
Abstract
Although the FOLFOX strategy has demonstrated benefits for tumor patients at advanced stages, chemoresistance remains a significant challenge to therapeutic efficacy. Thus, identifying strategies to overcome chemoresistance and enhance chemotherapy sensitivity is critical for optimizing HAIC-FOLFOX treatment. Comprehensive investigations of deubiquitinating enzymes (DUBs) across multiple bioinformatics cohorts and a local hepatocellular carcinoma (HCC) cohort identified ubiquitin-specific protease 1 (USP1) as a key regulator of HCC progression, correlating with poor survival outcomes. Functional assays demonstrated that USP1 overexpression promotes aggressive phenotypes in HCC cells, including enhanced proliferation, migration, and epithelial-mesenchymal transition (EMT), whereas USP1 inhibitor ML323 suppresses these effects and increases sensitivity to oxaliplatin and fluorouracil (5-FU), the primary agents in FOLFOX, both in vitro and in vivo. Mechanistic studies revealed that USP1 interacted with and stabilized the chromatin-remodeling factor lymphoid-specific helicase (HELLS) through deubiquitinating, thereby facilitating EMT and homologous recombination repair (HRR), thereby driving chemoresistance. Furthermore, USP1 promoted HELLS SUMOylation by stabilizing PIAS1, an E3 SUMO ligase, through deubiquitination and prevention of its ubiquitin-mediated degradation. Importantly, inhibition of SUMOylation significantly attenuated the aggressive effects mediated by USP1. In conclusion, this study highlights the USP1/PIAS1/HELLS deubiquitinating and SUMOylation axis as a critical driver of aggressiveness and DNA damage repair responses in HCC cells, offering a promising therapeutic strategy to suppress HCC progression and enhance the efficacy of FOLFOX-based chemotherapy.
Similar content being viewed by others


Introduction
Hepatocellular carcinoma (HCC) is the most common form of primary liver cancer, ranking as the seventh most prevalent cancer globally and the second leading cause of cancer-related mortality [1]. Early diagnosis of liver cancer is challenging due to its asymptomatic onset, leading to low rates of surgical resection and necessitating chemotherapy for many patients [2]. Although systemic chemotherapy has limited efficacy, localized treatments, such as transcatheter arterial chemoembolization (TACE) and hepatic arterial infusion chemotherapy (HAIC), remain commonly employed in HCC management. HAIC-FOLFOX, which includes fluorouracil, leucovorin, and oxaliplatin, has shown significant clinical benefits for patients with metastatic HCC [3]. Recent multicenter phase 3 randomized controlled trials (RCTs) indicate that HAIC combined with FOLFOX is more effective and safer than TACE [4]. Fluorouracil (5-FU) is a central component of various arterial chemotherapy regimens [5, 6]. Oxaliplatin, known for its broad-spectrum antitumor activity, lacks cross-resistance with other platinum-based compounds, offering enhanced DNA synthesis inhibition and cytotoxicity compared to cisplatin [7, 8]. Studies have demonstrated that oxaliplatin synergizes with fluorouracil (5-FU) to increase antitumor cytotoxicity [9]. A series of comparative studies has highlighted the efficacy of HAIC-FOLFOX over standard first-line treatments in managing HCC [4, 10, 11]. Compared to doxorubicin, systemic FOLFOX offers a survival advantage as a first-line treatment for advanced HCC, with minimal toxicity [3]. FOLFOX-based HAIC has emerged as a promising conversion strategy for patients with initially unresectable tumors, achieving surgical conversion in approximately 23.8% of such cases [10,11,12]. As a result, FOLFOX has become a leading global regimen for HAIC in liver cancer treatment [13]. However, the chemoresistance inherent in HCC, characterized by multidrug resistance to various anticancer agents, significantly impedes the effectiveness of HAIC-FOLFOX [14]. Therefore, improving chemotherapeutic sensitivity in HCC patients undergoing HAIC-FOLFOX treatment is of great significance.
The ubiquitin-proteasome system (UPS) regulates protein homeostasis by controlling protein quality. Ubiquitination involves the attachment of one or more ubiquitins to substrates, a process catalyzed by a series of enzymes: ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes (E2), and ubiquitin ligases (E3). Deubiquitination, in contrast, is the reverse process, where deubiquitinating enzymes (DUBs) remove ubiquitin from substrates. Deregulation of DUBs can contribute to various human diseases, including cancer. DUBs are classified into seven families: Ubiquitin-specific proteases (USPs), ovarian tumor proteases (OTUs), Jab1/MPN domain-associated metalloisopeptidases (JAMM/MPM + ), monocyte chemotactic protein-induced proteins (MCPIP), Ub C-terminal hydrolases (UCHs), Machado–Joseph domain proteases (MJDs), and Zinc finger UB-specific proteases (ZUP/ZUFSP). Among these, the USPs family is the most well-characterized, comprising 56 enzymes. Ubiquitin-specific protease 1 (USP1), a member of the USPs family with a highly conserved catalytic domain, plays a role in various biological processes by maintaining protein homeostasis. Recent studies have highlighted the role of USP1 in tumorigenesis and progression [15]. Furthermore, USP1 inhibitors combined with cisplatin have been suggested to enhance tumor growth inhibition [16, 17]. Our previous data suggest that USP1 is a potential therapeutic target for HCC due to its pivotal role in modulating mitochondrial fission and metabolic reprogramming [18]. However, the mechanisms underlying its role in tumor aggressiveness and FOLFOX therapy efficacy remain largely unclear.
In this study, we examined the expression profiles and clinical significance of USP1 in HCC and uncovered its dual role in regulating tumor progression and chemoresistance. Loss-of-function and gain-of-function assays were performed to explore the effects of USP1 on malignant behaviors, including proliferation, migration, and EMT, as well as its impact on chemoresistance to FOLFOX-based treatment. Mechanistically, mass spectrometry and molecular validations identified USP1 as a key regulator of HELLS stability through both deubiquitination and SUMOylation. We also assessed the therapeutic potential of combining the USP1-specific small molecule inhibitor ML323 with chemotherapy agents, both in vitro and in vivo, highlighting its ability to sensitize HCC cells to oxaliplatin and 5-FU. These findings offer mechanistic insights into the USP1/HELLS axis as a driver of HCC progression and chemoresistance, presenting a novel therapeutic strategy to enhance the efficacy of HAIC-FOLFOX-based chemotherapy in HCC patients.
Results
Identifying key DUBs in multiple HCC cohorts
To systematically investigate the expression patterns of DUBs, we included 88 HCC cohorts, comprising TCGA-LIHC, ICGC-LIRI-JP, CNHPP, and 85 GSE datasets. As demonstrated in Fig. 1A, a large number of DUBs exhibited differential expression in HCC tissues compared to non-tumor tissues. Thirteen DUBs exhibited a positive ratio exceeding 70% across the analyzed cohorts, including USP1, USP3, USP5, USP7, USP11, USP13, USP14, USP21, USP27X, USP34, USP36, USP39, and USP46. After evaluating their prognostic significance for overall survival, only USP1 and USP13 showed a statistically significant correlation with poor survival outcomes in the TCGA-LIHC and ICGC-LIRI-JP cohorts (Fig. 1B; Fig. S1A, B). We then investigated their associations with clinical parameters. Analysis of TCGA and TNM-plot datasets revealed that USP1 levels varied significantly between advanced/early pathological stages, histological grades, and metastatic/non-metastatic statuses (Fig. 1C, D). However, the differences associated with USP13 were not statistically significant. Additionally, analysis of single-cell sequencing dataset GSE149614 showed that USP1 was mainly enriched in HCC cells and samples at advanced stages, whereas USP13 exhibited no significant differences among cell types and stages (Fig. 1E). Therefore, our evaluation focused primarily on USP1. USP1 exhibited a significantly higher correlation with the proliferative marker MKI67 in HCC tissues compared to adjacent non-tumor liver tissues (Fig. 1F). Furthermore, according to the analysis in spatial transcriptomics dataset [19], USP1 expression in malignant region (Mal) was higher than boundary region (Bdy) or non-malignant region (nMal) (Fig. S2A). Spearman correlation analysis indicated the obviously positive correlation of USP1 expression with tumor cells in the spatial Transcriptomics data (Fig. S2B). Additionally, USP1 expression correlated with MKI67 levels in HCC cell lines, as analyzed in the CCLE and LIMORE databases (Fig. S2C). In our local cohort of 115 HCC cases, patients were classified into high and low USP1 expression groups based on the immunohistochemical score (Fig. 1G). As shown in Fig. 1H, high USP1 expression was significantly associated with poor patient survival. Stratification analysis of the TCGA-LIHC cohort consistently highlighted the prognostic value of USP1 across various subgroups at different stages and grades (Fig. S3A, B). These results suggest that USP1 may be a potential target and biomarker for liver cancer patients.

A Expression levels of DUB family members in 88 HCC cohorts. B Identification of DUBs with a high positivity ratio (>70%) and significant prognostic impact in the TCGA and ICGC cohorts. C Expression levels of USP1 and USP13 at different pathological stages and histological grades. D Comparative analysis of USP1 and USP13 expression in metastatic vs. non-metastatic tissues, assessed by TNM-PLOT. E Analysis of USP1 and USP13 expression in different cell type and stage in GSE141914. F Pearson correlation of USP1 with MKI67 in HCC and adjacent non-tumor liver tissues from 92 datasets. G Immunohistochemical staining of USP1 in 115 HCC tissues from the local cohort. H Kaplan-Meier survival analysis comparing high vs. low USP1 expression in HCC patients from the local cohort. *P < 0.05; **P < 0.01; ***P < 0.001; ***P < 0.0001; ns not significant.
USP1 Enhances Invasiveness of HCC Cells
We then performed loss-of-function and gain-of-function assays to evaluate the regulatory role of USP1 in HCC cells (Fig. S4A, B). Treatment with the USP1-specific inhibitor ML323 significantly inhibited HCC cell proliferation and colony formation, while USP1 knockdown using specific shRNAs also impaired cell proliferation (Fig. 2A, B). Additionally, genetic or pharmacologic depletion of USP1 significantly reduced the migration potential of HCC cells (Fig. 2C, D). Consistently, USP1 overexpression significantly enhanced the aggressive behaviors of Huh7 cells (Fig. 2E–G). Epithelial-mesenchymal transition (EMT) is a reversible process in which polarized epithelial cells lose adhesion and polarity, transforming into mesenchymal cells with invasive and migratory capabilities—key steps in cancer cell movement and invasion [20]. As shown in Fig. 2H, HCC cases with elevated USP1 expression had higher EMT scores. Additionally, GSEA and correlation analysis suggested the involvement of USP1 in the EMT process. As shown in Fig. 2I, J, immunofluorescence and western blotting assays revealed that USP1 positively modulated Vimentin and N-cadherin expression while negatively regulating E-cadherin expression. These findings suggest that USP1 promotes EMT and facilitates aggressive behaviors in HCC cells.

A Viability measured by CCK-8 in SK-Hep1 under ML323 treatment or shUSP1 knockdown. B Clonogenic proliferation assessed by colony-formation in SK-Hep1 under the indicated conditions. C Migration evaluated by Transwell in SK-Hep1 after ML323 or shUSP1. D Wound-healing migration in SK-Hep1 with shUSP1 and in Huh7 with USP1 overexpression. E Viability of Huh7 assessed by CCK-8 following USP1 overexpression. F Effects of USP1 overexpression on Huh7 colony formation. G Transwell migration of Huh7 with USP1 overexpression. H The association between USP1 profiles and EMT characteristics was evaluated using bioinformatics. EMT pathway scores were compared between high and low USP1 expression groups in the TCGA LIHC dataset. GSEA was performed to identify USP1-related pathways, and the correlation between USP1 and EMT-related markers in the TCGA dataset was analyzed. I Expression levels of EMT-related markers were analyzed by western blotting in HCC cells with USP1 overexpression or depletion. J Detection of EMT markers by immunofluorescence in HCC cells with different treatments. **P < 0.01; ***P < 0.001.
USP1 regulates chemotherapy sensitivity through mediating EMT and HRR in liver cancer
To further elucidate the role of USP1, we investigated its potential regulatory pathways using GSVA. USP1 showed highest correlation with DNA damage and DNA repair in both TCGA_LIHC and ICGC_LIRI datasets, among 14 oncogenic signatures (Fig. 3A; Fig. S5A). Consistently, GSEA and GO/KEGG enrichment analysis in TCGA_LIHC dataset indicated that cases with high USP1 expression were enriched in pathways related to DNA damage response, cellular response to chemical stress, DNA repair, DNA double-strand break repair, and DNA damage checkpoint signaling (Fig. S5B). Dysregulation of DNA damage repair can reduce tumor cell sensitivity to chemotherapeutic drugs [21, 22]. Based on these findings, we assessed the effects of USP1 depletion on treatment with oxaliplatin and 5-fluorouracil, the core drugs in HAIC-FOLFOX [13]. We then established baseline chemosensitivity using clonogenic assays. Huh7 showed marked sensitivity to oxaliplatin and 5-FU, whereas SK-Hep1 was relatively resistant (Fig. S6). ML323 potentiated oxaliplatin- and 5-FU–induced growth suppression in SK-Hep1, reducing CCK-8 viability and clonogenic survival compared with chemotherapy alone. By contrast, enforced USP1 expression in Huh7 attenuated the inhibitory effects of both drugs, increasing residual viability and colony number relative to vector controls (Fig. 3B, C). Consistent with our viability and clonogenic data, combining ML323 with oxaliplatin or 5-FU markedly increased the DNA-damage marker γ-H2AX (Fig. 3D, E). Conversely, USP1 overexpression reduced chemotherapy-induced γ-H2AX foci in Huh7 cells, indicating enhanced damage tolerance (Fig. S7A). Using the comet assay, we showed that combining ML323 with OXA or 5-FU significantly exacerbated DNA damage in HCC cells (Fig. 3F). The homologous recombination repair (HRR) pathway in double-strand break (DSB) repair is a complex process involving several proteins. A high frequency of HRR defects in tumors may underlie the effectiveness of cytotoxic therapy. Tumors with HRR defects exhibit high sensitivity to cross-linking agents, including cisplatin, carboplatin, and nitrosoureas, as well as ionizing radiation (IR) and topoisomerase I poison-induced DSBs. We further investigated the relationship between USP1 and the key HRR factor RAD51. Immunofluorescence experiments were performed to observe RAD51 recruitment after treatment with ML323 and chemotherapeutic agents. As shown in Fig. 3G, combining chemotherapeutic agents with ML323 significantly reduced RAD51 recruitment in the nuclei of HCC cells. However, USP1 overexpression increased RAD51 recruitment and partially reversed this reduction (Fig. S7B). Consistently, USP1 knockdown significantly reduced RAD51 protein expression, while ectopic USP1 upregulated RAD51 expression in HCC cells (Fig. 3H). These data suggest that USP1 depletion could enhance the therapeutic efficacy of HAIC-FOLFOX-related chemotherapeutic drugs by modulating HRR in HCC cells.

A GSVA was conducted to evaluate the correlation of USP1 expression with 14 types of oncogenic signatures in TCGA_LIHC dataset. B Viability of cells with USP1 depletion or overexpression treated with oxaliplatin (OXA), fluorouracil (5-FU) was assessed using the CCK-8 assay. C Colony formation assay was performed to validate the effects of indicated treatments on HCC cells. D Immunofluorescence assessment of γ-H2AX expression in different treatment groups, with quantitative data on the average number of foci per cell. E Western blot analysis of γ-H2AX levels in treatment groups. F Cells were harvested post-treatment for comet assay to evaluate DNA damage. Analysis includes quantification of the tail moment (n = 60). G Immunofluorescence analysis of RAD51 expression in different treatment groups, with quantitative data on the average number of foci per cell. H Western blotting to assess RAD51 protein levels in treated cells. **P < 0.01; ***, P < 0.001.
USP1 interacts with and co-localizes with HELLS
USP1, a protease that regulates ubiquitination by removing ubiquitin from substrates or cleaving ubiquitin chains, may mediate phenotypes by deubiquitinating specific functional proteins. 4D Label-free LC–MS/MS was used to identify differentially expressed proteins following ML323 treatment (Fig. 4A). Statistically significant proteins were enriched in pathways related to chemical response, toxicity, cellular response to chemical stress, DNA repair, G2/M DNA damage checkpoint, double-strand break repair, and DNA mismatch repair (Fig. 4B). HELLS was identified as the most downregulated protein induced by ML323 treatment, showing a 26.88-fold decrease (Fig. 4C). Similar to USP1, HELLS expression was significantly elevated in HCC tissues, particularly in cases at advanced stages or grades (Fig. 4D, E, Fig. S8A). High expression of HELLS was correlated with poor survival in HCC patients, with excellent prognostic performance confirmed by ROC analysis (Fig. S8B, C). Additionally, HELLS exhibited higher expression in malignant region than boundary region or non-malignant region (Fig. S9A), showing positive correlation with tumor cells in the spatial Transcriptomics data (Fig. S9B). Previous studies have suggested that HELLS regulates the expression of various tumor-related genes through epigenetic silencing, participating in EMT and HRR [23,24,25]. Furthermore, downregulation of HELLS increases pancreatic cancer sensitivity to platinum-based drugs [26]. We further validated whether HELLS is the key substrate contributing to USP1-mediated EMT and chemotherapy resistance. As shown in Fig. 4F, ML323 administration or shRNA treatment significantly downregulated HELLS protein expression, while ectopic USP1 induced HELLS expression. However, modulation of USP1 had less effect on HELLS at mRNA levels (Fig. 4G). Additionally, molecular docking analysis predicted the interaction between USP1 and HELLS (Fig. S9C). Consistently, immunofluorescence analysis revealed the co-localization of these proteins in HCC cells (Fig. 4H). The endogenous co-immunoprecipitation (Co-IP) analysis confirmed the interaction between the two proteins (Fig. 4I). To further determine the binding domain between USP1 and HELLS, we engineered three truncated USP1 mutants (UTM1, UTM2, UTM3) with FLAG-tags at their C-terminal and transfected HEK293T cells with these mutants and wild-type USP1 for 48 h (Fig. 4J). Co-IP assays revealed that wild-type USP1, UTM2, and UTM3, but not UTM1, interacted with HELLS, indicating that the C-terminal (401–785 aa) of USP1 is responsible for this interaction (Fig. 4K).

A Flowchart of 4D Label-free LC–MS/MS analysis. B Enrichment analysis of differentially expressed proteins identified by mass spectrometry. C Quantitative expression values of HELLS detected by mass spectrometry. D mRNA expression levels of HELLS in paired or unpaired normal/HCC tissues from the TCGA database. E Analysis of HELLS expression at different histologic grades and pathologic stages in HCC cases from the TCGA database. F, G Western blotting and RT-qPCR were performed to detect HELLS expression at the protein and mRNA levels, respectively. H USP1 and HELLS co-localization was validated by immunofluorescence. I Co-immunoprecipitation (Co-IP) assay was conducted to validate the USP1-HELLS interaction. J Structural models of wild-type USP1 (USP1-WT) and truncated USP1 mutants (USP1-UTMs). K Immunoprecipitation and immunoblotting were performed to detect the interaction between FLAG-USP1-WT and FLAG-USP1-UTMs in HEK293T cells. **P < 0.01; ***P < 0.001; ns not significant.
USP1 stabilizes HELLS protein expression via deubiquitination
Previous data indicated that USP1 interacted with HELLS and regulates its protein expression. As a deubiquitinase, USP1 is hypothesized to regulate HELLS protein levels through deubiquitination. Inhibiting USP1 activity or downregulating USP1 using ML323 or shRNAs significantly shortened the half-life of HELLS (Fig. 5A, B). Compared to the control group, USP1 significantly stabilized HELLS expression, as evidenced by reduced degradation in USP1-overexpressing cells (Fig. 5C). To determine whether USP1 stabilizes HELLS in a proteasome-dependent manner, we treated USP1-depleted cells with the proteasome inhibitor MG132. As demonstrated in Fig. 5D, E, MG132 reversed the inhibitory effects of ML323 or shRNAs on HELLS expression. In contrast, wild-type USP1, unlike the catalytically inactive mutant form (C90S), increased HELLS expression in Huh7 cells (Fig. 5F). We then examined the role of USP1 in modulating the ubiquitination levels of HELLS. Genetic or pharmacological depletion of USP1 significantly increased HELLS ubiquitination levels (Fig. 5G, H). USP1-WT, but not USP1-C90S, significantly downregulated HELLS ubiquitination in Huh7 cells (Fig. 5I, J). Different types of ubiquitin linkages confer distinct functions. Ubiquitination at K48 linkages is primarily associated with proteasome-mediated degradation, while K63 linkages are often linked to protein stability and signal transduction. We further investigated the type of polyubiquitination regulated by USP1 in HELLS. As shown in Fig. 5K, L, compared to K63, ML323 enhanced K48-linked ubiquitination of HELLS in HCC cells, while genetic depletion of USP1 had no significant effect on HELLS in K48-mutant HCC cells. These results suggest that USP1 stabilizes HELLS by inhibiting Lys48-linked polyubiquitination.

A–C Expression levels of HELLS were detected in cells exposed to cycloheximide for varying durations with different pre-treatments, assessed by Western blotting. D, E HELLS levels were analyzed in cells with USP1 silencing or ML323 treatment, followed by MG132 treatment, assessed by Western blotting. F HELLS levels were assessed by Western blotting in Huh7 cells transfected with wild-type USP1 plasmids or the catalytically inactive mutant form of USP1 (C90S) plasmids. G–J The effects of USP1 on HELLS ubiquitination levels were assessed by co-immunoprecipitation (Co-IP). K Co-IP analysis of HELLS ubiquitination in HEK293T cells transfected with HA-Ub, HA-Ub-K48, or HA-Ub-K63 plasmids. L HELLS protein levels were detected by Western blotting in HCC cells subjected to the indicated treatments. ***P < 0.001.
USP1 mediates EMT and DNA damage repair response by stabilizing HELLS protein abundance
The results above suggest that USP1 deubiquitinates and regulates HELLS expression. We further investigated whether USP1 regulates EMT and HRR by modulating HELLS expression in HCC cells. As shown in Fig. 6A, B, genetic or pharmacological depletion of USP1 negatively affected EMT marker expression, while overexpression of HELLS reversed these effects. Consistently, ectopic expression of HELLS restored the reduced immunofluorescence intensity of Vimentin, N-cadherin, and E-cadherin, as well as the diminished migration capacity caused by USP1 silencing or depletion (Fig. 6C–E). As indicated in Fig. 6F, G, the increase in γ-H2AX and decrease in RAD51 caused by ML323 or USP1 knockdown, when combined with chemotherapy drugs, was reversed by overexpressing HELLS. The comet assay confirmed that HELLS overexpression mitigated the DNA damage response induced by combining USP1 with OXA or 5-FU (Fig. 6H). Consistent with these findings, immunofluorescence observations of γ-H2AX and Rad51 foci further confirmed the modulatory roles of HELLS in USP1-mediated HRR in HCC undergoing chemotherapy (Fig. 6I, J). Previous studies suggest that USP1 enhances DNA damage repair in tumor cells by modulating the ubiquitination levels of FANCD2. We further investigated whether HELLS is involved in the USP1/FANCD2 axis, regulating the DNA damage response in HCC cells. As shown in Fig. S10A, B, overexpression of HELLS had no effect on FANCD2 downregulation induced by ML323 or USP1 knockdown. Our data demonstrate the critical role of HELLS in USP1-regulated EMT and DNA damage repair responses in HCC cells undergoing chemotherapy.

A–C Expression of HELLS, E-cadherin, N-cadherin, and Vimentin with different treatments was assessed by Western blotting and immunofluorescence. D, E Migration capacity was evaluated by Transwell assays. F, G Expression of γ-H2AX and RAD51 in cells with indicated treatments was analyzed by Western blotting. H Cells were harvested for comet assay following various treatments, with analysis including quantification of the tail moment (n = 60). I, J Expression of γ-H2AX and RAD51 in cells with specified treatments was further analyzed by immunofluorescent staining. **P < 0.01; ***P < 0.001.
USP1 promotes HELLS SUMOylation by stabilizing PIAS1 through deubiquitination
Previous data suggest that SUMOylation enhances the activity and localization of substrates. In HEK-293T cells, Co-IP assays showed that HELLS interacted with SUMO1, SUMO2, and SUMO3, with SUMO1 exhibiting the strongest interaction (Fig. 7A). Compared to the control group, USP1 significantly increased HELLS SUMOylation in both HEK-293T and Huh7 cells (Fig. 7B). Furthermore, we confirmed that USP1 enhanced SUMO1-mediated HELLS SUMOylation in Huh7 cells (Fig. 7C). These results suggest that USP1 regulates HELLS SUMOylation. To further elucidate the mechanism through which USP1 promotes HELLS SUMOylation, we analyzed the mass spectrometry data. Among the potential differential proteins, PIAS1, an E3 SUMO ligase, was significantly downregulated following ML323 treatment. Bioinformatics analyses indicated that PIAS1 was upregulated in HCC tissues with poor survival (Fig. S11A, B). As a deubiquitinase, USP1 is hypothesized to enhance HELLS SUMOylation by stabilizing PIAS1. We then investigated the role of USP1 in stabilizing PIAS1. Western blotting revealed that USP1 expression positively correlated with PIAS1 protein levels (Fig. 7D), whereas USP1 knockdown using shRNAs significantly decreased PIAS1 expression (Fig. 7E). Additionally, treatment with the proteasome inhibitor MG132 reversed the effects of USP1 depletion on PIAS1 levels, while the autophagy inhibitor chloroquine (CQ) had no effect (Fig. 7F). Cycloheximide chase assays showed accelerated PIAS1 degradation after USP1 knockdown and increased PIAS1 stability with USP1 overexpression (Fig. 7G). These findings indicate that USP1 stabilizes PIAS1 by inhibiting its proteasomal degradation. To determine whether USP1 regulated PIAS1 stability through deubiquitination, Co-IP assays showed that USP1 could directly interact with PIAS1 (Fig. 7H). Genetic depletion of USP1 increased PIAS1 ubiquitination levels, while overexpression of USP1 significantly reduced PIAS1 ubiquitination levels (Fig. 7I). These results demonstrate that USP1 stabilizes PIAS1 by removing ubiquitin chains, inhibiting its proteasomal degradation. Finally, we examined the functional consequences of USP1-mediated HELLS SUMOylation in liver cancer cells. USP1 overexpression promoted EMT, cell proliferation, and migration of HCC cells. However, treatment with the SUMOylation inhibitor TAK981 or PIAS1 knockdown significantly attenuated these effects, suggesting that the effects of USP1 were partially dependent on PIAS1-mediated SUMOylation (Fig. 7J–L). These results demonstrate that USP1 enhances HELLS SUMOylation by stabilizing PIAS1 through deubiquitination.

A–C HELLS SUMOylation (SUMO1/2/3) in HEK293T and Huh7 cells assessed by Co-IP. D, E Western blotting was performed to examine PIAS1 expression with USP1 knockdown or overexpression. F Western blotting was conducted to detect PIAS1 expression after USP1 knockdown or ML323 treatment with administration of MG132 or chloroquine (CQ). G Expression levels of PIAS1 were detected in cells exposed to cycloheximide for varying durations with different pre-treatments, assessed by Western blotting. H Co-IP assay was conducted to validate the USP1-PIAS1 interaction. I The effects of USP1 on PIAS1 ubiquitination levels were assessed by Co-IP in Huh7 and SK-Hep1 cells. J Expression levels of EMT-related markers were analyzed by western blotting in HCC cells with indicated treatments. K, L Transwell and colony formation assay was performed to validate the rescue effects of indicated treatments. ***P < 0.001; ****P < 0.0001.
The therapeutic efficiency in combing chemotherapy with ML323 and the clinical implications of USP1/HELLS axis
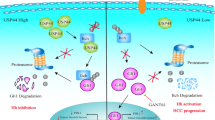
Based on the chemo-sensitizing effects of USP1 depletion observed in vitro, we used a xenograft tumor model in nude mice to investigate the combined anticancer potential of ML323 with OXA or 5-FU in vivo (Fig. 8A). As demonstrated in Fig. 8B, C, single administration of OXA or 5-FU reduced xenograft tumor volume compared to the control group. Interestingly, combining chemotherapy agents with ML323 further impeded tumor growth, enhancing the therapeutic effects of these drugs. Immunohistochemical staining showed that ML323 administration downregulated the expression of Ki67, RAD51, N-cadherin, HELLS, and vimentin, while increasing E-cadherin and γ-H2AX expression (Fig. 8D). Additionally, in the TCGA-LIHC and GSE27150 datasets, HCC patients with high expression of both USP1 and HELLS exhibited the worst overall survival (Fig. 8E). Our data suggest that USP1 stabilizes HELLS through the deubiquitination pathway, thereby promoting EMT and HRR-mediated chemotherapy resistance in HCC (Fig. 8F).

A Flowchart of the xenograft tumor model and drug administration. HCC cells were injected subcutaneously into nude mice (n = 5/group). Treatments included oxaliplatin (OXA, 5 mg/kg), fluorouracil (5-FU, 20 mg/kg), PBS, and combinations with ML323 (4 mg/kg). B Representative images of xenograft tumors after various treatments. C Volume and weight measurements of xenograft tumors in different treatment groups. D Representative immunohistochemical staining results for Ki67, E-cadherin, N-cadherin, Vimentin, RAD51, HELLS, and γ-H2AX in xenograft tissues. E Survival curves for patients in different subgroups stratified by USP1/HELLS expression levels, using data from the TCGA-LIHC and GSE14520 datasets. F Graphical summary of the molecular mechanisms by which the USP1/HELLS signaling axis regulates chemotherapy sensitivity in HCC. **P < 0.01, ***P < 0.001.
Discussion
Hepatocellular carcinoma often presents insidiously, resulting in low rates of early diagnosis and surgical resection. For advanced-stage HCC, localized therapies such as TACE and HAIC are commonly employed [27]. HAIC is widely used in Asia and endorsed by several regional clinical guidelines for treating intermediate to advanced HCC [28,29,30]. Oxaliplatin, a platinum-based chemotherapeutic agent with broad-spectrum antitumor activity, shows no cross-resistance with other platinum drugs. Its combination with 5-FU produces synergistic cytotoxic effects in tumor cells [7,8,9]. These findings have established the FOLFOX regimen, which includes fluorouracil, calcium folinate, and oxaliplatin, as a leading protocol for HCC treatment via HAIC globally [13]. The efficacy and safety of HAIC-FOLFOX in advanced HCC are well-established, offering a promising conversion strategy for initially unresectable cases to become amenable to curative surgery [12]. However, chemoresistance remains a significant obstacle to HAIC-FOLFOX success, severely limiting response rates and contributing to treatment failure in intermediate to advanced HCC.
The pivotal role of USP1 in tumorigenesis and cancer progression is well-documented, highlighting its potential as a therapeutic target [15]. Our study showed that elevated USP1 expression was significantly associated with poor survival outcomes in HCC patients. Furthermore, gain-of-function and loss-of-function assays showed that USP1 facilitated the aggressiveness of HCC cells, characterized by enhanced proliferative and migratory capacities. EMT, a highly conserved developmental process, plays a crucial role in driving tumor metastasis and therapy resistance [31, 32], both major contributors to cancer-related mortality [33]. Furthermore, USP1 modulates the expression of EMT markers, suggesting its regulatory role in the EMT process in HCC cells. Previous studies suggest that silencing USP1, combined with chemotherapy, significantly enhances drug sensitivity in cancer cells [16, 17]. We further evaluated the role of USP1 in regulating chemotherapy sensitivity and aggressiveness in HCC cells. Genetic downregulation or pharmacological inactivation of USP1 significantly increased HCC cell sensitivity to oxaliplatin and 5-FU, the core agents of the FOLFOX regimen. USP1 depletion also attenuated cell proliferation and migration, highlighting its role in promoting aggressive cancer phenotypes. Mechanistically, USP1 depletion reduced RAD51 expression, suggesting that downregulation of USP1 inhibited HRR post-chemotherapy, enhancing sensitivity to oxaliplatin and 5-FU. These results establish USP1 as a key modulator of both chemoresistance and cancer aggressiveness in HCC.
To elucidate the underlying mechanisms, we screened and validated USP1 substrates that mediate its pro-tumorigenic effects. HELLS, showing the highest fold change after ML323 treatment, was identified as a potential USP1 substrate through LC-MS/MS analysis. It is a chromatin remodeling factor and member of the SNF2 ATP-dependent chromatin remodeling complex family, which uses ATP hydrolysis to modify nucleosome structure and chromatin packaging. HELLS is implicated in DNA damage repair, genomic stability, and various cancer pathways [34]. Previous studies link HELLS to EMT and HRR in cancer cells [23,24,25]. Additionally, downregulation of HELLS increases pancreatic cancer sensitivity to platinum-based drugs [26]. The direct interaction between USP1 and HELLS was predicted by molecular docking and validated in HCC cells. Furthermore, we confirmed that USP1 inhibits Lys48-linked polyubiquitination of HELLS, stabilizing its protein levels.
Interestingly, our findings show that USP1 mediates a noncanonical SUMOylation process. Unlike the classical SUMOylation pathway, which directly involves SUMO E3 ligases, USP1 indirectly promotes HELLS SUMOylation by stabilizing PIAS1, a critical E3 SUMO ligase, through deubiquitination. This represents a novel mechanism in which a deubiquitinating enzyme controls SUMOylation, bridging the ubiquitination and SUMOylation pathways in a unique regulatory axis. This SUMOylation modification is essential for HELLS-mediated promotion of EMT and HRR. Furthermore, treatment with the SUMOylation inhibitor TAK981 significantly reduced USP1-driven proliferation and aggressiveness, confirming the functional importance of SUMOylation in USP1-mediated malignant phenotypes. Overexpression of HELLS partially rescued the inhibition of EMT and chemotherapy sensitivity induced by USP1 downregulation or inactivation, further confirming HELLS as a key downstream effector of USP1.
Our study clarifies the multifaceted role of USP1 in regulating HCC cell metastasis, proliferation, and chemotherapy sensitivity. By stabilizing HELLS through deubiquitination and SUMOylation, USP1 promotes EMT and HRR, which are critical for cancer cell survival under genotoxic stress. The repair of DSBs through HRR is a key determinant of radiotherapy and chemotherapy outcomes, with elevated HRR capacity contributing to resistance to agents such as ionizing radiation, cisplatin, and PARP inhibitors [35, 36]. Tumors overexpressing RAD51, a pivotal HRR protein, often exhibit resistance to treatment and poor patient survival [37]. RAD51 overexpression leads to hyper-recombination, facilitating cancer progression and enabling cells to resist DNA-damaging agents [38]. Notably, USP1 depletion reduced RAD51 expression by destabilizing HELLS, reversing HRR activity, and sensitizing HCC cells to FOLFOX components.
Emerging inhibitors targeting RAD51, such as CYT-0851, show promising therapeutic potential for advanced cancers, including solid tumors and hematological malignancies [39]. Similarly, targeting USP1 offers dual benefits by disrupting HELLS-mediated HRR and reversing EMT. This study is the first to report that USP1 regulates EMT and chemotherapy-related HRR by stabilizing HELLS expression and promoting its SUMOylation. These findings provide novel mechanistic insights and therapeutic opportunities, not only for HAIC-FOLFOX-based chemotherapy but also for radiotherapy and other DNA-damage-based treatments. However, our study has limitations, including the need for further validation of the signaling pathways and regulatory interactions in clinical HCC samples, particularly those resistant to chemotherapy. Further studies are required to uncover deeper molecular mechanisms through which USP1 promotes HCC progression via HELLS and to explore the broader implications of SUMOylation in this regulatory axis.
In summary, this study demonstrates that USP1 stabilizes HELLS through dual mechanisms of deubiquitination and SUMOylation, enhancing EMT and DNA damage repair in HCC cells. Targeting USP1, either through small-molecule inhibitors or modulating SUMOylation, offers a promising therapeutic strategy to counteract HCC progression and enhance the efficacy of FOLFOX-based chemotherapy.
Materials and methods
Clinical samples
HCC tissues were obtained from 115 patients who underwent surgery at the Affiliated Hospital of Nantong University (Nantong, Jiangsu, China) between January 2012 and October 2014. This study was approved by the Ethics Committee at the Affiliated Hospital of Nantong University (2023-L038) and conducted in accordance with the principles of the Helsinki Declaration. Informed consent was obtained from all participants in the study.
Cell culture and transfection
SK-Hep1 (#ZQ0030), Huh7 (#ZQ0025), SNU449(ZQ0787) and Hep3B(ZQ0024) cell lines were provided by Zhong Qiao Xin Zhou Biotechnology. Cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM, GIBCO, USA) supplemented with 10% fetal bovine serum (FBS, GIBCO, USA) and 1% penicillin/streptomycin solution, at 37 °C with 5% CO2. Plasmids, including USP1-shRNA-3/PTSB-SH-copGFP-2A-PURO, USP1-shRNA-4/PTSB-SH-copGFP-2A-PURO, OE-USP1/pTSB-CMV-copGFP-F2A-PURO, OE-HELLS/pTSB02-GFP-PURO, Myc-UBC9/pTSBX-CMV-MSC-EF1-copGFP-2A-PURO, His-SUMO1/pTSBX-CMV-MSC-EF1-copGFP-2A-PURO, His-SUMO2/pTSBX-CMV-MSC-EF1-copGFP-2A-PURO, and His-SUMO3/pTSBX-CMV-MSC-EF1-copGFP-2A-PURO were acquired from Transheep (Shanghai, China). Plasmid transfections were performed using Lipofectamine 3000 reagent (Invitrogen, CA, USA) following the manufacturer’s instructions. Cells transfected with plasmids were selected with puromycin (5 μg/mL, Sigma-Aldrich, CA, USA) for 30 days to establish stable transfections.
Cell counting Kit-8 and colony formation assays
Cell growth was assessed using the Cell Counting Kit-8 (CCK-8; Dojindo Laboratories, Kumamoto, Japan) according to the manufacturer’s protocol. Briefly, HCC cells were seeded in 96-well plates at a density of 1 × 10³ cells per well in 100 μL of medium. At specified time points (24, 48, and 72 h), 10 μL of CCK-8 solution was added to each well. After 2 h of incubation at 37 °C, absorbance was measured at 450 nm. Each experiment was performed independently three times. For colony formation assays, HCC cells were seeded at a density of 500 cells/well in six-well plates and incubated for 14 days. Cells were then fixed in 4% paraformaldehyde for 30 min and stained with 0.1% crystal violet.
Migration, invasion, and wound-healing assays
Transwell assays were used to evaluate the effects of ML323 treatment and USP1 knockdown or overexpression on cell migration. These assays were performed using 8-μm pore size Transwell chambers (Corning, Acton, MA, USA). A 200 μL suspension of cells was seeded in the upper chambers, and complete medium was added to the lower chambers. After 48 h of incubation, the cells in the chambers were fixed with paraformaldehyde (PFA) for 40 min at room temperature, stained with crystal violet, and analyzed using ImageJ software. In the wound healing assay, cells were incubated in 6-well plates. A wound was created using a 10 μL pipette tip. The wound size was examined randomly under a microscope.
Xenograft tumor assay
The Animal Care and Use Committee of Nantong University approved this animal study (P20230221-017). Twenty-five male BALB/c nude mice were obtained from the Experimental Animal Center of Nantong University and were randomly allocated to experimental groups. HCC cells (1 × 10⁷ cells in 200 μL of PBS) were subcutaneously injected into the flanks of the mice. Xenograft tumors were monitored every five days until sacrifice, with measurements of volume and weight. The maximal tumour size/burden was not exceeded and permitted by their ethics committee. The investigators were not blinded to the group allocation during experiments.
Western blotting
Proteins were extracted using RIPA buffer supplemented with protease inhibitors. After separation by SDS-PAGE, proteins were transferred to PVDF membranes, blocked in 5% milk for 2 h, and incubated overnight with primary antibodies (anti-USP1, Proteintech, Cat#14346-1-AP; anti-HA-tag, Proteintech, Cat#66006-2-Ig; anti-Flag tag, Abclonal, Cat#AE005; anti-β-actin, Proteintech, Cat#66009-1-Ig; anti-HELLS, Proteintech, Cat#11955-1-AP; anti-RAD51, Abcam, ab133534; E-cadherin, Proteintech, Cat# 20874-1-AP; N-cadherin, Proteintech, Cat# 22018-1-AP; Vimentin, Proteintech, Cat# 10366-1-AP; γ-H2AX, Santa Cruz Biotechnology, Cat#sc-517336) at 4 °C. This was followed by a 2 h incubation with HRP-conjugated secondary antibodies (Jackson, Cat#111-035-003, 115-035-003) at room temperature. Membranes were visualized using an Enhanced Chemiluminescence (ECL) kit (Millipore, MA, USA).
Immunohistochemistry assay
Human liver tissues and xenograft samples were subjected to standard procedures for fixation, embedding, and sectioning. Immunohistochemistry was performed using the MaxVision Kit (Maixin Biol), according to the manufacturer’s guidelines. The primary antibodies used were anti-Ki67, anti-N-cadherin, anti-E-cadherin, anti-Rad51, and anti-Vimentin. ImageJ software was used for image analysis and quantification. Each assay was conducted at least three times to ensure reliability.
Immunofluorescence (IF) assay
Cells were fixed and permeabilized using 4% formaldehyde and 0.25% Triton X-100. The cells were then blocked with 1% bovine serum albumin (BSA) for 1 h at room temperature. This was followed by a 12 h incubation with primary antibodies diluted in 1% BSA. After washing, the cells were incubated with fluorochrome-labeled secondary antibodies (ABclonal Technologies) and DAPI (CST). Finally, the cells were examined under a fluorescence microscope.
Immunoprecipitation and protein stability assay
Cell lysates were extracted using RIPA buffer for 30 min on ice and incubated overnight at 4 °C with a primary antibody against HELLS (11955-1-AP, ProteinTech). Samples were incubated with anti-rabbit Ig-IP beads (Rockland Immunochemicals, USA) for 3 h at 4 °C. Following centrifugation and PBS washing, proteins were immunoblotted after boiling in loading buffer (Bio-Rad) for 10 min. For protein half-life assays, cells were treated with 100 μg/mL CHX (Sigma, USA) and harvested at specified times for immunoblotting (IB) to detect CHX-induced protein degradation.
4D label-free LC–MS/MS
For the proteomics assays, SK-Hep1 cells were treated with ML323 or DMSO as indicated (3 vs. 3). Cells in each group were lysed in SDT buffer, heated in a boiling water bath for 15 min, and clarified by centrifugation at 14,000 g for 15 min. Protein concentration in the supernatant was measured by BCA and then stored at −80 °C. For quality control, 20 µg protein per sample was mixed with 6× loading buffer, boiled for 5 min, resolved on 12% SDS–PAGE at 250 V for 40 min, and stained with Coomassie Blue. Proteins were digested according to the (Filter-aided sample preparation) FASP workflow [40]. Briefly, 50–200 µg protein per sample was reduced with 100 mM DTT for 5 min at 100 °C, then transferred to 30 kDa MWCO ultrafiltration units (Sartorius) for FASP. Detergent, DTT, and other low-MW components were removed by repeated washes with UA buffer (8 M urea, 150 mM Tris-HCl, pH 8.5). Reduced cysteines were alkylated with 100 mM iodoacetamide in UA for 30 min in the dark. Filters were washed 3 times with UA and 2 times with 50 mM NH₄HCO₃, then proteins were digested overnight at 37 °C with 4 µg sequencing-grade trypsin in 40 µL 50 mM NH₄HCO₃. Peptides were collected by centrifugation, desalted on C18, and quantified by A₂₈₀ (extinction coefficient ε = 1.1 for a 0.1% [g/100 mL] solution, based on Trp/Tyr content).
Then the peptides were analyzed by ion-mobility–enabled 4D label-free LC–MS/MS at Shanghai GeneChem. Peptides were analyzed on a nanoElute UHPLC (Bruker) coupled to a timsTOF Pro with a CaptiveSpray source. Separation used a 25 cm × 75 µm IonOpticks column packed with 1.6 µm C18 with an integrated emitter and held at 50 °C in a column oven. The column was equilibrated with four column volumes, samples were loaded in 100% buffer A at 800 bar, and elution proceeded at 300 nL min⁻¹. Buffer A was 0.1% formic acid in water and buffer B was 0.1% formic acid in acetonitrile. The 90 min gradient was 2–22% B for 75 min, 22–37% B for 5 min, 37–80% B for 5 min, then hold at 80% B for 5 min. The timsTOF Pro ran in PASEF mode with mass range m/z 100–1700, ion mobility 1/K₀ from 0.75 to 1.40 V·s cm⁻², ramp time 100 ms, duty cycle locked at 100%, capillary voltage 1.5 kV, dry gas 3 L min⁻¹, and dry temperature 180 °C. PASEF acquisition used 10 MS/MS scans per cycle with total cycle time 1.16 s, charge range 0–5, active exclusion 0.5 min, scheduling target intensity 10,000, intensity threshold 2500, and CID collision energy 20–59 eV. Raw files were processed in MaxQuant v1.6.17.0 using Andromeda and searched against the UniProt human reference proteome selected for this project. The database search used trypsin/P specificity with up to two missed cleavages, a precursor mass tolerance of 10 ppm, and a fragment ion tolerance of 40 ppm. Carbamidomethylation of cysteine was set as a fixed modification, while protein N-terminal acetylation and methionine oxidation were set as variable modifications. Peptide spectrum matches and protein groups were controlled at a 1% false discovery rate using a target–decoy strategy. Label free quantification was enabled and protein abundance was taken from LFQ intensities. LFQ values were log2 transformed and median normalized across runs. Differential abundance between ML323 and DMSO was assessed with two sided Student t tests on log2 intensities. Proteins were considered differentially expressed when the absolute fold change was at least 2 and the p-value was less than 0.05. Pathway analysis used GSEA on all detected proteins with the signed t statistic as the ranking metric and GO, Reactome, and BIOCARTA gene-set collections. Enrichment results are shown as a ridgeline plot of the running enrichment score with NES and FDR annotations and as a bubble plot in which the x-axis is the enrichment score, color encodes NES, and point size reflects gene-set size.
Bioinformatics analysis in public datasets
Expression profiles for DUB genes were aggregated from 83 public databases. For the TCGA-LIHC analysis, all computations and plots were performed in R 4.2.1. Raw gene-level count matrices and clinical annotations were downloaded from TCGA after filtering low-abundance genes. Tumors were dichotomized into USP1-high and USP1-low groups based on the top and bottom 50% of USP1 expression. Differential expression was computed with “DESeq2” using the Wald test with Benjamini–Hochberg FDR correction. Variance-stabilizing transformation (VST) from “DESeq2” was used for visualization and correlation analyses. For pathway analysis, we ran pre-ranked GSEA in “clusterProfiler” using the “DESeq2” Wald statistic as the ranking metric, with gene sets obtained from MSigDB via “msigdbr” including Hallmark, GO Biological Process, and KEGG collections. Gene-set size was set to 15–500, permutations to 1000, and significance to FDR q < 0.05. Over-representation analyses of differentially expressed genes used “clusterProfiler” functions enrichGO and enrichKEGG with the same multiple-testing control. Time-dependent ROC curves were computed with “timeROC” and plotted using “ggplot2”. Survival analyses used “survival” and “survminer” for Kaplan–Meier curves, log-rank tests, and Cox models.
Comet assays
Comet analysis was performed using a comet assay kit (Beyotime, C2041S) following the manufacturer’s protocol. Lysates were prepared and cooled at 4 °C for at least 20 min prior to use. Pre-warmed 30 μL of 1% normal melting point agarose was spread on comet electrophoresis slides, covered with a coverslip, and left at 4 °C for 10 min to solidify before removing the coverslip. 0.7% low melting point agarose was melted in a boiling water bath for 5 min, then cooled for at least 20 min in a 37 °C water bath. 10 μL of cells (approximately 10⁴) were thoroughly mixed with the 0.7% low melting point agarose and rapidly pipetted as a 70 μL drop onto the first gel layer. Sections were placed in a refrigerator at 4 °C for 10 min, then soaked in lysate at 4 °C for 1 h, followed by immersion in alkaline electrophoresis solution for 30 min. Slides were electrophoresed at room temperature for 30 min at 25 V and neutralized with neutral buffer at 4 °C for 10 min. Samples were dried at 37 °C for 10 min, DAPI stained for 10 min, and images captured under a fluorescence microscope. Tail moments were analyzed using the Comet Analysis Software Project (CASP).
RT-qPCR
Total RNA was isolated using TRIzol reagent (Thermo Fisher Scientific, USA). cDNA synthesis was performed using the ReverTra Ace qPCR RT Kit (Toyobo, Japan). RT-qPCR was performed using the Fast SYBR Green Master Mix (Applied Biosystems, MA, USA) according to the manufacturer’s instructions. PCR conditions were as follows: initial polymerase activation at 95 °C for 30 s, followed by 40 cycles of denaturation at 95 °C for 5 s and annealing at 60 °C for 30 s. β-Actin was used as the internal control. Relative expression of the target gene was quantified using the 2-ΔΔCT method.
Statistical analysis
Statistical analyses were performed using IBM SPSS 26 and GraphPad Prism 10.1 (CA, USA). All data were from at least three replicate experiments. The χ² test and Student’s t-test were used to compare two groups, while ANOVA was employed for multiple group comparisons. P-values less than 0.05 were considered statistically significant.
Data availability
The dataset(s) supporting the findings of this study are included within the article.
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71:209–49.
Ikeda M, Morizane C, Ueno M, Okusaka T, Ishii H, Furuse J. Chemotherapy for hepatocellular carcinoma: current status and future perspectives. Jpn J Clin Oncol. 2018;48:103–14.
Qin S, Bai Y, Lim HY, Thongprasert S, Chao Y, Fan J, et al. Randomized, multicenter, open-label study of oxaliplatin plus fluorouracil/leucovorin versus doxorubicin as palliative chemotherapy in patients with advanced hepatocellular carcinoma from Asia. J Clin Oncol. 2013;31:3501–8.
Li Q-J, He M-K, Chen H-W, Fang W-Q, Zhou Y-M, Xu L, et al. Hepatic arterial infusion of oxaliplatin, fluorouracil, and leucovorin versus transarterial chemoembolization for large hepatocellular carcinoma: A randomized phase III trial. J Clin Oncol. 2022;40:150–60.
Ikeda M, Okusaka T, Ueno H, Takezako Y, Morizane C. A phase II trial of continuous infusion of 5-fluorouracil, mitoxantrone, and cisplatin for metastatic hepatocellular carcinoma. Cancer. 2005;103:756–62.
Yeo W, Mok TS, Zee B, Leung TWT, Lai PBS, Lau WY, et al. A randomized phase III study of doxorubicin versus cisplatin/interferon alpha-2b/doxorubicin/fluorouracil (PIAF) combination chemotherapy for unresectable hepatocellular carcinoma. J Natl Cancer Inst. 2005;97:1532–8.
Rottenberg S, Disler C, Perego P. The rediscovery of platinum-based cancer therapy. Nat Rev Cancer. 2021;21:37–50.
Raymond E, Chaney SG, Taamma A, Cvitkovic E. Oxaliplatin: a review of preclinical and clinical studies. Ann Oncol. 1998;9:1053–71.
Theile D, Grebhardt S, Haefeli WE, Weiss J. Involvement of drug transporters in the synergistic action of FOLFOX combination chemotherapy. Biochem Pharm. 2009;78:1366–73.
He M, Li Q, Zou R, Shen J, Fang W, Tan G, et al. Sorafenib plus hepatic arterial infusion of oxaliplatin, fluorouracil, and leucovorin vs sorafenib alone for hepatocellular carcinoma with portal vein invasion: A randomized clinical trial. JAMA Oncol. 2019;5:953–60.
Lyu N, Wang X, Li J-B, Lai J-F, Chen Q-F, Li S-L, et al. Arterial chemotherapy of oxaliplatin plus fluorouracil versus sorafenib in advanced hepatocellular carcinoma: A biomolecular exploratory, randomized, phase III trial (FOHAIC-1). J Clin Oncol. 2022;40:468–80.
Wang J, Zheng Z, Wu T, Li W, Wang J, Pan Y., et al. Hepatic arterial infusion chemotherapy as a timing strategy for conversion surgery to treat hepatocellular carcinoma: A single-center real-world study. J Hepatocell Carcinoma. 2022;9:999–1010.
Lyu N, Lin Y, Kong Y, Zhang Z, Liu L, Zheng L, et al. FOXAI: a phase II trial evaluating the efficacy and safety of hepatic arterial infusion of oxaliplatin plus fluorouracil/leucovorin for advanced hepatocellular carcinoma. Gut. 2018;67:395–6.
Pastorelli D, Cartei G, Zustovich F, Marchese F, Artioli G, Zovato S, et al. Gemcitabine and liposomal doxorubicin in biliary and hepatic carcinoma (HCC) chemotherapy: preliminary results and review of the literature. Ann Oncol. 2006;17:v153–7.
Das DS, Das A, Ray A, Song Y, Samur MK, Munshi NC, et al. Blockade of Deubiquitylating Enzyme USP1 Inhibits DNA Repair and Triggers Apoptosis in Multiple Myeloma Cells. Clin Cancer Res. 2017;23:4280–9.
Shang K, Zhang L, Yu Y, Xiao H, Gao Y, Yang L, et al. Disulfide-containing polymer delivery of C527 and a Platinum(IV) prodrug selectively inhibited protein ubiquitination and tumor growth on cisplatin resistant and patient-derived liver cancer models. Mater Today Bio. 2023;18:100548.
Sonego M, Pellarin I, Costa A, Vinciguerra GLR, Coan M, Kraut A, et al. USP1 links platinum resistance to cancer cell dissemination by regulating Snail stability. Sci Adv. 2019;5:eaav3235.
Bian S, Ni W, Zhou L, Tong Y, Dai C, Zhao X, et al. Ubiquitin-specific protease 1 facilitates hepatocellular carcinoma progression by modulating mitochondrial fission and metabolic reprogramming via cyclin-dependent kinase 5 stabilization. Cell Death Differ. 2024;31:1202–18.
Liu Y, Xun Z, Ma K, Liang S, Li X, Zhou S, et al. Identification of a tumour immune barrier in the HCC microenvironment that determines the efficacy of immunotherapy. J Hepatol. 2023;78:770–82.
Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9:265–73.
Buckley AM, Lynam-Lennon N, O’Neill H, O’Sullivan J. Targeting hallmarks of cancer to enhance radiosensitivity in gastrointestinal cancers. Nat Rev Gastroenterol Hepatol. 2020;17:298–313.
Kavanagh JN, Redmond KM, Schettino G, Prise KM. DNA double strand break repair: a radiation perspective. Antioxid Redox Signal. 2013;18:2458–72.
Kollárovič G, Topping CE, Shaw EP, Chambers AL. The human HELLS chromatin remodelling protein promotes end resection to facilitate homologous recombination and contributes to DSB repair within heterochromatin. Nucleic Acids Res. 2020;48:1872–85.
Law C-T, Wei L, Tsang FH-C, Chan CY-K, Xu IM-J, Lai RK-H, et al. HELLS Regulates Chromatin Remodeling and Epigenetic Silencing of Multiple Tumor Suppressor Genes in Human Hepatocellular Carcinoma. Hepatology. 2019;69:2013–30.
He X, Yan B, Liu S, Jia J, Lai W, Xin X, et al. Chromatin Remodeling Factor LSH Drives Cancer Progression by Suppressing the Activity of Fumarate Hydratase. Cancer Res. 2016;76:5743–55.
Hou X, Yang L, Wang K, Zhou Y, Li Q, Kong F, et al. HELLS, a chromatin remodeler is highly expressed in pancreatic cancer and downregulation of it impairs tumor growth and sensitizes to cisplatin by reexpressing the tumor suppressor TGFBR3. Cancer Med. 2021;10:350–64.
Iwamoto H, Shimose S, Shirono T, Niizeki T, Kawaguchi T. Hepatic arterial infusion chemotherapy for advanced hepatocellular carcinoma in the era of chemo-diversity. Clin Mol Hepatol. 2023;29:593–604.
Benson AB, D’Angelica MI, Abbott DE, Anaya DA, Anders R, Are C, et al. Hepatobiliary Cancers, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2021;19:541–65.
Kudo M, Matsui O, Izumi N, Iijima H, Kadoya M, Imai Y, et al. JSH Consensus-Based Clinical Practice Guidelines for the Management of Hepatocellular Carcinoma: 2014 Update by the Liver Cancer Study Group of Japan. Liver Cancer. 2014;3:458–68.
2022 KLCA-NCC Korea practice guidelines for the management of hepatocellular carcinoma. Clin Mol Hepatol. 2022; 28:583–705.
Brabletz T, Kalluri R, Nieto MA, Weinberg RA. EMT in cancer. Nat Rev Cancer. 2018;18:128–34.
Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol. 2017;14:611–29.
Zaorsky NG, Churilla TM, Egleston BL, Fisher SG, Ridge JA, Horwitz EM, et al. Causes of death among cancer patients. Ann Oncol. 2017;28:400–7.
Geiman TM, Tessarollo L, Anver MR, Kopp JB, Ward JM, Muegge K. Lsh, a SNF2 family member, is required for normal murine development. Biochim Biophys Acta. 2001;1526:211–20.
Chen G, Chen J, Qiao Y, Shi Y, Liu W, Zeng Q, et al. ZNF830 mediates cancer chemoresistance through promoting homologous-recombination repair. Nucleic Acids Res. 2018;46:1266–79.
Park S, Lee H, Lee B, Lee S-H, Sun J-M, Park W-Y, et al. DNA Damage Response and Repair Pathway Alteration and Its Association With Tumor Mutation Burden and Platinum-Based Chemotherapy in SCLC. J Thorac Oncol. 2019;14:1640–50.
Ward A, Khanna KK, Wiegmans AP. Targeting homologous recombination, new pre-clinical and clinical therapeutic combinations inhibiting RAD51. Cancer Treat Rev. 2015;41:35–45.
Gachechiladze M, Škarda J, Soltermann A, Joerger M. RAD51 as a potential surrogate marker for DNA repair capacity in solid malignancies. Int J Cancer. 2017;141:1286–94.
Preliminary Activity Seen with RAD51 Inhibitor. Cancer Discov. 2021; 11:OF1.
Wisniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat Methods. 2009;6:359–62.
Funding
This work was supported by grants from National Natural Science Foundation (82272839) and Postgraduate Research & Practice Innovation Program of Jiangsu Province (KYCX24_3605).
Author information
Authors and Affiliations
Contributions
Jie Gao, Nan Bai, Mingyu Liu, and Ninghua Yao contributed equally to this work. Wenjie Zheng, Hui Zhao, and Saiyan Bian conceived the ideas and supervised the research. Jie Gao, Nan Bai, and Mingyu Liu performed in vitro and in vivo experiments. Ninghua Yao, Chengchen Dai, Zhangzhi Tang, Weiting Chen, Xuyang He and Jiayu Shao contributed to the analysis of the data. Jie Gao prepared original draft. Wenjie Zheng revised the manuscript. All authors have read and approved the final version of this paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
This study was approved by the ethical committee of Affiliated Hospital of Nantong University (2023-L038) and was conducted in accordance with the Declaration of Helsinki. Informed consent was obtained from all participants in the study. The Animal Care and Use Committee of Nantong University approved the animal study (P20230221-017).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Gao, J., Bai, N., Liu, M. et al. Dual roles of USP1 in HELLS deubiquitination and SUMOylation drive EMT and FOLFOX-based chemoresistance. Oncogenesis 14, 49 (2025). https://doi.org/10.1038/s41389-025-00592-z
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41389-025-00592-z
