
Abstract
In stage 2–3 colon cancer (CC), postsurgery circulating tumor DNA (ctDNA) assessment is crucial for guiding adjuvant chemotherapy (ACT) decisions. While existing assays detect ctDNA and help identify high-risk persons with CC for recurrence, their limited sensitivity after surgery poses challenges in deciding on ACT. Additionally, a substantial portion of persons with CC fail to clear ctDNA after ACT, leading to recurrence. In this study, we performed whole-exome sequencing (WES) of ctDNA at different time points in participants with relapsed CC in two independent cohorts, alongside transcriptomic and proteomic analyses of metastases, to enhance comprehension of progression mechanisms. A plasma WES-based tumor-agnostic assay demonstrated higher sensitivity in detecting minimal residual disease (MRD) compared to current assays. Immune evasion appears to be the primary driver of progression in the localized CC setting, indicating the potential efficacy of immunotherapy for microsatellite stability in persons with CC. Organoid modeling further supports the promising potential of targeted therapy in eradicating MRD, surpassing conventional treatments.
Similar content being viewed by others

Main
Liquid biopsy has emerged as a highly valuable tool in guiding adjuvant chemotherapy (ACT) decisions for persons with stage 2–3 colon cancer (CC), where treatment decisions still rely on pathological staging despite a notable risk of mistreatment1. Retrospective studies have unequivocally demonstrated that the detection of circulating tumor DNA (ctDNA) after curative-intent surgery not only identifies persons at high risk of recurrence but also correlates with poorer disease-free survival (DFS)2,3,4,5,6,7,8,9. Moreover, ctDNA detected after ACT completion is also associated with high recurrence risk and worse DFS, which may indicate that a substantial proportion of these post-ACT ctDNA-positive persons did not benefit from standard ACT. Indeed, retrospective studies showed that up to 77% of ctDNA-positive persons who received ACT failed to achieve ctDNA negativity and all were subsequently diagnosed with recurrence7. More recently, prospective clinical trials such as DYNAMIC and PEGASUS, along with an observational study named GALAXY, corroborated and expanded upon these findings10,11,12,13,14. However, although first-generation commercial and academic assays for detecting ctDNA show remarkable specificity in identifying minimal residual disease (MRD) in persons who subsequently experience recurrence, their sensitivity is limited, particularly immediately after surgery when decisions for ACT are required, in contrast to the metastatic scenario15,16. The ideal assay would detect ctDNA in >90% of recurrence cases. Currently, the recurrence rates range from 10% to 30% in persons with undetectable ctDNA and from 30% to 70% in those with ctDNA positivity, which is promising but not strong enough for its implementation in clinical practice17.
MRD is defined by the presence of molecular hints of a tumor after its apparent surgical removal. Additionally, ctDNA detection not only evidences MRD but also precedes clinical relapse by several months2,3,4,5,6,7,8,9. If cure requires the eradication of all tumor cells capable of driving relapse, there are likely fewer of these cells at the time of MRD than when relapse is clinically evident by imaging. This represents an opportunity for targeting the molecular alterations found at this moment with a more specific approach18. The advent of emerging active drugs with diverse mechanisms of action opens up the possibility of disease relapse prevention through more precise and rational treatment of MRD. Numerous clinical trials are currently underway investigating various therapeutic interventions in ctDNA-positive persons with CC after surgery19. This advancement underscores the potential for liquid biopsy to transform the landscape of treatment decision-making strategies in localized CC.
A comprehensive understanding of the molecular mechanisms driving CC progression holds great potential for tailoring treatment strategies to more effectively eradicate MRD20. Furthermore, a substantial percentage of persons without ctDNA detection also experience recurrence, underscoring the need for advanced perspectives on ctDNA detection approaches aimed at increasing their sensitivity. In this study, two independent cohorts of participants diagnosed with relapsed CC were evaluated by whole-exome sequencing (WES) on longitudinal plasma samples and transcriptomics and proteomics analyses were also conducted on tissue specimens acquired during relapse onset. Ultimately, we sought to reveal mechanisms of tumor progression, identify pioneering therapeutic targets21 and guide the design of an innovative assay for sensitive detection of MRD through ctDNA analysis.
Results
Participant characteristics
We conducted a prospective study, enrolling 320 participants with stage 2 and 3 CC between 2015 and 2019 at Hospital Clínico Universitario in Valencia, Spain. Nested within these participants, considered as the discovery cohort, all individuals with recurrence (n = 25) who had plasma samples at relapse and tissue samples at baseline (primary tumor) available were selected for WES ctDNA analysis. Participants were predominantly male (15/25, 60%) and had a median age of 74 years (Supplementary Table 1). The median recurrence time was 13 months. Relapse sites were diverse, including one (21/25, 84%) or multiple (4/25, 16%) metastatic sites. Most participants (18/25, 72%) received ACT with either capecitabine (12/18, 67%) or capecitabine + oxaliplatin (CAPOX; 6/18, 33%).
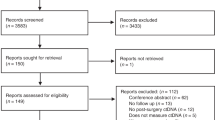
In the validation cohort, participants with relapsed CC were recruited between 2015 and 2022 at seven Danish hospitals; compared to the discovery cohort, the median age of the participants was 64 years and 40% (6/15) were male. The median recurrence time was 12 months. Among the participants, 67% (10/15) exhibited a solitary relapse site, while 33% (5/15) presented with multiple sites. Most participants (14/15, 93%) received ACT, with treatment regimens including CAPOX (7/14, 50%), fluorouracil + oxaliplatin (3/14, 21%), folinic acid + fluorouracil + oxaliplatin (1/14, 7%), capecitabine (2/4, 14%) or intravenous fluorouracil (1/14, 7%). A CONSORT (Consolidated Standards of Reporting Trials) diagram is provided in Fig. 1.

CONSORT diagram illustrating the enrolled cohort, detailing the different participant subgroups and sample collections at various time points, including baseline, postoperative and relapse stages. The discovery cohort is divided into relapse and nonrelapse participants, with associated tumor tissue and plasma samples analyzed for molecular characterization. PDOs were included as models on the basis of their molecular similarity to the discovery cohort. The validation cohort follows a similar structure, serving to confirm findings from the discovery cohort. The diagram also highlights the clinical inquiries addressed in the study.
Plasma ctDNA analysis reveals intratumor heterogeneity (ITH)
To demonstrate the capability of plasma ctDNA to provide a comprehensive representation of key genomic alterations in localized CC, WES was performed on paired plasma and tumor samples from individuals at baseline and relapse (Fig. 2). The analysis focused on identifying somatically acquired single-nucleotide variants (SNVs), small insertions and deletions (indels) and copy-number variants (CNVs).

Schematic representation illustrating the study workflow, depicting the analysis of ctDNA from plasma samples collected at different time points: baseline (pretreatment), postoperative and relapse stages. The figure highlights the transition of molecular alterations over time, represented by changes in the composition and abundance of mutations. A density plot visualizes the distribution and evolution of these alterations across different stages. PDOs were not sourced from participants within the discovery cohort; rather, they were selected as models on the basis of their molecular similarity to the participants in this cohort. Created with BioRender.com.
All participants in both cohorts exhibited at least one somatic mutation detected through plasma samples, both at baseline and during relapse. We conducted intraparticipant assessment, comparing point somatic mutations and CNVs present in tissue and plasma samples obtained simultaneously (Fig. 3a). In the discovery cohort, we observed a concordance of 26.6% for all somatic mutations detected in plasma versus tissue samples at baseline (23.2% in the validation cohort; Extended Data Fig. 1a), which dropped to 18.1% when comparing plasma and tissue samples collected at the point of relapse (Fig. 3b).

a, Comparative molecular landscape of pathogenic mutations and CNVs in paired tissue and plasma baseline samples from 12 participants with CC. Each box represents a mutated gene in a specific participant, divided into two parts. Left, results from the primary tumor. Right, results from plasma at baseline. Additionally, each box at a given collection moment is further divided into two parts. Left, point mutations. Right, CNVs. The y axis is organized by the number of point mutations for each gene across all participants. b, Percentage of concordance in somatic SNVs between primary tumor and plasma at baseline (n = 12 participants) and at relapse (n = 17 participants) in the participant cohort. Data are presented as the median values ± s.d. Concordance was determined by comparing each participant to themself at different stages. c, Percentage of concordance in CNVs between primary tumor and plasma at baseline (n = 12) and at relapse (n = 17 participants) across the participant cohort. Data are presented as the median values ± s.d. Similar to SNVs, concordance was calculated by comparing each participant to themself at different stages. d, Functional enrichment analysis based on REACTOME (n = 12 participants). Left, enriched signatures in genes with CNV loss in plasma compared to the primary tumor at baseline. Right, enriched signatures in genes with CNV gains in plasma relative to tissue. The gray bar corresponds to the −log(FDR) and the green line represents the number of genes overlapping in each REACTOME term.
The molecular profile (Fig. 3a) revealed tumor-specific mutations absent in paired plasma samples, while some point mutations were exclusive to plasma. In the discovery cohort, 33.9% of somatic mutations appeared only in plasma at baseline (49% in the validation cohort; Extended Data Fig. 1b), a pattern persisting at relapse (22.3% exclusive to ctDNA; Extended Data Fig. 2). Concordant mutations exhibited significantly higher variant allele frequency (VAF) than plasma-exclusive variants (n = 12, P = 2.2 × 10−16 in both cohorts according to a t-test).
CNV analysis showed 95.3% concordance between tumor and plasma at baseline (n = 12) and 90% at relapse (n = 17) (Fig. 3c), with no significant CNV concordance differences (n = 8, P = 0.1484 according to a Wilcoxon test). Baseline discordance involved 311 genes with loss and 380 with gain. Plasma copy-number losses were enriched in immune signaling pathways, while gains were linked to proliferative pathways (Fig. 3d).
These results not only highlight the advantage of plasma over tissue in analyzing ITH but also indicate a selective clonal process throughout the course of the disease, emphasizing the importance of plasma-based monitoring and revealing unique genetic signatures that could guide targeted therapeutic interventions for MRD eradication.
Immediate postoperative ctDNA status association with MRD
To assess whether performing WES on cell-free DNA (cfDNA) from plasma enhances MRD sensitivity detection compared to personalized assays based on a tumor-informed approach or custom panels, we conducted a WES tumor-agnostic (WES-TA) approach on plasma samples collected immediately after curative-intent surgery.
Following plasma sequencing at the postoperative time point, at least one somatic mutation was detected in 86.7% (13/15) and 100% (14/14) of participants in the respective cohorts. Additionally, WES data of postoperative plasma from 21 participants with CC who had not experienced relapse (from the discovery cohort) revealed only one participant classified as ctDNA positive, yielding a specificity of 95% for this technique.
To assess the clinical applicability of MRD detection using WES analysis within a tumor-informed framework, we focused on the 16 somatic mutations with the highest VAF present in the primary tissue exome of each participant similar to a bespoke commercial assay3,7. Our analysis to determine whether these mutations were discernible in postoperative plasma samples revealed that at least two of the candidate mutations were detected in plasma in 67% of participants (10/15) in the discovery cohort and 57% of participants (8/14) in the validation cohort (Supplementary Table 2). Conversely, when the selection of the 16 mutations with the highest VAF was based on the analysis of baseline plasma rather than primary tumor specimens (TAV16), a sensitivity of 67% (6/9) in the discovery cohort and 86% (12/14) in the validation cohort was obtained. However, if we considered that a participant with positive ctDNA was defined by the detection of only one mutation in plasma rather than two, the sensitivity increased to 89% (8/9) in the discovery cohort and 100% (14/14) in the validation cohort. These data suggest that, taking into account that the existing academic and commercial assays require positivity for only a variant in plasma4,10,11, our WES-TA approach increases sensitivity compared to other current assays while maintaining specificity by detecting a variant.
Additionally, the 16 candidate mutations selected from the WES of primary tumors differed from those identified through the plasma baseline approach. Comparing both sets, most participants (6/9, 67%) in the discovery cohort had no concordance, resulting in a median 0% concordance rate, while the validation cohort showed 6% concordance (Extended Data Fig. 3).
Notably, with the tumor-informed approach, 96% and 98% of selected variants were identified as unique to individual participants in the discovery and validation cohorts, respectively. Considering the plasma baseline, in contrast, 86% of the mutations were unique in the discovery cohort and 78% were unique in the validation cohort, supporting a key role for personalized assays in MRD detection.
These findings highlight the potential of using the WES-TA approach for monitoring MRD in localized CC, particularly concerning the identification of ctDNA positivity with the detection of only one variant in the plasma. Compared to current commercial and academic assays, this method offers greater sensitivity and equal specificity, suggesting possible uses for refining monitoring strategies and promoting precision medicine in this clinical setting.
Correlation of clonal evolution with tumor progression
To investigate temporal heterogeneity, we conducted WES on cfDNA at both baseline and relapse time points (Fig. 1). Mutations and CNV gains and losses are shown in Fig. 4a for the discovery cohort and Extended Data Fig. 4 for the validation cohort. Concordance of somatic variants between plasma samples at baseline and relapse was 61.7% in the discovery cohort and 50.5% in the validation cohort, higher than the concordance observed between primary tumor and ctDNA at relapse (Extended Data Fig. 5a,b; 27.7%, n = 12, P = 0.0015 according to a Wilcoxon test), reflecting both ITH and plasma’s better ability to capture clonal evolution. Furthermore, concordance between primary tumor and paired metastatic tissue was higher in participants with a single metastatic lesion compared to those with multiple lesions (multiple, n = 10, 29.75%; single, n = 7, 80.87%; P = 0.0068 according to a Wilcoxon test). A similar observation was made when comparing baseline plasma to metastatic tissue (multiple, n = 5, 25.55%; single, n = 3, 80.87%; P = 0.0357 according to a Wilcoxon test) and when comparing recurrence plasma to metastatic tissue (multiple, n = 10, 11.96%; single, n = 7, 32.66%; P = 0.0054 according to a Wilcoxon test). On the other hand, the concordance between baseline and relapse plasma was not significantly influenced by the presence of single or multiple metastases (multiple, n = 6, 57.56%; single, n = 6, 62.43%; P = 0.6991 according to a Wilcoxon test), further highlighting the tumor’s limitation in comprehensively capturing ITH.

a, Comparative molecular profiling of pathogenic mutations and CNVs in paired plasma baseline and relapse samples from 12 participants with CC. Each box signifies a mutated gene in an individual participant, with division into two parts separated by a line. Left, results obtained at baseline. Right, results from plasma at relapse. Similarly, each box corresponding to a collection moment is subdivided into two components. Left, point mutations. Right, CNVs. The y axis is organized on the basis of the number of point mutations for each gene across all participants. b, Evolutionary plot in the discovery cohort for seven paired participants (top) and the validation cohort for 14 paired participants (bottom), illustrating somatic mutations occurring at baseline, after surgery and at relapse. Colors indicate the presence of mutations over time, with gray representing mutations appearing at baseline but representing unselected subclones lost after surgery. The indications of the sampling time points are not drawn to time scale. Moving along the chromatic scale from green to purple signifies mutations persisting over time and considered clonal. Mutations emerging after surgery until relapse are represented in shades of red, indicating clones arising during tumor evolution in this period. Right, upset plot indicating the correspondence of colors with temporal points where the mutation was found. c, Spearman correlation (n = 7 participants; two-sided) between the B cell infiltration and mutational concordance between baseline and relapse plasma in the discovery cohort. Left, correlation between infiltrated B lymphocytes using RNA-seq deconvolution through the CIBERSORT pipeline versus the mutational concordance. Right, correlation of the intensity of CD20 positivity by IHC in the primary tissues versus mutational concordance. Representative images of some of the participants from CD20 IHC on the primary tissues are indicated. Colors are included for each of the different participants (points) to allow comparison to the validation data using IHC with CD20. The line represents the fitted relationship between the variables, while the shaded band corresponds to the 95% confidence interval around the regression estimate. d, Functional enrichment analysis by hallmark gene sets revealed enriched pathways in mutated genes in the discovery cohort (top; n = 7 participants) and the validation cohort (bottom; n = 14 participants). A one-sided hypergeometric test was used to assess whether the input gene set was significantly overrepresented in hallmark gene sets compared to a background set of genes. The P values were adjusted for multiple comparisons using the FDR correction, with a significance threshold of 0.05.
To gain a comprehensive understanding of tumor evolutionary dynamics in participants with CC, considering all detected mutations at each time point allowed us to discern alterations that diminished during the evolutionary process, those that endured over time and those that surfaced at the point of relapse, as illustrated in Fig. 4b. We detected acquired variants in ctDNA at the time of relapse, constituting 23% and 26.5% of somatic mutations in the discovery and validation cohorts, respectively.
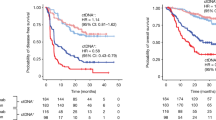
Notable individual heterogeneity was observed in the tempo of tumor evolution (Extended Data Fig. 6a,b). Some participants showed gradual changes, with mutations appearing at relapse absent in baseline plasma samples, regardless of chemotherapy. Transcriptomic deconvolution in primary tissues from the discovery cohort revealed that molecular similarity between baseline and relapse plasma correlated with activated B cells (Fig. 4c; n = 7, P = 0.0366 according to a Spearman correlation), confirmed by immunohistochemistry (IHC) in both cohorts (discovery, n = 7, P = 0.0215; validation, n = 14, P = 0.0460). Elevated infiltration at diagnosis was linked to reduced baseline–relapse concordance, indicating more rapid tumor evolution, while lower infiltration and higher similarity suggested slower evolution.
Our analysis showed that somatic mutations acquired during relapse in both the discovery and the validation cohorts were significantly associated with activation of the epithelial–mesenchymal transition (EMT) pathway (Fig. 4d). This was confirmed in the genomic profiles of relapsing tissue samples, where the EMT pathway was overrepresented in mutations acquired during relapse in metastatic lesions. Transcriptomic profiling revealed that participants with wild-type EMT genes did not undergo the transition, whereas participants with mutated genes shifted from an epithelial to a mesenchymal profile because of EMT pathway activation (Extended Data Fig. 6c). These participants carried high-impact mutations in FLNA, ITGB3, LAMC1, SLIT3 and TGFBR3 (Supplementary Table 3), known to activate the EMT pathway22,23,24,25,26.
Interestingly, we observed a significant enrichment in loss of heterozygosity in genes related to myogenesis (P = 6.759 × 10−5; MYH1, MYH2, MYH3, MYH4, MYH8 and CHRNB1) at baseline but not identified at relapse. On the other hand, our analysis of mutational signatures27 did not reveal any significant distinctions between these time points, as illustrated in Extended Data Fig. 7a,b.
Our findings suggest that tumoral evolution is more accurately captured through ctDNA analysis and also identify two evolution patterns associated with initial B cell infiltration into the primary tumor, which may contribute to immune evasion by tumor cells and subsequent cellular migration to other organs.
ctDNA parallel evolution analysis reveals tumor progression
To investigate the mechanisms underlying localized CC progression, we conducted an analysis of acquired somatic mutations at the time of relapse and their associated functions. Specifically, the number of mutations per gene present in plasma samples was examined at both baseline and relapse to investigate the parallel evolution of the tumor28.
In both the discovery and the validation cohorts, no significant differences in tumor mutational burden (TMB) at relapse compared to baseline were observed in either tissue or plasma (Extended Data Fig. 8a–c). A recent study suggested that TMB alone may not accurately predict responses to checkpoint inhibition29. In this context, the ratio of nonsynonymous to synonymous substitutions (dN/dS ratio) is a valuable metric for assessing the strength and mode of natural selection on protein-coding genes30.
We next examined the potential correlation between dN/dS and TMB, both at the time of diagnosis and at relapse. Our analysis revealed that, at diagnosis, no significant correlation could be established between these parameters in either tissue (Extended Data Fig. 8d) or plasma (Fig. 5a). In the discovery cohort, however, a noteworthy correlation between these variables emerged at the point of relapse, in both plasma and tissue samples (plasma, n = 12, P = 0.0228; tissue, n = 25, P = 0.0199, according to a Spearman correlation; Fig 5a and Extended Data Fig. 8d). This significant correlation at the time of recurrence was also observed in the validation cohort when comparing those parameters in plasma (n = 15, P = 0.0321, according to a Spearman correlation; Fig. 5a).

a, Spearman correlation (two-sided) between TMB and dN/dS in plasma at both baseline and relapse in the discovery cohort (left; n = 12) and the validation cohort (right; n = 15 participants). The P and ρ values are provided for each case. The line represents the fitted relationship between the variables, while the shaded band corresponds to the 95% confidence interval around the regression estimate. b, Volcano plot in the discovery cohort (left; n = 12 participants) and the validation cohort (right; n = 15 participants) illustrating genes significantly associated with a higher number of somatic mutations at relapse and baseline. The P-value threshold was set at 0.05 and the log2(fold change) range was between −0.6 and 0.6 (two-sided Wilcoxon test). The P values were adjusted for multiple comparisons using the FDR correction, with a significance threshold of 0.05. c, Functional enrichment analysis of all significant genes exhibiting a higher number of somatic mutations at relapse compared to the baseline stage in the discovery cohort (left; n = 12 participants) and the validation cohort (right; n = 15 participants). A one-sided hypergeometric test was used to assess whether the input gene set was significantly overrepresented in KEGG pathways compared to a background set of genes. The P values were adjusted for multiple comparisons using the FDR correction, with a significance threshold of 0.05. d, Comparative quantification of neoepitope abundance between paired metastatic and primary tumor samples (n = 13 participants). An asterisk denotes a statistically significant difference (P < 0.05) in neoepitope abundance between primary and metastatic tissues, as determined by a one-sided t-test. The analysis was based on the hypothesis that metastatic tissues exhibit a lower neoepitope abundance than primary tumors. The P value for the overall comparison between primary and metastatic tumors was <0.001. Individual P values for each participant were as follows: participant 13, 0.0132; participant 49, 0.0017; participant 63, 0.0068; participant 104, 0.0029; participant 107, 0.0219; participant 136, 0.9671; participant 185, 0.0001; participant 189, 0.8378; participant 204, 0.0001; participant 242, 0.0001; participant 243, 0.9945; participant 259, 0.0211; participant 261, 0.9997. e, Median protein quantification ratio of wild-type versus mutated metastasis samples identified by MS (n = 14 participants). The asterisk indicates a significant difference in protein ratio between primary and metastatic tissues based on the presence of the mutation at relapse according to a two-sided t-test analysis. Individual P values were as follows: PDIA3, 0.0014; HLA-A, 0.2091; HLA-B, 0.4807; HLA-C, 0.9548; HLA-DPB1, 0.6853; HLA-DQB1, 0.7936; HLA-DRB1, 0.0077; HLA-E, 0.2616; HSP90AA1, 0.7104; TAP1, 0.9010; CALR, 0.1663).
The correlation between TMB and dN/dS ratio during relapse suggests that tumors accumulate numerous pathogenic alterations, driven by positive evolutionary selection, leading to functions crucial for tumor progression. Our study focused on genes with significantly increased mutations at relapse compared to diagnosis, excluding participants with microsatellite instability (MSI) because of high TMB. In the discovery cohort, 115 genes met this criterion, with GOLGA6, HLA and PABP gene families notable in both cohorts (Fig. 5b). Kyoto Encyclopedia of Genes and Genomes (KEGG) functional enrichment analysis highlighted the involvement of hypermutated genes in immune evasion pathways, including antigen presentation and processing (Fig. 5c), which was consistently observed in the validation cohort.
To gain mechanistic insights, we performed a comparative analysis of neoepitope abundance between primary and metastatic tissues by integrating genomic and mass spectrometry (MS) data. Metastatic tissue samples exhibited significantly lower neoepitope abundance compared to their paired primary tissue samples (n = 13, P = 1.966 × 10−5 according to a Wilcoxon test; Fig. 5d). In addition, proteomic analysis in the discovery cohort revealed distinct proteomic levels for genes associated with the antigen presentation and processing pathway in metastatic tissues compared to their corresponding primary tissues. Specifically, PDIA3 was upregulated in metastatic tissues with mutations, while no such increase was observed in mutation-free metastases (n = 14, P = 0.0014 according to a t-test). Conversely, HLA-DRB1 showed reduced expression in metastases with mutations compared to primary tissues, with no reduction in wild-type metastases (n = 14, P = 0.0076, according to a t-test) (Fig. 5e).
This further supports our findings that enrichment of functional mutations in relapse samples preferentially targets specific genes, thereby promoting immune evasion and ultimately enhancing tumor fitness during relapse.
ctDNA profiling to guide targeted therapies
To evaluate the utility of ctDNA genotyping in selecting candidates for targeted therapy, we proceeded to identify potentially targetable mutations in participants from both cohorts.
Detection of inherited germline or acquired somatic variants could improve patient care. Plasma sequencing identified potentially pathogenic actionable mutations that were matched with OncoKB levels of evidence for targeted therapies (Table 1). In the discovery cohort, 75% (9/12) and 80% (20/25) of participants had clinically actionable mutations at baseline and relapse, respectively, primarily in the RAS, PI3K–AKT–mTOR and DNA damage repair pathways. Similar results were seen in the validation cohort, where 60% (9/14) and 73% (11/14) of participants had these mutations. Of the druggable mutations in the validation cohort (Supplementary Table 4), 62.5% were shared with the discovery cohort.
WES analysis of white blood cells (WBCs) facilitated the identification of pathogenic germline variants in 28% (7/25) of participants within the discovery cohort. These variants were found in genes associated with DNA damage repair (for example, CHEK2 and RAD54L), control of cell growth and division (for example, ATM and ERBB2) and tumor suppression and homologous recombination deficiency (for example, PALB2, BRCA1 and BRCA2), as detailed in Supplementary Table 5.
According to the MRD molecular alterations, 60% (9/15) of participants in the discovery cohort and 71% (10/14) in the validation cohort exhibited potentially actionable mutations after surgery. We selected some targeted drugs to evaluate their potential activity in the discovery cohort participants. We characterized 18 patient-derived organoids (PDOs) from participants with CC by WES, finally selecting the ones most molecularly matched with our participants’ characteristics. Three PDOs (CTO65, CTO119 and CTO147) were identified through hierarchical clustering and subsequently used for drug testing (Extended Data Fig. 9a).
In addition, a metastatic PDO corresponding to the 119 model (mCTO119) was evaluated, confirming that the selected mutations are driver alterations responsible for the metastasis development (observed VAF: TP53, 98.88%; FGFR2, 70%; KRASG12C, 55.83%).
CTO65 exhibited mutations in ARID1A, CHEK1/2, KRAS, PIK3CA and TP53; CTO119 carried mutations in FGFR2, KRASG12C and TP53; CTO147 featured mutations in PIK3CA and TP53 (Extended Data Fig. 9b). Various therapeutic agents targeting these alterations were tested, along with standard ACT agents for CC, fluorouracil and oxaliplatin, both individually and in combination. The PDO models showed sensitivity to molecularly matched therapies. CTO65 and CTO119 were more sensitive to the Wee1 inhibitor adavosertib because of TP53 and KRAS mutations compared to CTO147. CTO65 and CTO147 were more sensitive to the PI3K inhibitor alpelisib than CTO119. CTO119 exhibited a stronger response to the FGFR and KRAS-G12C inhibitors erdafitinib and adagrasib, respectively. All three PDOs showed growth inhibition with the MEK inhibitor trametinib, with CTO147 showing notable sensitivity despite lacking mutated KRAS (Extended Data Fig. 9c,d).
These data reinforce the necessity of conducting molecular studies for MRD to effectively eradicate disease and optimize patient treatment, thereby paving the way for further research in this field.
Discussion
The conventional tumor–node–metastasis staging system used for localized CC lacks precision1 and potentially results in patient overtreatment or undertreatment. While some persons may achieve a cure through surgery alone, other persons face a higher risk of relapse. Recent advancements in ctDNA analysis have contributed to identifying high-risk persons prone to relapse, yet many continue to have detectable ctDNA after surgery despite receiving standard ACT2,3,4,5,6,7,8,9,10,11,12,13,14. This highlights the inadequacy of current treatments for eliminating MRD and preventing recurrence7,8,9,10,11. To improve patient management, there is a need to enhance the sensitivity of ctDNA detection assays and deepen our understanding of colorectal cancer (CRC) progression mechanisms. Although ongoing clinical trials are exploring intensified cytostatic regimens, targeted therapies may hold promise for persons with persistent positive ctDNA following radical surgery19.
In this work, we performed ctDNA analysis of 40 participants with localized CC who underwent curative-intent surgery but subsequently experienced recurrence. The study, conducted across two institutions in Spain and Denmark, applied WES at different time points. Our objective was to surpass the constraints of custom panels by investigating whether a plasma-based WES approach could improve the sensitivity to detect MRD31. Additionally, we sought to uncover mechanisms underlying the progression of localized CC that could reveal potential therapeutic approaches for effectively eliminating MRD.
Our comprehensive analysis sheds light on the dynamic nature of ctDNA during CC recurrence, not only demonstrating the potential of plasma over tissue in analyzing ITH32 (Fig. 3) but also emphasizing the importance of plasma-based monitoring and revealing unique genetic signatures in the context of MRD.
In this study, we demonstrated that WES of ctDNA immediately after surgery (WES-TA approach) notably improves the sensitivity for detecting MRD by considering just one variant as indicative of ctDNA positivity. Sensitivity reached 86.7% and 100% in the discovery and validation cohorts, respectively, with a specificity of 95%, surpassing previous studies using personalized assays based on a tumor-informed or TA approach with custom panels2,3,4,5,6,7,8,9,10,11,12,13,14,33. To improve cost-effectiveness, we selected the top 16 variants with the highest VAF in the plasma baseline (TAV16) for postoperative monitoring. The TAV16 approach yielded sensitivity values similar to those obtained from the WES-TA approach when considering one mutation for ctDNA positivity (Supplementary Table 2). This finding, combined with the observation that the concordance between plasmas at different time points is stronger than that observed between primary tissue and plasma (Extended Data Fig. 5b), suggests that leveraging a personalized TA assay based on plasma WES at diagnosis rather than relying on primary tumor could be pivotal in developing a robust approach for monitoring MRD.
Postsurgery ctDNA genotyping detected 60% and 71% of participants with at least one potentially actionable variant in the discovery and validation cohorts, respectively, such as the ERBB2, PI3KCA and BRCA genes (Table 1). Crucially, none of them would have been detected through personalized assays targeting the 16 highest VAF variants at MRD diagnosis. This lack of detection reduces the possibility of effectively eliminating MRD and excludes these persons from the opportunity to receive experimental treatments within clinical trials. Moreover, enrolling persons in clinical trials remains a notable challenge, largely because of prolonged screening periods, the need for sequential tissue biopsies and time-consuming genotyping processes. ctDNA analysis presents a promising avenue to tackle these hurdles, offering superior accuracy in detecting genomic alterations compared to conventional tumor tissue analysis34,35,36.
The importance of a molecularly matched approach for MRD was also shown with our PDO models, which present molecular alterations similar to those found in MRD cases. This demonstrates that targeted treatments exhibit higher sensitivity than conventional ACT (Extended Data Fig. 9a), emphasizing the necessity of conducting molecular studies for MRD to inform optimal treatment strategies, thereby prompting further research.
Furthermore, WBC sequencing enables the identification of germline mutations, revealing a prevalence of 28% in the discovery cohort within genes associated with DNA damage repair, cell growth and division, tumor suppression and homologous recombination deficiency (Supplementary Table 5). These findings carry ethical implications for participants and their families and the importance of genetic counseling should be underlined37.
Notably, acquired mutations identified during relapse were associated with disruptions in the EMT pathway, pinpointing it as one of the mechanisms driving localized CC tumor progression and suggesting that alternative therapeutic avenues (Fig. 4d) such as treatment with WNT inhibitors could potentially inhibit tumor growth and metastasis38,39,40. The loss of heterozygosity of genes related to the myogenesis at baseline could disrupt cellular architecture, weakening cell–cell and cell–matrix interactions and, thus, facilitating cell migration and invasion, which are key features of EMT. However, the precise mechanisms underlying this relationship are not yet fully elucidated and further studies focusing on early events in the formation of myogenic precursors are needed41.
In persons with microsatellite stable (MSS) CC, those exhibiting increased clonal evolution, irrespective of whether they had received ACT, were characterized by higher activated B cell infiltration in the primary tumor (Fig. 4c). The selective pressure exerted by immune cells on primary tissue in MSS participants in our cohort paves the way for potential benefits from immunotherapy in eliminating MRD42. The concept of cell infiltration could also be contemplated in the neoadjuvant setting for MSS participants43. This phenomenon can be attributed to tumor evolution and the evolving characteristics of its microenvironment over time.
Although tumor progression was not linked to an increase in TMB29, changes in the dN/dS ratio (Fig. 5a) suggest that functional mutations rather than overall mutations are more important at relapse, highlighting the need for emerging biomarkers to identify persons who could benefit from immunotherapy. Positive evolutionary selection drives this phenomenon, leading to functions that contribute to tumor progression (Fig. 5b). These findings support the hypothesis that alterations in the antigen presentation and processing pathway have a key role in driving CC progression after surgery, leading to relapse (Fig. 5c). MS data further validate this, showing lower neoepitope abundance in metastatic tissues compared to primary tumors (Fig. 5d). This mechanism emphasizes the role of antigen presentation in tumor progression and identifies it as a potential therapeutic target, especially in localized CC42,43,44,45,46,47.
PDIA3 and HLA-DRB1 proteins were found to be particularly involved. The PDIA3 gene displayed a significant rise in activating mutations during relapse, resulting in elevated protein expression and implicating this gene in the suppression of antitumor immunity48. Conversely, increased suppressor mutations in the HLA-DRB1 gene had a pronounced effect on promoting a cold tumor environment, thereby suppressing immune system activation49. This further substantiates that the enrichment of functional mutations in relapse samples selectively favors specific genes, enhancing immune evasion and subsequently increasing tumor fitness during relapse (Fig. 5e).
WES provides a broader scope for detecting pathogenic mutations compared to tumor-informed approaches; it faces challenges in coverage and tumor fraction50, impacting false negatives and concordance with tissue samples31. Sensitivity for subclonal mutations improved in a validation cohort with higher coverage, highlighting the need for economic investment in identifying subclonal mutations through WES in localized tumors. Moreover, further validation with larger plasma-paired cohorts is essential to confirm our findings. Expanding the participant pool could improve tumor evolution characterization and allow for a more comprehensive assessment of the impact of ACT on individual participants’ mutational profiles51.
In conclusion, our study demonstrates that a WES-TA approach surpasses current commercial assays for detecting MRD. We identify immune evasion mechanisms as a primary driver of progression in the setting of localized CC, facilitated by a functional mutational burden at relapse. This suggests that immunotherapy could extend its efficacy to persons with MSS CC, thereby broadening treatment options and potentially facilitating the design of clinical trials for these participants. Lastly, organoid modeling adds to the excitement by confirming the promising potential of targeted therapy to exceed conventional treatments in eradicating MRD.
Methods
Participants and study design
The research ethics committees of the Hospital Clínico Universitario and Aarhus University approved the study protocol (institutional review board no. 2021/083). All participants provided written informed consent and the study was conducted in accordance with the Declaration of Helsinki. Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Through a liquid biopsy program within the Digestive Tumors Group at the Hospital Clínico Universitario, we prospectively enrolled 320 participants diagnosed with localized CC who had experienced recurrence between July 2015 and May 2021. Use of ACT was at the discretion of the treating clinician. As an inclusion criterion for the study, participants who had experienced relapse and had plasma at relapse and tissue at baseline available were selected. Additionally, 21 participants who had not experienced relapse and had sufficient quantities of plasma after surgery were also included in the analysis. In total, 25 participants in the discovery cohort were studied (Fig. 1). Primary tumor tissue, collected at diagnosis before any treatment, and plasma at relapse, collected from the participant a few days after the detection of disease progression on the computed tomography scan before starting treatment for advanced disease, were collected from all participants. Of the 25 participants with recurrence, 48% (n = 12) had plasma available preoperatively (considered as baseline time point) and 60% (n = 15) had plasma available postoperatively; matching tissue from the recurrence lesion was also available in 68% (n = 17) participants (Fig. 2). All peripheral blood from participants was collected in EDTA tubes at diagnosis, 6–8 weeks after surgery, before ACT and upon disease progression for ctDNA and WBC analysis. cfDNA was extracted from 4 ml of plasma for each participant. Tumor tissue DNA was extracted at diagnosis and relapse following macrodissection of samples to ensure >70% cellular content for subsequent DNA and RNA extraction. WES was performed on the collected samples. RNA deconvolution from primary tissue and proteomics from both primary (n = 25) and relapse (n = 17) tumor tissue of the discovery cohort were used to validate the findings from WES. To validate these results, we used an external cohort consisting of 15 participants with CC from seven hospitals in Denmark, in whom primary tissue, plasma at diagnosis and plasma at relapse were collected. Plasma was isolated within 2 h of blood collection by double centrifugation at 3,005g for 10 min and stored at −80 °C until DNA extraction. Postoperative plasma for analysis was available for 14 of the 15 participants (93%).
DNA extraction
In the discovery cohort, macrodissection of the formalin-fixed paraffin-embedded (FFPE) block with primary tissue was performed in each participant. Samples from relapses were also used if available. DNA was extracted using the AllPrep DNA/RNA FFPE kit (Qiagen) for tumoral DNA from FFPE cuts, Chemagic DNA blood kit (Chemagen) for germline DNA from matched WBCs and QIAamp circulating nucleic acid kit (Qiagen) for cfDNA from 4-ml plasma samples. All extraction protocols were performed according to the corresponding manufacturer’s instructions. Tumor DNA and WBCs were quantified using the QuantiFluor double-stranded DNA (dsDNA) system (Promega), whereas cfDNA quality and quantity were assessed with a cfDNA ScreenTape assay (Agilent). cfDNA samples were not accepted if cfDNA content was below 70%.
In the validation cohort, DNA was extracted from freshly frozen tumor tissue using the Puregene DNA purification kit (Gentra Systems) and from FFPE samples with the QiAamp DNA FFPE tissue kit (Qiagen). WBC DNA was extracted from the buffy coat using the Qiasymphony DNA mini kit (Qiagen). DNA from tumors and WBCs was quantified using the Qubit dsDNA BR assay kit (Thermo Fisher Scientific). cfDNA was purified from 4–8 ml of plasma using the QIAamp circulating nucleic acids kit (Qiagen) and quantified by droplet digital PCR (BioRad Laboratories) using assays targeting regions on Chr3 and Chr7, as described previously52.
WES
Discovery cohort
Libraries were prepared using 100-ng inputs of tumor DNA, 100 ng of WBC DNA and 10–40 ng of cfDNA. KAPA HyperPlus (Roche) with unique dual and molecular indices (UDI–UMI; Integrated DNA Technologies) was used for library preparation of tissue and WBC DNA, according to the instructions of KAPA HyperCap Workflow v3. The only modification applied was the use of 5 ml of 15 mM UDI–UMI at the ligation step instead of a universal adaptor and posterior precapture PCR with Illumina primer mix. KAPA HyperPrep (Roche) with the same adaptors was selected for cfDNA, with the following changes to the mentioned protocol: (1) the adaptor was diluted proportionally with the input DNA, with a maximum of 12 mM for 40 ng; (2) precapture PCR was performed with a total of 11 cycles; (3) post-PCR purifications were performed with 50 ml of KAPA HyperPure beads (Roche) and 10-min incubation with mixed beads; and (4) after precapture PCR and before ethanol cleanup, beads were eluted in 50 ml of Tris-HCl 10 mM pH8.0 for a second incubation with another 50 ml of beads. For the next step, precapture libraries were pooled as follows: 1,500 ng of four tumor DNA libraries, 1,500 ng of eight WBC DNA libraries and 1,000 ng of one cfDNA library. Capture of the exome was performed with KAPA HyperExome (Roche) following the manufacturer’s instructions and the same type of post-PCR purification for cfDNA exome samples. The quality of both precapture and postcapture libraries was determined with an HS D1000 ScreenTape assay (Agilent) from a 20-fold dilution of the library. Sequencing was performed on HiSeq 3000 (Illumina) or NovaSeq 6000 (Illumina) with 150-bp paired-end reads and an extended i7 read of 17 cycles for UMI reading. The median coverage obtained in the discovery cohort was 132× for WBCs, 194× for the primary tissue and 478×, 504× and 389× for the plasma at baseline, postoperative and relapse stages, respectively (Supplementary Tables 6–11 and Extended Data Fig. 10a).
Validation cohort
Tumor and normal DNA sequencing libraries were generated using xGen UDI–UMI adaptors (Integrated DNA Technologies) and the Twist library preparation enzymatic fragmentation kit 1.0 (Twist Bioscience). Libraries were prepared as described by the manufacturer. For normal and FrFr DNA, we used 50 ng of input and 10 min of fragmentation. For FFPE DNA, 200 ng of input and 6 min of fragmentation were used. All libraries were amplified with seven cycles of PCR. Libraries were quantified using a Qubit dsDNA BR assay kit (Thermo Fisher Scientific) and library size was estimated using a TapeStation D1000 (Agilent). Blood samples were collected in K2–EDTA 10-ml tubes (Becton Dickinson) from healthy controls and participants with CRC. Plasma sequencing libraries were prepared using cfDNA from 2 ml of plasma. cfDNA libraries were generated using xGen UDI–UMI adaptors (Integrated DNA Technologies) and a KAPA HyperPrep kit (Roche). Postligation cleanup was performed with AMPURE beads in a 1.4:1 ratio of beads to DNA to retain short fragments, while post-PCR cleanup was performed using a 1:1 ratio. The libraries were amplified with seven cycles of PCR. Libraries were quantified using a Qubit dsDNA BR assay kit (Thermo Fisher Scientific) and library fragment size was estimated using a TapeStation D1000 (Agilent). Libraries that did not show the usual bimodal fragment size distribution13 of cfDNA were excluded before sequencing. Tumor and WBC DNA libraries were captured using the next-generation sequencing human core exome (TWIST Bioscience, ~33 Mb) according to the manufacturer’s protocol. Target-enriched libraries were sequenced using the NovaSeq platform with 2× 150-bp paired-end sequencing. The median coverage obtained in the validation cohort for WBCs, primary tissue and plasma at baseline, postoperative and relapse stages was 58×, 95×, 844×, 1,022× and 1,003×, respectively (Supplementary Table 12 and Extended Data Fig. 10a).
FASTQ preprocessing, quality control and read mapping
Raw sample quality control was carried out by FastQC53 (version 0.11.8), whereas Cutadapt54 (version 2.10) was used for the adaptor removal and PrinSeq55 (version 0.20.4) was used to discards reads with a mean quality under Q30 in the FASTQ preprocessing step. Sequencing reads were mapped to the hg38 human reference genome using BWA56 (version 0.7.17) and BAM postprocessing was performed using Picard (version 2.18.6) and the Genome Analysis Toolkit (GATK)57 (version 4.2.0.0) best practices. Umi-tools58 (version 1.0.1) was used for UMI extraction and deduplication of reads from the same UMI family.
Variant calling and somatic variant prioritization
Variant calling of primary tumor, plasma and normal samples was performed using combined outputs from Mutect2 (GATK version 4.2.0.0) and Lofreq59 (version 2.1.5). For greater confidence, germline variants were also called by HaplotypeCaller (GATK) implemented in the Sarek pipeline (version 2.7.1). The minimum VAF in primary tumor variants was set to 5% and 0.01% in plasma samples. The final set of variants was yielded by intersecting outputs from the two callers, extracting variants detected in normal samples and annotation using Variant Effect Predictor60 (VEP, Ensembl version 102). The variants found within a blacklist of redundant mutations were considered sequencing artifacts and removed. This blacklist was created on the basis of the sequencing of 135 different plasmas. Clonal hematopoiesis of indeterminate potential variants were also removed in plasmas samples to avoid false positives. Somatic pathogenic variants were identified by annotation with COSMIC (version 94), OncoKB (version 1) and an in-house pathogenic mutations database. Only high-impact and moderate-impact somatic mutations were considered for the mechanistic analysis in this study. A manual review and curation process of the pathogenic mutations detected in each sample was carried out. Variant prioritization analysis, characterized mainly by the match of the pathogenic mutations with OncoKB levels of evidence, was performed to select specific targeted therapies.
Sequencing quality control
A sequencing quality control criterion was set, requiring sufficient sequencing coverage to ensure that all clonal alterations detected in plasma samples were supported by a minimum of three mutated reads. Tumor purity and cancer cell fraction (CCF) were estimated by the PureCN61 (version 2.0.2) software on the basis of copy number and mutational data. Clonal mutations were defined as those with CCF ≥ 0.9, with the remaining mutations classified as subclonal. Subsequently, the tumor fraction of clonal mutations in all available plasma samples in both cohorts was estimated. The sequencing coverage proved sufficient to meet the quality criterion, ensuring that all clonal mutations in the study’s plasma samples with a minimum of three mutated reads were identified.
The fraction of clonal and subclonal mutations from the primary tumor detected in the plasma was estimated, obtaining higher sensitivity values when detecting clonal than subclonal mutations in both cohorts. In the discovery cohort, a sensitivity of 29% was observed for clonal mutation identification, which decreased to 11% for subclonal mutations (P = 0.0005, according to Wilcoxon paired test). In the validation cohort, the sensitivity values were 32% and 29% for detection of clonal and subclonal mutations, respectively (P = 0.0413, according to Wilcoxon paired test). The sensitivity to detect subclonal mutations was higher in the validation than the discovery cohort (P = 0.0063, according to Wilcoxon test), given the significantly greater sequencing coverage in these samples.
TMB and tumor fraction estimation
TMB was estimated using nonsynonymous mutations with an impact on the protein (missense, frame-shift and small indel mutations). Participants with a TMB over 10 mutations per Mb were classified as having high TMB (Extended Data Fig. 10b). The tumor fraction was estimated as the number of mutated reads (overlapping the mutational compendium of the primary tumor) relative to the total number of reads overlapping the loci of the mutational in the plasma samples. (Extended Data Fig. 10c).
MRD detection
For increased confidence in variant calling in postsurgery plasma samples, a joint normal sample with a median depth of 3,474× was created, consisting of all available WBC samples. Paired variant calling was performed from this using a heuristic method and a statistical test using the number of aligned reads supporting each allele, based on VarScan2 (ref. 62) (version 2.4.4) software. A participant was considered positive if at least one somatic mutation was detected through this WES analysis.
To evaluate the clinical feasibility of MRD detection using WES analysis while optimizing cost-effectiveness, we focused on identifying somatic mutations with the highest VAF in the plasma baseline exome of each participant. The objective was to determine whether these mutations remained detectable in postoperative plasma samples. In evaluating sensitivity for MRD detection, we examined various numbers of candidate mutations (ranging from 15 to 20), adhering to the criterion of one or two detectable mutations necessary to classify a participant as ctDNA positive, consistent with the criteria of existing assays. Results from both the discovery cohort (88% sensitivity with one mutation, 67% with two mutations) and the validation cohort (100% with one mutation, 86% with two mutations) indicated that sensitivity did not improve with the selection of more than 16 candidate alterations. Therefore, we established the criterion of selecting 16 mutations (TAV16), aligning with the same number of mutations validated in commercial assays using a tumor-informed approach for the same purpose3,7. The TAV16 assay is protected by patent rights (EP25382212).
MS
A spectral peptide library was obtained using liquid chromatography (LC)–MS/MS by the Proteomics Service of the University of Valencia. LC–MS/MS was performed in a timsTOF fleX MS instrument (Bruker). The sample loaded in the Evotip pure was eluted to an analytical column (Endurance 8 cm × 100 µm, 3 µm; Evosep) by the Evosep One system and resolved with the 60 SPD chromatographic method defined by the manufacturer. The eluted peptides were ionized in a captive Spray with 1,600 V at 180 °C and quantitative analysis of individual samples was performed by parallel accumulation–serial fragmentation combined with data-independent acquisition. For every protein in the spectral library, a maximum of 20 peptides were quantified among those with a 95% confidence threshold and a false discovery rate (FDR) lower than 1%.
Transcriptomic and CIBERSORT analysis
Total RNA was isolated from primary tumor samples embedded in FFPE using an RNeasy FFPE Kit (Qiagen, 73504). The integrity of the extracted RNA was subsequently validated using Agilent Technologies TapeStation RNA analysis ScreenTape. Following the manufacturer’s guidelines, sequencing libraries were prepared using the NEBNext Ultra (TM) II Directional RNA library prep kit for Illumina module from New England Biolabs, in conjunction with the NEBNext poly(A) mRNA magnetic isolation module for mRNA enrichment. For quality control of sequences, a methodology akin to gene panels was applied. The filtered reads were then mapped to the human reference GRCh38 genome using STAR (version 2.7.3a)63. Isoform quantification was performed through RSEM (version 1.3.3)64 and subsequent processing involved Tximport (version 1.16.1)65 to summarize counts per gene. To derive the expression signal of immune cells infiltrated in our bulk RNA sequencing (RNA-seq) samples from the discovery cohort, the CIBERSORT package (version 1.0.4) was used. To characterize the transcriptomic profile of the primary and metastatic tissues as an epithelial or mesenchymal phenotype, R package imogimap66 was used. The provided EMT score is defined as the mean value of modified expressions. The EMT scores were calculated on the basis of a 76-gene expression signature reported by ref. 67 (Supplementary Table 1) and the metric mentioned on the basis of that gene signature68. For each sample, the score was calculated as a weighted sum of the 76 gene expression levels and the resulting scores were then mean-centered and normalized by s.d. as a function of the distribution of scores in all samples. Negative scores can be interpreted as indicating a mesenchymal phenotype, whereas positive scores indicate an epithelial phenotype.
IHC staining
All stainings were carried out on 3-μm paraffin-embedded slides using the DAKO autostainer, according to the manufacturer’s recommendations, with DAKO kit solutions (K8002, Dako). The CD20+ IHC staining was performed with the primary anti-CD20 antibody (clone L26; Dako). The slides were scanned using a slide scanner (3DHistech, P250).
The percentage and density of CD20+ B cells per sample were semiautomatically assessed using open-source digital analysis software QuPath (version 4.0.0)69 after tissue segmentation. The areas of interest selected included the entire invasive front of the tumor and all tertiary lymphoid structures related to the tumor, located at a maximum distance of 7 mm from the tumor.
Mutational signatures
Known mutation signatures from COSMIC (version 3.2 release) were matched with the somatic trinucleotide profile of each plasma and primary tumor sample using SigProfiler70 (version 1.1.3).
CNV detection
CNVs in primary tumor samples were called by a combination of CNVkit71 (version 0.9.7), VarScan (version 2.4.4) and FACETS72 (version 0.15) tools output in a paired tumor–normal mode with a 2,000-bp window size. A score-based CNV classification was performed to reduce false positives, considering size and requiring detection by at least two of the tools. CNVs in plasma samples were also called in a paired tumor–normal mode but using a combination of WisecondorX73 (within-sample copy-number aberration DetectOR, version 1.2.4) and CONTRA74 (version 2.0.8) software with a 100-kb window size. In plasma samples, a scored-based CNV classification was also used to determine real events.
Neoepitope abundance quantification
Protein sequences associated with somatic mutations identified from WES data of both primary and metastatic tumors were generated using pVACseq (version 4.4.1)75, a cancer immunotherapy pipeline designed to identify personalized variant antigens through cancer sequencing. The VCF files obtained from WES were annotated with the VEP tool to prepare the input for pVACseq. Mutated and corresponding wild-type protein sequences were subsequently generated using the ‘generate_protein_fasta’ command. HLA typing for each participant was performed using the nf-core/hlatyping (version 2.0.0)76 Nextflow pipeline, which provides a best-practice analysis for precision HLA typing from next-generation sequencing data using OptiType. The binding affinity of each identified neoepitope to major histocompatibility complex (MHC) molecules was predicted using NetMHCpan (version 4.1)77, which uses artificial neural networks to predict peptide binding for any MHC molecule with a known sequence, allowing for the selection of neoepitopes with the highest likelihood of presentation based on binding affinity. Neoepitope quantification from primary and metastatic tissue samples was conducted using MS data processed with MaxQuant (version 2.6.5)78, a quantitative proteomics tool for large-scale MS data analysis. One participant was excluded because of the absence of detected neoepitopes in the proteomic data and MSI participants were also not considered for the analysis. Lastly, the comparative abundance of neoepitopes in primary and metastatic tissues was analyzed using MSstats (version 4.8.7)79,80, a statistical toolkit for relative protein abundance analysis.
Drug sensitivity assay
The PDO model was trypsinized until a single-cell suspension was achieved and 3,000 cells per well were plated into 96-well plates. After 48 h, when organoids were formed, increasing doses of both standard chemotherapy and targeted therapies were added. Oxaliplatin and fluorouracil were provided by the HCUV Pharmacy Service. The targeted drugs tested were olaparib (AZD228; Selleckchem, S1060), trametinib (GSK1120212; Selleckchem, S2673), tazemetostat (EPZ-6438; Selleckchem, S7128), adavosertib (MK-1775; Selleckchem, S1525), RMC-4550 (Selleckchem, S8718) and alpelisib (BYL719; Selleckchem, S2814). Inhibitor drugs were diluted in DMSO such that assays contained vehicle control wells with a maximum concentration of 0.1% of DMSO. After 120 h of treatment, cell viability was measured using the CellTiterGlo3D assay following the manufacturer’s instructions. Luminescence was analyzed in a Fluoroskan Ascent FL (Thermo Fisher Scientific). Every assay was performed at least twice, with a technical triplicate for each condition.
Statistics and reproducibility
No statistical method was used to predetermine sample size. All individuals from both cohorts who had plasma samples at relapse and tissue samples at baseline (primary tumor) available were selected for the study and analyzed. MSI participants were excluded from the parallel evolution study because of their expected high TMB (Results). The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment. Data distribution was assumed to have equal variance but this was not formally tested. Normality was checked with the Shapiro–Wilk test. Qualitative variables are presented using frequencies and percentages while quantitative variables are expressed as the mean and s.d. if the normality assumption held true and the median and interquartile range otherwise. Comparison between continuous variables was carried out using a Student t-test if normality criteria were reached; otherwise, the Wilcoxon signed-rank test was used. The correlation between quantitative variables was assessed using Spearman’s ρ statistic. Software used for all analysis was R (version 4.0.1; R Core Team, 2021) and the cutoff for statistical significance was set at α = 0.05 in all tests. All tests were two-sided.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The raw sequencing data of the tissue and plasma samples from the discovery cohort that support the findings of this study were deposited to the European Genome–Phenome Archive (EGA) under controlled access with accession code EGAD50000000293. Data will be shared on reasonable request for academic or commercial use and within the limitations of the provided informed consent and under General Data Protection Regulation law. Data access requests must be submitted through the EGA platform’s request system. Requests will be reviewed by the Data Access Committee of INCLIVA Biomedical Research Institute and responses will be provided within approximately 2 weeks. Approved users must comply with data use agreements specifying permitted use and restrictions. External researchers (academic or commercial) interested in analyzing the Aarhus colorectal cancer dataset will need to contact the Data Access Committee by email to cla@clin.au.dk. Access to clinical data and processed sequencing data output files used in the article requires that the data requestor (legal entity) enter into Collaboration and Data Processing Agreements with the Central Denmark Region (the legal entity controlling and responsible for the data). Request for access to raw sequencing data requires that the purpose of the data reanalysis is approved by the Danish National Committee on Health Research Ethics. Upon reasonable request, the authors, on behalf of the Central Denmark Region, will enter into a collaboration with the data requestor to apply for approval. MS data were deposited to ProteomeXchange with primary accession code PXD061711. Source data are provided with this paper.
Code availability
Code and datasets used for the analysis and figures generation are available from GitHub (https://github.com/INCLIVA-bioinformatics/INCLIVA-CC-WES).
References
Argilés, G. et al. Localised colon cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 31, 1291–1305 (2020).
Tie, J. et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci. Transl. Med. 8, 346ra392 (2016).
Reinert, T. et al. Analysis of plasma cell-free DNA by ultradeep sequencing in patients with stages I to III colorectal cancer. JAMA Oncol. 5, 1124–1131 (2019).
Tie, J. et al. Circulating tumor DNA analyses as markers of recurrence risk and benefit of adjuvant therapy for stage III colon cancer. JAMA Oncol. 5, 1710–1717 (2019).
Tarazona, N. et al. Targeted next-generation sequencing of circulating-tumor DNA for tracking minimal residual disease in localized colon cancer. Ann. Oncol. 30, 1804–1812 (2019).
Tarazona, N. et al. Detection of postoperative plasma circulating tumour DNA and lack of CDX2 expression as markers of recurrence in patients with localised colon cancer. ESMO Open 5, e0008476 (2020).
Henriksen, T. V. et al. Circulating tumor DNA in stage III colorectal cancer, beyond minimal residual disease detection, toward assessment of adjuvant therapy efficacy and clinical behavior of recurrences. Clin. Cancer Res. 28, 507–517 (2022).
Taieb, J. et al. Prognostic value and relation with adjuvant treatment duration of ctDNA in stage III colon cancer: a post hoc analysis of the PRODIGE-GERCOR IDEA-France trial. Clin. Cancer Res. 27, 5638–5646 (2021).
Henriksen, T. V. et al. Unraveling the potential clinical utility of circulating tumor DNA detection in colorectal cancer—evaluation in a nationwide Danish cohort. Ann. Oncol. 35, 229–239 (2024).
Tie, J. et al. Circulating tumor DNA analysis guiding adjuvant therapy in stage II colon cancer. N. Engl. J. Med. 386, 2261–2272 (2022).
Lonardi, S. et al. The PEGASUS trial: post-surgical liquid biopsy-guided treatment of stage III and high-risk stage II colon cancer patients. Ann. Oncol. 34, S1268–S1269 (2023).
Kotani, D. et al. Molecular residual disease and efficacy of adjuvant chemotherapy in patients with colorectal cancer. Nat. Med. 29, 127–134 (2023).
Kataoka, K. et al. Survival benefit of adjuvant chemotherapy based on molecular residual disease detection in resected colorectal liver metastases: subgroup analysis from CIRCULATE-Japan GALAXY. Ann. Oncol. 35, 1015–1025 (2024).
Nakamura, Y. et al. ctDNA-based molecular residual disease and survival in resectable colorectal cancer. Nat. Med. 30, 3272–3283 (2024).
Vidal, J. et al. Plasma ctDNA RAS mutation analysis for the diagnosis and treatment monitoring of metastatic colorectal cancer patients. Ann. Oncol. 28, 1325–1332 (2017).
Grasselli, J. et al. Concordance of blood- and tumor-based detection of RAS mutations to guide anti-EGFR therapy in metastatic colorectal cancer. Ann. Oncol. 28, 1294–1301 (2017).
Taieb, J. ctDNA in early-stage CRC: tailoring the adjuvant therapy. Proceedings of the European Society for Medical Oncology Congress 2023 (eds André, F. & Haller, D. G.) (ESMO, 2023).
Luskin, M. R. et al. Targeting minimal residual disease: a path to cure? Nat. Rev. Cancer 18, 255–263 (2018).
Conca, V. et al. Waiting for the ‘liquid revolution’ in the adjuvant treatment of colon cancer patients: a review of ongoing trials. Cancer Treat. Rev. 126, 102735 (2024).
Keller, L. & Pantel, K. Unravelling tumor heterogeneity by single-cell profiling of circulating tumor cells. Nat. Rev. Cancer 19, 553–567 (2019).
Papaccio, F. et al. Proteotranscriptomic analysis of advanced colorectal cancer patient derived organoids for drug sensitivity prediction. J. Exp. Clin. Cancer Res. 42, 8 (2023).
Jiang, Z. et al. Targeting the SLIT/ROBO pathway in tumor progression: molecular mechanisms and therapeutic perspectives. Ther. Adv. Med. Oncol. 11, 1758835919855238 (2019).
Cheng, M. et al. FLNA promotes chemoresistance of colorectal cancer through inducing epithelial–mesenchymal transition and SMAD2 signaling pathway. Am. J. Cancer Res. 10, 403–423 (2020).
Bai, J. et al. Comprehensive analysis of LAMC1 expression and prognostic value in kidney renal papillary cell carcinoma and clear cell carcinoma. Front. Mol. Biosci. 9, 988777 (2022).
Zhu, C. et al. ITGB3/CD61: a hub modulator and target in the tumor microenvironment. Am. J. Transl. Res. 11, 7195–7208 (2019).
Nishida, J., Miyazono, K. & Ehata, S. Decreased TGFBR3/betaglycan expression enhances the metastatic abilities of renal cell carcinoma cells through TGF-β-dependent and -independent mechanisms. Oncogene 37, 2197–2212 (2018).
Pich, O. et al. The mutational footprints of cancer therapies. Nat. Genet. 51, 1732–1740 (2019).
Yates, L. R. et al. Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat. Med. 21, 715–759 (2015).
McGrail, D. J. et al. High tumor mutation burden fails to predict immune checkpoint blockade response across all cancer types. Ann. Oncol. 32, 661–672 (2021).
Martincorena, I. et al. Universal patterns of selection in cancer and somatic tissues. Cell 171, 1029–1041(2017).
Zviran, A. et al. Genome-wide cell-free DNA mutational integration enables ultra-sensitive cancer monitoring. Nat. Med. 26, 1114–1124 (2020).
Parikh, A. R. et al. Liquid versus tissue biopsy for detecting acquired resistance and tumor heterogeneity in gastrointestinal cancers. Nat. Med. 25, 1415–1421 (2019).
Martínez-Castedo, B. et al. Minimal residual disease in colorectal cancer. Tumor-informed versus tumor-agnostic approaches: unraveling the optimal strategy. Ann. Oncol. 36, 263–276 (2025).
Nakamura, Y. et al. Clinical utility of circulating tumor DNA sequencing in advanced gastrointestinal cancer: SCRUM-Japan GI-SCREEN and GOZILA studies. Nat. Med. 26, 1859–1864 (2020).
Gambardella, V. et al. Molecular profiling of advanced solid tumours. The impact of experimental molecular-matched therapies on cancer patient outcomes in early-phase trials: the MAST study. Br. J. Cancer 125, 1261–1269 (2021).
Nakamura, Y. et al. Targeted therapy guided by circulating tumor DNA analysis in advanced gastrointestinal tumors. Nat. Med. 31, 165–175 (2025).
Gimeno-Valiente, F. et al. Sequencing paired tumor DNA and white blood cells improves circulating tumor DNA tracking and detects pathogenic germline variants in localized colon cancer. ESMO Open 8, 102051 (2023).
Yaeger, R. et al. Adagrasib with or without cetuximab in colorectal cancer with mutated KRAS G12C. N. Engl. J. Med. 388, 44–54 (2023).
Fakih, M. G. et al. Sotorasib plus panitumumab in refractory colorectal cancer with mutated KRAS G12C. N. Engl. J. Med. 389, 2125–2139 (2023).
Desai, J. et al. Divarasib plus cetuximab in KRAS G12C-positive colorectal cancer: a phase 1b trial. Nat. Med. 30, 271–278 (2024).
Jeong, S., Lim, S., Schevzov, G., Gunning, P. W. & Helfman, D. M. Loss of Tpm4.1 leads to disruption of cell–cell adhesions and invasive behavior in breast epithelial cells via increased Rac1 signaling. Oncotarget 8, 33544 (2017).
Italiano, A. et al. Pembrolizumab in soft-tissue sarcomas with tertiary lymphoid structures: a phase 2 PEMBROSARC trial cohort. Nat. Med. 28, 1199–1206 (2022).
Chalabi, M. et al. Neoadjuvant immunotherapy leads to pathological responses in MMR-proficient and MMR-deficient early-stage colon cancers. Nat. Med. 26, 566–576 (2020).
de Gooyer, P. G. M. et al. Radiotherapy, atezolizumab, and bevacizumab to increase organ preservation in rectal cancer: the TARZAN study. Ann. Oncol. 35, S106–S118 (2024).
Chalabi, M. et al. Neoadjuvant Immunotherapy in locally advanced mismatch repair-deficient colon cancer. N. Engl. J. Med. 390, 1949–1958 (2024).
Cañellas-Socias, A. et al. Metastatic recurrence in colorectal cancer arises from residual EMP1+ cells. Nature 611, 603–613 (2022).
Pant, S. et al. Lymph-node-targeted, mKRAS-specific amphiphile vaccine in pancreatic and colorectal cancer: the phase 1 AMPLIFY-201 trial. Nat. Med. 30, 531–542 (2024).
Zhang, H. et al. PDIA3 correlates with clinical malignant features and immune signature in human gliomas. Aging (Albany NY) 12, 15392–15413 (2020).
Deng, H., Chen, Y., Wang, J. & An, R. HLA-DRB1: a new potential prognostic factor and therapeutic target of cutaneous melanoma and an indicator of tumor microenvironment remodeling. PLoS ONE 17, e0274897 (2022).
Bos, M. K. et al. Whole exome sequencing of cell-free DNA—a systematic review and Bayesian individual patient data meta-analysis. Cancer Treat. Rev. 83, 101951 (2020).
Mendelaar, P. A. J. et al. Whole genome sequencing of metastatic colorectal cancer reveals prior treatment effects and specific metastasis features. Nat. Commun. 12, 574 (2021).
Reinert, T. et al. Analysis of circulating tumour DNA to monitor disease burden following colorectal cancer surgery. Gut 65, 625–634 (2016).
Andrews S. FastQC: a quality control tool for high throughput sequence data (Babraham Bioinformatics, 2010); http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12 (2011).
Schmieder, R. & Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 27, 863–864 (2011).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Smith, T., Heger, A. & Sudbery, I. UMI-tools: modeling sequencing errors in unique molecular identifiers to improve quantification accuracy. Genome Res. 27, 491–499 (2017).
Wilm, A. et al. LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 40, 11189–11201 (2012).
McLaren, W. et al. The Ensembl Variant Effect Predictor. Genome Biol. 17, 122 (2016).
Riester, M. et al. PureCN: copy number calling and SNV classification using targeted short read sequencing. Source Code Biol. Med. 11, 1–13 (2016).
Koboldt, D. et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576 (2012).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 29, 15–21 (2013).
Soneson, C., Love, M. I. & Robinson, M. D. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res. 4, 1521 (2015).
Newman, A. M. et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 37, 773–782 (2019).
Bozorgui, B., Kong, E. K., Luna, A. & Korkut, A. Mapping the functional interactions at the tumor–immune checkpoint interface. Commun. Biol. 6, 462 (2023).
Byers, L. A. et al. An epithelial-mesenchymal transition (EMT) gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin. Cancer Res. https://doi.org/10.1158/1078-0432.CCR-12-1558 (2013).
Guo, C. C. et al. Dysregulation of EMT drives the progression to clinically aggressive sarcomatoid bladder cancer. Cell Rep. 27, 1781–1793 (2019).
Bankhead, P. et al. QuPath: open source software for digital pathology image analysis. Sci. Rep. 7, 16878 (2017).
Islam, S. M. A. et al. Uncovering novel mutational signatures by de novo extraction with SigProfilerExtractor. Cell Genom. 2, 100179 (2022).
Talevich, Eric et al. CNVkit: genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput. Biol. 12, e1004873 (2016).
Shen, R. & Venkatraman, E. S. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 44, e131 (2016).
Raman, L. et al. WisecondorX: improved copy number detection for routine shallow whole-genome sequencing. Nucleic Acids Res. 47, 1605–1614 (2019).
Li, J. et al. CONTRA: copy number analysis for targeted resequencing. Bioinformatics 28, 1307–1313 (2012).
Hundal, J. et al. pVACtools: a computational toolkit to identify and visualize cancer neoantigens. Cancer. Immunol. Res. 8, 409–420 (2020).
Ewels, P. et al. The nf-core framework for community-curated bioinformatics pipelines. Zenodo https://doi.org/10.5281/zenodo.1401039 (2022).
Jurtz, V. et al. NetMHCpan-4.0: improved peptide–MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. J. Immunol. 199, 3360–3368 (2017).
Tyanova, S., Temu, T. & Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 11, 2301–2319 (2016).
Choi, M. et al. MSstats: an R package for statistical analysis of quantitative mass spectrometry-based proteomic experiments. Bioinformatics 30, 2524–2526 (2014).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011).
Acknowledgements
We gratefully acknowledge the support of the INCLIVA BioBank (PT20/00029; B.0000768 ISCIII) as part of the Valencian Biobanking Network and the Spanish National Biobanks Network, the Precision Medicine Unit and the Bioinformatics Unit of the INCLIVA Biomedical Research Institute and especially all the participants and their families for their participation in this study. We acknowledge the Danish Cancer Biobank and Colorectal Cancer Research Biobank at Aarhus University Hospital for providing access to blood and tissue materials. We would like to express our sincere gratitude to J. C. Sanz from the Vall dHebron Institute of Oncology (Barcelona, Spain) for his valuable feedback and constructive criticism during the review process of this paper. Their insightful comments and suggestions greatly improved the quality of the manuscript.
This work was supported by Instituto de Salud Carlos III (PI21/00689 and PI21/00695), the Spanish Society of Oncology Medicine, the Spanish Cooperative Group for Digestive Tumor Therapy and the Spanish Association Against Cancer (AECC; GCAEC20030CERV). N.T. and V.G. are supported by Joan Rodés contracts JR20/00005 and JR21/00042, respectively. J. Martín-Arana is supported by a grant from Generalitat Valenciana (ACIF2020/381). F.G.-V. is supported by a Generalitat Valenciana fellowship program (APOSTD/2021/168). B.G.-M. is supported by a PFIS contract from Instituto de Salud Carlos III (FI22/00267). B.M.-C. is supported by a grant from Ministerio de Universidades (FPU21/00373). R.T.-M. is supported by a grant from the AECC (PRDVA172011TEBA). Part of the equipment used in this study was funded by Generalitat Valenciana and cofinanced with FEDER funds (OP FEDER of Comunitat Valenciana 2014–2020). This study was supported by the Novo Nordisk Foundation (NNF17OC0025052 and NNF22OC0074415, to C.L.A.) and the Danish Cancer Society (R231-A13845 and R257-A14700, to C.L.A.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
N.T. and A.C. designed and oversaw the study. N.T., F.G.-V., B.G.-M., R.T.-M., T.V.H. and B.M.-C. performed the translational experiments. V.G., S.R. and D.R. advised on the treatment of actionable variants. C.M.-C., K.A.G., C.L.A. and B.P. provided the material from tissue tumors for analysis. M.H., M.G.B., D.M., V.P., L.P.-S., J. Martín-Arévalo, D.C., S.G.-B., K.A.G., A.F., T.F. and A.E. recruited and followed the participants and created the database. J. Martín-Arana, J.A.C.-A., D.G.C. and F.G.-V. performed the statistical and bioinformatics analyses. F.G.-V. and J. Martín-Arana interpreted and analyzed the data presented and generated the figures and tables. N.T., A.C., J. Martín-Arana, F.G.-V., T.V.H. and C.L.A. primarily wrote the manuscript. All authors critically revised the manuscript for important intellectual content.
Corresponding authors
Ethics declarations
Competing interests
A.C. declares institutional research funding from Genentech, Merck Serono, BMS, MSD, Roche, Beigene, Bayer, Servier, Lilly, Natera, Novartis, Takeda, Astellas and Fibrogen and advisory board or speaker fees from Merck Serono, Roche, Servier, Takeda and Astellas. N.T. declares advisory board or speaker fees from Merck Serono, Servier, Pfizer, Natera and Guardant Health. M.H. declares advisory board and speaker fees from Servier. T.F. declares institutional research funding from Genentech, Adapt Immune, Roche, Beigene, Astelas, BMS, Daichii Sanyo and Amgen and speaker fees from Astrazeneca, Amgen, Bayer, BMS, Lilly, MSD and Servier. V.G. declares advisory board fees from Boehringer Ingelheim and institutional research funding from Bayer, Boehringer, Roche, Genentech, Merck Serono, Beigene, Servier, Lilly, Novartis, Takeda, Astelas, Fibrogen, Amcure, Natera, Sierra Oncology, AstraZeneca, Medimmune, BMS and MSD. S.R. declares personal fees as an invited speaker from Amgen, MSD and Servier, advisory board fees from Amgen, Servier and Sirtex and institutional funding from Ability Pharmaceuticals, Astellas, G1 Therapeutics, Hutchinson, Menarini, Mirati, Novartis, Pfizer, Pierre Fabre, Roche and Seagen. C.L.A. declares institutional research funding from Natera, C2i Genomics and BioRad Laboratories. V.P.M. reports consultancy for Johnson&Johnson and Baxter, has received honorarium for speaking at symposia and workshops by Johnson&Johnson, Medtronic and Braun Medical and has received support for attending meetings by Takeda. J. Martín-Arévalo reports consultancy for Baxter and has received honorarium for speaking at workshops by Johnson&Johnson and Medtronic. D.M. has received honorarium for speaking at symposia and workshops by Johnson&Johnson and Medtronic and support for attending meetings by Sanofi. S.G.-B., A.E. and L.P.-S. have received honorarium for educational courses by Johnson&Johnson, Marina Garcés Albir and Dixie Huntley. C.M.-C. declares advisory board or speaker fees from MSD, Astelas and BMS. The other authors declare no competing interests.
Peer review
Peer review information
Nature Cancer thanks Karuna Ganesh and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Molecular profiling of paired tissue and plasma comparison at baseline in the validation cohort.
a, Concordance analysis of primary tumor and plasma baseline somatic SNVs. The cohort’s median concordance is represented by a dot. b, Comparative molecular landscape of pathogenic mutations and CNV in paired tissue and plasma samples at baseline from 15 colorectal cancer CC patients. Each box illustrates a mutated gene in a specific patient, divided into two sections: the left section displays results from the primary tissue, and the right section depicts plasma at baseline. Similarly, each box at a given collection moment is subdivided into two parts, with the left indicating point mutations, and the right representing CNVs. The Y-axis is arranged by the number of point mutations for each gene across all patients.
Extended Data Fig. 2 Molecular landscape of the paired tissue-plasma comparison at relapse.
Comparative analysis of the molecular landscape, focusing on pathogenic mutations and CNVs, in paired tissue and plasma samples collected at the point of relapse from 17 CC patients. Each box within the representation signifies a mutated gene in an individual patient, and it is divided into two sections by a line. The left part corresponds to outcomes derived from the metastatic tissue, while the right part corresponds to plasma at the time of relapse. Similarly, each box corresponding to a collection moment is further divided into two components, with the left indicating point mutations, and the right representing CNVs. The Y-axis is arranged based on the number of point mutations for each gene across all patients.
Extended Data Fig. 3 Minimal residual disease concordance of candidate variants.
Median concordance of candidate variants when selecting the 16 somatic mutations with the highest VAF for MRD monitoring in primary tumor and plasma baseline samples within the discovery cohort (left, n = 12 patients) and the validation cohort (right, n = 14 patients). Two-sided Wilcoxon test; p-value = 0.047.
Extended Data Fig. 4 Molecular profiling of tumor evolution comparing plasma at both baseline and relapse in the validation cohort.
Comparative molecular landscape of somatic pathogenic mutations and CNVs in paired plasma samples at baseline and relapse from 15 CC patients. Each box within the representation signifies a mutated gene in an individual patient, divided into two sections by a line. The left segment corresponds to outcomes obtained in the plasma baseline, while the right segment corresponds to plasma at relapse. Similarly, each box corresponding to a collection moment is further divided into two components, with the left indicating point mutations and the right representing CNVs. The Y-axis is organized based on the number of point mutations for each gene across all patients.
Extended Data Fig. 5 Molecular landscape of the tumor evolution comparing tissue at baseline and plasma at relapse.
a, Comparative molecular profiling of pathogenic mutations and CNVs in paired tissue at baseline and plasma at relapse samples from 25 CC patients. Each box in the illustration denotes a mutated gene in an individual patient, bifurcated into two sections by a line. The left segment corresponds to findings derived from the primary tumor, while the right segment corresponds to plasma at relapse. Similarly, each box associated with a specific collection moment is further divided into two components: the left portion denotes point mutations, and the right portion represents CNVs. The Y-axis is organized based on the number of point mutations for each gene across all patients. b, Concordance comparison between the primary tumor and plasma at relapse (n = 25 patients) versus the concordance of plasma at both baseline and relapse (n = 12 patients) of somatic mutations across the discovery cohort (two-sided Wilcoxon test; p-value = 0.0015). Data are presented as median values +/- standard deviation. Concordance is calculated by comparing each patient with themselves at different stages.
Extended Data Fig. 6 Analysis of tumor evolution.
Evolutionary plot per patient in the a, discovery cohort (n = 7) and b, validation cohort (n = 14) illustrating somatic mutations occurring at baseline, post-surgery, and at relapse. The y-axis represents the accumulated number of mutations across the cohort. The presence of mutations over time is depicted by colors, where gray indicates mutations appearing at baseline but representing unselected subclones lost after surgery. Progressing up the chromatic scale from green to purple signifies mutations persisting over time, considered clonal. Conversely, mutations emerging after surgery until the patient’s relapse are depicted in shades of red, indicating clones arising due to tumor evolution during this period. c, EMT scores for metastatic and primary tissues. Distribution of EMT scores for primary tissues and metastatic tissues for each patient. Negative scores can be interpreted as indicating a mesenchymal phenotype, whereas positive scores indicate an epithelial phenotype.
Extended Data Fig. 7 Mutational signatures in the discovery cohort.
a, Identification of mutational signatures at relapse (n = 25 patients). Each bar represents an individual patient, with colors corresponding to different mutational signatures, as indicated in the legend. The upper panel provides patient metadata, including age, batch, tumor location, gender, MSI status, and stage. b, Comparative distribution of mutational signatures between plasma at both baseline and relapse (n = 12 patients). The distribution of mutational signatures at both time points is displayed, allowing visualization of changes in signature composition over time. Each bar represents an individual patient, with colors corresponding to different mutational signatures, as indicated in the legend. The upper panel provides patient metadata, including age, batch, tumor location, gender, MSI status, and stage.
Extended Data Fig. 8 Evaluation of TMB.
a, TMB comparison between tissue samples at baseline and relapse in the discovery cohort (n = 17; two-sided Wilcoxon test; p-value = 0.7910). The minimum values are the smallest number of TMB of the cohort. The first quartile above the whiskers represents the data point that separates the lowest 25% of the data from the rest. The center line per box plot represents the median value among the data points. The third quartile just on top of the box plot separates the lowest 75% of the data points from the highest 25%. The maximum value represents the highest TMB of the cohort. b, TMB comparison between plasma samples at both baseline and relapse in the discovery cohort (n = 12; two-sided Wilcoxon test; p-value = 0.9632). The minimum values are the smallest number of TMB of the cohort. The first quartile above the whiskers represents the data point that separates the lowest 25% of the data from the rest. The center line per box plot represents the median value among the data points. The third quartile just on top of the box plot separates the lowest 75% of the data points from the highest 25%. The maximum value represents the highest TMB of the cohort. c, TMB comparison between plasma samples at both baseline and relapse in the validation cohort (n = 15; two-sided Wilcoxon test; p-value = 0.1070). Each patient is individually compared across different stages. The minimum values are the smallest number of TMB of the cohort. The first quartile above the whiskers represents the data point that separates the lowest 25% of the data from the rest. The center line per box plot represents the median value among the data points. The third quartile just on top of the box plot separates the lowest 75% of the data points from the highest 25%. The maximum value represents the highest TMB of the cohort. d, Spearman correlation (two-sided) analysis between TMB and dN/dS in primary tissue (blue; n = 25; p-value = 0.0785) and relapse (red; n = 17; p-value = 0.0199) within the discovery cohort. P-values and rho scores are reported for each case. The line represents the fitted relationship between the variables, while the shaded band corresponds to the 95% confidence interval around the regression estimate.
Extended Data Fig. 9 Drug screening in PDOs models.
a. Dendrogram derived from hierarchical clustering to identify PDOs exhibiting molecular similarity to patients within our cohort. b, Landscape of actionable genes identified in each selected PDO model and their corresponding CC patient from the discovery cohort. c, Heatmap of Log-AUCs illustrating the responsiveness of three PDO models to various targeted therapies and conventional chemotherapy agents (dark shading indicating a favorable response, clear shading indicating no response). The left panel presents the actionable mutations identified in each PDO. For every PDO drug sensitivity assay, three biological replicates with three technical replicates each, were performed for each condition analyzed. d, Logarithmically transformed dose-response curves depicting the viability of PDO models (CTO65, CTO119, and CTO147) in response to escalating doses of standard chemotherapy agents and targeted therapy drugs. For every PDO drug sensitivity assay, three biological replicates with three technical replicates each, were performed for each condition analyzed. Data are presented as median values +/- standard deviation.
Extended Data Fig. 10 Comparison of sequencing statistics between the discovery and validation cohorts.
a, Sequencing coverage. The minimum values are the smallest number of coverage of the cohort. The first quartile above the whiskers represents the data point that separates the lowest 25% of the data from the rest. The center line per box plot represents the median value among the data points. The third quartile just on top of the box plot separates the lowest 75% of the data points from the highest 25%. The maximum value represents the highest coverage of the cohort. Two-sided Wilcoxon test; p-value: WBCs=7.8e-06; tissue=0.016; baseline plasma (PLASMA-BL) = 2.0e-05; post-operative plasma (PLASMA-PO) = 2.6e-08; relapse plasma (PLASMA) = 2.7e-07. b, Tumor fraction. The minimum values are the smallest number of tumor fraction of the cohort. The first quartile above the whiskers represents the data point that separates the lowest 25% of the data from the rest. The center line per box plot represents the median value among the data points. The third quartile just on top of the box plot separates the lowest 75% of the data points from the highest 25%. The maximum value represents the highest tumor fraction of the cohort. Two-sided Wilcoxon test; p-value: baseline plasma (PLASMA-BL) = 0.139; post-operative plasma (PLASMA-PO) = 0.014; relapse plasma (PLASMA) = 0.026. c, Tumor mutational burden. The minimum values are the smallest number of TMB of the cohort. The first quartile above the whiskers represents the data point that separates the lowest 25% of the data from the rest. The center line per box plot represents the median value among the data points. The third quartile just on top of the box plot separates the lowest 75% of the data points from the highest 25%. The maximum value represents the highest TMB of the cohort. Two-sided Wilcoxon test; p-value: baseline tissue=0.111; baseline plasma=0.318; post-operative plasma (PLASMA-PO) = 0.018; relapse plasma=0.074.
Supplementary information
Supplementary Tables 1–12
Supplementary Table 1: Participant characteristics. For quantitative variables, the Wilcoxon two-sided test was used. For categorical variables, Fisher’s exact test (two-sided) was applied for 2 × 2 comparisons, while the chi-square test (two-sided) was used for comparisons involving contingency tables larger than 2 ×2. Supplementary Table 2: MRD detection. Statistics obtained in the analysis of postsurgery plasma from the discovery cohort (left) and the validation cohort (right). The comparison of sensitivity of different MRDs with the different approaches is also shown. Sensitivity was calculated on the basis of whether one or two mutations are required to consider the participant positive for ctDNA. Supplementary Table 3: Descriptive list of the mutations found among the EMT pathway in the metastatic tissue. The CADD score represents the deleteriousness value of the variant. The score is the PHRED-like (−10log10(rank/total)) scaled C score ranking a variant relative to all possible substitutions of the human genome (8.6 × 109). This value ranges from 1 to 99, with a cutoff for deleteriousness above 15. LOF, loss of function; GOF, gain of function; Pred. Func., predicted function. Supplementary Table 4: Somatic targetable mutations in the validation cohort. Number and percentage of participants harboring actionable mutations in the validation cohort. The corresponding treatment recommendations from the OncoKB database are presented in the right column. Supplementary Table 5: Pathogenic germline mutations detected in the discovery cohort. Germline pathogenic mutations and their relation to hereditary cancer-predisposing syndrome and their implication in cancer. Supplementary Table 6: WBC sequencing statistics of the discovery cohort. Supplementary Table 7: Primary tumor sequencing statistics of the discovery cohort. Supplementary Table 8: Baseline plasma sequencing statistics of the discovery cohort. Supplementary Table 9: Postoperative plasma sequencing statistics of the discovery cohort. Supplementary Table 10: Metastatic tumor sequencing statistics of the discovery cohort. Supplementary Table 11: Relapse plasma sequencing statistics of the discovery cohort. Supplementary Table 12: Validation cohort sequencing statistics.
Source data
Source Data Fig. 3
Statistical source data.
Source Data Fig. 4
Statistical source data.
Source Data Fig. 5
Statistical source data.
Source Data Extended Data Fig. 1
Statistical source data.
Source Data Extended Data Fig. 2
Statistical source data.
Source Data Extended Data Fig. 3
Statistical source data.
Source Data Extended Data Fig. 4
Statistical source data.
Source Data Extended Data Fig. 5
Statistical source data.
Source Data Extended Data Fig. 6
Statistical source data.
Source Data Extended Data Fig. 7
Statistical source data.
Source Data Extended Data Fig. 8
Statistical source data.
Source Data Extended Data Fig. 9
Statistical source data.
Source Data Extended Data Fig. 10
Statistical source data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Martín-Arana, J., Gimeno-Valiente, F., Henriksen, T.V. et al. Whole-exome tumor-agnostic ctDNA analysis enhances minimal residual disease detection and reveals relapse mechanisms in localized colon cancer. Nat Cancer 6, 1000–1016 (2025). https://doi.org/10.1038/s43018-025-00960-z
Received:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/s43018-025-00960-z
