
Abstract
Heart failure (HF) is a growing global health issue. While most studies focus on cardiomyocytes, here we highlight the role of cardiac fibroblasts (CFs) in HF. Single-cell RNA sequencing of mouse hearts under pressure overload identified six CF subclusters, with one specific to the HF stage. This HF-specific CF population highly expresses the transcription factor Myc. Deleting Myc in CFs improves cardiac function without reducing fibrosis. MYC directly regulates the expression of the chemokine CXCL1, which is elevated in HF-specific CFs and downregulated in Myc-deficient CFs. The CXCL1 receptor, CXCR2, is expressed in cardiomyocytes, and blocking the CXCL1–CXCR2 axis mitigates HF. CXCL1 impairs contractility in neonatal rat and human iPSC-derived cardiomyocytes. Human CFs from failing hearts also express MYC and CXCL1, unlike those from controls. These findings reveal that HF-specific CFs contribute to HF via the MYC–CXCL1–CXCR2 pathway, offering a promising therapeutic target beyond cardiomyocytes.
Similar content being viewed by others


Main
The number of patients with heart failure (HF) is increasing worldwide, and HF is one of the most common causes of death due to aging1. Initially, cardiac hypertrophy acts as a compensatory response to hemodynamic overload and reduces wall stress. However, long-term overload induces cardiac dysfunction with myocardial remodeling, such as left ventricular dilatation and fibrosis, eventually leading to HF. Although there have been many studies on the mechanisms of HF, such as abnormal calcium handling2, adrenergic receptor overactivation3, apoptosis4 and DNA damage in cardiomyocytes5,6, the complex pathophysiology of HF is not yet fully understood.
Cardiomyocytes constitute only 20–30% of heart-forming cells in number, with over 70% being non-cardiomyocytes, such as endothelial cells, fibroblasts, smooth muscle cells, pericytes and blood cells7. This highlights the potential pivotal roles of non-cardiomyocytes in HF development. Among non-cardiomyocytes, cardiac fibroblasts (CFs) strongly interact with multiple cell types, including cardiomyocytes8. However, CFs have been studied primarily in terms of fibrosis, especially after myocardial infarction (MI). CFs are activated after MI and express many types of extracellular matrix proteins to stabilize scars9. Although significant fibrosis is also observed in failing hearts10 and has been reported to be involved in diastolic dysfunction by directly increasing left ventricular stiffness11, it is unknown whether CFs can affect cardiomyocytes or cause contractile dysfunction. In this study, a comprehensive gene expression analysis using single-cell RNA sequencing (scRNA-seq) revealed the dynamic heterogeneity of CFs in a murine HF model induced by pressure overload. One subpopulation of fibroblasts appears specifically in failing hearts and is critically involved in HF development.
Results
CF heterogeneity in HF
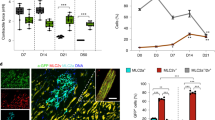
Investigating the change of cardiac fibroblasts during HF development, we pressure overloaded mouse hearts by transverse aortic constriction (TAC) to induce cardiac hypertrophy at 2 weeks (TAC2w) and HF at 12 weeks (TAC12w) (Fig. 1a). A heart with reduced contractile function after 12 weeks of TAC was defined as a ‘failing heart’. Mononuclear cells from mouse hearts of sham control (TAC0w), hypertrophic stage (TAC2w) and HF stage (TAC12w) underwent scRNA-seq12. Mononuclear cells were classified based on gene expression, using a dimensionality reduction method called t-distributed stochastic neighbor embedding (t-SNE) (Extended Data Fig. 1a,b). By defining anchors, we integrated the data from the hearts at the control, hypertrophic and HF stages, and each cell cluster was identified in terms of cell type by the expression of marker genes. CFs were identified as cells expressing fibroblast marker genes such as Pdgfra (platelet-derived growth factor receptor alpha), Tcf21 (transcription factor 21), Col1a1 (collagen type I alpha 1 chain) and Postn (periostin), and were roughly divided into two subclusters (Fig. 1b,c). We extracted data from the fibroblast population and reclassified them into six subclusters with distinct gene expression patterns (Fig. 1d). Notably, a fibroblast subcluster, subcluster 4, exclusively appeared in failing hearts, suggesting its involvement in HF development (Fig. 1d), and we named these CFs HF-specific fibroblasts (HF-Fibro), expressing Postn but not Acta2 (actin alpha 2, smooth muscle) or Chad (chondroadherin) (Extended Data Fig. 1c).

a, Experimental scheme using mice exposed to pressure overload. 0, day of TAC; 2 weeks, 2 weeks after TAC; 12 weeks, 12 weeks after TAC. On the day of TAC and 2 weeks and 12 weeks after TAC, non-cardiomyocytes were collected from the heart and scRNA-seq was performed. b, t-SNE visualization of unsupervised clustering of non-cardiomyocytes. Cells (dots) are colored according to the cell clusters and annotated using well-known marker genes. CE, capillary endothelial cell; MΦ, macrophage; SM, smooth muscle cell; AE, arterial endothelial cell; DC, dendritic cell; VE, venous endothelial cell; ME, MKI67 positive endothelial cell; NC, nerve cell; LE, lymphatic endothelial cell; BC, B cell; TC, T cell. c, Feature plot showing fibroblast marker gene expression on t-SNE. d, t-SNE reclassified CFs into six subpopulations according to their gene expression (left, colored according to the cell clusters; right, colored according to the time after TAC). The table shows the cell ratios of six clusters at each time course. e, GO analysis of genes specifically expressed in cluster 4, HF-specific CFs. f, Violin plot showing the expression of Myc and Tcf21 among the six clusters. g, Left: representative cardiac sections stained with azan. Scale bars, 1 mm. Right: representative gross cardiac appearance. h, Flow cytometry analyses of PDGFRα and MYC expression in sham and TAC12w hearts. Negative control, secondary antibody alone in each mouse. i, Immunofluorescence for collagen I and smFISH for MYC in sham and TAC12w hearts. The experiment was repeated independently three times with similar results. Scale bars, 20 μm. j, Western blot analysis of MYC and GAPDH expression in CFs of sham and TAC12w mice. The experiment was repeated independently three times with similar results.
To elucidate the characteristics of HF-Fibro, we analyzed their transcriptomic profiles. Gene Ontology (GO) analysis revealed that HF-Fibro express not only genes characteristic of fibroblasts, such as those encoding cell adhesion molecules and extracellular matrix, but also genes encoding defense and immune proteins. Next, we focused on transcription factors (TFs) that strictly regulate cell characteristics through gene expression and cell fate determinants (Fig. 1e). Eight TFs were enriched in HF-Fibro, and of these, expression of the proto-oncogene Myc (v-myc avian myelocytomatosis viral oncogene homolog) was restricted to HF-Fibro (Fig. 1f and Extended Data Fig. 1d), suggesting that MYC is the dominant regulator of HF-Fibro features.
MYC-positive CFs in failing hearts
To confirm the presence of MYC-positive CFs in failing hearts, mononuclear cells from sham and TAC mice were sorted by flow cytometry, using antibodies against PDGFRα and MYC. In the sham (TAC0w) mice, there were no double-positive cells for PDGFRα and MYC; however, a few double-positive cells were detected after 4 weeks of TAC and the number of these cells increased considerably after 8 weeks and 12 weeks (Fig. 1g,h and Extended Data Fig. 1e). To clarify the expression of MYC in CFs of failing hearts, immunostaining using anti-collagen I antibody and single-molecule fluorescence in situ hybridization (smFISH) for MYC were performed. MYC-positive CFs were distributed throughout the left ventricles of TAC12w mice but not in those of sham mice (Fig. 1i). Furthermore, we isolated CFs from sham and TAC12w hearts and performed western blot analysis using an anti-MYC antibody. MYC protein was detected only in the CFs of TAC12w hearts, but not in those of sham hearts (Fig. 1j). These results further ensure the HF-stage-specific presence of MYC-positive CFs identified by scRNA-seq analysis.
Myc overexpression aggravates HF
As Myc began to be expressed in CFs during hypertrophy and became highly expressed during HF, we examined the role of MYC in CFs in HF development using genetically modified mice. We generated CF-specific Myc overexpressing (OE) mice using Tcf21 iCre mice in which the Cre recombinase expression is induced in CFs by tamoxifen (TAX) administration, and R26StopFLMYC mice in which a STOP sequence flanked by loxPs between the CAG promoter and Myc gene was knocked into the Rosa26 locus13. By crossing Tcf21 iCre mice and R26StopFLMYC mice, we generated Tcf21 iCre–R26StopFLMYC mice (OE mice). TAX administration induced the expression of Cre recombinase, leading to MYC overexpression in CFs (Fig. 2a,b). Echocardiography revealed that cardiac systolic function was reduced by TAC in control mice and further reduced by TAC in OE mice (OE-TAC) (Fig. 2c,d and Extended Data Fig. 2a,b). Expression levels of HF marker genes such as Nppa (natriuretic peptide A), Nppb (natriuretic peptide B) and Myh7 (myosin, heavy polypeptide 7, cardiac muscle, beta) were significantly increased by TAC (WT-TAC) and even higher in OE-TAC mice (Fig. 2e). Histological analysis showed that cardiomyocyte size in WT-TAC mice was larger than in WT control mice and even larger in OE-TAC mice (Fig. 2f). Fibrosis was more pronounced in the hearts of WT-TAC mice than in WT control mice, and fibrosis in the perivascular region was slightly more pronounced in OE-TAC mice than in WT-TAC mice, but no difference was observed in the interstitial region (Fig. 2g). These results suggest that MYC in CFs has an important role in promoting HF, with a small contribution to fibrosis.

a, Experimental scheme for generating cardiac fibroblast-specific MYC-OE mice. 0, day of TAC; 12 weeks, 12 weeks after TAC. b, Western blot analysis of MYC and GAPDH expression in WT control, WT-TAC and OE-TAC mouse hearts. The experiment was repeated independently two times with similar results. c, Echocardiographic assessment of control and OE mice from before (0 week) to 12 weeks after TAC. d, Bar plots show cardiac function evaluated by echocardiography in control and OE mice before and after pressure overload (0–12 weeks). Means and standard errors are shown (TAC0w, control n = 6 and OE n = 5; TAC2w, 5 and 5; TAC4w, 6 and 5; TAC8w, 5 and 5; TAC12w, 4 and 5). Tcf21 iCre–R26StopFLMYC mice were used as control. *P < 0.05; P = 0.0014 (TAC12w); significance was determined using the two-sided Holm–Sidak test for mean comparisons. e, Relative mRNA expression of HF markers measured using RT-qPCR. Data are shown as mean ± s.d. *P < 0.05; P = 0.024 (control (n = 6) versus WT-TAC (n = 3), Nppa), P = 0.036 (WT-TAC versus OE-TAC (n = 5), Nppa), P = 0.0095 (control (n = 6) versus WT-TAC (n = 4), Nppb), P = 0.029 (WT-TAC versus OE-TAC (n = 4), Nppb), P = 0.49 (control (n = 4) versus WT-TAC (n = 4), Myh7), P = 0.029 (WT-TAC versus OE-TAC (n = 4), Myh7); significance was determined using the two-sided Mann–Whitney test for mean comparisons. f, Comparison of cardiomyocyte sizes among control, WT-TAC and OE-TAC mice. Cell size was evaluated using WGA staining. Scale bars, 50 μm. Representative histological data and quantitative analyses of the cell size in each mouse are shown (n = 100 cells each). The box plots show the median (center line) and first and third quartiles (box edges), while the whiskers go from each quartile to the minimum or maximum. *P < 0.05; P < 0.001 (control versus WT-TAC), P = 0.001 (WT-TAC versus OE-TAC); significance was determined using the two-sided Mann–Whitney test for mean comparisons. g, Comparison of fibrotic areas among the control, WT-TAC and OE-TAC mice. Fibrosis was evaluated in the perivascular and interstitial regions via azan staining. Scale bars, 300 μm. Representative histological data and quantitative analyses of the fibrotic areas in each mouse are shown (n = 5 each). Data are shown as mean ± s.d. *P < 0.05; NS, not significant; P = 0.0079 (control versus WT-TAC, perivascular), P = 0.016 (WT-TAC versus OE-TAC, perivascular), P = 0.0079 (control versus WT-TAC, interstitial), P = 1.0 (WT-TAC versus OE-TAC, interstitial); significance was determined using the two-sided Mann–Whitney test for mean comparisons.
Myc knockout in CFs ameliorates HF
To determine whether MYC in CFs is indispensable for HF development, we generated CF-specific MYC knockout (KO) mice by crossing MYC flox mice14 with Tcf21 iCre mice (Fig. 3a). TAX administration led to MYC deletion in CFs, resulting in significantly ameliorated cardiac dysfunction after TAC in KO mice (KO-TAC) compared with control mice (Fig. 3b–d and Extended Data Fig. 3a,b). HF marker gene expression was significantly suppressed in KO-TAC mice compared with that in WT-TAC mice (Fig. 3e). The heart size after TAC was smaller in KO-TAC mice than in WT-TAC mice (Extended Data Fig. 3b). Wheat germ agglutinin (WGA) staining of heart sections revealed that cardiomyocyte size was also significantly smaller in KO-TAC mice than in WT-TAC mice (Fig. 3f). Azan staining revealed no significant difference in fibrosis between KO-TAC mice and WT-TAC mice (Fig. 3g), suggesting that MYC in CFs is unlikely to contribute to fibrosis progression. Importantly, KO-TAC mice showed higher survival rates than control TAC mice (Fig. 3h). These results suggest that MYC expressed in CFs is a critical contributor to pressure-overload-induced HF development.

a, Experimental scheme for generating cardiac-fibroblast-specific Myc KO mice. 0, day of TAC; 12 weeks, 12 weeks after TAC. b, Relative mRNA expression of Myc in the heart as measured via RT-qPCR. Data are shown as mean ± s.d. *P < 0.05; P < 0.001 (control (n = 10) versus WT-TAC (n = 7)), P = 0.13 (control versus KO-TAC (n = 11)); significance was determined using the two-sided Mann–Whitney test for mean comparisons. c, Representative images of the echocardiographic assessment of control and KO mice before and after TAC (20 weeks). d, The bar plots show cardiac function evaluated by echocardiography in control and KO mice before and after pressure overload (0–20 weeks). Data are shown as mean ± s.e. (TAC0w, control n = 5 and KO n = 6; TAC2w, 4 and 6; TAC4w, 5 and 6; TAC8w, 4 and 6; TAC12w, 4 and 5; TAC14w, 4 and 5; TAC16w, 4 and 5; TAC20W, 3 and 4). Tcf21 iCre-MYC flox mice were used for control. *P < 0.05; P = 0.018 (TAC8w), P = 0.018 (TAC12w), P = 0.007 (TAC14w), P = 0.018 (TAC16w), P < 0.001 (TAC20w); significance was determined using the two-sided Holm–Sidak test for mean comparisons. e, Relative mRNA expression of HF markers measured using RT-qPCR (n = 4 each). Data are shown as mean ± s.d. *P < 0.05; P = 0.029 (control versus WT-TAC, Nppa), P = 0.029 (WT-TAC versus KO-TAC, Nppa), P = 0.029 (control versus WT-TAC, Nppb), P = 0.029 (WT-TAC versus KO-TAC, Nppb), P = 0.029 (control versus WT-TAC, Myh7), P = 0.029 (WT-TAC versus KO-TAC, Myh7); significance was determined using the two-sided Mann–Whitney test for mean comparisons. f, The size of cardiomyocytes in control, WT-TAC and KO-TAC mice was evaluated using WGA staining. Quantitative analyses of the cell size in each mouse are shown (n = 100 cells each). The box plots show the median (center line) and first and third quartiles (box edges), while the whiskers go from each quartile to the minimum or maximum. *P < 0.05; P < 0.001 (control versus WT-TAC), P < 0.001 (WT-TAC versus KO-TAC); significance was determined using the two-sided Mann–Whitney test for mean comparisons. g, Fibrotic areas in the control, WT-TAC and KO-TAC mice were evaluated at the perivascular and interstitial regions via azan staining. Scale bars, 300 μm. Representative histological data and quantitative analyses of the fibrotic areas in each mouse are shown (n = 5 each). Data are shown as mean ± s.d. *P < 0.05; P = 0.0079 (control versus WT-TAC, perivascular), P = 0.84 (WT-TAC versus KO-TAC, perivascular), P = 0.0079 (control versus WT-TAC, interstitial), P = 0.69 (WT-TAC versus KO-TAC, interstitial); significance was determined using the two-sided Mann–Whitney test for mean comparisons. h, Kaplan–Meier survival curves for control and KO mice after TAC were compared using log-rank tests (n = 11 (control) and 10 (KO)).
MYC in CFs regulates HF genes
To elucidate the molecular function of MYC in HF-Fibro, transcriptome analysis by RNA-seq was performed. Global gene expression analysis showed that CF-specific MYC deletion attenuated TAC-induced changes in the whole heart compared to WT-TAC mice (Fig. 4a and Extended Data Fig. 4a). Differentially expressed gene analysis showed that, compared with WT control mice, 1,261 genes were upregulated and 521 genes were downregulated in WT-TAC mice (Fig. 4b). GO term analysis showed that genes related to cardiac hypertrophy and HF were upregulated in WT-TAC mice compared with those in WT control mice (Fig. 4c and Extended Data Fig. 4b). On the other hand, 190 genes were upregulated and 296 genes were downregulated in KO-TAC mice compared with WT-TAC mice (Fig. 4b). GO term analysis showed that genes associated with cardiac hypertrophy and HF were reduced in KO-TAC mice compared with those in WT-TAC mice (Fig. 4d and Extended Data Fig. 4b). Gene set enrichment analysis (GSEA) showed negative enrichment of cardiac-hypertrophy-related genes and MYC-target genes in KO-TAC mice, while cardiac-fibrosis-related genes showed no significant difference between KO-TAC and WT-TAC mice (Fig. 4e–g and Extended Data Fig. 4c). These results also suggest that MYC regulates the expression of genes associated with cardiac hypertrophy and HF but not cardiac fibrosis.

a, Principal component analysis (PCA) visualization showing global gene expression in WT control, WT-TAC and KO-TAC mice (n = 3 each). b, Heat map showing the expression levels of differentially expressed genes in WT control, WT-TAC and KO-TAC mice. Left: differentially expressed genes in WT and WT-TAC mice. Right: differentially expressed genes between the WT-TAC and KO-TAC mice. c, GO analysis of differentially expressed genes. Genes were upregulated in WT-TAC mice compared with those in WT control mice. The red bars indicate GO terms associated with genes expressed in cardiomyocytes. The white bars represent other GO terms not specifically related to cardiomyocyte-expressed genes. d, GO analysis of differentially expressed genes. Genes were downregulated in KO-TAC mice compared with those in WT-TAC mice. The red bars indicate GO terms associated with genes expressed in cardiomyocytes. The white bars represent other GO terms not specifically related to cardiomyocyte-expressed genes. e, Top: GSEA of genes associated with MYC targets. Plots were prepared using the HALLMARK_MYC_TARGETS_V1 dataset (containing genes regulated by MYC) from the Molecular Signatures Database54. Bottom: GSEA of genes associated with the cardiac hypertrophy pathway. P values were derived from one-sided permutation tests with FDR correction. f, GSEA of genes associated with fibrosis. Genes associated with fibrosis were upregulated in WT-TAC mice compared with those in WT control mice. P values were derived from one-sided permutation tests with FDR correction. g, GSEA of genes associated with fibrosis. Genes associated with fibrosis were not significantly different between the KO and WT-TAC mice. P values were derived from one-sided permutation tests with FDR correction.
CXCL1 induces HF gene expression
To examine whether MYC-expressing CFs could induce cardiac hypertrophy and dysfunction through cellular interactions, we investigated factors secreted by these fibroblasts. We overexpressed MYC in cultured CFs and examined the effect of conditioned medium on cardiomyocytes of neonatal rats (Fig. 5a). The addition of medium conditioned from CFs overexpressing MYC induced HF marker gene expression in cardiomyocytes (Fig. 5b), suggesting paracrine effects on cardiomyocytes. Integrating single-cell and bulk RNA-seq data, we identified potential factors affecting cardiomyocytes. Focusing on overlapping genes (1) upregulated in WT-TAC mice, (2) downregulated in KO-TAC mice and (3) upregulated in HF-Fibro (subcluster 4), we identified two secreted factors, CXCL1 and TNFSF9 (Fig. 5c and Extended Data Fig. 5a). As MYC overexpression specifically upregulated Cxcl1 (chemokine (C–X–C motif) ligand 1) but not Tnfsf9 (tumor necrosis factor (ligand) superfamily, member 9) (Extended Data Fig. 5b), we focused on CXCL1. RNA-seq and quantitative PCR with reverse transcription (RT-qPCR) confirmed that Cxcl1, which was barely expressed in WT control mouse hearts, was upregulated in WT-TAC mouse hearts and downregulated in KO-TAC mouse hearts (Fig. 5d). To examine whether CXCL1 was a direct target of MYC, we performed chromatin immunoprecipitation (ChIP)-qPCR and confirmed that MYC binds to the promoter region, including E-box, of Cxcl1 (Fig. 5e and Extended Data Fig. 5c). Next, we examined whether CXCL1 acts directly on cardiomyocytes. RT-qPCR analysis showed that adding CXCL1 to cultured cardiomyocytes significantly increased HF marker gene expression (Fig. 5f and Extended Data Fig. 5d) and cardiomyocyte hypertrophy (Fig. 5g). Furthermore, we evaluated the contractile function of cardiomyocytes of neonatal rats, using a motion vector analysis system, the SI8000 Cell Motion Imaging System (SONY). The addition of CXCL1 to cultured cardiomyocytes significantly reduced contraction velocity (Fig. 5h). These results suggest that CXCL1 is a direct target of MYC in CFs and can act directly on cardiomyocytes.

a, Experimental setup. Culture medium conditioned by MYC overexpression in CFs using a retroviral vector was transferred to cultured neonatal rat cardiomyocytes (CM). b, Relative mRNA expression of HF markers in cardiomyocytes cultured in medium conditioned by MYC or GFP overexpression in CF. Data are shown as mean ± s.d. *P < 0.05; P = 0.036 (Nppa, GFP n = 3, MYC n = 5), P = 0.024 (Nppb, GFP n = 3, MYC n = 6), P = 0.024 (Myh7, GFP n = 3, MYC n = 6); significance was determined using the two-sided Mann–Whitney test for mean comparisons. c, Venn diagram of genes upregulated in WT-TAC mice versus WT control mice, downregulated in KO-TAC mice versus WT-TAC mice in RNA-seq data and upregulated in HF-specific CFs (cluster 4) versus other cluster 1–3 CFs in scRNA-seq data. A total of 21 common genes are listed. d, Top: genome browser view showing Cxcl1 gene expression in WT control, WT-TAC and KO-TAC mice. Bottom: relative mRNA expression of CXCL1 as measured by RT-qPCR (n = 4 each). Data are shown as mean ± s.d. *P < 0.05; P = 0.029 (control versus TAC), P = 0.029 (TAC versus KO); significance was determined using the two-sided Mann–Whitney test for mean comparisons. e, ChIP-qPCR showing MYC binding to the CXCL1 promoter region in CFs (n = 3 each). IgG, nonspecific IgG control; MYC, anti-MYC antibody. The red arrows indicate the region of the E-box in the CXCL1 promoter sequence, and the black arrows indicate primer positions in the Cxcl1 gene. Data are shown as mean ± s.d. *P < 0.05; P = 0.029 (IgG n = 4, MYC n = 3); significance was determined using the one-sided Mann–Whitney test for mean comparisons. f, Relative mRNA expression of HF markers determined by RT-qPCR after CXCL1 stimulation. Data are shown as mean ± s.d. *P < 0.05; P = 0.024 (Nppa, control n = 6, CXCL1 n = 3), P = 0.024 (Nppb, control n = 6, CXCL1 n = 3); significance was determined using the two-sided Mann–Whitney test for mean comparisons. g, Cardiomyocyte size in the control and CXCL1-stimulated conditions. Quantitative analyses of cell size are shown (n = 50 each, P < 0.001). Box plots show the median (center line) and first and third quartiles (box edges), while the whiskers go from each quartile to the minimum or maximum. *P < 0.05; significance was determined using the two-sided Mann–Whitney test for mean comparisons. h, Bar plots showing the effect of CXCL1 on the contractile properties of cardiomyocytes of neonatal rats. Contractile properties were analyzed using the SI8000 Cell Motion Imager. Each parameter was normalized by the value of the control well (n = 10 each, P < 0.001). Data are shown as mean ± s.d. *P < 0.05; significance was determined using the two-sided Mann–Whitney test for mean comparisons.
MYC–CXCL1–CXCR2 in HF pathogenesis
Although CXCL1 is a well-known chemokine that recruits inflammatory cells, its receptor, CXCR2, is widely expressed in a variety of cells, such as leukocytes, endothelial cells and smooth muscle cells15. As it is not known whether CXCR2 is expressed in cardiomyocytes, we examined its expression in cultured cardiomyocytes of neonatal rats and mice. Western blot analysis revealed that cardiomyocytes express CXCR2 (Fig. 6a), and RT-qPCR showed that blocking CXCR2 signaling with the neutralizing antibody inhibits the conditioned medium-induced increase in HF marker gene expression in cultured cardiomyocytes (Fig. 6b). In vivo, administration of the neutralizing antibody against CXCR2 in WT mice (anti-CXCR2 TAC mice) inhibited TAC-induced upregulation of HF markers and cardiomyocyte hypertrophy, and ameliorated cardiac dysfunction (Fig. 6c–e and Extended Data Fig. 6a,b), without affecting fibrosis (Fig. 6f), consistent with KO-TAC mice. Furthermore, we specifically knocked down CXCL1 in CFs by injecting an adeno-associated virus (AAV) vector with CXCL1 shRNA under the TCF21 promoter in TAC mice16, and specifically knocked down Cxcr2 (chemokine (C–X–C motif) receptor 2) in cardiomyocytes by injecting myoAAV2A vector with CXCR2 shRNA under the TnT promoter17. Echocardiograms revealed that at some time after TAC, knockdown of CF-specific CXCL1 significantly suppressed left ventricular remodeling and knockdown of cardiomyocyte-specific CXCR2 improved left ventricular dysfunction and remodeling (Fig. 6g and Extended Data Fig. 6c,d). Next, as it is well known that CXCR2 couples with Gαi/o as a G protein-coupled receptor, we assumed that CXCL1 might activate the Src–Ras–Raf–MEK–ERK pathway through CXCR2. To confirm this hypothesis, we examined the expression of p-ERK, an activated form of ERK, and found that CXCL1 induced activation of ERK, which was inhibited by the anti-CXCR2 antibody (Extended Data Fig. 6e).

a, Western blot analysis of CXCR2 expression in the negative control (NC), positive control (PC) and CM. Top: the NC is mouse embryonic fibroblasts, the PC is a human embryonic kidney cell line transfected with a Cxcr2 expression vector and the CM are cardiomyocytes of neonatal rats. Bottom: the NC is undifferentiated P19CL6 (mouse embryonic carcinoma) cells, the PC is a human embryonic kidney cell line transfected with a Cxcr2 expression vector and the CM are cardiomyocytes of mice. The experiment was repeated independently three times with similar results. b, Relative mRNA expression of HF markers in cardiomyocytes after addition of the medium conditioned by MYC overexpression in CFs, with and without a neutralizing antibody against CXCR2 (n = 3 each). Data are shown as mean ± s.d. *P < 0.05; P < 0.001 (GFP (n = 6) versus MYC (n = 10), Nppa), P = 0.0027 (MYC versus MYC plus anti-CXCR2 (n = 5), Nppa), P < 0.001 (GFP (n = 7) versus MYC (n = 10), Nppb), P = 0.0027 (MYC versus MYC plus anti-CXCR2 (n = 5), Nppb), P < 0.001 (GFP (n = 12) versus MYC (n = 9), Myh7), P = 0.0040 (MYC versus MYC plus anti-CXCR2 (n = 5), Myh7); significance was determined using the two-sided Mann–Whitney test for mean comparisons. c, Relative mRNA expression of HF markers measured by RT-qPCR in the hearts of control, TAC and TAC with neutralizing antibody against CXCR2. Data are shown as mean ± s.d. *P < 0.05; P < 0.001 (control (n = 10) versus TAC (n = 7), Nppa), P = 0.017 (TAC versus TAC plus anti-CXCR2 (n = 3), Nppa), P = 0.0020 (control (n = 10) versus TAC (n = 4), Nppb), P = 0.057 (TAC versus TAC plus anti-CXCR2 (n = 3), Nppb), P < 0.001 (control (n = 10) versus TAC (n = 6), Myh7), P = 0.038 (TAC versus TAC plus anti-CXCR2 (n = 4), Myh7); significance was determined using the two-sided Mann–Whitney test for mean comparisons. d, Bar plots showing cardiac function evaluated by echocardiography in TAC mice with (anti-CXCR2) and without neutralizing antibody against CXCR2 (control). Data are shown as mean ± s.d. (TAC0w, n = 8 (control) and 7 (anti-CXCR2); TAC2w, 6 and 6; TAC4w, 6 and 6; TAC8w, 6 and 7; TAC12w, 8 and 7; TAC14w, 5 and 6; TAC16w, 7 and 6). *P < 0.05; P = 0.036 (TAC12w), P = 0.0037 (TAC14w), P = 0.015 (TAC16w); significance was determined using the two-sided Holm–Sidak test for mean comparisons. e, Comparison of cardiomyocyte size between control and TAC mice with and without neutralizing antibody against CXCR2 (anti-CXCR2). Cell size was evaluated using WGA staining. Quantitative analyses of the cell size in each mouse are shown (n = 100 cells each). Box plots show the median (center line) and first and third quartiles (box edges), while the whiskers go from each quartile to the minimum or maximum. *P < 0.05; P < 0.001 (control versus TAC), P < 0.001 (TAC versus TAC + anti-CXCR2); significance was determined using the two-sided Mann–Whitney test for mean comparisons. f, Comparison of fibrotic areas between control and TAC mice with and without neutralizing antibody against CXCR2. Fibrosis was evaluated in the perivascular and interstitial regions by azan staining. Scale bars, 300 μm. Representative histological data and quantitative analyses of the fibrotic areas in each mouse are shown (n = 7 each). Data are shown as mean ± s.d. *P < 0.05; P < 0.001 (control versus TAC, perivascular), P = 0.54 (TAC versus TAC plus anti-CXCR2, perivascular), P < 0.001 (control versus TAC, interstitial), P = 0.38 (TAC versus TAC plus anti-CXCR2, interstitial); significance was determined using the two-sided Mann–Whitney test for mean comparisons. g, Bar plots showing cardiac function evaluated by echocardiography in control, and shRNA of CXCR2-injected mice (shCXCR2) after TAC. Data are shown as mean ± s.d. (n = 7 (control) and 5 (shCXCR2)). *P < 0.05; P = 0.025 (TAC8w); significance was determined using the two-sided Holm–Sidak test for mean comparisons. h, t-SNE plot of single-cell transcriptomes of human hearts (control: n = 1,673; HF: n = 29,496) (colored by cell clusters). Endo, endothelial cell; SM, smooth muscle cell; MC, myeloid cell; LC, lymphoid cell. i, t-SNE unsupervised clustering of fibroblasts in the human heart colored by cell clusters. j, Cell ratio of MYC-expressing CFs (control: n = 2; dilated cardiomyopathy (DCM): n = 4). Box plots show the median (center line) and first and third quartiles (box edges), while the whiskers go from each quartile to the minimum or maximum. k, Immunofluorescence for PDGFRα and smFISH for MYC and CXCL1 in human control hearts and DCM hearts. Scale bars, 20 μm. The experiment was repeated independently three times with similar results. The arrows indicate the colocalization of the MYC, CXCL1 and PDGFRα in the same cells. l, Bar plots showing the effect of CXCL1 on the contractile properties of iPSCM organoids. The plots show the rate of change of tissue contraction kinetics. Each parameter was normalized by the value of the control well (n = 14 each). Data are shown as mean ± s.d. *P < 0.05; P = 0.047; significance was determined using the two-sided unpaired t-test for mean comparisons.
Finally, to determine whether these findings might apply to human HF, we first examined whether MYC is also expressed in the CFs of patients with HF. We obtained cardiac biopsy samples from patients with HF undergoing left ventricular assist device (LVAD) insertion and from the hearts of patients without HF (control), which were then subjected to single-nucleus RNA-seq. Isolated nuclei were classified based on gene expression using t-SNE (Fig. 6h and Extended Data Fig. 7a–e). We extracted and classified the data from the fibroblast populations, and six subclusters with different gene expression patterns were identified (Fig. 6i and Extended Data Fig. 7f–j). The number of cells expressing MYC was not as high, but was higher in failing hearts (clusters 0, 1, 2, 3, 5; ~1.9%) than in control hearts (cluster 4; ~0.8%) (Fig. 6j and Extended Data Fig. 7i,j). We also performed smFISH and immunohistochemistry using formalin-fixed paraffin-embedded sections of human hearts to examine the expressions of MYC and CXCL1 (chemokine (C–X–C motif) ligand 1) in CFs. Although there were no MYC- or CXCL1-positive CFs in control hearts, MYC- and CXCL1-positive CFs were detected in failing hearts (Fig. 6k and Extended Data Fig. 8a). Our previous data showed expression of CXCR2 in human cardiomyocytes18 (Extended Data Fig. 8b), and western blot analysis showed expression of CXCR2 in human iPS-derived cardiomyocytes (iPSCM) (Extended Data Fig. 8c). RT-qPCR analysis showed that the addition of CXCL1 to iPSCM significantly increased expression levels of HF marker genes (Extended Data Fig. 8d). Functional analysis using human iPSCM organoids also showed that CXCL1 directly acts on human iPSCM and impairs the contractile function of their organoids (Fig. 6l). These results suggest that expression levels of MYC and CXCL1 also increase in CFs of human failing hearts and that CXCL1 may cause cardiac dysfunction via CXCR2 of human cardiomyocytes.
Discussion
Although CFs are thought to maintain cardiac homeostasis primarily by producing extracellular matrices, CFs communicate with other cell types, including cardiomyocytes, via direct interactions and paracrine signaling19,20. In response to diverse stresses under pathological conditions, CFs dynamically alter their phenotype, transitioning from resident fibroblasts to myofibroblasts and eventually matrifibrocytes after MI9,21. Notably, our scRNA-seq analysis identified heterogeneity in CFs across sham hearts, pressure-overload-induced hypertrophic hearts and failing hearts, revealing an HF-specific subpopulation of fibroblasts, HF-Fibro. HF-Fibro expressed Postn, which is expressed in fibroblasts activated by MI9, but not Acta2 or the osteochondral gene Chad (Extended Data Fig. 1c), suggesting that HF-Fibro are different from myofibroblasts or matrifibrocytes that appear in MI hearts.
The proto-oncogene Myc regulates cell proliferation and growth, and its role in tumorigenesis has been extensively studied22. In the cardiovascular field, Myc has been reported to be expressed in cardiomyocytes and induce their hypertrophic growth with fetal gene expression23, but its role in CFs remains unclear. MYC expression is intricately regulated transcriptionally and posttranscriptionally in response to various stimuli, including growth factors and cytokines24. In the heart, expression of the Myc gene has been reported to increase by a variety of stimuli, such as pressure overload24, catecholamines25, angiotensin II 26, and thyroid hormones27. The regulation of Myc expression in CFs of failing hearts is a subject for future research.
As CXCL1 is a chemokine, many studies have focused on its effects on blood cells28. CXCL1 expression has been reported to be upregulated in the heart of spontaneously hypertensive rats29 and induced by angiotensin II infusion30. The inhibition of CXCL1 was reported to reduce blood pressure and inhibit cardiomyocyte hypertrophy, and improve cardiac dysfunction by acting on blood cells in angiotensin II-infused mouse hearts31. The CXCL1 receptor CXCR2 is expressed on many other types of cells besides blood cells, and activation of CXCR2 triggers various intracellular signaling pathways32. We showed that Cxcl1 is expressed in HF-Fibro and that Cxcr2 is expressed in cardiomyocytes. CXCL1 directly upregulated HF-related genes in cardiomyocytes, and their upregulation in cardiomyocytes by culture medium conditioned with fibroblasts overexpressing MYC was inhibited by anti-CXCR2 antibody. In addition, not only anti-CXCR2 neutralizing antibody but also cardiomyocyte-specific CXCR2 knockdown ameliorated TAC-induced cardiac remodeling and dysfunction at some time after TAC. Because the increase in monocytes and macrophages, neutrophils and T cells in the heart after TAC was suppressed by the treatment with CXCR2 neutralizing antibody (Extended Data Fig. 9), immune cells may be involved in the pathogenesis of HF through activation of CXCL1–CXCR2 signaling, but taking all the results as a whole, CXCL1 induced by MYC in CFs might induce cardiac dysfunction at least partially via CXCR2 in cardiomyocytes. The lack of large differences may be due to the lack of high knockdown efficiency and high inter-mouse variability. Cell-specific knockout mice need to be generated to reach a clear conclusion, but this should be a future research project.
The single-nucleus RNA-seq analysis revealed that there were more MYC-positive CFs in human failing hearts than in normal hearts (Fig. 6j), but the number of MYC-positive CFs in human failing hearts was not large and it was difficult to show the CXCL1 expression in MYC-positive CFs in human failing hearts (Extended Data Fig. 7j). The reasons for this result are not clear at this time, but may be due to low expression levels of MYC and CXCL1, the small number of human samples, the limited area available for sampling, the lack of appropriate HF stages or causes that differ from mice and the fact that single-nuclear RNA-seq rather than scRNA-seq is biased. It is reported that the amount of mRNA in nuclei is much lower than that in whole cells, and the amount of mRNA in the nucleus and cytoplasm is different among cell types and various cellular conditions. These factors may result in differences in the diversity and proportion of cell types identified in the tissue from the same individual between scRNA-seq and snRNA-seq33. Therefore, we performed smFISH and immunohistochemistry using formalin-fixed paraffin-embedded sections of human hearts to examine the expressions of MYC and CXCL1 mRNA in CFs. There were MYC- and CXCL1-positive CFs in the human hearts of several types of HF such as dilated cardiomyopathy, dilated phase of hypertrophic cardiomyopathy and sarcoidosis but not of non-HF control (Fig. 6k and Extended Data Fig. 8a), suggesting a potential involvement of MYC- and CXCL1-positive CFs in human HF. In addition, we confirmed the expression of CXCR2 in human cardiomyocytes using single-nucleus RNA-seq data collected in our previous study18 (Extended Data Fig. 8b) and human iPSCM using western blot analysis (Extended Data Fig. 8c). We also showed that CXCL1 directly acts on human iPSCM and impairs the contractile function of their organoids (Fig. 6l and Extended Data Fig. 8d). Furthermore, a previous study on the plasma proteome reported that levels of CXCL1 were significantly higher in patients with HF than in non-HF controls31, suggesting that CXCL1 may play a role in the development of human HF. Taken together, these results suggest that the interaction between CFs and cardiomyocytes via CXCL1–CXCR2 may also be involved in HF pathogenesis in humans. Because the number of CFs expressing MYC and CXCL1 was lower in human hearts than in mouse hearts, further detailed studies using many human samples from various stages of HF are needed to confirm the involvement of HF-specific CFs in the pathogenesis of HF.
In this study, we identified a previously unrecognized CF population and uncovered a distinct role of CFs in the pathogenesis of HF. CFs dynamically change their subpopulations during HF development, communicate with cardiomyocytes, govern cardiomyocyte characteristics and regulate cardiac function. These findings highlight CFs and their cross-talk with cardiomyocytes as a potential therapeutic target in HF.
Methods
Mice
C57BL/6 mice were purchased from CLEA Japan, while R26StopFLMYC mice (stock number 020458, stock name: C57BL/6N-Gt(ROSA)26Sortm13(CAG-Myc,-CD2*)Rsky/J) were purchased from Jackson Laboratory. MYC flox mice were obtained from K. Nakayama (Kyushu University, Fukuoka, Japan)34. All mice were maintained under specific pathogen-free conditions at the animal facility of Keio University. Conditional deletion of Myc and conditional overexpression of Myc in CF were achieved by crossing MYC flox/flox homozygous mice with Tcf21 iCre hemizygous mice35 and R26StopFLMYC flox/flox homozygous mice with Tcf21 iCre hemizygous mice. MYCflox/flox;Tcf21 iCre (MYC-Tcf21-iCre) mice were used as MYC cardiac fibroblast-specific knockout (MYC KO) mice. R26StopFLMYC flox/flox mice; Tcf21 iCre (MYC-STOP-Tcf21-iCre) mice were used as MYC CF-specific overexpression (MYC OE) mice. All experiments involved male C57BL/6 mice aged 8–12 weeks, bred at the Keio University Animal Facility.
This study conformed to the ‘Guide for the Care and Use of Laboratory Animals’ published by the US National Institute of Health (NIH publication number 85e23, revised 1996), and the study protocol was approved by the Institutional Animal Care and Use Committee of Keio University School of Medicine.
scRNA-seq analysis
Non-cardiomyocytes were isolated from the left ventricular free wall after sham operation and at 2 weeks and 12 weeks after TAC. Echocardiography was performed to assess whether the heart was appropriately exposed to a pressure overload.
Non-cardiomyocyte extraction and scRNA-seq analysis were conducted as previously described12,36, and previously published scRNA-seq data were reanalyzed12.
Mice were euthanized by cervical dislocation, and hearts were perfused with 10 ml of ice-cold phosphate-buffered saline (PBS) via the right ventricle after incision of the left atrium. Hearts were excised, atria were removed and ventricles were minced in ice-cold PBS. The tissue was digested with type II collagenase (13.5 mg; Worthington Biochemical) and DNase I (2 mg; Qiagen) in F12 Ham’s medium (Thermo Fisher Scientific) for 30 min at 38 °C with stirring. After mechanical dissociation using an 18-G needle, digestion was stopped with ice-cold PBS. Following red blood cell lysis (RBC Lysis Buffer; Thermo Fisher Scientific), the suspension was filtered through 70-μm and 40-μm filters (pluriSelect). Cells were purified using Percoll (Cytiva) density gradient centrifugation and resuspended in CellCover (Anacyte Laboratories) for preservation.
Single-cell suspensions were processed using the Chromium Controller (10X Genomics), and cDNA libraries were sequenced on a HiSeq 3000 platform (Illumina). Data were processed with Cell Ranger (10X Genomics), and downstream analysis was performed in R 4.2.1 using Seurat 4.1.0, Monocle 3 v1.3.1 and circlize 0.4.15. After normalization and identification of variable features, datasets (TAC0w, TAC2w, TAC12w) were integrated using Seurat’s anchor-based approach. Clustering was performed using PCA and uniform manifold approximation and projection (UMAP) (dims = 1:30, resolution = 0.5). Major cell types were annotated by canonical markers, and subclustering was conducted for each lineage.
Immunohistochemistry
The murine hearts were fixed in 10% formalin neutral buffer solution (062-01661, Wako), embedded in paraffin and sectioned at 6-μm thickness. The sections were stained with hematoxylin and eosin and azan for routine histological examination under a light microscope. The sections were deparaffinized, blocked with a blocking buffer (ImmunoBlock; CTKN001, KAC) and then incubated with the following antibodies overnight at 4 °C under humidified conditions: rabbit monoclonal anti-PDGFRα antibody (prediluted; ab15501, Abcam) and chicken polyclonal anti-GFP antibody (1:100; NB100-1614, Novus Biologicals). For PDGFRα staining, a secondary antibody (goat anti-rabbit IgG [H + L] conjugated to Alexa Fluor 594 (1:200; A11012, Thermo Fisher Scientific)) was used, followed by incubation for 1 h at room temperature (20–25 °C). Rhodamine-conjugated WGA (1:200; RL-1022-5; Vector Laboratories) was used to visualize plasma membranes. Sections were mounted with DAPI (1:1,000; D1306, Thermo Fisher Scientific) and incubated for 10 min at room temperature. Images were obtained using a FV3000 confocal microscope (Olympus), an LSM 880 confocal microscope (Zeiss) and/or a BZ-9000 microscope (Keyence). Connective tissue was visualized via azan staining, and the fibrotic area was determined using ImageJ software (version 2.0.0-rc-69/1.52n, National Institutes of Health)37. Cardiomyocyte size was also analyzed using ImageJ software38.
smFISH
smFISH was performed with an RNAscope (Advanced Cell Diagnostics (ACD)) according to the manufacturer’s instructions using a probe against mouse MYC (NM_001177354.1, 413451, ACD), human MYC (NM_002467.4, 311761-C2, ACD) and human CXCL1 (NM_001511.4, 1256471-C3, ACD). RNA in situ hybridization was performed using an RNAscope 2.0 HD Brown Chromogenic Reagent Kit according to the manufacturer’s instructions (ACD). Target probes were designed using a previously described protocol39. Human tissues were fixed with G-Fix (Genostaff), embedded in paraffin on CT-Pro20 (Genostaff), using G-Nox (Genostaff) as a less toxic organic solvent than xylene, and sectioned at 4 μm. The probe was then hybridized for 2 h at 40 °C, followed by RNAscope amplification, and co-stained with Alexa Fluor 488-conjugated anti-PDGFRα antibody (1:100, 8871S; Cell Signaling Technology), anti-mouse collagen type 1 antibody (1:200, AB765P; Sigma-Aldrich) and DAPI to detect the cardiac fibroblasts and nuclei. Images were obtained using an LSM 880 confocal microscope (Zeiss).
Flow cytometry
Cardiomyocytes and non-cardiomyocytes were isolated using the following methods: mice were deeply anesthetized and intracardially perfused with 20 ml of ice-cold PBS to exclude blood cells. The heart was dissected, finely minced and then enzymatically digested with a cocktail of type II collagenase (LS004176; Worthington Biochemical) for 1 h at 38 °C with gentle agitation. After digestion, the tissue was triturated and passed through 70-µm and 40-µm cell strainers. Red blood cells were lysed using eBioscience 1xRBC Lysis Buffer (00-4333-57, Invitrogen, Thermo Fisher Scientific) for 5 min on ice. Single-cell suspensions were blocked using an Fc blocker (BUF041A, Bio-Rad Laboratories) for 10 min on ice. Cells were stained with a mixture of antibodies at 4 °C for 30 min. For intracellular staining, cells were fixed using a True-Nuclear Transcription Factor Buffer Set (424401, BioLegend) and stained with a mixture of antibodies at 4 °C for 30 min. Flow cytometry and sorting were performed using a FACSAria III instrument (BD Biosciences), BD FACS Melody (BD) and CytoFLEX S (Beckman Coulter). The following antibodies were used: rabbit monoclonal anti-MYC (ab32072, Abcam), anti-MYC antibody FITC (sc-40 FITC, Santa Cruz Biotechnology), anti-PDGFRα antibody APC (CD140a monoclonal antibody APC; 17-1401-81, Thermo Fisher Scientific), FITC anti-mouse CD3 (100204, BioLegend), APC anti-mouse/human CD11b (101211, BioLegend) and APC/Cyanine anti-mouse Ly-6G/Ly-6C (108424, BioLegend).
Western blot analysis
Cultured cells or murine left ventricular free wall was lysed in radioimmunoprecipitation buffer (08714-04, Nacalai Tesque) supplemented with a protease inhibitor (25955-11; Nacalai Tesque). Lysates were boiled with 4× Laemmli buffer (1610747; Bio-Rad Laboratories) for 5 min at 95 °C. Equal amounts of protein were combined with 12% precast polyacrylamide gels (Bio-Rad Laboratories) and transferred onto nitrocellulose membranes (GE Healthcare). Nitrocellulose membranes with proteins were washed with 0.05% Tween-20 in Tris-buffered saline (TBST), blocked (ImmunoBlock; CTKN001, KAC) for 30 min at room temperature (20–25 °C) and then incubated with primary antibody (goat polyclonal anti-GAPDH antibody (1:1,000; sc20357, Santa Cruz Biotechnology), rabbit monoclonal anti-MYC antibody (1:1,000; ab32072, Abcam), anti-CXCR2 antibody (1:1,000; PA1029; Boster Biological Technology), phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) antibody (1:1,000; 9101S, Cell Signaling Technology), p44/42 MAPK (Erk1/2) (137F5) antibody (1:1,000; 4695, Cell Signaling Technology) and CXCR2/CD182 Polyclonal Antibody (1:1000; Bioss)) at 4 °C overnight. After washing, the membranes were incubated with a secondary horseradish peroxidase (HRP) antibody (donkey anti-goat IgG-HRP (1:2,000; sc-2020; Santa Cruz Biotechnology) and donkey anti-rabbit IgG-HRP (1:2,000; NA934V; Sigma-Aldrich)) for 1 h at room temperature (20–25 °C). Membranes were detected using enhanced chemiluminescence (Chemi-Lumi One Super; Nacalai Tesque). Plasmid cDNA encoding CXCR2 (pCMV-CXCR2) was prepared by VectorBuilder and transfected into HEK293 cells using the FuGENE 6 transfection reagent (E2691, Promega) according to the manufacturer’s instructions. The HEK293 cell line was purchased from ATCC (CRL-1573; RRID: CVCL_0045).
Animal procedures and echocardiography
Tamoxifen was administered intraperitoneally at a treatment dose of 45 mg kg−1 for 5 consecutive days to activate the inducible MerCreMer protein, thereby inducing Cre recombinase activity. Male mice (8–12 weeks old) underwent TAC surgery to induce cardiac hypertrophy, HF or a sham operation40. TAC involved constricting the transverse aorta with a 7-0 silk suture parallel to a 27-gauge blunt needle, which was removed after constriction. Sham-operated mice underwent a similar surgical procedure without aortic constriction. Mice were anesthetized using 1.5% isoflurane inhalation and then anchored to a positioning platform in the supine position. Short-axis M-mode and Doppler echocardiographic measurements were performed using the Vevo 2100 Imaging System (FUJIFILM Visual Sonics) and SonoScape E2V (Shoei Japan). The interventricular septum, posterior wall and left ventricular internal end-systolic and end-diastolic diameters were measured using the leading-edge convention of the American Society of Echocardiography. The ejection fraction (EF) was calculated according to the following formula: EF (%) = ((LVEDV – LVESV)/LVEDV) × 100, where LVEDV is left ventricular end-diastolic volume and LVESV is left ventricular end-systolic volume. The heart rate was approximately 500–700 bpm and did not differ significantly among the groups during the echocardiographic assessments. In the echocardiographic procedure, Tcf21 iCre–R26StopFLMYC mice were used for the control of OE mice, and Tcf21 iCre-MYC flox mice were used for the control of KO mice.
Genotyping
DNA was isolated from the mouse tails using the HOTSHOT DNA preparation method41. PCR cycling conditions were as follows: 95 °C for 2 min; 36 cycles of 95 °C for 1 min, 58.8 °C for 30 s, 72 °C for 45 s and 72 °C for 5 min. All PCRs were performed using the GoTaq DNA Polymerase Kit (Promega). The primers are listed in Supplementary Table 1.
RNA extraction and RT-PCR
The mice were deeply anesthetized and killed by decapitation. A sternotomy was performed, cold PBS was perfused into the right and left ventricles and the hearts were extirpated. The cardiac tissue was immediately frozen in liquid nitrogen. Frozen heart tissues were homogenized using a homogenizer (MM400; Verder Scientific). These powders were dissolved in TRIzol reagent (Invitrogen, Thermo Fisher Scientific), and total RNA was extracted. Similarly, cultured mouse cardiomyocytes and CFs were washed with cold PBS and immediately dissolved in TRIzol reagent. Total RNA was reverse transcribed using ReverTra Ace qPCR RT Master Mix with gDNA Remover (Toyobo). mRNA expression was assessed via RT-qPCR using the Fast SYBR Green Master Mix (Applied Biosystems, Thermo Fisher Scientific). Samples were run on the StepOnePlus Real-Time PCR System (Applied Biosystems, Thermo Fisher Scientific), and data were analyzed using the delta-delta CT method. The housekeeping gene GAPDH was used as an internal control. The primer pair sequences were designed using Primer3Plus and are listed in Supplementary Table 2. Measurements were recorded in triplicate.
Adult mice CF culture
The following digestion media were prepared: Liberase TH/TM 50 µl per heart (5401135001/5401127001, Roche), HBSS 1 × 5 ml per heart (14025-092, Gibco, Thermo Fisher Scientific), DnaseI 2 µl per heart (314-08071, NIPPON GENE) and 10% Poloxamer 30 µl per heart (P5556, Sigma-Aldrich). Mice were deeply anesthetized and intracardially perfused with 20 ml of ice-cold PBS to exclude blood cells. The hearts were dissected and minced using fine scissors. The obtained cells were washed to remove contaminants with digestion media and then digested for 1 h at 37 °C with shaking at 100 rpm. After digestion, the cells were passed through a 70-µm cell strainer, washed three times with HBSS and centrifuged at 300g or 5 min at 25 °C. The cells were sown in a culture dish, and CFs were selectively attached to the cell culture dishes.
Neonatal ventricular cardiomyocyte culture
Neonatal ventricular cardiomyocytes from 1–2-day-old Sprague Dawley rats were subjected to Percoll gradient centrifugation and differential plating to enrich the cardiac myocyte population and reduce the number of non-cardiomyocytes42. Cardiomyocytes were cultured in a mixture of Dulbecco’s modified Eagle’s medium (DMEM, Sigma-Aldrich) and M199 (Sigma-Aldrich) with 10% fetal bovine serum (FBS, 26140-079, Gibco, Thermo Fisher Scientific). Stimulation involved 10 ng μl−1 CXCL1 (453-KC, R&D Systems).
Retrovirus production and treatment
For retrovirus production, Platinum E cells (1.2 × 106 cells per 60-mm dish) were seeded 1 day before transfection in DMEM supplemented with 10% FBS and 1% penicillin–streptomycin. On the day of transfection, when cells reached ∼80% confluence, DNA plasmids (pMXs-Puro-GFP retroviral vector and pMXs-Puro-MYC retroviral vector) were transfected using FuGENE 6 transfection reagent (E2691, Promega). After 24 h, adult mouse CFs were seeded in 6-well culture dishes for 1 h at a density of 1 × 105 cells per well. The medium was changed to DMEM supplemented with 10% FBS 24 h later, and polybrene was added to viral medium filtered through a 0.45-µm filter at 8 µg ml−1 concentration. The viral infection was repeated twice. At 24 h after the second infection, the viral medium was replaced with medium composed of DMEM and 10% FBS. At 3 days after the second infection, puromycin selection was performed by adding 10 mg ml−1 puromycin (ant-pr-5; InvivoGen) to each well.
RNA-seq sample preparation
Total RNA was extracted from whole hearts of sham, TAC-control, KO-TAC and OE-TAC mice using TRIzol Reagent (Invitrogen, Thermo Fisher Scientific), following the manufacturer’s protocol.
For the RNA-seq performed by Macrogen Japan, mRNA was isolated using the TruSeq Stranded mRNA LT Sample Prep Kit (Illumina), and libraries were prepared according to the TruSeq Stranded mRNA Sample Preparation Guide (Part number 15031047 Rev. E). Sequencing was performed on an Illumina NovaSeq 6000 platform using the NovaSeq 6000 S4 Reagent Kit with a paired-end 101-bp read length. HISAT2 was used for read alignment to the UCSC mm10 reference genome, and transcript abundance was estimated using StringTie to generate read counts and fragments per kilobase of exon per million mapped reads (FPKM) values. Differential gene expression analysis was performed using fold change and independent t-tests.
For another dataset, Rhelixa performed RNA-seq using the NEBNext Poly(A) mRNA Magnetic Isolation Module and NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (New England Biolabs), which applies the strand-specific dUTP method. Sequencing was performed using an Illumina NovaSeq 6000 system with 150-bp paired-end reads. Quality control, adapter trimming and low-quality read filtering were performed using FastQC and Trimmomatic. Reads were aligned using HISAT2, and gene expression was quantified by StringTie.
FPKM values were calculated, and differential gene expression was determined based on |fold change| ≥ 2 and P < 0.05 using independent t-tests.
Bioinformatic analysis of RNA-seq
Following sequencing, reads with Phred quality scores <20 and <35 bp after trimming were removed from further analysis using Trimgalore version 0.6.7. Quality-filtered reads were then aligned to the mouse reference genome GRCm38 (mm10) using the HISAT2 (v 2.2.1)43 aligner with default settings, and duplicates were marked using SAMtools version 1.4.1 (ref. 44). Aligned reads were quantified using StringTie version 2.2.1 (ref. 45) per gene ID against Gencode version M25 (ref. 46). Differential gene expression analysis was performed using the R package DESeq2 (version 1.38.3)47. Cutoff values of absolute fold change > 2.0 and false discovery rate (FDR) ≤ 0.1 were then used to select for differentially expressed genes between the control group and control TAC group. Fold change > 2.0 and FDR ≤ 0.3 were then used to select for differentially expressed genes between the control TAC group and KO-TAC group comparisons. GO enrichment and pathway analyses were performed using PANTHER and DAVID (v2021) gene functional annotation and classification tools to determine the molecular and biological functional categories48.
ChIP-qPCR analysis
For ChIP-qPCR sample preparation, CFs with or without MYC overexpression were cross-linked with 1% formaldehyde in PBS for 30 min and neutralized by the addition of glycine to a final concentration of 0.125 M for 5 min. CFs were collected and washed with cold PBS for ChIP49. For mouse heart sample preparation, ChIP was performed using ChIP-IT Express ChIP kits (Active Motif), following the manufacturer’s protocol. Briefly, cell lysates were sonicated (10 cycles of 30 s on–off) using a Bioruptor Pico sonicator (B01060010, Diagenode) to shear the DNA. Then, chromatin was incubated with indicated antibodies overnight at 4 °C. Pre-washed rinsed Dynabeads (protein G; 10003D, Thermo Fisher Scientific) were then added to the antibody-treated chromatin, and immunoprecipitation was performed on a rotator for 3 h at 4 °C. Antibodies used included an anti-MYC antibody (ab32072, Abcam), with rabbit IgG as the control.
Motion analysis of cardiomyocytes of neonatal rats
To evaluate the contractile function of CMs of neonatal rats, we used a motion vector analysis system, namely, the SI8000 Cell Motion Imaging System (SONY). This system enables quantitative analysis of the motion of synchronously beating CMs through video capture. The motion of each detection point was converted into a motion vector. The motion velocity within each region of interest was calculated based on the sum of the vector magnitudes. The maximum contraction velocity is considered to correspond to the contractile function of the myocardium. Video images were taken 15 min after drug administration, and the relative changes in contraction velocity were analyzed50.
AAV infection
The AAV vectors were prepared by VectorBuilder (https://en.vectorbuilder.com) according to established procedures51. Briefly, AAV vectors of serotypes 9 and MyoAAV2A (ref. 17) were generated in HEK293T cells, using triple-plasmid co-transfection for packaging. Viral stocks were obtained by CsCl2-gradient centrifugation. Titration of AAV viral particles was performed by real-time PCR quantification of the number of viral genomes, measured as CMV copy number. The viral preparations had a titer between 1 × 1012 and 5 × 1012 genome copies (GC) per ml. Viruses were administered in 100-μl saline via tail-vein injection. A total of 3 × 1011 GC doses of AAV9-Scramble shRNA (control vector) or 3 × 1011 GC doses of AAV9-shCxcl1, MyoAAV2A-shCxcr2 were administered to the mice 6 weeks after TAC surgery. Knockdown efficiency was evaluated by qPCR using the isolated cardiomyocytes (for the knockdown of Cxcr2) and fibroblasts (for the knockdown of Cxcl1).
Single-nucleus RNA-seq
Heart biopsy samples from patients with HF undergoing LVAD insertion and control subjects without HF were fresh frozen. We isolated nuclei using the Singulator 100 System (number 100-067-764, S2 Genomics) and a Singulator Nuclei Isolation Kit (100-060-817, S2 Genomics). Following the preparation of 5,000 nuclei to a concentration of 1,000 nuclei per µl, loading into the Chromium Controller (10x Genomics) and single-nucleus cDNA libraries were generated using a Chromium 3′v3 Chemistry Kit (PN-1000075, 10x Genomics). Libraries were sequenced on a NovaSeq 6000 System (Illumina) using a NovaSeq S4 Reagent Kit (200 cycles; 20027466, Illumina).
Single-nucleus RNA-seq analysis
Raw FASTQ files were processed for each sample using Cell Ranger software (ver 6.1.0, 10x Genomics) against the Cell Ranger GRCh38 human reference genome. Raw mapped counts were used as inputs for data processing with the Seurat R package (version 4.1.2)52. Data were processed using the CellBender software to exclude ambient RNA contamination53. We removed cells with detected genes <400, detected counts <5,000 and >20,000, and mitochondrial gene content >15%. Following the filtering step, we normalized read counts using the ‘NormalizeData’ function (10,000 default scale factor) for each dataset. Highly variable features were identified using the ‘FindVariableFeatures’ function, and integration was performed with the ‘FindIntegrationAnchors’ and ‘IntegrateData’ functions. The integrated data underwent dimensionality reduction and cluster detection. We performed linear regression using the ‘ScaleData’ function and a linear dimensionality reduction using the ‘RunPCA’ function. A total of 20 principal components were used for downstream graph-based, supervised clustering into distinct populations using the ‘FindClusters’ function, and UMAP dimensionality reduction was performed to project the cell population onto two dimensions using the ‘RunUMAP’ function. Cluster validation was carried out, and clusters with more than one marker gene recognizing a cell type overlapping within the same cluster, with expression values exceeding 1, were deemed doublet clusters and excluded from the analysis. After we retrieved the data for each cell type, we used the ‘FindVariableFeatures’ function to identify highly variable features for downstream analysis. We subclassified the PDGFRα-positive clusters as fibroblasts for subsequent analyses. We then used the ‘ScaleData’, ‘RunPCA’, ‘FindClusters’ and ‘RunTSNE’ functions on the subsetted cluster.
Patient recruitment and genetic analysis
All experiments using cells and tissues obtained from patients were approved by the Institutional Review Board of the University of Tokyo Hospital (approval numbers G-10032 and 11801). Detailed patient information is provided in Supplementary Table 3. All procedures were conducted in accordance with the Declaration of Helsinki, and written informed consent was obtained from all participants. Patients provided consent not only for tissue collection, but also for publication of their characteristics and data. Heart tissue was obtained within 24 h after death from noncardiac causes during autopsy or within 1 h after resection from patients during an LVAD transplantation procedure.
Functional analysis using human iPS-derived cardiomyocyte organoids
The culture device for microtissue formation was sculpted using polydimethylsiloxane; the device consists of two cylinders 500 µm in diameter and 1 mm high, spaced 500 µm apart, in a tub 2 × 3 mm square and 500 µm deep, and was manufactured with Ecoflex (Ecoflex 00-20, SMOOTH-ON). The precision aluminum mold f was fabricated using a 3D milling machine. The surfaces were dip coated with a thin layer of fluoropolymer (CYTOP, AGC). The uncured polymer solutions of Ecoflex were mixed, injected into the mold, defoamed in a vacuum chamber and allowed to cure at room temperature overnight. The devices were removed from the mold and irradiated with Excimer ultraviolet light (172 nm, E172-110, Excimer) for 15 s to hydrophilize the surfaces. The devices were coated with 3% BSA, and then tissue formation was performed with iPSCMs suspended in collagen gel. A solution of bovine acid-solubilized collagen I (FujiFilm Wako, final concentration 2.4 mg ml−1), 10X MEM medium (10%, v/v) and Matrigel (BD, final concentration 10%, v/v) were mixed on ice and neutralized with NaOH. Cellular density was adjusted to contain 5.0 × 104 cells per 3 µl of suspension per iPSCM organoid, and the solution was placed in molds and maintained at 37 °C for 45 min while maintaining saturated water vapor pressure. After gel formation, cells were cultured in DMEM medium containing 10% FBS, and the medium was changed every 2 days. The cell suspension gels contracted to bridge the columns and began to beat synchronously in about 48 h. Tissue contraction kinetics were evaluated after 1 week, and the effect of 10 µg ml−1 CXCL1 was assessed at day 10, with administration from day 7 to 10. After tissue preparation, the beating was filmed for 10 s using the Live Cell Imager SI8000 (SONY) to trace column deflection, and the deflection distance was quantified using the video analysis software KINOVEA (Kinovea). Force was estimated and calculated using the following equation.
Here F (µN) is the estimated tension, E (kPa) is Young’s modulus of the material, I is the cross-sectional secondary moment, L (mm) is the height of the displacement measurement point; x (mm) is the average height of the myocardial tissue attachment and δ (µm) is the pillar displacement. In this system, E = 83, I = 0.00307, L = 0.5 and substituting x = 0.5 yields
Statistical analysis
The data are presented as mean ± s.d. For comparisons between two groups, unpaired or Student’s two-tailed t-tests were performed as noted within the figure legends. Multiple-group comparisons were conducted using the Holm–Sidak test or Mann–Whitney test for mean comparisons. Unless stated otherwise, P < 0.05 was considered statistically significant. In the box plots, horizontal lines denote the medians, boxes depict the 25th–75th percentile and whiskers represent the minimum and maximum values.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The bulk RNA-seq data generated in this study have been deposited in the GEO repository under accession number GSE254172. The scRNA-seq data of mouse hearts were reported previously and have been deposited in the GEO repository under accession number GSE293319. The single-nucleus RNA-seq data for humans generated in this study have been deposited in the GEO repository under accession number GSE302337. All other data supporting the findings of this study are included in the article and Supplementary Information. Source data are provided with this paper.
References
Braunwald, E. The war against heart failure: the Lancet lecture. Lancet 385, 812–824 (2015).
Kho, C., Lee, A. & Hajjar, R. J. Altered sarcoplasmic reticulum calcium cycling—targets for heart failure therapy. Nat. Rev. Cardiol. 9, 717–733 (2012).
Mudd, J. O. & Kass, D. A. Tackling heart failure in the twenty-first century. Nature 451, 919–928 (2008).
Williams, R. S. Apoptosis and heart failure. N. Engl. J. Med. 341, 759–760 (1999).
Higo, T. et al. DNA single-strand break-induced DNA damage response causes heart failure. Nat. Commun. 8, 15104 (2017).
Nakada, Y. et al. DNA damage response mediates pressure overload-induced cardiomyocyte hypertrophy. Circulation 139, 1237–1239 (2019).
Pinto, A. R. et al. Revisiting cardiac cellular composition. Circ. Res. 118, 400–409 (2016).
Ko, T. et al. Cardiac fibroblasts regulate the development of heart failure via Htra3–TGF-β–IGFBP7 axis. Nat. Commun. 13, 3275 (2022).
Fu, X. et al. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J. Clin. Invest. 128, 2127–2143 (2018).
Aghajanian, H. et al. Targeting cardiac fibrosis with engineered T cells. Nature 573, 430–433 (2019).
Díez, J., González, A. & Kovacic, J. C. Myocardial interstitial fibrosis in nonischemic heart disease, part 3/4: JACC Focus Seminar. J. Am. Coll. Cardiol. 75, 2204–2218 (2020).
Katsuki, T. et al. Inhibition of Scarb1 on endothelial cells attenuates pressure overload-induced heart failure progression. JACC Basic Transl. Sci. https://doi.org/10.1016/j.jacbts.2025.05.003 (2025).
Calado, D. P. et al. The cell-cycle regulator c-Myc is essential for the formation and maintenance of germinal centers. Nat. Immunol. 13, 1092–1100 (2012).
Trumpp, A. et al. c-Myc regulates mammalian body size by controlling cell number but not cell size. Nature 414, 768–773 (2001).
Dhayni, K., Zibara, K., Issa, H., Kamel, S. & Bennis, Y. Targeting CXCR1 and CXCR2 receptors in cardiovascular diseases. Pharmacol. Ther. 237, 108257 (2022).
Francisco, J. et al. AAV-mediated YAP expression in cardiac fibroblasts promotes inflammation and increases fibrosis. Sci. Rep. 11, 10553 (2021).
Tabebordbar, M. et al. Directed evolution of a family of AAV capsid variants enabling potent muscle-directed gene delivery across species. Cell 184, 4919–4938.e22 (2021).
Nomura, S. et al. Cardiomyocyte gene programs encoding morphological and functional signatures in cardiac hypertrophy and failure. Nat. Commun. 9, 4435 (2018).
Vasquez, C. et al. Enhanced fibroblast–myocyte interactions in response to cardiac injury. Circ. Res. 107, 1011–1020 (2010).
He, K. et al. Long-distance intercellular connectivity between cardiomyocytes and cardiofibroblasts mediated by membrane nanotubes. Cardiovasc. Res. 92, 39–47 (2011).
Tallquist, M. D. & Molkentin, J. D. Redefining the identity of cardiac fibroblasts. Nat. Rev. Cardiol. 14, 484–491 (2017).
Baluapuri, A., Wolf, E. & Eilers, M. Target gene-independent functions of MYC oncoproteins. Nat. Rev. Mol. Cell Biol. 21, 255–267 (2020).
Xiao, G. et al. Inducible activation of c-Myc in adult myocardium in vivo provokes cardiac myocyte hypertrophy and reactivation of DNA synthesis. Circ. Res. 89, 1122–1129 (2001).
Komuro, I., Kurabayashi, M., Takaku, F. & Yazaki, Y. Expression of cellular oncogenes in the myocardium during the developmental stage and pressure-overloaded hypertrophy of the rat heart. Circ. Res. 62, 1075–1079 (1988).
Miki, N., Hamamori, Y., Hirata, K., Suematsu, M. & Kawashima, S. Transforming growth factor-beta 1 potentiated alpha 1-adrenergic and stretch-induced c-fos mRNA expression in rat myocardial cells. Circ. Res. 75, 8–14 (1994).
Sadoshima, J. I. & Izumo, S. Molecular characterization of angiotensin II-induced hypertrophy of cardiac myocytes and hyperplasia of cardiac fibroblasts critical role of the AT1 receptor subtype. Circ. Res. 73, 413–423 (1993).
Green, N. K., Gammage, M. D., Franklyn, J. A., Heagerty, A. M. & Sheppard, M. C. Regulation of β myosin heavy chain, c-myc and c-fos proto-oncogenes in thyroid hormone-induced hypertrophy of the rat myocardium. Clin. Sci. 84, 61–67 (1993).
Korbecki, J., Barczak, K., Gutowska, I., Chlubek, D. & Baranowska-Bosiacka, I. CXCL1: gene, promoter, regulation of expression, mRNA stability, regulation of activity in the intercellular space. Int. J. Mol. Sci. 23, 792 (2022).
Zhang, Y. L. et al. Selective blocking of CXCR2 prevents and reverses atrial fibrillation in spontaneously hypertensive rats. J. Cell. Mol. Med. 24, 11272–11282 (2020).
Kim, H. Y., Kang, Y. J., Song, I. H., Choi, H. C. & Kim, H. S. Upregulation of interleukin-8/CXCL8 in vascular smooth muscle cells from spontaneously hypertensive rats. Hypertens. Res. 31, 515–523 (2008).
Wang, L. et al. CXCL1–CXCR2 axis mediates angiotensin II-induced cardiac hypertrophy and remodelling through regulation of monocyte infiltration. Eur. Heart J. 39, 1818–1831 (2018).
Korbecki, J. et al. CXCR2 receptor: regulation of expression, signal transduction, and involvement in cancer. Int. J. Mol. Sci. 23, 2168 (2022).
Oh, J. M. et al. Comparison of cell type distribution between single-cell and single-nucleus RNA sequencing: enrichment of adherent cell types in single-nucleus RNA sequencing. Exp. Mol. Med. 54, 2128–2134 (2022).
Onoyama, I. et al. Fbxw7 regulates lipid metabolism and cell fate decisions in the mouse liver. J. Clin. Invest. 121, 342–354 (2011).
Tani, H. et al. Direct reprogramming improves cardiac function and reverses fibrosis in chronic myocardial infarction. Circulation 147, 223–238 (2023).
Macosko, E. Z. et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161, 1202–1214 (2015).
Kallikourdis, M. et al. T cell costimulation blockade blunts pressure overload-induced heart failure. Nat. Commun. 8, 14680 (2017).
Schneider, C. A., Rasband, W. S. & Eliceiri, K. W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012).
Wang, F. et al. RNAscope: a novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J. Mol. Diagn. 14, 22–29 (2012).
Sano, M. et al. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature 446, 444–448 (2007).
Truett, G. E. et al. Preparation of PCR-quality mouse genomic DNA with hot sodium hydroxide and tris (HotSHOT). Biotechniques 29, 52–54 (2000).
Katsumata, Y. et al. Endogenous prostaglandin D2 and its metabolites protect the heart against ischemia–reperfusion injury by activating Nrf2. Hypertension 63, 80–87 (2014).
Pertea, M., Kim, D., Pertea, G. M., Leek, J. T. & Salzberg, S. L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 11, 1650–1667 (2016).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Mudge, J. M. & Harrow, J. Creating reference gene annotation for the mouse C57BL6/J genome assembly. Mamm. Genome 26, 366–378 (2015).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Thomas, P. D. et al. PANTHER: making genome-scale phylogenetics accessible to all. Protein Sci. 31, 8–22 (2022).
Mardis, E. R. ChIP-seq: welcome to the new frontier. Nat. Methods 4, 613–614 (2007).
Ito, M. et al. Characterization of a small molecule that promotes cell cycle activation of human induced pluripotent stem cell-derived cardiomyocytes. J. Mol. Cell. Cardiol. 128, 90–95 (2019).
Ruozi, G. et al. AAV-mediated in vivo functional selection of tissue-protective factors against ischaemia. Nat. Commun. 6, 7388 (2015).
Stuart, T., Srivastava, A., Madad, S., Lareau, C. A. & Satija, R. Single-cell chromatin state analysis with Signac. Nat. Methods 18, 1333–1341 (2021).
Fleming, S. J. et al. Unsupervised removal of systematic background noise from droplet-based single-cell experiments using CellBender. Nat. Methods 20, 1323–1335 (2023).
Liberzon, A. et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740 (2011).
Acknowledgements
We thank Y. Suzuki, Department of Computational Biology and Medical Sciences, Graduate School of Frontier Sciences, the University of Tokyo, for next-generation sequencing support; K. Miyashita and K. Kinouchi, Department of Endocrinology, Metabolism and Nephrology, Keio University, for experimental support; and K. Nakayama, Anticancer Strategies Laboratory, Tokyo Medical and Dental University Institute for Advanced Study, for transferring the mice. This work was supported by grants from the Grant-in-Aid for Scientific Research (JSPS KAKENHI) program with grant numbers JP16H05304 (S. Yuasa), JP16K15415 (S. Yuasa), JP18K08047 (S. Yuasa), JP19H03622 (S. Yuasa), JP20H03678 (S. Yuasa), JP20K08461 (S. Yuasa), JP20K08193 (S. Yuasa), JP24K02452 (S. Yuasa), JP21J12663 (J.K.), JP23KJ0353 (J.K.), JP23K23798 (H.H.), JP23K18581 (H.H.), JP22H00471 (S.N.), JP21H05045 (I.K.), JP24K23940 (I.K.) and JP25H01050 (S.N.). Additional support came from the SENSHIN Medical Research Foundation (J.K., H.H., S. Yuasa), Keio University Grant-in-Aid for Encouragement of Young Medical Scientists (J.K.), Ushioda Memorial Foundation for Keio Doctoral Student Research Support Programme (J.K.), Daiwa Securities Health Foundation (J.K.), Grant for Basic Research of the Japanese Circulation Society (2020) (J.K.), Japan Heart Foundation Research Grant (J.K.), Japanese Heart Failure Society Basic Research Grant (J.K.), Sakakibara Heart Foundation Grant (J.K.), Cardiovascular Innovative Conference Grant (J.K.), Novartis Pharma Grants for Basic Research 2022 (J.K.), The Bayer Scholarship for Cardiovascular Research (J.K.), Mochida Memorial Foundation for Medical and Pharmaceutical Research Grants (J.K., H.H.), MSD Life Science Foundation (J.K., H.H.), Takeda Science Foundation (H.H.), Chugai Foundation for Innovative Drug Discovery Science: C-FINDs (S. Yuasa), The Mitsubishi Foundation (S. Yuasa), Astellas Foundation for Research on Metabolic Disorders (S. Yuasa), UTEC-Utokyo FSI Research Grant Programme (S.N.), JST FOREST Programme (grant number JPMJFR210U) (S.N.) and Japan Agency for Medical Research and Development (P18km0405209, JP21ek0109543, JP22ama121016, JP22ek0210172, JP22ek0210167, JP22bm1123011, JP23tm0724607, JP23gm4010020, JP223fa627011, JP22ek0109617, JP23tm0524009, JP23tm0524004, JP23jf0126003, JP24ek0109755, JP24ek0210205, JP24jf0126011, JP25bk0104192, JP25ek0109795 (S.N. and I.K.)).
Author information
Authors and Affiliations
Contributions
J.K., H.H., D.K., I.K. and S. Yuasa designed the experiments. J.K., T. Katsuki, M. Katoh, T. Ko, M. Ito, M. Katagiri, M. Kubota, S. Yamada, T.N., Y.A., T. Kouka, K.K., M. Kimura, S.I. and H.H. collected the data. J.K., T. Katsuki, D.K., M. Katoh, T. Ko, M. Ito and S. Yuasa analyzed the data. S. Yuasa, T. Katsuki, S.N., I.K., K.F. and M. Ieda supervised the study. J.K., H.H., I.K. and S. Yuasa wrote the paper.
Corresponding author
Ethics declarations
Competing interests
K.F. is a founding scientist funded by the SAB of Heartseed Co., Ltd. The other authors, J.K., H.H., T. Katsuki, D.K., M. Katoh, T. Ko, M. Ito, M. Katagiri, M. Kubota, S. Yamada, T.N., Y.A., T. Kouka, K.K., M. Kimura, S.I., S.N., I.K., S. Yuasa and M. Ieda, declare no competing interests.
Peer review
Peer review information
Nature Cardiovascular Research thanks Leslie Leinwand and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Single-cell RNA-seq of non-cardiomyocytes in the murine heart.
Related to Fig. 1. a: t-distributed stochastic neighbour embedding (t-SNE) visualisation of unsupervised clustering of non-cardiomyocytes. The cells (dots) are coloured according to the cell clusters, and the numbers represent cluster numbers. b: Non-cardiomyocytes classification based on gene expression using t-SNE. Non-cardiomyocytes were defined as cells expressing each marker gene. Marker genes include Car4 (capillary endothelium), Cd14 (macrophage), Acta2 (smooth muscle cells), Efnb2 (arterial endothelium), Cd80 (dendritic cells), Vwf (vein endothelium), Mki67 (Mki67 positive endothelium), Plp1 (nerves), Mmrn1 (lymphatic endothelium), Cd79a (B cells), and Cd3g (T cells). c: Violin plot depicting HF-Fibro (cluster 4) expression of Postn but not Acta2 or Chad. d: Violin plot showing the expression of transcription factors in six clusters. e: FACS analyses of PDGFRα and MYC expression in sham (TAC0w), TAC4w, TAC6w, and TAC 8w hearts.
Extended Data Fig. 2 Generation of cardiac fibroblast-specific MYC overexpression mice.
Related to Fig. 2. a: Bar plots illustrating left ventricle chamber size and wall thickness by echocardiography in control and OE mice pre- and post-pressure overload (TAC0w, n = 6 [control] and 5 [OE]; TAC2w, 5 and 5; TAC4w, 6 and 5; TAC8w, 5 and 5; TAC12w, 4 and 5). Tcf21 iCre-R26StopFLMYC mice were used as controls. b: Kaplan–Meier survival curves for control and OE mice after TAC were compared using the Gehan-Breslow-Wilcoxon test (n = 10 [control] and 20 [OE]).
Extended Data Fig. 3 Generation of cardiac fibroblast-specific MYC knockout mice.
Related to Fig. 3. a: FACS analyses of MYC-expressed fibroblasts in MYC KO hearts of TAC 12w. Negative control, secondary antibody alone. b: Bar plots showing left ventricle chamber size and wall thickness by echocardiography in control and MYC KO mice pre- and post-pressure overload (TAC0w, n = 5 [control] and 6 [KO]; TAC2w, 4 and 6; TAC4w, 5 and 6; TAC8w, 4 and 6; TAC12w, 4 and 6; TAC14w, 4 and 5; TAC16w, 4 and 5; TAC20w, 3 and 4). Tcf21 iCre-MYC flox mice were used as controls. *, p < 0.05; p = 0.018 (LVDs, TAC20w); significance was determined using the two-sided Holm-Sidak test for mean comparisons. Source data are provided as a Source Data file.
Extended Data Fig. 4 RNA-seq of WT control mice, WT-TAC mice, and KO-TAC mice.
Related to Fig. 4. a: Principal component analysis visualisation (PC1-PC2) showing global gene expression in hearts of WT control, WT-TAC, and KO-TAC mice (n = 3 each). b: Gene ontology (GO) analysis of differentially expressed genes. Left, Genes downregulated in WT-TAC mice vs. WT control mice. Right, Genes upregulated in KO-TAC mice vs. WT-TAC mice. c: GSEA of fibrosis-associated genes, indicating upregulation in WT-TAC mice vs. WT control mice and no significant difference between KO and WT-TAC mice.
Extended Data Fig. 5 Identifying secreted factors, such as CXCL1, as a direct target of MYC.
Related to Fig. 5. a: GO analysis bar graph for 21 candidate genes, revealing Cxcl1 and Tnfsf9 as intercellular signalling molecules. b, Relative mRNA expression of Cxcl1 and Tnfsf9 in CFs overexpressing MYC compared to that in the control (GFP). Data are shown as mean ± SD. *, p < 0.05; p = 0.0079 (CXCL1); significance was determined using the two-sided Mann-Whitney test for mean comparisons. Source data are provided as a Source Data file. c: ChIP-qPCR showing MYC at Cdh1, as a negative control, and Snail1, as a positive control, in MYC-overexpressing CF. IgG, nonspecific IgG control; MYC, anti-MYC antibody. d: Relative mRNA expression of the HF marker Myh7 after stimulation with CXCL1, measured by RT-qPCR. Data are shown as mean ± SD. *, p < 0.05, n.s., not significant; p = 0.26 (Control n = 6, CXCL1 n = 3); significance was determined using the two-sided Mann-Whitney test for mean comparisons. Source data are provided as a Source Data file.
Extended Data Fig. 6 Effect of CXCR2 neutralising antibody and targeted knockdown of CXCL1 in CFs and CXCR2 in cardiomyocytes.
Related to Fig. 6. a: Experimental scheme of TAC mice injected with a neutralising antibody against CXCR2. b: Bar plots showing left ventricle chamber size and wall thickness measured by echocardiography in control and anti-CXCR2 mice after TAC. Mean and standard error are shown (TAC0w, n = 8 [control] and 7 [anti-CXCR2]; TAC2w, 6 and 6; TAC4w, 6 and 6; TAC8w, 6 and 7; TAC12w, 8 and 7; TAC14w, 5 and 6; TAC16w, 7 and 6). *, p < 0.05; p = 0.040 (LVDs, TAC16w); significance was determined using the two-sided Holm-Sidak test for mean comparisons. Source data are provided as a Source Data file. c: Bar plots showing left ventricle chamber size and wall thickness measured by echocardiography in control and shCXCR2 mice after TAC. Mean and standard error are shown (n = 6 [control] and 5 [shCXCR2] for TAC0w, TAC2w, TAC4w, TAC6w, TAC8w, and TAC10w). *, p < 0.05; p = 0.015 (LVDd, TAC10w), p = 0.0020 (LVDs, TAC10w); significance was determined using the two-sided Holm-Sidak test for mean comparisons. Source data are provided as a Source Data file. d: Bar plots showing cardiac function, left ventricle chamber size, and wall thickness measured by echocardiography in control and shRNA of Cxcx1 injected mice (shCXCL1) after TAC. Mean and standard error are shown (n = 7 [control] and 5 [shCXCL1] for TAC0w, TAC2w, TAC4w, TAC6w, TAC8w, and TAC10w). *, p < 0.05; p = 0.0064 (LVDd, TAC10w); significance was determined using the two-sided Holm-Sidak test for mean comparisons. Source data are provided as a Source Data file. e: Left panel, Western blot analysis of p-ERK, ERK, and GAPDH expression in cardiomyocytes from neonatal rats treated with CXCL1 alone or in combination with the anti-CXCR2 antibody. Representative data are shown. Right panel, quantification of Western blot optical densities, with p-ERK/ERK ratios displayed. *, p < 0.05; significance was determined using the two-sided Mann-Whitney test for mean comparisons. Source data are provided as a Source Data file.
Extended Data Fig. 7 Single-nucleus RNA-seq analysis of patients with HF.
Related to Fig. 6. a: t-distributed stochastic neighbour embedding (tSNE) plot of human heart single-cell transcriptomes. The cells (dots) are coloured according to the cell clusters, and the numbers represent cluster numbers. b: Classification of whole heart cells based on gene expression using tSNE dimensionality reduction, identifying marker genes for cardiomyocytes, cardiac fibroblasts, endothelial cells, smooth muscle cells, myeloid cells, and lymphoid cells. c: tSNE plot of human heart single-cell transcriptomes. The cells (dots) are coloured according to the patient type. d: Feature plot displaying MYC expression on t-SNE of all cells in human heart. e: Violin plot showing the expression of MYC of all cells in human heart. f: t-SNE unsupervised clustering of fibroblasts in the human heart coloured by patient type. g: Gene Ontology analysis of differentially expressed genes. Genes were upregulated in human DCM CFs compared with those in human control CFs. h: Gene Ontology analysis of differentially expressed genes. Genes were downregulated in human DCM CFs compared with those in human control CFs. i: Feature plot visualising expression of MYC on t-SNE of fibroblasts. j: Violin plot showing the expression of MYC and CXCL1 in the six clusters.
Extended Data Fig. 8 Expressions of MYC and CXCL1 in patients with HF and the effect of CXCL1 on human cardiomyocytes.
Related to Fig. 6. a: Immunofluorescence for PDGFRα and single-molecule fluorescence in situ hybridization for MYC and CXCL1 in human dilated phase HCM (dHCM) and sarcoidosis hearts. Scale bars, 20 μm. Arrows indicate the colocalization of the MYC, CXCL1, and PDGFRα in the same cells. b: CXCR2 expression data using single-cell RNA-seq from our previous report. Horizontal axis, TNNT2 expression; vertical axis, CXCR2 expression. Black squares, cardiomyocytes from human control hearts; blue triangles, cardiomyocytes from human hearts of dilated cardiomyopathy. c: Western blot analysis of CXCR2 and GAPDH expression in iPSCM. d: Relative mRNA expression of HF markers in iPSCM (n = 3–5 each). Data are shown as mean ± SD. *, p < 0.05; p = 0.029 (Nppa), p = 0.036 (Nppb); significance was determined using the two-sided Mann-Whitney test for mean comparisons. Source data are provided as a Source Data file.
Extended Data Fig. 9 CXCR2 neutralising antibody suppresses TAC-induced increase in cardiac immune cells.
FACS analyses of CD3, CD11b, and Ly6G expression in mice hearts of sham (TAC0w), TAC4w, TAC8w, TAC12w, and TAC 12w with neutralising antibody against CXCR2.
Supplementary information
Supplementary Information (download PDF )
Supplementary Fig. 1.
Supplementary Tables 1–3 (download XLSX )
Supplementary Table 1: Genotyping primers. Supplementary Table 2: RT-qPCR primers. Supplementary Table 3: Patient information.
Source data
Source Data Fig. 1 (download XLSX )
Statistical source data for Fig. 1.
Source Data Fig. 2 (download XLSX )
Statistical source data for Fig. 2.
Source Data Fig. 3 (download XLSX )
Statistical source data for Fig. 3.
Source Data Fig. 4 (download XLSX )
Statistical source data for Fig. 4.
Source Data Fig. 5 (download XLSX )
Statistical source data for Fig. 5.
Source Data Fig. 6 (download XLSX )
Statistical source data for Fig. 6.
Source Data Extended Data Fig. 2 (download XLSX )
Statistical source data for Extended Data Fig. 2.
Source Data Extended Data Fig. 3 (download XLSX )
Statistical source data for Extended Data Fig. 3.
Source Data Extended Data Fig. 5 (download XLSX )
Statistical source data for Extended Data Fig. 5.
Source Data Extended Data Fig. 6 (download XLSX )
Statistical source data for Extended Data Fig. 6.
Source Data Extended Data Fig. 7 (download XLSX )
Statistical source data for Extended Data Fig. 7.
Source Data Extended Data Fig. 8 (download XLSX )
Statistical source data for Extended Data Fig. 8.
Source Data Figs. 1, 2 and 6 and Extended Data Figs. 6 and 8 (download PDF )
Unprocessed gels and western blots for Figs. 1j, 2b and 6a and Extended Data Figs. 6e and 8c.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Komuro, J., Hashimoto, H., Katsuki, T. et al. Heart failure-specific cardiac fibroblasts contribute to cardiac dysfunction via the MYC–CXCL1–CXCR2 axis. Nat Cardiovasc Res 4, 1135–1151 (2025). https://doi.org/10.1038/s44161-025-00698-y
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s44161-025-00698-y
This article is cited by
-
MYC–CXCL1–CXCR2 Axis in Heart Failure-Specific Cardiac Fibroblasts: a Potential Novel Therapeutic Target Beyond Cardiomyocytes
Journal of Cardiovascular Translational Research (2025)
