles30,31. Crucially, exogenous BMP2 rescued METTL7A knockdown’s inhibitory effects, and piR48444 knockdown upregulated METTL7A/BMP2 in multiple MSCs, confirming piR48444 suppresses osteogenesis by blocking METTL7A-mediated BMP2 m6A methylation and translation.
Our study reveals a critical interaction between METTL7A and eIF4E, regulating osteogenic differentiation in SHED. While METTL7A was previously shown to bind eIF4E in cancer cells32, we demonstrate its novel role in osteogenesis, where eIF4BP1 downregulation in piR48444-knockdown SHED likely releases eIF4E to promote differentiation, consistent with known eIF4E regulation by eIF4BP133. We confirmed direct binding of METTL7A to eIF4E in SHED and established their functional synergy, i.e., eIF4E overexpression enhanced osteogenesis and BMP2 expression, while METTL7A knockdown attenuated eIF4E’s pro-osteogenic effects, and vice versa. Collectively, this demonstrates a METTL7A-eIF4E-BMP2 axis driving osteogenic differentiation, although the specific binding sites warrant further investigation.
While our findings establish the novel piR48444-METTL7A-eIF4E-BMP2 axis in MSCs osteogenesis, several limitations warrant consideration: (1) The precise structural basis of METTL7A-eIF4E binding remains unresolved, necessitating further structural studies to map interaction sites; (2) While we demonstrated METTL7A-mediated m6A modification of BMP2 mRNA, the specific m6A reader proteins (e.g., YTHDF1/3) involved in this regulatory cascade were not investigated; (3) The in vivo bone repair models focused on SHED, and additional validation in other MSC types would strengthen translational relevance; (4) The potential off-target effects of piR48444 antagomir therapy require systematic evaluation before clinical translation; (5) While we linked piR48444 to aging- and inflammation-related bone loss, the upstream regulators of piR48444 expression in these pathological contexts remain unexplored. These limitations highlight important directions for future research to fully exploit this pathway for bone regeneration therapies.
In summary, our study demonstrates that piR48444 acts as a negative regulator of MSC osteogenic differentiation by directly targeting METTL7A. Mechanistically, METTL7A functions as an m6A writer to modify BMP2 mRNA, thereby enhancing its stability and promoting translation through interaction with eIF4E. By suppressing METTL7A expression, piR48444 disrupts this regulatory axis, impairing BMP2-dependent osteogenesis. These findings position piR48444 as both a promising therapeutic target for enhancing MSC-based bone regeneration and a potential biomarker for osteoporosis, particularly in contexts of inflammatory and aging-related bone loss.
Materials and methods
Cell isolation and culture
Stem cells from defoliated deciduous teeth (SHED) were isolated from the deciduous teeth pulp tissue of 6 male patients aged 6–8 years old, as described previously34. Periodontal ligament stem cells (PDLSCs) were collected as per the previous report35. BMSCs and human adipose mesenchymal stem cells were purchased from Oricell (Guangzhou, China). This study has been approved by the Ethics Committee of the School and Hospital of Stomatology, Guangzhou Medical University (LCYJ2021017). Informed written consent was obtained from the parents of each patient. All ethical regulations relevant to human research participants were followed. BMSCs and culture medium were purchased from OriCell (Guangzhou).
Alizarin red staining
SHED (26,000 cells/well) were seeded at 48-well culture plates and cultured with osteogenic medium (50 μg/mL vitamin C (Solarbio, cat#IA0530, China), 0.01 μM dexamethasone (Solarbio, cat#D8040, China), and 10 mM β-glycerophosphate (Solarbio, cat#G9422, China)) for 14 days. Cells were fixed with 4% paraformaldehyde and stained with Alizarin Red S solution (1%, pH 4.2) (Solarbio, cat#G1452, China) for 10 min. The staining was visualized using a stereomicroscope (Leica EZ4HD, Leica, Germany). For quantitative analysis, the alizarin red-stained mineralized matrix was dissolved in 200 μL 10% hexadecylpyridinium chloride monohydrate (Aladdin, cat#H108697, China) for 1 h. The optical density of the supernatant (100 μL) was measured using a microplate reader (Biotech, USA) at 562 nm.
PiRNA and mRNA sequencing
PiRNA and mRNA were sequenced by Origingene (Shanghai, China) with the NovaSeq 6000-PE150 platform. Fastq data were aligned to the hg38 genome using STAR. Transcripts per million RNA were used to evaluate gene expression, calculated using RNA-Seq by the Expectation Maximization software package. Differential expression of piRNAs and mRNAs was calculated by edgeR and DESeq in the R package from Bioconductor (http://master.bioconductor.org/). PiRNA target genes were predicted from RNA sequence data in the miRanda database. Pathway enrichment analysis was performed using the clusterProfiler package in R. The sequencing data have been deposited in the GSA database (HRA005430) (https://ngdc.cncb.ac.cn/gsa-human/s/uA6JG2dT).
RT-qPCR analysis
According to the manufacturer’s instructions, the piRNAs were extracted from SHED using the MolPure® Cell/Tissue miRNAs Kit (Yeasen, cat#19331ES50, China). The piRNAs were extracted from plasma using SteadyPure Blood, Serum, and Plasma Small RNA Extraction Kit (Accurate Biology, cat#AG21030, China). The piRNAs were further reversed by Tailing reaction using miRNAs 1st strand cDNA synthesis kit (Accurate Biology, cat#AG11717, China). The extraction of mRNAs was performed as follows: cells were lysed with Trizol (Accurate Biology, AG21102, China) and extracted with isopropanol. Total RNA (500 ng) was transcribed with reverse transcriptase (Accurate Biology, cat#AG11701, China). Quantitative PCR was performed using SYBR® Green Premix Pro Taq HS qPCR Kit (Accurate Biology, cat#AG11701, China) on an AriaMx Real-time quantitative PCR machine (Agilent, USA). The fold change relative to the control group was measured by the 2−∆∆Ct method. The primers used are listed in Table S1.
PiR48444 expression detected in osteoporosis patients and healthy controls
The bioinformatic analysis of piR48444 in the mononuclear cells from peripheral blood samples (3 healthy controls and 3 osteoporosis patients) (GSE159121) was performed as described above.
We also analyzed the expression of piR48444 in the serum of a total of 30 participants, including 15 patients with osteoporosis and 15 age- and gender-matched healthy controls, who were enrolled at Guangdong Province Second General Hospital and included in this study. Individuals who had thyroid dysfunction, metabolic diseases such as diabetes, hyperlipidemia, or hyperuricemia, autoimmune diseases such as systemic lupus erythematosus, rheumatoid arthritis, cancer, or serious diseases, drug therapy including hormones within one year, or organ transplantation were excluded. Written informed consent was obtained from all participants. This study was approved by the ethical committee of the Guangdong Province Second General Hospital (approved number: GD2H-KY IRB-AF-SC.07-01.2). All ethical regulations relevant to human research participants were followed. The blood samples were collected in anticoagulant tubes. The samples were then centrifuged at 1200 rpm for 30 min. The plasma was frozen and stored at −80 °C.
RNA separation
To detect the piR48444 location, RNA separation was performed using the Cytoplasmic & Nuclear RNA Purification Kit (Norgen Biotech, cat#21000, Canada) according to the manufacturer’s instructions. In brief, SHED were lysed and centrifuged at 15,000 rpm for 3 min. The supernatant (cytoplasm) and pellet (nucleus) fractions were added to buffer SK and applied to spin columns. Then RNAs were washed and eluted. The expression level of piR48444 was detected in the cytoplasm and nucleus. GAPDH and U6 were used as internal controls.
PiR48444 probe detection
SHED were cultured with PM and OM medium for 7 days. Cell slides were fixed in 4% PFA for 20 min, and washed in PBS (pH 7.4) on a decolorization shaker. Protease K (5 μg/mL) was added for digestion at 40 °C for 5 min. After washing with pure water and PBS, pre-hybridization was performed. The probe-containing hybridization solution was added and mixed overnight in the incubator. The non-specific hybrids were further washed. The branch probe was hybridized at 40 °C for 45 min. After washing, the signal probe (1:400 dilution) was incubated at 40 °C for 3 h. After washing, DAPI staining was conducted. Microscopic photography was scanned using the Pannoramic 250 FLASH (3DHISTECH, Hungary).
PiRNA pulldown assay
To determine the direct binding proteins of piR48444, a piRNA pulldown was conducted. Biotin-modified piR48444 was purchased from Igeobio (China). Biotin-modified piRNA and control were transfected into SHED with EZ trans-Lipo (Life-iLab, cat#AC04L071, China). PiRNA pulldown assay was performed using a miRNA pulldown kit (Besinbio, cat#Bes5109, China). In brief, cells were lysed with lysis buffer, which included proteinase/RNAase inhibitor, DTT, for 20 min. Lysed cells were incubated with blocked Streptavidin magnetic beads for 4 h. The beads were washed 5 times and then eluted with elution buffer for 2 h at 37 °C. Western blot analysis was further performed.
Plasmid construction and lentivirus preparation
The 3’UTR sequences and mutant sequences of METTL7A and BMP2 (200 bp upstream and 200 bp downstream of the predicted binding sites) were synthesized and cloned into pmirGLO Dual-Luciferase miRNAs Target Expression Vector (Promega, USA) by Generay (China). The lentivirus plasmids, including the control plasmid, METTL7A overexpression, and METTL7A knockdown plasmid, were purchased from Genery (China). The plasmids, including the scramble plasmid, the piR48444 plasmid for overexpression (cat#VB210513-1332hry), and the piR48444 knockdown plasmid (cat#VB210513-1357qek), were purchased from VectorBuilder (China).
For lentiviral transduction, HEK293T cells were co-transfected with expression plasmids, including control, pCDH-METTL7A, pCDH-METTL7A shRNA, scramble, pLV-piR48444, or pLV-piR48444 sponge, along with the packaging plasmid pMD2.VSVG, pMDLg/pRRE, and pRSV-REV using EZ trans transfection regent (Life-iLab, cat#AC04L092, China). Fresh lentiviral supernatant was collected after 2 days and used for infection.
Alkaline phosphatase (ALP) staining
SHED or PDLSCs (26,000 cells/well) were seeded in 48-well culture plates and cultured in an osteogenic medium for 7 days. Meanwhile, BMSCs were cultured using an osteogenic differentiation medium (OriCell, cat#HUXMX-90021, Guangzhou). Then, the cells were fixed with paraformaldehyde and stained using an ALP staining kit (Beyotime, cat# C3206, China). The staining was visualized with a stereomicroscope using LAS EZ software (Leica, Germany). ALP staining intensity was further analyzed using ImageJ 1.5.1j8 (NIH, USA).
The repair of nude mice calvarial defects
SHED were infected with scramble or piR48444 shRNA lentivirus and induced with osteogenic medium for 7 days. SHED-derived cell sheets induced with 20 μg/mL vitamin C (Solarbio, cat#IA0530, China) for 7 days and osteogenic medium for 7 days were prepared. SHED were infected with control or piR48444 shRNA lentivirus and induced with osteogenic medium for 7 days. SHED (105/sample) was mixed with 15% Gelma hydrogel (EFL-Tech Co., Ltd, cat#EFL-GM60, China) and light solidified for 40 s. Five-week-old male nude mice were purchased from Southern Medical University, Guangzhou, China (6 mice/group, 1 defect/mouse) and were used for the calvarial bone defect repair study. After anesthesia with pentobarbital sodium (50 mg/kg), the cranium surface was exposed through a skin incision, and a circular and full-thickness bone defect with a 3 mm diameter was generated using a trephine drill. The bone fragments were taken out. Cell sheets or hydrogel-incorporated SHED were filled in the skulls, and the incisions were sutured. After 2 months, calvarial bones were collected, and the healing level was analyzed using Micro-CT. The animal experiment was conducted in accordance with the guidelines approved by the Laboratory Animal Ethics Committee of Guangdong Huawei Testing Co., Ltd., Guangzhou, China (approved number: 202202002). We have complied with all relevant ethical regulations for animal use.
PiR48444 antagomir injected in LPS-treated mice
PiR48444 antagomir used in vivo was synthesized by Guangzhou IGE Biotechnology Co., Ltd (China). Wild-type C57BL/6J mice (16 mice/group, including 8 male mice and 8 female mice) were injected intraperitoneally with LPS (8 mg/kg) twice 1 week, for 2 weeks. Mice were weighed before LPS injection. After 3 weeks, control siRNA and piR48444 (30 μg) were administered intravenously once a week for 4 weeks. Femurs were collected for micro-CT analysis or H&E staining.
PiR48444 antagomir injected in aging mice
Control siRNA and piR48444 antagomir (30 μg) were administered intravenously once a week for 4 weeks in 18-month-old mice (12 mice/group, including 6 male mice and 6 female mice). Femurs were collected for micro-CT analysis and H&E staining.
Micro-CT
Micro-CT scanning was performed to evaluate the bone formation in the skull defect model using Bruker Sky1172 Skyscan (Kontich, Belgium). In brief, a total of 100 slices (1 mm) was measured for 3D reconstruction and quantification of trabecular and cortical bone. The X-ray tube was operated at 96 kV and 65 μA using a 0.5 mm Al filter. Scanning was performed by 180 ° rotation around the vertical axis, camera exposure time of 1300 ms, rotation step of 0.6 °, frame averaging of 2, and random movement of 10. 3D images were made and data viewer software (Skyscan, Kontich, Belgium) was used for images and linear analysis. Relative bone formation parameters including BV/TV, BMD, Tb.N, Tb.Th, and Tb.Sp were further analyzed.
Hematoxylin and eosin (H&E) staining
Calvaria bones were fixed with 4% paraformaldehyde for 2 days. Tissues were decalcified with EDTA decalcification solution (Sangon Biotech, cat#E671001-0500, China) for 18 days. Then slices of bone tissue at 4 μm thickness were cut. The slices were further performed H&E staining by dewaxing, hematoxylin staining, eosin staining, and dehydration.
Masson staining
The Masson staining was performed according to previously reported36. The dehydrated tissue sections were immersed overnight in Bouin’s solution. After rinsing, sections were stained with hematoxylin solution for 5 min. Then sections were differentiated with 0.8–1% hydrochloric acid alcohol. After that, sections were stained with ponceau acid fuchsin solution for 5 min. Sections were treated with a phosphomolybdic acid solution for about 5 min and stained with an aniline blue solution for 5 min. Sections were further treated with 1% glacial acetic acid for 1 min and dehydrated with 95% alcohol. Finally, tissue sections were dehydrated with absolute alcohol and treated with xylene, then mounted in neutral balsam.
Immunohistochemistry (IHC)
The IHC was performed per the previous report37. Tissue sections were deparaffinized and rehydrated. Antigen retrieval was performed by EDTA (pH 9.0), endogenous peroxidase activity was blocked by 3% hydrogen peroxide and treated for 25 min at room temperature, and 3% BSA was used for serum sealing. The sections were incubated with rabbit polyclonal METTL7A (1: 150, Abclonal, cat#A8201, China) and BMP2 (1:150, Abclonal, cat#A0231, China) overnight at 4 °C. After washing with PBS, the sections were further incubated with HRP-labeled Goat anti-rabbit IgG (Abcam, cat#ab6721, 1:200, China). Finally, DAB development and nuclear counterstaining were performed.
Western blot analysis
Total protein (20 μg) was added to a 10% SDS-polyacrylamide gel. After electrophoresis, the protein was transferred to PVDF membranes (Millipore, USA) and blocked for 1 h with a blocking buffer (Beyotime, China). Then PVDF membranes were incubated with the primary antibodies against METTL7A (Proteintech, cat#17092-1-AP, 1:5,000, China), BMP2 (Proteintech, cat#6383-1-Ig, 1:5000, China), PIWIL1 (Zenbio, cat#R382334, 1:5000, China), PIWIL2 (SAB, cat#25385, 1:5000, USA), PIWIL4 (SAB, cat#31113, 1:5000, USA) and GAPDH (Proteintech, cat#10494-1-AP, 1:10,000, China) overnight at 4 °C. The membranes were further incubated with horseradish peroxidase-conjugated secondary antibody for 1 h and reacted with ECL (Millipore, USA) for 1 min. Finally, photographs were taken using the Tanon-5200 system (Tanon, China).
Luciferase reporter assay
We further confirmed the METTL7A as a target gene of piR48444 by luciferase reporter assay METTL7A/BMP2 3’UTR (100 ng) with NC (50 nM), METTL7A/BMP2 3’UTR (100 ng) with piR48444 (50 nM), and the METTL7A/BMP2 mutant 3’UTR plasmid (100 ng) with piR48444 (50 nM) were co-transfected into HEK293T cells by lipofectamine 3000 (Thermo Fisher Scientific Inc., cat#L3000015, USA). To evaluate the effect of CDS on BMP2 expression, the wild-type or mutant-1/-2 CDS of BMP2 was cloned as pmirGLO-BMP2-WT and pmirGLO-BMP2-Mut-2/-3 and co-transfected with or without METTL7A-OE plasmid. After 48 h, the luciferase reporter assay was performed according to the manufacturer’s instructions using Luc-Pair™ Duo-Luciferase Assay Kit 2.0 (Genecopedia, cat#LF001, USA). In brief, cells were lysed and incubated with 100 μL Fluc work solution for 5 min, and the fluorometric value was measured by Varioskan® Flash (Thermo Fisher, USA). The lysis solution was supplemented with 100 μL Rluc and incubated for 5 min, after which the fluorometric value was further detected.
Immunofluorescence staining
SHED were induced with osteogenic differentiation medium for 7 days. Cells were treated with 4% paraformaldehyde for 20 min. 0.5% Triton X-100 was used to treat cells for 20 min. Samples were blocked with 3% BSA for 1 h and incubated overnight with METTL7A antibody (Abclonal, cat#A8201, China, 1:150). After washing, samples were incubated with a secondary antibody (Proteintech, SA00013-2, China, 1:150) for 1 h. Images were taken with a Confocal microscope (Leica SP8, Germany).
Protein extraction from cytoplasm and nucleus
According to the manufacturer’s instructions, the protein separation was performed with MinuteTM Cytoplasmic and Nuclear Extraction Kit for Cells (Invent technologies, cat#SC-003, USA).
RNA immunoprecipitation (RIP)
pcDNA3.1-FLAG-METTL7A plasmid was purchased from Genery (China). The plasmid was transfected into SHED. After 48 h, RIP assay was performed per the manufacturer’s instructions (Bersinbio, cat#bes5101, China). Cells were lysed for 30 min. DNAse was added to remove DNA. The lysate was incubated with 2 μg of FLAG antibody (Proteintech, cat#20543-1-AP, China) in a shaker for 16 h. Then, balanced protein A/G magnetic beads were added and mixed for 1 h. Polysome washing buffer and elution buffer were added to elute RNA. Finally, RNA was extracted and RT-qPCR was performed.
Methylated RNA immunoprecipitation (MeRIP)
According to the manufacturer’s instructions, the MeRIP assay was performed using a MeRIP kit (BerinBio, cat#Bes5203, China). Total RNA was extracted with Trizol regent (Accurate Biology, cat#AG21102, China) and fragmented with fragmentation buffer at 94 °C for 3 min. After extraction, 50 μL of fragmented RNA was used as input. RNA (400 μL) was used for immunoprecipitation with 4 μg m6A antibody (Synaptic systems, cat#202003, Germany) at 4 °C for 4 h. The treated protein A/G magnetic beads were mixed with antibodies for 1 h at 4 °C. After digestion with proteinase K at 55 °C for 30 min, the input and immunoprecipitated RNA were extracted with Phenol-Chloroform-Isoamylol (25:24:1). The m6A level of BMP2 was measured by RT-qPCR; primers were listed in Table S2.
RNA stability
Cells were transfected with METTL7A-OE, METTL7A-KD, and control plasmids and incubated with actinomycin D (MCE, cat#17559, USA) at 5 μg/mL. Cells were then collected at 0, 3, and 6 h. RNAs were isolated for RT-qPCR to evaluate RNA stability.
Protein synthesis
BMP2 protein synthesis of SHED transfected with METTL7A-OE, METTL7A-KD, and control plasmids was confirmed by incubation of cells with 100 μg/mL cycloheximide (CHX) (MCE, cat#HY-12320, USA) for 6 h. The expression of BMP2 was measured using Western blot analysis.
Protein degradation
BMP2 protein degradation of SHED transfected with METTL7A-OE, METTL7A-KD, and control plasmids was detected by treatment of SHED with 5 μM MG132 (MCE, cat#HY-13259, China) for 6 h. Protein isolation and WB were further performed.
The predication of protein-interaction
The protein structure files of METTL7A and eIF4E were downloaded from the Alphafold protein structure database (https://alphafold.com/). Then the structure files were uploaded to ClusPro (ttps://cluspro.org/) for protein-protein docking. The docking results were further analyzed and visualized with PyMol molecular graphics software.
Co-immunoprecipitation (Co-IP)
Flag-METTL7A and control plasmids were purchased from Genery Biotechnology (China). Flag-METTL7A and control plasmids (10 μg) were transfected into SHED. After 3 days, 107 cells were collected and lysed for 30 min. Lysed cells were centrifuged at 12,000 × g for 10 min. The supernatant was collected. Then, 25 μL balanced Anti-Flag Nanobody Magarose Beads (AlpaLifeBio, KTSM1338, China) were added to the supernatant and rotated at 4 °C for 3 h. The beads were washed five times with PBST and eluted with 1× SDS loading buffer at 95 °C for 10 min. The protein was detected with a flag (1:10,000) (Proteintech, cat#20543-1-AP, China) and eukyrotic translation initiation factor 4E (eIF4E, 1:5000, Abclonal, cat#A25608, China) antibody.
Statistics
Prism version 8.5 was used for the statistical analysis. Data are expressed as mean ± SD. A two-tailed Student’s t-test was used for two-group comparisons. The comparison of multiple groups was performed by one-way analysis of variance (ANOVA) or two-way ANOVA compared to every other group. Statistical significance was set at p < 0.05, which was considered a significant difference.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Numerical source data underlying all graphs in the manuscript can be found in the supplementary data file/figshare link with the identifier https://doi.org/10.6084/m9.figshare.31042333.
References
Ding, D. C., Shyu, W. C. & Lin, S. Z. Mesenchymal stem cells. Cell Transpl. 20, 5–14 (2011).
Rosenbaum, A. J., Grande, D. A. & Dines, J. S. The use of mesenchymal stem cells in tissue engineering: a global assessment. Organogenesis 4, 23–27 (2008).
Hou, S. Y., Zhang, H. Y., Quan, D. P., Liu, X. L. & Zhu, J. K. Tissue-engineered peripheral nerve grafting by differentiated bone marrow stromal cells. Neuroscience 140, 101–110 (2006).
Lau, N. C. et al. Characterization of the piRNA complex from rat testes. Science 313, 363–367 (2006).
Vagin, V. V. et al. A distinct small RNA pathway silences selfish genetic elements in the germline. Science 313, 320–324 (2006).
Zhang, J., Chen, S. & Liu, K. Structural insights into piRNA biogenesis. Biochim. Biophys. Acta Gene Regul. Mech. 1865, 194799 (2022).
Wang, X., Ramat, A., Simonelig, M. & Liu, M. F. Emerging roles and functional mechanisms of PIWI-interacting RNAs. Nat. Rev. Mol. Cell Biol. 24, 123–141 (2023).
Han, H. et al. piRNA-30473 contributes to tumorigenesis and poor prognosis by regulating m6A RNA methylation in DLBCL. Blood 137, 1603–1614 (2021).
Chen, L. et al. Site-specific expression pattern of PIWI-interacting RNA in skin and oral mucosal wound healing. Int. J. Mol. Sci. 21, 521 (2020).
Dai, P. et al. A translation-activating function of MIWI/piRNA during mouse spermiogenesis. Cell 179, 1566–1581.e16 (2019).
Wang, A. et al. Identification and Comparison of piRNA expression profiles of exosomes derived from human stem cells from the apical papilla and bone marrow mesenchymal stem cells. Stem Cells Dev. 29, 511–520 (2020).
Della Bella, E. et al. Differential regulation of circRNA, miRNA, and piRNA during early osteogenic and chondrogenic differentiation of human mesenchymal stromal cells. Cells 9, 398 (2020).
Liu, J., Chen, M., Ma, L., Dang, X. & Du, G. piRNA-36741 regulates BMP2-mediated osteoblast differentiation via METTL3 controlled m6A modification. Aging 13, 23361–23375 (2021).
Chen, G. Y. et al. PiRNA-63049 inhibits bone formation through Wnt/degrees beta-catenin signaling pathway. Int. J. Biol. Sci. 17, 4409–4425 (2021).
Nakajima, K. et al. Comparison of the bone regeneration ability between stem cells from human exfoliated deciduous teeth, human dental pulp stem cells and human bone marrow mesenchymal stem cells. Biochem. Biophys. Res. Commun. 497, 876–882 (2018).
Kunwong, N. et al. Optimization of poly (lactic-co-glycolic acid)-bioactive glass composite scaffold for bone tissue engineering using stem cells from human exfoliated deciduous teeth. Arch. Oral. Biol. 123, 105041 (2021).
La Greca, A. et al. PIWI-interacting RNAs are differentially expressed during cardiac differentiation of human pluripotent stem cells. PloS ONE 15, e0232715 (2020).
Li, D., Taylor, D. H. & van Wolfswinkel, J. C. PIWI-mediated control of tissue-specific transposons is essential for somatic cell differentiation. Cell Rep. 37, https://doi.org/10.1016/j.celrep.2021.109776 (2021).
Garcia-Lopez, J. et al. Diversity and functional convergence of small noncoding RNAs in male germ cell differentiation and fertilization. RNA 21, 946–962 (2015).
Gou, L. T. et al. Pachytene piRNAs instruct massive mRNA elimination during late spermiogenesis. Cell Res. 24, 680–700 (2014).
Barckmann, B. et al. Aubergine iCLIP Reveals piRNA-Dependent Decay of mRNAs involved in germ cell development in the early embryo. Cell Rep. 12, 1205–1216 (2015).
Rojas-Rios, P., Chartier, A., Pierson, S. & Simonelig, M. Aubergine and piRNAs promote germline stem cell self-renewal by repressing the proto-oncogene Cbl. EMBO J. 36, 3194–3211 (2017).
Goh, W. S. et al. piRNA-directed cleavage of meiotic transcripts regulates spermatogenesis. Genes Dev. 29, 1032–1044 (2015).
Lee, E., Kim, J. Y., Kim, T. K., Park, S. Y. & Im, G. I. Methyltransferase-like protein 7A (METTL7A) promotes cell survival and osteogenic differentiation under metabolic stress. Cell Death Discov. 7, 154 (2021).
Jiang, X. et al. The role of m6A modification in the biological functions and diseases. Signal. Transduct. Target Ther. 6, 74 (2021).
An, Y. & Duan, H. The role of m6A RNA methylation in cancer metabolism. Mol. Cancer 21, 14 (2022).
Zhang, T. et al. m(6)A mRNA modification maintains colonic epithelial cell homeostasis via NF-kappa B-mediated antiapoptotic pathway. Sci. Adv. 8, eabl5723 (2022).
Liu, Z. K., Chen, Y. Q. & Shen, T. Evidence based on an integrative analysis of multi-omics data on METTL7A as a molecular marker in pan-cancer. Biomolecules 13, 195 (2023).
Xie, Q. et al. piRNA-14633 promotes cervical cancer cell malignancy in a METTL14-dependent m6A RNA methylation manner. J. Transl. Med. 20, 51 (2022).
Li, A. et al. ZNF677 suppresses renal cell carcinoma progression through N6-methyladenosine and transcriptional repression of CDKN3. Clin. Transl. Med. 12, e906 (2022).
Jin, Y., Han, X., Wang, Y. & Fan, Z. METTL7A-mediated m6A modification of corin reverses bisphosphonates-impaired osteogenic differentiation of orofacial BMSCs. Int. J. Oral. Sci. 16, 42 (2024).
Briata, P. et al. LncRNA EPR-induced METTL7A1 modulates target gene translation. Nucleic Acids Res. 50, 7608–7622 (2022).
Bartish, M. et al. The role of eIF4F-driven mRNA translation in regulating the tumour microenvironment. Nat. Rev. Cancer 23, 408–425 (2023).
Zhao, X. D. et al. Metformin enhances osteogenic differentiation of stem cells from human exfoliated deciduous teeth through AMPK pathway. J. Tissue Eng. Regen. Med. 14, 1869–1879 (2020).
Li, W., Tian, W., Wu, Y. & Guo, S. A novel magnetic manipulation promotes directional growth of periodontal ligament stem cells. Tissue Eng. Part A 29, 620–632 (2023).
da Silva Brum, I. et al. Performance of nano-hydroxyapatite/beta-tricalcium phosphate and xenogenic hydroxyapatite on bone regeneration in rat calvarial defects: histomorphometric, immunohistochemical and ultrastructural analysis. Int. J. Nanomed. 16, 3473–3485 (2021).
Li, Z. et al. Leptin-deficient ob/ob mice exhibit periodontitis phenotype and altered oral microbiome. J. Periodontal Res. 58, 392–402 (2023).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (U22A20159), the Science and Technology Program of Guangzhou (2023B03J1240, 202201010073, 202201020116, 202201020156) and First-class major construction project 02-408-240603071.
Author information
Authors and Affiliations
Contributions
Conceptualization, Z.Z., R.J., J.P., and L.W.; Formal analysis, Z.Z., X.L., J.P., and L.W.; Funding, Z.Z., W.Q., J.P., and L.W.; Investigation, Z.Z., X.L., W.Q., H.C., S.Y., T.X., W.X., Z.F., G.P., Y.Z., W.H., F.L., and Y.J.; Supervision, L.W., J.P., and R.J.; Writing-original draft preparation, Z.Z. and X.L.; Writing-review and editing, R.J., J.P., and L.W.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Akira Nifuji and Tomoyuki Iwata for their contribution to the peer review of this work. Primary handling editors: Bibekanand Mallick and Joao Valente. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zheng, Z., Li, X., Qin, W. et al. PiR48444 inhibits MSC osteogenic differentiation and bone regeneration via targeting METTL7A/eIF4E-mediated BMP2 m6A methylation. Commun Biol 9, 337 (2026). https://doi.org/10.1038/s42003-026-09583-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-026-09583-1
