
Abstract
Axonal myelination finely tunes action potential conduction to control precise timing in neural circuits. Little is known about how dynamic myelinating oligodendrocytes are in the adult brain primarily due to limited approaches for their investigation at the cellular level over time in their native environment. This protocol describes optical imaging approaches that allow specific label-free detection of compact myelin which, when combined with genetically encoded fluorescence reporters and small molecule dyes, permits high-resolution longitudinal and fixed sample imaging of myelin and oligodendrocytes in live mice, in live organotypic slices, and in postmortem tissues. Data generated with these approaches can be used to test fundamental questions related to myelin development, plasticity, maintenance, and repair.
Similar content being viewed by others


Introduction
Myelin is a lipid-rich membrane that provides metabolic support to neurons1, is essential for rapid axonal conduction velocity, and decreases action potential failure rate, therefore fine-tuning neural circuits2. In the central nervous system, myelin is produced by oligodendrocytes, which arise from brain resident oligodendrocyte precursor cells (OPCs) throughout life3,4,5. Myelination continues into late adulthood6,7, and myelin plasticity (i.e. the addition of new myelin sheaths or structural alterations to existing sheaths) has been associated with learning and memory8,9,10,11,12,13,14. Consequently, myelin loss with age or in neurodegenerative disorders is thought to contribute to cognitive decline15,16,17.
Despite recent advances, many questions remain regarding oligodendrocyte lineage cell dynamics, including cellular and molecular mechanisms governing OPC fate decisions, successful oligodendrocyte differentiation and myelination, myelin sheath plasticity, and myelin loss; and how these dynamic processes contribute to neural plasticity and function. However, approaches to investigate oligodendrocyte and myelin dynamics at the single-cell level in the intact central nervous system remain limited.
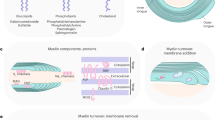
In this protocol, we describe approaches to use a combination of label-free imaging techniques, small molecule dyes, and fluorescent reporters to study oligodendrocyte and myelin dynamics with intravital and ex vivo imaging (Fig. 1). We detail how to implant a cranial window for chronic intravital imaging, how to generate organotypic slice cultures for longitudinal imaging of the oligodendrocyte lineage ex vivo, how to inject or apply small molecule dyes such as sulforhodamine 101 (SR101) or fluoromyelin for fluorescent labeling of oligodendrocytes and myelin sheaths, and how to image compact myelin in a label-free manner, using spectral confocal reflectance microscopy (SCoRe). Additionally, we describe different transgenic mouse lines that allow detailed labeling, imaging, and fate mapping of the oligodendrocyte lineage which can be combined with the approaches outlined above. Recent work has implemented these approaches to discover that oligodendrocytes and their myelin sheaths are not static structures in vivo. This highlights the need for further use of these technologies to investigate developmental myelination, myelin plasticity during adulthood, and myelin degeneration in aging and disease.

a Schematic of the imaging hardware. A confocal microscope can be used to image both label-free SCoRe and fluorescent proteins or dyes up to ~200 µm below the pial surface. A multiphoton microscope can be used to image fluorescent proteins or dyes up to ~500 µm below the pial surface. b SCoRe, fluorescent proteins, and small molecule dyes can be used to image OPCs, oligodendrocytes and myelin in vivo. c SCoRe, fluorescent proteins, and small molecule dyes can also be combined with immunohistochemical staining in fixed tissue. d Label-free and fluorescent imaging of oligodendrocyte lineage cells and myelin can be used to probe mechanisms governing OPC proliferation and differentiation into myelinating oligodendrocytes, as well as myelin sheath plasticity.
Methods
Microscopes
For SCoRe imaging, a laser scanning confocal microscope with at least 2 lasers spaced 75 nm apart and tunable detectors should be used (for example, the imaging in this study was performed with Leica SP5 and Leica SP8 microscopes). Genetically encoded fluorophores or small molecule dyes can be imaged with either a laser scanning confocal microscope or a laser scanning multiphoton microscope (for example, the data in this study were acquired with Brucker multiphoton microscopes equipped with MaiTai or InSight X3 femtosecond pulsed lasers (Spectra Physics)). Cranial windows should be imaged with upright microscopes, while slice cultures should be imaged with inverted microscopes. For slice culture imaging, we used an InverterScope objective inverter (LSM Tech) mounted on our upright microscopes.
Transgenic mice
All animal procedures were approved by the Institutional Animal Care and Use Committee at Yale University and Dartmouth College. When appropriate, mice were deeply anesthetized by either an intraperitoneal injection of 100 mg/kg ketamine and 10 mg/kg xylazine or by 2% isoflurane inhalation and euthanized by cervical dislocation or perfusion-fixation. We used Cnp-mEGFP18 mice (JAX: 026105) to label myelinating oligodendrocytes with membrane tethered EGFP, Cspg4-CreER5 mice (JAX: 008538) for conditional labeling of Cspg4 expressing cells including OPCs and their progeny, and Plp-CreERT19 mice (JAX: 005975) for conditional labeling of oligodendrocytes. Cspg4-CreER and Plp-CreERT mice were crossed with either Ai920 (JAX: 007909) floxed cytosolic tdTomato Cre reporter mice or mT/mG21 (JAX: 007586) membrane tethered EGFP Cre reporter mice. Thy1-YFP22 (JAX: 003782) mice were used to label subsets of cortical pyramidal neurons and subcortical neuronal populations.
Equipment
Dissecting scissors
Dissecting forceps
Dissecting microscope
Double edged razor blade
Iodine swabs
Ethanol swabs
Coverglass, 3mm-4mm diameter
Dental Drill
Drill Bit
Glass Bead Sterilizer
Autoclave
Tissue Slicer or Vibratome
Microscope Stage-top Incubator
Reagents
Sterile phosphate buffered saline (PBS)
Sterile artificial cerebrospinal fluid (ACSF)
Sterile saline
Ketamine
Xylazine
Carprofen
Eye ointment
Dental Cement
70% Ethanol
Cyanoacrylate glue
Fluoromyelin
SR101
4% paraformaldehyde
Cranial window surgery
-
1.
Autoclave instruments before beginning the surgery. A glass bead sterilizer can be used to sterilize instruments between surgeries on the same day.
-
2.
Anesthetize the mouse using a 100 mg/kg ketamine and 10 mg/kg xylazine intraperitoneal injection or 2% isoflurane. If using isoflurane, maintain anesthesia with 1.5-1.8% isoflurane throughout the surgical procedure.
-
3.
Administer analgesia, 5 mg/kg carprofen.
-
4.
Shave the head with a razor blade or clippers.
-
5.
Sterilize the shaved area with iodine and ethanol swabs.
-
6.
Apply eye ointment.
-
7.
Cut the skin to expose the skull.
-
8.
Gently remove the connective tissue on the skull with a razor blade to dry the skull. Air puffs can help to dry the skull.
-
9.
Glue the edges of the skin to the skull.
-
10.
Glue a headplate or nut onto the skull.
-
11.
Apply dental cement to hold the headplate or nut in place and cover the exposed skull (except where the craniotomy will be performed).
-
12.
Headfix the mouse using the headplate or nut.
-
13.
Drill a circle slightly wider than the glass coverslip diameter (e.g. for a 3 mm coverslip, drill a 3.5 mm diameter circle). Sterile ACSF can be applied to check skull thickness but should be dried off again before continuing to drill.
-
14.
To check skull thickness, lightly tap the center of the circle; if flex is observed the skull cap is ready to be removed for the craniotomy.
-
15.
Apply sterile ACSF and gently lift the skull to expose the brain. ACSF should now always be present to avoid the brain drying out.
-
16.
If needed, remove the dura. A 30 G needle can be used to pierce the dura to assist in removal.
-
17.
Remove excess ACSF.
-
18.
Gently place the coverslip over the exposed brain, while removing excess ACSF.
-
19.
Holding down the coverslip, remove all excess ACSF and glue the edges of the coverslip to the skull.
-
20.
Administer analgesia, 5 mg/kg carprofen.
-
21.
Perform post operative monitoring daily for 48 h following surgery. Administer analgesia, 5 mg/kg carprofen at 24 and 48 h.
Cranial windows can be imaged acutely immediately following the surgery, or starting 3 weeks after the surgery, to allow for the surgery-induced inflammation to resolve.
Organotypic slice culture
Here, we briefly describe how to prepare organotypic brain slice cultures. For a detailed step-by-step protocol, see Hill et al.23 and Sherafat et al.24.
-
1.
Place tissue culture inserts in 6-well plates, add 1 ml of media, and place in an incubator.
-
2.
Deeply anesthetize postnatal day 8-10 mice and decapitate them with scissors.
-
3.
Dissect the forebrain and cerebellum.
-
4.
Place the dissected tissue in ice cold and oxygenated artificial cerebral spinal fluid.
-
5.
Cut 300 µm thick coronal forebrain slices or sagittal cerebellar slices with a vibratome or manual tissue chopper and place the slices in cold oxygenated artificial cerebral spinal fluid to dissect the regions of interest.
-
6.
Place the dissected regions of interest on tissue culture inserts
-
7.
Change the culture media the following day, and every 2-3 days thereafter.
-
8.
Image the slices in glass bottom culture dishes with an inverted fluorescence microscope equipped with a stage-top incubator.
It is important to culture slices for at least one week before live imaging and experimentation to allow for recovery from the dissection.
Preparing fixed tissue slices
-
1.
Anesthetize the mouse using a 100 mg/kg ketamine and 10 mg/kg xylazine intraperitoneal injection or 2% isoflurane.
-
2.
Perfuse-fix the mouse with 4% PFA.
-
3.
Dissect the brain and submerge the tissue in 4% PFA for 24 h for post fixation.
-
4.
Wash in PBS.
-
5.
Cut 50-150 µm thick slices with a vibratome. Tissue sections can be stored at 4 °C in PBS with 0.01% sodium azide or stored at −20 °C in cryostorage solution (1% Polyvinylpyrrolidone, 30% sucrose, and 30% ethylene glycol in 0.2 M sodium phosphate buffer (pH 7.4)).
When storing slices in cryostorage solution, wash the slices three times in PBS before proceeding with staining and imaging.
Imaging oligodendrocyte lineage cells and myelin with genetically encoded fluorescent proteins
Several mouse lines have been generated to allow for the visualization of oligodendrocyte lineage cells, providing a range of options to label cells with different reporter proteins at different stages in the lineage (Fig. 2). When selecting a mouse line, it is important to consider the fluorophore (e.g. ECFP, mEGFP, EYFP, DsRed, tdTomato, or mKate2) to ensure that it can be excited, and the emitted light collected, with the available hardware. Moreover, cellular localization of the fluorophore can impact detection. For instance, in fine OPC processes, membrane anchored calcium indicators allow for the detection of more calcium transients than cytoplasmic calcium indicators25. Similarly, as the cytoplasm is extruded during myelin compaction, compact myelin is likely easier to visualize with fluorophores targeted to the membrane (e.g. Cnp-mEGFP, Plp-CreERT:mT/mG, Cspg4-CreER:mT/mG) rather than with cytoplasmic labels26 (e.g. Plp-DsRed26, Mobp-EGFP27, Cspg4-CreER:Ai9) (Fig. 2b), although it should be noted that membrane-tethered fluorescent proteins can also be excluded during myelin compaction28. It is also important to consider how fluorophore expression is regulated. For example, fluorophores in mouse lines such as NG2-mEGFP29, NG2-DsRed30, or NG2-EYFP31 are constitutively expressed in oligodendrocyte precursor cells, while fluorophores in lines such as Cnp-mEGFP, Plp-DsRed, or Mobp-EGFP are constitutively expressed in differentiating and mature oligodendrocytes, and their myelin sheaths. If needed, temporal control can be achieved with tamoxifen-inducible expression, using mouse lines such as Cspg4-CreER (to label OPCs and new oligodendrocytes) or Plp-CreERT (to label existing oligodendrocytes) crossed to mouse lines with floxed fluorophores (e.g. tdTomato, mT/mG) (Fig. 2). Using a low tamoxifen concentration can help achieve sparse labelling if required, for instance when analyzing properties of individual cells. Here, we describe how to image genetically encoded fluorophores acutely and longitudinally in the intact brain (Figs. 2 and 4), in slice cultures (Fig. 5), or in fixed brain slices (Figs. 6 and 7).
-
1.
Perform a cranial window surgery on, prepare slice cultures with, or perfuse-fix mice expressing genetically encoded fluorescent proteins (see above for step-by-step details for these procedures).
-
2.
Place the sample on the microscope.
-
a.
For intravital imaging, anesthetize a mouse that has a cranial window with a 100 mg/kg ketamine and 10 mg/kg xylazine intraperitoneal injection or 2% isoflurane. If using isoflurane, maintain anesthesia with 1.5–1.8% isoflurane throughout the imaging session. Place the mouse on the microscope and headfix it using a headplate or nut. Level the window to maintain consistency longitudinally.
-
b.
For slice culture imaging, place the slice culture insert into a glass bottom 6-well plate and add the plate to a stage top incubator.
-
c.
For fixed tissue imaging, mount the slices and add a glass coverslip.
-
a.
-
3.
Add water and lower the objective (for example 20× 1.0 NA) onto the window or slice culture.
-
4.
Choose an imaging position.
-
5.
Capture a z-stack and/or time lapse images as needed.
-
6.
For longitudinal imaging, take a low zoom image of the surface of the brain or the slice to capture vascular patterns or other landmarks in the tissue.
-
7.
Take a landmark image using a camera on the microscope or a cell phone through the eyepiece to relocate the imaging position.
-
8.
Repeat steps 2 to 5 for each imaging session, or as required depending on the experimental design.

a Conditional expression of a membrane-anchored fluorescent protein in an OPC (left) and an oligodendrocyte (right). White arrows indicate cell soma. b Oligodendrocytes labeled with both a conditionally expressed cytoplasmic fluorescent protein and a constitutive membrane-tethered fluorescent protein. Arrows indicate an oligodendrocyte cell soma. c Longitudinal imaging of two OPCs differentiating. The top OPC successfully differentiates into a CNP+ oligodendrocyte while the bottom cell eventually dies. The yellow arrow indicates the newly differentiated Cnp-mEGFP+ oligodendrocyte and the white arrows indicate new myelin sheaths.
Fluorescent proteins can be imaged on either a confocal microscope (down to ~200 µm below the pial surface) or with a multiphoton microscope (down to ~500 µm below the pial surface). Using a confocal microscope will generate higher resolution images, but if the window quality diminishes over time and the fluorescent signal becomes dim, switching to a multiphoton microscope can help. Note differences in objective lenses, microscope optics, etc. when transitioning between different microscopes.
Intravital imaging of genetically encoded fluorescent reporters in mice or zebrafish has been used to investigate OPC motility and migration29,32,33,34, proliferation29,33,35, and differentiation dynamics (Fig. 2c)7,29,35,36,37,38, as well as other potential OPC functions such as synapse engulfment39 or axonal remodeling40. Intravital imaging of fluorescent proteins and calcium indicators in zebrafish has allowed the investigation of mechanisms regulating myelination during early development28,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57, while recent work in mice has demonstrated that myelin sheaths in the cortex remain plastic during adulthood6,7, and can remodel in response to learning14 or sensory deprivation58. In addition, several groups have used genetically encoded fluorophores to image demyelination and remyelination in live mouse or zebrafish demyelination models59,60,61,62,63,64,65,66,67,68, as well as myelin degeneration with aging6,63. Thus, this technique is a powerful tool to study oligodendrocyte and myelin dynamics in vivo. However, there are limitations to using genetically encoded fluorescent proteins, including the need for a genetically tractable model organism, possible phototoxicity due to fluorescence bleaching generating reactive oxygen species, or insufficient fluorophore intensity for intravital imaging. Some of these limitations can be overcome by label-free imaging methods or small molecule dyes, as we describe below.
Imaging compact myelin with SCoRe
SCoRe allows for the label-free visualization of compact myelin using a standard confocal microscope69 (Fig. 3). As compact myelin is a lipid-rich multi-lamellar structure, it has a higher refractive index than the surrounding brain tissue, and therefore, reflects incident light. Individual wavelengths lead to reflection in different locations across a myelin sheath, creating a patchy pattern of myelin (Fig. 3d). By combining multiple laser wavelengths and collecting the reflected light with a tunable detection system, whole compacted myelin sheaths can be visualized (Fig. 3). This can be achieved by using a white-light broadband laser and collecting reflectance from wavelengths spaced 10 nm apart. However, white-light lasers are not widely available, and this is time consuming and generates large data files. In addition, wavelengths spaced ~10 nm apart generate similar reflectance pattern, and therefore, using a white-light laser does not necessarily yield more contiguous myelin signal than combining the reflectance signal from three wavelengths spaced ~75 nm apart: 488 nm, 561 nm and 633 nm69 (Fig. 3e). Thus, depending on the number of available laser lines and detectors, we recommend using two to six wavelengths for SCoRe. The reflectance from individual wavelengths can be collected either simultaneously or sequentially, depending on the number of detectors available. Similarly, reflectance can be acquired either simultaneously with or sequentially to fluorescence from exciting genetically encoded fluorescent proteins, small molecule dyes, or fluorescent antibodies. While SCoRe imaging has been performed with multiple types of lasers (e.g. argon, helium-neon, continuous-wave diode or diode-pumped solid-state), how the physical properties of the laser beam impact SCoRe detection has not been systematically investigated, and the imaging parameters should therefore be optimized for each imaging setup. SCoRe can be used to detect compact myelin in vivo through a cranial window (Figs. 3 and 4), or ex vivo in, for instance, organotypic slices (Fig. 5) or fixed brain slices (Figs. 6 and 7).
-
1.
Place the sample on the confocal microscope
-
a.
For intravital imaging, anesthetize a mouse that has a cranial window with a 100 mg/kg ketamine and 10 mg/kg xylazine intraperitoneal injection or 2% isoflurane. If using isoflurane, maintain anesthesia with 1.5-1.8% isoflurane throughout the imaging session. Place the mouse on the microscope and headfix it using a headplate or nut. Level the window to avoid unwanted reflection from the glass coverslip and maintain consistency longitudinally.
-
b.
For slice culture imaging, place the slice culture insert into a glass bottom 6-well plate and add the plate to a stage top incubator.
-
c.
For fixed tissue imaging, mount the slices in PBS and add a glass coverslip, ensuring there is enough PBS to keep the tissue hydrated, but not so much that the coverslip might drift. This step is critical to maintain refractive index differences across the tissue as many mounting media components will normalize the refractive index and thus diminish the SCoRe signal.
-
a.
-
2.
Add water and lower the objective (for example 20× 1.0 NA) onto the window or slice culture.
-
3.
Choose an imaging position.
-
4.
Use two to six laser lines, and center the tunable detectors around the laser wavelengths: for example, the 488 nm, 561 nm, 633 nm laser lines, with detectors centered at 486-491 nm, 559-564 nm and 631-636 nm to collect the reflected light. Laser intensities should be significantly lower than traditionally used for fluorescence imaging and all laser intensities and detector gains should be adjusted to equalize the signals from each wavelength.
-
5.
Capture a z-stack and/or time series images as needed.
-
6.
After the image is acquired, the individual SCoRe channels can all be pseudo colored with the same color to reconstitute the full myelin sheath (Fig. 3).
-
7.
For longitudinal imaging, take a low zoom image of the surface of the brain or slice to capture vascular patterns or other landmarks in the tissue.
-
8.
Take a landmark image using a camera on the microscope or a cell phone through the eyepiece to relocate the position.
-
9.
Repeat steps 1 to 6 for each imaging session, or as required depending on the experimental design.

a Schematic of intravital SCoRe imaging. b Multi-lamellar compact myelin has a high refractive index and therefore reflects incident light. c Intravital image of cortical layer 1. Myelin can be visualized with the reflection of incident light across multiple wavelengths. d Individual wavelengths lead to reflection in different locations across a myelin sheath. Combining multiple laser lines allows the whole sheath to be detected. e Boxed area in (d). Combining 3 wavelengths ~75 nm apart is sufficient to detect the whole myelin sheath. f Fluorescence intensity of individual wavelengths across a 12 µm segment of myelin. g Overlay of the reflected light collected from four laser lines: 458 nm, 488 nm, 555 nm, and 633 nm. The SCoRe overlay can be used to detect patterns of compact myelin in the mouse cortex throughout life.

a A single OPC captured through a cranial window in a Cspg4-CreER:Ai9 mouse imaged every 5 min revealing motility of some processes (yellow arrowheads) but not others (white arrowheads). b Longitudinal intravital imaging of oligodendrocyte generation (yellow arrow) in the cerebral cortex. c single myelin sheaths exhibit length plasticity (yellow arrowheads) while other sheaths are stable over the same time interval (white arrowheads). Notice that the segment associated with extension has fluorescent membrane labeling but no SCoRe signal suggesting that segment of the myelin sheath is not compact at these time points.

a Images of oligodendrocytes and their myelin sheaths captured from a live cerebellar organotypic slice culture. b Example time series of longitudinal imaging of oligodendrocytes and their myelin sheaths in hippocampal organotypic slice cultures. The images on the top show a single oligodendrocyte (white arrows) captured over 100 days. The middle images show the generation of a new oligodendrocyte (yellow arrows). The bottom images show myelin sheath extension (yellow arrowheads).

a Left: tiled image of a single z-plane from a fixed Cnp-mEGFP mouse coronal forebrain slice. Myelin can be visualized with both SCoRe and mEGFP fluorescence. Right: maximum projection of upper cortical layers, and single z-plane of the boxed area in (a). SCoRe signal overlaps with EGFP for myelin sheaths parallel to the imaging plane, but not for myelin sheaths orthogonal to the imaging plane. b Left: Tiled image of a single sagittal cerebellar z-plane from a fixed Cnp-mEGFP mouse. Myelin can be visualized with both SCoRe and mEGFP fluorescence. Right: maximum projection of the boxed area. SCoRe signal overlaps with EGFP in the granule cell layer (GCL) and white matter (WM), and both signals are absent from the molecular layer (ML), an unmyelinated area.

a, b Combining SCoRe with Thy1-YFP and antibody labeling against Caspr allows the visualization of compact myelin and nodes of Ranvier along sparsely genetically labeled axons. c, d Breaks in SCoRe signal flanked by Caspr immunolabeling indicate a node of Ranvier. e SCoRe overlaps with antibody labeling against MBP. f SCoRe (white arrows) is excluded from the Ankyrin-G+ axon initial segment (AIS; red arrows). g SCoRe can be used to trace myelination patterns (t-SCoRe) along the axon (t-axon) of sparsely genetically labeled neurons.
The detected SCoRe signal is from light reflected by the top and bottom of the myelin sheath, where multiple compact membrane wraps are perpendicular to the incident light, rather than the lateral sides of the myelin sheath69 (Fig. 3b). This also means that myelin sheaths traveling through the z axis cannot be resolved with SCoRe.
For fixed tissue imaging, once imaging is complete, the microscope slide can be flooded with PBS to allow careful removal of the coverslip. Then, tissue sections can be stored in PBS (short term) or re-mounted normally in glycerol-based mounting medium.
SCoRe has been used to demonstrate that cortical myelination continues into late adulthood in the mouse cortex (Fig. 3), examine myelin sheath plasticity in the adult cortex (Fig. 4), and investigate myelin pathology or degeneration with age6,63. In addition, SCoRe can be used to investigate hypomyelination70, hypermyelination71, myelin loss and regeneration in murine demyelination models63,72,73,74, or myelination in healthy or diseased human tissue69,75,76. Beyond a label-free myelin imaging approach, SCoRe can also be used to assess myelin compaction in transgenic mice exhibiting compaction defects69,75. When combined with genetically encoded fluorescent proteins, SCoRe can be used to distinguish different stages in the oligodendrocyte lineage77, or different phases of myelin deposition and compaction, or myelin decompaction and loss63.
If imaging both genetically encoded fluorescent proteins and SCoRe, use a confocal microscope. If the window quality diminishes over time, fluorescent proteins can also be imaged on a multiphoton microscope. If imaging SCoRe on a confocal microscope and fluorescent proteins on a multiphoton microscope, use the landmarks from steps 7-8 to find the same location and image the fluorescent reporters to later combine with the SCoRe images. Note differences in objective lenses, microscope optics, etc. when transitioning between different microscopes.
Intravital myelin imaging with fluoromyelin
Fluoromyelin is a water-soluble dye that selectively labels myelin. Fluoromyelin is commonly used to label myelin in fixed tissue but has also been applied directly to cultured cells78, the murine brain69 or spinal cord79, murine digits80, or whole zebrafish81 for live imaging of myelin. Fluoromyelin can easily be combined with SCoRe as well as with genetically encoded fluorescent proteins (Fig. 8), although care should be taken to when selecting the fluoromyelin and fluorescent protein wavelengths to ensure adequate separation.
-
1.
Following the craniotomy, and prior to implanting the cranial window, apply 20 µL of a 50% ACSF dilution of the stock solution from the bottle to the cortical surface for 45 min. Wash with ACSF thoroughly. To ensure good tissue penetration, remove the dura before applying fluoromyelin.
-
2.
Immediately after the surgery, while the mouse is still anesthetized, place the mouse on the microscope and headfix it using a headplate or nut. Level the window.
-
3.
Add water and lower the objective (for example 20× 1.0 NA) onto the window.
-
4.
Choose an imaging position.
-
5.
Capture a z-stack or time series images, as required by the experimental question.

Intravital imaging of the cortex of a Cnp-mEGFP mouse following topical fluoromyelin application. Myelin can be visualized with SCoRe, EGFP, and fluoromyelin. The yellow arrowheads indicate an example of two myelin sheaths separated by a node of Ranvier (white arrow). The orange arrows indicate an oligodendrocyte soma and processes which are labelled by mEGFP but not SCoRe or fluoromyelin.
Fluoromyelin washes out after a few hours and can therefore only be used for acute imaging immediately following the cranial window surgery or potentially in preparations where molecules can be locally applied to a chronic cranial window82,83,84.
To combine fluoromyelin imaging with SCoRe imaging, use a confocal microscope. If imaging fluoromyelin and genetically encoded fluorescent proteins or fluoromyelin only, use either a confocal microscope (to image up to 200 µm from the pial surface) or a multiphoton microscope (to image up to 500 µm from the pial surface), although it is important to note that labeling efficiency diminishes with depth after topical application of fluoromyelin.
Intravital imaging of oligodendrocytes and myelin with sulforhodamine 101
Although traditionally used as an astrocyte label, sulforhodamine 101 (SR101) diffuses through astrocyte-oligodendrocyte gap junctions, and can therefore be used to label both oligodendrocytes and their myelin sheaths85,86,87 (Fig. 9). If combining SR101 with genetically encoded fluorescent proteins (Fig. 9), it is important to consider whether the fluorophore emission spectrum can be separated from that of SR101.
-
1.
Administer SR101 to the mouse ~140 min before imaging, to allow diffusion into oligodendrocytes.
-
a.
Following the craniotomy, and prior to implanting the cranial window, apply 20 µL of 50 µM SR101 prepared in sterile ACSF directly onto the cortical surface for 5-10 min. Wash with ACSF for 10 min. To ensure good tissue penetration, remove the dura before applying SR101.
-
b.
As an alternative to applying SR101 during the cranial window surgery, inject 100 µL of 5 mM SR101 dissolved in sterile saline with an intravenous injection.
-
a.
-
2.
Anesthetize a mouse that has a cranial window with a 100 mg/kg ketamine and 10 mg/kg xylazine intraperitoneal injection or 2% isoflurane. If using isoflurane, maintain anesthesia with 1.5-1.8% isoflurane throughout the imaging session. Place the mouse on the microscope and headfix it using a headplate or nut. Level the window.
-
3.
Add water and lower the objective (for example 20× 1.0 NA) onto the window.
-
4.
Choose an imaging position.
-
5.
Capture a z-stack or timelapse images, as required by the experimental question.
-
6.
For longitudinal imaging, take a low zoom image of the surface of the tissue to capture vascular patterns or other landmarks in the tissue.
-
7.
Take a landmark image using a camera on the microscope or a cell phone through the eyepiece to relocate the position.
-
8.
Repeat steps 1 to 5 for each imaging session, or as required depending on the experimental design.

a SR101 labels both astrocytes (magenta only) and oligodendrocytes (magenta and green; indicated by the white arrows) in upper cortical layers. b, c Injecting a fluorescent dye intravenously (yellow) can help distinguish SR101+ astrocytes, whose endfeet contact blood vessels, from SR101+ oligodendrocytes. d SR101 also labels myelin sheaths.
SR101 washes out from the tissue in less than 24 h, although some residue may remain. For repeated imaging, SR101 should be administered by intravenous injection prior to each imaging session.
To combine SR101 imaging with SCoRe imaging, use a confocal microscope. If imaging SR101 only, use either a confocal microscope (to image up to 200 µm from the pial surface) or a multiphoton microscope (to image up to 500 µm from the pial surface).
Injecting a fluorescent dye intravenously to label the vasculature can help distinguish SR101+ astrocytes from SR101+ oligodendrocytes, as astrocyte endfeet are associated with blood vessels, while oligodendrocytes are not85 (Fig. 9).
Troubleshooting
-
1.
There is bright uneven SCoRe signal near the brain surface: level the window and adjust the z focus to be below the coverslip. Reflection does occur above the pial surface, but ideally, SCoRe below the pial surface should come into focus at the same time within the field of view.
-
2.
There are reflective debris on the brain surface: be careful during the cranial window placement that no dust or debris is under the coverslip. Debris will obscure SCoRe and fluorescence imaging below in addition to impacting tissue health.
-
3.
There is low signal to noise during SCoRe imaging in fixed tissue: the tissue is mounted in glycerol based mounting medium, try mounting the tissue in PBS to maintain refractive index differences.
-
4.
There is bright reflective signal from the surface coverslip during SCoRe imaging in fixed tissue: adjust the z focus so the field of view is on the tissue and well below the cover slip. If taking a tiled image, ensure that the sample is level so that the SCoRe signal is even throughout, and reflection is not detected from objects outside the tissue.
-
5.
There is low SR101 signal to noise: the incubation time is too long, the dye concentration is too high, or the sample was imaged too soon after dye application.
-
6.
SR101 does not penetrate into the tissue: the dye is not fresh. Make the solution immediately before use and store the powder in aliquots at −80 °C before use.
-
7.
The cranial window becomes cloudy over time: longevity of cranial windows improves as the technique is mastered. Avoiding damage to the brain and ensuring secure attachment of the nut/headplate and cover slip are key to a long-lasting cranial window. If the cloudy appearance is occurring shortly after surgery (1 or 2 weeks), wait until 3 weeks after surgery before imaging, acute inflammation is normal.
-
8.
There are motion artifacts during intravital imaging: check that the anesthesia delivery is normal, that the mouse head is secure, and that the microscope is stable (e.g. the air table is floating; the microscope tensioner is tightened, if there is one). If using inhaled anesthetic such as isoflurane, wait for the mouse to adjust to the anesthesia and for the mouse’s breathing to be regular before imaging.
-
9.
The fluorescence is unusually dim during intravital imaging: check for water evaporation or an air bubble underneath the lens if using a water immersion lens.
-
10.
The images appear warped or dimmer than in other locations across the window: you may be on the edge of the window. Imaging positions closer to the center tend to be clearer and stable for longitudinal imaging.
-
11.
There is blood under the coverslip or a shadowy appearance: bleeds can occur during the cranial window surgery and obscure optical access to the brain. It is best practice to master cranial window surgery and avoid bleeds before acquiring data. Rinsing the brain with ACSF prior to cover glass placement can clear minor amounts of blood.
Discussion
In this protocol, we described methods for intravital or ex vivo imaging of oligodendrocyte lineage cells and myelin in mice using genetically encoded fluorescent proteins, small molecule dyes, or label-free SCoRe. Despite recent advances, several outstanding questions remain in the myelin field, including cellular and molecular mechanisms regulating OPC fate decisions, differentiating oligodendrocyte survival, successful formation of myelin sheaths and integration into the neural circuit, and myelin plasticity. In addition, it is still unclear whether and how myelin remodeling impacts neural circuitry and plasticity. Finally, myelin loss is increasingly linked to neurodegenerative disorders88,89,90,91,92,93, and is thought to contribute to cognitive decline with aging15,16,17, but little is known about the distinct steps leading to this degeneration. Although valuable insights into these mechanisms have been gained by visualizing myelin and oligodendrocyte lineage cells in fixed tissue, this approach is limited by the lack of temporal information regarding processes that happen over time. Combining both fixed tissue and live imaging using the tools and procedures we described in this protocol has great potential to help address these questions, as this would provide in situ data with temporal resolution.
Genetically encoded fluorescent proteins have been widely used to study oligodendrocyte lineage and myelin dynamics. However, their use is limited to genetically tractable model organisms such as mice or zebrafish. In contrast, using small molecule dyes or label-free approaches widens the scope of possible model organisms to larger mammals, including rats, rabbits94, pigs, or non-human primates. In contrast to other label-free methods to image myelin such as optical coherence microscopy, third harmonic generation or coherent anti-stokes Raman scattering microscopy, which require specialized optical equipment, SCoRe only requires a standard confocal laser-scanning microscope with at least two lasers, making it widely accessible95. However, as noted above SCoRe does not resolve myelin sheaths in z: this can be circumvented by combining SCoRe with genetically encoded fluorescent reporters or dyes. Like for most intravital imaging, tissue penetration is limited, with SCoRe imaging possible down to 400 µm with visible wavelengths from the pial surface, and fluorescent dye or reporter imaging possible down to 200 µm (confocal microscope) or 500-1000 µm (multiphoton microscope). However, recent work suggests that using longer excitation wavelengths (1300 nm to 1700 nm) allows SCoRe imaging in deeper brain regions96. In summary, the methods we described in this protocol can be used to address many questions related to myelin development, myelin plasticity and its impact on neural plasticity, and myelin degeneration with aging and disease.
Data availability
All data in support of the findings of this paper are available within the article or as supplementary material.
References
Nave, K.-A. Myelination and support of axonal integrity by glia. Nature 468, 244–252 (2010).
Pajevic, S., Basser, P. J. & Fields, R. D. Role of myelin plasticity in oscillations and synchrony of neuronal activity. Neuroscience 276, 135–147 (2014).
Rivers, L. E. et al. PDGFRA/NG2 glia generate myelinating oligodendrocytes and piriform projection neurons in adult mice. Nat. Neurosci. 11, 1392–1401 (2008).
Young, K. M. et al. Oligodendrocyte dynamics in the healthy adult CNS: evidence for myelin remodeling. Neuron 77, 873–885 (2013).
Zhu, X. et al. Age-dependent fate and lineage restriction of single NG2 cells. Development 138, 745–753 (2011).
Hill, R. A., Li, A. M. & Grutzendler, J. Lifelong cortical myelin plasticity and age-related degeneration in the live mammalian brain. Nat. Neurosci. 21, 683–695 (2018).
Hughes, E. G., Orthmann-Murphy, J. L., Langseth, A. J. & Bergles, D. E. Myelin remodeling through experience-dependent oligodendrogenesis in the adult somatosensory cortex. Nat. Neurosci. 21, 696–706 (2018).
McKenzie, I. A. et al. Motor skill learning requires active central myelination. Science 346, 318–322 (2014).
Xiao, L. et al. Rapid production of new oligodendrocytes is required in the earliest stages of motor-skill learning. Nat. Neurosci. 19, 1210–1217 (2016).
Pan, S., Mayoral, S. R., Choi, H. S., Chan, J. R. & Kheirbek, M. A. Preservation of a remote fear memory requires new myelin formation. Nat. Neurosci. 23, 487–499 (2020).
Shimizu, T. et al. Oligodendrocyte dynamics dictate cognitive performance outcomes of working memory training in mice. Nat. Commun. 14, 6499 (2023).
Steadman, P. E. et al. Disruption of oligodendrogenesis impairs memory consolidation in adult mice. Neuron 105, 150–164.e6 (2020).
Fields, R. D. Change in the Brain’s White Matter. Science 330, 768–769 (2010).
Bacmeister, C. M. et al. Motor learning drives dynamic patterns of intermittent myelination on learning-activated axons. Nat. Neurosci. 25, 1300–1313 (2022).
Wang, F. et al. Myelin degeneration and diminished myelin renewal contribute to age-related deficits in memory. Nat. Neurosci. 23, 481–486 (2020).
Peters, A. et al. Neurobiological bases of age-related cognitive decline in the rhesus monkey. J. Neuropathol. Exp. Neurol. 55, 861–874 (1996).
Peters, A. & Kemper, T. A review of the structural alterations in the cerebral hemispheres of the aging rhesus monkey. Neurobiol. Aging 33, 2357–2372 (2012).
Deng, Y. et al. Direct visualization of membrane architecture of myelinating cells in transgenic mice expressing membrane-anchored EGFP. Genesis 52, 341–349 (2014).
Doerflinger, N. H., Macklin, W. B. & Popko, B. Inducible site-specific recombination in myelinating cells. Genesis 35, 63–72 (2003).
Madisen, L. et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 13, 133–140 (2010).
Muzumdar, M. D., Tasic, B., Miyamichi, K., Li, L. & Luo, L. A global double-fluorescent Cre reporter mouse. Genesis 45, 593–605 (2007).
Feng, G. et al. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron 28, 41–51 (2000).
Hill, R. A., Medved, J., Patel, K. D. & Nishiyama, A. Organotypic slice cultures to study oligodendrocyte dynamics and myelination. J. Vis. Exp. https://doi.org/10.3791/51835 (2014).
Sherafat, A., Hill, R. A. & Nishiyama, A. Organotypic slice cultures to study oligodendrocyte proliferation, fate, and myelination. Methods Mol. Biol. 1791, 145–156 (2018).
Lu, T.-Y. et al. Norepinephrine modulates calcium dynamics in cortical oligodendrocyte precursor cells promoting proliferation during arousal in mice. Nat. Neurosci. 26, 1739–1750 (2023).
Hirrlinger, P. G. et al. Expression of reef coral fluorescent proteins in the central nervous system of transgenic mice. Mol. Cell. Neurosci. 30, 291–303 (2005).
Gong, S. et al. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature 425, 917–925 (2003).
Snaidero, N. et al. Myelin membrane wrapping of CNS axons by PI(3,4,5)P3-dependent polarized growth at the inner tongue. Cell 156, 277–290 (2014).
Hughes, E. G., Kang, S. H., Fukaya, M. & Bergles, D. E. Oligodendrocyte progenitors balance growth with self-repulsion to achieve homeostasis in the adult brain. Nat. Neurosci. 16, 668–676 (2013).
Zhu, X., Bergles, D. E. & Nishiyama, A. NG2 cells generate both oligodendrocytes and gray matter astrocytes. Development 135, 145–157 (2008).
Karram, K. et al. NG2-expressing cells in the nervous system revealed by the NG2-EYFP-knockin mouse. Genesis 46, 743–757 (2008).
Piller, M., Werkman, I. L., Brown, R. I., Latimer, A. J. & Kucenas, S. Glutamate signaling via the AMPAR sSubunit GluR4 regulates oligodendrocyte progenitor cell migration in the developing spinal cord. J. Neurosci. 41, 5353–5371 (2021).
Kirby, B. B. et al. In vivo time-lapse imaging shows dynamic oligodendrocyte progenitor behavior during zebrafish development. Nat. Neurosci. 9, 1506–1511 (2006).
Yang, L., Chen, M., Zhang, J., Ren, D. & Hu, B. Hypoxia delays oligodendrocyte progenitor cell migration and myelin formation by suppressing Bmp2b signaling in Larval Zebrafish. Front. Cell. Neurosci. 12, 348 (2018).
Marisca, R. et al. Functionally distinct subgroups of oligodendrocyte precursor cells integrate neural activity and execute myelin formation. Nat. Neurosci. 23, 363–374 (2020).
Hill, R. A., Patel, K. D., Goncalves, C. M., Grutzendler, J. & Nishiyama, A. Modulation of oligodendrocyte generation during a critical temporal window after NG2 cell division. Nat. Neurosci. 17, 1518–1527 (2014).
Thornton, M. A. et al. Long-term in vivo three-photon imaging reveals region-specific differences in healthy and regenerative oligodendrogenesis. Nat. Neurosci. 27, 846–861 (2024).
Almeida, R. & Lyons, D. Oligodendrocyte development in the absence of their target axons in vivo. PLOS ONE 11, e0164432 (2016).
Auguste, Y. S. S. et al. Oligodendrocyte precursor cells engulf synapses during circuit remodeling in mice. Nat. Neurosci. 25, 1273–1278 (2022).
Xiao, Y., Petrucco, L., Hoodless, L. J., Portugues, R. & Czopka, T. Oligodendrocyte precursor cells sculpt the visual system by regulating axonal remodeling. Nat. Neurosci. 25, 280–284 (2022).
Djannatian, M. et al. Two adhesive systems cooperatively regulate axon ensheathment and myelin growth in the CNS. Nat. Commun. 10, 4794 (2019).
Vagionitis, S. et al. Clusters of neuronal neurofascin prefigure the position of a subset of nodes of Ranvier along individual central nervous system axons in vivo. Cell Rep. 38, 110366 (2022).
Li, J., Miramontes, T. G., Czopka, T. & Monk, K. R. Synaptic input and Ca2+ activity in zebrafish oligodendrocyte precursor cells contribute to myelin sheath formation. Nat. Neurosci. 27, 219–231 (2024).
Mensch, S. et al. Synaptic vesicle release regulates myelin sheath number of individual oligodendrocytes in vivo. Nat. Neurosci. 18, 628–630 (2015).
Nawaz, S. et al. Actin filament turnover drives leading edge growth during myelin sheath formation in the central nervous system. Dev Cell 34, 139–151 (2015).
Almeida, R. G. et al. Myelination induces axonal hotspots of synaptic vesicle fusion that promote sheath growth. Curr. Biol. 31, 3743–3754.e5 (2021).
Hines, J. H., Ravanelli, A. M., Schwindt, R., Scott, E. K. & Appel, B. Neuronal activity biases axon selection for myelination in vivo. Nat. Neurosci. 18, 683–689 (2015).
Czopka, T., ffrench-Constant, C. & Lyons, D. A. Individual oligodendrocytes have only a few hours in which to generate new myelin sheaths in vivo. Dev. Cell 25, 599–609 (2013).
Gronseth, J. R. et al. Synaptic vesicle release regulates pre-myelinating oligodendrocyte-axon interactions in a neuron subtype-specific manner. Front. Cell. Neurosci. 18, 1386352 (2024).
Baraban, M., Koudelka, S. & Lyons, D. A. Ca2+ activity signatures of myelin sheath formation and growth in vivo. Nat. Neurosci. 21, 19–23 (2018).
Koudelka, S. et al. Individual neuronal subtypes exhibit diversity in CNS myelination mediated by synaptic vesicle release. Curr. Biol. 26, 1447–1455 (2016).
Krasnow, A. M., Ford, M. C., Valdivia, L. E., Wilson, S. W. & Attwell, D. Regulation of developing myelin sheath elongation by oligodendrocyte calcium transients in vivo. Nat. Neurosci. 21, 24–28 (2018).
Fedder-Semmes, K. N. & Appel, B. The Akt-mTOR pathway drives myelin sheath growth by regulating cap-dependent translation. J. Neurosci. 41, 8532–8544 (2021).
Hughes, A. N. & Appel, B. Oligodendrocytes express synaptic proteins that modulate myelin sheath formation. Nat. Commun. 10, 4125 (2019).
Yang, M. L. et al. CNS myelination requires cytoplasmic dynein function. Dev. Dyn. 244, 134–145 (2015).
Almeida, R. G. et al. Myelination of neuronal cell bodies when myelin supply exceeds axonal demand. Curr. Biol. 28, 1296–1305.e5 (2018).
Nelson, H. N. et al. Individual neuronal subtypes control initial myelin sheath growth and stabilization. Neural Dev. 15, 12 (2020).
Yang, S. M., Michel, K., Jokhi, V., Nedivi, E. & Arlotta, P. Neuron class–specific responses govern adaptive myelin remodeling in the neocortex. Science 370, eabd2109 (2020).
Bottes, S. & Jessberger, S. Live imaging of remyelination in the adult mouse corpus callosum. Proc. Natl. Acad. Sci. 118, e2025795118 (2021).
Morris, A. D. & Kucenas, S. A Novel Lysolecithin Model for Visualizing Damage in vivo in the Larval Zebrafish Spinal Cord. Front. Cell Dev. Biol. 9, 654583 (2021).
Snaidero, N. et al. Myelin replacement triggered by single-cell demyelination in mouse cortex. Nat. Commun. 11, 4901 (2020).
Orthmann-Murphy, J. et al. Remyelination alters the pattern of myelin in the cerebral cortex. eLife 9, e56621 (2020).
Chapman, T. W., Olveda, G. E., Bame, X., Pereira, E. & Hill, R. A. Oligodendrocyte death initiates synchronous remyelination to restore cortical myelin patterns in mice. Nat. Neurosci. 26, 555–569 (2023).
Chapman, T. W., Kamen, Y., Piedra, E. T. & Hill, R. A. Oligodendrocyte maturation alters the cell death mechanisms that cause demyelination. J. Neurosci. 44, e1794232024 (2024).
Mezydlo, A. et al. Remyelination by surviving oligodendrocytes is inefficient in the inflamed mammalian cortex. Neuron 111, 1748–1759.e8 (2023).
Romanelli, E. et al. Myelinosome formation represents an early stage of oligodendrocyte damage in multiple sclerosis and its animal model. Nat. Commun. 7, 13275 (2016).
Auer, F., Vagionitis, S. & Czopka, T. Evidence for myelin sheath remodeling in the CNS revealed by in vivo imaging. Curr. Biol. 28, 549–559.e3 (2018).
Olveda, G. E., Barasa, M. N. & Hill, R. A. Microglial phagocytosis of single dying oligodendrocytes is mediated by CX3CR1 but not MERTK. Cell Rep. 43, 114385 (2024).
Schain, A. J., Hill, R. A. & Grutzendler, J. Label-free in vivo imaging of myelinated axons in health and disease with spectral confocal reflectance microscopy. Nat. Med. 20, 443–449 (2014).
Michell-Robinson, M. A. et al. Hypomyelination, hypodontia and craniofacial abnormalities in a Polr3b mouse model of leukodystrophy. Brain 146, 5070–5085 (2023).
Li, A. M., Hill, R. A. & Grutzendler, J. Intravital imaging of neocortical heterotopia reveals aberrant axonal pathfinding and myelination around ectopic neurons. Cereb. Cortex 31, 4340–4356 (2021).
Bacmeister, C. M. et al. Motor learning promotes remyelination via new and surviving oligodendrocytes. Nat. Neurosci. 23, 819–831 (2020).
Gonsalvez, D. G. et al. Imaging and quantification of myelin integrity after injury with spectral confocal reflectance microscopy. Front. Mol. Neurosci. 12, 275 (2019).
Call, C. L. & Bergles, D. E. Cortical neurons exhibit diverse myelination patterns that scale between mouse brain regions and regenerate after demyelination. Nat. Commun. 12, 4767 (2021).
Craig, G. A. et al. Reflective imaging of myelin integrity in the human and mouse central nervous systems. Front. Cell. Neurosci. 18, 1408182 (2024).
Sadler, G. L. et al. Lipid metabolism is dysregulated in the motor cortex white matter in amyotrophic lateral sclerosis. Metabolites 12, 554 (2022).
Bame, X. & Hill, R. A. Mitochondrial network reorganization and transient expansion during oligodendrocyte generation. Nat. Commun. 15, 6979 (2024).
Monsma, P. C. & Brown, A. FluoroMyelinTM Red is a bright, photostable and non-toxic fluorescent stain for live imaging of myelin. J. Neurosci. Methods 209, 344–350 (2012).
Romanelli, E. et al. Cellular, subcellular and functional in vivo labeling of the spinal cord using vital dyes. Nat. Protoc. 8, 481–490 (2013).
He, C. et al. 3-photon microscopy of myelin in mouse digital skin excited at the 1700-nm window. J. Biophotonics 13, e202000321 (2020).
Zhu, X.-Y. et al. Development of zebrafish demyelination model for evaluation of remyelination compounds and RORγt inhibitors. J. Pharmacol. Toxicological Methods 98, 106585 (2019).
Roome, C. J. & Kuhn, B. Chronic cranial window with access port for repeated cellular manipulations, drug application, and electrophysiology. Front Cell Neurosci. 8, 379 (2014).
Heo, C. et al. A soft, transparent, freely accessible cranial window for chronic imaging and electrophysiology. Sci. Rep. 6, 27818 (2016).
Hou, S. S. et al. Shallow-angle intracranial cannula for repeated infusion and in vivo imaging with multiphoton microscopy. Neurophotonics 12, 025001 (2025).
Hill, R. A. & Grutzendler, J. In vivo imaging of oligodendrocytes with sulforhodamine 101. Nat. Methods 11, 1081–1082 (2014).
Wasseff, S. K. & Scherer, S. S. Cx32 and Cx47 mediate oligodendrocyte:astrocyte and oligodendrocyte:oligodendrocyte gap junction coupling. Neurobiol. Dis. 42, 506–513 (2011).
Hagos, L. & Hülsmann, S. Unspecific labelling of oligodendrocytes by sulforhodamine 101 depends on astrocytic uptake via the thyroid hormone transporter OATP1C1 (SLCO1C1). Neurosci. Lett. 631, 13–18 (2016).
Depp, C. et al. Myelin dysfunction drives amyloid-β deposition in models of Alzheimer’s disease. Nature 618, 349–357 (2023).
Franklin, R. J. M. Why does remyelination fail in multiple sclerosis?. Nat. Rev. Neurosci. 3, 705–714 (2002).
Calabrese, M. et al. Exploring the origins of grey matter damage in multiple sclerosis. Nat. Rev. Neurosci. 16, 147–158 (2015).
Chen, J.-F. et al. Enhancing myelin renewal reverses cognitive dysfunction in a murine model of Alzheimer’s disease. Neuron 109, 2292–2307.e5 (2021).
Prins, N. D. & Scheltens, P. White matter hyperintensities, cognitive impairment and dementia: an update. Nat. Rev. Neurol. 11, 157–165 (2015).
Bartzokis, G. Age-related myelin breakdown: a developmental model of cognitive decline and Alzheimer’s disease. Neurobiol. Aging 25, 5–18 (2004).
Ehinger, B., Zucker, C. L., Bruun, A. & Adolph, A. In vivo staining of oligodendroglia in the rabbit retina. Glia 10, 40–48 (1994).
Hill, R. A. & Grutzendler, J. Uncovering the biology of myelin with optical imaging of the live brain. Glia 67, 2008–2019 (2019).
Xia, F. et al. In vivo label-free confocal imaging of the deep mouse brain with long-wavelength illumination. Biomed. Opt. Express 9, 6545–6555 (2018).
Acknowledgements
This work was supported by the National Institutes of Health R00NS099469, R01NS122800, R01NS122800-S1, and the Esther A. & Joseph Klingenstein Fund and Simons Foundation to R.A.H., a Postdoctoral Fellowship FG-2307-42173 from the National Multiple Sclerosis Society to Y.K., and an American Heart Association grant, 23PRE1018862, to X.B.
Author information
Authors and Affiliations
Contributions
All authors performed experiments and contributed to data in the manuscript. Y.K., M.E.D and R.A.H. wrote the first draft of the paper and all authors contributed to editing. R.A.H. originally conceived of and supervised the study.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kamen, Y., Doty, M.E., Ciolkowski, M.E. et al. Label-free and fluorescence imaging of oligodendrocytes and myelin. npj Imaging 3, 33 (2025). https://doi.org/10.1038/s44303-025-00098-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s44303-025-00098-8
