
Abstract
The APOE-ε4/ε4 genotype is the strongest genetic risk factor for sporadic Alzheimer’s disease, though the relative risk is diminished in individuals with African ancestry. Through analysis of phased APOE alleles, we identify a 19 bp deletion approximately 1.1 kb distal to the APOE 3′UTR in a SPI1 microglial transcription factor binding site. The deletion is present in 60% of African American APOE-ε4 homozygotes and reduces Alzheimer’s disease odds ratio relative to individuals without the deletion. The deletion also delays Alzheimer’s disease onset in APOE-ε4/ε4 cases with local African ancestry at APOE. The All of Us dataset confirms reduced Alzheimer´s disease risk associated with the deletion and identifies additional variants between APOE and APOC1 that disentangle APOE-ε4 neurological and lipid-related phenotypes. Functional assays reveal that the 19 bp deletion abolishes SPI1 repression at this region. Collectively, these findings describe a protective allele at APOE in African Americans that mediates APOC1 expression, reducing relative Alzheimer´s disease risk.
Similar content being viewed by others


Introduction
Alzheimer’s disease (AD) is the most common form of dementia and affects nearly 7 million Americans age 65 or older, with an increasing prevalence in global populations1. AD pathology is characterized by the accumulation of amyloid beta (Aβ) plaques and tau neurofibrillary tangles. The Apolipoprotein E (APOE) locus is the strongest genetic risk factor for sporadic AD2, consistently replicating across genome-wide association studies (GWAS) for AD3,4,5. Unlike many AD GWAS signals that appear to be driven by a single nucleotide polymorphism (SNP) or haplotype in intergenic regions, the APOE GWAS signal is largely explained by two missense variants, rs429358 (p.C112R) and rs7412 (p.R158C). These variants define three major APOE isoforms (ε2, ε3, and ε4). From a practical standpoint, the reference genome haplotype (APOE-ε3) changes to APOE-ε4 with a T-to-C change at rs429358, while APOE-ε3 converts to APOE-ε2 with a C-to-T change at rs7412.
Even though the APOE isoforms were first defined in studies of lipid metabolism6,7, APOE is also mechanistically linked to Aβ clearance. For instance, APOE null mice have reduced Aβ deposition in the brain8, while APOE-ε4 binds more readily with Aβ9. Interestingly, while three amyloid-lowering therapies have been approved by the FDA for AD, the clinical benefit is less obvious in APOE-ε4 carriers, who have additional Amyloid Related Imaging Abnormalities (ARIA) complications10, underscoring the need for therapies that effectively target APOE-ε4.
The absolute risk of developing dementia is higher for African Americans and individuals of African descent relative to those of European ancestry11,12,13. Both genetics and environment are likely contributing factors. For example, pathogenic variants in ATP-binding cassette, subfamily A, member 7 (ABCA7) are more common among individuals with African ancestry14. Multiple GWAS studies with samples of individuals with African ancestry have replicated increased risk of AD associated with APOE and ABCA7 variation, along with other hits15. Those of African ancestry are more likely to harbor APOE-ε4 relative to those of European ancestry16. Despite the fact that APOE genotype status emerges as a global risk factor for AD, individuals with African ancestry and the APOE-ε4/ε4 genotype have considerably lower odds of developing AD relative to those genetically more similar to APOE-ε4 homozygotes of European ancestry. Local African ancestry at APOE can dampen AD risk in Caribbean Hispanics17, Puerto Ricans, and African Americans18. Conversely, APOE-ε4 is associated with a greater increase in AD risk among East Asian populations19,20. Together, this evidence suggests that APOE-ε4 haplotype variants modulate AD risk in different global populations, and that a protective variant may exist on haplotypes of African descent.
In addition to genotype status, variation in APOE alters isoform-specific risk of AD. The rs405509 variant in the promoter of APOE is linked to increased risk of AD in APOE-ε4 samples of East Asian ancestry21, whereas the APOE R145C variant is associated with increased risk of AD in African Americans and particularly in those with APOE-ε3/ε4 genotype22. In contrast, the APOE V236E and R251G alleles, inherited on the APOE-ε4 background, confer decreased risk for AD23. The impact of noncoding variants at the APOE locus is less clear, as the region contains several genes in close proximity with related functions, many of which have AD risk SNPs in linkage disequilibrium (LD) with APOE-ε4 and APOE-ε2 alleles.
Long-read sequencing technology has enabled phasing of complete chromosome sequences, allowing for the detection of multiple cis-acting variants on the same chromosomal strand rather than relying on estimates from population data. Here, we leveraged 78 phased long-read genomes from diverse reference populations24,25,26 to identify APOE-ε4 allele variants in individuals of African ancestry to determine a potential mechanism for their reduced relative risk of AD. Our efforts identified a common 19 bp deletion that lies within a microglial SPI1 transcription factor binding site of APOE-ε4 carriers of African descent. Importantly, we find that this 19 bp deletion is frequent in individuals with African-derived APOE-ε4 alleles who do not develop AD and that this deletion acts as a transcriptional repressor in vitro, thereby conferring protection for AD.
Results
We surmised that the fully phased chromosomes sequenced from 1000 Genomes Project samples25 would permit the identification of potential protective alleles in cis with the well-established APOE-ε4 risk allele. Our search prioritized insertions, deletions, and structural variants as opposed to single nucleotide variants (SNVs), which we reasoned would have been identified in previous association studies. We also focused on variants with a high minor allele frequency in individuals with African ancestry, but relatively low minor allele frequency in individuals with European ancestry. In evaluating long-read sequence data from the Human Pangenome Reference Consortium (HPRC24, 43 long-read samples; 86 alleles) and the Human Genome Structural Variation Consortium (HGSVC26, 35 long-read samples including 6 trios; 64 alleles), we identified five individuals of African descent with an APOE-ε4 allele. We compared these haplotypes with non-African individuals with an APOE-ε4 genotype, Africans with an APOE-ε3 allele, and non-Africans with an APOE-ε3 allele along with the GRCh38 and CHM13 reference genomes. We selected a ~ 75 kb window before or after APOE to align each phased chromosome.
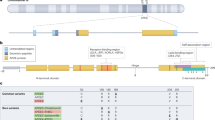
We identified a 19 bp deletion (rs148353395) ~ 1.1 kb past the 3′UTR of APOE that met our criteria (Fig. 1a), present in three individuals as heterozygotes (HG03486mat, HG02486mat, and HG02011-h2). The minor allele frequency is 0.1239 in African samples and 0.1248 in African Americans, but only 0.0005 in Europeans (Fig. 1b). Therefore, the deletion is expected to often be observed on APOE-ε4 haplotypes among individuals of African descent. We examined the frequency of the deletion in all 2,504 samples from 26 populations collected as part of the 1000 Genomes Project and found the deletion at frequencies ranging from 0.066 in African Ancestry in Southwest US (ASW) to 0.217 in Gambian in Western Division (GWD) samples (Fig. 1c). None of the 505 European ancestry samples carried the 19 bp deletion allele. Among APOE-ε4 homozygotes, the deletion allele frequency is 0.750 in GWD, Yoruba in Ibadan, Nigeria (YRI), and Esan in Nigeria (ESN) populations (Fig. 1d).

a Alignment of phased long-read alleles. b gnomAD minor allele frequency of the 19 bp SNP. c Relative frequency of the deletion in 1000 Genomes Project populations ordered by relative abundance. * = African Ancestry in Southwest US. d Number of alleles with the deletion in APOE-ε4 homozygous individuals from the 1000 Genomes Project populations. e UCSC genome browser track showing the location of the deletion allele relative to APOE and transcription factor binding sites on the GRCh37 build. f Presence of the deletion and strong (red) and modest (orange) predicted SPI1 binding sites along with sequence conservation in the mouse genome.
Notably, rs148353395 is present in a Spi-1 proto-oncogene (SPI1) binding site based on the University of California Santa Cruz (UCSC) hg19 genome browser transcription factor ChIP-seq cluster track from ENCODE data (Fig. 1e). The region is in a DNAseI hypersensitivity site in myeloid cell lines such as K562 cells. SPI1 is also known as PU.1 – a well-established transcription factor in macrophage lineages including microglia27. Forced expression of PU.1 reliably generates iPSC-derived microglia28. Importantly, SPI1 falls within a GWAS locus for AD4,5,29,30 and variants in this locus that lower SPI1 expression are protective for AD31. The strongest predicted SPI1 binding site is ~40 bp from the 19 bp deletion (Fig. 1f)32.
The Alzheimer’s Disease Sequencing Project (ADSP; release ng00067.v16) from the National Institute on Aging Alzheimer’s Disease Data Storage Site (NIAGADS) has amassed substantial genetic resources to uncover genetic factors implicated in AD, including in African American individuals. To test whether the 19 bp deletion variant is protective against AD in individuals of African ancestry, we performed logistic regression analysis in 1,522 AD cases and 3,929 controls of African or African American ancestry from the ADSP. We removed related individuals and controlled for age, sex, cohort, sequencing center, sequencing platform, and APOE-ε2 and APOE-ε4 status. The results revealed significant protection against AD (OR = 0.70; 95% CI 0.58–0.83; p = 5.7 × 10-5) for the 19 bp deletion. In APOE-ε4 homozygotes, the deletion allele frequency was 0.55 in controls (n = 121) and 0.41 in AD cases (n = 195). In APOE-ε3/ε4 heterozygotes, the deletion allele frequency was 0.28 in controls (n = 1249) and 0.22 in AD cases (n = 702). Odds ratios comparing those with and without APOE-ε4/ε4 varied by 19 bp genotype, decreasing from 3.81 (p = 3.27 × 10-22; 95% CI 2.90–4.99; n = 2,479) for those with two reference alleles to 2.58 (p = 1.94 × 10-20; 95% CI 2.11–3.16; n = 2,523) for heterozygotes for the deletion and 2.50 (p = 6.11 × 10-12; 95% CI 1.92–3.24; n = 2,457) for 19 bp deletion homozygotes (Fig. 2a).

a Genotype distribution (top) and odds ratio (bottom) from African American ADSP individuals. b Stratification of cases and controls by local African ancestry at APOE with the percent allele distribution (top) and genotype distribution (Bottom). c, d Forest plot of odds ratios and 95% confidence intervals for the rs148353395 19 bp deletion, rs7412 the APOE-ε2 defining SNP, and rs429358, the APOE-ε4 defining SNP to APOE-ε3 homozygous individuals, adjusting for effects of the other variants in the model, sex, age, sequencing center, sequencing platform, and cohort in c, samples with local African ancestry at APOE (n = 7,248) and d all ADSP Samples (n = 38,994). e Kaplan-Meier survival analysis of age of onset for different genotype combinations for APOE-ε3, APOE-ε4, and the rs148353395 19 bp deletion for individuals of African local ancestry (n = 2,002) at APOE. *p < 0.05 by log rank test versus APOE-ε4/ε4 individuals without the 19 bp deletion.
Importantly, the contribution of African ancestry across the genome can vary widely amongst African American individuals. Local ancestry estimation at loci such as APOE is therefore critical in ensuring that appropriate comparisons are made17. We used two methods to restrict our analysis to those with African ancestry at the APOE locus. The first was based on previous published methodology33 in 16,905 ADSP individuals also demonstrated a protective effect against AD for the 19 bp deletion (Fig. 2b). We then replicated these findings with an alternative approach based on a random forest model trained on 1000 Genomes Project samples and applied to the entire cohort of 48,804 ADSP individuals after filtering (see methods). Using logistic regression in the subset of these individuals with local African ancestry and again correcting for age, sex, cohort, sequencing center, sequencing platform and the APOE-ε2 and APOE-ε4 defining SNPs, we found that the rs148353395 19 bp deletion was protective against AD (OR = 0.76; 95% CI 0.66–0.88; p = 0.0002; n = 7,248; Fig. 2c). Including all samples (regardless of local ancestry), rs148353395 was as protective (OR = 0.73; 95% CI 0.66–0.82; p = 1.00 × 10-8) as rs7412, the APOE-ε2 defining SNP (OR = 0.68; 95% CI 0.62–0.74; p = 5.59 × 10-18; n = 38,944; Fig. 2d). The deletion was not significant in Non-Hispanic White samples [minor allele frequency (MAF) = 0.004; OR = 0.75; 95% CI = 0.51–1.11; p = 0.15].
We applied a survival analysis approach to investigate whether the rs148353395 19 bp deletion impacted age of AD onset among individuals with local African ancestry at APOE. Kaplan Meier curves illustrated a 3-year median delay in onset for APOE-ε4/ε4 individuals heterozygous or homozygous for the 19 bp deletion compared to APOE-ε4/ε4 individuals without the 19 bp deletion (p = 0.01 for 1 copy of the deletion, p = 0.02 for 2 copies by log-rank Mantel-Cox test). There was no significant delay in age of onset in APOE-ε3/ε4 individuals with or without the deletion (1 year median delay; p = 0.11; Fig. 2e). These findings reinforce the notion that the 19 bp deletion is protective on a background of APOE-ε4 alleles of African descent.
The NIH All of Us version 8 dataset contains ~380,179 samples that underwent whole genome sequencing (WGS), 255,622 of which were coupled with electronic health record (EHR) information, enabling powerful connections between genetic variants and phenotypic traits, including in diverse populations34. To address whether the 19 bp deletion modulates odds of AD and other traits in this independent dataset and potentially affects other traits, we performed a phenotype-wide association study (PheWAS) on these samples, which included over 47,143 samples of African ancestry. After correcting for age, sex, the first 3 principal components of genetic ancestry, and removing related samples (as completed in the All of Us publication34), we found that the APOE-ε4 defining SNP, rs429358, conferred the most significant phenotypic association with hyperlipidemia (OR = 1.21; p = 1.06 × 10-91) and hypercholesterolemia (OR = 1.25; p = 1.76 × 10-59). Despite only 656 cases, the rs429358 odds ratio for AD was 2.73 (p = 2.14 × 10-53; Fig. 3a, b). The dementia odds ratio also increased, as did mild cognitive impairment, vascular dementia, and memory loss (Supplementary Data 1). As expected, PheWAS results were inverted for rs7412, the SNP that defines the APOE-ε2 genotype, which was associated with a strongly protective effect for hyperlipidemia (OR = 0.71, p = 4.30 × 10-160) and hypercholesterolemia (OR = 0.64, p = 3.59 × 10-124; Supplementary Data 2). rs7412 was significantly associated with reduced odds for AD (OR = 0.55; p = 4.88 × 10-6) after Bonferroni correction for 1,816 traits. Odds for other neurological disorders like Parkinson’s disease (OR = 1.05; p = 0.37) or amyotrophic lateral sclerosis (OR = 1.08; p = 0.66) was not elevated for APOE-ε4 carriers, though APOE-ε4 carriers did have a nominally reduced risk of open angle glaucoma (OR = 0.86; p = 0.0073). Odds for hypertriglyceridemia was elevated in both APOE-ε2 and APOE-ε4 carriers, as has been observed previously for APOE-ε235, while odds for liver disease was reduced in APOE-ε4 carriers (Fig. 3a)36.

a–c Phenome-wide association study (PheWAS) Manhattan plot for 1816 traits grouped into related categories on the x-axis compared with significance on the y-axis for a, rs429358 (the APOE-ε4 defining SNP), b rs7412 (the APOE-ε2 defining SNP) and c rs148353395 (the 19 bp deletion). d Forest plot of the PheWAS odds ratio, standard error, and whether Bonferroni significance was reached for the conditions which passed Bonferroni significance for at least the APOE-ε4 or APOE-ε2 defining SNP and had at least 50 cases for rs7412 (green), rs429358 (blue), and rs148353395 (yellow). Also shown are the odds ratio for rs148353395 after adjusting for rs7412 and rs429358 (in purple) and for rs429358 after adjusting for rs148353395 (in red). Significance is denoted by a filled square for conditions which passed Bonferroni correction (p < 2.75 × 10-5), nominal significance (p < 0.05) with a filled triangle, and an unfilled circle for conditions which did not reach significance. The n for each trait and variant can be found in Supplementary Data 1-5. Delirium dementia*: full phenotype name is: “Delerium dementia and amnestic and other cognitive disorders”. Source data are provided as a Source Data file.
We next performed a PheWAS for all samples to examine the impact of the 19 bp deletion. This analysis revealed that rs148353395 variant carriers had significantly elevated hyperlipidemia (OR = 1.18, p = 3.55 × 10-13) and hypercholesterolemia (OR = 1.24, p = 2.88 × 10-10) at levels similar to rs429358 (Supplementary Data 3). However, the increased odds ratio for AD was no longer significant (OR = 1.32, p = 0.17; Fig. 3c). Since the 19 bp deletion is in cis with the APOE-ε4 variant in most individuals (Table 1), we performed conditional analysis adding APOE genotype as a covariate. Adjusting for APOE-ε4 and APOE-ε2, the odds of developing AD for 19 bp deletion carriers versus non-carriers dropped (OR = 0.56, p = 0.0058) consistent with our findings from the ADSP data (Fig. 3d, Supplementary Data 4). Lipid traits also dropped to neutral risk, such as hyperlipidemia (OR = 1.00, p = 0.87). Conversely, adding the 19 bp deletion as a covariate to PheWAS for rs429358 increased the associated risk of AD (OR = 2.92; Supplementary Data 5). In the 4073 APOE-ε4 homozygotes, the odds ratio of developing AD for 19 bp deletion carriers was 0.57 (p = 0.20; Supplementary Data 6).
A PheWAS analysis restricted to samples of African ancestry, as inferred from principal component analysis, revealed reduced risk for AD and related neurological traits (Supplementary Fig. 1, Supplementary Data 7-9). The effect of the 19 bp deletion on AD was not significant before adjusting for APOE-ε4 and APOE-ε2 (OR = 1.29, p = 0.287, Supplementary Data 9) or after adjusting for APOE-ε4 and APOE-ε2 (OR = 0.54, p = 0.053, Supplementary Data 10). Performing a local ancestry determination with the random forest model described for ADSP samples revealed similar findings for the 19 bp deletion from individuals of local African ancestry at APOE (Supplementary Data 11,12).
To visualize the different phenotypic effects of rs429358, rs7412, and rs148353395, we generated a forest plot that included odds ratios for each of the top 20 All of Us phenotypes based on significance in rs429358 (Fig. 3d). Here, levels of the top traits had a mirror-like effect: conditions were increased in APOE-ε4 and decreased in APOE-ε2, and vice versa. Individuals with the 19 bp deletion predominantly had neutral risk for neurodegenerative traits, except for lipid-related traits that mimicked the effect for rs429358. Female-only or male-only analysis revealed increased odds for hyperlipidemia for female APOE-ε4 and increased relative protection for female APOE-ε2 carriers, but negligible sex-related differences for AD and other neurological phenotypes (Supplementary Fig. 2, Supplementary Data 13-18).
To evaluate the functional consequences of the 19 bp deletion, we cloned the APOE 3′UTR and a ~ 400 bp region downstream of APOE containing the 19 bp deletion into a psiCheck2.2 luciferase reporter construct (Fig. 4a). We transfected this reporter into HMC3 human brain-derived microglia cells and SH-SY5Y human neuronal-like cells. Unexpectedly, we found that addition of the genomic sequence with the SPI1 binding site (important in microglial biology27) distal to the APOE 3′UTR reduced expression of the luciferase reporter, thereby acting as a transcriptional repressor (p = 0.0016, Fig. 4b). This repression was abolished in the context of the 19 bp deletion (p = 0.8252, Fig. 4b). Furthermore, this repression was not present after the SPI1 binding site was mutated (p = 0.9108, Supplementary Fig. 3). This regulatory module was not present in SH-SY5Y cells (Supplementary Fig. 3), rather the presence of the extended 3′UTR or 19 bp deletion significantly decreased luciferase expression relative to the 3′UTR only sequence (p = 0.0002 and <0.0001, respectively). In C8-D1A murine astrocytic cells, we observed no regulatory impact of the APOE 3′UTR on luciferase activity (Supplementary Fig. 3). As expected, these data suggest that the functional impact of the 19 bp deletion and SPI1 site differs between glial and neuronal and astrocytic cells.

a Schematic of the luciferase experimental design and constructs. The APOE 3′UTR (APOE 3′UTR only) plus ~400 bp of downstream DNA containing the 19 bp deletion region and SPI1 binding site were cloned into a psiCheck2.2 dual luciferase reporter construct. b Renilla:Firefly luciferase expression data in HMC3 cells after delivery of APOE SPI1 WT sequence (p = 0.0016 relative to the APOE 3′UTR only) or delivery of the 19 bp deletion [p = 0.8252; one-way ANOVA, n = 12 samples/group (3 biological replicates, 4 technical replicates), ± SD]. c–e HMC3 gene expression following SPI1 overexpression. c Effect of SPI1 overexpression on c APOE expression (p = 0.3831, F = 1.789, unpaired two-tailed t-test, n = 12 control and 12 SPI1 biological replicates derived from the average of 4 technical replicates/sample, ± SEM), d APOC1 expression (p = <0.0001, F = 2.987, unpaired two-tailed t-test, n = 11 control and 12 SPI1 biological replicates derived from the average of 4 technical replicates/sample, ± SEM), and e lncRNA ENSG00000280087 expression (p = 0.0170, F = 5.414, unpaired two-tailed t-test, n = 12 control and 10 SPI1 biological replicates derived from the average of 4 technical replicates/sample, ± SEM). f–h HMC3 gene expression following Aβ 1:42 treatment. f Effect of Aβ 1:42 treatment on APOE expression (p = 0.2098, F = 1.243, unpaired two-tailed t-test, n = 12 control and 12 Aβ biological replicates derived from the average of 4 technical replicates/sample, ± SEM), g APOC1 expression (p = 0.0015, F = 4.657, unpaired two-tailed t-test, n = 11 control and 12 Aβ biological replicates derived from the average of 4 technical replicates/sample, ± SEM) and h, lncRNA ENSG00000280087 expression (p = 0.0255, F = 2.502, unpaired two-tailed t-test, n = 12 control and 12 Aβ biological replicates derived from the average of 4 technical replicates/sample, ± SEM) in HMC3 cells.
Given the luciferase suppression associated with presence of the intact SPI1 binding site, we predicted that APOE expression would decrease after SPI1 delivery. Accordingly, we administered a construct expressing SPI1 to HMC3 cells. We surprisingly found no change in APOE levels (p = 0.3831, F = 1.789) but instead detected significantly decreased expression of the adjacent gene, APOC1 (p < 0.0001, F = 2.987), and significantly increased levels of a long non-coding RNA, ENSG00000280087, that spans the APOE-APOC1 intergenic interval and includes the 19 bp deletion site (p = 0.0170, F = 5.414, Fig. 4e, f). This model is consistent with recent findings demonstrating sequential regulation of first APOC1 followed by APOE after CRISPR-mediated regulation of the intergenic region between APOE and APOC137. These results were corroborated when delivering Aβ oligomers to model AD pathology; specifically, we found significant decrease in APOC1 (p = 0.0015, F = 4.657), no change in APOC (p = 0.2098, F = 1.243), and elevation of ENSG00000280087 expression (p = 0.0255, F = 2.502, Fig. 4g-i). Interestingly, in C8-D1A astrocytic cells, Aβ treatment significantly decreased APOC1 (p = 0.0173, F = 4.434) and ENSG00000280087 expression (p = 0.0048, F = 1.025), while SPI1 treatment significantly increased ENSG00000280087 expression (p = 0.0002, F = 2.371). Of note, the GTEx consortium38 lists rs148353395 as an expression quantitative trait locus for APOE in the cortex (p = 2.90 × 10-7) and spinal cord (p = 0.02), though with the caveat that only four samples heterozygous for the deletion were assessed.
Since the 19 bp deletion is located between the coding region of APOE and APOC1, the effect on APOC1 prompted us to evaluate the intergenic genomic region between APOE and APOC1 for additional variants that could mediate the interaction between APOE and APOC1 (Table 2, Supplementary Fig. 4a, b). Linkage disequilibrium (LD) calculations revealed modest LD between rs429358 and the 19 bp deletion (r2 = 0.48 in African ancestry, Fig. 5a, b). Further LD analysis in the 150 kb region around APOE in long-read genomes uncovered several other SNPs in high LD with the 19 bp deletion, including rs10424663 (r2 = 0.77), and rs435380 (r2 = 0.73). We did not detect structural variants from any of the phased long-read genomes; however, several SNPs were more abundant in samples of African ancestry including rs438811 (MAF 0.515 in African ancestry versus 0.220 in European ancestry individuals from gnomAD). In contrast to rs148353395, individuals with the rs438811 variant had an increased odds ratio of developing neurological phenotypes such as AD (OR 1.73, SE = 0.06, p = 3.66 × 10-19; Fig. 5c,d; Supplementary Data 19), while there was no associated effect on lipid traits like hyperlipidemia (OR = 0.99; p = 0.31). Similar findings were detected for a 4 bp insertion (rs11568822) in the 5′UTR of APOC1 (MAF 0.289 in African ancestry versus 0.213 in European ancestry individuals; Supplementary Fig. 4, Supplementary Data 20). Conversely, a variant (rs72654473) in LD with rs7412 was similarly associated with reduced risk of lipid-related traits, but without the expected protective effect on neurological conditions (Fig. 5e,f, Supplementary Data 21). Therefore, while APOE-ε4 and APOE-ε2 variants affect both lipid and neurological traits, variants between APOE and APOC1 appear to decouple these traits and contribute disproportionately to either neurological or lipid manifestations (Supplementary Fig. 5).

a Pairwise linkage disequilibrium (LD) plot of Pearson correlation (r2) between SNPs present in the intergenic region between APOE and APOC1 with a minor allele frequency of >1% and >0.2 r2 in samples of African ancestry (n = 32,886 individuals). b LD map for the same SNPs in samples of European ancestry (n = 104,107 individuals). c Manhattan PheWAS multi-ancestry plot for rs438811. d Corresponding forest plot of odds ratios and standard error for rs438811 of the top 20 most significant phenotypes. e Manhattan plot for rs72654473. f Corresponding forest plot of odds ratios and standard error for rs72654473 of the top 20 most significant All of Us phenotypes. All values and ns available in Supplemental Tables 19 and 21.
Discussion
By capitalizing on recent efforts to generate phased long-read genomes, we were able to identify a protective 19 bp deletion that is common in African APOE-ε4 carriers. The location of the 19 bp deletion – only 1.1 kb past the 3′UTR – initially raised the possibility that it had a direct effect on APOE regulation. However, we found that the intergenic enhancer element distal to APOE acts to negatively regulate APOC1, which in turn can affect APOE levels. Regulation of APOE is a highly orchestrated event. For instance, Huang et al. found that knockdown of SPI1 increases APOE expression, while conversely, over-expression of flag-tagged SPI1 decreases levels of APOE31. These results were corroborated in another study whereby Spi1 knockdown in mice led to increased Apoe expression39. SPI1 impacts expression of several targets from upregulation or knockdown of SPI1 in RNA-seq datasets from mice40. Notably, Huang and colleagues also determined that SPI1 did not associate with the APOE locus based on chromatin accessibility studies in CD14+ myeloid cells31, demonstrating the contextual importance of the regulatory relationship between SPI1 and APOE. Overexpression of SPI1 in our HMC3 data did not alter APOE expression, and this may have occurred because sufficient SPI1 is already present to repress APOE or that the effect is mediated through APOC1.
APOC1 and APOE are intrinsically linked. Several studies have examined the impact of APOC1 on cognition. One study found that Apoc1-/- knockout mice have deficits on memory tasks41, while another determined that Apoc1 overexpression is also linked to impaired function on memory tasks42. These data demonstrate the importance of Apoc1 homeostatic regulation in mice. Interestingly, while non-human primates contain two copies of APOC1; one copy has become a pseudogene in humans43. It should be noted that the APOE-ε4 allele is the only allele present in non-human primates and rodents, further supporting the case that the APOE-ε3 and APOE-ε2 alleles may carry evolutionary benefits to humans. Of note, the reference APOE-ε4 gibbon genome contains a 20 bp deletion that overlaps with the human APOE-ε4 19 bp deletion sample (GRCh38 chr19:44910519-44910538 del CTTGTTTCTCCCCCAACCCC), though it is unclear whether this deletion affects cognition or APOC1 regulation. Since only one reference allele is listed per non-human primate genome, it remains possible that a similar deletion allele is common in other non-human primates in addition to gibbons.
There are several examples of possible antagonistic pleiotropy at the APOE locus, where individuals with APOE-ε4 are protected from primary open-angle glaucoma relative to APOE-ε3 carriers44,45, while having increased risk for certain viral infections including SARS-CoV-246. Notably, mice challenged with a murine version of SARS-CoV-2 fared far worse when harboring humanized APOE-ε4 or APOE-ε2, relative to APOE-ε347. In this study, 100% of male mice on the APOE-ε4 background succumbed to SARS-CoV-2 infection compared to only 30% of male mice on the APOE-ε3 background; risk of human mortality from SARS-CoV-2 infections was also higher for APOE-ε4/ε4 and APOE-ε2/ε2 homozygotes47. Given that human life expectancy has only exceeded ~40 years in the last century, for most of human history any effect of the APOE-ε4 on cognitive function would have an insignificant role relative to the APOE-ε4 advantage in terms of reproductive fitness48. Even in present day humans, the role of the APOE-ε4 allele in preventing infections such as hepatitis C may remain a more important evolutionary consideration than late-stage cognitive effects36. As African populations exhibit greater sequence diversity given human population history, by representing diverse populations we can better capture natural protective and risk alleles.
Other searches for protective alleles at the APOE locus have identified variants including rs10423769 > 2 Mb from APOE that are protective against AD49. This variant was found beyond a more local topologically associated domain that surrounds APOE18. Additionally, distal enhancer elements have been identified including one 3.3 kb downstream and another 15 kb downstream of human APOE50,51. The 19 bp deletion, rs148353395, is listed in a previous publication by Nuytemans et al.52, where the authors tested for variants at the APOE locus to determine whether variants had enhancer function through a massively parallel reporter assay (MPRA), and enhancer activity of PCR amplicons corresponding to different segments of ~850 bp. This work found that segment 25, which includes the 19 bp deletion as well as another SNP, rs75627662, ~120 bp distal to the 19 bp deletion, demonstrated enhancer activity, but only in the human microglia line HMC3, and not in human neuronal SH-SY5Y or astrocyte U-118 cell lines. Therefore, the segment encompassing the deletion allele has enhancer activity exclusively in microglia. Our findings that the 19 bp deletion only mitigates SPI1 repression in human HMC3 cells, and not neuronal SH-SY5Y or astrocytic C8.D1A cells, further supports the microglia-specific function of this deletion, even though APOE has prominent functions in astrocytes53,54. Further studies detected increased expression of APOE-ε4 from the ancestral European versus African haplotype in single cell sequencing data, including 2.16-fold increased expression (the largest change that was detected) in microglia55. It is unknown whether the African American samples harbored the 19 bp deletion allele.
In the coming years, the NIH All of Us database is poised to make seminal genetic discoveries, especially in populations such as those of African descent historically under-represented by previous GWAS endeavors56. The large-scale linkage of EHR and WGS data enables the discovery of connections between phenotypic traits and genetic variants. We posit that another protective trait likely drove selection at APOE, as we clearly observe evidence supporting decreased odds for hyperlipidemia and hypercholesterolemia in APOE-ε4 to APOE-ε3 and APOE-ε2 carriers, corroborating established connections57. Other traits, such as those linked to high cholesterol or lipid levels like coronary artery disease, are also impacted. As mentioned, the 19 bp deletion is found nearly exclusively on the APOE-ε4 background which carries the highest risk for hyperlipidemia and hypercholesterolemia. While these traits are still elevated, their effect is dampened considerably in those harboring the 19 bp deletion, effectively reducing relative AD risk.
Importantly, many variants are in linkage disequilibrium with the 19 bp deletion and could have regulatory roles themselves that are difficult to rule out, such as rs43538058, which was previously linked to AD risk in samples of African ancestry58. As more long-read genomes become available it will be important to replicate long-read findings in larger cohorts, which may uncover other indels or structural variants. It is probable that not only one causal variant exists at the APOE/APOC1 locus, rather, multiple evolutionary attempts to move away from the ancestral APOE-ε4 genotype have likely taken place. Several studies have identified additional risk factors at the APOE locus, including a variable length poly-T polymorphism in TOMM4059,60. The fact that variants in high LD with APOE-ε2 or APOE-ε4 in between APOE and APOC1 can have effects exclusively on lipid or neurological traits confirms previous observations that a singular genetic event is causally not responsible for these phenotypes61.
Overall, these findings underscore the benefits of including diverse participants in research studies. Information gleaned from this study provides further insight into population-specific risk and protective alleles in AD and may assist in the development of targeted therapeutics.
Methods
1000 Genomes Project sample analysis
Reads that mapped to the APOE locus including the 19 bp deletion (GRCh38 co-ordinates chr19:54,691,726-54,692,180) were extracted from CRAM files for each individual from the 1000 Genomes Project25. HPRC24 and HGSVC26 phased long-read genomes were used for detecting variants on the APOE-ε4 background. We analyzed phased variant calls from the HPRC and HGSVC (214 haplotypes) with the vcftools (0.1.17) hap-r2 tool to identify SNVs within ±75 kb of APOE in LD with the 19 bp deletion.
Alzheimer’s disease sequencing project (ADSP) analysis
ADSP sequencing and phenotype data were downloaded from the NIAGADS data storage site (ADSP Umbrella Study version ng00067.v16). This dataset includes 58,507 samples with information about race, ethnicity, age, sex, AD status, Braak stage, and other features. Sequences for case-control analysis were used separated by reported race or ethnicity. The “Status of Alzheimer’s Disease Diagnosis” field (ADSP release 5) was used to ascribe AD status (0 = control, 1 = case). Samples listed as NA and samples flagged for relatedness and siblings were excluded. In total, 48,804 unique samples from the ADSP Case-Control study that contained genotype information for rs7412, rs429358, and rs148353395 (the 19 bp deletion) were used for analysis. Genotypes for rs148353395 were calculated based on GRCh38 position chr19:44910531 extracted from combined VCF files, and compared to APOE genotype from whole genome sequence data. Logistic regression analyses were performed to calculate odds ratios based on reference or deletion genotypes, adjusted for age, sex, sequencing center, sample cohort, and sequencing platform. 95% confidence intervals were calculated by the Wald method. Local African ancestry was previously calculated33 using 16,905 whole genome sequences as part of NIAGADS release ng00067.v8. To accommodate additional samples present in ng00067.v16, we developed an APOE/APOC1 local ancestry prediction model using genotype data from 2,373 individuals with known ancestry from the 1000 Genomes Project. To avoid potential effects of mixed ancestry62 we excluded the 1000 Genome Project individuals African Caribbean in Barbados (ACB) and African Ancestry in Southwest US (ASW) populations. Using the 204 SNPs from the region between APOE and APOC1 (chr19: 44903121-44921336) common between variant calling from the 1000 Genomes Project and the ADSP, we used a Random Forest classifier (scikit-learn v1.4) model to identify individuals with local African ancestry (AFR). This allowed us to create a model with 96.61% accuracy, with a 50% threshold for determining African or non-African local ancestry at APOE/APOC1. We trained the model on 80% of the dataset (n = 1,884) and used 20% for testing (n = 472). The location and relative weight of each variant is provided in Supplementary Data 22.
To test whether the 19 bp deletion had an effect on age of onset in individuals with local African Ancestry at APOE, we generated Kaplan-Meier survival curves and performed log rank tests to compare APOE-ε4 homozygous or APOE-ε3ε4 heterozygous individuals with or without the 19 bp deletion. Local African ancestry at APOE was determined by the random forest method described above. The log-rank (Mantel-Cox) test was used to evaluate differences in age of onset.
NIH All of Us PheWAS analysis
Data were selected and analyzed from the NIH All of Us database33. The study population of 380,179 included 255,622 with both WGS and EHR data, including 154,387 European ancestry and 47,168 African ancestry samples. Genetic ancestry was determined by principal component analysis of 56,671 high quality SNVs by All of Us. Local ancestry at APOE was calculated with the same Random Forest model as above, though this resulted in only 51,129 individuals with ancestry prediction including 8,654 with African ancestry, due to our strict filtering on variant call quality across the 204 sites. We further cleaned the data by removing individuals with a high degree of kinship determined and flagged by All of Us33, any individuals without male or female as their sex, individual with less than 20 different phecodes in their EHR records, and any samples from individual PheWAS with a Genome Quality (GQ) score of less than 20 for the locus in question. We utilized the R PheWAS package63 which performed allele frequency calculations, tests of Hardy-Weinberg equilibrium, Bonferroni correction, allelic p-value, allelic odds ratio (OR), and translated ICD-10 to ICD-9 electronic health record codes. Additionally, we used the R PheWAS option to sum duplicate phecodes into individual phenotypes, and employed a minimum code count of two for every condition to be considered a case, while still excluding single code counts from being denoted as controls. The PheWAS program phecode 290.11 was used for Alzheimer’s disease diagnosis. We used age, sex, and the first three principal components of genetic ancestry as covariates to correct for any effects they may introduce.
SNPs with a minor allele frequency greater or equal to 1% from 1000 Genomes Project samples in dbSNP release 155 were extracted from the intergenic region between APOE and APOC1 along with rs429358 and rs7412. To calculate pairwise LD between each pair of SNPs, the Pearson correlation coefficient (r²) was used via the python program scipy.stats.pearsonr. Individual samples with missing or invalid SNP values were excluded; only individuals with complete SNP information were included in the analysis. For visualization in the LD heatmap, a filtering step was applied whereby SNPs that did not show a squared correlation coefficient (r²) greater than 0.2 with any other SNP were excluded from the plot.
Cell culture
HMC3 human microglia cell lines (ATCC CRL-3304) were grown in EMEM media and seeded at 3.34 × 104 cells plated in 12-well plates for luciferase assays. SH-SY5Y cells (obtained from ATCC, CRL-2266) were grown in DMEM/F12 and seeded at 2 × 105 cells in 24-well plates for luciferase assays. C8-D1A murine astrocyte cells, a generous gift from Dr. Jonathan R. Weinstein, were grown in DMEM media and seeded at 2 × 104 cells per well in 48-well plates for luciferase assays. For all RT-qPCR experiments, HMC3, and C8-D1A cells were plated at 70% confluency (2.5 × 104 cells/well, and 1.2 × 105 cells/well, respectively) in 12 well plates.
Luciferase assay
Different variants of the APOE 3′UTR and extended SPI1 region were cloned into the 3′UTR region of a Renilla luciferase transcript in a psiCheck2.2 dual luciferase reporter construct (Promega). The first construct contained only the 143 bp APOE 3′UTR (“APOE 3′UTR only”). The remaining constructs all contained the 143 bp APOE 3′UTR, and an extended sequence containing either the SPI1 binding site or the 19 bp deletion spanning GRCh38 chr19:44,910,240-44,910,647 (“APOE SPI1 WT” and “APOE 19 bp Deletion”, 408 bp and 389 bp, respectively), as shown in Fig. 4a. We cloned different variations of the SPI1/19 bp deletion fragment into the 424 bp section of this construct, including a 19 bp deletion (405 bp, “3′UTR 19 bp Deletion”), the deletion of the SPI1 sequence (409 bp, “APOE SPI1 Deletion”), both the 19 bp deletion and the deletion of the SPI1 sequence (390 bp, “APOE 19 bp and SPI1 Deletion”), and a sequence containing a mutated SPI1 site (424 bp, “APOE SPI1 Mutation”), as shown in Supplementary Fig. 3). One day post-passage, the HMC3 and SH-SY5Y cells were assessed for viability and subsequently transfected with each dual-luciferase reporter construct using Lipofectamine 3000 Transfection Reagent (Invitrogen L3000015).
Native media was replaced 1.5 h after transfection for HMC3 and C8-D1A cells, and was not replaced for SH-SY5Y cells. 24 h after transfection, media was removed, and cells were washed with 1× PBS. Cells were then lysed in 1× Passive Lysis Buffer and evaluated using a Dual Luciferase Reporter Assay (Promega), detected on a Modulas Microplate Luminometer (Turner Biosystems). Four technical replicates were completed for each of the three experiments.
Luciferase data analysis
Relative luciferase expression was obtained by normalizing Renilla luciferase to Firefly luciferase as an internal control. The resulting data points were then normalized against the average of the APOE Extended 3′UTR data for each experiment, and significance was assessed relative to the APOE 3′UTR only using a one-way ANOVA in GraphPad PRISM 10.
RT-qPCR assay
Twenty-four hours post-passage, cells were transfected with SPI1 or treated with Aβ. For the SPI1 transfections, cells received 250 ng of an SPI1 construct via Lipofectamine 3000 transfection, per manufacturer’s instructions (n = 10–12/group). Briefly, the SPI1 expression plasmid containing the human SPI1 transcript variant 1 driven by a CMV promoter was obtained from Origene (RC217488). For the Aβ treatment, lyophilized amyloid beta 1:42 protein (Aβ 1:42; Tocris, cat. #1428) was obtained and reconstituted at a concentration of 100 μM. The reconstituted Aβ was incubated at 37 °C for 48 h to allow for fibrilization and aggregation64. Cells were exposed to fibrilized Aβ 1:42 at a concentration of 1 μM for a total of 6 hours prior to collection.
Twenty-four hours following SPI1 transfection or 6 h following Aβ, treated cells and untreated controls were washed once with 1× sterile PBS and collected in 600 μl of RNA Lysis Buffer (NEB, part of T2010S). Following collection, RNA was extracted using the Monarch Total RNA Miniprep Kit (NEB, T2010S), per manufacturer’s instructions. RNA concentration and quality were determined using a Nanodrop One Spectrophotometer. Samples with 260/280 values lower than 1.8 and concentrations lower than 16.6 ng were omitted from further downstream analysis. RNA was reverse transcribed into cDNA using the First Strand cDNA Synthesis (NEB, E6560) kit. Briefly, 100 ng of RNA was converted into cDNA using the “Easy Protocol” with d(T)23 VN. For RT-qPCR, primer sequences were either obtained from prior publications for APOE54 or generated using NCBI Primer Blast. The APOC1 primer was designed to detect all 4 transcript variants. RT-qPCR reactions were run using Luna Universal qPCR Master Mix (NEB, M3003) and undiluted cDNA from the samples. UBE2D2 was utilized as a housekeeping gene. The assay was run using a CFX384 Touch Real-Time PCR Detection System (BioRad, CA).
RT-qPCR data analysis
Samples with ∆CT values ± 2 standard deviations of the mean were omitted. The subsequent data was analyzed using the ∆∆CT method. Significance was assessed via a student’s 2 tailed t-test GraphPad PRISM 10. Primers are listed in Supplementary Table 1.
Ethics statement
The use of human study participants was conducted in accordance with criteria set forth by the Declaration of Helsinki. Samples used from the ADSP and All of Us were collected with ethics approval from the relevant institutional review boards as part of the data use agreements for the respective sources of data. We complied with the data use agreements.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
ADSP (release ng00067.v16) and All of Us (Version 8) data is available upon access approval through the following websites: dss.niagads.org and https://workbench.researchallofus.org/login, respectively. Data used to generate PheWAS output and forest plots are included within the Supplementary Data file. Source data are provided with this paper.
Code availability
Code for running PheWAS analysis is available on Github [github.com/ValdmanisLab/AllofUs_PheWAS] and archived at Zenodo under accession code 17595118. Code for running local African ancestry analysis is available on Github [github.com/ValdmanisLab/LocalAncestry_APOE-APOC1_AFR] and archived at Zenodo under accession code 17595120.
References
2024 Alzheimer’s disease facts and figures. Alzheimers Dement 20, 3708–3821 (2024).
Corder, E. H. et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923 (1993).
Genin, E. et al. APOE and Alzheimer disease: a major gene with semi-dominant inheritance. Mol. Psychiatry 16, 903–907 (2011).
Bellenguez, C. et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 54, 412–436 (2022).
Lambert, J. C. et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 45, 1452–1458 (2013).
Havel, R. J. & Kane, J. P. Primary dysbetalipoproteinemia: predominance of a specific apoprotein species in triglyceride-rich lipoproteins. Proc. Natl. Acad. Sci. USA 70, 2015–2019 (1973).
Zannis, V. I. et al. Proposed nomenclature of apoE isoproteins, apoE genotypes, and phenotypes. J. Lipid Res. 23, 911–914 (1982).
Bales, K. R. et al. Lack of apolipoprotein E dramatically reduces amyloid beta-peptide deposition. Nat. Genet. 17, 263–264 (1997).
Strittmatter, W. J. et al. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. USA 90, 1977–1981 (1993).
van Dyck, C. H. et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 388, 9–21 (2023).
Potter, G. G. et al. Cognitive performance and informant reports in the diagnosis of cognitive impairment and dementia in African Americans and whites. Alzheimers Dement 5, 445–453 (2009).
Barnes, L. L. & Bennett, D. A. Alzheimer’s disease in African Americans: risk factors and challenges for the future. Health Aff. (Millwood) 33, 580–586 (2014).
Mayeda, E. R., Glymour, M. M., Quesenberry, C. P. & Whitmer, R. A. Inequalities in dementia incidence between six racial and ethnic groups over 14 years. Alzheimers Dement 12, 216–224 (2016).
Reitz, C. et al. Variants in the ATP-binding cassette transporter (ABCA7), apolipoprotein E 4,and the risk of late-onset Alzheimer disease in African Americans. JAMA 309, 1483–1492 (2013).
Kunkle, B. W. et al. Novel Alzheimer disease risk loci and pathways in African American individuals using the African genome resources panel: a meta-analysis. JAMA Neurol. 78, 102–113 (2021).
Corbo & Scacchi, R. M. R. Apolipoprotein E (APOE) allele distribution in the world. Is APOE*4 a ‘thrifty’ allele? Ann. Hum. Genet 63, 301–310 (1999).
Blue, E. E., Horimoto, A., Mukherjee, S., Wijsman, E. M. & Thornton, T. A. Local ancestry at APOE modifies Alzheimer’s disease risk in Caribbean Hispanics. Alzheimers Dement 15, 1524–1532 (2019).
Rajabli, F. et al. Ancestral origin of ApoE epsilon4 Alzheimer disease risk in Puerto Rican and African American populations. PLoS Genet 14, e1007791 (2018).
Liu, M., Bian, C., Zhang, J. & Wen, F. Apolipoprotein E gene polymorphism and Alzheimer’s disease in Chinese population: a meta-analysis. Sci. Rep. 4, 4383 (2014).
Farrer, L. A. et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 278, 1349–1356 (1997).
Choi, K. Y. et al. APOE Promoter Polymorphism-219T/G is an Effect Modifier of the Influence of APOE epsilon4 on Alzheimer’s Disease Risk in a Multiracial Sample. J Clin Med 8 (2019).
Le Guen, Y. et al. Association of African Ancestry-Specific APOE Missense Variant R145C With Risk of Alzheimer Disease. JAMA 329, 551–560 (2023).
Le Guen, Y. et al. Association of Rare APOE Missense Variants V236E and R251G With Risk of Alzheimer Disease. JAMA Neurol. 79, 652–663 (2022).
Liao, W. W. et al. A draft human pangenome reference. Nature 617, 312–324 (2023).
Genomes Project, C. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Ebert, P. et al. Haplotype-resolved diverse human genomes and integrated analysis of structural variation. Science (2021).
Kierdorf, K. et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat. Neurosci. 16, 273–280 (2013).
Sonn, I. et al. Single transcription factor efficiently leads human induced pluripotent stem cells to functional microglia. Inflamm. Regen. 42, 20 (2022).
Kunkle, B. W. et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat. Genet 51, 414–430 (2019).
Marioni, R. E. et al. GWAS on family history of Alzheimer’s disease. Transl. Psychiatry 8, 99 (2018).
Huang, K. L. et al. A common haplotype lowers PU.1 expression in myeloid cells and delays onset of Alzheimer’s disease. Nat. Neurosci. 20, 1052–1061 (2017).
Minderjahn, J. et al. Mechanisms governing the pioneering and redistribution capabilities of the non-classical pioneer PU.1. Nat. Commun. 11, 402 (2020).
Horimoto, A. et al. Admixture mapping implicates 13q33.3 as ancestry-of-origin locus for Alzheimer disease in Hispanic and Latino populations. HGG Adv. 4, 100207 (2023).
All of Us Research Program Genomics, I Genomic data in the All of Us Research Program. Nature 627, 340–346 (2024).
de Beer, F. et al. Apolipoprotein E2 (Lys146->Gln) causes hypertriglyceridemia due to an apolipoprotein E variant-specific inhibition of lipolysis of very low density lipoproteins-triglycerides. Arterioscler Thromb. Vasc. Biol. 20, 1800–1806 (2000).
Wozniak, M. A. et al. Apolipoprotein E-epsilon 4 protects against severe liver disease caused by hepatitis C virus. Hepatology 36, 456–463 (2002).
Morris, J. A. et al. Discovery of target genes and pathways at GWAS loci by pooled single-cell CRISPR screens. Science 380, eadh7699 (2023).
Consortium, G. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020).
Pimenova, A. A. et al. Alzheimer’s-associated PU.1 expression levels regulate microglial inflammatory response. Neurobiol. Dis. 148, 105217 (2021).
Kim, B. et al. Effects of SPI1-mediated transcriptome remodeling on Alzheimer’s disease-related phenotypes in mouse models of Abeta amyloidosis. Nat. Commun. 15, 3996 (2024).
Berbee, J. F. et al. Apolipoprotein CI knock-out mice display impaired memory functions. J. Alzheimers Dis. 23, 737–747 (2011).
Abildayeva, K. et al. Human apolipoprotein C-I expression in mice impairs learning and memory functions. J. Lipid Res 49, 856–869 (2008).
O’Bleness, M., Searles, V. B., Varki, A., Gagneux, P. & Sikela, J. M. Evolution of genetic and genomic features unique to the human lineage. Nat. Rev. Genet 13, 853–866 (2012).
Margeta, M. A. et al. Association of APOE With Primary Open-Angle Glaucoma Suggests a Protective Effect for APOE epsilon4. Invest Ophthalmol. Vis. Sci. 61, 3 (2020).
Margeta, M. A. et al. Apolipoprotein E4 impairs the response of neurodegenerative retinal microglia and prevents neuronal loss in glaucoma. Immunity 55, 1627–1644 e7 (2022).
Wang, C. et al. ApoE-Isoform-Dependent SARS-CoV-2 Neurotropism and Cellular Response. Cell Stem Cell 28, 331–342 e5 (2021).
Ostendorf, B. N. et al. Common human genetic variants of APOE impact murine COVID-19 mortality. Nature 611, 346–351 (2022).
Trumble, B. C. et al. Apolipoprotein-epsilon4 is associated with higher fecundity in a natural fertility population. Sci. Adv. 9, eade9797 (2023).
Rajabli, F. et al. A locus at 19q13.31 significantly reduces the ApoE epsilon4 risk for Alzheimer’s Disease in African Ancestry. PLoS Genet 18, e1009977 (2022).
Grehan, S., Tse, E. & Taylor, J. M. Two distal downstream enhancers direct expression of the human apolipoprotein E gene to astrocytes in the brain. J. Neurosci. 21, 812–822 (2001).
Shih, S. J. et al. Duplicated downstream enhancers control expression of the human apolipoprotein E gene in macrophages and adipose tissue. J. Biol. Chem. 275, 31567–31572 (2000).
Nuytemans, K. et al. Identifying differential regulatory control of APOE varepsilon4 on African versus European haplotypes as potential therapeutic targets. Alzheimers Dement (2022).
Chen, Y. et al. The multifaceted roles of apolipoprotein E4 in Alzheimer’s disease pathology and potential therapeutic strategies. Cell Death Discov. 11, 312 (2025).
Arnaud, L. et al. APOE4 drives inflammation in human astrocytes via TAGLN3 repression and NF-kappaB activation. Cell Rep. 40, 111200 (2022).
Griswold, A. J. et al. Increased APOE epsilon4 expression is associated with the difference in Alzheimer’s disease risk from diverse ancestral backgrounds. Alzheimers Dement 17, 1179–1188 (2021).
Martin, A. R. et al. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat. Genet 51, 584–591 (2019).
Mack, S. et al. A genome-wide association meta-analysis on lipoprotein (a) concentrations adjusted for apolipoprotein (a) isoforms. J. Lipid Res 58, 1834–1844 (2017).
Fu, M. et al. Improving genetic risk modeling of dementia from real-world data in underrepresented populations. Commun. Biol. 7, 1049 (2024).
Deters, K. D. et al. TOMM40-APOE haplotypes are associated with cognitive decline in non-demented Blacks. Alzheimers Dement 17, 1287–1296 (2021).
Li, G. et al. TOMM40 intron 6 poly-T length, age at onset, and neuropathology of AD in individuals with APOE epsilon3/epsilon3. Alzheimers Dement 9, 554–561 (2013).
Fullerton, S. M. et al. Apolipoprotein E variation at the sequence haplotype level: implications for the origin and maintenance of a major human polymorphism. Am. J. Hum. Genet 67, 881–900 (2000).
Yang, H. C., Chen, C. W., Lin, Y. T. & Chu, S. K. Genetic ancestry plays a central role in population pharmacogenomics. Commun. Biol. 4, 171 (2021).
Carroll, R. J., Bastarache, L. & Denny, J. C. R. PheWAS: data analysis and plotting tools for phenome-wide association studies in the R environment. Bioinformatics 30, 2375–2376 (2014).
Aloi, M. S. et al. The pro-inflammatory microRNA miR-155 influences fibrillar beta-Amyloid(1)(-42) catabolism by microglia. Glia 69, 1736–1748 (2021).
Acknowledgements
This work was supported by R01NS122766, funded by the National Institute of Neurological Disorders and Stroke (NINDS) and the National Institute on Aging (NIA), to P.N.V. by R21AG089267 from the NIA to E.E.B., and by U01AG058589 from the NIA to E.M.W. J.N.B. was supported by National Institutes of Health Blueprint and Brain Research Through Advancing Innovative Neurotechnologies (BRAIN) Initiative Diversity Specialized Predoctoral to Postdoctoral Advancement in Neuroscience (D-SPAN) Award K00NS125830 funded by NINDS. Data for this study were prepared, archived, and distributed by the National Institute on Aging Alzheimer’s Disease Data Storage Site (NIAGADS) at the University of Pennsylvania (U24-AG041689), funded by the NIA. Full NIAGADS acknowledgment details are provided as a Supplementary Note. We thank the late Deborah Nickerson for her initial inspiration for this project. We thank Dr. Jonathan R. Weinstein for the generous gift of the C8-D1A murine astrocytic cell line. We gratefully acknowledge All of Us participants for their contributions, without whom this research would not have been possible. We also thank the National Institutes of Health’s All of Us Research Program for making available the participant data examined in this study.
Author information
Authors and Affiliations
Contributions
Conceptualization: J.N.B., E.E.B., and P.N.V. Experimental design and completion: J.N.B., T.B., E.N., E.J.K., E.L., S.C., A.S., S.N.S., Y.Y., I.P. B.L., E.M.W and P.N.V. Data and statistical analysis/interpretation: J.N.B., T.B., E.J.K., E.L., E.N., N.E.H., E.E.B., E.M.W., and P.N.V. Bioinformatics: E.N., E.J.K., and P.N.V. Manuscript original draft: J.N.B. and P.N.V. All authors contributed to final manuscript review and editing.
Corresponding author
Ethics declarations
Competing interests
B.L., I.P., S.C., and A.S. are employees of Cajal Neuroscience. All other authors declare no competing financial interests.
Peer review
Peer review information
Nature Communications thanks Sudeshna Das, Jennifer Yokoyama and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Brutman, J.N., Busald, T., Nizamis, E. et al. A common 19 bp APOE enhancer deletion is protective against Alzheimer’s disease in African Americans. Nat Commun 17, 2237 (2026). https://doi.org/10.1038/s41467-026-68808-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-68808-3
