
Abstract
Gastric Squamous Cell Carcinoma (GSCC) is a rare but aggressive subtype of gastric cancer with unique histopathology, whose etiology remains poorly understood. Here, we perform genomics analyses of twenty GSCC samples and find that epigenetic regulation genes are among the most frequently mutated genes, including Enhancer of zeste homolog 2 (EZH2). Ezh2 loss induces squamous feature both in gastric organoids in vitro and in vivo mouse model. Ezh2 deficiency, together with Trp53 and Pten loss, both of which are also frequently mutated in GSCC, give rise to full-blown GSCC in mice. Mechanistically, we find that Ezh2 could repress the expression of Transcription factor AP-2 gamma (Tfap2c), a transcription factor with the ability to initiate epidermal squamous differentiation, through H3K27 methylation. Disruption of Tfap2c reduces the squamous characteristics of the Ezh2 loss-driven GSCC and reverses its resistance to chemo treatment. Our findings elucidate key molecular mechanisms underlying GSCC pathogenesis and identify potential therapeutic targets for this aggressive malignancy.
Similar content being viewed by others


Introduction
Gastric cancer (GC), a leading malignancy, is responsible for about one million deaths per year1,2. GC is known for its heterogeneity, including the histopathology, the infections of Helicobacter pylori and Epstein-Barr virus (EBV), and genetic alterations, which complicates its diagnosis and treatment3. Most of the GC are adenocarcinoma, which can be grouped into four molecular subtypes: EBV-infected (EBV), microsatellite instability (MSI), genomically stable (GS), and chromosomal instability (CIN)4. Each of these subtypes has their unique pathogenesis and might respond differently to current treatments. Besides gastric adenocarcinoma (GAC), there are many other types of gastric cancers with distinct histopathology, such as medullary carcinoma, which is characterized with irregular nests of polygonal tumor cells and associated with EBV infection, Paneth cell carcinoma, which is majorly composed of malignant Paneth cells, and carcinosarcoma, which displays histological features of both carcinoma and sarcoma5,6,7. Among them, gastric squamous cell carcinoma (GSCC), purely composed of squamous tumor cells, is a very rare type of GC8. It is proposed to originate from the gastric mucosa, but its molecular pathogenesis is still poorly understood9,10. While GSCC consists of only 0.04–0.4% of total GC and so far, only about 100 cases have been reported9,11,12,13, gastric adenosquamous carcinoma (GASC), a mixture of adenocarcinoma and squamous tumor cells, is relatively common14,15. Most of both GSCC and GASC are diagnosed at late stages, often have worse prognosis than GAC16,17,18, and seem to be less responsible for current standard treatments. Therefore, new diagnostic and treatment strategies need to be established for a better understanding of the molecular mechanisms underlying the process of GSCC tumorigenesis.
Current understanding of genetic aberrations in GSCC remains limited due to insufficient acquisition of high-quality biospecimens attributable to its rarity. In this work, formalin-fixed paraffin-embedded (FFPE) tissue samples from 20 histologically confirmed GSCC patients are collected at our institution between 2017 and 2024 for whole exome sequencing (WES) and targeted bisulfite sequencing (TBS) analysis. Frequent genetic alterations in GSCC primarily include TP53 mutations, abnormalities in the PTEN/PI3K pathway, and alterations in epigenetic regulatory genes. Notably, our study identifies a high frequency of EZH2 loss in GSCC patients. Accordingly, we generate a primary and orthotopic GSCC mouse model and find that EZH2 loss promotes GSCC probably through regulating the expression of TFAP2C, a transcription factor critical for squamous cell differentiation.
Results
The molecular characteristics of GSCC
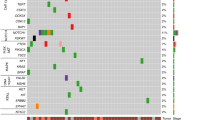
To elucidate the genomic and epigenomic characteristics of GSCC, we obtained 20 tumor samples from West China Hospital of Sichuan University (Supplementary Fig. 1a and Supplementary Data. 1). Hematoxylin and Eosin (H&E) staining confirmed the predominantly squamous malignant cell composition of all specimens, and Immunohistochemistry (IHC) staining indicated the expression of the squamous carcinoma canonical diagnostic marker P40 (Supplementary Fig. 1b). The median age at diagnosis for these GSCCs was 57 years, with a female-to-male ratio of 1:3 (Supplementary Data. 1). Laser capture microdissection (LCM) was utilized to isolate pure tumor regions from GSCC specimens for subsequent genomic sequencing analysis. Through whole-exome sequencing analysis, we identified TP53 mutations in 60% (12/20) of GSCC patients, consistent with its status as one of the most frequently mutated genes across various cancer types18. Comparative analysis with the TCGA gastric adenocarcinoma (STAD) cohort revealed significantly elevated mutation frequencies in epigenetic regulatory genes among GSCC samples: KMT2A (40% vs. 29%), KMT2B (55% vs. 16%), KMT2C (60% vs. 18%), and KMT2D (40% vs. 19%). Furthermore, key components of the PTEN/PI3K pathway, including PIK3CA, MTOR, TSC1/2, and AKT 1/2, demonstrated high mutation frequencies in GSCC patients (Fig. 1a). Notably, EZH2 mutations were identified in 5 of 20 patients (Fig. 1a, b). Copy number analysis revealed chromosome 7q deletions encompassing the EZH2 locus in 3 cases, with one patient (GSCC6) demonstrating both a 7q deletion and a missense mutation in EZH2 (Fig. 1a and Supplementary Fig. 1c). Furthermore, we performed targeted bisulfite sequencing (TBS) of the CpG island within the EZH2 promoter region using LCM-isolated tumor and paired normal tissues from 7 GSCC patients. Tumor regions showed increased methylation levels than adjacent normal tissues (Supplementary Fig. 1d). These findings collectively demonstrate that EZH2 undergoes functional loss in GSCC through genetic or epigenetic mechanisms, including mutations, deletions, and increased promoter methylation (Fig. 1c).

a Curated list of frequently altered genes in 20 GSCC patients. b Protein domain structure of EZH2 with mutations identified in GSCC patients. c Summary of mutations, deletions, and methylation levels of EZH2 in 20 GSCC patients. d Boxplot showing the expression levels of EZH2 in squamous carcinoma gene signature high (n = 58) and low (n = 313) samples in the TCGA-STAD cohort. Two-tailed Wilcoxon rank-sum test. The boxes indicate the median value, interquartile range, with whiskers extending from the box boundaries to upper/lower quartile ± 1.5 interquartile range. The p-value was determined by the two-tailed unpaired Wilcoxon rank-sum test. e GSEA showed that the GO_KERATINIZATION gene sets were significant enriched in GC patients with high expression of squamous carcinoma gene signatures (n = 58) compared to low expression ones (n = 313) in the TCGA-STAD cohort. Gene set enrichment significance was assessed by the unpaired two-tailed permutation test. f Survival curves of GC patients stratified by high and low expression of squamous carcinoma gene signature in the TCGA-STAD cohort. The cut-off values were determined by maximally selected rank statistics. The p-value was determined by the log-rank test. g The scatter plot showing the negative correlation between expression levels of squamous signature genes and EZH2 in the TCGA-STAD cohort. The p-value was determined by the two-tailed ttest. r, Pearson’s correlation coefficient. The shaded band around the regression line indicates the 95% confidence interval (CI) of the linear fit. h Representative IF staining of EZH2 (red) and P40 (green) in GAC tumor tissues (left) and GSCC tumor tissues (right). Scale bar, 100 µm. Source data are provided as a Source Data file.
We next sought to determine whether EZH2 expression is associated with squamous differentiation in gastric tumors. Given that some gastric cancers, though diagnosed as adenocarcinoma, contain more or less tumor cells with squamous differentiation19,20. We stratified The Cancer Genome Atlas GC cohort (TCGA-STAD) patients into Squahi (n = 58) and Squalow (n = 313) groups, utilizing a gene signature derived from the TCGA esophageal carcinoma cohort (ESCC versus other subtypes) (Supplementary Data. 2). We found that EZH2 expression was significantly reduced in Squahi patients compared to Squalow patients (Fig.1d). Gene Set Enrichment Analysis (GSEA) revealed that the GO_KERATINIZATION gene set was significantly positively enriched in the upregulated genes of Squahi GC patients compared to Squalow ones (Fig. 1e). In accordance with prior reports17,21, Squahi GC patients exhibited a dramatically reduced overall survival compared to the Squalow ones (Fig.1f and Supplementary Data. 2). Differential expression analysis between Squahi and Squalow patients revealed 1499 upregulated and 1446 downregulated genes, respectively (p < 0.05 and absolute log2fold-change ≥0.5, Supplementary Data. 2). Notably, the KERATIN genes, such as KRT5, KRT6A, KRT6B, and KRT14, were upregulated in the Squahi GC patients compared to the Squalow ones. Concurrently, adenocarcinoma typical marker genes, including MUCINs gene family, LGR5, and TFF1, were downregulated in the Squahi patients22,23,24. The reduced levels of ERBB2 observed here were consistent with those in lung squamous cell carcinoma, comparing to lung adenocarcinoma25. These data suggest that GSCC, like other squamous cell carcinomas, would be less dependent on the ERBB2 signaling pathway. MKI67 was downregulated in the Squahi patients, which might be due to the more differentiation status of those samples, for we selected those with significant squamous differentiation properties and differentiated tumors generally expressed lower levels of MKI6726 (Supplementary Fig. 1e). Of note, the expression levels of EZH2 exhibited a striking negative correlation with squamous signature genes (Fig. 1g). Moreover, our analysis revealed a complete absence of EZH2 expression in P40 positive GSCC patients, in contrast to its significantly elevated expression levels observed in gastric adenocarcinoma (GAC) patients (Fig. 1h). Collectively, these data strongly argued that EZH2 was generally downregulated in GSCC.
Ezh2 deficiency gives rise to squamous features in vitro and in vivo
To better understand the potential biological function of EZH2 in GSCC. First, we cultured gastric epithelial organoids from normal gastric tissue of CAS9-EGFP mice, sgScr or sgEzh2 were introduced into these organoids by lentivirus infection. Western blot analysis confirmed the successful knockout of Ezh2, as evidenced by a significant reduction in H3K27 trimethylation (H3K27me3) levels following Ezh2 deletion (Fig. 2a, b). Morphologically, Ezh2 deficient organoids displayed a significantly increased size, and altered morphology compared to controls (Fig. 2c and Supplementary Fig. 2a). Furthermore, compared to wild-type ones, Ezh2 deficient organoids exhibited a higher proportion of CK14 and P40 positive cells, indicating the acquisition of squamous epithelial characteristics upon Ezh2 loss (Fig. 2c and Supplementary Fig. 2b, c).

a Western blot analysis of EZH2 in sgScr (Cas9-sgScramble), sgEzh2-1(Cas9-sgEzh2-1), and sgEzh2-2(Cas9-sgEzh2-2) gastric organoids (−1 and −2 represent two independent sgRNAs targeting Ezh2). Representative blot (a) of n = 2 technical replicates. b Western blot analysis of H3K27me3 in sgScr, sgEzh2-1, and sgEzh2-2 gastric organoids. Representative blot (b) of n = 2 technical replicates. c Representative bright-field(top), H&E(middle), CK14 and P40 IF staining(bottom) images of sgScr, sgEzh2-1, and sgEzh2-2 mouse gastric organoids. Scale bars, 50 µm (top) and 20 µm (middle and bottom). d The heatmap showing the differential expression genes (p < 0.05 and absolute log2 fold-change > 0.5, p-value was determined by unpaired two-tailed Wald test) between sgScr, sgEzh2-1 and sgEzh2-2 gastric organoids, measured by RNA-seq analyses. e Statistical graphs showing the diameter of TP (Trp53-/-; sgPten) and TPE (Trp53-/-; sgPten; sgEzh2) gastric organoids. f Statistical graphs showing the organoids formation rate of TP and TPE gastric organoids (n = 3 biological replicates). g The bright-field image of subcutaneous tumors of TP (n = 3 mice) and TPE (n = 3 mice), Scale bar, 1 cm. h Tumor weight of subcutaneously transplanted TP (n = 3 mice) and TPE (n = 3 mice). i H&E staining of subcutaneous tumor tissues of TP and TPE mice. Scale bar, 20 µm. j Representative images of CK14(left), CK5(middle) and P40(right) IF staining of subcutaneous tumor tissues of TP and TPE mice. Scale bars, 20 µm and 50 µm. Data are shown as means ± SD, p-value was determined by unpaired two-tailed t test(e, f, h). Source data are provided as a Source Data file.
To comprehensively characterize the molecular features of Ezh2 deficient gastric organoids, we conducted RNA-seq analysis to profile the transcriptomic differences of gastric organoids with sgScr and sgEzh2. Ezh2 deficiency induced substantial alterations in gene expression (633 upregulated genes versus 506 downregulated genes) (Fig. 2d and Supplementary Data 3). A series of keratin genes, which represent the squamous differentiation, and Hox family genes, known downstream targets of Ezh2, were significantly upregulated in Ezh2-deficient organoids (Fig. 2d). These transcriptomic alterations indicated that Ezh2 loss induces squamous features. GSEA showed that the chromatin segregation and MYC target pathways were significantly positively enriched in the upregulated genes of sgEzh2 organoids compared to sgScr ones (Supplementary Fig. 2d). Squamous signature genes were also enriched in Ezh2-deficient organoids compared to the control (Supplementary Fig. 2e).
To investigate whether EZH2 deficiency drives the development of gastric squamous cell carcinoma, the OPCM (Organoid-initiated Precision Cancer Model) strategy was applied27,28,29. Firstly, based on the WES data of the 20 GSCC patients, we identified that the combination of EZH2, TP53, and PTEN deficiency has strong relevance to the GSCC patients. Hence, we first introduced Pten-targeting sgRNA into Trp53-/-; Cas9-EGFP gastric organoids. After confirming successful Pten mutation (Supplementary Fig. 2f, g), we subsequently transduced Ezh2-targeting sgRNA and scramble sgRNA, a non-targeting control sgRNA that does not bind any coding regions, tagging with red fluorescence. The T7 endonuclease I assay validated successful Ezh2 mutation (Supplementary Fig. 2f). Consistently, TPE (Trp53-/-; sgPten; sgEzh2) organoids showed significantly increased dimensions and proliferation capacity compared to TP (Trp53-/-; sgPten) ones (Fig. 2e, f). Importantly, TPE organoids also presented squamous features, as demonstrated by CK5 and CK14 IF staining (Supplementary Fig. 2h, i).
Furthermore, premalignant TP and TPE organoids were subcutaneously transplanted into recipient mice to assess their tumorigenic potential. Ezh2 loss dramatically accelerated tumor growth (Fig. 2g, h). Pathological analysis revealed that the TPE tumor displays a disorganized arrangement with significant atypia. The cells exhibit large, vesicular nuclei, high nuclear-to-cytoplasmic ratios, and prominent nucleoli, consistent with the pathological features of typical squamous cell carcinoma. In contrast, mice with TP organoids developed hyperplasia (Fig. 2i). Further, IF staining of markers for squamous cell carcinoma, such as CK14, CK5 and P40, confirmed that TPE organoids formed GSCC (Fig. 2j). Therefore, Ezh2 deficiency cooperates with p53 and Pten loss to promote GSCC tumorigenesis in vivo.
Ezh2 deficiency drives gastric squamous cell carcinoma progression in an orthotopic mouse model
Next, we transplanted the TP and TPE gastric organoids into the submucosal layer of the recipients’ stomach. The recipient mice were monitored weekly and sacrificed once their body weights had decreased to around 80% of the original level. Notably, mice transplanted with TPE organoids exhibited shorter overall survival compared to the control group (TPE-1: median 167.5 days after transplantation, p = 0.005; TPE-2: median 133.5 days after transplantation, p = 0.0016) (Fig. 3a). Biopsy dissection confirmed the development of lesions in the stomach originating from TPE gastric organoids, characterized by specific expressions of CAS9 and mCherry (Fig. 3b and Supplementary Fig. 3a, b). Furthermore, Ezh2 deficiency significantly augmented tumor growth (Fig. 3c). The loss of EZH2, accompanied by reduction in H3K27me3, was confirmed by IHC staining and western blotting in TP and TPE tumors (Fig. 3d–g). Pathological analyses revealed that the TPE tumors displayed large areas of keratin pearls, and concentric layers of keratin deposition, surrounded by hyperplasia of differentiated keratinocyte-like cells (Fig. 3h, i). In addition, many squamous cell carcinoma markers, such as P40, CK14, CK5/6, and CK5, exhibited remarkable expression in TPE tumors (Fig. 3h and Supplementary Fig. 3d). In contrast, TP tumors exhibited more representative adenocarcinoma cell features according to the histomorphology analysis and the expressions of CK20, MUC1, MUC5AC, and CK7 as indicated by IHC staining (Supplementary Fig. 3c, d). Further, TPE tumors exhibited elevated distal metastatic capabilities, particularly in peritoneal lymph nodes and livers, whereas no detectable metastases were observed in TP ones (Fig. 3j–l and Supplementary Fig. 3e–g). Taken together, we generated a primary, orthotopic, and genetically defined GSCC mouse model. This model faithfully replicates the clinical features observed in GSCC patients, encompassing both histological and pathological characteristics.

a Survival curve of mice orthotopically transplanted with TP, TPE-1, and TPE-2 organoids (n = 6 mice). All curves were analyzed by log-rank (Mentel-Cox) test (− 1 and − 2 represent two independent sgRNAs targeting Ezh2). b Representative bright-field (top) and red fluorescence(bottom) images of gastric cancer transduced with TP, TPE-1 and TPE-2 organoids. Scale bar, 1 mm. c Statistical graphs showing the tumor weight of mice orthotopically transplanted with TP, TPE-1 and TPE-2 organoids (n = 6 mice). d Representative IHC staining of EZH2 in gastric tumor sections of TP (left), TPE-1 (middle), and TPE-2(right) mice. Scale bar, 20 µm. e Western blot analysis of EZH2 in TP, TPE-1 and TPE-2 mouse gastric tumor organoids. Representative blot (e) of n = 2 biological replicates. f Representative IHC staining of H3K27me3 in gastric cancer sections of TP (left), TPE-1 (middle), and TPE-2(right) mice. Scale bar, 20 µm. g Western blot analysis of H3K27me3 in TP, TPE-1, and TPE-2 gastric tumor organoids. Representative blot (g) of n = 2 biological replicates. h Representative H&E, P40, CK14, CK5/6, and CK5 IHC staining of TP (top), TPE-1 (middle), and TPE-2(bottom) tumor section from mice. Scale bar, 50 µm. i Statistical graphs of keratin pearl area percentages in tumor tissues from TP, TPE-1, and TPE-2 mice (n = 4 mice). j Representative bright-field (left) and red-fluorescence(right) images of liver from TP (top) and TPE (bottom) tumor-bearing mice. Scale bars, 2 mm(top) and 1 mm(bottom). k H&E staining of liver sections from TP (top) and TPE (bottom) tumor-bearing mice. Scale bar, 50 µm. l Representative IHC staining of CAS9, CK14, CK5/6 and P40 in liver sections from TPE tumor-bearing mice. Scale bar, 50 µm. Data are shown as means ± SD, p-value was determined by unpaired two-tailed t test(c, i). The samples derive from the same experiment, and that gels were processed in parallel for quantitative comparisons (e.g.,). Source data are provided as a Source Data file.
Multi-omics analyses reveal the squamous feature driven by EZH2 deficiency
To delineate the molecular characteristics of GSCC, we conducted RNA sequencing (RNA-seq) analysis of TP and TPE tumor organoids. The transcriptome analysis revealed TP and TPE tumor organoids formed distinct clusters in principal component analysis (Supplementary Fig. 4a). Notably, TPE tumor organoids exhibited a greater number of upregulated genes compared to the TP groups (Fig. 4a and Supplementary Data. 3). Among these upregulated genes, HOX family, known targets of EZH2, were significantly enriched, consistent with observations in Ezh2-knockout normal organoids (Figs. 2d and 4a). Several pathways associated with keratin metabolism, TP63 targets, and SUZ12 targets were significantly enrichment in TPE organoids, indicating more aggressive and malignant characteristics of the TPE tumors30,31,32,33 (Supplementary Fig. 4b). Meanwhile, squamous signature genes were significantly positively enriched in upregulated genes of TPE tumors compared to TP ones (Fig. 4b). Quantitative calculations also revealed that TPE tumors exhibited significantly elevated expression levels of gastric squamous signature genes compared to TP ones (Fig. 4c).

a The heatmap showing the differential expression genes (p < 0.05 and absolute log2 fold-change > 0.5, p-value was determined by the unpaired two-tailed Wald test) between TP (n = 2 mice) and TPE (n = 2 mice) tumor organoids. b GSEA showing that the squamous signature genes were significantly positively enriched in the upregulated genes of TPE organoids compared to TP ones. Gene set enrichment significance was assessed by the unpaired two-tailed permutation test. c Box plots illustrating the gastric squamous signature scores in TP (n = 2 mice) and TPE (n = 2 mice) tumor organoids, measured by RNA-seq analyses. The boxes indicate the median value, interquartile range, with whiskers extending from the box boundaries to upper/lower quartile ± 1.5 interquartile range. p-value was determined by the two-sided Likelihood ratio test. d Pie chart depicting the distribution of EZH2 binding peaks across annotated genomic regions in TP gastric organoids. e Average intensity curves (top) and tornado plots (bottom) showing the binding intensity of EZH2 (left) and H3K27me3modification levels in these corresponding regions between TP and TPE organoids (right). Scale bars denote BPM (bins per million mapped reads) for CUT&Tag signal. f Pie chart (left) showing the proportion of genes with reduced H3K27me3 modification levels in TPE gastric organoids compared to TP ones, categorized by the presence or absence of EZH2 binding. Pie chart (right) displaying the proportion of transcriptional changes in genes with both EZH2 binding and reduced H3K27me3 levels in TPE gastric organoids compared to TP ones. g Gene ontology enrichment results of overlapping genes associated with EZH2 binding, significantly reduced H3K27me3 modification levels, and up-regulated expression in TPE gastric organoids compared to the TP group. p-values were determined by the unpaired one-tailed hypergeometric test. h The network showing protein-protein interactions among EZH2-bound squamous signature genes with both reduced H3K27me3 and up-regulated expression levels in TPE vs. TP organoids, analyzed using the STRING database. Source data are provided as a Source Data file.
Given that EZH2, the catalytic subunit of the PRC2/EED-EZH2 complex, is essential for H3K27 di- and tri-methylation34,35, we performed Cleavage Under Targets and Tagmentation (CUT&Tag) to investigate the corresponding EZH2-binding genomic regions and H3K27me3 levels in TP and TPE premalignant gastric organoids. The results demonstrated that Ezh2 knockout significantly influenced H3K27me3 modification, causing substantial changes (Supplementary Fig. 4c, d). In accordance with EZH2’s enzymatic activity, Ezh2-deficient gastric organoids exhibited a reduction in the number of modified peaks and the average signal intensities of H3K27me3. Specifically, Ezh2-deficient organoids displayed 1294 significantly reduced H3K27me3 peaks compared to TP organoids (Supplementary Fig. 4e, f and Supplementary Data. 4). Upon further analysis of EZH2 direct binding sites revealed their preferential distribution in distal intergenic (31.12%), intron (29.13%), and promoter regions (34.31%), consistent with previous reports36,37,38 (Fig. 4d and Supplementary Data. 4). These EZH2-directly regulated regions showed a significant reduction in H3K27me3 modifications in TPE organoids compared to TP organoids (Fig. 4e). Particularly, 97.1% of genes showing reduced H3K27me3 modification levels in TPE organoids compared to TP organoids were EZH2-bound. Among these EZH2-bound genes, 57.8% were upregulated in TPE tumors, while only 18% were downregulated (Fig. 4f).
Functional analysis revealed significant enrichment of Gene Ontology terms related to epidermal development, skin morphogenesis, keratinocyte differentiation, and keratinization processes (Fig. 4g). Notably, beyond known EZH2 target genes, we identified that TFAP2C, as a crucial regulator of surface ectoderm differentiation, occupies a central position in the squamous regulatory network, and may interact with ZIC1, ZIC3, FOXD3, BMP4, TFAP2A, TFAP2B, and DLX3 (Fig. 4h). Then, by integrating EZH2 binding strength, H3K27me3 modification levels, and RNA expression abundance into a composite score, we identified Tfap2c as the top-ranked gene among EZH2-bound targets (Fig. 5a). EZH2 bound the region within 5 kb of the transcriptional start site (TSS) of Tfap2c, maintaining a high level of H3K27me3 decoration to suppress its expression (Fig. 5b). Moreover, TPE tumors showed higher expression of Tfap2c at both mRNA and protein levels compared to TP ones (Fig. 5c, d). In addition, TFAP2C positive cells were accompanied by P40 positive in only TPE tumors (Fig. 5e and Supplementary Fig. 5a, b). Furthermore, multiplex Immunohistochemistry (mIHC) analysis of GSCC patient specimens revealed that EZH2 is exclusively expressed with both P40 and TFAP2C specifically within tumor regions (Fig. 5f and Supplementary Fig. 5c). These results indicate that Tfap2c plays a crucial role in promoting GSCC in the context of Ezh2 deficiency.

a The scatter plot showing the integrated ranking of the EZH2 regulated genes, The integrated score combines EZH2 binding intensity, H3K27me3 modification levels, and RNA expression abundance. b Integrative Genomics Viewer showing the EZH2 binding and H3K27me3 modification levels at the Tfap2c locus in TP and TPE gastric organoids. c Box plot showing the relative expression levels of Tfap2c in TP (n = 3 mice) and TPE (n = 3 mice). d Western blot analysis of TFAP2C in TP, TPE-1, and TPE-2 mice gastric tumor organoids. Representative blot (d) of n = 2 biological replicates. The samples derive from the same experiment and that gels were processed in parallel for quantitative comparisons. e IF staining of P40 (red), TFAP2C (green), and EZH2(white) in TPE tumors. Scale bar, 50 µm. f mIHC staining of P40 (red), TFAP2C (green), and EZH2 (white) in GSCC16 tumor tissue and normal tissue. Scale bars, 200 µm and 20 µm. g Statistical graphs showing the P40 and TFAP2C fluorescence intensity of TPE tumor organoids with sgScr, sgTfap2c-1 and sgTfap2c-2 (n = 4 biological replicates) (− 1 and − 2 represent two sgRNAs targeting Tfap2c). h Representative images of H&E and TFAP2C, MUC1 and MUC5AC IHC staining of TPE tumors with sgScr, sgTfap2c-1 and sgTfap2c-2. Scale bar, 20 µm. i Statistical graphs showing the percentages of keratin pearl area of tumors in TPE tumor organoids with sgScr, sgTfap2c-1 and sgTfap2c-2 (n = 4 mice). j Representative IF staining of TFAP2C (green) and P40 (red) in TPE-sgScr and TPE-sgTfap2c tumor tissues. Scale bar, 50 µm. Data are shown as means ± SD, p-values were determined by unpaired two-tailed t test(c, g, i). Source data are provided as a Source Data file.
TFAP2C is negatively regulated by EZH2 and plays a crucial role in maintaining the characteristics of squamous cell carcinoma
To elucidate the functional roles of TFAP2C in GSCC pathogenesis, we systematically evaluated its expression patterns across two independent TCGA cohorts: the Stomach Adenocarcinoma (STAD) cohort and the Esophageal Cancer cohort. Notably, the expression level of TFAP2C was significantly elevated in EZH2-loss GC patients compared to those with EZH2-intact ones (Supplementary Fig. 5d and Supplementary Data. 5). And consistently, the expression level of TFAP2C was significantly increased in esophageal squamous cell carcinoma patients compared to esophageal adenocarcinoma patients (Supplementary Fig. 5e and Supplementary Data. 5). Furthermore, GSEA showed that GO_KERATINOCYTE_DIFFERENTIATION, GO_KERATIN_FILAMENT, GO_KERATINIZATION, and GO_REGULATION_OF_KERATINOCYTE_DIFFERENTIATION pathways genes were significantly positively enriched in the upregulated genes of TFAP2Chi GC patients compared to TFAP2Clow ones (Supplementary Fig. 5f).
To further investigate the function of TFAP2C in GSCC, we disrupted Tfap2c with CRISPR-Cas9 in TPE tumor organoids, the disruption efficiency of Tfap2c was validated by T7 endonuclease I mismatch detection assay (T7E1) (Supplementary Fig. 5g). In addition, western blot analysis showed a dramatic reduction in TFAP2C protein levels after Tfap2c knockout (Supplementary Fig. 5h). IF staining showed that Tfap2c-knockout tumor organoids exhibited negative expression of P40, whereas TPE tumors displayed strong positive expression of P40 (Fig. 5g and Supplementary Fig. 5i).
Furthermore, TPE-sgScr and TPE-sgTfap2c tumor organoids were transplanted into the mouse stomach. Histopathological analysis revealed a dramatic reduction in keratin pearls and elevated expression of adenocarcinoma markers, MUC1 and MUC5AC, in Tfap2c disruption tumors, in contrast to TPE tumors, which maintained typical squamous cell carcinoma characteristics (Fig. 5h, i). Consistent with histopathological analyses results, tumor tissues with Tfap2c deletion exhibited nearly complete loss of P40 expression (Fig. 5j). In conclusion, these data revealed that Tfap2c, negatively regulated by Ezh2, plays an important role in maintaining the pathological and molecular features of squamous cell carcinoma.
To investigate the molecular mechanism underlying Tfap2c mediated squamous characteristics in GSCC, we performed CUT&Tag of TFAP2C in TPE gastric organoids. Initially, TFAP2C was specifically bound to the promoter regions (32.47%), intronic regions (30.31%) and distal intergenic regions (31.45%) (Fig. 6a and Supplementary Data. 4). Notably, high levels of TFAP2C binding were detected in both the distal intergenic and promoter regions of genes associated with squamous differentiation, including Trp63, Krt5, and Krt14 (Fig. 6b). RNA-seq analysis showed a notable transcriptional alteration between TPE tumor organoids with or without sgTfap2c (Fig. 6c). Importantly, GSEA showed that DRAWICHE_SQUAMOUS_CELL_CARCIMOMA_UP pathway genes were negatively enriched in sgTfap2c organoids compared to TPE organoids (Fig. 6d). Specifically, 1275 genes, directly bound by TFAP2C, exhibited significant downregulated expression levels upon Tfap2c knockout (Fig. 6e). These TFAP2C-regulated genes displayed a remarkable enrichment of Gene Ontology terms related to epithelial cell proliferation, the Wnt signaling pathway, keratinocyte proliferation, and the keratinization process (Fig. 6f). Quantitative calculations revealed that TPE tumor organoids with sgTfap2c exhibited significantly downregulated expression levels of gastric squamous signature genes compared to TPE ones (Fig. 6g). Besides, we found that disrupting Tfap2c reduced its resistance to chemotherapy (Supplementary Fig. 6a–c). Understanding of the molecular mechanisms underlying the pathogenesis might lead to the identification of potential therapeutic targets and drugs for GSCC. Indeed, we found that many drugged and druggable genes were upregulated in the TPE tumors, compared to the TP tumors, while downregulated by Tfap2c loss (Fig. 6h). And these genes were enriched in multiple kinase activity regulation pathways, such as the CDK pathway, the MAPK pathway, and the JAK-STAT pathway (Fig. 6i), indicating that these genes and pathways may serve as potential therapeutic targets for GSCC.

a Pie chart showing the distribution of TFAP2C binding peaks in annotated regions of the genome in TPE gastric organoids. b Integrative Genomics Viewer showing the TFAP2C binding levels at ΔNp63, Krt5, and Krt14 gene loci in TPE gastric organoids. c The heatmap showing the differential expression genes (p < 0.05 and absolute log2 fold-change > 0.5, p-value was determined by the unpaired two-tailed Wald test) between TPE-sgScr, TPE-sgTfap2c−1, and TPE-sgTfap2c−2 gastric tumor organoids. d GSEA showing the negative enrichment of the DARWICHE_SQUAMOUS_CELL_CARCINOMA_UP gene set in TPE-sgTfap2c−1, TPE-sgTfap2c−2 compared to TPE- sgScr gastric tumor organoids. Gene set enrichment significance was assessed by the unpaired two-tailed permutation test. e Pie chart showing the proportion of genes with reduced expression levels in TPE-sgTfap2c gastric tumor organoids compared to TPE-sgScr ones, categorized by the presence or absence of TFAP2C binding. f GO analysis of overlapping genes in Fig. 6e. P-values were determined by the unpaired one-tailed hypergeometric test. g The box plots showing the gastric squamous signature scores in TPE-sgScr, TPE-sgTfap2c-1 and TPE-sgTfap2c-2 tumor organoids (n = 3 biological replicates). The boxes indicate the median value, interquartile range, with whiskers extending from the box boundaries to upper/lower quartile ± 1.5 interquartile range. P-values were determined by the two-sided Likelihood ratio test. h The Venn diagram showing the overlapping genes between the top 1000 upregulated genes in TPE tumor organoids, and the 1000 downregulated genes in TPE-sgTfap2c, relative to TP and TPE-sgScr groups, respectively. P-value was determined by a hypergeometric distribution test. i Gene ontology enrichment analysis of overlapping genes in Fig. 6h. P-values were determined by the unpaired one-tailed hypergeometric test. Source data are provided as a Source Data file.
Discussion
GSCC is a rare but aggressive malignancy, and its molecular features and mechanisms underlying its genesis have not yet been fully elucidated. With whole exome sequencing of 20 GSCC samples, we start to reveal the mutations associated with this disease. It seems that GSCC shares many common mutated genes with other GCs, such as p53 and PTEN/PI3K pathway genes39,40,41,42. Surprisingly, we find that multiple epigenetic regulator genes, especially those involved in histone methylation, are frequently altered in GSCC. This different genomic spectrum further suggests that GSCC is a distinct subtype of GC and thus needs further studies to understand its etiology. Our data, though limited by the small number of patients in the cohort, provides a start to explore the molecular mechanisms of this disease.
Given the extreme difficulty to obtain the clinic samples of GSCC, we have created an in vivo model of GSCC to study its biology. With a strategy with genetically engineered premalignant organoids, recently developed by our and other laboratories28,43,44,45, we generate both subcutaneous and in situ models of GSCC in mice. These models have been developed from premalignant gastric epithelial cells, driven by mutations associated with GSCC, representing the whole process of its tumorigenesis and progression, and thus would be a standard system to investigate the biology of this malignancy. And we have also developed GSCC organoids from these mouse models, which would be a useful tool for large-scale drug screening. These GSCC organoids and animal models, faithfully recapitulating the pathological and molecular characteristics of human disease, would be of value for both basic and translational studies.
With these models, we validate EZH2, a frequently mutated epigenetic regulator genes in GSCC, as a tumor suppressor gene. We show that EZH2 disruption, together with p53 and PTEN loss, gives rise to full-blown GSCC in mice. These results are striking, for EZH2 has been generally proposed to be oncogenic and therefore a therapeutic target in numerous solid cancers, including GAC46,47,48,49,50. Mechanistically, EZH2 directly represses the expression of TFAP2C through H3K27 methylation. TFAP2C is a transcription factor of the AP-2 family and has been shown to be sufficient for skin squamous differentiation by inducing p63 expression51. In GSCC, EZH2 loss leads to upregulation of TFAP2C, which, in turn, is responsible for the malignancy of GSCC. The validation of EZH2 as a GSCC tumor suppressor, the mechanism studies would not only shed light on the understanding of molecular pathology but also facilitate developing potential treatment of this disease.
Methods
Human specimens
This study has obtained all required approvals from China’s Ministry of Science and Technology related to the export of genetic information and materials associated with this research. This study was approved by the Ethical Research Committee on Biomedical Research, West China Hospital of Sichuan University (No.20221046). All participants provided written informed consent prior to participating in the study. The final 20 GSCC samples were obtained from West China Hospital of Sichuan University, with recruitment conducted between 2017 and 2024. All patients were pathologically confirmed to be GSCC by pathologists in West China Hospital. As the information on sex or gender was not relevant in this study, no related analysis was carried out. The characters and related application of clinical patients included in this study was shown in Supplementary Data.1.
Mice
All these mice were bred and kept in the specific pathogen-free animal facility at Sichuan University with autoclaved food, bedding and water. Animals were housed at room temperature (23 ± 2 °C) at a humidity of 30–70% on a 12 h light/12 h dark cycle(6:00–18:00). All animal procedures were approved by the Animal Care and Use Committee of Sichuan University. BALB/cA-nu mice were purchased from Beijing HuaFukang Biological Technology Co. Ltd (6–8 weeks, male, approximately 18–20 g weight). Trp53-/- mice and Cas9-EGFP mice were purchased from Jackson Laboratories (Cat# 002101 and Cat# 026179, respectively). In the mouse experiments, the tumor volumes did not exceed the maximal permitted tumor volume of 1000 mm3.
Gastric organoid genome editing
Mouse gastric organoids culture method was as the same as in our previous report52. sgRNAs were designed on the website of DNA2.0 Gene Design & Synthesis (https://www.atum.bio/pipeline/dna) and cloned into the lentiviral constructs U6-sgRNA-EFS-mCherry-P2A-puro. sgRNA sequences were listed in Supplementary Data. 6. HEK 293 T cells were used to package lentivirus. After organoid dissociation with TrypLETM (GIBCO, Cat# 12604-028) and collected, then resuspended with lentivirus in 24-wells plate, The plate was centrifuged at 1000 g at 32 oC for 1 h, then incubated for 2–3 h at 37 oC. Polybrene (4 ng/mL) is added to enhance the efficiency of the lentiviral infection. After incubation, cells were resuspended with Matrigel and cultured as described above. To validate targeted mutations, the genome DNA was isolated from infected organoids, and the T7E1 (Vazyme, EN303-01) assay was performed with the PCR product for each sgRNA, T7E7 PCR primers were listed in Supplementary Data. 6. Western blotting was also performed to confirm the sgRNA’s work efficiency.
Subcutaneous and orthotopic transplantation
For the subcutaneous mouse model: male adult BALB/c-nu immunodeficient mice (~6–8 weeks old) were obtained from the Huafukang company. Organoids were crushed by the tip and collected in a new sterile EP tube. Then, the collected organoids were centrifuged at 2000 × g for 5 min. Approximately 2*10^5 cells were injected subcutaneously in the left flank of BALB/c-nu immunodeficient mice. The diameters of the tumor were measured using calipers.
For the orthotopic mouse model: male adult BALB/c-nu immunodeficient mice (~6–8 weeks old) were obtained from the Huafukang company. Organoids were crushed by the tip and collected in a new sterile EP tube. Then, the collected organoids were centrifuged at 2000 ×g for 5 min. Approximately 2*10^5 cells for each mouse. Then, isoflurane inhalation anesthetized mice. Mice were then placed in a supine position, and the extremities secured to a clean platform with tape. Scissors cut open 1 cm of the skin of the abdomen. The stomach was carefully pulled out with forceps. A 25–50 μL cell suspension of organoids, admixed with 100% Matrigel (Corning), was directly injected into the submucosa of mice. The stomach was carefully put back into the abdominal cavity, and the wound was sutured with suture and disinfected with Iodophor. The recipient mice were put back into the cage and waited for natural recovery.
Western blotting
Western blotting for histone methylation was conducted using acid-extracted proteins from tumor organoids. Antibodies were Histone H3 [3-C4] (Hangzhou Huabio Biotechnology, EM30605, 1:5000), H3K27me3 (Cell Signaling Technology, 9733S, 1:1000). Organoids and tumor organoids were extracted in RIPA buffer (Beyotime, Cat# P0013) supplemented with protease inhibitors (Beyotime, Cat# P1045), followed by SDS–PAGE gel electrophoresis and blotted onto PVDF membranes (Millipore). Primary antibodies were diluted in 5% non-fatty milk and incubated overnight at 4 °C. HRP-conjugated secondary antibodies were applied at 1:10000 dilution. Images were obtained by NcmECL Ultra Reagent (NCM biotech). Primary antibodies were EZH2 (Cell Signaling Technology, 5246S, 1: 1000), PTEN (Cell Signaling Technology, 9559S, 1:1000), TFAP2C (Abclonal, A7739, 1: 1000).
H&E, IHC and Immunofluorescence staining
Fresh tumor tissues were fixed in 4% paraformaldehyde overnight at room temperature, gradient dehydration, embedded in paraffin, cut into 5 μm. H&E staining were performed according to the standard protocol. A two-step detection kit (ZSGB-BIO, Cat# PV-9001 and Cat# PV-9002) was used for IHC. IF staining was performed according to the standard protocol. Primary antibodies were H3K27me3 (Abclonal, A2363,1:200), Ezh2 (D2C9) XP® Rabbit mAb (Cell Signaling Technology, 5246S, 1:50 for IHC, 1:1000 for IF), Anti-AP2 gamma/TFAP2C antibody [3B5] (Abcam, ab110635,1:50 for IHC, 1:500 for IF), Cytokeratin 5Monoclonal antibody(2C2) (Invitrogen, MA5-17057, 1:500 for IHC, 1:1000 for IF), Anti-Cytokeratin 14 antibody (Abcam, ab7800, 1:500), Cas9 (S. pyogenes) (E7M1H) XP® Rabbit mAb (Cell Signaling Technology, 19526S, 1:400), Anti-Cytokeratin 7 antibody [EPR17078] - Cytoskeleton Marker (Abcam, ab181598, 1:4000), CK20 (Abcam, ab53120,1:100), anti-P40-DeltaNp63antibody [EPR17863-47] (Abcam, ab203826, 1:1000 for IHC, 1:50 for IF), Cytokeratin 5/6 Monoclonal antibody (Thermo Fisher Scientific, MA5-12429, 1:200), Anti-MUC1 antibody [EP1024Y] (Abcam, ab45167, 1:400), MUC5AC monoclonal antibody (45M1) (Thermo Fisher Scientific, MA5-12178, 1:400).
RT-qPCR
Total RNA was extracted from TP/TPE premalignant and tumor organoids for transcriptome profiling. Total RNA extraction using with Trizol reagent (Thermo Fisher Scientific, Cat# 15596026) and complementary DNA (cDNA) synthetization using HiScript-III RT SuperMix kit (Vazyme, Cat# R323-01) were performed under the manufacturer’s instructions. Real-time quantitative PCR (RT-qPCR) was performed using Fast SYBR Green MasterMix (Vazyme, Cat# Q711-02/03) on the StepOne Real-Time PCR System (Thermo Fisher Scientific, Applied Biosystems QuantStudio 3). qRT-PCR primer was list in Supplementary Data 6.
Organoid proliferation assay
Organoids were digested into single cells by TrypLETM (GIBCO, Cat# 12605-028). 3000 cells were seeded in 96-well plates. After 5 days culture, organoids were photographed (Olymp IX73P2F). The size of organoids was measured using Image J (Version 1.51).
In vitro treatment
Organoids were digested with TrypLETM(GIBCO, Cat# 12605-028), 3000 organoids were seeded in 96-well plates culture overnight. Then 5-FU was added at the indicated concentrations into organoids. Cell Counting Kit-8(CCK8) assay tested cell viability at 72 h after treatment.
WES library preparation from FFPE samples
Squamous cell carcinoma components of GSCC patients were collected by PALM microbeam according to P40 IHC staining. DNA extraction was performed with GeneRead DNA FFPE Kit (Cat# 180134) according to the manufacturer’s instructions. The exome sequences were efficiently enriched from 0.4 μg genomic DNA using Agilent Sure Select Human All Exon V6 (Agilent USA, Catalog #5190-8864) according to the manufacturer’s protocol. Firstly, qualified genomic DNA was randomly fragmented to an average size of 180–280 bp by Covaris LE220R-plus (Covaris, USA). Remaining overhangs were converted into blunt ends via exonuclease polymerase activities. Secondly, DNA fragments were end repaired and phosphorylated, followed by A-tailing and ligation at the 3’ends with paired-end adapters. DNA fragments with ligated adapter molecules on both ends were selectively enriched in a PCR reaction. After the PCR reaction, libraries hybridize with a liquid phase with biotin-labeled probe, then use magnetic beads with streptomycin to capture the exons of genes. Captured libraries were enriched in a PCR reaction to add index tags to prepare for sequencing. Products were purified using the AMPure XP system (Beckman Coulter, Beverly, USA), libraries were analyzed for size distribution by the Agilent 5400 system AATI) (Agilent, USA) and quantified by real-time PCR (Life Technologies, USA) (1.5 nM). The qualified libraries were pooled and sequenced on Illumina platforms with a PE150 strategy in Novogene Bioinformatics Technology Co., Ltd (Beijing, China), according to effective library concentration and the data amount required.
Targeted bisulfite sequencing library construction
Tumor cells and adjacent normal cells from GSCC paraffin-embedded samples were collected using the MMI CellCut Plus Laser Microdissection System. Genomic DNA was extracted using the TIANamp FFPE DNA Kit (DP331) (Tiangen Biotech Co. Ltd.). CpG islands in the EZH2 promoter region were selected based on the following criteria: (1) minimum length of 200 bp (2) GC content ≥50%, and (3) observed/expected CpG ratio ≥0.60. Three regions meeting these criteria were selected and sequenced. DNA methylation analysis was performed using MethylTarget® (Genesky Biotechnologies Inc., Shanghai, China), a next-generation sequencing (NGS)-based targeted CpG methylation analysis method. Genomic regions of interest were simulated for bisulfite conversion using geneCpG software, and primers were designed using Methylation Primer software. Bisulfite conversion was carried out using the EZ DNA Methylation™-GOLD Kit (Zymo Research) following the manufacturer’s protocol. Multiplex PCR was performed using optimized primer sets. PCR amplicons were diluted and further amplified with indexed primers. Products (170–270 bp) were separated by agarose gel electrophoresis and purified using the QIAquick Gel Extraction Kit (QIAGEN).
RNA-seq library construction
Total RNA was extracted from organoids for transcriptome profiling. RNA-seq libraries were constructed using an Illumina stranded mRNA sample preparation kit (NEB, E7770) according to the manufacturer’s protocol. RNA integrity was assessed using the Bioanalyzer 2100 system (Agilent Technologies, CA, USA). Messenger RNA was purified from total RNA using poly-T oligo-attached magnetic beads. After fragmentation, the first strand cDNA was synthesized using random hexamer primers, followed by the second strand cDNA synthesis. The library was ready after end repair, A-tailing, adapter ligation, size selection, amplification, and purification. The library was checked with Qubit and real-time PCR for quantification and a bioanalyzer for size distribution detection. After library quality control, different libraries were pooled based on the effective concentration and targeted data amount, then subjected to Illumina sequencing. The basic principle of sequencing is “Sequencing by Synthesis”, where fluorescently labeled dNTPs, DNA polymerase, and adapter primers are added to the sequencing flow cell for amplification. As each sequencing cluster extends its complementary strand, the addition of each fluorescently labeled dNTP releases a corresponding fluorescence signal. The sequencer captures these fluorescence signals and converts them into sequencing peaks through computer software, thereby obtaining the sequence information of the target fragment.
CUT&Tag sequencing library construction
Premalignant organoids of TP and TPE were edited, cultured and prepared, with 1*10^5 collected for each assay. TP and TPE were used for H3K27me3 CUT-Tag, TP for EZH2 CUT-Tag. TPE for TFAP2C CUT-Tag. Organoids were collected in 1.5 μL EP tube. The CUT&Tag assay was performed using a NovoNGS CUT&Tag 2.0 High-Sensitivity kit (Novoprotein Scientific, N259-YH01-01A). 10 μL ConA beads were added into cells incubated 10 min at 25 °C. The supernatant was removed, and added primary antibody buffer and incubated overnight at 4 °C. The supernatant was removed and added secondary antibody buffer for 1 h at 25 °C. Beads were washed 3 times. The chromatin was combined with ChiTag TM2.0 Transposome, then fragmented using Tagmentation Buffer for 1 h at 37 °C. ChIP DNA was purified using DNA Clean Beads and amplified using the PCR system from this kit. PCR conditions: 72 °C for 3 min, 98 °C for 30 s, followed by 10–25 cycles of 98 °C for 15 s, 60 °C for 15 s and 72 °C for 10 s, 72 °C for 2 min. Amplified libraries were purified with the DNA Clean Beads. The purified ChIP DNA was used for library preparation.
Somatic variant calling
WES sequencing was performed by the DNBSEQ-T7 platform. FASTQ data was aligned to the reference human genome (hg19) using BWA-MEM, followed by a standard workflow. During pre-processing, the MarkDuplicates, FixMateInformation, BaseRecalibrator, and ApplyBQSR steps were performed sequentially using the Genome Analysis Toolkit (GATK; v4.4.0.0), producing analysis-ready BAM files. Subsequently, during the mutation calling step, approximately 400 normal tissue and peripheral blood genomic datasets from healthy individuals, as provided by the GATK database (https://storage.googleapis.com/gatk-best-practices/somatic-b37/Mutect2 -exome-panel.vcf), were used as controls. The “--panel-of-normals” command was applied to filter out potential false-positive mutations. Following this, the LearnReadOrientationModel, GetPileupSummaries, CalculateContamination, and FilterMutectCalls steps were employed to identify allele frequencies of somatic variants at the exon locus in the tumor BAM files. The variant caller was run with dbSNP (version 138) data for known polymorphic sites. VCF files were then converted into MAF format using the Vcf2maf tool, and annotated with data from the Ensembl Variant Effect Predictor (VEP; release version 95) using the hg19 database. Common variants were filtered based on ExAC_nonTCGA VCF data. To increase sensitivity in identifying somatic variants at the genomic locus, variants with a mutant frequency of 1, a minimum sequencing depth <10, and fewer than 3 alternative alleles were excluded from the final analysis.
Copy number variation identification
In the copy number variation (CNV) detection step, peripheral blood samples were used as controls. CNV analysis was performed using CNVkit with the target-avg-size parameter set to 500 bp. Genomic segments with sequencing coverage >50 and p-value < 0.01 (t test) were retained for analysis. Segments with the copy number (CN) ≥3 were classified as amplifications, whereas those with CN <2 were defined as deletions. Genomic data visualization was conducted using the R packages Maftools and ggplot2.
Targeted bisulfite sequencing analysis
Targeted bisulfite sequencing was performed on the Illumina NovaSeq platform according to the manufacturer’s protocols. Sequencing was performed with a 2 × 150 bp paired-end mode. After generating FASTQ data, raw reads underwent quality control and low-quality read filtering using fastp. The resulting high-quality reads were then aligned to the EZH2 promoter region (Chr7: 148,883,700–148,885,800, GRCh38) using Bismark v0.24.2 with Bowtie2 under default parameters. CpG methylation levels were quantified using the bismark_methylation_extractor function in Bismark.
CUT&Tag analysis
The Illumina NovaSeq 6000 platform was employed to perform genomic sequencing. Following quality control by the fastp standard workflow, the Bowtie2 software was utilized to construct an index for the mouse genome (mm10) and align paired-end clean data of 150 bp length using the options “--local --very-sensitive-local --no-unal --no-mixed --no-discordant --phred33 -I 10 -X 700”. The SAMtools program was utilized to convert sam files to sorted bam files. Picard’s MarkDuplicates tool was used to remove duplicates with the option “REMOVE_DUPLICATES=true”. Normalized bw files were generated using deepTools with the option “bamCoverage -bs = 1 --normalizeUsing BPM” for further visualization in the Integrated Genome Viewer (IGV). Peak calling was performed using the MACS2 software with the option “macs2 callpeak --broad -q 1e-2 -f BAMPE -g mm --keep-dup all”. ChIPseeker was employed to identify the genomic distribution of the peaks, with the TSS region “tssRegion” set as (−3000, 3000) for annotation. The “narrowPeak” files were transformed into GrangesList forms, and the peaks were converted into consensus counts. Data normalization and differential comparison were performed using DESeq2. Regions of differential modification were identified for downstream analyses using a p-value cutoff of 0.05 (Wald test).
RNA-seq analysis
Transcriptome sequencing was conducted using the Illumina NovaSeq 6000 platform with 150 bp paired-end sequencing. After removing the adapters, ploy-N, and low-quality reads, clean data were aligned against the reference mouse genome (mm10) using STAR _2.6.0. Following this, raw counts were normalized and significance scores, as well as log2-fold change values, were calculated using the DESeq2 (v.1.22.2). The differentially expressed genes were used to perform Gene Ontology enrichment analyses with the R package clusterProfiler (v. 3.10.1) (RRID:SCR_016884). Furthermore, Gene Set Enrichment Analysis (GSEA) was employed to identify significantly enriched pathways. Data visualization was conducted using the R packages ggpubr, ggplot2, pheatmap, and Vennerable.
Referred GC patient transcriptome data analysis and signature gene definition
We implemented the DESeq2 standard pipeline to identify genes exhibiting significantly elevated expression levels in esophageal squamous cell carcinoma relative to esophageal other subtype samples (p < 0.05 and log2foldchange > 1) from the TCGA cohort. These genes were subsequently designated as the squamous signature genes. Transcriptome data and corresponding clinical information of GC patients were obtained from the Cancer Genome Atlas Stomach Adenocarcinoma (TCGA-STAD) project. The cut-off values of signature high or low groups in survival analysis were determined by maximally selected rank statistics. Survival significant levels were determined by the log-rank test. In order to elucidate the molecular characteristics of GSCC, we preformed differential expression analysis between signature high or low GC patients using DESeq2. The significantly up-regulated genes (Squahi vs. Squalow, p < 0.05 and log2fold-change >1) were subsequently designated as the GSCC signature genes. GSEA was implemented to identify significantly enriched pathways between genes or signature high and low patients.
Quantification and statistical analysis
RT-PCR, organoids numbers and sizes, tumor weights and positive cell numbers, metastasis number assay, and in vitro treatment were analyzed for statistical significance using unpaired two-tailed t-tests by 9.0 GraphPad Prism. In addition, survival analyses were performed by 9.0 GraphPad Prism (log-rank test). The numbers of independent experiments, the p-values and samples are indicated in the Fig. legends. Data are represented as mean ± standard deviation (SD). Blinding was performed for tumor measurements and analysis of organoid numbers and sizes. No data were excluded from the study. Statistical analyses of bioinformatics were performed as described in the Fig. legends.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The RNA-seq and CUT&Tag sequencing data in this study deposited in the Gene Expression Omnibus (GEO) database: GSE254318, (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE254318). The WES raw data have been deposited in the Genome Sequence Archive (GSA) database in National Genomics Data Center, China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (GSA database: HRA011323, https://ngdc.cncb.ac.cn/gsa-human/browse/HRA011323). Due to data privacy regulations related to patient consent for data sharing, access to the FASTQ files can be requested through the “Request Data” process on the GSA website or by contacting the corresponding author. The data are available for non-commercial academic research purposes only. The approximate response time for access requests is about 2 weeks. Once access has been granted, the data will be available for download for 3 months. The remaining data are available within the Article, Supplementary Information, or Source data are provided in this paper.
Code availability
Our analysis code has been uploaded into the GitHub (https://github.com/AilingZhong/GSCC_Project).
References
Morgan, E. et al. The current and future incidence and mortality of gastric cancer in 185 countries, 2020-40: A population-based modelling study. EClinicalMedicine 47, 101404 (2022).
Wong, M. et al. Global Incidence and Mortality of Gastric Cancer, 1980-2018. JAMA Netw. Open 4, e2118457 (2021).
Karimi, P., Islami, F., Anandasabapathy, S., Freedman, N. & Kamangar, F. Gastric cancer: descriptive epidemiology, risk factors, screening, and prevention. Cancer Epidemiol. Biomark. Prev. 23, 700–713 (2014).
Cancer Genome Atlas Research, N Comprehensive molecular characterization of gastric adenocarcinoma. Nature 513, 202–209 (2014).
Uprak, T., Akmercan, A., Coşkun, M. & Attaallah, W. Gastric medullary carcinoma: clinicopathological features and prognosis-a single-center study. ANZ J. Surg. 91, 2425–2429 (2021).
Kim, J., Kim, G. & Kim, K. Paneth Cell Carcinoma of the Stomach]. Korean J. Gastroenterol. 80, 34–37 (2022).
Cirocchi, R. et al. Gastric carcinosarcoma: A case report and review of the literature. Oncol. Lett. 4, 53–57 (2012).
Chen, Y. et al. Clinicopathological Characteristics, Treatment, and Prognosis of 21 Patients with Primary Gastric Squamous Cell Carcinoma. Gastroenterol. Res. Pract. 2016, 3062547 (2016).
Hwang, S. et al. Primary squamous cell carcinoma of the stomach: A case report. Oncol. Lett. 8, 2122–2124 (2014).
González-Sánchez, J. A., Vitón, R., Collantes, E. & Rodríguez-Montes, J. A. Primary Squamous Cell Carcinoma of the Stomach. Clin. Med. Insights Oncol. 11, 1179554916686076 (2017).
Bonnheim, D., Sarac, O. & Fett, W.J.T.A.J.O.G. Primary squamous cell carcinoma of the stomach. Am. J. Gastroenterol. 80, 91–94 (1985).
Tokuhara, K., Nakano, T., Inoue, K., Nakane, Y. & Kwon, A. H. Primary squamous cell carcinoma in the gastric remnant. Surg. Today 42, 666–669 (2012).
Jakubik, J. et al. An unusual non‑metastatic, mismatch repair‑deficient primary gastric squamous cell carcinoma presenting as a large, exophytic, bleeding tumor: A case report. Oncol. Lett. 25, 82 (2023).
Aly, R. et al. Primary gastric adenosquamous carcinoma: A case report. Gastroenterol. Res. 13, 155–157 (2020).
Chen, Y.-Y. et al. Adenosquamous carcinoma of the stomach and review of the literature. Pathol. Oncol. Res. 21, 547–551 (2015).
Li, H. S. et al. Clinicopathologic characteristics, survival, and treatments for gastric adenosquamous carcinoma: a population-based study. Curr. Oncol. 27, e527–e536 (2020).
Akce, M. et al. Gastric squamous cell carcinoma and gastric adenosquamous carcinoma, clinical features and outcomes of rare clinical entities: a National Cancer Database (NCDB) analysis. J. Gastrointest. Oncol. 10, 85–94 (2019).
Comprehensive molecular characterization of gastric adenocarcinoma. Nature 513, 202–209 (2014).
Zhu, F. H. et al. The histopathological types and distribution characteristics of gastric mixed tumors. Front. Oncol. 12, 873005 (2022).
Shin, J. & Park, Y. S. Unusual or uncommon histology of gastric cancer. J. Gastric Cancer 24, 69–88 (2024).
Dong, C. et al. The clinicopathological features and prognostic factors of gastric squamous cell carcinoma. Medicine 95, e4720 (2016).
Xiangfei, W. et al. LGR5 regulates gastric adenocarcinoma cell proliferation and invasion via activating Wnt signaling pathway. Oncogenesis 7, 57 (2018).
Barker, N. et al. Lgr5(+ve) stem cells drive self-renewal in the stomach and build long-lived gastric units in vitro. Cell Stem Cell 6, 25–36 (2010).
Kirikoshi, H. & Katoh, M. Expression of TFF1, TFF2 and TFF3 in gastric cancer. Int. J. Oncol. 21, 655–659 (2002).
Hong, L. et al. Molecular landscape of ERBB2 alterations in 3000 advanced NSCLC patients. NPJ Precis. Oncol. 8, 217 (2024).
Liang, Y. et al. Prognostic significance of grade of malignancy based on histopathological differentiation and Ki-67 in pancreatic ductal adenocarcinoma. Cancer Biol. Med. 21, 416–432 (2024).
Na, F. et al. KMT2C deficiency promotes small cell lung cancer metastasis through DNMT3A-mediated epigenetic reprogramming. Nat. Cancer 3, 753–767 (2022).
Wang, M. et al. Acquired semi-squamatization during chemotherapy suggests differentiation as a therapeutic strategy for bladder cancer. Cancer Cell 40, 1044–1059 (2022).
Wan, X. et al. Primary and orthotopic murine models of nasopharyngeal carcinoma reveal molecular mechanisms underlying its malignant progression. Adv. Sci. 11, e2403161 (2024).
Gupta, R. A. et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464, 1071–1076 (2010).
Piunti, A. et al. Polycomb proteins control proliferation and transformation independently of cell cycle checkpoints by regulating DNA replication. Nat. Commun. 5, 3649 (2014).
Jiang, Y. et al. Reciprocal inhibition between TP63 and STAT1 regulates anti-tumor immune response through interferon-γ signaling in squamous cancer. Nat. Commun. 15, 2484 (2024).
Trink, B., Osada, M., Ratovitski, E. & Sidransky, D. p63 transcriptional regulation of epithelial integrity and cancer. Cell Cycle 6, 240–245 (2007).
Margueron, R. & Reinberg the polycomb complex PRC2 and its mark in life. Nature 469, 343–349 (2011).
Cao, R. et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 298, 1039–1043 (2002).
Kim, J. et al. Polycomb- and methylation-independent roles of EZH2 as a transcription activator. Cell Rep. 25, 2808–2820 (2018).
Li, F. et al. Ezh2 programs T(FH) differentiation by integrating phosphorylation-dependent activation of Bcl6 and polycomb-dependent repression of p19Arf. Nat. Commun. 9, 5452 (2018).
Velichutina, I. et al. EZH2-mediated epigenetic silencing in germinal center B cells contributes to proliferation and lymphomagenesis. Blood 116, 5247–5255 (2010).
Wang, K. et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat. Genet. 46, 573–582 (2014).
Wang, K. et al. KDM4C-mediated senescence defense is a targetable vulnerability in gastric cancer harboring TP53 mutations. Clin. Epigenet. 15, 163 (2023).
Saito-Adachi, M. et al. Oncogenic structural aberration landscape in gastric cancer genomes. Nat. Commun. 14, 3688 (2023).
Mina, S. et al. PTEN deletion is rare but often homogeneous in gastric cancer. J. Clin. Pathol. 65, 693–698 (2012).
O’Rourke, K. P. et al. Transplantation of engineered organoids enables rapid generation of metastatic mouse models of colorectal cancer. Nat. Biotechnol. 35, 577–582 (2017).
Roper, J. et al. In vivo genome editing and organoid transplantation models of colorectal cancer and metastasis. Nat. Biotechnol. 35, 569–576 (2017).
Pan, X. et al. Identifying a confused cell identity for esophageal squamous cell carcinoma. Signal Transduct. Target. Ther. 7, 122 (2022).
Souroullas, G. et al. An oncogenic Ezh2 mutation induces tumors through global redistribution of histone 3 lysine 27 trimethylation. Nat. Med. 22, 632–640 (2016).
Zhang, L. et al. EZH2 engages TGFβ signaling to promote breast cancer bone metastasis via integrin β1-FAK activation. Nat. Commun. 13, 2543 (2022).
Verma, A. et al. EZH2-H3K27me3 mediated KRT14 upregulation promotes TNBC peritoneal metastasis. Nat. Commun. 13, 7344 (2022).
Varambally, S. et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 419, 624–629 (2002).
Xu, J. et al. EZH2 promotes gastric cancer cells proliferation by repressing p21 expression. Pathol. Res Pr. 215, 152374 (2019).
Matulay, J. T. et al. The impact of squamous histology on survival in patients with muscle-invasive bladder cancer. Urol. Oncol. 37, 353 e317–353.e324 (2019).
Lu, Z. et al. Dissecting the genetic and microenvironmental factors of gastric tumorigenesis in mice. Cell Rep. 41, 111482 (2022).
Acknowledgements
We thank Dr. Yuquan Wei for generous support. We thank all the CC-LY lab members for discussion and technical support. We thank Fangfang Wang of Institute of hematology of West China Hospital for technical support. We thank the Core Facilities of West China Hospital and the Chengdu OrganoidMed Medical Laboratory for technical support. This work was supported by grants from the National Natural Science Foundation of China (82330087 [C.C.]; T2221004 [C.C.], 82130007 [Y.L.], 82470188 [Y.L.], 82170171 [C.C.], 82303880 [M.W.], 82300185 [Q.Z.], 82300186, [B.W.]); the National Science and Technology Major Project of China (2023ZD0500500 [Y.L.], 2024YFF0507400 [C.C.]); the Sichuan Science and Technology Program (2025NSFTD0030 [Y.L.], 2023ZYD0057 [Y.L.], 2025ZNSFSC0047 [C.C.]; the Frontiers Medical Center, Tianfu Jincheng Laboratory Foundation (TFJC2023010004 [C.C.]); the 1.3.5. Project for Disciplines of Excellence, West China Hospital, Sichuan University (ZYGD22012 [Y.L.]); National Clinical Research Center for Geriatrics, West China Hospital, Sichuan University (Z2024JC001 [C.C.]); the 1.3.5. Project for Disciplines of Excellence, West China Hospital, Sichuan University (ZYGD22012 [Y.L.]); Chengdu Science and Technology Project (2024-YF05-00535-SN,[J.X.]); Natural Science Foundation of Sichuan Province of China (2024NSFSC1696, [J.X.], 2024NSFSC1699, [B.W.], 2023NSFSC1904 [M.W.]); Postdoctoral Fellowship Program of CPSF (GZB20230466 [Q.Z.]); China Postdoctoral Science Foundation (2023M732465 [Q.Z.]); the Post-Doctor Research Project, West China Hospital, Sichuan University (2023HXBH018 [M.W.], 2023HXBH094 [Q.Z.]); the China Postdoctoral Science Foundation under Grant Number (2023M732457 [B.W.], 2023TQ0226 [B.W.], 2024T170604 [Q.Z.], 2023M732465[Q.Z.]); the Post-Doctor Research Project of Sichuan University (2024SCU12033, [B.W.]).
Author information
Authors and Affiliations
Contributions
Conceptualization: C.C.; Methodology: M.Z., A.Z., H.L., Lei.Zhao, Y.W., Zhenghao.Lu, Lanxin.Zhang, X.P., Zuoyu.Liang., L.G., X.C., J.W., S.D., X.W., M.W., Jingyao.Chen, J.D., S.C., T.P., Jingyang.Cai, L.C., Y.Y., C.X., M.Y., R.Q., C.Y., and S.L.; Investigation: M.Z., A.Z., H.L., and L.Z.; Resources: C.Z., H.D., Y.W., L.C., J.H., B.H., S.Y., F.N., L.G., Y.L., and C.C.; Writing-Original Draft: M.Z., A.Z., H.L., and C.C.; Writing-Review & Editing: M.Z., A.Z., H.L., L.Z., Y.L., and C.C.; Funding Acquisition: B.W., Q.Z., M.W., J.X., Y.L., and C.C.; Supervision: M.Z., A.Z., H.L., L.Z., and C.C.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Tatsuhiro Shibata, Makoto Yamagishi and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, M., Zhong, A., Liu, H. et al. EZH2 loss promotes gastric squamous cell carcinoma. Nat Commun 16, 6032 (2025). https://doi.org/10.1038/s41467-025-61024-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-61024-5
