
Abstract
Osteoporosis is a metabolic bone disease characterized by a disruption in the balance between bone resorption and formation. The specific role and mechanism of stomatin (STOM), a major component of lipid rafts, in regulating bone physiology remain unclear. This research identifies increased STOM expression in bone tissue of osteoporosis patients and ovariectomized mice. STOM-deficient mice show higher bone mass under both normal conditions and after ovariectomy. Furthermore, we clarify that STOM is a positive regulator of osteoclast differentiation. Through transcriptomic and bioinformatics approaches, our research reveals that inhibiting STOM increases antioxidant proteins and suppresses ROS-mediated transcriptional pathways in osteoclasts. Mechanistically, STOM interacts with Prdx1, promoting its degradation through the lysosomal pathway and boosting intracellular ROS production, thereby activating osteoclastogenesis. Ultimately, targeted inhibition of macrophage STOM expression in mice alleviates ovariectomized-induced bone loss. Overall, these findings show STOM directly regulates osteoclasts, suggesting that targeting it may modulate pathological bone remodeling.
Similar content being viewed by others


Introduction
Osteoporosis (OP) is a systemic bone disease characterized by low bone mass, destruction of bone microstructure, increased bone fragility, and decreased exercise tolerance1,2. Statistics indicate that the global population affected by OP has reached 250 million, with a projected increase due to aging. Osteoporotic fractures, a severe consequence of OP, are a leading cause of motor dysfunction and disability in the elderly. Approximately 9 million individuals worldwide suffer fragility fractures due to OP annually, with 50% facing the risk of disability3. The current scenario in many countries presents significant challenges in the prevention and treatment of OP, characterized by high prevalence and low treatment rates4,5. While the etiology and pathogenesis of OP remain a focus of research, the specific molecular mechanisms are yet to be fully understood6. This endeavor holds crucial scientific significance.
Both prokaryotic and eukaryotic cell membrane systems exhibit discrete functional membrane microdomains (FMMs), commonly referred to as lipid microdomains or lipid rafts7,8. These nanoscale liquid-ordered domains in the plasma membrane are rich in glycosphingolipids, sphingomyelins, cholesterol, and specific proteins. They play a crucial role in cell signaling, facilitating extracellular and intracellular communication9,10,11. Previous research has demonstrated the significant involvement of lipid rafts in nuclear factor kappa-B (NF-κB) signaling and osteoclast function. Specifically, the receptor activator of nuclear factor-κB ligand (RANKL)-induced expression of tetraspanin CD9 within the membrane microstructural domains of lipid rafts promotes osteoclast fusion12. Furthermore, the activation of FGFR2 leads to the recruitment of E3 ubiquitin ligase Cbl in raft microstructural domains, triggering PI3K ubiquitination and proteasomal degradation, ultimately resulting in the down-regulation of osteoblast survival13. Galectin-9, a β-galactoside-binding lectin, has also been found to stimulate osteoblast proliferation by promoting the aggregation of lipid rafts at membranes, followed by the phosphorylation of the c-Src/ERK signaling pathway14. The emerging understanding of the lipid rafts in bone-associated cell differentiation and intracellular signal transduction is becoming more apparent.
The SPFH (STOM, Prohibitin, Flotillin, and HflK/C) family is a group of membrane proteins found in various biofilm systems and are known as key components in lipid rafts15,16. Due to the oligomeric nature of SPFH, it is believed to play a crucial role in scaffolding for the formation and organization of FMMs17. Studies have shown increased expression of the flotillin component of SPFH during osteoclast differentiation, with RANK facilitating the translocation of TRAF6 to rafts containing Src. Disruption of these rafts hindered TRAF6 translocation and Akt activation by RANK ligands in osteoclasts, leading to reduced osteoclast survival18. However, the specific role of other SPFH members in regulating bone physiology and their mechanisms of action remain unclear.
STOM, a lipid raft-associated integral membrane protein from the SPFH superfamily, is found in the plasma membrane and intracellular vesicles19. It plays a role in intracellular lipid transport and regulates mechanosensory ion channels20,21. STOM’s α-helical fragments can induce membrane curvature by aggregating lipid rafts22. Research indicates that STOM proteins form molecular assemblies within lipid rafts, facilitating cell fusion. Wu et al. demonstrated that in adipocyte-like cells, increased STOM promotes the growth and enlargement of lipid droplets by facilitating the fusion of lipid droplets23. Lee et al. suggested that STOM contributes to macrophage fusion and potentially influences osteoclast activation24. Nonetheless, the exact mechanism through which STOM affects bone metabolism requires further investigation.
In our study, we initially observed alterations in the expression and localization of STOM in individuals and mice with OP. Subsequently, we generated STOM systemic knockout (STOM −/−) mice and osteoclast lineage-specific STOM knockout (Ctsk-Cre; STOM f/f) mice to explore the impacts and underlying mechanisms of STOM knockout on the differentiation of bone-related cells. Our findings provided insights into the involvement of lipid raft-associated STOM in bone metabolism and underscored the potential therapeutic significance of targeted intervention of STOM in the management of OP.
Results
STOM expression is elevated in OP patients and OVX mice
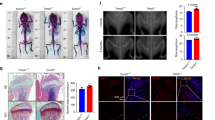
To investigate the expression and localization of STOM in human tissues, we analyzed data from the GEO database focusing on OP patients (GSE147287). Our findings indicated that STOM was predominantly expressed in bone marrow cells such as mesenchymal cells (MSCs), neutrophils, and macrophages in OP patients (Fig. 1A, B). Pseudotemporal analysis revealed that STOM expression is elevated during the later stages of macrophage lineage differentiation. In contrast, STOM expression is significantly increased during the early stages of MSC lineage differentiation, followed by a slight downregulation in the later stages. Nevertheless, there is an overall trend of increased expression when compared to the early stages (Fig. 1C–F). In addition, we examined bone tissue from patients with OP and those without. We utilized the co-marker CD44/CD73/CD90 to identify the location of human bone tissue MSCs25,26, while CD68 was employed to label human bone tissue macrophages27. The results indicated that STOM expression was elevated in both cell types in OP patients (Fig. 1G, H and Supplementary Fig. 1A). The elevated STOM levels were positively correlated with bone loss in OP patients, suggesting a potential role of STOM in the progression of OP. Subsequent experiments using OVX mice also showed a significant reduction in bone mass and increased STOM expression in bone tissues, consistent with the human histological findings (Fig. 1I). We collected human femoral neck tissue and mouse distal femur bone tissue. The results of the Western blot analysis indicated that, compared to the Non-OP group, the expression levels of STOM in the bone tissue of both OP patients and OVX mice were significantly increased (Fig. 1J). Overall, our results indicate that STOM expression is upregulated during OP progression.

A Cell types and clusters in OP samples from dataset GSE147287; B Expression levels and cluster distribution of STOM; C Branches and states in macrophage pseudo-time analysis; D Dynamic changes of STOM on the pseudo-time axis; E Branches and states in pseudo-time analysis of MSCs; F Dynamic changes of STOM on the pseudo-time axis; G Immunofluorescence co-staining of CD44/CD73/CD90 and STOM in bone tissue of OP and Non-OP individuals with fluorescence quantitative analysis. The red arrow indicated four-positive cells. The fluorescence images of the femoral neck tissues from ten patients in each group were quantified. CD44/CD73/CD90-positive cells in each image were identified as the subjects of the study, and the average fluorescence intensity of their STOM was measured (n = 10 biologically independent samples); H CD68 and STOM immunofluorescence co-staining and fluorescence quantitative analysis in bone tissue of OP and Non-OP individuals. The red arrow indicated double-positive cells. The fluorescence images of the femoral neck tissues from ten patients in each group were quantified. In each image, specific CD68-positive cells were chosen as subjects for analysis, and the average fluorescence intensity of STOM was quantified (n = 10 biologically independent samples); I Immunofluorescence staining showing changes in STOM expression in mouse bone tissue. Fluorescence quantifies the average intensity of STOM across the entire image (n = 7 biologically independent samples); J Western blot analysis demonstrated the expression of STOM in the bone tissue of both OP patients and OVX mice (n = 6 biologically independent samples). The data were represented as mean ± SD. Statistical significance was determined by two-tailed Student’s t test. OP osteoporosis, Non-OP Non-osteoporosis. Source data are provided as a Source Data file.
Knockout of STOM affects osteoclastogenesis and osteogenesis in vivo
To investigate the regulatory effects of STOM on bone biology, we generated STOM −/− mice. The successful generation of STOM −/− mice was confirmed through tail gene identification (Supplementary Fig. 1B). The gross appearance of STOM −/− mice compared to WT mice is illustrated in Supplementary Fig. 1C. We found that there was no significant difference in body weight between WT mice and STOM −/− mice of the same sex. Subsequent immunofluorescence staining of bone tissues from STOM −/− mice showed nearly complete absence of STOM expression (Supplementary Fig. 1D). Tissue western blot analysis confirmed the absence of STOM expression in the liver, kidney, and heart of STOM −/− mice (Supplementary Fig. 1E). Micro-CT scanning of 12-week-old WT mice and STOM −/− mice revealed significant differences in bone histological parameters, including increased bone mineral density (BMD), bone volume (BV), trabecular volume to total bone volume ratio (BV/TV), number of trabeculae (Tb. N) and trabecular thickness (Tb. Th) in the distal femur of STOM −/− mice, along with a decrease in bone trabecular spacing (Tb. Sp). These findings suggest that STOM knockout leads to an up-regulation in bone volume in the mice (Fig. 2A–C). Furthermore, under physiological conditions, the knockout of the STOM gene resulted in increased bone mass in the mice, which was consistent across both female and male mice, and no significant changes were observed in the behavior of the mice. Morphological differences in tissues and organs between the two groups of mice were highlighted by H&E staining (Supplementary Fig. 1G). Fig. 2D shows the histomorphology of the distal femur joints of WT and STOM −/− mice, demonstrating denser bone volume in the marrow cavity of the femur in STOM −/− mice.

A Micro-CT imaging reveals differences in bone mass between WT and STOM-/- mice; B Quantitative analysis of BMD (n = 7 biologically independent samples); C Quantitative statistics of trabecular morphological parameters, including BV, BV/TV, Tb. N, Tb. Sp and Tb. Th (n = 7 biologically independent samples); D Histological staining with H&E of mouse femur (n = 5 biologically independent samples); E, F Alizarin red and von Kossa staining of mouse femur (n = 5 biologically independent samples); G Immunofluorescence staining and quantification analysis for col1a1 in bone tissue of WT and STOM-/- mice. Fluorescence quantifies the average intensity of col1a1 across the entire image (n = 5 biologically independent samples); H Immunofluorescence staining and quantification analysis for osterix in bone tissue of WT and STOM-/- mice. Fluorescence quantifies the average intensity of osterix across the entire image (n = 5 biologically independent samples); I: ALP staining (n = 3 biologically independent samples); J Alizarin red staining (n = 3 biologically independent samples); K TRAcP staining of the distal femur and proximal tibia in mice and quantitative analysis of osteoclasts (n = 5 biologically independent samples); L Osteoclastic induction of bone marrow cells was conducted using WT and STOM-/- mice, followed by staining with TRAcP staining (n = 3 biologically independent samples). M The bone plate resorption assay was utilized to assess the osteoclastic resorption function of both WT and STOM-/- mice in vitro (n = 3 biologically independent samples). The data were represented as mean ± SD. Statistical significance was determined by two-tailed Student’s t test. WT wild type, STOM-/- STOM systemic knockout, BMD bone mineral density, BV bone volume, BV/TV trabecular volume to total bone volume ratio, Tb. N number of trabeculae, Tb. Sp bone trabecular spacing, Tb. Th trabecular thickness. Source data are provided as a Source Data file.
Changes in osteoblasts and osteoclasts activity play a crucial role in skeletal bone mass alterations, and we further investigated differences in osteoblasts and osteoclasts expression in the bone tissues of the two mouse groups. Alizarin red and von Kossa staining showed reduction in osteogenesis and calcification within bone tissue of STOM −/− mice compared to WT mice (Fig. 2E, F), while immunofluorescence staining indicated a significant decrease in expression of osteoblast-specific markers col1a1 and osterix within the bone tissue of STOM −/− mice (Fig. 2G, H). ALP and alizarin red staining reveals that STOM knockout inhibits osteoblast differentiation in vitro (Fig. 2I, J). Interestingly, we also observed that STOM knockout significantly suppressed osteoclastogenesis in the distal femur and proximal tibia of mice compared to WT mice (Fig. 2K). Serological analysis indicated that the serum bone turnover marker CTX-1 was significantly reduced in STOM −/− mice (Supplementary Fig. 1F). Additionally, the expression levels of ACP5 and CTSK in the bone tissue of STOM −/− mice were also significantly lower when compared to WT mice (Supplementary Fig. 1H, I). By extracting primary bone marrow cells from STOM −/− mice to induce osteoclast differentiation, we observed that, compared to WT mice, osteoclast differentiation was significantly inhibited in vitro following STOM knockout, and the osteoclastic resorption function was markedly diminished (Fig. 2L, M). Given that STOM −/− mice ultimately exhibited increased bone mass, we hypothesized that this could be attributed to a more pronounced effect of inhibiting osteoclast resorption compared to the attenuation of osteogenesis after STOM knockout, resulting in the up-regulation of bone mass in mice.
Subsequently, to investigate whether the overexpression of STOM in the bone marrow would influence bone mass in mice, we designed an adeno-associated virus (AAV) aimed at enhancing STOM levels in the bone marrow. At 8 weeks of age, mice were injected with intramedullary STOM-overexpressing AAV (AAV-STOM). After an 8-week intervention period, mouse femurs were collected for analysis (Supplementary Fig. 2A). Compared to the Sham group, the intramedullary injection of AAV-STOM significantly enhanced the expression of STOM in the bone marrow cavity of the mice (Supplementary Fig. 2D). However, Micro-CT analysis revealed a significant reduction in the bone density of the distal femur (Supplementary Fig. 2B, C). H&E staining indicated that the trabecular bone area in the medullary cavity of AAV-STOM-injected mice was significantly diminished, while TRAcP staining demonstrated that AAV-STOM intramedullary injection markedly promoted osteoclast activation in vivo (Supplementary Fig. 2E, F). Additionally, we isolated mouse bone marrow cells and induced their differentiation into osteoclasts using M-CSF and RANKL. Consistent with previous findings, TRAcP staining suggested that AAV-STOM facilitated osteoclast differentiation in vitro (Supplementary Fig. 2G). qRT-PCR analysis further indicated that AAV-STOM not only enhanced the expression of Stom but also significantly increased the expression of osteoclast-related markers Nfatc1, Ctsk, Acp5, and Mmp9 (Supplementary Fig. 2H). These results suggest that STOM may contribute to bone loss by activating osteoclasts.
Systemic and osteoclast-specific knockout of STOM both mitigate OVX-induced bone loss though inhibiting osteoclasts
Additionally, OVX surgery was performed on WT and STOM −/− mice to investigate the impact of STOM knockout on OP. Following an 8-week modeling period, mouse femurs were harvested. The Micro-CT results indicated that STOM −/− mice exhibited a significant attenuation of OVX-induced bone loss compared to WT mice (Fig. 3A, B). Histomorphological evaluation through H&E staining of intramedullary trabecular bone structures validated the Micro-CT scan findings (Fig. 3C). In vivo TRAcP staining further suggested that STOM knockout effectively alleviated osteoclastogenesis in bone tissue of OVX mice (Fig. 3D). Serological assay analysis also demonstrated a significant reduction in the expression of the serum bone resorption marker CTX-1 in STOM −/− mice (Fig. 3E).

A Micro-CT was used to detect changes in bone mass in WT and STOM-/- mice following OVX; B Bone parameters analysis (n = 7 biologically independent samples); C H&E staining (n = 5 biologically independent samples); D Representative images of TRAcP staining with quantification of the number of osteoclasts (n = 5 biologically independent samples); E CTX-1 levels in mouse serum (n = 5 biologically independent samples); F–H Representative Micro-CT images and quantitative analysis of BMD, BV, BV/TV, Tb. N, Tb. Th and Tb. Sp in the distal femurs (n = 7 biologically independent samples); I Representative images of distal femurs sections in each group stained with H&E (n = 5 biologically independent samples); J TRAcP staining and quantitative analysis of osteoclasts were conducted on distal femurs of STOMf/f and Ctsk-Cre; STOMf/f mice (n = 5 biologically independent samples). The data were represented as mean ± SD. Statistical significance was determined by two-tailed Student’s t test. WT wild type, STOM-/- STOM systemic knockout, BMD bone mineral density, BV bone volume, BV/TV trabecular volume to total bone volume ratio, Tb. N number of trabeculae, Tb. Sp bone trabecular spacing, Tb Th, trabecular thickness. Source data are provided as a Source Data file.
To further elucidate the role of STOM in osteoclasts and physiological bone remodeling, we developed a transgenic mouse model that specifically lacks STOM in the osteoclast lineage. This model was created by crossing STOM f/f mice with Ctsk-Cre mice, resulting in specific STOM gene depletion in Ctsk-positive osteoclasts (Ctsk-Cre; STOM f/f) (Supplementary Fig. 3A, B). We found that, compared to the STOMf/f group, the bone density of Ctsk-Cre; STOM f/f mice was significantly increased. The trabecular bone volume and number in these mice were higher than those in the STOM f/f group, while the Tb. Sp was lower (Fig. 3F–H). Histological immunostaining revealed that in Ctsk-Cre; STOM f/f mice, a significantly larger area of trabecular bone was observed in the medullary cavity, and the number of TRAcP+ osteoclasts on the bone surface was reduced (Fig. 3I, J). Immunofluorescence staining of Col1a1 and Osterix in bone tissue demonstrated that, following the conditional knockout of STOM in the osteoclast lineage, the osteogenesis-related indicators exhibited minimal changes when compared to the STOM f/f mice (Supplementary Fig. 3C, D). More importantly, after constructing a mouse model of postmenopausal OP through OVX surgery, the rate of bone loss in Ctsk-Cre; STOM f/f mice was significantly slowed, and osteoclast activation was similarly reduced. These in vivo data indicate that the depletion of STOM in osteoclast-lineage cells inhibits osteoclastic bone resorption and increases bone mass.
STOM promotes osteoclast activation and bone resorption in vitro
Building upon previous research that investigated the effects of STOM intervention on bone in vivo, we delved into the impact of STOM on bone marrow macrophages (BMMs)-induced osteoclast differentiation in vitro. Bone marrow cells were extracted from both WT and STOM −/− mice and differentiated into macrophages using M-CSF. Immunofluorescence staining highlighted the deficiency of STOM in macrophages from STOM −/− mice (Fig. 4A). Western blot and qRT-PCR analyses demonstrated that the expression levels of osteoclast-related markers MMP9, ACP5, CTSK, and NFATc1 in macrophages were significantly reduced following STOM knockout (Fig. 4B, C and Supplementary Fig. 4A), a finding that aligns with the results obtained from immunofluorescence staining (Supplementary Fig. 4B). Osteoclast differentiation was induced using RANKL and M-CSF. Results from western blot and qRT-PCR assay indicated that knockout of STOM also led to reduced expression of osteoclast-related markers (Fig. 4D, E and Supplementary Fig. 4C). Immunofluorescence staining further validated this by showing decreased expression of osteoclast-related markers ACP5 and NFATc1 (Fig. 4F).

A Immunofluorescence staining was utilized to assess the expression levels of STOM in BMMs extracted from both WT and STOM-/- mice; B Western blot analysis was performed to detect the expression levels of STOM and osteoclast-related proteins in macrophages (n = 3 biologically independent samples); C qRT-PCR was used to detect the expression levels of STOM and osteoclast-related genes (n = 3 biologically independent samples); D Bone marrow cells from WT and STOM-/- mice were induced to differentiate into osteoclasts, and the expression of proteins related to osteoclast activation was assessed using western blot (n = 3 biologically independent samples); E qRT-PCR was used to detect the expression levels of osteoclast-related genes (n = 3 biologically independent samples); F Immunofluorescence staining was utilized to assess the expression levels of ACP5 and NFATc1 in osteoclasts (n = 3 biologically independent samples); G Western blot analysis were conducted to investigate changes in STOM expression levels during osteoclast differentiation (n = 3 biologically independent samples); H Western blot analysis was used to evaluate the impact of siSTOM on STOM expression (n = 3 biologically independent samples), NC (negative control) transfects cells solely with a non-targeting siRNA sequence; I Western blot analysis was used to evaluate the impact of OE-STOM on STOM expression (n = 3 biologically independent samples), NC (negative control) involves the transfection of cells solely with an empty vector; J TRAcP staining and quantitative analysis after siSTOM and OE-STOM interfere with osteoclast differentiation (n = 3 biologically independent samples), NC (negative control) involves the transfection of cells solely with an empty vector; K Bone resorption assay and quantitative analysis after siSTOM and OE-STOM interfere with osteoclast differentiation (n = 3 biologically independent samples); L Immunofluorescence co-staining of ACP5, CTSK and NFATc1 with F-actin (n = 3 biologically independent samples); M qRT-PCR reveals the expression levels of osteoclast-related genes after siSTOM and OE-STOM interfere with osteoclast differentiation (n = 3 biologically independent samples). The data were represented as mean ± SD. Statistical significance was determined by one-way ANOVA. Source data are provided as a Source Data file.
Furthermore, our study observed an increase in STOM expression during osteoclast differentiation (Fig. 4G and Supplementary Fig. 4D). Subsequently, transfection reagents small interfering RNA (siSTOM) and overexpression plasmid (OE-STOM) were developed to respectively knock down and overexpress STOM in macrophages. Western blot analysis confirmed the effectiveness of these transfection reagents (Fig. 4H, I and Supplementary Fig. 4E, F). Subsequently, siSTOM3, siSTOM4, and OE-STOM were selected to further investigate the impact of STOM interference on osteoclastic differentiation. TRAcP staining indicated that inhibiting STOM reduced osteoclast activation (Fig. 4J), and bone resorption assays confirmed that STOM inhibition decreased osteoclastic resorption (Fig. 4K), whereas STOM overexpression had the opposite effect. Immunofluorescence analysis results showed that inhibiting STOM suppressed the expression of osteoclast-associated markers NFATc1, ACP5, and CTSK, while overexpression enhanced their expression (Fig. 4L). These findings were consistent with the results of qRT-PCR results (Fig. 4M), supporting a direct regulatory role of STOM in osteoclast activation.
STOM promotes ROS production by regulating intracellular oxidation levels
To further elucidate the specific mechanisms by which STOM regulates osteoclast function, we extracted primary BMMs from WT and STOM −/− mice. Both groups of cells were induced to differentiate towards macrophages using M-SCF and we performed whole transcriptome RNAseq. The heatmap revealed 970 differentially expressed genes (1.5-fold up- or down-regulated) in the macrophages of the STOM −/− group compared with the Ctrl group (Fig. 5A). Differential expression analysis showed that 522 genes were down-regulated and 448 genes were up-regulated in STOM −/− cells. Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis indicated that the differential genes were mainly classified under terms such as rheumatoid arthritis and hematopoietic cell lineage (Fig. 5B). Gene Set Enrichment Analysis (GSEA) demonstrated significant enrichment of the differential genes in osteoclast differentiation and response to oxidative stress (Fig. 5C, D). Visualization of genes associated with these pathways showed that Mmp9, Nfatc1, Atp6v0d2, and Csf1 were down-regulated in STOM −/− macrophages, while antioxidant genes like Sod1, Sod2, Hmox1 and Nqo1 were up-regulated, suggesting a role of oxidative stress in mediating STOM’s effects (Fig. 5E). Western blot and qRT-PCR results showed increased expression of intracellular antioxidant markers SOD1, SOD2, NQO1, and HO-1 in BMMs after STOM knockout, consistent with the sequencing results (Fig. 5F, G and Supplementary Fig. 4G). Conversely, we found that the overexpression of STOM via plasmid transfection led to a downregulation in the expression of these antioxidant proteins (Supplementary Fig. 4H). Subsequent flow cytometry analysis on primary macrophages from both groups of mice indicated a notable decrease in ROS levels within BMMs following STOM knockout (Fig. 5H and Supplementary Fig. 4I). When we employed siSTOM to intervene in osteoclast differentiation, we observed that osteoclast differentiation was inhibited and intracellular ROS levels were significantly reduced. Furthermore, by employing OE-STOM to modulate intracellular STOM levels, we observed an amplified effect on osteoclast activation, which was accompanied by a marked increase in ROS levels (Fig. 5I, J and Supplementary Fig. 4J). Interestingly, the use of the ROS inhibitor N-Acetylcysteine (NAC) to intervene in the process of OE-STOM amplified osteoclast activation resulted in a significant reduction of intracellular ROS and a notable inhibition of osteoclast formation (Fig. 5K, L and Supplementary Fig. 4K).

A Heat map reveals all differential expressed genes between WT and STOM-/- mice derived BMMs; B Enrichment analysis of differentially expressed genes in KEGG pathways was performed through the hypergeometric test with one-sided test and Benjamini-Hochberg adjustment; C The GSEA analysis of differentially expressed genes in osteoclast differentiation was performed with a one-sided test, and FDR correction was applied; D The GSEA analysis of differentially expressed genes in response to oxidative stress was performed with a one-sided test, and FDR correction was applied; E The heat map illustrates the expression levels of genes associated with osteoclastic differentiation and response to oxidative stress; F Western blot analysis demonstrates the expression of oxidative stress-related proteins in the macrophages of both WT and STOM-/- mice (n = 3 biologically independent samples), statistical significance was determined by two-tailed Student’s t test; G qRT-PCR demonstrates the expression of oxidative stress-related genes in the macrophages of both WT and STOM-/- mice (n = 3 biologically independent samples), statistical significance was determined by two-tailed Student’s t test; H Flow cytometry analysis to detect intracellular ROS levels in the macrophages of both WT and STOM-/- mice (n = 3 biologically independent samples), statistical significance was determined by two-tailed Student’s t test; I Flow cytometric analysis of ROS (n = 3 biologically independent samples), statistical significance was determined by one-way ANOVA; J TRAcP staining was utilized to assess the impact of replenishing STOM on osteoclast differentiation following the knockdown of STOM in the cells (n = 3 biologically independent samples), statistical significance was determined by one-way ANOVA; K Flow cytometric analysis of ROS (n = 3 biologically independent samples), statistical significance was determined by one-way ANOVA; L After OE-STOM mediates osteoclast activation, the ROS inhibitor NAC is subsequently employed to intervene in this process, and osteoclastic differentiation is assessed using TRAcP staining (n = 3 biologically independent samples), statistical significance was determined by one-way ANOVA; M Analysis of ATP production (n = 3 biologically independent samples), statistical significance was determined by one-way ANOVA; N TEM for the detection of intracellular substructures; O and Q MitoSOX immunofluorescence staining and quantitative analysis (n = 3 biologically independent samples), statistical significance was determined by one-way ANOVA; P and R JC-1 immunofluorescence staining and quantitative analysis (n = 3 biologically independent samples), statistical significance was determined by one-way ANOVA. The data were represented as mean ± SD. WT wild type, STOM-/- STOM systemic knockout. Source data are provided as a Source Data file.
Subsequently, WT mouse BMMs were extracted and siSTOM and OE-STOM were utilized to modulate intracellular STOM expression, inducing macrophage differentiation resembling osteoclasts. ATP assays demonstrated that STOM inhibition hindered RANKL-induced osteoclast energy production, while ATP levels were elevated in cells overexpressing STOM (Fig. 5M). Mitochondria were identified as a key source of ROS generation28, with transmission electron microscopy (TEM) revealing an increase in intracellular mitochondria post-RANKL intervention, and STOM inhibition mitigating macrophage mitochondrial accumulation and mitochondrial swelling (Fig. 5N). Conversely, STOM overexpression led to mitochondria with blurred cristae. Examination of mitochondrial superoxide formation during osteoclast differentiation via mitoSOX staining and flow cytometry analysis demonstrated an increase in mitochondrial superoxide levels, which were notably reduced upon STOM inhibition (Fig. 5O, Q and Supplementary Fig. 4L). Furthermore, the mitochondrial membrane potential (MMP) during osteoclast differentiation was evaluated using JC-1 staining. The results indicated a significant increase in MMP following RANKL intervention. There was a notable decrease in MMP after STOM inhibition, while STOM overexpression yielded the opposite effect (Fig. 5P, R and Supplementary Fig. 4M).
STOM targets Prdx1 degradation to regulate oxidative stress and osteoclastic differentiation
To investigate the mechanisms underlying STOM’s regulation of oxidative stress, STOM-interacting proteins were identified through protein immunoprecipitation combined with mass spectrometry (IP/MS). The study revealed that STOM exhibited increased binding to Prdx1, a 23 kDa redox protein predominantly located in the cytoplasm and mitochondria29, regardless of RANKL intervention (Fig. 6A, B). Molecular docking technique showed that Prdx1 chain bands Thr18, Thr90, and Asp28 form hydrogen bonding interactions with Asp140, Lys93, and Arg144 of protein STOM (Fig. 6C). Co-immunoprecipitation (Co-IP) further validated the binding interaction between Prdx1 and STOM (Fig. 6D). We subsequently conducted direct point mutations on STOM and Prdx1, guided by the molecular docking results. Our findings confirmed a mutual binding relationship between the Thr18 residue of the Prdx1 protein chain and the Asp140 residue of the STOM protein (Supplementary Fig. 5A).

A IP/MS results revealing putative proteins binding to STOM; B IP/MS results revealing putative proteins binding to STOM under RANKL intervention; C Molecular docking of STOM and Prdx1 proteins; D Co-IP analysis (n = 3 biologically independent samples); E Western blot showing the expression level of intracellular Prdx1 after overexpression of STOM with or without RANKL intervention (n = 3 biologically independent samples), statistical significance was determined by two-tailed Student’s t test; F Western blot indicating the expression level of intracellular Prdx1 after intervention with MG-132 or HCQ (n = 3 biologically independent samples), statistical significance was determined by one-way ANOVA; G Immunofluorescence co-staining of Lamp1 and Prdx1 protein (n = 3 biologically independent samples); H Immunofluorescence co-staining of Lyso-Marker and Prdx1 protein (n = 3 biologically independent samples); I Western blot analysis demonstrates the primary mechanism of lysosome-mediated degradation of Prdx1 (n = 3 biologically independent samples), statistical significance was determined by two-tailed Student’s t test, NC (negative control) transfects cells solely with a non-targeting siRNA sequence; J ROS flow cytometric analysis (n = 3 biologically independent samples), statistical significance was determined by one-way ANOVA; K qRT-PCR revealing intracellular levels of antioxidant-related genes such as Prdx1, Sod1, Sod2, Nqo1 and Hmox1 (n = 3 biologically independent samples), statistical significance was determined by one-way ANOVA; L TEM imaging revealing intracellular sub-structures (n = 3 biologically independent samples); M TRAcP staining and quantification of osteoclasts (n = 3 biologically independent samples), statistical significance was determined by one-way ANOVA; N qRT-PCR showing intracellular expression levels of Acp5, Ctsk, Nfatc1, and Mmp9 genes (n = 3 biologically independent samples), statistical significance was determined by one-way ANOVA; O Bone resorption analysis and quantification (n = 3 biologically independent samples), statistical significance was determined by one-way ANOVA. The data were represented as mean ± SD. Source data are provided as a Source Data file.
Western blot analysis demonstrated that elevated STOM expression resulted in reduced levels of Prdx1 in the presence or absence of RANKL (Fig. 6E and Supplementary Fig. 5B). To investigate the degradation mechanism of Prdx1, cells were treated with proteasome and lysosomal inhibitors (MG-132 and HCQ). The results showed that proteasome inhibition did not prevent STOM-induced Prdx1 degradation, whereas lysosomal inhibition blocked the degradation process (Fig. 6F and Supplementary Fig. 5C). Immunofluorescence analysis indicated that following the overexpression of STOM, there was a reduction in the expression of Prdx1 in the cells. This reduction was accompanied by an increase in the expression of the lysosomal-associated membrane protein Lamp1 (Fig. 6G). Concurrently, there was also an increase in lysosomal activation and its binding to Prdx1 (Fig. 6H). Endogenous lysosomal degradation primarily occurs through three forms of autophagy: macroautophagy, chaperone-mediated autophagy, and endosomal microautophagy. In our study, we utilized the macroautophagy blocker 3-MA, along with the endosomal microautophagy blocker siVps4a and the chaperone-mediated autophagy blocker siLamp2a. Western blot analysis revealed that both 3-MA and siLamp2a interventions inhibited the degradation of Prdx1. These findings suggest that STOM-mediated macroautophagy and molecular chaperone-mediated autophagy are responsible for the rapid degradation of Prdx1 (Fig. 6I and Supplementary Fig. 5F).
Subsequently, an overexpression plasmid of Prdx1 was constructed and its overexpression efficiency was confirmed through western blot and qRT-PCR (Supplementary Fig. 5D, E). Flow cytometry results and qRT-PCR indicated that Prdx1 overexpression inhibited STOM-mediated ROS surge and notably increased the expression levels of antioxidant-related genes Hmox1, Nqo1, Sod1, and Sod2 (Fig. 6J, K). TEM results indicate that following the overexpression of Prdx1, intracellular mitochondrial swelling is alleviated, and the blurriness of cristae is diminished (Fig. 6L and Supplementary Fig. 5G). Moreover, it was observed that Prdx1 overexpression rescued STOM-mediated osteoclast activation and bone resorption (Fig. 6M and O), with a significant decrease in the expression of osteoclast-related genes Acp5, Nfatc1, Ctsk, and Mmp9 (Fig. 6N). These findings suggest a potential role for Prdx1 in mediating STOM-induced oxidative stress and osteoclast activation.
Targeted knockdown of macrophage STOM alleviates OVX-mediated bone loss
Our previous animal model with STOM knockout demonstrated that inhibiting STOM mitigated osteoclast-mediated bone loss. Subsequently, we developed an AAV-shSTOMF4/80 construct targeted specifically to macrophages. We assessed the therapeutic potential of bone-targeted STOM inhibition in alleviating OVX-induced bone loss (Fig. 7A). Mice in the OVX group received injections of normal saline into the bone marrow cavity as a control, whereas mice in the experimental group were injected with AAV-NC and AAV-shSTOMF4/80 vectors into the bone cavity. After 8 weeks of AAV treatment, mice were euthanized, and their femurs were examined. Following sectioning and immunofluorescence staining of femoral sections from mice in the AAV-shSTOMF4/80 group, we observed that the red fluorescence, indicating the site of virus transfection, overlapped with the F4/80 position. This finding suggests the specificity of AAV targeting towards macrophages (Fig. 7B). Following the injection of AAV-NC and AAV-shSTOMF4/80 into the medullary cavity of OVX mice, primary macrophages and bone marrow-derived stem cells (BMSCs) were isolated from each group of mice. Western blot analysis revealed that AAV-NC did not affect the expression of STOM in the bone marrow of the mice, whereas AAV-shSTOMF4/80 effectively inhibited STOM expression in the macrophages of OVX mice, but did not influence STOM expression in the BMSCs (Supplementary Fig. 6A, B). These findings were further corroborated by tissue immunofluorescence staining of STOM in the bone tissue of mice (Fig. 7F).

A Flowchart of animal experiments. Created in BioRender59; B Tissue ZsRed (red) co-staining with F4/80 (green) immunofluorescence (n = 7 biologically independent samples); C Micro-CT reveals the changes of bone mass in each group of mice after AAV-NC and AAV-shSTOMF4/80 interventions (n = 7 biologically independent samples); D BMD analysis (n = 7 biologically independent samples); E Analysis of bone parameters, including BV, BV/TV, Tb. N, Tb. Sp, and Tb. Th (n = 7 biologically independent samples); F Bone tissue was co-stained with F4/80 (green) and STOM (orange) immunofluorescence (n = 5 biologically independent samples); G H&E staining and quantitative analysis of bone surface (n = 5 biologically independent samples); H TRAcP staining and quantitative analysis of osteoclasts (n = 5 biologically independent samples); I CTX-1 levels in mouse serum (n = 5 biologically independent samples); J Bone tissue was co-stained with CTSK (green) and STOM (orange) immunofluorescence, and quantification analysis for CTSK. Fluorescence quantifies the average intensity of CTSK across the entire image (n = 5 biologically independent samples). The data were represented as mean ± SD. Statistical significance was determined by one-way ANOVA. BMD bone mineral density, BV bone volume, BV/TV trabecular volume to total bone volume ratio. Tb. N number of trabeculae, Tb. Sp bone trabecular spacing, Tb. Th trabecular thickness. Source data are provided as a Source Data file.
Micro-CT revealed that OVX exacerbated trabecular parameters such as BMD, BV, BV/TV, and Tb. N, all of which were significantly improved by STOM knockdown (Fig. 7C–E). H&E staining also demonstrated a significant increase in the surface area of trabecular bone in mice belonging to the AAV-shSTOMF4/80 group (Fig. 7G). Western blot results indicated that, compared to OVX mice, the expression of STOM in the liver, kidney, and muscle bone tissues of mice treated with AAV-shSTOMF4/80 did not exhibit significant changes (Supplementary Fig. 6C). After H&E staining of the mouse liver, kidney, and muscle in each group, there was minimal difference observed in histomorphology (Supplementary Fig. 6D). Tests of mouse serum alanine transaminase (ALT), aspartate aminotransferase (AST), and urea revealed that, compared to OVX mice, there were no statistically significant differences in the changes of each index value among the mice in the AAV-shSTOMF4/80 intervention group (Supplementary Fig. 6E–G). This suggests the in vivo safety of intramedullary injection of AAV-shSTOMF4/80. Furthermore, the increased number of osteoclasts and serum CTX-1 levels observed in the OVX group were reversed by AAV-STOM treatment (Fig. 7H, I). The expression of the osteoclast marker CTSK in the distal femoral tissue was significantly reduced following the intramedullary injection of AAV-shSTOMF4/80 (Fig. 7J). In conclusion, our results suggest that inhibiting macrophage lineage STOM expression protects trabecular bone in osteoporotic mice and underscores the potential role of anti-osteoclast efficiency in OP treatment.
Given the crucial role of STOM in regulating osteoclast activation, we further examined whether modulating STOM influences disease progression in a mouse model of rheumatoid arthritis (RA), which is another orthopedic condition characterized by osteoclast activation. We established a collagen-induced arthritis (CIA) model to simulate RA. On the day following the second immunization, we injected AAV-NC and AAV-shSTOMF4/80 into the tail vein of the mice and harvested the samples 50 days post-modeling (Supplementary Fig. 7A). Our results indicated that, following collagen injection, the paws of the mice in the CIA group exhibited a significant increase in thickness. The paw thickness of mice treated with AAV-NC was comparable to that of the CIA group, whereas treatment with AAV-shSTOMF4/80 significantly reduced the swelling of the paws in CIA mice (Supplementary Fig. 7B). Quantitative analysis of arthritis score further verified this therapeutic effect (Supplementary Fig. 7C). Through Micro-CT and three-dimensional (3D) reconstruction evaluation, it was observed that the joint surfaces of the paws of CIA mice were damaged. Furthermore, AAV-shSTOMF4/80 intervention was found to alleviate both bone erosion and hyperplasia on the joint surfaces of the paws in CIA mice (Supplementary Fig. 7D). AAV-shSTOMF4/80 significantly reduced the expression of STOM in CIA mice following tail vein injection (Supplementary Fig. 7E). H&E staining of hind paw sections revealed significant synovial hyperplasia, joint surface destruction, and inflammatory cell infiltration in CIA mice. In contrast, the mice in the AAV-shSTOMF4/80 intervention group exhibited almost no pathological manifestations (Supplementary Fig. 7F). Obviously, AAV-shSTOMF4/80 intervention also significantly inhibited the excessive activation of osteoclasts in the paws of CIA mice (Supplementary Fig. 7G).
Targeted knockdown of STOM in bone marrow adipocytes mitigates bone loss induced by OVX
The skeletal system is a complex structure, and various cells in the bone marrow play a crucial role in bone reconstruction. Numerous studies have demonstrated that bone marrow adipocytes play a crucial role in regulating bone density30,31. In our previous H&E staining concerning WT and STOM −/− mice, we found a significant reduction of bone marrow adipocytes in STOM −/− mice. Furthermore, we investigated the impact of intervening with STOM on bone marrow adipocytes and examined whether targeted intervention in the expression of STOM within adipocytes affects bone quality. We extracted primary BMSCs from STOM −/− mice and WT mice, subsequently inducing their differentiation into bone marrow adipocytes. Our results indicated that the knockout of the STOM significantly inhibited adipocyte formation in vitro (Fig. 8A), leading to a reduction in the expression of adipocyte-related marker genes, including Pparg, Cebpa, and Fabp4 (Fig. 8B). Furthermore, we employed siRNA and overexpression plasmids to manipulate the adipocyte differentiation process, revealing that the overexpression of STOM notably promoted adipocyte differentiation (Fig. 8C–E).

A Oil Red O staining was employed to assess the differentiation potential of BMSCs derived from WT and STOM-/- mice into adipocytes (n = 3 biologically independent samples). Adipo refers to the differentiation of BMSCs into bone marrow adipocytes through the use of adipogenic medium; B qRT-PCR was employed to quantify the intracellular levels of adipocyte markers, specifically Pparg, Cebpa, and Fabp4 (n = 3 biologically independent samples); C qRT-PCR was employed to assess the impact of siSTOM and OE-STOM on Stom expression in BMSCs (n = 3 biologically independent samples), NC (negative control) involves the transfection of BMSCs solely with an empty vector; D Intracellular STOM (red) and F-actin (green) immunofluorescence co-staining (n = 3 biologically independent samples); E Oil Red O staining demonstrates the impact of siSTOM and OE-STOM interventions on the differentiation of bone marrow adipocytes (n = 3 biologically independent samples); F Micro-CT reveals the changes of bone mass in each group of mice after AAV-NC and AAV-shSTOMFABP4 interventions (n = 7 biologically independent samples); G BMD analysis (n = 7 biologically independent samples); H Analysis of bone parameters, including BV, BV/TV, Tb. N, Tb. Sp, and Tb. Th (n = 7 biologically independent samples); I Bone tissue was co-stained with Perilipin (green) and STOM (red) immunofluorescence. Quantification of fluorescence was performed to determine the average STOM fluorescence intensity in perilipin-positive cells within each image (n = 5 biologically independent samples); J H&E staining and quantitative analysis of bone surface (n = 5 biologically independent samples). The data were represented as mean ± SD. Statistical significance was determined by one-way ANOVA. WT wild type, STOM-/- STOM systemic knockout, BMD bone mineral density, BV bone volume, BV/TV trabecular volume to total bone volume ratio, Tb. N number of trabeculae, Tb. Sp bone trabecular spacing, Tb. Th trabecular thickness. Source data are provided as a Source Data file.
The design of AAV intended to target bone marrow adipocytes and achieve STOM gene knockout is complicated by the distinct biological properties of bone marrow adipose tissue compared to peripheral adipose tissue, as well as the absence of specific molecular biomarkers for bone marrow adipocytes32,33. We attempted to utilize an AAV targeting STOM, which carries the FABP4 promoter, to intervene in OVX mice through intramedullary injection, aiming to knock down the expression of STOM in adipocytes. Interestingly, we discovered that AAV-shSTOMFABP4 intervention could partially reverse bone loss in OVX mice (Fig. 8F–H). Moreover, this intervention method effectively reduced STOM expression in mouse bone marrow adipocytes and significantly diminished the number of adipocytes in the medullary cavity (Fig. 8I, J). Our findings suggest that STOM-mediated activation of bone marrow adipocytes may also contribute to OP; however, the specific mechanisms underlying this process warrant further investigation.
Discussion
OP has been increasing in prevalence in recent decades due to the growing ageing population, affecting huge population worldwide and thus posing a major challenge to healthcare34. Human bone undergoes continuous remodeling maintained by osteoblast-mediated bone formation and osteoclast-mediated bone resorption35,36. Bone is in a constant state of balance between loss and rebuilding. Osteoblasts and osteoclasts mutually constrain regulation to maintain normal bone function. Imbalance of osteoblast and osteoclast coupling is the pathophysiological basis of OP37. Delving into the pathogenic mechanism of OP and identifying new intervention targets can offer fresh insights and theoretical foundations for enhancing OP prevention and treatment, ultimately reducing the incidence of osteoporotic fractures.
Lipid rafts are cholesterol- and sphingolipid-rich microstructural domains in the plasma membrane. The components and structural features of lipid rafts are favorable for protein interactions and conformational transformations, and can be involved in signal transduction, cellular protein function, and ion channel activity38,39. The occurrence of some infectious diseases, cardiovascular diseases and tumors may be closely related to lipid raft dysfunction40,41. Previous studies have shown that major molecules of lipid rafts are involved in the differentiation of bone-associated cells, and that osteogenic and osteoclast functions are heavily dependent on the expression and integrity of raft membrane microstructural domains42,43. In our study, we found that the lipid raft major molecule protein STOM, an ancient, widely expressed oligomeric membrane protein and mostly confined to membrane structural domains, is increased in expression during OP progression. Previous literature reported that STOM, with its unique molecular topology, promotes cell-cell fusion by forming molecular assemblies. However, the specific effects of STOM on bone physiology are unclear.
In our study, we obtained STOM −/− mice by CRISPR/Cas9 technology. Our in vivo experiments revealed that the knockout of STOM led to a significant impairment in osteoclast and osteoblast formation in mouse bone tissue. Interestingly, despite this, the overall bone mass of STOM −/− mice was found to be upregulated. This led us to hypothesize that STOM may play a critical role in inhibiting osteoclast activity. To test this hypothesis, we generated conditional knockout mice that lack STOM in the osteoclast cell line. Interestingly, these mice exhibited increased bone mass under physiological conditions, and the formation of osteoclasts was significantly inhibited. Additionally, upon the induction of OVX, osteoclast formation was similarly obstructed.
We induced osteoclasts on primary BMMs from both WT and STOM −/− mice, and observed a significant inhibition in osteoclast formation in the STOM −/− group. Inhibiting the differentiation of BMMs into osteoclasts and reducing the bone resorption function of osteoclasts could be a key strategy in preventing and managing OP. In our study, our results suggest that STOM acts as a positive regulator of osteoclast differentiation to influence bone homeostasis. While there are various drugs available for treating OP, such as the RANKL monoclonal antibody (denosumab), which inhibits osteoclast activation and increases bone density, long-term use of denosumab may pose risks such as hypocalcemia and osteonecrosis of the jaw44. Therefore, further investigation into the mechanisms of STOM-mediated osteoclast overactivation and functional enhancement under OP conditions is crucial for the development of effective strategies for preventing and treating OP.
Further, we performed high-throughput RNA-seq analysis of BMMs isolated from WT and STOM −/− mice. The study confirmed that knockout of STOM led to down-regulation of STOM and osteoclast-related marker genes. Additionally, the expression of several antioxidant genes, including Sod1, Sod2, Nqo1 and Hmox1, was significantly increased. Many bone diseases associated with osteoclast activation are linked to redox instability, with dysregulation of intracellular oxidative stress playing a key role in osteoclast activation45,46. Mitochondria generate ATP and ROS during oxidative phosphorylation, and the excess ROS from the mitochondrial electron transport chain triggers various signaling pathways, such as NF-κB and MAPK, in osteoclasts47,48. Furthermore, our study demonstrated that overexpression of STOM promoted cellular and mitochondrial ROS production in osteoclasts, as well as increased intracellular ATP levels following RANKL intervention.
Prdx1, a member of the peroxide-reducing protein antioxidant enzyme family, plays a crucial role in reducing H2O2 and alkyl hydroperoxides to provide antioxidant protection49,50. Our study revealed that STOM interacts with Prdx1, leading to the degradation of Prdx1 through the lysosomal pathway. Prdx1 has a significant paralog, Prdx2, known for scavenging H2O2 generated by RANKL and eliminating RANKL-induced Ca2+ oscillations51. Macrophages with Prdx2 knockdown exhibited higher ROS levels compared to WT mice, resulting in increased osteoclast differentiation52. Numerous studies have discussed the involvement of STOM in the regulation of oxidative stress signaling. In a study by Wu et al., it was demonstrated that reducing STOM levels can mitigate oxidative stress induced by sepsis53. Additionally, STOM-like protein-2 has been found to offer neuroprotection to neurons through the regulation of AMPK/Nrf2 signaling54. Our findings suggest that STOM plays a role in mediating oxidative stress. Interestingly, our findings showed that restoring Prdx1 levels in STOM-overexpression macrophages effectively suppressed ROS production and mitigated osteoclast activation, highlighting Prdx1 as a crucial downstream effector of STOM-mediated oxidative stress.
Ovariectomy is commonly used as a standardized method to induce bone loss due to estrogen withdrawal55. Our previous animal experiments demonstrated that reducing STOM expression helped alleviate bone loss caused by OVX. By utilizing an AAV vector targeting STOM, our in vivo results indicated that inhibiting macrophage STOM was successful in improving bone trabecular microstructure and reducing overactive osteoclasts. These results highlight the potential of targeting STOM as a therapeutic intervention for treating osteoporotic bone loss in vivo. Our additional experiments also indicated that the targeted inhibition of macrophage STOM may hold potential for the treatment of RA. The differentiation and metabolism of adipocytes in the bone marrow act in regulating bone mass. Additionally, we investigated the impact of intervening STOM on bone marrow adipocytes and found that STOM acts as an accelerator of both bone marrow adipocyte differentiation and the induction of OP.
In summary, our results demonstrated that inhibition of STOM delayed RANKL-induced osteoclastogenesis in vitro and effectively attenuated OVX-induced bone loss in vivo. Mechanistically, STOM mediated enhanced oxidative stress and ROS production within osteoclasts, at least in part by targeting prdx1 degradation, ultimately leading to enhanced osteoclast differentiation.
Methods
Ethics statement
Our research adheres to all pertinent ethical regulations. The study has received approval from the Ethics Committee of the First Affiliated Hospital of Soochow University (approval no.2024-698). Additionally, the animal care protocols and experiments have been reviewed and approved by the Animal Ethics Committee of Soochow University (SUDA20240911A36). All animals underwent welfare testing and were ultimately euthanized using isoflurane. All animal experiments complied with the ARRIVE guidelines for reporting animal experiments.
Drugs and reagents
Recombinant RANKL and M-CSF were obtained from Amizona Biotechnology Co., Ltd (Hangzhou, China). siRNA and plasmid targeting STOM were obtained from GenePharma (Suzhou, China). α-MEM and fetal bovine serum (FBS) were purchased from Thermo Fisher Scientific (St. Louis, USA). The primary antibodies utilized in the experiment are presented in the Table S1. Secondary antibody was purchased from Multisciences (Hangzhou, China). Phosphate-buffered saline (PBS) was purchased from Biosharp (Anhui, China). ATP Assay Kit were purchased from Beyotime (Shanghai, China). BCIP/NBT Alkaline Phosphatase Color Development Kit and Alizarin Red S Staining Kit for Osteogenesis were purchased from Beyotime. Oil Red O Staining Kit was purchased from Amizona Biotechnology Co., Ltd.
Single-cell analysis
OP samples from the GEO database GSE147287 dataset were selected for data analysis. OE biotech Co., Ltd (Shanghai, China) conducted the analysis using the official 10x genomics software Cell Ranger to assess data quality and obtain key quality control statistics such as cell number and median gene value. Subsequently, the Seurat software package was employed for further quality control processing, retaining high-quality cells based on specific criteria. Doublet cells were identified and removed using DoubletFinder software. Following quality control, data normalization was performed using the NormalizeData function in the Seurat package. The data was visualized in two dimensions using UMAP for nonlinear dimensionality reduction. Marker gene identification was carried out using the FindAllMarkers function in the Seurat package, with visualization of related genes through VlnPlot and FeaturePlot functions. Cell differentiation trajectories were inferred using the Monocle2 package, involving steps such as conversion to a CellDataSet object, screening genes for cell sorting, dimensionality reduction clustering, and differentiation trajectory inference.
Acquisition of human tissue specimens
Human bone tissues were collected from patients who underwent total hip arthroplasty due to hip disease at the First Affiliated Hospital of Soochow University. The specimens were divided into two groups based on bone mineral density, with one group having a T-score below -2.5 standard deviations (OP group) and the other group having a T-score above -1.0 standard deviations (Non-OP group). Each group consisted of ten samples, with the femoral neck tissues being cut into small pieces of 3 × 3 mm, fixed with 10% formalin, and used for experiments. The Ethics Committee of the First Affiliated Hospital of Soochow University has approved this study. All tissues were obtained through surgical procedures, with written informed consent obtained in advance. The basic information regarding the patients, including their gender and age, was presented in Table S2. The research was conducted in accordance with the principles outlined in the Declaration of Helsinki.
Construction of STOM −/− and Ctsk-Cre; STOM f/f mice
STOM −/− mice with a C57BL/6 J background were acquired from Shanghai Model Organisms (Shanghai, China). CRISPR/Cas9 technology was utilized in this study to induce mutations through non-homologous recombination repair, resulting in a frameshift mutation in the STOM gene and loss of function. The methodology involved obtaining Cas9 mRNA and gRNA via in vitro transcription, microinjecting them into fertilized eggs of C57BL/6 J mice to generate F0 mice, and subsequently mating positive F0 mice with C57BL/6 J mice to produce positive F1 mice. The STOM-floxed mice were constructed using CRISPR/Cas9 technology in GemPharmatech Co., Ltd (Jiangsu, China). The Ctsk-Cre mice were provided by Shanghai Model Organisms. STOM-floxed mice were crossed with Ctsk-Cre mice separately to generate osteoclast-specific STOM knockout (Ctsk-Cre; STOM f/f mice) mice, and the STOM-floxed littermates were used as controls. All animals were housed in the Animal Experimentation Centre of Soochow University, where they were provided with a suitable environment (20 ± 5 °C, 55% ± 5% humidity, 12-hour light/dark cycle) and fed standard food (SSFZP01-001, Suzhou Shuangshi Laboratory Animal Feed Technology Co., Ltd., Jiangsu, China).
OVX-induced OP mouse model
8-week-old C57BL/6 J female mice from the Animal Experiment Centre of Soochow University were acclimatized for one week before surgery under suitable temperature and light conditions. OVX mice were anesthetized with isoflurane, had their ventral region shaved and disinfected, muscle tissues incised, and ovaries removed bilaterally to induce OP56. Mice were kept for 8 weeks post-surgery before being sacrificed for evaluation. The same method was used to ovariectomize STOM −/− and Ctsk-Cre; STOM f/f mice in order to create OP model. For the AAV-shSTOMF4/80 therapeutic intervention, 8-week-old female C57BL/6 J mice were randomly assigned to 4 groups: Sham group, OVX group (underwent OVX surgery followed by intramedullary injection of saline), AAV-NC group (underwent OVX surgery followed by intramedullary injection of AAV-NC), and AAV-shSTOMF4/80 group (underwent OVX surgery followed by intramedullary injection of AAV-shSTOMF4/80). Mice were injected with an appropriate dose of AAV via the medullary cavity one day after surgery. Then, mice were euthanised 8 weeks post-surgery, and femurs and sera were collected from each group for additional measurements. The animal intervention methods employed for AAV-shSTOMFABP4 are consistent with those utilized for AAV-shSTOMF4/80.
The design and construction of AAV
AAV was produced by Hanheng Biotechnology Co., Ltd (Shanghai, China). First, an AAV vector is prepared. An appropriate vector is selected, and PCR primers for the target fragment are designed. The vector is then digested using a suitable restriction enzyme, and the resulting linearized vector is purified from agarose gel. Target fragment PCR is performed according to the designed primers, and the target fragment of the correct size is recovered through agarose gel electrophoresis. The linearized vector and the target fragment are then connected using either homologous recombination or T4 ligation. The bacterial solution is spread onto a plate and incubated for 12 to 16 h. Single clones are selected for colony verification, and positive clones with confirmed colony verification are chosen for sequencing. Successfully sequenced clone samples undergo plasmid extraction. Next, the AAV packaging and quality testing process is described. A three-plasmid AAV system is employed for virus packaging, which includes a vector plasmid carrying the target gene or shRNA, the pAAV-RC vector plasmid, and the pHelper vector plasmid. After extracting the three plasmid vectors with high purity and ensuring they are endotoxin-free, Hanheng’s Lipofiter™ transfection reagent is used to co-transfect AAV-293 cells with the three plasmids. Cell pellets are collected 72 h post-transfection. Column purification is performed to obtain a high-titer AAV preservation solution, and various quality indicators of the AAV are assessed according to stringent quality standards. The design sequence of AAV is presented in Table S3.
Micro-CT scanning
Following euthanasia of experimental mice, mouse femurs were collected and fixed in 10% formalin. After two days, the microstructure of the distal femur was scanned using a SkyScan 1176 Micro-CT instrument (Kontich, Belgium). Subsequently, 2D and 3D raw images were reconstructed using DataViewer and NRecon software. Structural parameters within the region of interest were analyzed using CTAn software (Kontich, Belgium). The bone-related parameters examined included BMD (g/cm3), BV (mm3), BV/TV (%), Tb. N (1/mm), Tb. Sp (mm) and Tb. Th (mm).
ELISA testing
Mouse serum was obtained from blood samples through centrifugation at 3000 × g for 20 min following a 1-hour incubation at room temperature. The level of CTX-1 in mouse serum samples was detected using the Mouse CTX-1 ELISA Kit (Elabscience, Wuhan, China), with the working solution prepared as per the kit instructions.
Morphological analysis of bone tissue
Following Micro-CT scanning, mouse femurs were decalcified using a 10% EDTA (Sigma-Aldrich, Sydney, Australia) solution for one month until the tissue reached a softened state. Subsequently, the decalcified tissue was embedded in paraffin. The femurs were then sectioned into 6 µm-thick slices for hematoxylin and eosin (H&E) as well as TRAcP staining. The staining procedures were conducted in accordance with established protocols, and the outcomes were visualized. H&E staining revealed morphological alterations, and bone volume was quantitatively assessed. Osteoclasts, identified by TRAcP-positive cells displaying red staining with well-defined borders, were observed. The number of TRAcP-positive multinucleated osteoclasts in specific regions of each experimental group was quantified using ImageJ software (Bethesda, USA). Staining was examined under an Axiovert 40 C microscope (Zeiss, Germany).
Tissue immunofluorescence staining
Tissue sections were deparaffinized in xylene, rehydrated using a gradient of alcohol, and then underwent antigen retrieval with citrate solution. Subsequently, the sections were incubated in a humid chamber at room temperature for 1 h, followed by two washes with PBS and overnight incubation at 4 °C with the appropriate concentration of primary antibody. After aspiration of the primary antibody and another wash with PBS, a fluorescent secondary antibody was added dropwise and incubated at room temperature in a humid chamber for 1 h, protected from light. For tissues requiring staining for two target proteins, a new primary antibody was applied and incubated overnight at 4 °C. After washing off the secondary antibody with PBS, the samples were incubated for 15 min in a humid chamber with DAPI (Beyotime), shielded from light. Finally, the samples were blocked with an anti-fade mounting medium and stored by drying at 37 °C. The expression of target proteins was visualized under a laser scanning microscope (LSM 510, Zeiss, Germany).
Cell culture and osteoclast induction
BMMs were prepared following the methodology outlined in our previous study57. In summary, BMMs were isolated from the femoral bone marrow of 8-week-old C57BL/6 J mice. Then, cells were seeded at a density of 1 × 105 cells per well on a 24-well plate and cultured in α-MEM medium supplemented with M-CSF (50 ng/mL) for 24 h. The cells were then treated with a combination of M-CSF (50 ng/mL) and RANKL (50 ng/mL) to induce differentiation towards osteoclasts, with osteoclast formation observed by the fourth day.
Cell transfection
BMMs were transfected with siRNAs and plasmids targeting STOM to modulate intracellular STOM levels through knockdown and overexpression. The sequences of the STOM transfection reagents are detailed in Table S4. BMMs were cultured in six-well plates with strict control over cell numbers to maintain a cell density of approximately 60% during transfection. The specified amount of siRNA or plasmid was added to serum-free cell culture medium following the manufacturer’s instructions. Lipofectamine 3000 (Thermo Fisher Scientific) was then added at room temperature to create a stable transfection complex. Subsequently, the cells were treated with fresh medium and the appropriate amount of the transfection complex was introduced.
Induction of bone marrow osteoblasts and adipocytes
The femoral and tibial cavities of 8-week-old C57BL/6 J mice were flushed with α-MEM to extract BMSCs. The cells were cultured in dishes using α-MEM supplemented with 10% FBS and penicillin/streptomycin antibiotics, with medium changes occurring every 48 h. Once the cells reached 70%–80% confluence, they were subcultured. Using third-generation BMSCs, the cells were induced into osteogenesis and adipogenesis using respective induction kits for osteogenesis (Procell, Wuhan, China) and adipogenesis (Amizona Biotechnology Co., Ltd).
TRAcP staining
Following washing with PBS, the cells were fixed using 4% paraformaldehyde (Beyotime) and then stained with the TRAcP staining kit (Amizona Biotechnology Co., Ltd), following the manufacturer’s protocol. Mature osteoclasts were identified as fused cells containing three or more nuclei. Images were captured using an inverted microscope (Zeiss, Dresden, Germany). The quantification of TRAcP positive multinucleated osteoclasts was performed using ImageJ software (Bethesda, USA).
Bone resorption assays
BMMs were seeded at a density of 1 × 105 cells per well on a 24-well plate coated with bone slices (Amizona Biotechnology Co., Ltd), and mature osteoclasts were generated by stimulation with 50 ng/ml RANKL and 50 ng/ml M-CSF. Various interventions were employed to disrupt the osteoclast differentiation process. After seven days, bone slices were harvested, and the bone resorption pits were visualized using an FEI Quanta 250 scanning electron microscope (Hillsboro, USA). ImageJ software was utilized to analyze four regions of the bone slice in the bone resorption experiment.
Western blot
Cell samples were collected and processed, with cellular proteins extracted using RIPA lysate (Beyotime). The proteins were then separated using SDS-PAGE (Epizyme, Shanghai, China) and transferred to a nitrocellulose membrane (Beyotime). Following blocking with Blocking Buffer (NCM biotech, Suzhou, China) for a minimum of 1 h, the membranes were exposed to primary antibodies overnight at 4 °C. Subsequently, the membranes were washed with Tris-buffered saline Tween (TBST, Beyotime) and incubated with the respective secondary antibody at room temperature for 1 h. Protein bands were visualized using High-sig enhanced chemiluminescence (Tanon, Shanghai, China) with Tanon 5200 Multi (Shanghai, China), and quantification of protein levels was performed using ImageJ software.
High‑throughput transcriptome sequencing
In this study, we investigated the changes in transcriptional expression in primary BMMs following STOM knockout. BMMs were separately obtained from WT and STOM −/− mice. RNA sequencing analysis was conducted by Azenta Co., Ltd (Suzhou, China) on the Illumina HiSeqTM 2500 platform. Raw data in fastq format were processed using a custom perl script, and paired-end reads were aligned to the reference genome with Hisat2 v2.0.5, resulting in FPKM values. Differential expression analysis was performed using the DESeq2 R package, with differentially expressed genes identified based on a significance level of P < 0.05 and thresholds |Log2 Fc | >1.5. Subsequent analyses included KEGG and GSEA pathways to explore key biological processes and pathways.
Cell immunofluorescence staining
BMMs were seeded at a density of 1 × 105 cells per well in 24-well plates coated with coverslips and cultured in α-MEM for 24 h before undergoing various interventions. Following this, all cells on coverslips were fixed with 4% paraformaldehyde for 15 min, permeabilized with 0.2% Triton X-100 (Beyotime) for 20 min, and then blocked with QuickBloock™ blocking buffer (Beyotime) for 60 min. Subsequently, primary antibodies were added and incubated at 4 °C for 12 h. The cells were then washed thrice with PBS and incubated with a fluorescent secondary antibody Alexa Fluor 488 or Alexa Fluor 647 (Beyotime) for another 1 h. Coverslips were mounted on microscope slides in a fluorescent antifade solution containing DAPI (Beyotime). Imaging was performed using a confocal microscope (Zeiss, Germany).
Co-Immunoprecipitation
Cells were collected and lysed with pre-cooled RIPA lysate (Beyotime), followed by the addition of 1 μg of the corresponding antibody to the lysate, and incubated at 4 °C overnight. Subsequently, 10 μl of pre-treated protein A/G-beads (Beyotime) were added to the cell lysate and incubated at 4 °C with shaking for 4 h. After centrifugation, the supernatant was removed, and another 15 μl of loading buffer (Beyotime) was added before heating again at 100 °C for 8 min in a metal water bath. The following steps for detection were identical to those used in the Western blotting method.
Quantitative reverse transcription polymerase chain reaction (qRT-PCR)
Total RNA was extracted from samples using TRIzol reagent (NCM biotech) followed by chloroform extraction. The RNA-containing upper clear liquid layer was then mixed with an equal volume of isopropanol. Subsequently, the RNA was reverse transcribed into cDNA using a thermal cycler (Thermo Fisher Scientific). A mixture of TB Green reagent (TaKaRa, Japan), RNase-free H2O (TaKaRa, Japan), and specific forward and reverse primers (sequences refer to Table S5) was utilized. Gene expression levels of Ctsk, Acp5, Nfatc1, Mmp9, Sod1, Sod2, Nqo1, Hmox1, Prdx1, and Stom were measured using a LightCycler® 480 instrument (Roche Diagnostics International Ltd., Switzerland). The housekeeping gene GAPDH was used for normalization, and fold change was calculated using the comparative 2−ΔΔCq method.
TEM scanning
Cells were collected and immersed in glutaraldehyde solution, then stored at 4 °C. Following fixation, the supernatant was removed from the sediment, and the sample was rinsed with PBS. Subsequently, the samples underwent treatment with a 1% osmotic acid solution and dehydration with an ethanol gradient. The samples were then immersed in acetone for 20 min before embedding. Sectioning was carried out using an ultrathin microtome (Leica Microsystems, Germany) to obtain 70–90 nm sections. After staining with lead citrate solution and 50% ethanol-saturated uranyl acetate solution for 5–10 min, the sections were dried and examined under a TEM (Hitachi H-7650, Japan).
ROS detection
Following various treatments of osteoclasts, levels of ROS were assessed using a ROS Assay kit. Specifically, cells were exposed to the prepared DCFH-DA solution for 30 min and subsequently washed with PBS thrice, as per the provided guidelines. Subsequently, the cells were analyzed using flow cytometry (BD Biosciences, USA). FlowJo software version 10.6.2 (Tree Star, San Carlos, CA, USA) were utilized for flow cytometry data processing, and the results are presented as the mean fluorescence intensity (MFI).
Mitochondrial superoxide assay
We utilize siSTOM or OE-STOM to intervene in the osteoclast differentiation process. Following treatment of the cells, immunofluorescence staining was conducted using the Mitochondrial Superoxide Assay Kit with MitoSOX Red (Beyotime). According to the protocol, MitoSOX Red should be diluted to a ratio of 1:1000, and then applied to the cells for a duration of 30 min. After this incubation period, the cells were washed and subsequently examined under a confocal microscope. As an alternative, cells may be harvested and analyzed using flow cytometry. FlowJo software were utilized for data processing, and the results are presented as the MFI.
JC-1 staining
For the treated cells, add the pre-prepared JC-1 staining (Beyotime) working solution and incubate in a cell culture incubator at 37 °C for 20 min. Following incubation, wash the cells twice with JC-1 staining buffer. Then, add fresh cell culture medium to the well plate and observe the cells using a laser confocal microscope.
IP/MS protein profiles
BMMs were transfected with STOM overexpression plasmids and cultured under RANKL-stimulated or non-stimulated conditions. For immunoprecipitation analysis, cells were lysed in ice-cold RIPA buffer containing protease inhibitors and incubated with 1 μg of anti-STOM antibody at 4 °C overnight. Protein A/G magnetic beads were then added to the lysates and rotated at 4 °C for 4 h. Following magnetic separation, the bead-bound complexes were washed three times with lysis buffer and resuspended in loading buffer. Protein denaturation was achieved by boiling at 100 °C for 8 min. The samples were harvested and subjected to western blot analysis. During electrophoresis, the samples were shifted down 1 cm for harvesting, and the adhesive strip was cut. The protein strips were then cut and transferred into 1.5 mL centrifuge tubes, rinsed twice with ultrapure water, and sent to OE biotech Co., Ltd for further experiments and analyses.
Molecular docking and point mutation experiment
Download the sequence of Prdx1 from the UniProt website and utilize the AlphaFold3 online platform to predict its 3D structure. Subsequently, download the 3D structure of STOM from the Protein Data Bank. Use PyMOL to evaluate the predicted protein structure and select the conformation with the highest predicted structure ranking as the docking structure file for Prdx1 and STOM. Molecular docking was performed using HDOCK software, with all docking parameters set to their default values, employing a global blind docking scheme. The docking results were visualized in 3D using PyMOL. Based on the results of molecular docking, Genechem Co., Ltd. (Shanghai, China) assisted in the design of plasmids associated with mutations in the amino acid sites related to Prdx1 and STOM.
Establish of CIA model
The CIA mouse model was established following standardized procedures58. Initially, bovine type II collagen (2 mg/mL, CHONDREX, USA) was thoroughly mixed with complete Freund’s adjuvant (CHONDREX, USA) in a 1:1 ratio. Subsequently, 0.1 mL of this collagen-adjuvant mixture was injected intradermally at the base of the tail for the initial immunization. On day 21, DBA/1 mice received a second intradermal injection of 0.1 mL of a 1:1 mixture of bovine type II collagen and incomplete Freund’s adjuvant (CHONDREX, USA) as a booster immunization. The CIA mice were then randomly divided into three groups, each administered the same dose of saline, AAV-NC, or AAV-shSTOMF4/80 via tail vein injection. On day 50, all groups of mice were sacrificed to collect the hind paws for further evaluation.
Statistical analysis
The data were statistically analyzed using GraphPad Prism 8.0 software. Data conforming to a normal distribution were presented as mean ± standard deviation (SD). Student’s t-test was used to compare two groups, while one-way ANOVA was used for data from three or more groups. The experiments were repeated independently at least three times with similar results unless otherwise mentioned, and the micrographs were representative data from individual samples (the n was provided in figure legends). No sample size calculations were performed, but the number of mice analyzed in animal studies was determined in agreement with the standards in the field. The investigators were not blinded to the experimental allocation or outcome assessment. A p-value of less than 0.05 was considered statistically significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data supporting the findings described in this manuscript are available in the article and in the supplementary information and the source data are provided with this paper. Previously published data used in this study is GSE147287: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE147287. The RNA sequencing data generated in this study have been deposited in the NCBI Sequence Read Archive under accession code PRJNA1235203. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (https://proteomecentral.proteomexchange.org) via the iProX partner repository with the dataset identifier PXD061779. Source data are provided with this paper.
Code availability
We did not develop custom code for our single-cell analysis; instead, we utilized standard code for the analysis. The link to the specific standard workflow is as follows: https://satijalab.org/seurat/articles/pbmc3k_tutorial.html.
References
Schousboe, J. T., et al. Simultaneous automated ascertainment of prevalent vertebral fracture and abdominal aortic calcification in clinical practice: role in fracture risk assessment. J. Bone Miner Res., https://doi.org/10.1093/jbmr/zjae066 (2024).
Kim, K. et al. Sestrin2 inhibits RANKL-induced osteoclastogenesis through AMPK activation and ROS inhibition. Free Radic. Biol. Med 211, 77–88 (2024).
Khired, Z. A. et al. Osteoporosis knowledge, attitudes, and practices among female princess nourah university students in Riyadh, Saudi Arabia. Arch. Osteoporos. 16, 1 (2021).
Baccaro, L. F., Conde, D. M., Costa-Paiva, L. & Pinto-Neto, A. M. The epidemiology and management of postmenopausal osteoporosis: a viewpoint from Brazil. Clin. Inter. Aging 10, 583–591 (2015).
Wang, Y., Tao, Y., Hyman, M. E., Li, J. & Chen, Y. Osteoporosis in china. Osteoporos. Int 20, 1651–1662 (2009).
Keen, R. W. Burden of osteoporosis and fractures. Curr. Osteoporos. Rep. 1, 66–70 (2003).
Lingwood, D. & Simons, K. Lipid rafts as a membrane-organizing principle. Science 327, 46–50 (2010).
Yu, M., Cui, Y., Zhang, X., Li, R. & Lin, J. Organization and dynamics of functional plant membrane microdomains. Cell Mol. Life Sci. 77, 275–287 (2020).
Simons, K. & Toomre, D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 1, 31–39 (2000).
Shelby, S. A., Castello-Serrano, I., Wisser, K. C., Levental, I. & Veatch, S. L. Membrane phase separation drives responsive assembly of receptor signaling domains. Nat. Chem. Biol. 19, 750–758 (2023).
Li, Q., Luo, X. & Muallem, S. Functional mapping of Ca2+ signaling complexes in plasma membrane microdomains of polarized cells. J. Biol. Chem. 279, 27837–27840 (2004).
Ishii, M. et al. RANKL-induced expression of tetraspanin CD9 in lipid raft membrane microdomain is essential for cell fusion during osteoclastogenesis. J. Bone Min. Res. 21, 965–976 (2006).
Dufour, C. et al. FGFR2-Cbl interaction in lipid rafts triggers attenuation of PI3K/Akt signaling and osteoblast survival. Bone 42, 1032–1039 (2008).
Tanikawa, R. et al. Interaction of galectin-9 with lipid rafts induces osteoblast proliferation through the c-Src/ERK signaling pathway. J. Bone Min. Res. 23, 278–286 (2008).
Rungaldier, S. et al. Structure-function analysis of human stomatin: A mutation study. PLoS One 12, e0178646 (2017).
Skryabin, G. O. et al. Stomatin is highly expressed in exosomes of different origin and is a promising candidate as an exosomal marker. J. Cell Biochem 122, 100–115 (2021).
Wessel, A. K. et al. Escherichia coli SPFH membrane microdomain proteins HflKC contribute to aminoglycoside and oxidative stress tolerance. Microbiol Spectr. 11, e0176723 (2023).
Hada, N. et al. Receptor activator of NF-κB ligand-dependent expression of caveolin-1 in osteoclast precursors, and high dependency of osteoclastogenesis on exogenous lipoprotein. Bone 50, 226–236 (2012).
Ma, C. et al. Structural insights into the membrane microdomain organization by SPFH family proteins. Cell Res. 32, 176–189 (2022).
Fricke, B. et al. Epithelial Na+ channels and stomatin are expressed in rat trigeminal mechanosensory neurons. Cell Tissue Res. 299, 327–334 (2000).
Appelman, M. D. et al. The lipid raft component stomatin interacts with the Na(+) taurocholate cotransporting polypeptide (NTCP) and modulates bile salt uptake. Cells 9, 986 (2020).
Sedensky, M. M. et al. A stomatin and a degenerin interact in lipid rafts of the nervous system of Caenorhabditis elegans. Am. J. Physiol. Cell Physiol. 287, C468–C474 (2004).
Wu, S. C. et al. Stomatin modulates adipogenesis through the ERK pathway and regulates fatty acid uptake and lipid droplet growth. Nat. Commun. 13, 4174 (2022).
Lee, J. H. et al. Lipid raft-associated stomatin enhances cell fusion. Faseb j. 31, 47–59 (2017).
Ullah, I., Subbarao, R. B. & Rho, G. J. Human mesenchymal stem cells - current trends and future prospective. Biosci. Rep. 35, e00191 (2015).
Rahimzadeh, A. et al. Biotechnological and biomedical applications of mesenchymal stem cells as a therapeutic system. Artif. Cells Nanomed. Biotechnol. 44, 559–570 (2016).
Holness, C. L. & Simmons, D. L. Molecular cloning of CD68, a human macrophage marker related to lysosomal glycoproteins. Blood 81, 1607–1613 (1993).
Yang, Y. et al. Mitochondria and mitochondrial ROS in Cancer: Novel targets for anticancer therapy. J. Cell Physiol. 231, 2570–2581 (2016).
Kim, S. et al. The antioxidant enzyme Peroxiredoxin-1 controls stroke-associated microglia against acute ischemic stroke. Redox Biol. 54, 102347 (2022).
Hawkes, C. P. & Mostoufi-Moab, S. Fat-bone interaction within the bone marrow milieu: Impact on hematopoiesis and systemic energy metabolism. Bone 119, 57–64 (2019).
Xu, K. et al. SOD3 regulates FLT1 to affect bone metabolism by promoting osteogenesis and inhibiting adipogenesis through PI3K/AKT and MAPK pathways. Free Radic. Biol. Med. 212, 65–79 (2024).
Lecka-Czernik, B. et al. Marrow adipose tissue: Skeletal location, sexual dimorphism, and response to sex steroid deficiency. Front Endocrinol. (Lausanne) 8, 188 (2017).
Salamanna, F. et al. Role of bone marrow adipocytes in bone metastasis development and progression: a systematic review. Front Endocrinol. (Lausanne) 14, 1207416 (2023).
Yu, B. & Wang, C. Y. Osteoporosis and periodontal diseases - An update on their association and mechanistic links. Periodontol 2000 89, 99–113 (2022).
Rachner, T. D., Khosla, S. & Hofbauer, L. C. Osteoporosis: now and the future. Lancet 377, 1276–1287 (2011).
Raisz, L. G. Pathogenesis of osteoporosis: concepts, conflicts, and prospects. J. Clin. Invest 115, 3318–3325 (2005).
Phromnoi, K. et al. Anti-osteoporosis effect of perilla frutescens leaf hexane fraction through regulating osteoclast and osteoblast differentiation. Molecules 27, 824 (2022).
Levental, I., Levental, K. R. & Heberle, F. A. Lipid Rafts: Controversies resolved, mysteries remain. Trends Cell Biol. 30, 341–353 (2020).
Simons, K. & Sampaio, J. L. Membrane organization and lipid rafts. Cold Spring Harb. Perspect. Biol. 3, a004697 (2011).
Luo, W. et al. SREBP signaling is essential for effective B cell responses. Nat. Immunol. 24, 337–348 (2023).
Sha, Y. L. et al. B3GALT4 remodels the tumor microenvironment through GD2-mediated lipid raft formation and the c-met/AKT/mTOR/IRF-1 axis in neuroblastoma. J. Exp. Clin. Cancer Res 41, 314 (2022).
Ryu, J. et al. Proteomic analysis of osteoclast lipid rafts: the role of the integrity of lipid rafts on V-ATPase activity in osteoclasts. J. Bone Min. Metab. 28, 410–417 (2010).
Matsubara, T. et al. Cbp recruitment of Csk into lipid rafts is critical to c-Src kinase activity and bone resorption in osteoclasts. J. Bone Min. Res. 25, 1068–1076 (2010).
Kim, A. S., Girgis, C. M. & McDonald, M. M. Osteoclast recycling and the rebound phenomenon following denosumab discontinuation. Curr. Osteoporos. Rep. 20, 505–515 (2022).
Dong, Y., et al. A clinical-stage Nrf2 activator suppresses osteoclast differentiation via the iron-ornithine axis. Cell Metab, https://doi.org/10.1016/j.cmet.2024.03.005 (2024).
Sun, X. et al. The Nrf2 activator RTA-408 attenuates osteoclastogenesis by inhibiting STING dependent NF-κb signaling. Redox Biol. 28, 101309 (2020).
Brookes, P. S., Yoon, Y., Robotham, J. L., Anders, M. W. & Sheu, S. S. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 287, C817–C833 (2004).
Marques-Carvalho, A., Sardão, V. A., Kim, H. N. & Almeida, M. ECSIT is essential for RANKL-induced stimulation of mitochondria in osteoclasts and a target for the anti-osteoclastogenic effects of estrogens. Front Endocrinol. (Lausanne) 14, 1110369 (2023).
Liu, W. et al. PRDX1 activates autophagy via the PTEN-AKT signaling pathway to protect against cisplatin-induced spiral ganglion neuron damage. Autophagy 17, 4159–4181 (2021).
Moretton, A. et al. A metabolic map of the DNA damage response identifies PRDX1 in the control of nuclear ROS scavenging and aspartate availability. Mol. Syst. Biol. 19, e11267 (2023).
Dalla Rizza, J., Randall, L. M., Santos, J., Ferrer-Sueta, G. & Denicola, A. Differential parameters between cytosolic 2-Cys peroxiredoxins, PRDX1 and PRDX2. Protein Sci. 28, 191–201 (2019).
Sadvakassova, G. et al. Active hematopoiesis triggers exosomal release of PRDX2 that promotes osteoclast formation. Physiol. Rep. 9, e14745 (2021).
Wu, K. & Wang, L. Stomatin-knockdown effectively attenuates sepsis-induced oxidative stress and inflammation of alveolar epithelial cells by regulating CD36. Exp. Ther. Med. 23, 69 (2022).
Wang, M., Li, C. & Shi, W. Stomatin-like protein-2 confers neuroprotection effect in oxygen-glucose deprivation/reoxygenation-injured neurons by regulating AMPK/Nrf2 signalling. J. Drug Target 28, 600–608 (2020).
Li, B. et al. Puerarin improves OVX-induced osteoporosis by regulating phospholipid metabolism and biosynthesis of unsaturated fatty acids based on serum metabolomics. Phytomedicine 102, 154198 (2022).
Shen, G. et al. Foxf1 knockdown promotes BMSC osteogenesis in part by activating the Wnt/β-catenin signalling pathway and prevents ovariectomy-induced bone loss. EBioMedicine 52, 102626 (2020).
Li, Y. et al. Urolithin B suppressed osteoclast activation and reduced bone loss of osteoporosis via inhibiting ERK/NF-κB pathway. Cell Prolif. 55, e13291 (2022).
Ge, G. et al. Punicalagin ameliorates collagen-induced arthritis by downregulating M1 macrophage and pyroptosis via NF-κB signaling pathway. Sci. China Life Sci. 65, 588–603 (2022).
Lab, J. https://BioRender.com/b49m861 (2025).
Acknowledgements
Fig. 7A, Supplementary Figs. 2A, 3A and 7A in the manuscript were created using the online platform www.biorender.com, and all images are appropriately licensed. This work was supported by the National Natural Science Foundation of China (82472525 and 82272567 to D.C.G.), Jiangsu Medical Research Project (ZD2022014 to D.C.G.), Program of Suzhou Health Commission (GSWS2022002 to D.C.G.), National and Local Engineering Laboratory of New Functional Polymer Materials (SDGC2205 to D.C.G.), Foundation of National Center for Translational Medicine (Shanghai) SHU Branch (SUITM-202403 to D.C.G.), the Project of MOE Key Laboratory of Geriatric Diseases and Immunology (No. KJS2502 to D.C.G.), the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD to D.C.G.), National Key Research and Development Program of China, Ministry of Science and Technology (2023YFC2509903 to H.L.Y.) and Medical Research Project of Jiangsu Provincial Health and Wellness Commission (MQ2024038 to K.C.). We express our gratitude to Professor Zaixiang Tang from Soochow University for his meticulous guidance and assistance in applying statistical methods in our manuscript.
Author information
Authors and Affiliations
Contributions
H.Q.T.: Writing – original draft, Software, Methodology, Visualization, Data curation, Conceptualization. K.C.: Software, Data curation, Methodology. Q.F.W.: Software, Data curation. P.F.Z.: Software, Methodology. M.C.: Software, Methodology. W.X.C.: Software, Methodology. C.Y.: Resources. Y.G.: Software. H.L.Y.: Methodology, Supervision, Funding acquisition. Q.Z: Writing – review & editing, Supervision. S.J.L.: Writing – review & editing, Resources, Conceptualization. D.C.G.: Writing – review & editing, Methodology, Supervision, Resources, Funding acquisition, Conceptualization.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Danilo Segovia and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tao, H., Chen, K., Wang, Q. et al. Targeting lipid raft-related stomatin to ameliorate osteoporosis in preclinical models. Nat Commun 16, 5495 (2025). https://doi.org/10.1038/s41467-025-60032-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-60032-9
