
Abstract
Directed collective cell migration is essential for morphogenesis, and chemical, electrical, mechanical and topological features have been shown to guide cell migration in vitro. Here we provide in vivo evidence showing that endogenous electric fields drive the directed collective cell migration of an embryonic stem cell population—the cephalic neural crest of Xenopus laevis. We demonstrate that the voltage-sensitive phosphatase 1 is a key component of the molecular mechanism, enabling neural crest cells to specifically transduce electric fields into a directional cue in vivo. Finally, we propose that endogenous electric fields are mechanically established by the convergent extension movements of the ectoderm, which generate a membrane tension gradient that opens stretch-activated ion channels. Overall, these findings establish a role for electrotaxis in tissue morphogenesis, highlighting the functions of endogenous bioelectrical stimuli in non-neural contexts.
Similar content being viewed by others

Main
The cephalic neural crest of Xenopus laevis is induced at the interface between the neural plate and the non-neural ectoderm, from where they directionally and collectively migrate by forming stereotypical paths known as streams1 (Extended Data Fig. 1a). The migration of neural crest cells is required for the formation of several tissues across vertebrates and their ability to invade other tissues has been related to cancer metastasis1 and, hence, the potential of understanding how these cells migrate. Chemical and mechanical cues have been proposed to influence neural-crest-directed collective cell migration (dCCM)2,3. Yet, in vitro experiments have also demonstrated the ability of neural crest cells to directionally migrate by following extrinsically imposed electric fields, in a process termed as electrotaxis (or galvanotaxis)4,5,6. In addition, seminal works have detected the presence of electric fields in embryos7,8,9 and found that the application of external electric fields led to developmental defects10. In this scenario, electrotaxis emerges as an alternative or complementary mechanism to explain neural crest dCCM in vivo. Still, whether and how endogenous electric fields emerge to drive dCCM via electrotaxis in living organisms remains elusive.
Electric fields emerge along the neural crest migratory path
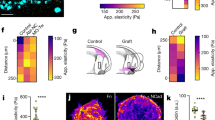
To map extracellular electric currents along the migratory path of neural crest cells, we used ultrasensitive vibrating probes11 (Extended Data Fig. 1b–d). Vibrating probe measurements (calibration and controls are shown in Extended Data Fig. 1e–h and Supplementary Note 1) were performed from the neural fold (in the dorsal part of the embryo) towards the flanking non-neural ectoderm (ventral), and at stages in which the neural crest cells are entering their migratory phase (pre-migratory) (Fig. 1a and Supplementary Note 1). These measurements detected the presence of outward currents in the neural fold and inward currents in the flanking non-neural ectoderm, with measured average current densities ranging from 0.5855 µA cm−2 (neural fold) to −0.2334 µA cm−2 (flank ectoderm) (Fig. 1b). On the basis of calculations derived from Ohm’s law12 (Supplementary Note 1), this pattern of extracellular current densities suggests that subectodermal endogenous electric fields, ranging from 8 to 45 mV mm−1, emerge in the migratory path of the neural crest. The presence of these subectodermal electric currents was directly corroborated by impaling the space between the ectoderm and the neural crest with glass microelectrodes (Fig. 1c and Supplementary Note 1). Transepithelial potentials (TEP) obtained with this technique confirmed the existence of subectodermal currents that elicit the presence of subectodermal electric fields ranging from 7 to 37 mV mm−1 (Fig. 1d). Further temporal analysis showed that outward currents (and, in turn, electric fields) appear at early non-migratory stages, reaching a maximum at stages in which neural crest cells are preparing to migrate, and that these values experience a slight decrease when migration starts (Extended Data Fig. 2a,b). Altogether, these results indicate that the neural crest is in contact with a subectodermal electric field that emerges in its migratory path from early stages of migration, with the cathode (−pole) in the neural fold and the anode (+pole) in the flanking non-neural ectoderm (Fig. 1e).

a, Scheme of an X. laevis embryo at pre-migratory stages (left) and cross-sectional cartoon showing the vibrating probe measurements (right). Pos, position; A, anterior; P, posterior; M, middle; L, lateral; D, dorsal; V, ventral. b, Vibrating probe current density measurements. The solid lines represent the mean and the shade, the standard errors. Reference was set as >1 mm away from the embryo. Two-tailed Mann–Whitney U-test, ****P < 0.0001, n = 7 embryos. The calculated electric field (EF) is shown in the inset. c, Schematic showing glass microelectrodes impaling positions in the subectodermal space beneath the neural fold and flank ectoderm. The reference electrode is >1 mm away from the embryo. d, TEP measurements. The solid lines represent the mean and the shade, the standard errors. Two-tailed paired t-test, ***P = 0.0001, n = 11 embryos. The calculated EF is shown in the inset. e, Schematic showing the endogenous EFs in the migratory path of the neural crest as observed from current densities and TEP measurements. f–i, Ex vivo electrotaxis assay. Time-colour-coded trajectories of neural crests migrating in the absence of EFs (f), under EFs (100 mV mm−1) (g) and after EF reversal (h), 4 h in each case. The arrows indicate the direction of migration. Scale bar, 100 μm. i, FMI. The red lines represent the mean and the error bars, the standard deviation. Two-tailed t-test for no EF versus EF, two-tailed Mann–Whitney U-test for no EF versus EF reversal, ****P < 0.0001, nNo EF = 27, nEF = 33 clusters. j–m, Application of exogenous EFs (100 mV mm−1) in vivo, with treatments as indicated. j,l, Lateral views of embryos hybridized against neural crest markers c3 (j) and sox8 (l). Scale bar, 200 μm. k,m, Normalized stream displacement. The red lines represent the mean and the error bars, the standard deviation. k, Two-tailed t-test, ****P < 0.0001, nNo EF = 36, nAntiparallel EF = 31 embryos. m, Two-tailed t-test with Welch’s correction, *P = 0.0110, nNo EF = 42, nParallel EF = 39 embryos. f–h,j,l, Representative examples from three independent experiments; CI = 95%.
Neural crest cells undergo anodal collective electrotaxis
To dissect whether endogenous electric fields influence neural crest dCCM, we generated an ex vivo electrotaxis assay capable of reproducing the recorded levels of endogenous electric fields in a controlled microenvironment (Extended Data Fig. 3a,c,d and Supplementary Note 1). In the absence of an electric field, cells within cultured neural crest clusters tend to radially disperse by performing random migration (Fig. 1f). However, when an electric field was applied, neural crest cells persistently and collectively migrated towards the anode with enhanced collectiveness (Fig. 1g and Supplementary Video 1), as previously proposed for cultured monolayers13,14,15,16. In addition, reverting the polarity of the electric field led to a switch in the direction of migration towards the new anodal position (Fig. 1h and Supplementary Video 1). These observations were quantitatively confirmed by computing the forward migration index (FMI) as a readout of the directionality displayed by clusters in each condition (Fig. 1i). Note that although FMIs closer to 0 represent random migration, FMIs near 1 or −1 account for a directional response. The localization of the polarity marker Rac1 was analysed to control the effect of electric field application in cell polarity (Extended Data Fig. 3e). Further experiments showed that the directional response of neural crest cells to electric fields is more effective in clusters than in isolated cells (Extended Data Fig. 3f–h and Supplementary Video 2). Furthermore, the directionality, displacement and velocity of neural crest clusters increased as a function of the electric field strength (Extended Data Fig. 4a–d and Supplementary Video 3), as it has been proposed for other cell types13,14,17,18. This is relevant as the recorded endogenous electric fields ranged from ~8 to 50 mV mm−1 on average (Fig. 1b,d), suggesting that cells can reply to varying levels of endogenous currents when migrating in vivo. By using an adapted electrotactic device that allows to extrinsically apply antiparallel or parallel endogenous-like levels of electric fields to embryos (Extended Data Fig. 3b,c and Supplementary Note 1), we also tested the influence of applied currents in neural crest dCCM. Although the neural crests of control embryos migrated by following stereotypical streams, a hallmark of dCCM2, the application of an electric field that runs from ventral to dorsal (hereafter referred as antiparallel) impaired the formation of neural crest streams, as observed by in situ hybridization (Fig. 1j,k). Conversely, the application of a parallel electric field was sufficient to enhance neural crest dCCM (Fig. 1l,m). This was consistent with ex vivo observations, showing that cluster directionality, velocity and displacement increased as a function of the electric field strength (Extended Data Fig. 4a–d). Altogether, these results indicate that endogenous-like electrical currents influence the collective and directional migration of neural crest clusters towards anodal positions.
Membrane tension gradients underlie electric field emergence
We next investigated the mechanism underlying the endogenous electric field formation. Previous work has shown that membrane stretching activates ion channels to allow for ion translocation19. In addition, stress measurements in amphibians predict increased stress/tension in dorsal tissues in the vicinity of the neural fold and lower values towards the ventral region, where the non-neural ectoderm is located20,21,22. Thus, we hypothesized that the endogenous electric fields emerge from a differential ionic movement due to a higher stretching of neural fold membranes, via the activity of mechanosensitive (or stretch-activated) ion channels. To test this hypothesis, we first confirmed that there was a tension build-up in the membrane of neural fold cells from non-migratory to pre-migratory stages, by using membrane laser ablations experiments and cell–cell junction recoil velocity analysis23 (Extended Data Fig. 5a–c and Supplementary Video 4). Ablation experiments also revealed that membrane tension gradients appear along the neural crest migratory path of wild-type embryos at pre-migratory stages, with higher tension being reported in the neural fold and lower values in the flanking ectoderm (Extended Data Fig. 5b,c and Supplementary Video 4). Consequently, we sought to test the relevance of these tension gradients for electric field formation and neural crest dCCM. It has been proposed that anisotropic membrane tensions can arise in the neural plate owing to the activity of the planar cell polarity (PCP) pathway24,25. Thus, we analysed the impact of PCP inhibition in the formation of this dorsoventral membrane tension gradient and endogenous electric field formation. PCP was inhibited by targeting the injection of DshDEP+, a known PCP inhibitor25, into the neural fold (Fig. 2a and Extended Data Fig. 5d,e). DshDEP+ injection in the neural fold led to a strong decrease in neural fold membrane tension, as revealed by laser ablation experiments (Fig. 2b, Extended Data Fig. 5c and Supplementary Video 4), and caused a drastic reduction in the neural fold outward currents (Fig. 2c,d). These values resemble those recorded in the ventral non-neural ectoderm of wild-type control embryos (Fig. 1a,b), indicating a depletion of endogenous electric fields. According to our initial hypothesis, we then tested whether the activity of mechanosensitive ion channels is required for neural fold outward currents. For this, live recordings were performed before, during and after GsMTx4 inoculation, a peptide toxin that specifically inhibits stretch-activated channels in Xenopus and in other species26,27. Our dynamic measurements reported that shortly after GsMTx4 inoculation, the outward currents of the neural fold decreased when compared with the control inoculation (Fig. 2e,f), resembling inward currents observed in ventral non-neural ectoderm of control embryos (Fig. 1a,b). Altogether, these datasets support the idea that PCP mediates the formation of dorsoventral membrane tension gradients and consequently the emergence of the endogenous electric fields in the dorsal part of Xenopus embryos. Considering these results, our next step was to explore the relevance of endogenous electric field depletion for neural crest dCCM in vivo. Targeted injections of DshDEP+ to the neural fold were sufficient to inhibit neural crest dCCM in vivo, as observed by in situ hybridization analysis (Extended Data Fig. 5f). This was quantitatively analysed by performing graft experiments in which wild-type neural crest cells (with fluorescently tagged nuclei) were transplanted into host wild-type embryos or in embryos in which DshDEP+ injection was targeted into the neural fold (Fig. 2g). Our in vivo analyses of cell trajectories revealed that control neural crest cells persistently and directionally migrated when grafted into wild-type hosts (Fig. 2h,j,k and Supplementary Video 5). However, the directionality of wild-type neural crest grafted into embryos treated with DshDEP+ in the neural fold was reduced, as shown by their poor FMI and by the absence of stereotypical streams (Fig. 2i–k and Supplementary Video 5). These datasets indicate that the subectodermal electric fields detected in the migratory path of cranial neural crests are relevant for their dCCM.

a, Scheme of DshDEP+ neural-fold-targeted injection and laser ablation. b, Recoil velocity plots. The red lines represent the mean and the error bars, the standard deviation; two-tailed t-test with Welch’s correction; ****P < 0.0001; nControl = 21, nDshDEP+ = 19 membranes. c,d, Scheme (c) and plot (d) of vibrating probe measurement in DshDEP+-treated embryos. The solid lines represent the mean and the shade, the standard errors. Paired t-test, **P = 0.0019, n = 14 embryos. Reference probe was away from the embryo. e,f, Scheme (e) and plot (f) of electric current measurements in embryos treated with GsMTx4 (5 µM). The solid line represents the mean and the shade, the standard errors. Two-tailed Wilcoxon matched-pairs test of time-matched treatments, ****P < 0.0001, n = 6 embryos. g, Neural crest graft assays. h,i, Time-colour-coded neural crest trajectories in vivo (lateral view). Scale bar, 200 μm. j, FMI. The red lines represent the median and the error bars, the interquartile ranges. Two-tailed Mann–Whitney U-test, ****P < 0.0001, nControl = 36, nDshDEP+ = 25 cells. k, Percentage of embryos displaying streams. The bars represent the mean and the error bars, the standard deviations. Two-tailed Fisher’s exact test, ***P = 0.0002, nControl = 19, nDshDEP+ = 18 embryos. h,i, Representative examples from at least three independent experiments; CI = 95%.
Electric fields depolarize neural crest cells
To gain further insights into the mechanism of electrotaxis, we next assessed whether neural crest cells are electrically responsive to endogenous electric fields by using a genetically encoded voltage indicator (GEVI)28,29,30, hereafter referred as GEVI Marina. Briefly, the voltage-sensing domain of this GEVI is linked to a mutated form of GFP, in which an increase in fluorescence reports cell membrane depolarization and, in turn, electrical responses (Extended Data Fig. 6a,b). We first calibrated the GEVI Marina levels to achieve optimal sensitivity by combining microinjections and ex vivo electrotaxis assays (concentration is described in the Methods). At optimal levels, we detected a robust signal of the GEVI, indicating that the membranes of neural crest cells are depolarized at wild-type levels of endogenous electric fields (Extended Data Fig. 6c). Nonetheless, these endogenous levels of GEVI Marina were lowered to a minimum when the neural crest clusters were cultured ex vivo, in the absence of electric fields. However, when an electric field of identical magnitude to those recorded in vivo was applied, the signal of GEVI Marina was restored (Extended Data Fig. 6d,e). This confirmed that GEVI Marina can sense voltage changes and report the depolarization or repolarization of neural crest cell membranes at voltage ranges detected within the embryos. In this context, we next depleted endogenous electric fields with targeted injections of DshDEP+ into the neural fold (Extended Data Fig. 5d,e). This treatment led to a consistent reduction in the GEVI Marina’s signal, suggesting the repolarization of treated neural crest membranes compared with wild-type animals, in which the membranes are depolarized by endogenous electric fields (Extended Data Fig. 6f,g,i). Remarkably, the introduction of a parallel electric field was sufficient to depolarize the neural crest membranes in vivo (Extended Data Fig. 6h,i). These results confirm that neural crest cells are electrically sensitive to voltage levels detected in endogenous electric fields.
Vsp1 is a specific sensor of electric fields
We next explored how neural crest cells sense and transduce endogenous electric fields into a directional cue. Ion channels and other proteins, which are mostly described as part of general cell polarity and motility programs, have been proposed to mediate electrotaxis in vitro31,32,33,34. Yet, the mechanism used by neural crest cells to transduce electric fields into a directional response in vivo remains elusive. To address this, we searched for candidate electrosensors in RNA libraries generated from isolated wild-type neural crests (Extended Data Fig. 7a). After confirming the quality of our libraries (Extended Data Fig. 7b), we generated a list of candidates composed of ion channels and pumps whose roles have been linked to bioelectrics in other systems (Extended Data Fig. 7c)12,34. Although each one of these candidates may have the potential to mediate the response of neural crest cells to electrical inputs and eventually electrotaxis, we focused on the voltage-sensitive phosphatase 1 (originally described as transmembrane phosphoinositide 3-phosphatase and tensin homolog 2 (tpte2.L) and referred here as vsp1)35,36. Vsp1 became a candidate due to its versatile structure, featuring a channel-like transmembrane domain and a cytosolic catalytic domain with phosphatase activity35. This peculiar protein structure suggested that Vsp1 could not only sense but also transduce electrical stimuli into a cascade that could eventually lead to electrotaxis. The localization of Vsp1 to the cell membrane, where it is expected to operate37, was confirmed by expressing an X. laevis Vsp1-GFP fusion construct (Extended Data Fig. 7d) and vsp1 mRNA expression in the neural crest was validated by polymerase chain reaction (PCR) experiments, amplifying its coding sequence from neural crest tissue-specific libraries and in situ hybridization (Extended Data Fig. 7e). Note that the paralogous gene vsp2 (tpte2.S) is not detected at these stages37. We next tested whether Vsp1 is required for neural crest electrotaxis ex vivo and in vivo by using a combination of knockdown approaches: a catalytically inactive form of Vsp1 (Xl-Vsp1-C301S), which was previously shown to reduce its phosphatase activity in X. laevis36; a CRISPR-Cas13-based approach38, which targets Cas13 to vsp1 mRNA for its degradation (Vsp1-Cas13); and an antisense oligo, which targets a splicing site within vsp1 RNA (Vsp1-SSMO). Our knockdown experiments showed that although control clusters directionally migrated in the ex vivo electrotaxis setup, all Vsp1 knockdown approaches caused a loss of cluster directionality (Fig. 3a–c and Supplementary Video 6). A similar result was observed in vivo where control neural crest cells directionally migrated, but Vsp1 knockdown impaired the formation of stereotypical streams in all conditions (Extended Data Fig. 7f–i,l). In addition, the co-injection of Vsp1-SSMO or Vsp1-Cas13 with vsp1 full-length mRNA was sufficient to rescue stream formation, confirming the specificity of the observed phenotypes (Extended Data Fig. 7j–l). The efficiency of Vsp1-Cas13 and Vsp1-SSMO was controlled by PCR and immunofluorescence (Extended Data Fig. 7m–o). The observed effects were quantitatively analysed by performing graft experiments in which neural crest cells (with fluorescently tagged nuclei) from control, Xl-Vsp1-C301S, Vsp1-SSMO or Vsp1-Cas13 embryos were transplanted into wild-type untagged host embryos (Fig. 3d). Our analyses of cell trajectories revealed that although control cells persistently and directionally migrated, the directionality of Xl-Vsp1-C301S-, Vsp1-SSMO- and Vsp1-Cas13-injected cells was reduced in vivo, as shown by a low FMI and the absence of stereotypical streams (Fig. 3e,f and Supplementary Video 7). Our result also shows that even though the directionality of cells within Vsp1 knockdown clusters was affected, these cells are still motile in vivo. This is also the case ex vivo, where cells within clusters radially disperse and migrate, but fail to bias their migration to applied electric fields as a collective (Fig. 3a–c and Supplementary Video 6), suggesting that Vsp1 is required for collective electrotaxis but not for cell motility. We confirmed these observations by analysing the effect of Vsp1 knockdown in single-neural-crest motility parameters. The injection of Xl-Vsp1-C301S, Vsp1-SSMO or Vsp1-Cas13 did not affect the cell directionality and cell velocity (Extended Data Fig. 8a,b and Supplementary Video 8). Furthermore, since chemotaxis has been proposed to guide neural crest migration39, we exposed the control and knockdown clusters to a previously described chemotaxis assay, which assesses the response of neural crest clusters to the chemokine stromal cell-derived factor 1 (SDF-1, also known as CXCL12)39 (Extended Data Fig. 8c). Our results showed that Xl-Vsp1-C301S, Vsp1-SSMO or Vsp1-Cas13 injections did not affect the neural crest response towards SDF-1 (Extended Data Fig. 8d,e and Supplementary Video 9), confirming a specific requirement of Vsp1 for an electrical response. Complementarily, we explored whether the Vsp1 activity modulates the sensitivity of neural crest cells to electric currents. For this, we first explored the minimum value of electric fields to which neural crest clusters are sensitive to and determined that below 5 mV mm−1, the neural crest elicited a poor response. Then, we injected a validated mutant form of Vsp1, which is more sensitive to voltage (Xl-Vsp1-R152Q-GFP)36,40, and analysed its impact in neural crest electrotaxis ex vivo. Xl-Vsp1-R152Q-GFP expression increased the number of neural crest clusters displaying a persistent and directional response under suboptimal electric fields (5 mV mm−1) (Extended Data Fig. 9a–c and Supplementary Video 10), confirming that Vsp1 activity modulates the sensitivity of neural crest cells to electric currents. Together, these datasets indicate that even though not being required for neural crest motility or chemotaxis, Vsp1 is essential for the response of the neural crest to electric fields and, in turn, for collective electrotaxis in vivo. Since the role of Vsp1 in electrotaxis has not been previously described, we predict that its study can have major implications across research fields. Indeed, by accessing publicly available databases, we found that orthologous VSP are expressed in several mammalian cell types. These cell lines are widely used as wound healing and cancer models and have been shown to electrotax in vitro (Supplementary Table 2), suggesting a requirement of Vsp1 in these biological contexts.

a, Cartoon depicting a typical electrotaxis experiment. b, Time-colour-coded cluster trajectories ex vivo, showing the impact of Vsp1 knockdowns in the migration of neural crest clusters (the conditions are indicated). The arrows indicate the direction of migration. Scale bar, 100 μm. c, FMI. The red lines represent the mean and the error bars, the standard deviation. Two-tailed Student’s t-test, ****PXl-Vsp1-C301S < 0.0001 (with Welch’s correction), ****PVsp1-SSMO < 0.0001 and ****PVsp1-Cas13 < 0.0001 (with Welch’s correction), nControl = 26, nXl-Vsp1-C301S = 20, nControl = 28, nVsp1-SSMO = 33, nControl Cas13 = 35 and nVsp1-Cas13 = 33 clusters. d, Neural crest graft assays. e, Time-colour-coded neural crest trajectories in vivo (lateral view). Scale bar, 200 μm. f, FMI quantifications. The red lines represent the median and the error bars, the interquartile ranges. Two-tailed Mann–Whitney U-test, ****P < 0.0001 (for all comparisons), nControl = 29, nXl-Vsp1-C301S = 43, nControl = 26 and nVsp1-SSMO = 39 cells, nControl = 36 and nVsp1-Cas13 = 34 cells. b,e, Representative examples from at least three independent experiments; CI = 95%.
Endogenous electric fields drive neural crest migration
Finally, we performed rescue experiments to corroborate the relevance of endogenous electric fields for electrotaxis in vivo. For this, we assessed whether the introduction of a parallel exogenous electric field of identical magnitude and direction to the ones recorded in vivo would rescue the neural crest directionality, after depleting the endogenous electric field. As we previously observed (Fig. 2g–k and Extended Data Fig. 5f), PCP inhibition in the neural fold disrupted the neural crest dCCM in vivo (Fig. 4a,c,e,f). Remarkably, the introduction of parallel electric fields was sufficient to rescue the dCCM of wild-type neural crests in embryos with depleted PCP activity in the neural fold (Fig. 4b,d–f). Nonetheless, the application of an exogenous electric field failed to rescue dCCM of Vsp1 knockdown neural crest cells, further confirming the requirement of Vsp1 in the electrotactic response of neural crest clusters to electric fields (Fig. 4b,d–f). These experiments confirm the relevance of endogenous electric fields for neural crest electrotaxis, as by purely reintroducing an electrical gradient was sufficient to restore the PCP-inhibited dCCM in vivo. Combined, our datasets support the idea that endogenous subectodermal electric fields promote collective neural crest electrotaxis, and that Vsp1 is a key component of a specific electrosensitive cascade by which neural crest cells transduce these endogenous electric currents into dCCM (Fig. 4g).

a,b, Schemes of DshDEP+ neural-fold-targeted injection (a) and parallel EF (100 mV mm−1) application (b) in embryos. c,d, In situ hybridization (lateral views) against sox8 showing the results of the indicated treatments: no parallel EFs (c) and parallel EFs (100 mV mm−1) (d). Scale bar, 200 μm. e, Percentage of embryos displaying streams. The bars represent the mean and the error bars, the standard deviations. Two-tailed Fisher’s exact test: ****PControl vs. DshDEP < 0.0001; PControl vs. DshDEP + EFs = 0.1176; **PDshDEP vs. DshDEP + EFs = 0.008; ****PControl vs. DshDEP + Xl-Vsp1-C301S + EFs < 0.0001; **PDshDEP + EFs vs. DshDEP + Xl-Vsp1-C301S + EFs = 0.0058; ****PControl vs. DshDEP + Vsp1-Cas13 + EFs < 0.0001; **PDshDEP + EFs vs. DshDEP + Xl-Vsp1-Cas13 + EFs = 0.0083. f, Normalized stream displacement. The red lines represent the mean and the error bars, the standard deviation. Two-tailed Student’s t-test: ***PControl vs. DshDEP = 0.0002; PControl vs. DshDEP + EFs = 0.0570; ****PDshDEP+ vs. DshDEP + EFs < 0.0001; **PControl vs. DshDEP + Xl-Vsp1-C301S + EFs = 0.0052; ***PDshDEP+ + EFs vs. DshDEP + Xl-Vsp1-C301S + EFs = 0.0004; **PControl vs. DshDEP + Vsp1-Cas13 + EFs = 0.0074; **PDshDEP + EFs vs. DshDEP + Xl-Vsp1-Cas13 + EFs = 0.0013; nControl = 43, nDshDEP+ = 46, nDshDEP+ + EFs = 22, nDshDEP+ + Xl-Vsp1-C301S + EFs = 20, nDshDEP+ + Xl-Vsp1-Cas13 + EFs = 17 embryos. c,d, Representative examples from at least three independent experiments; CI = 95%. g, Schematic summarizing the mechanism of neural crest collective electrotaxis in vivo: PCP-driven membrane stretch allows for ion mobilization in the neural fold. This, in addition to physiological inward currents in the flanking non-neural ectoderm, allows the establishment of an electric field along the migratory path of neural crest cells (cyan). Then, the activity of Vsp1 is required in the neural crest to specifically transduce these electric stimuli into dCCM. SACs, stretch-activated channels.
Outlook
A large body of evidence supports the idea that electrotaxis directs single and collective cell migration in vitro12,13,14,17,18. Some hypotheses have been proposed to explain the establishment of endogenous electric fields41, although no experimental demonstration was provided so far. Here we provide evidence indicating that endogenous electric fields emerge along the migratory path of neural crest cells by a mechanism involving the PCP-driven mechanical stretching of the neural fold cell membranes. Furthermore, the depletion of endogenous electric fields proved their relevance for neural crest dCCM. In combination with voltage sensors in the neural crest, these electric field depletion experiments demonstrated the sensitivity of neural crest to voltage ranges detected in endogenous electric fields. This invites research to investigate whether a similar mechanism drives electric field formation and dCCM in other biological systems. Our data also show a requirement of Vsp1 in the neural crest to convert endogenous electric fields into collective electrotaxis towards the subectodermal anode. Complementarily, we also observed that electrotaxis emerged at suboptimal levels of electric fields when overexpressing an active form of Vsp1. Yet, more experiments are necessary to confirm or rule out whether these effects were due to the presence of a larger amount of Vsp1 molecules available for activation or due to the use of the more sensitive form of Vsp1 per se. Another question arising from our study is how do subectodermal electric fields activate Vsp1. Previous reports revealed that Vsp1 is activated on membrane depolarization35,36, suggesting that endogenous electric fields first depolarize neural crest membranes and as a response, Vsp1 becomes active. Indeed, our GEVI experiments support this idea as GEVI reports that electric fields depolarize neural crest cells (Extended Data Fig. 6). Given that in addition to Vsp1, we found several stretch-activated ion channels in the neural crest, one possibility is that these proteins allow membrane depolarization on exposure to electric fields, operating upstream of Vsp1 in the cascade of events that mediate the response of neural crest to endogenous electric fields. Thus, studying whether and how these proteins, or other candidates in our list, play a role in mediating electrotaxis and establishing their hierarchical roles in relation to Vsp1 activity will provide new insights about the mechanism of electrotaxis. Another question is related to the downstream signals by which Vsp1 mediates electrotaxis. It has been described that VSP dephosphorylates several species of phosphoinositides35,36—molecules required for electrotactic-directed migration33. Therefore, Vsp1 may be allowing neural crest cells to polarize and directionally respond to electric fields via phosphoinositide dephosphorylation. This idea is in agreement with our active-Rac1 data (Extended Data Fig. 3a), as Rac1 is known to interact with phosphoinositides to mediate cell polarity2. Although further research will unveil the detailing of Vsp1 involvement in electrotaxis, our current results contribute to advance the accelerating field of electrotransduction31,34,42 by positioning Vsp1 as an electrosensor/transducer that has the potential to operate in several other biological contexts (Supplementary Table 2).
Our data also show that electric fields already form at the early non-migratory stages of neural crest development (Extended Data Fig. 2). Since mechanical inputs have been shown to be essential to trigger the onset of dCCM in vivo43,44, an interesting possibility is that these long-range electric fields would guide neural crest migration only once its substrate has stiffened. In this context, studying the mechanism underlying the interplay between mechanical and electrical cues in vivo can lead to a more comprehensive understanding of dCCM in convoluted microenvironments. Therefore, studying the potential of other candidates in integrating electrical cues with other biasing signals will provide further insights into the mechanisms governing directed migration in multicue environments. Given the wide relevance of bioelectricity and directed cell migration in a variety of biological processes such as embryogenesis, tissue repair, fibrosis and cancer16,43,45,46,47,48,49, our results have the potential to deeply impact the research across these fields. Moreover, our research can influence in vitro organ production50, as this field may benefit from the inclusion of endogenous electric fields and from considering mechanoelectrical interplay in their protocols.
Methods
All animal procedures were reviewed and approved by the Ethics Committee and Animal Welfare Body of the Instituto Gulbenkian de Ciência (IGC) and complied with the Portuguese (Decreto-Lei number 113/2013) and European (Directive 2010/63/EU) legislation.
Frog manipulation and embryo generation
X. laevis embryos (wild-type strain) were obtained by in vitro fertilization51. Briefly, the ovulation of mature females was induced by the injection of human chorionic gonadotropin (Chorulon). Oocytes were then fertilized using a sperm solution containing 500 µl of Marc’s modified Ringer (MMR) 0.1× medium (10 mM NaCl, 0.2 mM CaCl2·2H2O, 0.2 mM KCl, 0.1 mM MgCl2·6H2O and 0.5 mM HEPES (pH 7.1–7.2)). Embryos were then maintained in 0.1× MMR at 12–23 °C and staged by following established tables52.
Microinjection and pharmacologic modulations
All the mRNA and DNA microinjections were performed by using calibrated glass needles that were mounted onto a cell microinjector (MDI, PM1000), programmed to deliver 10 nl in a pulse of 0.2 s. After de-jelly, embryos were transferred into 5% Ficoll (Sigma, P7798)/0.45× MMR (w/v). To target the neural crest, dorsal and ventral animal blastomeres were injected at 8 cells; the neural fold was targeted by injecting a dorsal animal blastomere at 16 cells. Transcripts for injection were generated in vitro by using the mMESSAGE mMACHINE SP6 or T7 kits (Thermo Fisher, AM1340 or AM1344).
Then, 250 pg of nuclear RFP, membrane RFP and membrane GFP or 1 ng of DshDEP+ mRNA were injected per blastomere. The plasmids for Xl-Vsp1-C301S (Addgene, 51882), Xl-Vsp1-R152Q-GFP (Addgene, 51884) and Vsp1-GFP (Addgene, 51883) were previously reported36 and injected at 750 pg (DNA), 500 pg (RNA) and 750 pg or 1,500 pg (RNA) per blastomere, respectively. Marina-T2A-nls-mCherry (GEVI Marina)28 (Addgene, 74216) was modified to introduce a membrane localization sequence (as shown below) and transcribed by using a T7 kit (as shown above) from a PCR template that was generated by using primers 1 and 2 (Supplementary Table 1). The PCR conditions were as follows: (1) 98 °C for 30 s; (2) 98 °C for 10 s; (3) 30 s at 66 °C; (4) 72 °C for 1 min (34 cycles, steps (2)–(4)); and (5) 72 °C for 2 min. GEVI Marina mRNA was injected at ~350 pg per blastomere. Vsp1 splicing morpholino (Supplementary Table 1) was injected at a concentration of 100 µM. Cas13d-GFP mRNA was injected at 800 pg and the guide for vsp1 3′ UTR at 600 pg per blastomere.
GsMTx4 (Smartox Biotechnology, 08GSM001) was used at 5 µM in 0.1× MMR.
GEVI Marina subcloning
Marina-T2A-nls-mCherry (GEVI Marina)28 (Addgene, 74216) was modified, replacing nls-mCherry by a membrane-targeted reporter Lyn-mCherry (Lyn, tyrosine-protein kinase Lyn isoform A from Mus musculus, NP_001104566.1). Briefly, Lyn domain together with mCherry was amplified by using pSP72 mRFP43 as a template and primers 1 and 2 (Supplementary Table 1) containing the restriction sites for XmaI and NotI. The PCR conditions were as follows: (1) 98 °C for 30 s; (2) 98 °C for 10 s; (3) 30 s at 72 °C; (4) 72 °C for 45 s (24 cycles, steps (2)–(4)); and (5) 72 °C for 2 min. The amplified product was then double digested with XmaI and NotI, purified and ligated using T7 DNA Ligase (NEB, M0202) into Marina-T2A-nls-mCherry digested with the same enzymes. The appropriate ligation was confirmed by sequencing (Eurofins). The transcription of Marina-T2A-Lyn-mCherry was performed using mMachine T7 kit (AM1344) and a PCR template amplified with primers 3 and 4 (Supplementary Table 1). The PCR conditions were as follows: (1) 98 °C for 30 s; (2) 98 °C for 10 s; (3) 30 s at 66 °C; (4) 72 °C for 1 min (24 cycles, steps (2)–(4)); and (5) 72 °C for 2 min.
Neural crest dissection, culture and grafts
Dissections
Cephalic neural crests were dissected from embryos at stage 16 or 17 and cultured ex vivo as previously described53. Briefly, after removing the vitellin membrane, embryos were held in modelling clay. Then, a hair knife (eyebrow glued into a glass pipette) was used to remove the epidermis and explant neural crest clusters.
Culture
Clusters were cultured in glass-bottom dishes coated with 62.5 μg ml−1 fibronectin (Sigma-Aldrich, F1141) and filled with Danilchik’s for Amy 1× medium (53 mM NaCl, 5 mM Na2CO3, 4.5 mM potassium gluconate, 32 mM sodium gluconate, 1 mM MgSO4·7H2O, 1 mM CaCl2·2H2O and 0.1% (w/v) bovine serum albumin adjusted to pH 8.3 with 1 M Bicine).
Grafts
Neural crests from donor and host were explanted as mentioned earlier. Using the hair knife, the donor neural crest was grafted into the host embryo and held in place with a piece of coverglass (~1 × 1 mm2). Once the host embryos healed (~60 min), the glass was removed, and the embryos were kept in 0.3× MMR. For time-lapse imaging, the embryos were mounted on agarose dishes with ~1.2 mm lanes filled with 3% (w/v) methyl cellulose in phosphate-buffered saline (PBS).
Vibrating probe and glass microelectrode measurements in vivo
Details for both approaches are presented in Supplementary Note 1. Briefly, extracellular net electric current density was measured using a non-invasive vibrating voltage probe11,46,54,55. TEP was measured by impaling a glass microelectrode across the ectoderm of embryos46,56,57.
Electrotaxis assay ex vivo
The application of exogenous electric fields was performed in a custom-made polydimethylsiloxane-based electrotaxis chamber (Supplementary Note 1 and Extended Data Fig. 3a). Once prepared, the surface of the electrotactic chambers was activated by coating with 62.5 μg ml−1 of fibronectin (Sigma-Aldrich, F1141). The chambers were filled with Danilchik’s for Amy media and the neural crest clusters were plated and allowed to adhere for about 20 min. Then, using electric insulating grease (Dow Corning, DC4), a roof made of coverglass (number 1.5) was attached to the polydimethylsiloxane sides to cover the clusters and electrically seal the electric field tunnel. The electric fields were channelled through the chamber tunnel via chloridized silver electrodes and agar salt bridges, composed of agarose 1.5% in Steinberg’s solution (w/v) (58 mM NaCl, 0.67 mM KCl, 1.3 mM MgSO4·7H2O, 0.44 mM CaNO3·4H2O and 4.6 mM Tris base at pH 7.4). The required voltages were provided by a direct-current power supply (Apelex, 117240 or BK Precision, 9132B) and measured in the electrotactic chamber by a voltmeter. The Apelex power supply was fine-tuned by a resistance decade box (Tenma, 72-7270).
We also controlled the current in the system’s circuit with an ammeter in series. We obtained approximate values of 0.02 and 0.20 mA for a stimulation electric field of 10 and 100 mV mm−1, respectively. Knowing the cross-sectional area of the electrotactic chamber (Supplementary Note 1), we calculated the respective current densities applied to the neural crest clusters as 833 and 8,333 μA cm−2. The lower end of these values is in a similar order of magnitude to the estimated current density in vivo (445 μA cm−2; calculations are provided in Supplementary Note 1).
Variations in pH and temperature were not observed in our electrotactic systems under an electric field of 100 mV mm−1 for 4 h (Extended Data Fig. 3c,d). We measured the pH with a pH meter before and immediately after the application of 100 mV mm−1 for 4 h. To assess the temperature, we used a probe (PC sensor, TEMPer1F) placed at the entrance of the electric field tunnel. Then, the temperature was recorded before and along the application of an electric field.
Application of electric fields in vivo
To apply parallel or antiparallel exogenous electric fields (100 mV mm−1) along the migratory path of neural crest cells, we used the same power supply, electrodes and agar bridges described in the ex vivo setup, but in a different electric chamber (Supplementary Note 1 and Extended Data Fig. 3b). Electric fields were applied from stage 17 to stages 21 and 22, and the embryos were immediately fixed and processed for in situ hybridization.
Motility, protrusion and chemotaxis assays
For motility, the neural crest clusters were plated into a plastic-bottom dish (Falcon) coated with fibronectin (125 μg ml−1). After adhesion for 1 h, cells were live imaged. For chemotaxis, heparin-acrylic beads (Sigma-Aldrich) were soaked overnight with 3.3 μg ml−1 human SDF-1 (Sigma-Aldrich) at 4 °C. Beads were fixed to a fibronectin-coated glass-bottom dish by using grease lanes. Explants were located in front of the beads and imaged. Protrusions were imaged in the same dish but without a bead.
In situ hybridization
For Xenopus, in situ hybridization embryos were hybridized with digoxigenin-labelled antisense probes against neural crest markers (c3 (ref. 58) or sox8 (ref. 59)) and against vsp1 by following step-by-step protocols53. To synthesize a vsp1 antisense probe, a vsp1 fragment of 600 bp was amplified using primers 5 and 6 (Supplementary Table 1). Probes were transcribed with a Riboprobe in vitro Transcription System (Promega), according to the manufacturer’s instructions.
Immunofluorescence
Neural crest explants were fixed for 25 min with 3.7% formaldehyde in 0.1% PBS Tween-20 (v/v) at room temperature, rinsed and permeabilized with 0.1% PBS Triton X-100 (v/v) for 3 min. Explants were then blocked with 4% bovine serum albumin in PBS (v/v) for 30 min at room temperature and incubated overnight at 4 °C with primary antibody (mouse active Rac1-GTP at 1:4,000 in 4% bovine serum albumin; New East Biosciences 26903). To detect Vsp1, explants were blocked for 30 min with 10% normal goat serum in PBS (v/v) and incubated with anti-VSP/TPTE at 1:100 in 10% normal goat serum (antibody (N432/21), Antibodies 75-485). Excess primary antibodies were removed by washing with 0.1% PBS Tween-20, and the explants were incubated with anti-mouse Alexa Fluor 555 or anti-mouse Alexa Fluor 647 secondary antibodies at 1:350 (Invitrogen) and DAPI (1:750) for 2 h at room temperature. Excess secondary was washed 3 times in PBS Tween-20 0.1%, fixed for 5 min and mounted with Mowiol (Millipore, 475904) for confocal microscopy.
Reverse-transcription PCR
RNA extraction from heads (stage 22) was performed using the RNeasy Mini kit (Qiagen, 74104). cDNA was generated with SuperScript IV (Invitrogen, 18090050) using Oligo (dT) Primer 50 µM (Invitrogen, AM5730G) in all cases, except for the validation of the splicing morpholino, which was performed using Random Hexamer Primers (Invitrogen, N8080127). Amplification by PCR was performed using the Q5 Hot Start High-Fidelity DNA Polymerase (NEB, M0493), with the primers indicated in Supplementary Table 1. The PCR steps were as follows: (1) 98 °C for 30 s; (2) 98 °C for 10 s; (3) 30 s (annealing) at 63 °C (primers 7 and 8), 64 °C (primers 9 and 10), 55 °C (primers 11 and 12); (4) 72 °C for 20 or 30 s (32–35 cycles, steps (2)–(4)); (5) 72 °C for 2 min. Amplicons were resolved in agarose 0.8–2% (w/v) gels.
For semi-quantitative analysis, after background subtraction, values were normalized to the control of each condition and the ratio between vsp1 and elf1α of the normalized values was determined and compared.
RNA-sequencing procedures and analysis
RNA quality was assessed in an HS RNA Screen Tape Analysis (Agilent Technologies), and an mRNA library was prepared using SMART-Seq2 (ref. 60). Illumina libraries were generated with the Nextera protocol61. Library quality was assessed in a fragment analyser (AATI). Sequencing was carried out in NextSeq500 Sequencer (Illumina) using 75 SE high-throughput kit. Sequences were extracted in the FASTQ format using bcl2fastq v. 2.19.1.403 (Illumina).
After discarding ribosomal contamination, sequences were mapped against the reference genome of X. laevis using the annotation XENLA_9.2_Xenbase.gtf (v. 9.2) (https://ftp.xenbase.org/pub/Genomics/JGI/Xenla9.2/). Gene expression tables were imported into R v. 3.6.3 to normalize the gene expression with the trimmed mean of M values procedure62,63 by using the NOISeq R package (v. 2.30.0)64.
CRISPR-Cas13d to downregulate Vsp1
Design and preparation of the guide
To design the guide RNA (gRNA) for the 3′ UTR of vsp1, we used an open platform (https://cas13design.nygenome.org)65,66. The DNA template to generate the gRNA was produced by the fill-in PCR technique38 using the gRNA universal primer in combination with the primers indicated in Supplementary Table 1 (numbers 13 and 14). The PCR conditions were as follows: (1) 98 °C for 30 s; (2) 98 °C for 10 s; (3) 30 s at 65 °C; (4) 72 °C for 30 s (34 cycles, steps (2)–(4)); and (5) 72 °C for 2 min. A 72 bp amplicon was generated and purified, and 500 ng was used for T7 transcription.
Cloning and preparation of Cas13d mRNA
Two nuclear localization signals were removed from Cas13d (Addgene, 109049)67 by using the Gibson Assembly Cloning Kit (New England Biolabs, E5510) with primers 15–18 (Supplementary Table 1). The PCR conditions were as follows: (1) 98 °C for 30 s; (2) 98 °C for 10 s; (3) 30 s at 72 °C; and (4) 72 °C for 3 min (29 cycles, steps (2)–(4)). Cas13d was then subcloned into pCS2+ and fused to msfGFP67, yielding a pCS2+Cas13d-msfGFP plasmid by using Gibson Assembly and primers 19–22 (Supplementary Table 1). The PCR conditions were as follows: (1) 98 °C for 30 s; (2) 98 °C for 10 s; (3) 30 s at 65 °C (primers 19 and 20) and 72 °C (primers 21 and 22); (4) 72 °C for 2 min (29 cycles, steps (2)–(4)); and (5) 72 °C for 2 min. The pCS2+Cas13d-msfGFP plasmid was digested with NotI and transcribed with SP6.
Vsp1-GFP and GEVI Marina imaging and analysis
Neural crest cells injected with Vsp1-GFP36 were plated into fibronectin-coated glass-bottom dishes (WPI, FD35), allowed to adhere and imaged by confocal microscopy.
GEVI Marina is a genetically encoded fluorescent voltage indicator that has been used to directly report voltage variations and electrical activity in cells28,29,30. The construct of this reporter has a voltage-sensing domain35 linked to a mutated variant of super ecliptic pHluorin that directly reports the membrane potential28. In addition, the construct has an internal reporter (mCherry) for normalization (Extended Data Fig. 6a). This is an important feature as it permits to readily reference the intensity of the voltage indicator against the expression levels of the reporter (mCherry). For GEVI Marina fluorescence analysis, the normalized pHluorin to membrane mCherry fluorescence intensity was calculated (Extended Data Fig. 6b). Regions of interest (ROIs) were analysed using the ROI manager tool in Fiji (https://imagej.net/software/fiji/; v. 1.54f). The nuclei mCherry fluorescence was subtracted using the Image Calculator tool in Fiji.
Cryosectioning
Embryo sectioning was performed as previously described43,44. Briefly, neural-fold-injected embryos were fixed for 2 h at room temperature and washed twice with phosphate buffer 1× (NaH2PO4·H2O 0.2 M and K2HPO4 0.2 M, pH 7.4) for 5 min in each wash. Embryos were passed through sucrose 15% in phosphate buffer (w/v) for 2 h at room temperature. Embryos were then embedded and orientated in a gelatine solution (gelatine 8% in 15% sucrose in phosphate buffer, w/v) and incubated for 1 h at 42 °C. Gelatine blocks were flash frozen at −80 °C in pre-cooled heptane and sectioned in 25 μm slices in a cryostat (Leica, CM3050 S).
Laser ablation assay
Recoil velocity as a proxy of membrane tension build-up or stretching was assessed using laser ablation experiments23. After an initial (intact) image was acquired, membranes were photoablated using the MicroPoint system (Andor) fitted with a 350 nm pulsed laser and 365/435 dye or using the Ablate! system (3i Marianas) fitted with an attenuatable 355 nm pulsed laser. The systems were controlled by Metamorph (Molecular Devices) or SlideBook 6 (3i) software, respectively. Images were acquired using HC PL APO ×63/1.30-numerical-aperture (NA) glycerol immersion objective (Leica) or a PL APO 60×/1.20-NA water-immersion objective (Nikon) and 488 and 561 nm laser lines with iXon Ultra electron-multiplying charge-coupled device (Andor) or Prime 95b scientific complementary metal–oxide–semiconductor (Teledyne Photometrics) cameras. Junctions were ablated using one pulse of 5–10% laser. Typically, three ablations were performed per embryo to minimize potential effects due to laser damage. Ablated membranes were mediolaterally oriented, perpendicular to the neural fold. For analysis, the right and left vertexes of fluorescently labelled membranes were tracked for up to 20 s after photoablation using the Manual Tracking plug-in in Fiji.
Microscopy and time-lapse imaging
In situ hybridization
Embryos were mounted on an agarose dish with small depressions to facilitate acquisition. USB Dino-Eye eyepiece camera (Dino-Lite, AM7025X) or Flexacam C1 or C3 (Leica) mounted onto stereoscopes were used at ×2.5 or ×3.2 magnifications, using DinoCapture v. 2.0 (Dino-Lite) or Leica’s built-in software.
Immunofluorescence
A z-stack series was acquired on a Leica Stellaris 5 upright system using an HC PL APO ×63/1.40-NA oil-immersion objective (Leica) and the 405 and 568 nm laser lines. The system was controlled by LAS X (Leica).
Graft experiments
Grafted embryos were imaged using 488 and 561 nm laser lines on an Imager Z2/ApoTome.2 system (Zeiss) equipped with an Orca Flash 4.0 v2 complementary metal–oxide–semiconductor camera (Hamamatsu) or on an SP5 or Stellaris 5 confocal system (Leica). Water-immersion ×10 objectives were used in both systems: N-Achroplan ×10/0.30 NA (Zeiss) and HC PL APO ×0/0.30 NA (Leica). Microscopes (both upright setups) were controlled by ZEN v. 3.1 (Zeiss), LAS AF (Leica) or LAS X (Leica).
Electrotaxis
Clusters were imaged on a Nikon HCS microscope, equipped with a Zyla 4.2 scientific complementary metal–oxide–semiconductor camera (Andor) and using a PL APO ×10/0.45-NA objective (Nikon). The system was controlled by NIS-Elements (Nikon) to acquire time-lapse microscopy images every 5 min.
Motility
ApoTome system (Zeiss) used an EC Plan-Neofluar ×10/0.30-NA objective (Zeiss) or Stellaris 5 with an HC PL APO ×10/0.40-NA objective (Leica). The systems were controlled by ZEN v. 3.1 (Zeiss) or LAS X (Leica) to acquire time-lapse images every 2.5 or 5 min.
Protrusions
SP5 confocal system with an HC PL APO ×40/0.80-NA water-immersion objective (Leica) was used in the time-lapse mode every 30 or 60 s.
Chemotaxis
SP5 or Stellaris 5 confocal systems or a Thunder Imager 3D Cell Culture system (Leica) was used with the HC PL APO ×10/0.40-NA objective (Leica) in the time-lapse mode every 5 min.
Vsp1-GFP imaging
An inverted confocal system (Zeiss, LSM 980) controlled by ZEN Blue v. 3.3 (Zeiss) was used with the 488 and 561 nm laser lines and the C Plan-Apochromat ×63/1.40-NA oil-immersion objective (Zeiss).
GEVI Marina imaging
An upright (Stellaris 5) confocal system was used with the 488 and 561 nm laser lines. The HC PL APO ×63/1.40-NA oil-immersion objective (Leica) was used for ex vivo cluster imaging and the HC PL APO ×10/0.30-NA or ×25/0.95-NA water-immersion objectives (Leica) were used for in vivo imaging.
Neural-fold-targeted injection imaging
The Stellaris 5 confocal system with the HC PL APO ×10/0.30-NA water-immersion objective (Leica) was used to image the full embryo. Sections were imaged using the LSM 980 confocal system with the LCI Plan-Apochromat ×25/0.80-NA oil-immersion objective (Zeiss).
Cell motility analysis
Cluster and cell migration
All the tracks were performed using the Manual Tracking plug-in in Fiji. FMI and velocities were computed using the Chemotaxis and Migration Tool v. 2.0 (ibidi). The frequency of angles of the clusters/cells in relation to the electric field vector was computed using Rozeta v. 2.0 (freeware developed by J. Pazera).
Cell protrusions
Maximum protrusion area was measured in Fiji using the freehand selection tool. Protrusion dynamics correspond to the time for a complete cycle of protrusion expansion and retraction. The maximum protrusion area was determined at the maximum protrusion expansion frame.
Neural crest migration analysis
The displacement of neural crest cells was derived from the ratio between the stream length and dorsoventral length of the embryo. All the ratios were normalized against the maximum length of the control stream. Lengths were obtained in Fiji using the measurement tool.
Image and video treatment
Substacking, z projections (maximum intensity), rotations, time-colour-coded projections, look-up tables and time-lapse videos were obtained using Fiji. General treatment, including the adjustment of contrast and brightness, resizing, pseudocolouring, addition of scales and overlay of text in images and videos, were conducted in Fiji, Photoshop 2021 (Adobe) or Illustrator 2021 (Adobe). In Extended Data Fig. 6d,f–h, the nuclear area and membrane of mCherry were segmented and separated by using the Fiji image calculator subtraction plug-in. Time-lapse images from grafts were registered using the StackReg plug-in in Fiji. In few in situ hybridization microphotographs, the background was pseudocoloured in Photoshop and in some time-colour-coded projections, autofluorescent debris was coloured in Fiji, both for clarity purposes and without interfering with the sample itself.
Statistics and reproducibility
No statistical methods were used to pre-determine the sample sizes, but our sample sizes are similar to those reported in previous publications2,44. Data collection and analysis were not performed blind to the conditions of the experiments. The experimenters were not blinded due to the nature of the experimental procedures, that is, unviable, uninjected or misinjected embryos, cells or clusters needed to be excluded in advance. After this selection, embryos were randomly allocated to the specified experimental conditions, and all the experiments were repeated at least three independent times. When possible, data collection started either with control or with treatment conditions at random. Datasets were tested for normality using the d’Agostino–Pearson, Shapiro–Wilk or Kolmogorov–Smirnov tests in Prism10 (v. 10.2.2, GraphPad). When the distributions proved to be normal, significance values were calculated with a Student’s t-test (two tailed) in Prism10. For unequal variances, Welch’s correction was applied to the Student’s t-test. For instances other than normal distribution, significance values were calculated with Mann–Whitney or Wilcoxon tests (two tailed). Multiple comparisons were carried out using a one-way analysis of variance, with Tukey’s or Dunnett’s multiple comparisons tests for parametric datasets and with Kruskal–Wallis with Dunn’s correction for non-parametric datasets (Prism10). Only multiple comparisons showing P < 0.05 were processed for individual comparisons, and the significance values were calculated as described above. A 95% confidence interval (CI) was used in all the analyses and statistical detailing is provided in all the figure legends. Data outliers were identified using the statistical Grubbs’ test and/or the ROUT method (Prism10).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data supporting the findings of this study are available within the article and its Supplementary Information. Any other information is available from the corresponding author upon request. Source data are provided with this paper.
References
Etchevers, H. C., Dupin, E. & Le Douarin, N. M. The diverse neural crest: from embryology to human pathology. Development 146, dev169821 (2019).
Bajanca, F. et al. In vivo topology converts competition for cell-matrix adhesion into directional migration. Nat. Commun. 10, 1518 (2019).
Shellard, A. & Mayor, R. Collective durotaxis along a self-generated stiffness gradient in vivo. Nature 600, 690–694 (2021).
Mehta, A. S. et al. Physiological electric fields induce directional migration of mammalian cranial neural crest cells. Dev. Biol. 471, 97–105 (2021).
Stump, R. F. & Robinson, K. R. Xenopus neural crest cell migration in an applied electrical field. J. Cell Biol. 97, 1226–1233 (1983).
Nuccitelli, R. & Smart, T. Extracellular calcium levels strongly influence neural crest cell galvanotaxis. Biol. Bull. 176, 130–135 (1989).
Metcalf, M. E. M., Shi, R. & Borgens, R. B. Endogenous ionic currents and voltages in amphibian embryos. J. Exp. Zool. 268, 307–322 (1994).
Shi, R. & Borgens, R. B. Three-dimensional gradients of voltage during development of the nervous system as invisible coordinates for the establishment of embryonic pattern. Dev. Dyn. 202, 101–114 (1995).
Jaffe, L. F. & Stern, C. D. Strong electrical currents leave the primitive streak of chick embryos. Science 206, 569–571 (1979).
Metcalf, M. E. M. & Borgens, R. B. Weak applied voltages interfere with amphibian morphogenesis and pattern. J. Exp. Zool. 268, 323–338 (1994).
Reid, B., Nuccitelli, R. & Zhao, M. Non-invasive measurement of bioelectric currents with a vibrating probe. Nat. Protoc. 2, 661–669 (2007).
McCaig, C. D., Rajnicek, A. M., Song, B. & Zhao, M. Controlling cell behavior electrically: current views and future potential. Physiol. Rev. 85, 943–978 (2005).
Li, L. et al. E-cadherin plays an essential role in collective directional migration of large epithelial sheets. Cell. Mol. Life Sci. 69, 2779–2789 (2012).
Zajdel, T. J., Shim, G., Wang, L., Rossello-Martinez, A. & Cohen, D. J. SCHEEPDOG: programming electric cues to dynamically herd large-scale cell migration. Cell Syst. 10, 506–514.e3 (2020).
Cohen, D. J., James Nelson, W. & Maharbiz, M. M. Galvanotactic control of collective cell migration in epithelial monolayers. Nat. Mater. 13, 409–417 (2014).
Wolf, A. E., Heinrich, M. A., Breinyn, I. B., Zajdel, T. J. & Cohen, D. J. Short-term bioelectric stimulation of collective cell migration in tissues reprograms long-term supracellular dynamics. PNAS Nexus 1, pgac002 (2022).
Allen, G. M., Mogilner, A. & Theriot, J. A. Electrophoresis of cellular membrane components creates the directional cue guiding keratocyte galvanotaxis. Curr. Biol. 23, 560–568 (2013).
Cho, Y., Son, M., Jeong, H. & Shin, J. H. Electric field-induced migration and intercellular stress alignment in a collective epithelial monolayer. Mol. Biol. Cell 29, 2292–2302 (2018).
Richardson, J., Kotevski, A. & Poole, K. From stretch to deflection: the importance of context in the activation of mammalian, mechanically activated ion channels. FEBS J. https://doi.org/10.1111/febs.16041 (2021).
Benko, R. & Brodland, G. W. Measurement of in vivo stress resultants in neurulation-stage amphibian embryos. Ann. Biomed. Eng. 35, 672–681 (2007).
Chen, X. & Wayne Brodland, G. Multi-scale finite element modeling allows the mechanics of amphibian neurulation to be elucidated. Phys. Biol. 5, 015003 (2008).
Jacobson, A. G. & Gordon, R. Changes in the shape of the developing vertebrate nervous system analyzed experimentally, mathematically and by computer simulation. J. Exp. Zool. 197, 191–246 (1976).
Scarpa, E., Finet, C., Blanchard, G. B. & Sanson, B. Actomyosin-driven tension at compartmental boundaries orients cell division independently of cell geometry in vivo. Dev. Cell 47, 727–740.e6 (2018).
Davey, C. F. & Moens, C. B. Planar cell polarity in moving cells: think globally, act locally. Development 144, 187–200 (2017).
Nishimura, T., Honda, H. & Takeichi, M. Planar cell polarity links axes of spatial dynamics in neural-tube closure. Cell 149, 1084–1097 (2012).
Suchyna, T. M. et al. Identification of a peptide toxin from Grammostola spatulata spider venom that blocks cation-selective stretch-activated channels. J. Gen. Physiol. 115, 583–598 (2000).
Koser, D. E. et al. Mechanosensing is critical for axon growth in the developing brain. Nat. Neurosci. 19, 1592–1598 (2016).
Platisa, J., Vasan, G., Yang, A. & Pieribone, V. A. Directed evolution of key residues in fluorescent protein inverses the polarity of voltage sensitivity in the genetically encoded indicator ArcLight. ACS Chem. Neurosci. 8, 513–523 (2017).
Bando, Y., Grimm, C., Cornejo, V. H. & Yuste, R. Genetic voltage indicators. BMC Biol. 17, 71 (2019).
Lee, S., Kang, B. E., Song, Y. K. & Baker, B. J. A trafficking motif alters GEVI activity implicating persistent protein interactions at the membrane. Biophys. Rep. 2, 100047 (2022).
Nakajima, K. et al. KCNJ15/Kir4.2 couples with polyamines to sense weak extracellular electric fields in galvanotaxis. Nat. Commun. 6, 8532 (2015).
Sun, Y. et al. Infection-generated electric field in gut epithelium drives bidirectional migration of macrophages. PLoS Biol. 17, e3000044 (2019).
Zhao, M. et al. Electrical signals control wound healing through phosphatidylinositol-3-OH kinase-γ and PTEN. Nature 442, 457–460 (2006).
Cortese, B., Palamà, I. E., D’Amone, S. & Gigli, G. Influence of electrotaxis on cell behaviour. Integr. Biol. 6, 817–830 (2014).
Murata, Y., Iwasaki, H., Sasaki, M., Inaba, K. & Okamura, Y. Phosphoinositide phosphatase activity coupled to an intrinsic voltage sensor. Nature 435, 1239–1243 (2005).
Ratzan, W. J., Evsikov, A. V., Okamura, Y. & Jaffe, L. A. Voltage sensitive phosphoinositide phosphatases of Xenopus: their tissue distribution and voltage dependence. J. Cell. Physiol. 226, 2740–2746 (2011).
Ratzan, W., Rayaprolu, V., Killian, S. E., Bradley, R. & Id, S. C. K. The voltage sensing phosphatase (VSP) localizes to the apical membrane of kidney tubule epithelial cells. PLoS ONE 14, e0209056 (2019).
Kushawah, G. et al. CRISPR-Cas13d induces efficient mRNA knockdown in animal embryos. Dev. Cell 54, 805–817.e7 (2020).
Theveneau, E. et al. Collective chemotaxis requires contact-dependent cell polarity. Dev. Cell 19, 39–53 (2010).
Hossain, I. et al. Enzyme domain affects the movement of the voltage sensor in ascidian and zebrafish voltage-sensing phosphatases. J. Biol. Chem. 283, 18248–18259 (2008).
Borgens, R. B., Robinson, K. R., Vanable, J. W. & McGinnis, M. E. Electric Fields in Vertebrate Repair: Natural and Applied Voltages in Vertebrate Regeneration and Healing (Alan R. Liss, 1989).
Zhao, S., Mehta, A. S. & Zhao, M. Biomedical applications of electrical stimulation. Cell. Mol. Life Sci. 77, 2681–2699 (2020).
Barriga, E. H., Franze, K., Charras, G. & Mayor, R. Tissue stiffening coordinates morphogenesis by triggering collective cell migration in vivo. Nature 554, 523–527 (2018).
Marchant, C. L., Malmi-Kakkada, A. N., Espina, J. A. & Barriga, E. H. Cell clusters softening triggers collective cell migration in vivo. Nat. Mater. 21, 1314–1323 (2022).
Levin, M. Bioelectric signaling: reprogrammable circuits underlying embryogenesis, regeneration, and cancer. Cell 184, 1971–1989 (2021).
Ferreira, F., Luxardi, G., Reid, B. & Zhao, M. Early bioelectric activities mediate redox-modulated regeneration. Development 143, 4582–4594 (2016).
Friedl, P. & Gilmour, D. Collective cell migration in morphogenesis, regeneration and cancer. Nat. Rev. Mol. Cell Biol. 10, 445–457 (2009).
Chernet, B. T. & Levin, M. Transmembrane voltage potential of somatic cells controls oncogene-mediated tumorigenesis at long-range. Oncotarget 5, 3287–3306 (2014).
Kennard, A. S. & Theriot, J. A. Osmolarity-independent electrical cues guide rapid response to injury in zebrafish epidermis. eLife 9, e62386 (2020).
Murphy, S. V. & Atala, A. 3D bioprinting of tissues and organs. Nat. Biotechnol. 32, 773–785 (2014).
Sive, H. L., Grainger, R. M. & Harland, R. M. Xenopus laevis in vitro fertilization and natural mating methods. Cold Spring Harb. Protoc. https://doi.org/10.1101/pdb.prot4737 (2007).
Nieuwkoop, P. D. & Faber, J. Normal Table of Xenopus Laevis (Daudin): A Systematical & Chronological Survey of the Development from the Fertilized Egg till the End of Metamorphosis (North-Holland, 1967).
Barriga, E. H., Shellard, A. & Mayor, R. in Neural Crest Cells: Methods and Protocols, Methods in Molecular Biology Vol. 1976 (eds Schwarz, Q. & Wiszniak, S.) 135–152 (Humana Press, 2019).
Shipley, A. M. & Feijó, J. A. in Fertilisation in Higher Plants: Molecular and Cytological Aspects 235–250 (Springer, 1999).
Bastos, A. C., Quevedo, M. C., Karavai, O. V. & Ferreira, M. G. S. Review—on the application of the scanning vibrating electrode technique (SVET) to corrosion research. J. Electrochem. Soc. 164, C973–C990 (2017).
Hotary, K. B. & Robinson, K. R. Endogenous electrical currents and voltage gradients in Xenopus embryos and the consequences of their disruption. Dev. Biol. 166, 789–800 (1994).
Bastos, A. E. P., Costa, P. F., Varderidou-Minasian, S., Altelaar, M. & Lima, P. A. Feeding cycle alters the biophysics and molecular expression of voltage-gated Na+ currents in rat hippocampal CA1 neurones. Eur. J. Neurosci. 49, 1418–1435 (2019).
McLin, V. A., Hu, C. H., Shah, R. & Jamrich, M. Expression of complement components coincides with early patterning and organogenesis in Xenopus laevis. Int. J. Dev. Biol. 52, 1123–1133 (2008).
O’Donnell, M., Hong, C. S., Huang, X., Delnicki, R. J. & Saint-Jeannet, J. P. Functional analysis of Sox8 during neural crest development in Xenopus. Development 133, 3817–3826 (2006).
Macaulay, I. C. et al. Separation and parallel sequencing of the genomes and transcriptomes of single cells using G&T-seq. Nat. Protoc. 11, 2081–2103 (2016).
Baym, M. et al. Inexpensive multiplexed library preparation for megabase-sized genomes. PLoS ONE 10, e0128036 (2015).
Robinson, M. D. & Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25 (2010).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Tarazona, S. et al. Data quality aware analysis of differential expression in RNA-seq with NOISeq R/Bioc package. Nucleic Acids Res. 43, e140 (2015).
Wessels, H. H. et al. Massively parallel Cas13 screens reveal principles for guide RNA design. Nat. Biotechnol. 38, 722–727 (2020).
Guo, X. et al. Transcriptome-wide Cas13 guide RNA design for model organisms and viral RNA pathogens. Cell Genom. 1, 100001 (2021).
Abudayyeh, O. O. et al. RNA targeting with CRISPR-Cas13. Nature 550, 280–284 (2017).
Acknowledgements
We thank A. P. Ramos (Instituto Gulbenkian de Ciência (IGC)), E. Theveneau (Center for Integrative Biology Toulouse) and B. Steventon (Cambridge University) for helpful comments on the paper. In addition, we also thank J. Mata for technical assistance, E. Theveneau for plasmids, and the IGC’s Advanced imaging (PPBI-POCI-01-0145-FEDER-022122), Genomics (LISBOA-01-0246-FEDER-000037), Bioinformatics and Aquatic animal facilities. M.Z. thanks R. R. Jiang. We thank A. Shipley (Applicable Electronics) and E. Karplus (Science Wares) for SVET hardware and software support, and A. Bastos and P. A. Lima from Sea4Us (Portugal) for help with microelectrode experiments. This work was supported by grants from the European Research Council Starting Grant (ERC-StG) under the European Union’s Horizon 2020 research and innovation programme, grant agreement number 950254 (to E.H.B.); The European Molecular Biology Organization (EMBO) Installation Grant, project number 4765 (to E.H.B.); EMBO Young Investigator program, project number 5248 (to E.H.B.); EMBO postdoctoral fellowship, ALTF 27-2020 (to F.F.); La Caixa Junior Leader Incoming, number 94978 (to E.H.B.); and Fundação para a Ciência e a Tecnologia (FCT) postdoctoral fellowship, 2020.00759.CEECIND (to S.M.). E.H.B. acknowledges support from the IGC, Fundação Calouste Gulbenkian (FCG), start-up grant I-411133.01, and from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy (EXC 2068, 390729961), Cluster of Excellence Physics of Life of TU Dresden.
Author information
Authors and Affiliations
Contributions
E.H.B. conceptualized the study with inputs from F.F. and M.Z. E.H.B. and F.F. developed the research and F.F., S.M. and E.H.B. performed the experiments. F.F. and E.H.B. wrote the article and prepared the figures. All the authors edited the text. E.H.B. supervised the study and provided the experimental resources.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Materials thanks Yasushi Okamura and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Neural crest development and vibrating probe setup and calibration.
a, Scheme showing the transition of Xenopus laevis embryos from stage 17.5 to stage 23. We refer to these stages as pre- or early migratory stage and migratory, respectively. Streams containing groups of neural crest cells (or clusters) are observed at migratory stages as a sign of directed collective cell migration (dCCM). b, Overview of the vibrating probe setup. c, Zoom-in of the dashed red rectangle shown in (b) showing the measuring chamber with an embryo. M, middle; L, lateral; D, dorsal; V, ventral. d, Upper panel is a zoom-in of the yellow rectangle shown in (d), displaying a vibrating probe in static mode. Scale bar, 50 µm. d, Bottom panel shows the probe in vibrating mode as observed while located in the neural fold position (pos). Neural crest (cyan). Scale bar, 200 µm. e–g, Calibration and validation with an artificial source. e, Example results of vibrating probe calibration with an artificial source at the indicated values (details in Supplementary Note 1). Inset in the top left corner shows a glass microelectrode (point source) and vibrating probe. Scale bar, 150 µm. f,g, Validation results. Reference is recorded when the probe is > 1 mm away from the artificial source and measurements are taken at the indicated distance. g, Current density as a function of the distance to the artificial source using observed in (f) and theoretical data calculated using Equation 1 (Supplementary Note 1). Inset in (g) shows the results of fitting both observed and theoretical results to a linear regression. Note the exponential drop of current density with distance indicating a successful calibration. Two-tailed Pearson’s r2, ****p < 0.0001. h, In vivo probe validation using the blastopore of a Xenopus laevis embryos as a natural source of outward currents and the ventral ectoderm as a point of inward currents (or ‘sink’).
Extended Data Fig. 2 Extracellular current measurements from non-migratory to migratory stages.
a, Cartoon showing temporal vibrating probe measurements along the migratory path of the neural crest from non-migratory to migratory stages (st13, st15, st17, st20 and st22). Drawing also shows the positions where the measurements were taken from the neural fold to the flanking non-neural ectoderm. b, Results from vibrating probe current density measurements in the neural fold and flank non-neural ectoderm (as indicated). Dots represent mean and shade the standard errors. Two-tailed paired t-test (st13–20) or two-tailed Wilcoxon matched-pairs test (st22), **pst13 = 0.0017, **pst15 = 0.0042, *pst17 = 0.0339, ***pst20 = 0.0008, **pst22 = 0.0078, n = 35 embryos.
Extended Data Fig. 3 Ex vivo electrotactic setup, impact in polarity and response of neural crest clusters versus single cells.
Exploded view drawings of the design and assembly steps of the electrotactic chamber used ex vivo (a) and for the electric chamber used for in vivo experiments (b). Order of assembly is bottom-up, except for the agar bridges that close the circuit just prior to EF application. Further details in Methods. Charge flows from anode (+ pole) to cathode (− pole). Scale bar, 10 mm. (c) and (d) device controls. (c) Temperature and (d) pH controls when applying a 100 mV mm−1 electrical stimulus for 4 h. Central tendencies are median and errors interquartile ranges. c, Kruskal-Wallis’ test, n = 4 each case; d, Two-tailed Wilcoxon matched-pairs test, n = 4. 4 independent experiments. e, Immunofluorescence against activated Rac1 during electrotaxis, 1 h of exposure to EF in each case. Arrows indicate the direction of migration. Scale bar, 30 μm. f, Time color-coded trajectories of clusters and isolated neural crest cells migrating in electric fields of 100 mV mm−1. White arrows depict clusters trajectories. Note that single cell representative panel has been zoomed in to visualize the single cell. Scale bars, 100 μm. g, Rose plots showing the angle frequencies of migration in relation to the electric field vector (anode and cathode are indicated). h, Forward migration index (FMI). Red lines represent mean and error bars standard deviation. Two-tailed t-test with Welch’s correction, ****p < 0.0001, nClusters = 18, nSingle cells = 43. e,f, Representative examples from three independent experiments; CI = 95%.
Extended Data Fig. 4 Electrotactic response of neural crest clusters in a physiological range of electric field strength.
a, Time colour-coded trajectories of neural crest clusters migrating under the indicated conditions. Arrows indicate trajectories. Scale bar, 100 μm. Representative examples from three independent experiments; CI = 95%. b–d, Quantification of migratory parameters. Red lines represent median and error bars interquartile range. nNo EF = 27, n10 mV mm−1 = 32, n50 mV mm−1 = 18, n100 mV mm−1 = 44 clusters. b, Forward migration index (FMI). No EF vs. 10 mV mm−1, two-tailed t-test with Welch’s correction, *p = 0.0253; No EF vs. 50 mV mm−1, two-tailed t-test, ****p < 0.0001; No EF vs. 100 mV mm−1, two-tailed t-test with Welch’s correction, ****p < 0.0001; 10 vs. 50 mV mm−1, two-tailed t-test, ****p < 0.0001; 50 vs. 100 mV mm−1’, two-tailed t-test with Welch’s correction, *p = 0.0436. c, Velocity. No EF vs. 10 mV mm−1, two-tailed t-test, p = 0.0967; No EF vs. 50 mV mm−1, two-tailed t-test, ***p = 0.0005; No EF vs. 100 mV mm−1, two-tailed Mann Whitney U-test, ****p < 0.0001; 10 vs. 50 mV mm−1, two-tailed t-test, **p = 0.0065; 50 vs. 100 mV mm−1, two-tailed Mann Whitney U-test, *p = 0.0212. d, Net displacement. No EF vs. 10 mV mm−1, two-tailed t-test, *p = 0.0201; No EF vs. 50 mV mm−1, two-tailed t-test, ****p < 0.0001; No EF vs. 100 mV mm−1, two-tailed Mann Whitney U-test, ****p < 0.0001; 10 vs. 50 mV mm−1, two-tailed t-test; **p = 0.0010; 50 vs. 100 mV mm−1, two-tailed Mann Whitney U-test, *p = 0.0171.
Extended Data Fig. 5 Estimation of membrane tension and DshDEP+ controls.
a, Scheme of laser ablations in the neural fold and flank ectoderm. b, Tension estimated from the neural fold and in the flank ectoderm, stages as indicated. Red lines represent mean and error bars standard deviation. Two-tailed t-test with Welch’s correction, ****pNeuralFoldStage13 vs. NeuralFoldStage17 < 0.0001, two-tailed t-test ****pNeuralFoldStage17 vs. FlankEctoderm < 0.0001, nNeuralFoldStage13= 15 nNeuralFoldStage17 = 20, nFlankEctoderm = 17 cell membranes. Scale bar, 15 μm. c, Representative images of the neural fold membrane recoil velocity as a readout of stored tension (stages and treatments as indicated). Membrane tension was estimated from the recoil velocity of the membrane junctions (red dots) that were adjacent to the ablation point (red arrowhead). d, Scheme of neural fold-targeted injection. e, Neural fold-tagged embryo with membrane GFP (green) at stage 17 on the left (neural crest, cyan). Red dashed line shows the transverse plane of a cryosection presented in the right panel; DshDEP+ and membrane GFP (green) distribution can be detected mostly in the neural fold. Scale bar, 200 μm. AP, anteroposterior; nf, neural fold; np, neural plate; nc, neural crest; nt, notochord. f, Lateral views of representative embryos displaying in situ hybridisations against sox8, a neural crest marker. Scale bar, 200 μm. Representative examples from at least three independent experiments; CI = 95%.
Extended Data Fig. 6 Neural crest is electrically sensitive to applied and endogenous electric fields ex vivo and in vivo.
a, Genetically encoded voltage indicator Marina-T2A-lyn-mCherry (GEVI Marina) construct. b, GEVI Marina signals is normalized to the membrane marker mCherry, which is encoded by the same construct. c, In vivo confocal projection of an embryo expressing GEVI Marina in the neural crest (fire lookup table). Scale bar, 150 μm. Orange square and arrow indicate the region from where images in f–h were taken. d, Ex vivo confocal projections of neural crest clusters expressing both GEVI Marina (fire lookup table) and membrane as well as nuclear mCherry (gray scale). Conditions as indicated (electric field of 100 mV mm−1). Scale bar 50 μm. e, Quantification of the normalized fluorescence intensity (electric field of 100 mV mm−1). Red lines represent mean and error bars standard deviation. Two-tailed Student’s t-test with Welch’s correction, ****p < 0.0001, nNo EF = 22 and nEF = 40 cells. f–h, In vivo confocal projections of embryos expressing GEVI Marina in the neural crest (fire lookup table) and membrane as well as nuclear mCherry (gray scale). Conditions as indicated (electric field of 100 mV mm−1). Scale bar, 30 μm. Projections are neural crest leading edges from a region similar to the indicated with an orange square in c. i, Quantification of the normalized fluorescence intensity (electric field of 100 mV mm−1). One-way ANOVA with Dunnet’s multiple comparisons test, ****p < 0.0001 for both comparisons, nwild type endogenous EF = 14, nDshDEP+-no endogenous EF = 13, and napplied EF = 14 membranes. Red lines represent mean and error bars standard deviation. c,d,f,g,h, Representative confocal projections from three independent experiments; CI = 95%.
Extended Data Fig. 7 Detection of Vsp1 in Xenopus cranial neural crest and knockdown specificity and effectiveness controls.
a, Scheme and workflow of RNA-seq experiments from a single neural crest explant dissected at pre-migratory stages (~st17) and processed for SMART-seq2 RNA-seq. b, Heatmap showing relative expression levels. Note that neural crest markers are enriched (warm colors) in our libraries, unlike surrounding tissues markers (cold colors), confirming library purity (N stands for independent experiments). c, List of represented ion channels found by RNA-seq after TMM (Trimmed Mean of M-values). The voltage-sensitive phosphatase vsp1 (tpte2.L) is highlighted in red. d, Vsp1-GFP locates in the membranes of neural crest cells ex vivo. e, Representative bands of a PCR against vsp1 or sox8 (a neural crest marker) and embryo hybridized with a probe against vsp1. One side of the embryo injected with Vsp1-Cas13, showing a decreased vsp1 signal. f–k, Lateral views of embryos displaying the result of in situ hybridization against sox8 (a neural crest marker). Scale bar, 200 μm. l, Percentage of embryos displaying streams. Bars represent mean and error bars standard deviation. Two-tailed Fisher’s exact test, ****p < 0.0001 and **pVsp1-SSMO vs. Vsp1-SSMO + Vsp1-GFP = 0.0011, nControl = 25, nXl-Vsp1-C301S = 24, nControl = 35, nVsp1-SSMO = 35, nVsp1-SSMO + Vsp1-GFP = 22, nControl Cas13 = 24, nVsp1-Cas13 = 37 and nVsp1-Cas13 + Vsp1-GFP = 47 embryos. m, Representative gel showing that Vsp1-SSMO specifically targets the splicing site of vsp1. Primers spam a splicing site; hence, the presence of a band means that Vsp1-SSMO precluded splicing, and in turn knocking down vsp1. n, Representative gel showing that Vsp1-Cas13 effectively downregulates vsp1 transcripts. o, Normalized ratio intensity of vsp1/ef1α. Bars represent mean and error bars standard deviation. Two-tailed Mann Whitney U-test, ***p = 0.0001; nControl = 7, nVsp1-Cas13 = 10 gel bands. p–r, Representative confocal projections of immunofluorescence against Vsp1, showing that Vsp1-SSMO and Vsp1-Cas13 downregulates Vsp1 protein in the membranes of neural crest cells ex vivo. s, Quantification of the fluorescence intensity profile of lines overlaying cell membranes. Solid lines represent mean and shades the 95% confidence interval. Brown-Forsythe and Welch ANOVA with Dunnett’s multiple comparison against control, ****p < 0.0001 for both comparisons, n = 18 cell membranes. d,e,f–k,m,n,p–r, Representative examples from three independent experiments; CI = 95%.
Extended Data Fig. 8 Impact of vsp1 knockdown on neural crest cells motility and chemotaxis ex vivo.
a, Overlay of isolated neural crest cells at 0 min (t1) and 60 min (t2) of migration. White lines depict cell trajectories. Scale bar, 30 μm. b, Quantification of single cells normalized velocity. Red lines represent mean and error bars standard deviation. One-way ANOVA with Dunnett's multiple comparisons test, nControl = 14, nXl-Vsp1-C301S = 19, nVsp1-SSMO = 9 and nVsp1-Cas13 = 9 cells. c, Scheme of a chemotaxis assay. d, Top panels show clusters after 5 min of migration towards SDF-1 and bottom panels show clusters arriving to the SDF-1 beads after 3–4 h (red arrows depict the direction of migration). Scale bar, 100 μm. e, Forward migration index (FMI) quantifications. Red lines represent mean and error bars standard deviation. One-way ANOVA with Dunnett's multiple comparisons test, nControl = 14, nXl-Vsp1-C301S = 9, nVsp1-SSMO = 7 and nVsp1-Cas13 = 8 clusters. a,d, Representative examples from three independent experiments; CI = 95%.
Extended Data Fig. 9 Vsp1 gain of function promotes electrotaxis at suboptimal voltages.
a, Electrotaxis assay ex vivo. b, Time colour-coded trajectories ex vivo. Control, neural crest injected with nuclear RFP; Xl-Vsp1-R152Q, neural crest injected with nuclear RFP and Xl-Vsp1-R152Q-GFP. Scale bar, 100 μm. c, FMI quantifications. Red lines represent mean and error bars standard deviation. Two-tailed t-test, **p = 0.0069, nControl = 47, nXl-Vsp1-R152Q = 39 clusters. b, Representative examples from at least three independent experiments; CI = 95%.
Supplementary information
Supplementary Information
Supplementary Note 1, Tables 1 and 2, captions for Supplementary Videos 1–10 and refs. 1–35.
Supplementary Video 1
Neural crest cells migrate towards the anode of electric fields ex vivo. Representative time-lapse images of neural crest clusters in the absence (top panel) or presence (bottom panel) of electric fields (EFs) for ~7 h. The cluster in the bottom panel was exposed to EFs of 100 mV mm−1 for ~3.5 h, with polarity reversal for another ~3.5 h. Time-lapse setting was 1 picture every 5 min. Note how cells within the cluster spread in the absence of EFs and the consistent anodal migration in the presence of EFs. Neural crest tagged with nuclear RFP (magenta) and membrane GFP (cyan). Scale bar, 100 μm (Fig. 1).
Supplementary Video 2
Neural crest cells electrotact as a collective but not as single cells ex vivo. Representative time-lapse images of a neural crest cluster and an isolated neural crest cell exposed to electric fields of 100 mV mm−1. Time-lapse setting was 1 picture every 5 min; 12 frames are shown at 7 frames per second. Note the anodal-directed migration of the cluster and the random migration of the single cell. Neural crest tagged with nuclear RFP (magenta) and membrane GFP (cyan). Scale bar, 50 μm (Extended Data Fig. 3).
Supplementary Video 3
Neural crest cells electrotaxis ex vivo under a range of electric fields. Representative time-lapse images of neural crest clusters exposed to electric fields of 10, 50 or 100 mV mm−1. Time-lapse setting was 1 picture every 5 min; 48 frames are shown at 7 frames per second. Note the dose dependency of the efficiency of anodal migration. Neural crest cells tagged with nuclear RFP (cyan). Scale bar, 100 μm (Extended Data Fig. 4).
Supplementary Video 4
Membrane ablation and junction recoil analyses. Representative time-lapse images of laser ablations (solid red line) in the membranes of neural folds injected with non-migratory (st13), control (pre-migratory st17) and DshDEP+ (pre-migratory st17), as well as the flank ectoderm (pre-migratory st17). Laser ablations at the centre of mediolaterally oriented membrane junctions (that is, perpendicular to the neural fold). Time-lapse setting was 1 picture every 2 s; 6 frames are shown at 4 frames per second. Note that the membrane junction recoil in the control neural fold is higher than that in non-migratory, DshDEP+ neural folds and in flank ectoderm (red dots). Neural fold and flank ectoderm cells tagged with membrane GFP (grey). Scale bar, 15 μm (Fig. 2 and Extended Data Fig. 5).
Supplementary Video 5
PCP in the neural fold controls the directionality of neural crest cells in vivo. Representative time-lapse images showing the lateral views of embryos in which wild-type neural crest explants (nuclear RFP in cyan) were grafted into control embryos (injected with membrane GFP in the neural fold, shown in grey) or into embryos in which DshDEP+ injection was targeted to the neural fold (in grey). Time-lapse setting was 1 picture every 5 min; 25 frames are shown at 7 frames per second. Insets show the ROI outlining the length of a stream. Note that the wild-type neural crest directionally migrates in GFP but not in DshDEP+ hosts with targeted neural folds. Scale bar, 200 μm (Fig. 2).
Supplementary Video 6
Knockdown of vsp1 impairs neural crest electrotaxis ex vivo. Representative time-lapse images of neural crest clusters injected with control, Xl-Vsp1-C301S, Vsp1-Cas13 and Vsp1-SSMO exposed to electric fields of 100 mV mm−1. Time-lapse setting was 1 picture every 5 min; 34 frames are shown at 7 frames per second. Note the anodal migration of the control cluster and radial spreading as well as random walk of Xl-Vsp1-C301S Vsp1-Cas13 and Vsp1-SSMO clusters. All treatments were tagged with nuclear RFP (magenta), and Cas13 was tagged with GFP (Cas13-GFP shown in green). Scale bar, 100 μm (Fig. 3).
Supplementary Video 7
Knockdown of vsp1 affects neural crest directionality in vivo. Representative time-lapse images showing the lateral views of wild-type embryos in which control-, Xl-Vsp1-C301S-, Vsp1-SSMO- or Vsp1-Cas13-injected neural crest explants were grafted. Time-lapse setting was 1 picture every 7.5 min; 35 frames are shown at 7 frames per second. Insets show the ROI outlining the length of a stream. Note the persistent migration of control neural crests in contrast to the random migration of cells within Vsp1 knockdown clusters in vivo. Neural crest tagged with nuclear RFP construct in cyan. Embryo and eye (ellipse) outlined in solid white lines. Scale bar, 200 μm (Fig. 3).
Supplementary Video 8
Knockdown of vsp1 does not affect neural crest cells motility ex vivo. Representative time-lapse images showing the migration of isolated control-, Xl-Vsp1-C301S-, Vsp1-Cas13- or Vsp1-SSMO-injected cells. Time-lapse setting was 1 picture every 5 min; 14 frames are shown at 7 frames per second. Note that no major differences were found between conditions. All the treatments were tagged with nuclear RFP (magenta) and membrane GFP (cyan) and Cas13 was tagged with GFP (Cas13-GFP (cyan)). Scale bar, 30 μm (Extended Data Fig. 8).
Supplementary Video 9
Knockdown of vsp1 does not alter neural crest chemotaxis towards SDF-1 ex vivo. Representative time-lapse images of control-, Xl-Vsp1-C301S- and Vsp1-Cas13-injected neural crest clusters exposed to SDF-1-coated beads. Time-lapse setting was 1 picture every 5 min; 36 frames are shown at 7 frames per second. Note the similar directional migration (towards the SDF-1 bead) in all the conditions. Neural crest tagged with nuclear RFP (magenta), membrane GFP (cyan) or Cas13-GFP (cyan; for Vsp1-Cas13 condition). Scale bar, 100 μm (Extended Data Fig. 8).
Supplementary Video 10
Xl-Vsp1-R152Q allows for neural crest electrotaxis under suboptimal electric fields ex vivo. Representative time-lapse images of control (neural crest injected with nuclear RFP) and Xl-Vsp1-R152Q (neural crest injected with nuclear RFP and Xl-Vsp1-R152Q-GFP) neural crest clusters exposed to suboptimal electric fields of 5 mV mm−1. Time-lapse setting was 1 picture every 5 min; 72 frames are shown at 7 frames per second. Note the random migration of the control cluster and the anodal migration of the Xl-Vsp1-R152Q cluster. Neural crest tagged with nuclear RFP construct in cyan; fusion GFP of the Xl-Vsp1-R152Q-GFP construct in green. Scale bar, 100 μm (Extended Data Fig. 9).
Source data
Source Data Fig. 1
Statistical source data.
Source Data Fig. 2
Statistical source data.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 4
Statistical source data.
Source Data Extended Data Fig./Table 1
Statistical source data.
Source Data Extended Data Fig./Table 2
Statistical source data.
Source Data Extended Data Fig./Table 3
Statistical source data.
Source Data Extended Data Fig./Table 4
Statistical source data.
Source Data Extended Data Fig./Table 5
Statistical source data.
Source Data Extended Data Fig./Table 6
Statistical source data.
Source Data Extended Data Fig./Table 7
Statistical source data and unprocessed PCR blots.
Source Data Extended Data Fig./Table 7
Statistical source data and unprocessed PCR blots.
Source Data Extended Data Fig./Table 8
Statistical source data.
Source Data Extended Data Fig./Table 9
Statistical source data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ferreira, F., Moreira, S., Zhao, M. et al. Stretch-induced endogenous electric fields drive directed collective cell migration in vivo. Nat. Mater. 24, 462–470 (2025). https://doi.org/10.1038/s41563-024-02060-2
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41563-024-02060-2
