
Abstract
Calcium salt deposition in the kidney induces epithelial-to-mesenchymal transition (EMT) in renal tubular epithelial cells, which is the pathological basis for the progression to renal fibrosis in patients with renal stones; however, effective drugs to prevent and treat this disease have not been adequately investigated. In this study, we conducted a comprehensive analysis of fibrosis-related core genes by utilizing bioinformatics on RNA-seq data, along with web database information. Additionally, we designed both in vivo and in vitro experiments to elucidate the mechanisms and signaling pathways through which Desmodium styracifolium polysaccharides (Ds) mitigate renal fibrosis induced by nephrolithiasis. Renal fibrosis is present in both patients afflicted with calcium oxalate (CaOx) stones and in model rats. RNA-seq analysis and network database examination identified TGF-β as a fibrosis-related core gene. Moreover, Ds were found to accumulate in the kidneys of these model rats, effectively reducing crystalline deposits, mitigating renal injury, and alleviating renal fibrosis. Ds effectively attenuated nano-CaOx-induced HK-2 damage and delayed the EMT process by interfering with TGF-β expression and secretion and inhibiting the activation of the TGF-β/Smad pathway in vitro. Ds may emerge as a potential therapeutic option for the clinical treatment of crystalline renal fibrosis.

Similar content being viewed by others

Introduction
Kidney stones represent one of the most prevalent urological conditions globally, characterized by high incidence and recurrence rates, resulting in a significant societal and economic burden. Among these, calcium oxalate (CaOx) stones represent the predominant type, constituting approximately 70–80% of renal calculi cases. Notably, 35% of individuals with CaOx stones are accompanied by renal injury, and 29% are accompanied by chronic kidney disease, such as renal fibrosis1. Recent investigations have elucidated that the majority of renal injuries induced by oxalate and CaOx crystals are resistant to complete repair, posing a substantial risk of progressing into renal fibrosis2. Therefore, understanding the mechanism underlying fibrosis induced by crystalline renal injury and decelerating the progression toward chronic kidney disease in stone-afflicted patients represents a pressing clinical imperative.
Renal fibrosis is the end result of a combination of progressive chronic kidney diseases that predispose to tubular fibrosis, glomerulosclerosis, renal artery stenosis, and chronic inflammatory cell infiltration3. Presently, it is widely accepted that the mechanism underlying renal fibrosis is intricately linked to processes such as EMT, the signaling cascade of transforming growth factor-β (TGF-β), and the accumulation of extracellular matrix (ECM). These processes are triggered by chronic hypoxia and oxidative stress3. Regrettably, due to the absence of effective treatments for renal fibrosis, patients progress to end-stage renal disease (ESRD), necessitating dialysis or kidney transplantation3,4. Consequently, investigating the connection between kidney stones and renal fibrosis, as well as identifying strategies to mitigate or reverse renal fibrosis, holds promise for introducing novel approaches to clinical prevention and treatment of kidney stones5.
Renal fibrosis is a widespread occurrence in patients afflicted with diverse forms of renal stones, encompassing CaOx, phosphate, and other stone compositions. Prolonged oxidative injury or obstructions caused by crystal deposition within the tubules induce intrarenal inflammation and fibrosis, ultimately resulting in the functional impairment of certain renal units and the potential progression to end-stage renal disease6. The phenotypic transformation of renal tubular epithelial cells constitutes a pivotal mechanism in the evolution of renal fibrosis7,8. Conversely, the loss of this epithelial phenotype amplifies adherence to crystals, thereby fostering the initiation and advancement of stone formation. Li et al.9 identified that the differentiation of renal tubular epithelial cells heightens the adhesion of CaOx monohydrate crystals to MDCK cells.
Throughout the process of renal fibrosis, transforming growth factor-β, also known as TGF-β1, is acknowledged as a principal orchestrator governing EMT, ECM, and the transition of macrophages into myofibroblasts (MMT). Simultaneously, the activation of the TGF-β signaling pathway stands out as a pivotal instigator of renal fibrosis10. Nevertheless, the availability of drugs capable of effectively impeding the progression and reversing renal fibrosis remains exceedingly scarce. Consequently, the exploration and development of drugs targeting the inhibition of EMT present a promising therapeutic approach to curbing crystal-induced renal fibrosis and safeguarding renal function.
As a traditional Chinese medicine, Desmodium styracifolium has a long history of use in China for the therapy of a variety of diseases, including kidney stones. Due to the complexity of the composition of Desmodium styracifolium, the current research on Desmodium styracifolium in the treatment of kidney stones is not sufficiently in-depth, especially the research on its active ingredients is fewer. The available studies mainly focus on Fu-Fang-Jin-Qian-Cao herbal granules (multiple herbal components)11,12, Desmodium styracifolium extract13,14, flavonoids fraction15, and extracts of different polarities16. However, there is no study on polysaccharides, an essential active ingredient in Desmodium styracifolium, against kidney stone-induced fibrosis.
In this study, we will investigate the potential role of Desmodium styracifolium polysaccharides (Ds) in inhibiting oxidative stress and renal fibrosis induced by nano-COM crystals, and comprehensively analyze the possibility of the dual pathway of Ds to inhibit crystalline renal injury and fibrosis for the treatment of stones, as well as further explore the molecular mechanism and the potential signaling pathways.
Results
Structural characterization of Ds
The functional groups of Ds were analyzed using FTIR (Fig. S1). The vibrational region detected between 3200 cm−1 and 3600 cm−1 corresponds to the hydroxyl group characteristic band, which represents the polysaccharide’s characteristic peak. The fluctuations observed at 2925 cm−1 represent the absorption peaks of C-H bonds. These include stretching vibrations of C-H, C-H2, and C-H3. The absorption peak at 1622 cm−1 is indicative of the bending vibration of C=O and suggests the presence of uronic acid in Ds17.In addition, the vibrations in the range of 1000–1200 cm−1 are caused by CO-C and C-O-H of the pyranose ring, which proves that Ds contains pyranose. The α-configuration of glucans was responsible for the vibrations within the 860–830 cm–1 range18.
The monosaccharide composition of Ds was quantified using high-performance anion-exchange chromatography (HPAEC) (Fig. S2A, B). The results revealed that Ds mainly consisted Glc and Gal with minor amounts of Ara, Man, Gal-UA, Glc-UA, Xyl, and Rha monosaccharides, as displayed in Table S1. The composition percentages were 89.75%, 3.36%, 1.49%, 1.37%, 1.30%, 1.25%, 0.75% and 0.73%, respectively.
Presence of renal fibrosis in stone kidney tissue
Renal cortex tissue was obtained from three patients (no other kidney disease, such as hydronephrosis, severe urinary tract infection, renal atrophy, etc.) who underwent ureterscope (URS). Before stone removal, we obtained renal cortex tissue from calyces using BIGopsy Backloading Biopsy Forcepy19. Normal renal cortex tissue sample were obtained from three kidney cancer patients who underwent nephrectomy at the first affiliate hospital of Guangzhou Medical University (Table S2). Renal cortex tissue was excised from normal adjacent tissues >5 cm distance from the tumor tissue, and subsequent pathological examination confirmed the absence of tumor invasion.
The fibrosis marker protein α-SMA exhibited notably heightened expression levels in the kidney tissues of patients with renal stones (Fig. 1A). Subsequently, we corroborated this phenomenon using a rat kidney stone model, wherein we observed a significantly elevated MASSON-stained collagen volume fraction in the stone group in comparison to the control group of rat kidney tissues (Fig. 1B). These findings suggest the occurrence of differing degrees of fibrosis in the stone kidneys.

A Immunofluorescence double staining and quantification of relative fluorescence intensity of α-SMA in kidney tissues from clinical kidney stone patients. (Green: CK-18, epithelial cell marker; Red: α-SMA, Blue: DAPI). Magnification is×400, Scale bar = 20 μm. Data are mean ± SD, n = 3, one-way ANOVA. B Masson staining of rat kidneys and calculation of the percentage of collagen volume fraction in the kidney stone model (collagen blue area/total tissue area × 100%); Magnification is ×200, Scale bar = 100 μm. Data are mean ± SD, n = 3, one-way ANOVA. C Volcano plots displaying differential gene expression between the normal control and kidney stone model groups in rats. n = 3. D Venn plots illustrating the overlap between differential genes in the rat model and fibrosis-related genes from databases. E Diagram depicting the protein interactions among differential genes, with connecting lines indicating interactions between the proteins. F Cytoscape calculation of core genes, with darker colors indicating higher gene scores. Compared with the Normal or Control group, ** indicates P < 0.01, *** indicates p < 0.001, **** indicates p < 0.0001.
Screening of pivotal genes associated with renal fibrosis induced by kidney stones
To further elucidate the molecular mechanisms underpinning renal fibrosis resulting from kidney stones, we conducted a transcriptome sequencing analysis of RNA expression differences in the kidney tissues of kidney stone model rats compared to normal rats. This data was subsequently analyzed using bioinformatics techniques. In the stone model group, we identified 1396 significantly altered differential genes compared to the control group (Fig. 1C). Subsequently, we retrieved a gene set containing the keyword “fibrosis” from five databases, namely GeneCards20, OMIM21, DrugBank22, TTD23, and PharmGkb24, and intersected it with above 1396 differential genes, resulting in 11 differential genes (Fig. 1D). Following this, we conducted protein interaction analysis (Fig. 1E) and excluded two proteins with no interactions, leaving us with nine genes for further analysis. Using the Cytoscape algorithm in Cytohub software (Fig. 1F), we determined that TGF-β1 emerged as the core gene in this context. In our previous study, we analyzed single-cell RNA data of three Randall’s plaque and three normal renal papillae tissue, and screened differentially expressed genes among all cell types between Randall’s plaque and normal renal papillae25. Our analysis of single-cell sequencing data revealed that TGF-β1 was significantly highly expressed in renal tissues of renal stone patients (Fig. S3A, D).
Ds accumulates within the kidney and affords protection to renal function
The distribution and metabolism of Ds within organisms were initially investigated in normal mice. FTIR spectral analysis confirmed the successful labeling of DS by ICG, as evidenced by the corresponding distinct absorption peaks and some peak shifts of ICG (Fig. S4). The in vivo distribution of ICG-labeled DS was monitored using an animal in vivo imaging system at intervals of 1 h, 2 h, 4 h, 8 h, and 12 h (Fig. 2A). These observations revealed that the highest fluorescence intensity was detected in the kidneys at the 4-h mark (Fig. 2B), after which the fluorescence intensity gradually diminished. Immunofluorescence staining further confirmed this finding (Fig. 2C, F). Furthermore, we conducted a comparative analysis of the metabolic disparities of Ds in the kidneys of normal and stone-model mice. Notably, the fluorescence intensity in the kidneys of the stone-model group was markedly higher than that in the normal mice (Fig. 2D). This finding was subsequently validated through immunofluorescence staining of frozen sections, confirming the enrichment of Ds within the kidneys of stone-model mice (Fig. 2E, G). The formed stones predispose to a high expression of renal injury factors and adhesion proteins. These changes enhance DS adsorption and residency within renal tubules. In addition, concurrent crystal formation exacerbates tubule blockage, further promoting DS retention.

A Representative ICG intensity of in vivo images at different time points after i.p. injection of ICG-Ds. n = 3. B Enrichment of the Ds in various organs of mice at different time points. n = 3. C Immunofluorescence staining of mouse kidneys at different time points. (Green: CK-18; Red: ICG-Ds); Magnification is ×200, Scale bar = 100 μm. n = 3. D Fluorescence signal of ICG-Ds obtained from normal (NC+Ds) and modeling mice (DC+Ds), respectively, after ICG-Ds was i.p. injected at 4-h time point. n = 3. E Immunofluorescence staining of mouse kidneys at 4-h time point. n = 3. F, G Quantification of ICG fluorescence intensity. Data are mean ± SD, n = 3, one-way ANOVA. Compared with the 1 h group, ** indicates p < 0.01; and ns indicates p > 0.05, with no statistical difference. Compared with the NC+Ds group, *** indicates p < 0.001.
Hematoxylin and eosin (H&E) and periodic acid-Schiff (PAS) staining results revealed that the kidneys of the control group were morphologically intact, with no swelling or damage to the glomeruli and tubules. In contrast, the kidneys in the model group displayed compromised integrity, marked by various degrees of tubular swelling and crystal deposition. Conversely, the kidneys in the Ds-treated group demonstrated preserved structural integrity, and there was a notable reduction in tubular swelling and damage compared to the model group (Fig. 3A, D).

A H&E staining analysis of rat kidney. B Polarization analysis. Magnification is ×400, Scale bar = 20 μm. C MASSON staining of rat kidney. D PAS staining of rat kidney. Magnification is ×200, Scale bar = 100 μm. E Total area count of crystals. Data are mean ± SD, n = 3, one-way ANOVA. F Crystal number count. Data are mean ± SD, n = 3, one-way ANOVA. G Collagen volume fraction. Data are mean ± SD, n = 3, one-way ANOVA. Compared with the EG + AG Group, *** indicates p < 0.001, **** indicates p < 0.0001.
Creatinine and urea nitrogen are established biomarkers for assessing renal function. Our results revealed that Ds effectively decreased serum creatinine and urea nitrogen levels in a rat model of kidney stones (Fig. S5), suggesting its reno-protective potential. Additionally, evaluation of essential ion level and H&E staining of major organs (Figs. S6–S10) further confirmed the favorable biosafety profile of Ds.
Polarization microscopy was employed to observe crystal deposition within the renal tissues. The renal tubules in the model group contained crystals of varying sizes. In contrast, the Ds treatment group exhibited a substantial reduction in both the size and quantity of crystals (Fig. 3B, E, F). Additionally, MASSON staining unveiled a notable increase in the collagen volume fraction within the kidneys of the model group in comparison to the control group. The areas expressing collagen were primarily concentrated around the region surrounding the damaged renal tubules. Conversely, the collagen volume fraction in the kidneys of the rat was markedly reduced in the Ds treatment group, accompanied by a significant decrease in collagen fiber staining around the renal tubules (Fig. 3C, G).
Immunohistochemistry was employed to assess changes in oxidative stress damage, inflammation, and adhesion factors following DS treatment. The results indicated that in the kidney stone model rats, kidney injury factor (KIM-1), monocyte chemoattractant protein-1 (MCP-1), adhesion-related molecules (CD44 and OPN), and oxidative stress-related molecules (NOX-1 and NOX-4) were up-regulated to varying degrees. Simultaneously, antioxidant-related molecules (NRF-2 and SOD-1) were down-regulated in these rats (Fig. 4A, B, F). The results of single-cell sequencing showed that in the epithelial cells of renal tubules of patients with renal stones, kidney injury factor KIM-1, MCP-1 (Fig. S3B), adhesion-related molecules (CD44 and OPN) (Fig. S3C), and oxidative stress-related molecules (NOX-1) (Fig. S3E) were up-regulated, and antioxidant-related molecules (NRF-2) (Fig. S3E) were down-regulated. Notably, Ds treatment was effective in reversing these alterations in renal stone risk factors induced by the kidney stone in rats model, ultimately reducing renal damage (Fig. 4A, B, F).

A–C Immunohistochemical staining of rat kidney (CD44, KIM-1, MCP-1, OPN, NOX-1, NOX-4, NRF-2, SOD-1, FN, N-cadherin, Vimentin, α-SMA, TGF-β). n = 3. D Relative quantification of fluorescence intensity in rat kidney immunofluorescence staining (E-cadherin). Magnification is ×200, Scale bar = 100 μm. Data are mean ± SD, n = 3, one-way ANOVA. E Rat kidney immunofluorescence staining (E-cadherin). Magnification is ×200, Scale bar = 100 μm. n = 3. F, G Rat kidney immunohistochemical staining relative quantification. Data are mean ± SD, n = 3, one-way ANOVA. Compared with the EG + AG Group, * indicates p < 0.05, ** indicates p < 0.01, *** indicates p < 0.001, **** indicates p < 0.0001.
Furthermore, fibrosis-related proteins (FN, N-cadherin, Vimentin, α-SMA, and TGF-β) were obviously up-regulated, and the epithelial cell marker E-cadherin in the model rats were obviously down-regulated (Fig. 4C–E, G). Which suggests that there may be a phenomenon of functional transformation of epithelial cells (EMT) in the renal stone model rats. The results of single-cell sequencing showed that in the epithelial cells of renal tubules of patients with renal stones, fibrosis-related proteins (FN, Vimentin, and TGF-β) (Fig. S3D) were up-regulated, and the epithelial cell marker E-cadherin were down-regulated (Fig. S3E). Conversely, Ds treatment effectively reduced the alterations in fibrosis-associated factors in rat model.
Ds efficiently mitigates cellular damage induced by nano-COM
In order to better simulate the model of crystallization-induced renal injury and fibrosis, we prepared nano-scale nano-COM crystals in vitro by direct precipitation. Scanning electron microscope images of CaOx crystals are shown in Fig. 5A, and the crystals exhibit a rhombic shape. By using Nano Measurer software (v1.2.5), the diameter of the prepared crystals was counted to be about 244 ± 77 nm (Fig. 5B). Figure 5C shows the XRD spectrum of the synthesized nano-COM crystals, diffraction peaks with crystal face spacing d = 0.593, 0.365, 0.296, 0.249, and 0.235 nm were detected, which were attributed to the (−101), (020), (−202), (112), and (130) crystal faces of the COM, respectively (PDF Card No. 20-231), which indicated that the synthesized crystals were pure phase COM crystal.

A Scanning electron microscope images of CaOx crystals. B Diameter of the prepared crystals. C XRD spectra of the nano-COM crystals. D Effect of gradient Ds on the cell viability of HK-2 cells and NRK-52E cells. Data are mean ± SD, n = 5, one-way ANOVA. E Effect of gradient nano-COM on the cell viability of HK-2 cells. Data are mean ± SD, n = 3, one-way ANOVA. F Gradient Ds protects the cell viability of HK-2 cells. Data are mean ± SD, n = 6, one-way ANOVA. G Phase-contrast microscopy observation. Magnification is ×100, Scale bar = 200 μm. n = 3. H Transmission electron microscopy observation; red arrows indicate endocytosed crystals and blue arrows indicate mitochondria. Magnification is ×2000, Scale bar = 5 μm; ×7000, Scale bar = 2 μm. Compared with the Control Group, ** indicates p < 0.01, **** indicates p < 0.0001. Compared with the COM Group, #### indicates p < 0.0001.
Toxicity testing revealed that concentrations of Ds below 150 μg/mL had no significant impact on HK-2 cells and NRK-52E cells (Fig. 5D). The cytotoxicity of nano-COM displayed a positive correlation with the crystal concentration (Fig. 5E). The optimal therapeutic effect was observed when the concentration of Ds was set at 80 μg/mL (Fig. 5F). Microscopic examination unveiled that a substantial quantity of nano-COM crystals adhered to the cell surfaces in the modeling group. Consequently, the cells adopted an elongated, spindle-shaped morphology, and there was a noticeable reduction in cell numbers. In contrast, the Ds-treated group showed an obvious increase in the number of cells and a markedly decrease in the proportion of spindle-shaped cells (Fig. 5G).
Transmission electron microscopy unveiled that a substantial number of crystals adhered to the cell membrane, and numerous crystals were endocytosed into the cells in the modeling group. This led to swollen and ruptured mitochondria, as well as distorted nuclei. Conversely, in the Ds treatment group, there was a reduction in the crystals adhering to the cell membrane, along with a significant decrease in the number of cellular endocytosed crystals. Additionally, the morphology of mitochondria appeared more intact, with a noticeable decrease in the proportion of swollen mitochondria compared to the modeling group (Fig. 5H). The evaluation of oxidative stress-related parameters indicated that nano-COM crystals induced oxidative stress in HK-2 cells, as evidenced by an elevation in intracellular ROS generation, a decline in mitochondrial membrane potential (Δψm), an increase in the number of dead cells, and heightened levels of LDH and MDA. Conversely, these aforementioned indicators demonstrated significant improvement following Ds treatment (Fig. 6A–F). The results from the Western blot assay on NRF-2 and KEAP-1 further corroborated these findings (Fig. 6G, H).

A ROS, JC-1, and PI staining of HK-2 cells. In JC-1 staining, red represents JC-1 aggregates and green represents JC-1 monomers. Magnification is ×200, Scale bar = 100 μm. n = 3. B LDH kit quantification. Data are mean ± SD, n = 4, one-way ANOVA. C MDA kit quantification. Data are mean ± SD, n = 3, one-way ANOVA. D Relative quantification of ROS fluorescence. Data are mean ± SD, n = 3, one-way ANOVA. E Relative Fluorescence intensity ratio of mitochondrial membrane potential. Data are mean ± SD, n = 3, one-way ANOVA. F Counting of PI-stained cells. Data are mean ± SD, n = 3, one-way ANOVA. G Immunoblotting analysis of NRF-2 and KEAP-1. n = 3. H Semi-quantitative chart of KEAP-1 and NRF-2 expression. Data are mean ± SD, n = 3, one-way ANOVA. Compared with the COM Group, * indicates p < 0.05, ** indicates p < 0.01, *** indicates p < 0.001, **** indicates p < 0.0001.
Ds delays the EMT process in HK-2 cells triggered by nano-COM
In the preliminary stage of this study, TGF-β was found to be a key factor in kidney stones leading to fibrosis, and the cell morphology suggested that HK-2 cells might have undergone EMT (Fig. 5G). Subsequently, we delved into investigating whether nano-COM induced EMT in HK-2 cells. The results show that nano-COM stimulation of cells at graded concentrations and durations triggers expression of TGF-β and N-cadherin from epithelial cells (Fig. 7D, E, G–I). Cellular immunofluorescence analysis revealed the co-expression of the epithelial marker protein CK-18 and the mesenchymal cell marker Vimentin in HK-2 cells following nano-COM stimulation. This co-expression of both epithelial and mesenchymal cell markers indicated the occurrence of EMT in HK-2 cells (Fig. 7A, B). Additionally, the expression of the mesenchymal cell marker protein Vimentin was significantly elevated in these cells, and their morphology changed to a long spindle shape. Conversely, in the Ds treatment group, the cell morphology remained largely unchanged, and the expression of Vimentin was significantly lower than that in the modeling group. This observation indicates that Ds effectively suppressed the EMT process in HK-2 cells induced by nano-COM stimulation.

A Immunofluorescence double staining of HK-2 cells (Green: CK-18; Red: Vimentin; Blue: DAPI). Magnification is ×400, Scale bar = 20 μm. n = 3. B Relative quantification of fluorescence expression of Vimentin. Data are mean ± SD, n = 3, one-way ANOVA. C Detection of TGF-β expression in culture media by Elisa Kit. Data are mean ± SD, n = 4, one-way ANOVA. D TGF-β protein expression in HK-2 cells stimulated by gradient concentrations of nano-COM. n = 3. E Nano-COM induces changes in TGF-β and N-cadherin protein expression in HK-2 cells at gradient times. n = 3. F Changes in the expression of fibrosis-related proteins and TGF-β/Smad pathway-related proteins. n = 3. G–J Semi-quantitative statistical graphs of TGF-β, E-cadherin, N-cadherin, Vimentin, Smad2/3, P-Smad2, and P-Smad3. Data are mean ± SD, n = 3, one-way ANOVA. K–M RT-qPCR analysis of TGF-β, E-cadherin, and Vimentin. Data are mean ± SD, n = 3, one-way ANOVA. Compared with the COM Group, * indicates p < 0.05, ** indicates p < 0.01, *** indicates p < 0.001, **** indicates p < 0.0001.
The secretion of the TGF-β protein was assessed in the culture medium of nano-COM-stimulated HK-2 cells utilizing an Elisa kit. The outcomes indicated that nano-COM stimulation markedly increased the secretion of TGF-β protein. Conversely, the secretion of TGF-β protein from the Ds treatment group was significantly lower than that from the modeling group, signifying the effectiveness of Ds in inhibiting the secretion of TGF-β protein (Fig. 7C). Subsequently, we examined the key proteins involved in the major signaling pathways associated with TGF-β-induced fibrosis. The Western blot results revealed that nano-COM stimulation led to a decrease in the expression of the epithelial phenotypic protein E-cadherin in HK-2 cells. Simultaneously, there was a corresponding increase in the expression of mesenchymal phenotypic proteins, including N-cadherin and Vimentin. The expression of TGF-β protein, a key activator of fibrosis, was also elevated. Moreover, the expression of phosphorylated Smad2 and Smad3 (P-Smad2 and P-Smad3) was found to be up-regulated. These findings suggest that following stimulation of HK-2 cells with nano-COM, the cells expressed TGF-β and concurrently activated the TGF-β/Smad pathway, leading to EMT in renal tubular epithelial cells (Fig. 7F). Remarkably, the epithelial phenotypic proteins in Ds-protected HK-2 cells remained at high levels compared to the modeling group. Conversely, mesenchymal phenotypic proteins were at low levels, and the expressions of TGF-β, P-Smad2, and P-Smad3 were noticeably reduced. This observation suggests that Ds effectively inhibited the overexpression of TGF-β in HK-2 cells induced by nano-COM stimulation. Additionally, it inhibited the activation of the TGF-β/Smad pathway, consequently suppressing the EMT process in HK-2 cells (Fig. 7F–J). These findings were further supported by subsequent RT-qPCR results (Fig. 7K–M). Our experimental results indicate that Ds effectively reduces the expression and secretion of TGF-β in HK-2 cells due to nano-COM stimulation, thereby inhibiting the activation of the TGF-β/Smad pathway and, consequently, the EMT process in HK-2 cells.
Ds inhibits cellular EMT through regulation of TGF-β expression and secretion
To further delve into the therapeutic mechanism of Ds, we used human recombinant protein TGF-β to stimulate HK-2 cells, and explored whether Ds could inhibit EMT in HK-2 cells induced by human recombinant protein TGF-β. HK-2 cells were cultured for 48 h in the presence of recombinant TGF-β. The cell morphology was observed using phase-contrast microscopy, and it was observed that the recombinant TGF-β notably induced a morphological transformation of HK-2 cells, causing them to adopt a long shuttle-like shape. However, Ds effectively maintained the elliptical cell morphology of HK-2 cells (Fig. 8A). Cellular immunofluorescence analysis revealed that HK-2 cells stimulated with human recombinant TGF-β displayed high expression of the mesenchymal cell marker Vimentin, while Ds effectively maintained low levels of vimentin in HK-2 cells (Fig. 8B). Subsequently, we conducted a Western blot assay for EMT markers and observed that recombinant TGF-β significantly reduced the epithelial marker protein E-cadherin, elevated the mesenchymal phenotypic protein N-cadherin, and increased the protein level of TGF-β in HK-2 cells (Fig. 8C, D). Intriguingly, Ds effectively attenuated the rise in fibrosis-associated proteins induced by human recombinant TGF-β in HK-2 cells, while reducing the cellular TGF-β protein expression level. This was further supported by RT-qPCR results (Fig. 8E), confirming that Ds inhibited fibrosis in HK-2 cells induced by human recombinant TGF-β. Additionally, we constructed a TGF-β overexpression plasmid, and the TGF-β-expressing vector was subsequently transfected into HK-2 cells. TGF-β overexpression was confirmed by western blotting after 48 h transient transfection (Fig. S11A). The results showed that transient transfection up-regulated TGF-β protein expression in HK-2 cells, along with increased expression of the EMT marker N-cadherin (Fig. S11B). TGF-β and N-cadherin expression levels were significantly reduced in the group treated with Ds (Fig. S11B). Ds counteracted the effects of overexpressed TGF-β, possibly by reducing TGF-β protein expression and secretion through interference with TGF-β mRNA transcription or translation.

A Cell morphology observation. Magnification is ×100, Scale bar = 200 μm. n = 3. B Immunofluorescence staining of HK-2 cells (Green: CK-18; Red: Vimentin; Blue: DAPI) and relative quantification of Vimentin fluorescence expression. Magnification is × 200, Scale bar = 100 μm. Data are mean ± SD, n = 3, one-way ANOVA. C Immunoblotting analysis of E-cadherin, N-cadherin, and TGF-β. n = 3. D Semi-quantitative statistical graphs of proteins. Data are mean ± SD, n = 3, one-way ANOVA. E RT-qPCR analysis of TGF-β, E-cadherin, and Vimentin. Data are mean ± SD, n = 3, one-way ANOVA. Compared with the TGF-β Group, * indicates p < 0.05, ** indicates p < 0.01, *** indicates p < 0.001, **** indicates p < 0.0001.
In subsequent functional rescue experiments, cells were stimulated with human recombinant TGF-β for 1 h prior to Ds treatment, after which changes in damage- and fibrosis-related markers were assessed. Phase-contrast microscopy revealed that the cell morphology in the Ds treatment group following the addition of human recombinant TGF-β differed from that of the Ds treatment alone group. It displayed more mesenchymal cell characteristics, with cells adopting a long spindle shape, similar to the COM group (Fig. 9A). Subsequently, we assessed pertinent injury markers, including intracellular ROS generation, mitochondrial membrane potential, and the count of viable and non-viable cells. It was observed that the TGF-β pretreatment led to an elevation in intracellular ROS production, a disruption in mitochondrial membrane potential equilibrium, and an increase in the count of non-viable cells compared to the COM + Ds group (Fig. 9B–E). This suggests that TGF-β pretreatment counteracts some of the therapeutic effects of Ds. Additionally, fibrosis-related parameters in the cells were assessed. Cellular immunofluorescence revealed that in the group pre-treated with TGF-β, the cells expressed a higher level of the mesenchymal cell phenotypic protein Vimentin, along with corresponding alterations in cellular morphology, in contrast to the COM + Ds group (Fig. 9F). These findings were consistent with the results obtained from the Western blot analysis (Fig. 9G, I). The cellular oxidative stress-related protein NRF-2 was analyzed, and the results showed that NRF-2 protein was significantly down-regulated in the COM + Ds + TGF-β group compared to the COM + Ds group (Fig. 9G, H). This indicates that Ds’s protective effect against oxidative stress was partially reversed. Similar results were also observed in the RT-qPCR data (Fig. 9J, K). Consequently, we concluded that human recombinant protein TGF-β partially counteracted the protective effect of Ds on HK-2 cells in vitro.

A Cell morphology of HK-2 cells observation by phase-contrast microscope. Magnification is ×100, Scale bar = 200 μm. n = 3. B ROS, JC-1, and PI staining of HK-2 cells. Magnification is ×100, Scale bar = 200 μm; ×200, Scale bar = 100 μm. n = 3. C Relative quantification of ROS fluorescence. Data are mean ± SD, n = 3, one-way ANOVA. D Relative Fluorescence intensity ratio of mitochondrial membrane potential. Data are mean ± SD, n = 3, one-way ANOVA. E Counting of PI-stained cell. Data are mean ± SD, n = 3, one-way ANOVA. F Immunofluorescence double staining of HK-2 cells (Green: CK-18; Red: Vimentin; Blue: DAPI) and relative quantification of Vimentin fluorescence intensity; Magnification is ×400, Scale bar = 20 μm. Data are mean ± SD, n = 3, one-way ANOVA. G Immunoblotting analysis of NRF-2 and Vimentin. n = 3. H, I Semi-quantitative statistical graphs of proteins. Data are mean ± SD, n = 3, one-way ANOVA. J, K RT-qPCR analysis of E-cadherin and Vimentin. Data are mean ± SD, n = 3, one-way ANOVA. Compared with the COM group, ** indicates p < 0.01, *** indicates p < 0.001, **** indicates p < 0.0001. Compared with the COM + Ds Group, ## indicates p < 0.01, ### indicates p < 0.001, #### indicates p < 0.0001, and ns indicates p > 0.05, with no statistical difference.
Discussion
In the genesis of nephrolithiasis, renal inflammation and oxidative stress-induced damage stand as pivotal contributors26,27. Furthermore, direct interaction between CaOx crystals and renal tubular epithelial cells causes cellular injury2. The internalization of crystals within renal tubular epithelial cells can perturb mitochondrial function, thereby culminating in heightened production of ROS (Fig. 5). This phenomenon engenders disruptions in lysosomal homeostasis, protease leakage, and prompts the activation of inflammatory factors, including the NLRP3 inflammasome, ultimately resulting in cellular injury3,4,28,29. Persistent oxidative stress and ongoing inflammatory stimuli precipitate the functional deterioration of renal units, ultimately progressing to end-stage renal disease and considerably compromising patients’ quality of life30. Kidney injury factor (KIM-1) and monocyte chemotactic protein-1 (MCP-1) exhibited significant upregulation in the stone model rats (Fig. 4A). This observation underscores that renal stones provoke renal injury and inflammation. Furthermore, the heightened expression of adhesion molecules such as cluster of differentiation 44 (CD44) and osteopontin (OPN) (Fig. 4A) intensifies the aggregation and adhesion of CaOx crystals, thereby amplifying the process of stone formation31. Wang et al.32 demonstrated that carboxymethylated Ds inhibited the adhesion and endocytosis of CaOx crystals dihydrate (COD) in HK-2 and reduced the cellular damage induced by COD.
Furthermore, oxidative stress plays an indelible role in the development of kidney stones33. The KEAP-1/NRF-2 system plays a crucial role in maintaining cellular redox homeostasis and metabolic balance34. In addition, the KEAP-1/NRF-2 pathway is a major regulator of the cellular protective response in response to endogenous and exogenous stresses induced by ROS and electrophilic electrons35. The primary regulatory mechanism of NRF-2 involves Keap-1, a repressor protein that negatively regulates and dynamically interacts with NRF-2. In the resting state, NRF-2 in the cytoplasm binds to KEAP-1, which directs it to the Cul-3-Rbx-1 E3 ubiquitin ligase complex, promoting ubiquitination and degradation of NRF-2. Upon exposure to oxidative stress, NRF-2 is released from KEAP-1 and translocates to the nucleus. Once in the nucleus, it activates the transcription of genes encoding antioxidant proteins. This mechanism protects cells from oxidative damage36. In this current investigation, we have observed that Ds proved efficacious in mitigating crystal deposition within rat renal tissues (Fig. 3), subsequently reducing renal injury, and preserving the redox homeostasis primarily governed by NRF-2 (Figs. 4A, B and 6).
Recent studies have indicated varying degrees of renal fibrosis in patients with different clinical renal stone types37. Renal fibrosis is recognized as a contributing factor to the progression of renal stone patients to CKD38. The phenotypic transformation of renal tubular epithelial cells stands out as a pivotal mechanism in the progression of renal fibrosis7,8. Epithelial cells undergo a transformation from an epithelial phenotype to a mesenchymal phenotype via the EMT pathway. This transformation is marked by the downregulation or absence of the adhesion protein E-cadherin in epithelial cells, along with an upregulation of fibrous matrix proteins, including α-SMA, fibronectin, and collagen (Figs. 4 and 7)7,39. Marcia et al.40 showed a significant change in the morphology of renal tubular epithelial cells from cobblestone-like monolayer to spindle-shaped cells after stimulation of HK-2 with oxalate, CaOx, and TGF-β. Li et al.9 found that the differentiation of renal tubular epithelial cells promotes the adhesion of COM crystals to MDCK cells. In addition, the maintenance of epithelial phenotype of renal tubular epithelial cells plays an important role in the stabilization of renal function37. Loss of epithelial phenotype of renal tubular epithelial cells increases adhesion to crystals and promotes stone occurrence and development.
In renal fibrosis, TGF-β is a key regulator of EMT, ECM accumulation, and MMT; meanwhile, activation of the transforming growth factor TGF-β signaling pathway is a key driver of renal fibrosis10,41. Recent studies have shown that TGF-β is abnormally expressed in patients with kidney stones and in animal models. Deng et al.42 found that peripheral serum TGF-β expression levels were significantly higher in patients with idiopathic hypercalciuria kidney stones and in patients with non-idiopathic hypercalciuria kidney stones than in the normal group. Oka et al.43 tested samples from kidney biopsy patients undergoing kidney stone surgery for renal fibrosis. The results found that kidney stone patients with high TGF-β levels had a twofold risk of renal fibrosis. Wang et al.44 found significantly elevated serum and urinary TGF-β levels in a rat model of hereditary hypercalciuric urolithiasis. Hakan et al.45 found that the concentration of TGF-β in the serum of patients with obstructive ureteral stones was significantly higher than that of healthy controls. Chen et al.46 found significantly higher levels of TGF-β in the urine of patients with ureteral stone-induced urinary tract obstruction. In addition, our sequencing results further validate that TGF-β plays an important role in renal fibrosis caused by kidney stones. The results of single-cell sequencing of kidney tissues from patients and sequencing of animal transcriptomes showed that the content of TGF-β mRNA in the tissues of patients with kidney stones was significantly higher than that in normal kidney tissues. Further analysis of the results of animal transcriptomes suggested that TGF-β was a key factor in renal fibrosis due to kidney stones (Figs. 1 and S3).
TGF-β signaling is mainly mediated through the classical Smad pathway and non-Smad pathway mediated by the TGF-β receptor (TGF-βR). It has been shown that the classical Smad pathway is activated by phosphorylation of regulatory Smads (Smad2 and Smad3, R-Smads) due to their recruitment to the TGF-β receptor and phosphorylation in response to stimulation by the TGF-β. Subsequently, activated R-Smads heterodimerize with Smad4 and translocate to the nucleus, where they co-activate the expression of downstream pro-fibrotic proteins47, thus regulating the process of renal fibrosis development; whereas Smad7 effectively inhibits the expression of Smad2/348, thus inhibiting the process of renal fibrosis. Geng et al.49 found that overexpression of TGF-β significantly induced the phosphorylation of Smad2/3, which directly activated the accumulation of fibronectin and collagen, thereby promoting the progression of endometrial metastatic lesions. The downstream pathways of TGF-β include mitogen-activated protein kinase (ERK, JNK, and P38) signaling pathway, which is over-activated under continuous stimulation of TGF-β, thus promoting the TGF-β-induced EMT process in renal tubular epithelial cells50,51.
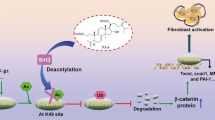
Transforming growth factor-β promotes renal fibrosis through direct and indirect effects at different cell types10. Inhibition of TGF-β has been shown to reduce fibroblast activation, decrease collagen deposition, and effectively inhibit fibrosis in animal models of kidney disease52. Sung et al.53 showed that inhibition of Smad signaling suppressed the EMT process and the accumulation of ECM in renal fibrosis. Cui et al.54 found that capsaicin could ameliorate hepatic fibrosis by activating PPAR-γ and inhibiting TGF-β/Smad pathway. Investigators found that tissue samples from patients with fibrotic nephropathy and mice displayed persistently activated (i.e., phosphorylated) Smad2 and Smad355,56, and found that in vivo and in vitro overexpression of EndoA2 significantly inhibited UUO- or TGF-β-induced Smad2/3 phosphorylation and renal tubular epithelial cell EMT, suggesting that EndoA2 prevents renal fibrosis by targeting the TGF- β/Smad signaling pathway to prevent renal fibrosis. In the present study, Ds inhibited the activation of the TGF-β/Smad pathway and suppressed the EMT process in renal tubular epithelial cells by interfering with the expression and secretion of TGF-β induced by nano-COM and human recombinant protein TGF-β (Fig. 10). In addition, overexpression experiments suggesting that Ds may reduce TGF-β protein expression and secretion by interfering with either the transcription or translation of TGF-β mRNA (Fig. S11).

Ds effectively attenuated nano-COM-induced HK-2 damage and delayed the EMT process by interfering with TGF-β expression and secretion and inhibiting the activation of the TGF-β/Smad pathway (drawn by Figdraw).
The oxidative stress hypothesis plays an important role in the development of kidney stones57. Meanwhile, TGF-β is the only isoform that can be directly activated by ROS, for a unique methionine residue at the amino acid position 253 of its latency-associated peptide is required for oxidation-triggered conformational change58. In addition, ROS can induce other TGF-β activators, such as TSP-159 and MMPs60 to activate all three isoforms in an indirect manner. Desong et al.61 found that curcumin inhibited ROS levels in hepatocytes through activation of PPAR-α, and suppressed TGF-β/Smad signaling, thereby effectively reducing the occurrence of EMT.
Through in vitro and in vivo experiments, we found that Nano-COM crystals stimulate HK-2 cells to produce a large amount of ROS, which was accompanied by mitochondrial damage and membrane potential disruption, elevation of lipid peroxidation indexes MDA and LDH, and a disruption of the balance of oxidative stress-related proteins NRF-2 and KEAP-1, as well as an increase in the cellular adherence to the crystals and endocytosis; the same phenomenon of oxidative stress was also demonstrated in the kidney of the kidney stone model rats.
Meanwhile, with the prolongation of Nano-COM crystal stimulation time, the secretion of TGF-β protein by HK-2 cells increased, along with EMT-related changes, a decrease in epithelial cell phenotype, and an increase in mesenchymal cell phenotype. In the kidneys of kidney stone model rats, the expression of renal fibrosis-related proteins was increased, and renal fibrosis appeared in the kidney tissues; and the treatment of Ds could inhibit the EMT of HK-2 cells as well as renal fibrosis by effectively inhibiting the generation of ROS, as well as interfering with the expression and secretion of TGF-β. In addition, exogenous human recombinant TGF-β protein partially reversed the therapeutic effect of Ds, suggesting that Ds relies on interfering with TGF-β expression and secretion to inhibit the EMT process (Fig. 10).
In summary, we investigated the inhibitory effect and molecular mechanism of Ds on renal fibrosis caused by kidney stones through in vivo and in vitro studies, combined with clinical specimens and animal models, and validated using transcriptome sequencing and molecular biology experiments. Ds can reduce the oxidative damage induced by nano-COM; and also, by interfering with the expression and secretion of TGF-β, inhibiting the activation of TGF-β/smad pathway, and suppressing renal fibrosis caused by nano-COM crystals. Thus, Ds may be a potential pharmaceutical agent for preventing CaOx stones and stone-induced fibrosis.
Materials and methods
Materials
Human renal proximal tubular epithelial cells (HK-2), Rat renal tubular epithelial cells (NRK-52E) were purchased from the Shanghai Cell Bank of the Chinese Academy of Sciences (Shanghai, China). The following materials were also purchased: fetal bovine serum and cell culture medium (DMEM/F12 and DMEM) (Gibco, California, USA), Penicillin, and streptomycin (Solarbio, Beijing, China). Indocyanine Green (ICG) were purchased from Macklin Biochemical Technology Co., Ltd. (Shanghai, China). The mitochondrial membrane potential assay kit (JC-1), Calcein AM/PI, and RIPA Lysis Buffer were purchased from the Shanghai Beyotime Bio-Tech Co., Ltd. (Shanghai, China). BCA kit was purchased from Thermo Fisher Scientific (Massachusetts, USA). NC membranes were purchased from Pall Corporation (New York, USA). The cell proliferation assay kit (CCK-8) and reactive oxygen species (ROS) were purchased from KeyGEN Biotechnology Co. (Nanjing, China). Transforming growth factor-β1 was purchased from Peprotech US (New Jersey, USA). Human TGF-β1 precoated ELISA Kit was purchased from Shenzhen DAKEWE BIOTECH Co., Ltd. (Shenzhen, China). Ultra-Sensitive TM SP (mouse/rabbit) IHC Kit and MaxvisionTM3 HRP-Polymer (Mouse/Rabbit) IHC Kit were purchased from MXB biotechnologies Co., Ltd. (Fuzhou, China). Cy3 Goat Anti-Rabbit IgG (H + L) and FITC Goat Anti-Mouse IgG (H + L) were purchased from ABclonal Co., Ltd. (Wuhan, China). DAPI reagent was purchased from Guangzhou Exon Biotechnology Co., Ltd. (Guangzhou, China). The TGF-β overexpression plasmid and the empty vector were purchased from GENECHEM (Shanghai, China). Invitrogen Lipofectamine 2000 was purchased from Thermo Fisher Scientific Inc. (Shanghai, China).
Extraction, purification, and characterization of Ds
Fresh samples of Desmodium styracifolium (Osb.) Merr were washed, dried at a temperature of 60 °C, and pulverized in a high-speed pulverizer. The resulting powder was then sieved and extracted with distilled water at a ratio of 1:10 (w/v) to obtain the polysaccharides62,63. The resulting supernatant was filtered and concentrated at a temperature of 60 °C using a rotary evaporator and subsequently precipitated with four times the volume of anhydrous ethanol. The obtained crude polysaccharide was then centrifuged for 10 min. The dried crude polysaccharide that was obtained was dissolved in water and then deproteinized using the Sevag method. Subsequently, a petroleum ether reagent was used to extract the fat. Next, a macroporous resin, AB-8, was employed to remove the pigment, followed by hydrodialysis (3000 Da) and concentration. Finally, the purified polysaccharide was obtained through a process of freeze-drying. Fourier transform infrared spectrometry (FTIR) was employed to determine the spectra of Ds utilizing a Nicolet iZ-10 spectrometer (Thermo Nicolet, USA). Monosaccharide composition was analyzed by high-performance anion-exchange chromatography (HPAEC).
Preparation and characterization of nano-COM
x50 mL of K2Ox (0.60 mol/L) solution was rapidly mixed with 50 mL of CaCl2 (0.60 mol/L) solution in a clean beaker. The reaction was allowed to fully react by stirring with a magnetic stirrer for 6 min, then ethylene glycol was added to promote the precipitation. The precipitated crystals were collected by repeated washing with anhydrous ethanol and purified water and dried in a drying oven for 24 h. The prepared crystals were characterized by scanning electron microscopy (SEM) and X-ray Powder Diffraction (XRD). The diameter of the prepared crystals was statistically measured by using Nano Measurer software (v1.2.5).
Specimens and study approval
Kidney tissue collection from clinical patients: All operations were performed with patients’ consent before surgery and approved by the Medical Ethics Committee of the First Affiliated Hospital of Guangzhou Medical University (NO.202091).
C57BL/6 mice and Sprague-Dawley (SD) rats were utilized and sourced from the Guangdong Experimental Medical Laboratory Animal Center. These procedures were carried out with the approval of the Medical Ethics Committee of the First Affiliated Hospital of Guangzhou Medical University. We have complied with all relevant ethical regulations for animal use.
In vivo distribution of Ds
Fluorescent labeling of Ds: Indocyanine Green (ICG) is a commonly utilized near-infrared fluorescent dye that is frequently employed to label a multitude of biomolecules, including polysaccharides, due to its favorable optical properties and biocompatibility64. 20 mg of Ds aqueous solution was mixed with 2 mg of ICG, and stirred overnight in a dark environment. Then, added anhydrous ethanol to the solution’s volume fraction of 80% for alcohol precipitation, and afterwards, remove the supernatant by centrifugation. After washing and precipitating with alcohol multiple times, the ICG-Ds complex was obtained and stored away from light.
Mice share similar enzyme systems with rats and humans in many basic biochemical processes, particularly in major pathways of drug metabolism, such as the cytochrome P450 system. Therefore, to facilitate the observation of small animal imaging systems and to reduce the amount of fluorescently labeled Ds, we used 6–8-week-old male C57BL/6 mice to study the distribution and metabolism of Ds in vivo. Differences in the in vivo distribution of Ds in normal and stone modeling mice were observed by small animal imaging system. Normal mouse group: normal mice were injected intraperitoneally with 200 μL of saline daily for 1 week. Subsequently, ICG-Ds was given intraperitoneally at a dose of 200 mg/kg body weight. Glyoxylic acid stone modeling group: stone model was constructed in mice by intraperitoneal injection of glyoxylic acid 70 mg/kg/d daily for 1 week. Subsequently, ICG-Ds was given intraperitoneally at a dose of 200 mg/kg body weight. Fluorescence expression was monitored at 1, 2, 4, 8, and 12 h utilizing a small animal imaging system.
Animal experiment
The rats selected for this study were male SD rats, aged 6–8 weeks, with a weight ranged from 180 to 200 g. The rats were randomly divided into three groups. The experimental procedures were conducted as follows: (A) Normal Control Group (Control): Providing with a standard diet. (B) Stone Group (EG + AG Group): providing with a standard diet, drinking 1% ethylene glycol by volume for 28 days, and treated accordingly with 1% ammonium chloride by gavage for the first 3 days of the first and third weeks of the experiment. (C) Ds Treatment Group (EG + AG+Ds Group): treated with 100 mg/kg body weight of Ds by i.p. daily for 28 days in parallel with the corresponding treatment in the stone group. During the experimental period, all rats were fed ad libitum on regular food and kept at 25 °C with light/dark cycling conditions. Rats were executed on day 28, and tissue specimens were collected. Subsequently, serum of rats was collected to examine biochemical indices.
Histology and immunohistochemical staining
Tissue specimens were fixed with 4% paraformaldehyde, dehydrated with gradient ethanol, transparent with xylene, embedded in paraffin, and cut into 5-μm paraffin sections. Routine paraffin sections were subjected to Hematoxylin-eosin (H&E) staining, Masson trichrome staining, and PAS staining.
Paraffin-embedded sections underwent antigen retrieval utilizing sodium citrate solution. Subsequently, they were incubated overnight at 4 °C with the anti-KIM-1(1:50; ABCAM), CD44 (1:200; ABCAM), MCP-1 (1:200; Proteintech), OPN (1:200; ABCAM), NOX-1 (1:200; ABCAM), NOX-4 (1:200. Proteintech), NRF-2 (1:200; ABCAM), SOD-1 (1:200; Proteintech), Fibronectin (1:200; Proteintech), N-cadherin (1:200; ABCAM), Vimentin (1:200; ABCAM), α-SMA (1:200; Proteintech), TGF-β (1:200; Proteintech) antibody. According to the manufacturer’s protocol using the Ultra-Sensitive TM SP (mouse/rabbit) IHC Kit (KIT-9730; MXB biotechnologies) and MaxvisionTM3 HRP-Polymer (Mouse/Rabbit) IHC Kit (KIT-5220; MXB biotechnologies), perform the analysis.
Immunofluorescence staining
Cryosections were embedded in optimal cutting temperature (OCT) compound and sliced into 5-μm sections. Frozen sections were subjected to incubation with Vimentin (1:200; ABCAM), E-cadherin (1:200; ABCAM), and CK-18 (1:500; Proteintech) antibodies. Subsequently, they were incubated with secondary antibodies labeled Cy3 Goat Anti-Rabbit IgG (H + L) (AS007) (1:500; ABclonal) and FITC Goat Anti-Mouse IgG (H + L) (1:500; AS001; ABclonal) simultaneously. Cell nuclei were stained with DAPI reagent with fluorescence quencher (F-0045; Guangzhou Exon biotechnology). E-cadherin and CK-18 antibodies has been shown to have good specificity (Fig. S12).
Transcriptome sequencing and core factor screening
Rat kidney tissues were submitted to Gene Denovo (Gene Denovo, Guangzhou, China) for transcriptome sequencing. We retrieved gene sets related to “fibrosis” from five databases (Gene Cards, OMIM, Drug Bank, TTD, PharmGkb) and intersected them with the differentially expressed genes identified through RNA-seq analysis in both control and stone-model rats. This process resulted in 11 intersected genes. Subsequently, these 11 differential genes underwent protein interaction analysis, excluding two proteins that did not exhibit any interactions. The remaining nine genes were subjected to interaction scoring and further analyzed using the Cytoscape algorithm within the Cytohub software. These analyses revealed that TGF-β stood out as the core gene.
Cell culture, transfection, and grouping
HK-2 cells and NRK-52E cells were cultured in DMEM/F12 and DMEM medium containing 10% fetal bovine serum, penicillin-streptomycin antibiotics, and placed in a constant temperature incubator at 37 °C and 5% CO2. In the cell injury assay, the treatment time was 24 h, while in the EMT assay, the treatment time extended to 48 h. Phosphorylated proteins were extracted after 1 h of cell treatment.
Transient transfection was carried out using LipofectamineTM 2000 (Invitrogen, Carlsbad, CA, USA) reagent according to the manufacturer’s recommendations. Cells were seed into six-well plates 1 day before transfection. Cells were maintained in serum-free medium during transfection, and replaced with fresh culture medium 6 h later. After 48 h expression, cells were subject to different treatments.
Cell groupings are described below: (1) Control group: which was treated with serum-free medium; (2) Control + Ds group: which was treated with serum-free medium containing 80 μg/mL Ds;(3) COM group: which was treated with serum-free medium containing 200 μg/mL nano-COM;(4) COM + Ds group: serum-free medium containing 200 μg/mL nano-COM and 80 μg/mL Ds solution was added and incubated;(5) TGF-β group: which was treated with serum-free medium containing recombinant TGF-β;(6) TGF-β + Ds group: serum-free medium containing recombinant TGF-β and 80 μg/mL Ds solution was added and incubated;(7) TGF-β + COM + Ds group: TGF-β was introduced to HK-2 cells for 1 h before the administration of Ds and nano-COM; (8) Vector group: transfected with empty vector; (9) Vector +Ds group: transfected with empty vector and 80 μg/mL Ds solution was added and incubated; (10) TGF-β overexpression group: transfected with TGF-β expression vector; (11) TGF-β overexpression + Ds group: transfected with TGF-β expression vector and 80 μg/mL Ds solution was added and incubated.
CCK-8 cell viability detection
Pharmacotoxicity assessment
Cell suspensions (1.0 × 105cells/mL) were inoculated in 96-well plates at 100 μL per well and incubated for 24 h. Serum-free medium containing Ds at concentrations of 10, 20, 40, 60, 80, 100, 120, and 150 μg/mL was co-cultured with two types of renal cells (HK-2 and NRK-52E) for 24 h. Upon reaching the incubation time 10 μL of CCK-8 reagent was added to each well, and the cells were incubated for 2 h. After incubation, the absorbance at 450 nm was measured using a multifunctional enzyme marker.
Nano-COM cytotoxicity assessment
Serum-free medium containing nano-COM at concentrations of 25, 50, 100, 150, 200, 250, 300, and 400 μg/mL was co-cultured with HK-2 cells. After incubation, the absorbance at 450 nm was measured.
Pharmacoprotective assessment
200 μg/mL nano-COM solution was selected for damaged HK-2 cells to construct the damage cell model, and the concentrations of 20, 40, 60, 80, 100, and 120 μg/mL Ds solution were added and co-incubated for 24 h. After incubation, the absorbance at 450 nm was measured.
Cell morphology observation
2 mL of cell suspension was inoculated in 6-well culture plates for 24 h, and after the treatment time was reached, the cell morphology was observed using a phase-contrast microscope.
Cell injury detection
Detection of intracellular reactive oxygen species (ROS) level: After reaching the treatment time, the supernatant was then removed, and the cells were washed three times with PBS. Subsequently, 500 μL of DCFH-DA (S0033M; Beyotime), diluted in serum-free medium at a 1:1000 ratio, was added to the cells. The cells were then placed in a 37 °C incubator for 30 min, and images were captured using fluorescence microscopy.
Detection of mitochondrial membrane potential (ΔΨm): After reaching the treatment time, the supernatant was then removed, and the cells were washed three times with PBS. The JC-1 assay working solution was prepared according to the kit instructions (C2006, Beyotime), and each group of cells was added with 1 mL of JC-1 assay working solution, incubated at 37 °C for 30 min away from light. At the end of the incubation, the cells were washed with serum-free medium for 3 times to remove the excessive JC-1 assay working solution, and then each well was added with 1 mL of serum-free medium. The ΔΨm was observed under a fluorescence microscope, and the fluorescence intensity was counted by ImageJ software.
Live/dead cell staining (C2015M, Beyotime), malondialdehyde (MDA) (S0131M, Beyotime) level, and lactate dehydrogenase (LDH) (C0017, Beyotime) release assays were carried out following the instructions provided in the kit.
Transmission electron microscopy (TEM)
Upon reaching the designated incubation time, the cells were washed once with a PBS solution. Subsequently, they were treated with an electron microscope fixative, gently scraped off from the culture dish using a cell scraper, and then centrifuged at 3000 rpm at 25 °C for 3 min to remove the supernatant. The cells were then immersed in an appropriate volume of fresh electron microscope fixative and left to fix at room temperature for 2 h. Afterward, they were transferred to a 4 °C environment for overnight storage and later observed and captured using transmission electron microscopy.
Western blot analysis
Cell lysis was performed using RIPA Lysis Buffer (P0013B; Beyotime), and protein concentrations were determined using a BCA kit (A53225; Thermo Fisher Scientific). Protein concentrations were normalized to a uniform concentration using 2× Dual-color Loading buffer (DTT) (FD003-25; Fdbio science). The proteins were then separated using a 10% SDS-PAGE gel and transferred to NC membranes (66485; Pall). Subsequently, membranes were blocked with 5% skimmed milk or bovine serum albumin to prevent nonspecific binding. Primary antibodies were applied and incubated overnight at 4 °C. Protein bands were visualized using a ChemiDoc XRS instrument (Bio-Rad) after incubation with fluorescent secondary antibodies for 1 h. ImageJ software was used to analyze all bands and normalize the expression of target proteins to GAPDH. NRF-2 (1:1000; ABCAM), KEAP-1(1:1000; ABCAM), TGF-β (1:1000; Proteintech), N-cadherin (1:1000; ABCAM), E-cadherin (1:1000; ABCAM), Smad2/3 (1:1000; CST), P-Smad2 (1:1000; CST), P-Smad3 (1:1000; CST), Vimentin (1:1000; ABCAM), GAPDH (1:10000; Proteintech).
Real-time qPCR analysis
Total RNA was extracted from HK-2 cells using Trizol Reagent (Zomanbio). TGF-β were amplified by the following pairs of primers: 5′-CAGGAGTCATCAGTGTGGT-3′; 5′-GGAGGATTATCGTTGGTGTCAG-3′. N-cadherin: 5′-CAATTCCTGGCGATACCTCAG-3′; 5′-GCACAACTCCGGTGACATCAA-3′. Vimentin:5′-TCTACGAGGAGGAGATGCGG-3′; 5′-GGTCAAGACGTGCCAGAGAC-3′. GAPDH:5’-TGTGTCCGTCGTGGATCTGA-3′; 5′-TTGCTGTTGAAGTCGCAGGAG-3′. The mRNA expression was determined by real-time quantitative PCR (qRT-PCR). The mRNA levels were normalized to the GAPDH mRNA levels in the corresponding samples.
Elisa assay
The cell culture medium was collected following the protocol outlined in the Human TGF-β1 Precoated ELISA Kit (2201-2; DAKEWE). Samples from the kit were prepared in advance, kit reagents were appropriately warmed according to the specified requirements, and the experimental procedures were meticulously conducted in accordance with the instructions provided with the kit.
Detection of BUN and creatinine
Animal creatinine and urea nitrogen assays were performed using Beckman Coulter creatinine (OSR6678) and Beckman Coulter urea nitrogen reagent (472482) kits at the AU5800 Series Clinical Chemistry Analyzers platform in the Laboratory Department of the First Hospital of Guangzhou Medical University.
Single-cell sequencing data analysis
scRNA-seq data analysis were analyzed as previously described25. Briefly, low-quality reads and adapter sequences were filtered from the FASTQ data65. The cell barcode whitelist was identified with UMI-tools, and clean data were mapped to the human genome (Ensemble version 91) using STAR66. Cells with over 4800 genes or fewer than 200, or with >5% of UMIs mapped to mitochondrial genes, were discarded. Seurat (version 3.1.0) was used for normalization, with Harmony for batch correction67. PCA was performed with the top 2000 variable genes, and tSNE was constructed with the first 15 components. Graph-based clustering identified cell clusters, and marker genes were determined using the FindAllMarkers function (P value < 0.05, log2(Fold change) > 0.25, min.pct > 0.15).
Statistics and reproducibility
All experiments were independently repeated at least three times, and statistical methods are described in the figure legends. The results of this study are presented as mean ± standard deviation (mean ± SD). Differences between two groups were analyzed using Student’s t-test, while comparisons among multiple groups were performed using one-way ANOVA. A p value less than 0.05 was considered statistically significant. The following symbols were defined: ns p > 0.05, * p < 0.05, ** p < 0.01, *** p < 0.001, and **** p < 0.0001. Data and graphics were processed using GraphPad Prism 9.00 (GraphPad Software, La Jolla, CA, USA).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
RNA sequencing data used in this study are accessible with the following link: https://ngdc.cncb.ac.cn/gsa/browse/CRA027937. All data supporting the findings of this study are available in the article and its Supplementary Information. Uncropped Western blot images are provided in the Supplementary Information. RNA-seq data and the raw data underlying the graphs and charts are available in Supplementary Data 1. Additional data and materials are available from the corresponding author upon reasonable request.
References
Evan, A. P. Physiopathology and etiology of stone formation in the kidney and the urinary tract. Pediatr. Nephrol. 25, 831–841 (2010).
Sun, X. Y., Ouyang, J. M. & Yu, K. Shape-dependent cellular toxicity on renal epithelial cells and stone risk of calcium oxalate dihydrate crystals. Sci. Rep. 7, 7250 (2017).
Liu, Y. Cellular and molecular mechanisms of renal fibrosis. Nat. Rev. Nephrol. 7, 684–696 (2011).
Boor, P., Ostendorf, T. & Floege, J. Renal fibrosis: novel insights into mechanisms and therapeutic targets. Nat. Rev. Nephrol. 6, 643–656 (2010).
Webster, A. C., Nagler, E. V., Morton, R. L. & Masson, P. Chronic Kidney Disease. Lancet 389, 1238–1252 (2017).
Boonla, C. et al. Fibrosis and evidence for epithelial-mesenchymal transition in the kidneys of patients with staghorn calculi. BJU Int. 108, 1336–1345 (2011).
Liu, Y. Epithelial to mesenchymal transition in renal fibrogenesis: pathologic significance, molecular mechanism, and therapeutic intervention. J. Am. Soc. Nephrol. 15, 1–12 (2004).
Kalluri, R. & Neilson, E. G. Epithelial-mesenchymal transition and its implications for fibrosis. J. Clin. Investig. 112, 1776–1784 (2003).
Li, S. et al. L-carnitine protects renal tubular cells against calcium oxalate monohydrate crystals adhesion through preventing cells from dedifferentiation. Kidney Blood Press. Res. 41, 582–592 (2016).
Meng, X. M., Nikolic-Paterson, D. J. & Lan, H. Y. TGF-beta: the master regulator of fibrosis. Nat. Rev. Nephrol. 12, 325–338 (2016).
Liu, W. R. et al. Fu-Fang-Jin-Qian-Cao herbal granules protect against the calcium oxalate-induced renal EMT by inhibiting the TGF-β/smad pathway. Pharm. Biol. 58, 1115–1122 (2020).
Chen, W. et al. Metabolomic analysis reveals a protective effect of Fu-Fang-Jin-Qian-Chao herbal granules on oxalate-induced kidney injury. Biosci. Rep. 39, BSR20181833 (2019).
Mi, J. et al. Evaluation of antiurolithic effect and the possible mechanisms of Desmodium styracifolium and Pyrrosiae petiolosa in rats. Urol. Res. 40, 151–161 (2012).
Hou, J. et al. Exploring the therapeutic mechanism of desmodium styracifolium on oxalate crystal-induced kidney injuries using comprehensive approaches based on proteomics and network pharmacology. Front. Pharmacol. 9, 620 (2018).
Zhou, J. et al. Total flavonoids of Desmodium styracifolium attenuates the formation of hydroxy-L-proline-induced calcium oxalate urolithiasis in rats. Urolithiasis 46, 231–241 (2018).
Xiang, S. et al. Antilithic effects of extracts from different polarity fractions of Desmodium styracifolium on experimentally induced urolithiasis in rats. Urolithiasis 43, 433–439 (2015).
Gong, Y. et al. Structure features and in vitro hypoglycemic activities of polysaccharides from different species of Maidong. Carbohydr. Polym. 173, 215–222 (2017).
Wang, L., Chen, L., Li, J., Di, L. & Wu, H. Structural elucidation and immune-enhancing activity of peculiar polysaccharides fractioned from marine clam Meretrix meretrix (Linnaeus). Carbohydr. Polym. 201, 500–513 (2018).
Taguchi, K. et al. Fatty acid-binding protein 4 downregulation drives calcification in the development of kidney stone disease. Kidney Int. 97, 1042–1056 (2020).
Stelzer, G. et al. The GeneCards Suite: from gene data mining to disease genome sequence analyses. Curr. Protoc. Bioinform. 54, 1.30.31–33 (2016).
Amberger, J. S., Bocchini, C. A., Schiettecatte, F., Scott, A. F. & Hamosh, A. OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 43, D789–D798 (2015).
Wishart, D. S. et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 46, D1074–D1082 (2018).
Zhou, Y. et al. TTD: Therapeutic Target Database describing target druggability information. Nucleic Acids Res. 52, D1465–D1477 (2024).
Hewett, M. et al. PharmGKB: the Pharmacogenetics Knowledge Base. Nucleic Acids Res. 30, 163–165 (2002).
Liu, Z. et al. Landscape of microenvironment in Randall’s plaque by single-cell sequencing. Preprint at bioRxiv https://doi.org/10.1101/2021.05.30.444880 (2021).
Sun, X. Y., Ouyang, J. M., Gan, Q. Z. & Liu, A. J. Renal epithelial cell injury induced by calcium oxalate monohydrate depends on their structural features: size, surface, and crystalline structure. J. Biomed. Nanotechnol. 12, 2001–2014 (2016).
Liu, Y. et al. Inhibition of autophagy-attenuated calcium oxalate crystal-induced renal tubular epithelial cell injury in vivo and in vitro. Oncotarget 9, 4571–4582 (2018).
Wynn, T. A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 214, 199–210 (2008).
Lopez-Novoa, J. M. & Nieto, M. A. Inflammation and EMT: an alliance towards organ fibrosis and cancer progression. EMBO Mol. Med. 1, 303–314 (2009).
Chaiyarit, S. & Thongboonkerd, V. Changes in mitochondrial proteome of renal tubular cells induced by calcium oxalate monohydrate crystal adhesion and internalization are related to mitochondrial dysfunction. J. Proteome Res. 11, 3269–3280 (2012).
Peng, H., Ouyang, J. M., Yao, X. Q. & Yang, R. E. Interaction between submicron COD crystals and renal epithelial cells. Int. J. Nanomed. 7, 4727–4737 (2012).
Wang, Z. et al. Carboxymethylated Desmodium styracifolium polysaccharide reduces the risk of calcium oxalate kidney stone formation by inhibiting crystal adhesion and promoting crystal endocytosis. J. Cell. Physiol. 239, e31272 (2024).
Khan, S. R. Reactive oxygen species, inflammation and calcium oxalate nephrolithiasis. Transl. Androl. Urol. 3, 256–276 (2014).
Deng, J. W. et al. Mechanism of porphyra yezoensis polysaccharides in inhibiting hyperoxalate-induced renal injury and crystal deposition. J. Agr. Food Chem. 72, 6372–6388 (2024).
Kansanen, E., Jyrkkänen, H. K. & Levonen, A. L. Activation of stress signaling pathways by electrophilic oxidized and nitrated lipids. Free Radic. Bio Med. 52, 973–982 (2012).
Martinez, V. D. et al. Frequent concerted genetic mechanisms disrupt multiple components of the NRF2 inhibitor KEAP1/CUL3/RBX1 E3-ubiquitin ligase complex in thyroid cancer. Mol. Cancer 12, 124 (2013).
Li, S. et al. Peroxisome proliferator-activated receptor gamma modulates renal crystal retention associated with high oxalate concentration by regulating tubular epithelial cellular transdifferentiation. J. Cell. Physiol. 234, 2837–2850 (2019).
Steiger, S. et al. Anti-transforming growth factor beta IgG elicits a dual effect on calcium oxalate crystallization and progressive nephrocalcinosis-related chronic kidney disease. Front. Immunol. 9, 619 (2018).
Liu, Y. New insights into epithelial-mesenchymal transition in kidney fibrosis. J. Am. Soc. Nephrol. 21, 212–222 (2010).
Convento, M. B. et al. Calcium oxalate crystals and oxalate induce an epithelial-to-mesenchymal transition in the proximal tubular epithelial cells: Contribution to oxalate kidney injury. Sci. Rep. 7, 45740 (2017).
Sato, M., Muragaki, Y., Saika, S., Roberts, A. B. & Ooshima, A. Targeted disruption of TGF-beta1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J. Clin. Investig. 112, 1486–1494 (2003).
He, D. et al. Calcium ions promote primary renal epithelial cell differentiation into cells with bone-associated phenotypes via transforming growth factor-β1-induced epithelial-mesenchymal transition in idiopathic hypercalciuria patients. Mol. Med. Rep. 11, 2199–2206 (2015).
Oka, A. A. G., Suwitra, K. & Soebadi, D. M. Obstructive nephropathy of kidney stone: the role of caspase-3, transforming growth factor-β and tumor necrosis factor-α in kidney fibrosis. BJU Int. 117, 6–6 (2016).
He, D. et al. The Wnt11 signaling pathway in potential cellular EMT and osteochondral differentiation progression in nephrolithiasis formation. Int. J. Mol. Sci. 16, 16313–16329 (2015).
Vuruskan, H. et al. Elevated plasma concentrations of transforming growth factor-beta 1 in patients with unilateral ureteral obstruction. Urol. Res. 33, 465–469 (2005).
Chen, X. et al. Urinary TGF-1 has a supplementary value in predicting renal function recovery post unilateral ureteral obstruction. Int. Urol. Nephrol. 47, 33–37 (2015).
Li, Y. et al. Inhibitory effects of peroxisome proliferator-activated receptor gamma agonists on collagen IV production in podocytes. Mol. Cell. Biochem. 405, 233–241 (2015).
Lan, H. Y. Smad7 as a therapeutic agent for chronic kidney diseases. Front. Biosci. 13, 4984–4992 (2008).
Geng, X. Q. et al. Ganoderic acid hinders renal fibrosis via suppressing the TGF-beta/Smad and MAPK signaling pathways. Acta Pharm. Sin. 41, 670–677 (2020).
Hung, T. W. et al. Pentraxin 3 activates JNK Signaling And Regulates The Epithelial-to-mesenchymal Transition In Renal Fibrosis. Cell. Physiol. Biochem. 40, 1029–1038 (2016).
Rhyu, D. Y. et al. Role of reactive oxygen species in TGF-beta1-induced mitogen-activated protein kinase activation and epithelial-mesenchymal transition in renal tubular epithelial cells. J. Am. Soc. Nephrol. 16, 667–675 (2005).
Border, W. A., Okuda, S., Languino, L. R., Sporn, M. B. & Ruoslahti, E. Suppression of experimental glomerulonephritis by antiserum against transforming growth factor beta 1. Nature 346, 371–374 (1990).
Sung, W. J. et al. Antifibrotic effect of synthetic Smad/Sp1 chimeric decoy oligodeoxynucleotide through the regulation of epithelial mesenchymal transition in unilateral ureteral obstruction model of mice. Exp. Mol. Pathol. 95, 136–143 (2013).
Choi, J. H. et al. Capsaicin Inhibits Dimethylnitrosamine-induced Hepatic Fibrosis By Inhibiting The TGF-beta1/Smad pathway via peroxisome proliferator-activated receptor gamma activation. J. Agric. Food Chem. 65, 317–326 (2017).
Inazaki, K. et al. Smad3 deficiency attenuates renal fibrosis, inflammation,and apoptosis after unilateral ureteral obstruction. Kidney Int. 66, 597–604 (2004).
Kim, J. H., Kim, B. K., Moon, K. C., Hong, H. K. & Lee, H. S. Activation of the TGF-beta/Smad signaling pathway in focal segmental glomerulosclerosis. Kidney Int. 64, 1715–1721 (2003).
Huang, H. S., Ma, M. C., Chen, C. F. & Chen, J. Lipid peroxidation and its correlations with urinary levels of oxalate, citric acid, and osteopontin in patients with renal calcium oxalate stones. Urology 62, 1123–1128 (2003).
Jobling, M. E. et al. Isoform-specific activation of latent transforming growth factor β (LTGF-β) by reactive oxygen species. Radiat. Res. 166, 839–848 (2006).
Hayashi, H., Sakai, K., Baba, H. & Sakai, T. Thrombospondin-1 is a novel negative regulator of liver regeneration after partial hepatectomy through transforming growth factor-beta1 activation in mice. Hepatology 55, 1562–1573 (2012).
Wang, H. & Kochevar, I. E. Involvement of UVB-induced reactive oxygen species in TGF-beta biosynthesis and activation in keratinocytes. Free Radic. Biol. Med. 38, 890–897 (2005).
Kong, D. et al. Curcumin blunts epithelial-mesenchymal transition of hepatocytes to alleviate hepatic fibrosis through regulating oxidative stress and autophagy. Redox Biol. 36, 101600 (2020).
Yang, Y. et al. Structural characterization and antioxidant activities of one neutral polysaccharide and three acid polysaccharides from Ziziphus jujuba cv. Hamidazao: a comparison. Carbohydr. Polym. 261, 117879 (2021).
Wang, Y. et al. Abelmoschus manihot polysaccharide fortifies intestinal mucus barrier to alleviate intestinal inflammation by modulating Akkermansia muciniphila abundance. Acta Pharm. Sin. B 14, 3901–3915 (2024).
Wang, Y. et al. Polysaccharide nanodonuts for photochemotherapy-amplified immunogenic cell death to potentiate systemic antitumor immunity against hepatocellular carcinoma. Adv. Funct. Mater. 33, 2208486 (2023).
Chen, S. F., Zhou, Y. Q., Chen, Y. R. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, 884–890 (2018).
Smith, T., Heger, A. & Sudbery, I. UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res. 27, 491–499 (2017).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019).
Acknowledgements
This work was granted by the National Natural Science Foundation of China (No. 82370766), the Guangdong Basic and Applied Basic Research Foundation (No. 2020A1515011461), the Science and Technology Plan Project of Guangzhou (No. 202201020511), the Guangzhou Clinical High-tech Major Technology Program (No. 2024C-GX21), the start-up fund of Nanshan Scholarship of Guangzhou Medical University, and the Nanshan Emerging Talent Program of the First Affiliated Hospital of Guangzhou Medical University.
Author information
Authors and Affiliations
Contributions
Bang-Xian Yu: investigation, writing—original draft. Jun Long: investigation. Ye-Ping Liang: investigation and data curation. Quan Zhang: formal analysis. Yang Liu: data curation, formal analysis. Guo-Hua Zeng: funding acquisition. Yong-Da Liu: project administration and funding acquisition. Xin-Yuan Sun: conceptualization, methodology, funding acquisition, resources, supervision, writing-review and editing. All authors read and approved the final manuscript. All data were generated in-house, and no paper mill was used. All authors agree to be accountable for all aspects of the work, ensuring integrity and accuracy.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editor: Ophelia Bu. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yu, BX., Long, J., Liang, YP. et al. Therapeutic potential of Desmodium styracifolium polysaccharide in attenuating nano-calcium oxalate induced renal injury and fibrosis. Commun Biol 8, 1330 (2025). https://doi.org/10.1038/s42003-025-08757-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-08757-7
