
Abstract
Monomethylation of histone 3 lysine 4 (H3K4me1) marks enhancers in mammals. However, the function of H3K4me1 in plants remains largely unclear. Here, we present the genome-wide distribution of H3K4me1 in diverse species across evolution, revealing a distinctive H3K4me1 distribution pattern in land plants. To explore the function of H3K4me1 in plants, we identified an H3K4me1-specific reader protein, Early heading date 3 (Ehd3), and solved the structure of Ehd3 in complex with the H3K4me1 peptide, revealing a unique binding module differing from the previously reported PHD finger proteins. We further identified an Ehd3-binding protein, SET domain group 724 (SDG724), and the deletion of either Ehd3 or SDG724 caused similar defects in plant phenotype and changes in transcriptome and epigenome profiles. Both Ehd3 and SDG724 are enriched at chromatin regions marked by H3K4me1 but not H3K4me2 or H3K4me3. Ehd3 activates the H3K36 methyltransferase SDG724, and H3K36me2/me3 are colocalized with H3K4me1 in the genomes of the examined land plants. Collectively, our results reveal that H3K4me1 directs the establishment of H3K36me2 and H3K36me3 in land plants.
Similar content being viewed by others


Introduction
Histone post-translational modifications (PTMs) contribute to transcriptional regulation and play essential roles in diverse biological processes, including development and response to environmental cues1,2. These modifications, including methylation, acetylation, phosphorylation, and ubiquitination, are dynamically established and removed by specific writer and eraser enzymes, respectively, and are recognized by distinct reader proteins3,4. Methylation of histone 3 lysine 4 (H3K4) is among the most extensively studied PTMs that confers active or repressive transcription depending on its status (mono-, di-, or trimethylation)5. H3K4 trimethylation (H3K4me3) is distributed around the transcription start site (TSS) and is associated with gene activation in yeast6, animals7,8, and plants9. H3K4 dimethylation (H3K4me2) is diversely distributed in eukaryotes: it is enriched at the TSS and associated with gene activation in yeast6 and humans7,8, whereas it is located in the gene body region and linked to gene repression in plants9. H3K4 monomethylation (H3K4me1) is enriched over the gene body in yeast6 and plants9, but tends to be concentrated around the TSS in mammals7,8. In addition, H3K4me1 is associated with other PTMs that mark distinct states of enhancers in mammals. Active enhancers are enriched with H3K4me1 and histone 3 lysine 27 acetylation (H3K27ac)10. During the intermediate state, primed enhancers primarily exhibit enrichment of H3K4me111. Poised enhancers in pluripotent cells are marked by H3K4me1 and H3K27me312. However, the deposition of H3K4me1 at enhancers is not conserved in plants13 and the role of H3K4me1 in plants has yet to be elucidated.
Reader proteins possess specific domains that recognize H3K4 methylation, including the plant homeodomain (PHD), double chromodomain, tandem Tudor domain, and zinc finger CW3. To date, a number of structures of PHD finger–H3K4me2/me3 complexes have been determined in animals and plants, whereas the PHD finger in complex with H3K4me1 has not been reported in plants and exclusive readers for H3K4me1 are lacking in animals14. The structures of CW or Tudor domains complexed with H3K4me1 have been reported in plants, including the CW domain of H3K36 methyltransferase SET DOMAIN GROUP 8 (SDG8)15 and the Tudor domain of RNA-directed DNA methylation 15 (RDM15)16 in Arabidopsis. Nevertheless, the biological function of H3K4me1 decoding in plants remains largely uncharacterized.
Here, we identified a distinct distribution pattern of H3K4me1 in land plants that differs from that observed in yeast and animals. A newly identified PHD finger domain-containing protein, Early heading date 3 (Ehd3), specifically recognizes H3K4me1 and recruits the H3K36 methyltransferase SDG724 to facilitate H3K36me2/me3 deposition. The colocalization of H3K4me1 and H3K36me2/me3 is conserved in the examined land plants, revealing a specific role of H3K4me1 in directing H3K36me2/me3 deposition in plants.
Results
H3K4me1 exhibits a distinct genome-wide profile in land plants
To determine the function of H3K4me1 in plants, we analyzed the genome-wide distributions of H3K4me1/me2/me3, and the correlation between H3K4 methylations and gene transcription by conducting chromatin immunoprecipitation followed by sequencing (ChIP-seq) and RNA-sequencing (RNA-seq) assays in a variety of plant species, comprising the unicellular aquatic Chlamydomonas reinhardtii, the early land plant Physcomitrium patens, and the flowering plants Arabidopsis thaliana and Oryza sativa. By integrating previously published ChIP-seq and RNA-seq data for the unicellular yeast Saccharomyces cerevisiae, the invertebrate Drosophila melanogaster, and the vertebrates Mus musculus and Homo sapiens17,18,19,20,21, we analyzed the genome-wide H3K4me1/me2/me3 profiles across diverse organisms (Fig. 1 and Supplementary Fig. 1).

a Heatmaps showing the individual gene distribution of H3K4me1, together with their associated transcription level (log2-transformed). All genes are sorted based on enrichment of H3K4me1. Gradient colors indicate levels of H3K4me1 enrichment and transcription, respectively. b Integrative genomic distribution of H3K4me1. c Scatter plots showing the correlation between H3K4me1 level and gene transcription level. The species (from left to right) are Saccharomyces cerevisiae, Drosophila melanogaster, Mus musculus, Homo sapiens, Chlamydomonas reinhardtii, Physcomitrium patens, Arabidopsis thaliana, and Oryza sativa, respectively. The transcribed genes (RPKM > 1) enriched with the H3K4me1 modification were divided into 100 groups based on the levels of their transcription and modification. The average values of each group of modifications and transcription were calculated for analysis. The heatmaps and plots present the region from 3 kb upstream of the transcription start site (TSS) to 3 kb downstream of the transcription termination site (TTS). For scatter plots, Spearman’s rank correlation coefficient (rho) indicates the correlation between the methylation level and gene transcription level. The p-values were determined based on two-sided Spearman’s rank correlation test. Publicly available data were downloaded and analyzed for Saccharomyces cerevisiae (ref. 17), Drosophila melanogaster (GSE47281 and SRR1197331), Mus musculus (ref. 18), Homo sapiens (refs. 19,20), and Arabidopsis thaliana (ref. 21). One replicate of ChIP-seq data from each sample was selected for analysis.
We observed that H3K4me1 exhibited particularly distinct characteristics across the examined species. In animals, comprising Drosophila melanogaster, Mus musculus, and Homo sapiens, H3K4me1 was enriched near the TSS and gradually decreased along the gene body. In contrast, H3K4me1 was mainly enriched in gene bodies in Chlamydomonas reinhardtii, Physcomitrium patens, Arabidopsis thaliana, and Oryza sativa as previously reported22 (Fig. 1a, b). In yeast and animals, a positive correlation was observed between H3K4me1 and gene transcription (Fig. 1c). In Chlamydomonas reinhardtii, a weak correlation between H3K4me1 and gene transcription was detected. In contrast, the land plants Physcomitrium patens, Arabidopsis thaliana, and Oryza sativa displayed a distinctive relationship between H3K4me1 and gene transcription: a high percentage of genes displayed a positive correlation between H3K4me1 and gene transcription, but the most highly expressed genes exhibited lower levels of H3K4me1 (Fig. 1c). Thus, land plants displayed a unique genome-wide distribution pattern for H3K4me1, and a distinct correlation between H3K4me1 and gene transcription.
Unlike animals, in which two peaks around the TSS were observed, H3K4me2 was distributed over the entire gene body in land plants (Supplementary Fig. 1a, b). Consistent with a previous study, a positive correlation between H3K4me2 level and gene transcription level was observed in yeast and animals (Supplementary Fig. 1c). However, H3K4me2 was negatively correlated with gene transcription in land plants, consistent with our previous report9 (Supplementary Fig. 1c). The H3K4me3 distributions in all investigated species were similar, peaking close to the TSS (Supplementary Fig. 1d, e). Moreover, H3K4me3 consistently displayed a positive correlation with gene transcription in all species examined, highlighting the conserved association of H3K4me3 with gene activation in eukaryotes (Supplementary Fig. 1f).
Ehd3, a PHD finger domain-containing protein, specifically recognizes H3K4me1
The unique distribution pattern and distinct correlation with gene transcription suggested that H3K4me1 may play a unique role in land plants. Given that the epigenetic information encoded by histone PTMs is interpreted by specialized reader proteins, we thus investigated the function of H3K4me1 in land plants by searching for its specific reader proteins. There are multiple plant homeodomain (PHD) domain-containing proteins in plants and PHD domains are widely reported to recognize H3K4 methylations, thus we analyzed the PHD domain-containing proteins in Chlamydomonas reinhardtii and Physcomitrium patens, aiming to discover the potential H3K4me1 readers conserved in land plants but not in Chlamydomonas reinhardtii. We identified 52 and 107 proteins containing at least one PHD domain in Chlamydomonas reinhardtii and Physcomitrium patens, respectively. Notably, this expansion in Physcomitrium patens was particularly pronounced for proteins containing multiple PHD domains (n ≥ 2) (Supplementary Fig. 2a). Proteins containing triple-tandem PHD domains were presented in both Chlamydomonas reinhardtii and Physcomitrium patens, however, those with double-tandem PHD were unique in Physcomitrium patens (Supplementary Fig. 2b). Therefore, our searching focused on the proteins containing double-tandem PHD domains. Our phylogenetic analysis showed that a protein (A0A2K1L966) evolved a double-tandem PHD domain in Physcomitrium patens, which was absent in Chlamydomonas reinhardtii but conserved in land plants (Fig. 2a). For in-depth functional study, we selected a homolog of A0A2K1L966, Ehd3, which is known to regulate flowering time in rice23 (Fig. 2b). To explore the histone modification decoding mode of Ehd3, we purified the full-length recombinant Ehd3 protein and performed an in vitro binding assay using a histone peptide microarray with diverse histone PTMs. Ehd3 exhibited a strong preference for binding to H3K4me1 over other modifications (Supplementary Fig. 2c). A biotinylated histone peptide pulldown assay similarly showed that Ehd3 interacted exclusively with the H3K4me1 peptide, but not with other tested modifications, including H3K4me0, H3K4me2, and H3K4me3 (Fig. 2c and Supplementary Data 1). Further investigation showed that individual PHD1, PHD2, or PHD3 failed to bind to H3K4me1, whereas the tandem PHD finger domain (PHD2–PHD3) was essential and sufficient for recognition of H3K4me1 (Fig. 2d). Isothermal titration calorimetry (ITC) analysis further confirmed robust interaction between Ehd3 and the H3K4me1 peptide with a dissociation constant (Kd) of 1.7 μM (Fig. 2e and Supplementary Data 1–2). In summary, Ehd3 specifically recognized H3K4me1 through its tandem PHD finger domain.

a Neighbor-joining dendrogram (left) and domain architecture (right) of Ehd3 homologous proteins in plants. The neighbor-joining dendrogram was constructed based on the tandem plant homeodomain (PHD) amino acid sequence alignment. Ac Arabian coffee, Ca Capsicum annuum, Gm Glycine max, Rc Rosa chinensis, Dc Daucus carota, At Arabidopsis thaliana, Zm Zea mays, Os Oryza sativa, Si Setaria italica, Ta Triticum aestivum, Bd Brachypodium distachyon, Pp Physcomitrium patens, Cr Chlamydomonas reinhardtii. UniProt (https://www.uniprot.org) entries for each protein are given after the species abbreviation. b Schematic representation of the full-length Ehd3 protein. The individual PHD finger domains are indicated by different colored boxes. c Immunoblotting analysis of histone peptide pulldown using the full-length Ehd3 fused with glutathione-S-transferase (GST). d Schematic representation of different truncated Ehd3 proteins fused with GST (left) and immunoblotting analysis of H3K4me1-peptide pulldown (right). Roman numerals indicate the corresponding truncations. e Isothermal titration calorimetry results showing the binding affinities of the full-length Ehd3 with different H3K4 methylated peptides. NDB no detectable binding. Source data are provided.
Structure of Ehd3 tandem PHD finger domain in complex with H3K4me1 peptide
To clarify the molecular mechanism underlying Ehd3 recognition of H3K4me1, we solved the crystal structure of Ehd3417–547 bound to a H3K4me1 peptide (A1RT-Kme1-QTARKSTG) at atomic resolution (1.5 Å, Supplementary Data 3). Ehd3418–540 adopts an L-shaped overall architecture and the peptide binds across an acidic groove formed by the PHD2 and PHD3 domains (Fig. 3a). Both PHD2 and PHD3 of Ehd3 adopt interleaved topologies chelating two Zn2+ ions (Supplementary Fig. 2d, e), while the H3K4me1 peptide adopts an extended conformation (Supplementary Fig. 2f, g). Partial atoms of Gln5, Arg8, and Lys9 are invisible in the map, suggesting flexible disposition of these residues (Supplementary Fig. 2f, g). The side chains of Ala1 and Arg2 of the H3K4me1 peptide reside in two surface channels separated by Asp493 of Ehd3. The monomethylated Lys4 inserts into a deep pocket adjacent to the Arg2-binding channel, forming a ‘Y’-shaped conformation with Ala1 and Arg2 that anchor the binding interface (Fig. 3b). The main-chain amino group of Ala1 forms two stable hydrogen bond (H-bond) interactions with the main-chain carbonyl groups of Pro512 and Gly514, respectively, whereas the methyl group of Ala1 forms a hydrophobic interaction with the aromatic ring of Pro512 (Supplementary Fig. 2h). The main-chain amide group and carbonyl group of Arg2 form two water-mediated H-bonds and one direct H-bond interaction with Met491 and Asp493, respectively. Via the guanidyl group, the side chain of Arg2 forms three stable H-bond interactions with Tyr446, Cys492, and Asp496 (Fig. 3c and Supplementary Fig. 2i, j).

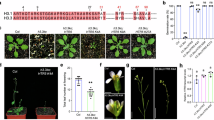
a Overall structure (upper panel) and electrostatic surface view (lower panel) of the Ehd3 tandem plant homeodomain (PHD) finger domain in complex with the H3K4me1 peptide. PHD2 and PHD3 of the Ehd3 tandem PHD finger domain are shown in cyan and green, respectively, and the H3K4me1 peptide is in yellow. Blue and red shades on the surface indicate positive and negative charges, respectively. b Electrostatic surface showing the details of the Ehd3 tandem PHD finger domain–H3K4me1 complex. c Detailed interaction between the Ehd3 tandem PHD finger domain and H3R2. Hydrogen bonds are indicated by dashed lines. d Detailed interaction of the Ehd3 tandem PHD finger domain and H3K4me1. Hydrogen bonds are indicated by dashed lines. e Ehd3 tandem PHD finger domain cut-away view of H3K4me1 recognition. f Simulated interactions between the Ehd3 tandem PHD finger domain and H3K4me3. g Isothermal titration calorimetry results showing the binding affinities between the wild-type or site-mutated Ehd3 tandem PHD finger domain with the H3K4me1 peptide. NDB, no detectable binding. h Representative image (upper panel) and heading date (lower panel) of wild-type rice ‘Nipponbare’ (NiP), ehd3-1, ehd3-4, HA-Ehd3/ehd3-1-1, and HA-Ehd3/ehd3-1-2 plants grown under long-day (LD) and short-day (SD) photoperiods. i Representative image (upper panel) and heading date (lower panel) of wild-type NiP, ehd3-1, and HA-Ehd3mu/ehd3-1-1 plants grown under LD and SD photoperiods. Scale bar, 20 cm. Values are the mean ± standard deviation of 20 individual plants. Statistical significances were determined with one-sided Ordinary one-way ANOVA test followed two-sided Tukey’s Honestly Significant Difference test: ** adjusted p-value < 0.01; ns (not significant). Source data are provided.
In contrast to trimethylated Lys4-binding pockets present in many PHD proteins, which are composed of several conserved aromatic residues within a single PHD domain (Supplementary Fig. 3), the monomethylated Lys4-binding pocket of Ehd3 is formed by residues from both PHD2 (His441, Tyr471, and Cys475) and PHD3 (Asp483, Asp486, and Ile489) (Fig. 3d and Supplementary Fig. 2j, k). The side-chain primary amino group of monomethylated Lys4 forms H-bond interactions with the main-chain carbonyl group of PHD2 Cys475 and the side-chain carbonyl group of PHD3 Asp486. The side-chain methyl and alkyl groups of Lys4 hydrophobically interact with the side chains of PHD2 Tyr471 and His441, respectively. The main chains of Lys4 and PHD3 Ile489 form a H-bond and the side chain of Ile489 hydrophobically interacts with the side-chain alkyl groups of Lys4. The phenolic hydroxyl group of PHD2 Tyr471 forms a H-bond interaction with the side-chain carbonyl group of PHD3 Asp483 to stabilize the binding pocket (Fig. 3d and Supplementary Fig. 2j). As a result, the side chain of monomethylated Lys4 perpendicularly and compactly inserts into the pocket, which harbors a hydrophobic environment created by PHD2 and a polar environment created by PHD3 (Fig. 3e). Our modeling study suggests that the introduction of a second or third methyl group to Lys4 will lead to a clash with Cys475 and/or Asp486, which will prevent the binding of Ehd3 to H3K4me2 or H3K4me3, implying the H3K4me1 specificity of Ehd3 (Fig. 3f). In addition to the monomethylated Lys4, the side-chain imidazole ring of PHD2 His441 forms one stable H-bond interaction with the main-chain carbonyl group of Gln5 of the peptide. Residues 6–9 of the peptide mainly form direct or water-mediated H-bond interactions with symmetry-related Ehd3 molecules, whereas residues 10–12 of the peptide are completely disordered in the structure (Supplementary Fig. 2j, l, m). Consistent with the structural observation, mutation of H3R2-interacting residues (Tyr446 and Asp496) or H3K4me1 binding-pocket residues (His441, Tyr471, Asp486, and Ile489) either abolished or markedly reduced the binding affinity of the Ehd3 tandem PHD finger to the H3K4me1 peptide (Fig. 3g and Supplementary Data 2), which further confirmed that both H3R2-binding and H3K4me1-binding residues are critical for H3K4me1 recognition by Ehd3. The dissociation constant of the wild-type Ehd3 tandem PHD to H3K4me1 peptide was comparable to that of the full-length Ehd3 (Figs. 2e and 3g), excluding the potential contribution of PHD1 to specific recognition of H3K4me1.
Given that Double PHD Finger 3b (DPF3b) is a well-studied epigenetic factor that recognizes histone modifications through a tandem PHD finger domain24, we generated a sequence alignment of the tandem PHD of Ehd3 and DPF3b. Most H3K4me1-binding sites in Ehd3 were not conserved in DPF3b (Supplementary Fig. 3a). The tandem PHD finger domain in Ehd3 forms a narrow and sealed pocket, whereas DPF3b relies on a single PHD finger domain to create an open pocket for H3K14ac binding25 (Fig. 3e and Supplementary Fig. 3c, d). In addition, we compared the reported structures of the H3K4me3-binding PHD finger domains in the Bromodomain PHD finger transcription factor (BPTF)26, PHD finger protein 2 (PHF2)27, YNG128, and Inhibitor of growth protein 2 (ING2)29 with that of Ehd3–H3K4me1. The interaction between the PHD finger domain and H3K4me3 is highly conserved in these proteins and the critical residues are not conserved in the three PHD finger domains of Ehd3 (Supplementary Fig. 3b, e–h). In most cases, a single PHD finger domain is sufficient for recognition of histone modifications, highlighting a novel binding mode of the tandem PHD finger domain within Ehd3 to specifically recognize H3K4me1.
Ehd3–H3K4me1 interaction is required for proper timing of flowering in rice
To further investigate the functional significance of Ehd3–H3K4me1 binding in vivo, we first generated CRISPR/Cas9 mutants targeting Ehd3 in the ‘Nipponbare’ (NiP) background. Two single guide RNAs (sgRNAs) targeting the second exon of Ehd3 were designed. We selected mutants with two independent transgene-free mutations, one with a 1-bp deletion at target site 1 and a 9-bp deletion at target site 2 (ehd3-1), and the other with a 4-bp deletion at target site 1 and a 1-bp insertion at target site 2 (ehd3-4), for further analysis (Supplementary. Fig. 4a). Mutations in the ehd3-4 allele resulted in a net 3-bp deletion and amino acid changes within the residues 52–92 of Ehd3. The ehd3-1 and ehd3-4 mutant plants displayed a delayed-flowering phenotype under both natural long-day (LD) and short-day (SD) photoperiods (Supplementary Fig. 4b), consistent with the reported phenotypes of ehd3 mutants in the Oryza sativa subsp. japonica ‘Tohoku IL9’ background23,30. We then generated the plants HA-Ehd3/ehd3-1-1 and HA-Ehd3/ehd3-1-2, expressing Ehd3 fused with the 4×HA tag driven by its native promoter in the ehd3-1 mutant background (Supplementary Fig. 4c). The delayed-flowering phenotype of ehd3-1 was fully rescued upon the expression of HA-Ehd3 (Fig. 3h), indicating that the delayed-flowering phenotype is attributable to mutations in Ehd3.
In addition, we generated transgenic plants expressing a mutated version of Ehd3 by substituting two critical Ehd3–H3K4me1-interacting residues, Tyr471 and Asp496, with Ala under the control of its native promoter in the ehd3-1 mutant background (Supplementary Fig. 4c). The transgenic line (HA-Ehd3mu/ehd3-1-1) carrying these site-specific mutations exhibited a delayed-flowering phenotype similar to that of the ehd3-1 mutant (Fig. 3i), suggesting that the mutation of critical residues responsible for Ehd3–H3K4me1 interaction within Ehd3 failed to rescue the phenotype of the ehd3-1 mutant. All of the aforementioned findings showed that binding to H3K4me1 is critical for the biological function of Ehd3 in rice.
Ehd3 physically interacts with the H3K36 methyltransferase SDG724
To further investigate the role of H3K4me1 through Ehd3 in plants, we identified the co-factors of Ehd3 by immunoprecipitation followed by mass spectrometry (IP-MS). Transgenic plants were generated harboring Enhanced Green Fluorescent Protein (EGFP) fused to Ehd3 driven by the rice Ubiquitin promoter (Supplementary Fig. 4d). Among the candidates for Ehd3-binding proteins identified in the IP-MS experiment, the H3K36-specific methyltransferase SDG724 was identified31 (Fig. 4a and Supplementary Data 4). Therefore, transgenic plants expressing Yellow Fluorescent Protein (YFP) fused to SDG724 driven by the Ubiquitin promoter were generated (Supplementary Fig. 4e). Ehd3 was among the SDG724-binding candidates identified by IP-MS (Fig. 4a and Supplementary Data 5), suggesting the possible interaction between Ehd3 and SDG724. While transgenic plants expressing YFP alone driven by the Ubiquitin promoter were served as the control, neither Ehd3 nor SDG724 was detected in the IP-MS experiment (Supplementary Data 6). Yeast two-hybrid, glutathione-S-transferase (GST) pulldown assays, co-immunoprecipitation, and bimolecular fluorescence complementation (Fig. 4b–e and Supplementary Fig. 4f) assays consistently confirmed the physical interaction between Ehd3 and SDG724 in rice. Furthermore, the Arabidopsis homolog of rice Ehd3, Q1JPM3 (named as AtEHD3), also interacted with the homolog of rice SDG724, SDG7, suggesting a potentially conserved interaction between H3K4me1 readers and H3K36 methyltransferases in plants (Supplementary Fig. 4g, h).

a Scatter plot representing mass spectrometry results of EGFP-Ehd3 and YFP-SDG724 immunoprecipitation analyses. The x-axis and y-axis indicate the log10-abundance of proteins in IP-MS assays. b Yeast two-hybrid assay showing the interaction between Ehd3 and SDG724 in yeast cells. AS1 and AS2 were used as the positive control. c Glutathione-S-transferase (GST) pulldown assay showing the interaction between Ehd3 and SDG724. Ehd3 was pulled down by GST-SDG724 but not GST. GST, GST-SDG724, and Ehd3 are marked by asterisks in the gel. d Co-immunoprecipitation assay showing the interaction of Ehd3 and SDG724 in vivo. Proteins from rice protoplasts expressing Myc-Ehd3 and HA-SDG724 were extracted. Myc-Ehd3 and HA-SDG724 were detected by western blot using anti-Myc and anti-HA antibodies, respectively. e Bimolecular fluorescent complementation assay showing the interaction of Ehd3 and SDG724 in rice protoplasts. Scale bar, 10 μm. f Heading date (left) and representative images (right) of wild-type rice ‘Nipponbare’ (NiP), ehd3-1, sdg724-1, and ehd3 sdg724 plants grown under long-day (LD) and short-day (SD) photoperiods. Scale bar, 20 cm. Values are the mean ± standard deviation of 20 individual plants. Statistical significances were determined with one-sided Ordinary one-way ANOVA test followed two-sided Tukey’s Honestly Significant Difference test: ** adjusted p-value < 0.01; ns (not significant). g Heatmap showing the log2(fold change) of differentially expressed genes (DEGs) in the ehd3-1, sdg724-1, and ehd3 sd724 mutants compared with the wild-type NiP. The union of DEGs was divided into two clusters using the k-means algorithm (the Lloyd-Forgy method). Up- and down-regulated genes are indicated in red and blue, respectively. h Scatter plots showing the highly correlated DEGs (p-value ≤ 0.05 and |fold change| ≥ 1.5) in the ehd3-1, sdg724-1, and ehd3 sdg724 mutants. Correlations between different mutants were evaluated using two-sided Pearson’s correlation coefficient (cor) test. i Scatter plots comparing H3K4me1, H3K36me1, H3K36me2, and H3K36me3 levels between the wild-type NiP and the ehd3-1, sdg724-1, and ehd3 sdg724 mutants. Each dot represents the histone methylation level of one peak corrected with MAnorm. The transition from deep blue to cyan indicates the increase in dot density. Source data are provided.
Deletion of Ehd3 or SDG724 leads to similar changes in plant phenotype, transcriptome, and epigenome
Both Ehd3 and SDG724 promote flowering under SD and LD photoperiods31 (Fig. 4f). To investigate the genetic relationship between Ehd3 and SDG724, we crossed the ehd3-1 and sdg724-1 mutants to generate the ehd3 sdg724 double mutant. The ehd3 sdg724 double mutant displayed a similar late-flowering phenotype to that of each single mutant under both SD and LD photoperiods (Fig. 4f), implying that Ehd3 and SDG724 promote flowering through the same genetic pathway.
To evaluate how Ehd3 and SDG724 regulate gene transcription at the genome scale, we conducted RNA-seq analyses of the wild-type NiP and the ehd3-1, sdg724-1, and ehd3 sdg724 mutants. Hundreds of differentially expressed genes (DEGs) were identified in the ehd3-1, sdg724-1, and ehd3 sdg724 mutants compared with NiP (Supplementary Fig. 5a and Supplementary Data 7). Nearly half of the up- and down-regulated genes among the single and double mutants were overlapped (Supplementary Fig. 5b, c). The overlapping DEGs displayed similar transcription changes in the ehd3-1, sdg724-1, and ehd3 sdg724 mutants (Fig. 4g). Furthermore, pairwise scatterplot analyses indicated a strong correlation in the DEGs among the ehd3-1, sdg724-1, and ehd3 sdg724 mutants (Fig. 4h), implying cooperative regulation of gene transcription by Ehd3 and SDG724.
Given that Ehd3 is an H3K4me1 reader and SDG724 is an H3K36 methyltransferase, we investigated the global distributions of H3K4me1/me2/me3, H3K36me1/me2/me3, and H3 control in NiP and the ehd3-1, sdg724-1, and ehd3 sdg724 mutants via ChIP-seq analysis. Gene-by-gene scatterplots and heatmaps revealed no alterations in the distributions of H3K4me2/me3, similarly subtle changes for H3K4me1 and H3K36me1, and similarly pronounced changes in H3K36me2/me3 distributions in the ehd3-1, sdg724-1, and ehd3 sdg724 mutants compared with those of NiP (Fig. 4i and Supplementary Fig. 6a, b). As a control, the H3 distribution in these mutants was comparable to that in NiP (Supplementary Fig. 6c, d). Therefore, Ehd3 cooperates with SDG724 to regulate H3K36me2/me3 distributions, gene transcription, and plant development.
The Ehd3 homologs containing tandem PHD finger domain recognize H3K4me1 in land plants
In mammals, H3K4me1 marks enhancers but fewer H3K4me1 reader proteins have been identified to date14. The present analysis revealed the prevalence of Ehd3 homologs in both plants and animals (Fig. 5a). Distinct domain architecture and protein length were observed among Ehd3 homologs across plant and animal species (Fig. 5a). Sequence alignment of the tandem PHD finger domains within Ehd3 homologs in land plants revealed a remarkable conservation of residues responsible for recognition of H3K4me1, particularly His441 and Tyr471 (Supplementary Fig. 7a). The tandem PHD finger domains within Ehd3 homologs in various species were predicted with AlphaFold (Fig. 5b–e and Supplementary Fig. 7b–g)32; the interleaved topology and residues crucial for H3K4me1 binding were largely identical to those of Ehd3 in land plants, including Zea mays, Triticum aestivum, Arabidopsis thaliana, and Daucus carota (Fig. 5b–e). However, the tandem PHD finger domains in Physcomitrium patens showed only partial similarity, and the overall conformations of those in Chlamydomonas reinhardtii, Penaeus vannamei, Elysia chlorotica, Mus musculus, and Homo sapiens (Supplementary Fig. 7b–g) differed significantly from the tandem PHD finger domain within Ehd3.

a Neighbor-joining dendrogram (left) and protein domain architecture (right) of the tandem plant homeodomain (PHD) domain-containing proteins in higher eukaryotes. The dendrogram was constructed based on the Ehd3 tandem PHD sequence alignment. The diverse protein domains are displayed in different colored boxes as indicated. Mm Mus musculus, Pd Phyllostomus discolor, Fc Felis catus, An Aotus nancymaae, Hs Homo sapiens, Ec Elysia chlorotica, Ob Octopus bimaculoides, Pv Penaeus vannamei, Zm Zea mays, Si Setaria italica, Os Oryza sativa, Ta Triticum aestivum, Bd Brachypodium distachyon, Ac Arabian coffee, Ca Capsicum annuum, Dc Daucus carota, Gm Glycine max, Rc Rosa chinensis, At Arabidopsis thaliana, Pp Physcomitrium patens, Cr Chlamydomonas reinhardtii. UniProt (https://www.uniprot.org) entries of the proteins are marked behind the species abbreviations. Structure superimposition of the tandem PHD in A0A1D6LDK6 (b) A0A3B6RKR3 (c) AtEHD3 (d) A0A162A0W2 (e) with Ehd3. The structure of Ehd3 is colored in cyan. f ITC results showing the binding affinities of homologous tandem PHD in Chlamydomonas reinhardtii, Physcomitrium patens, Arabidopsis thaliana, and Mus musculus with H3K4me0 and methylated H3K4 peptides. NDB no detectable binding. Source data are provided.
The ITC analysis for the tandem PHD finger domains within Ehd3 homologs in Chlamydomonas reinhardtii, Physcomitrium patens, Arabidopsis thaliana, and Mus musculus indicated their diverse binding affinities towards H3K4 methylations. Specifically, the tandem PHD finger domains in Mus musculus showed binding affinity exclusively with the H3K4me0 peptide (PHD23, Kd = 6.5 μM; PHD56, Kd = 13.5 μM) (Fig. 5f). The single PHD finger domain in Chlamydomonas reinhardtii failed to bind to any methylated H3K4. In contrast, the tandem PHD finger domain in Physcomitrium patens exhibited a stronger affinity for H3K4me1, with even greater binding observed in Arabidopsis thaliana, similar to that in Oryza sativa (Fig. 5f). Collectively, the tandem PHD finger domain initiates H3K4me1 recognition in land plants, and with highly exclusive affinity in flowering plants.
H3K4me1 directs H3K36me2 and H3K36me3 deposition in land plants
Genome-wide occupancies of Ehd3 and SDG724 were analyzed by ChIP-seq using the EGFP-Ehd3 and YFP-SDG724 transgenic plants. 15,967 Ehd3-occupied genes and 17,070 SDG724-occupied genes were identified, respectively. Among them, 10,325 genes showed significant co-occupancy by Ehd3, SDG724 and H3K4me1 modification (Fig. 6a and Supplementary Data 8–9). Gene-by-gene analysis confirmed that Ehd3 and SDG724 were preferentially enriched at genes marked with H3K4me1, but not H3K4me2 or H3K4me3, compared with the random control genes, supporting the hypothesis that Ehd3 specifically recognizes H3K4me1 in vivo (Fig. 6b, c, Supplementary Fig. 8a, b, and Supplementary Data 8–9). Importantly, the enrichment of SDG724 at its target genes was dramatically abolished when Ehd3 was deleted, highlighting an essential role of Ehd3 in recruiting SDG724 to H3K4me1-marked genes (Supplementary Fig. 8c–f). Furthermore, gene-by-gene heatmaps revealed that loss of either Ehd3 or SDG724 caused similar changes in H3K36me2/me3 distributions within the genes enriched of H3K4me1 (Fig. 6d). In detail, H3K36me2 occupancy decreased at the 3′ regions, whereas H3K36me3 decreased across the gene body in the ehd3-1, sdg724-1, and ehd3 sdg724 mutants compared with NiP (Fig. 6d). We thus examined whether the introduction of site-mutated Ehd3 can rescue the genome-wide occupancy of H3K36me2/me3 in the HA-Ehd3mu/ehd3-1-1 transgenic plants. Comparative analysis revealed that the site-mutated Ehd3 (Ehd3mu), which lost its binding capacity to H3K4me1, failed to recover the H3K36me2/me3 profiles in the HA-Ehd3mu/ehd3-1-1 plants, while the wild-type Ehd3 did in the HA-Ehd3/ehd3-1-1 plants (Supplementary Fig. 9a). Gene-by-gene analysis further confirmed that the site-mutated Ehd3 failed to rescue the aberrant H3K36me2/me3 distribution patterns in the ehd3-1 mutant background (Supplementary Fig. 9b, c). Together, all these results proved that Ehd3-binding to H3K4me1 is necessary for SDG724 to establish H3K36me2/me3 in planta, supporting that Ehd3 recognizes H3K4me1 and recruits SDG724 to H3K4me1-marked chromatin to promote the deposition of H3K36me2 and H3K36me3 in rice (Fig. 6e and Supplementary Fig. 9d).

a Venn diagram showing the overlap of H3K4me1-, Ehd3-, and SDG724-enriched genes. p-values were determined by one-sided Fisher’s exact test. b Heatmaps showing Ehd3 and SDG724 levels within H3K4me1-enriched genes from 3 kb upstream of the transcription start site (TSS) to 3 kb downstream of the transcription termination site (TTS). An equal number of genes lacking H3K4me1 were randomly selected as controls. All genes are sorted by H3K4me1 enrichment (n = 17,228). c Violin plots and box plots showing Ehd3 and SDG724 levels within H3K4me1-enriched, H3K4me2 (without H3K4me1)-enriched, or H3K4me3 (without H3K4me1)-enriched genes. Controls were genes lacking H3K4me1, H3K4me1/H3K4me2, or H3K4me1/H3K4me3. The box plot center line indicates the median, and edges correspond to the 75th and 25th percentiles, respectively. Outliers are hidden and the whiskers extend to a maximum of 1.5 times the interquartile range from the quartiles. d Heatmaps showing changes in H3K36me2/me3 levels within H3K4me1-enriched genes (n = 17,228), compared with the wild-type rice ‘Nipponbare’ (NiP), from 3 kb upstream of the TSS to 3 kb downstream of the TTS in the mutants. All genes are sorted by H3K4me1 enrichment in the wild-type NiP. e Integrative Genomics Viewer (IGV) images of ChIP-seq data showing distributions of Ehd3, SDG724, H3K4me1/me2/me3, and H3K36me2/me3 along the representative gene in the wild-type NiP. f Heatmaps showing individual gene distributions of H3K4me1/me2/me3 and H3K36me1/me2/me3 from 3 kb upstream of the TSS to 3 kb downstream of the TTS in the wild-type NiP. All genes (n = 55,801) are sorted by H3K4me1 enrichment. g IGV images of ChIP-seq data showing the H3K4me1- and H3K36me2/me3-enrichment profile in representative region in Chlamydomonas reinhardtii, Physcomitrium patens, Arabidopsis thaliana, and Oryza sativa. h Histone methyltransferase assay showing that Ehd3 stimulates the H3K36 methyltransferase activity of SDG724. H3K4me0 and H3K4me1 mononucleosomes were used as the substrates. Glutathione-S-transferase (GST) and truncated SDG725 (containing the CW and SET domains) were used as negative and positive controls, respectively. Gradient colors indicate enrichment levels (b, d, f) or log2-transformed fold changes (d). Publicly available data were analyzed for Arabidopsis thaliana (refs. 21,41). Source data are provided.
Different from human, correlation analyses showed that H3K4me1 was positively correlated with H3K36me2 (R = 0.53) and H3K36me3 (R = 0.63), but not with other tested histone modifications, implying that H3K4me1 is associated with H3K36me2/me3 in rice33,34,35,36,37,38 (Supplementary Fig. 10a). SDG724 is required for deposition of H3K36me2/me3 at H3K4me1-enriched genes, supporting the positive correlation between H3K4me1 and H3K36me2/me3 in rice (Fig. 6e and Supplementary Fig. 10a). In addition, gene-by-gene heatmaps showed that genes enriched with H3K4me1 exhibited significant enrichment of H3K36me2/me3 (Fig. 6f and Supplementary Data 8–9), supporting the observation that H3K4me1 specifically colocalizes with H3K36me2/me3 in rice. To investigate if the colocalization of H3K4me1 and H3K36me2/me3 is conserved in plants, we performed ChIP-seq analysis of H3K36me2/me3 in Chlamydomonas reinhardtii and Physcomitrium patens, and integrated published H3K36me2/me3 ChIP-seq data for Saccharomyces cerevisiae, Drosophila melanogaster, Mus musculus, Homo sapiens, and Arabidopsis thaliana21,39,40,41,42 (Fig. 6g and Supplementary Fig. 10b, c). In contrast to the diverse patterns of H3K4me1 and H3K36me2/me3 distributions in Chlamydomonas reinhardtii, Drosophila melanogaster, Mus musculus, and Homo sapiens, the distributions of H3K4me1 and H3K36me2/me3 displayed high similarity in Physcomitrium patens, Arabidopsis thaliana, and Oryza sativa (Fig. 6g and Supplementary Fig. 10b, c). Moreover, the published ChIP-seq data22 also presented similar distributions of H3K4me1 and H3K36me3 in other land plants including Eutrema salsugineum, Populus trichocarpa, Phaseolus vulgaris, Asparagus officinalis, Brachypodium distachyon, Setaria viridis, Sorghum bicolor, and Zea mays (Supplementary Fig. 10d). Therefore, a distinct evolutionary trend for colocalization of H3K4me1 and H3K36me2/me3 in land plants was implied.
To investigate the molecular mechanism linking H3K4me1 and H3K36me2/me3, a histone methyltransferase assay was performed to test the activity of SDG724 and SDG725, which are H3K36-specific methyltransferases in rice, by using H3K4me0- or H3K4me1-modified mononucleosomes. The CW domain of Arabidopsis SDG8, a homolog of rice SDG725, is responsible for binding to H3K4me115, and the critical residues within the CW domain of SDG8 involved in H3K4me1 binding are conserved in SDG725 (Supplementary Fig. 10e), supporting the hypothesis that SDG725 is able to recognize H3K4me1 through its CW domain. Thus, the truncated rice H3K36 methyltransferase SDG725 containing the CW and SET domains, and GST were used as positive and negative controls, respectively. When incubated with H3K4me0 nucleosomes, neither SDG724 nor SDG725 showed obvious methyltransferase activity (Fig. 6h and Supplementary Fig. 10f). However, when using H3K4me1-modified nucleosomes as the substrate, the activity of SDG725 greatly increased. Importantly, the SDG724 activity obviously increased in the presence of Ehd3, indicating that Ehd3 is required to stimulate the catalytic activity of SDG724 (Fig. 6h and Supplementary Fig. 10f). Collectively, recognition of H3K4me1-modified nucleosomes by the interacting reader protein (Ehd3) or internal reading domain (the CW domain of SDG725) is essential for the activation of rice H3K36 methyltransferases, leading to H3K36me2/me3 deposition on chromatin. Taken together, all these results revealed that H3K4me1 directs H3K36me2/me3 deposition in land plants.
Discussion
In this study, we identified a specific H3K4me1 reader protein, Ehd3, and suggested a crucial role of H3K4me1 in directing proper H3K36me2/me3 deposition in rice. Mechanistically, Ehd3 recognizes H3K4me1 through its tandem PHD finger domain and recruits the H3K36 methyltransferase SDG724 to H3K4me1-enriched chromatin regions (Fig. 7). Moreover, interaction with Ehd3 enhances the catalytic activity of SDG724 in H3K36 methylation on H3K4me1-modified nucleosomes. The genomic colocalization of H3K4me1 and H3K36me3 across the genome is conserved in land plants. Collectively, our findings illustrate an evolutionary tendency for the recognition of H3K4me1 and its linkage to H3K36me2/me3 in land plants. The Ehd3 homolog in Chlamydomonas reinhardtii, lacking the tandem PHD finger domain, neither recognizes methylated H3K4 nor exhibits an obvious correlation between H3K4me1 and H3K36me2/me3. During the transition to terrestrial habitats, the Ehd3 homolog in Physcomitrium patens evolved a tandem PHD finger domain that acquired the ability to recognize H3K4 methylation, particularly H3K4me1, coinciding with the emergence of colocalization of H3K4me1 and H3K36me2/me3. In flowering plants, such as Arabidopsis thaliana and Oryza sativa, the tandem PHD finger domains exhibit an exclusive affinity for H3K4me1 and the Ehd3 homologs show a conserved interaction with H3K36 methyltransferases, corresponding to the conserved colocalization of H3K4me1 and H3K36me2/me3 observed in land plants. All these findings revealed a previously unknown mechanism that H3K4me1 functions in directing H3K36me2/me3 deposition in land plants, emphasizing the central role of histone modification recognition in decoding and transmitting epigenetic information.

Upper panel: In animals, H3K4me1 marks various enhancer states. Within a given tissue, a subset of poised enhancers are characterized by H3K4me1 and H3K27me3. Prior to activation, enhancers existing in a primed state marked only with H3K4me1. Active enhancers, which lack nucleosomes, are adjacent to nucleosomes marked with H3K4me1 and H3K27ac. Specific transcription factors link the active enhancers and target genes, mediated by cohesin and other protein complexes. Lower panel: In plants, H3K4me1 directs the establishment of H3K36me2/me3. The tandem plant homeodomain (PHD) finger domain-containing protein Ehd3 specifically recognizes H3K4me1 and interacts with the H3K36 methyltransferase SDG724. This interaction stimulates the catalytic activity of SDG724, facilitating the deposition of H3K36me2/me3, which results in the colocalization of H3K4me1 and H3K36me2/me3 in plants.
The crystal structure of the Ehd3 tandem PHD finger domain in complex with H3K4me1 reveals a unique binding mode distinct from all previously reported H3K4me1/me2/me3 recognition mechanisms15,16,43. The SDG8 CW domain binds to H3K4me1 with a pocket formed by one polar, two aromatic, and two hydrophobic residues15. The MORC3 CW domain binds to H3K4me1/me3 by two hydrophobic and two polar residues43. RDM15 specifically binds to H3K4me1 through the Tudor domain by an aromatic cage and a specific H-bond network16. The unshielded and exposed side of the H3K4me1 binding pockets in SDG8, MORC3, and RDM15 allows for the binding of H3K4me2 and H3K4me3, indicating a flexible mechanism for methylated H3K4 recognition. In contrast, the tandem PHD finger domain of Ehd3 forms a tight and narrow pocket only capable of accommodating H3K4me1, thereby restricting the binding of a greater number of methyl groups (Fig. 3). The PHD finger domain belongs to one of the most extensive families of domains present in reader proteins44, and recognizes multiple histone modifications ranging from H3K4 methylation and H3K14 acetylation24 to H3R2 methylation45. Most PHD finger domain structures are complexed with either an H3K4me2 or H3K4me3 peptide, exhibiting the conserved structural features that a single PHD finger domain forms an aromatic pocket for binding to a particular methylated lysine26,27,28,29,46,47,48. However, the H3K4me1 binding pocket of Ehd3 is formed by the tandem PHD finger domain, which comprises two PHD fingers. A set of proteins indeed harbors a tandem PHD finger domain, but the recognition mechanisms for these proteins remain largely unclear. DPF3b and MOZ proteins recognize histones through their tandem PHD finger domains24,25,49,50. In these proteins, the first PHD finger binds to H3K14ac24,25 and H3K14ac/H3K14 crotonylation49,50, respectively, whereas the second PHD finger interacts with unmodified H3K4, which represents a recognition mechanism distinct from Ehd3–H3K4me1.
In this study, we observed that Ehd3 binds to H3K4me1 via its tandem PHD finger domain and interacts with the H3K36 methyltransferase SDG724 to deposit H3K36me2/me3 in the genome of rice. In Arabidopsis, the triple mutant atx1/2/r7, which lacked H3K4me1 deposition, exhibited a concomitant loss of both H3K4me1 and H3K36me351. This observation supports the notion that H3K4me1 plays a critical role in directing H3K36me3 deposition in land plants. In addition, SDG725 is one of the H3K36 methyltransferases in rice and its CW domain recognizes methylated H3K4 with a preference for H3K4me1 over nonmethylated H3K4 or H3K4me2/me315. We noted that SDG725 exhibited strong activity to catalyze H3K36 methylation when using H3K4me1 mononucleosomes as the substrate (Fig. 6h and Supplementary Fig. 10f). Collectively, H3K4me1 readout is essential for H3K36 methyltransferases in land plants to deposit H3K36me2/me3, either through interactions with specific reader proteins or by employing their own H3K4me1-recognizing domain.
In yeast and animals, RNA polymerase II (RNAPII) recruits H3K36 methyltransferases to establish transcription elongation-coupled H3K36me3, and the Set2–Rpb1 interacting (SRI) domain within H3K36 methyltransferases is responsible for interaction with the C-terminal domain (CTD) of RNAPII52,53. In contrast, we detected no SRI domains within the known H3K36 methyltransferases in higher plants (Supplementary Fig. 11a, b). Interestingly, the Arabidopsis H3K4me2-specific demethylase LDL3 possesses an SRI domain and interacts with the CTD-phosphorylated RNAPII54. Structure prediction by AlphaFold 3.055 indicated that, similar to those within yeast Set2, the SRI domains within LDL3 homologs in land plants, including Physcomitrium patens, Arabidopsis thaliana, and Oryza sativa, display a conserved structure of a left-turned three-helix bundle (Supplementary Fig. 11c, d). We generated rice CRISPR/Cas9 mutants targeting OsLDL3 (LOC_Os10g38850) in the NiP background. Two sgRNAs were designed to target the first exon of OsLDL3. One identified mutant, named osldl3, exhibited a 3-bp deletion at target site 1 and a 1-bp deletion at target site 2 (Supplementary Fig. 11e). Similar to the Arabidopsis ldl3 mutant54, deletion of rice OsLDL3 resulted in reduced H3K4me1 and increased H3K4me2 levels (Supplementary Fig. 11f). In addition, GST pulldown assays showed that Ser-2- or Ser-5-phosphorylated RNAPII was pulled down by the rice OsLDL3 SRI domain, but not by the H3K36 methyltransferases SDG724 or SDG725 (Supplementary Fig. 11g). The SRI domain of OsLDL3 especially interacted with the Ser-2-phosphorylated CTD of RNAPII, which marked transcription elongation, but not the nonphosphorylated CTD, indicating that RNAPII directs the transcription elongation-coupled recruitment of OsLDL3 (Supplementary Fig. 11h and Supplementary Data 1–2). The foregoing observations suggest that, in plants, H3K4me1 deposition might be coupled with RNAPII, as RNAPII recruits H3K4me2 demethylase but not H3K36 methyltransferase. In yeast, H3K36me2/me3 suppress cryptic transcription initiation via Rpd3S-dependent histone deacetylation56,57,58, while in animals this suppression is mediated by Dnmt3b-dependent DNA methylation59. The Rpd3S complex asymmetrically engages with H3K36me3 nucleosomes through the chromodomains of the Eaf3-A/B subunits, positioning the catalytic center of Rpd3 towards the histone H4 N-terminal tail for deacetylation60. Dnmt3b recognizes H3K36me3 through the PWWP domain and binds to gene body regions to facilitate de novo DNA methylation61. Rpd3S-dependent histone deacetylation and Dnmt3b-dependent DNA methylation establish a repressive chromatin environment, which inhibits spurious entries of RNAPII and ultimately ensures the fidelity of transcription initiation. Plants and animals establish H3K36 methylation through different mechanisms; however, whether H3K36 methylation inhibits cryptic transcription initiation in plants remains to be elucidated.
As sessile organisms, plants must rapidly respond to environmental changes for survival, which is mainly achieved through dynamic regulation of gene transcription62. Thus, plants may undergo evolutionary adaptation to develop a more efficient mechanism to regulate the coordination among diverse histone modifications, thereby influencing the chromatin state and dynamically regulating gene transcription. Notably, H3K4 and H3K36 methylation are strongly associated with biotic and abiotic stresses in plants63,64,65,66,67,68,69. The present transcriptome data indicated that a set of stress-responsive genes were dysregulated in the ehd3-1, sdg724-1, and ehd3 sdg724 mutants (Supplementary Fig. 5b, c and Supplementary Data 7). Thus, deposition of H3K36me2/me3 directed by H3K4me1 potentially plays an important role in plant environmental adaptation.
Methods
Plant material and growth conditions
The background rice material used in this study was Oryza sativa subsp. japonica ‘Nipponbare’ (NiP). Genomic DNA-edited mutants ehd3-1, ehd3-4, and osldl3 were generated using the CRISPR/Cas9 system and obtained from BIORUN (Wuhan, China). The sdg724-1 mutant was described previously31. The ehd3 sdg724 double mutant was produced through a genetic cross between the single mutants ehd3-1 and sdg724-1. The rice seedlings were cultured on Murashige and Skoog (MS) solid medium (comprising 2.2 g/L MS medium (M0222; Duchefa, Haarlem, The Netherlands), 3% (w/v) sucrose, and 0.4% (w/v) phytagel, adjusted to pH 5.8) under a 14-h photoperiod (30 °C light/28 °C dark) in an artificial climate chamber. Rice plants were grown in paddy fields at two distinct locations: Shanghai, under long-day (LD) conditions, and Sanya, under short-day (SD) conditions. For all experiments, 20 plants were used to evaluate the heading date, defined as the date at which the panicle emerged from the flag leaf. Arabidopsis seedlings were grown on MS solid medium (consisting of 4.9 g/L MS medium (M0255, Duchefa), 1% (w/v) sucrose, and 0.5% (w/v) agarose, adjusted to pH 5.7) under a 16-h photoperiod at 22 °C in an artificial climate chamber. Chlamydomonas reinhardtii strains were cultured in Tris-acetate-phosphate (TAP) medium70 on a rotary shaker at 25 °C in an artificial climate chamber. Physcomitrium patens were propagated on BCD medium71 under a 16-h photoperiod at 25 °C in an artificial climate chamber.
Vector construction and transgenic plants
To generate the Ubi::EGFP-Ehd3 construct, the Ehd3 cDNA sequence was amplified by PCR and subsequently cloned into the pRHVnGFP vector72. For the Ubi::YFP-SDG724 construct, the SDG724 cDNA sequence was amplified by PCR and cloned into the pU1301 plant expression vector. For the Ubi::SDG724-4×HA construct, the SDG724 cDNA sequence was amplified by PCR and cloned into the pRHVcHA vector72. For the pEhd3::4×HA-Ehd3 and pEhd3::4×HA-Ehd3mu constructs, the native Ehd3 promoter (2,005-bp upstream of ATG), the coding sequence for 4×HA, and the genomic DNA sequence of either wild-type Ehd3 or the mutated Ehd3 (Y471A and D496A) were amplified by PCR and cloned into the pCAMBIA1300 vector (CAMBIA, https://cambia.org). The primers used in cloning are listed in Supplementary Data 10.
The Ubi::EGFP-Ehd3 and Ubi::YFP-SDG724 constructs were transformed into NiP to generate transgenic overexpression plants, designated as EGFP-Ehd3/NiP and YFP-SDG724/NiP. The Ubi::SDG724-4×HA construct was transformed into the sdg724-1 single mutant to obtain SDG724-HA/sdg724−1-1, then SDG724-HA/ehd3 sdg724−1 was obtained by a genetic cross with the ehd3 sdg724 double mutant. In addition, HA-Ehd3/ehd3-1−1 and HA-Ehd3mu/ehd3-1-1 plants were generated by transforming the pEhd3::4×HA-Ehd3 and pEhd3::4×HA-Ehd3mu constructs into the ehd3-1 mutant. Genetic transformations were conducted by BIORUN and stable transgenic plants were selected using hygromycin B (Roche, Mannheim, Germany). The stable transgenic plants were confirmed by western blot using Rabbit anti-GFP (ab290, Abcam, Cambridge, UK, 1:2000), Rabbit anti-HA (ab9110, Abcam, 1:2000), Rabbit anti-H3 (ab1791, Abcam, 1:2000), or Mouse anti-Actin (M20009, Abmart, 1:2000) antibodies. In total, we obtained eight EGFP-Ehd3/NiP lines, five YFP-SDG724/NiP lines, two HA-Ehd3/ehd3-1 lines, and two HA-Ehd3mu/ehd3-1 lines. EGFP-Ehd3/NiP-1 and YFP-SDG724/NiP-1 were selected for the following experiments. In addition, HA-Ehd3/ehd3-1-1, HA-Ehd3/ehd3-1-2, and HA-Ehd3mu/ehd3-1-1, which demonstrated comparable abundance of HA-Ehd3 and HA-Ehd3mu proteins (Supplementary Fig. 4c), were selected for subsequent analysis.
Phylogenetic analysis
PHD domain encoding genes were identified using Phytozome database (https://phytozome-next.jgi.doe.gov/). For each identified gene, a representative annotated protein sequence was analyzed using the SMART database (http://smart.embl-heidelberg.de/) to characterize domain architectures. Homologs in different species were identified using BLAST tool searches against the UniProtKB database (https://www.uniprot.org/). Multiple sequence alignment and neighbor-joining dendrograms were constructed using MEGA version 10.2.673.
Protein expression and purification
The coding sequences of Ehd3 (1–563 amino acids (aa), 281–563 aa, 281–405 aa, 406–563 aa, 406–473 aa, 473–563 aa, and 417–547 aa), OsLDL3 (1263–1832 aa), SDG724 (1–394 aa), SDG725 (1240–1882 aa), and Arabidopsis Q1JPM3 (named as AtEHD3) were amplified by PCR using primers (Supplementary Data 10) and subsequently cloned into the pGEX-6P-1 vector (GE Healthcare, Milwaukee, WI, USA) to produce glutathione-S-transferase (GST)-fusion proteins. In addition, DNA fragments encoding Ehd3 (1–563 aa and 417–547 aa), the Ehd3 417–547 aa mutants (H441A, Y446A, Y471A, D486A, I489A, and D496A), OsLDL3 (1744–1828 aa), SDG725 (1240–1898 aa), A0A2K3D188 (173–217 aa), A0A2K1L966 (364–473 aa), AtEHD3 (342–474 aa), Q8BRH4 (338–468 aa), and Q8BRH4 (911–1043 aa) were amplified by PCR using primers (Supplementary Data 10) and cloned into the pSUMO vector74. The coding sequence of Arabidopsis SDG7 was amplified by PCR using primers (Supplementary Data 10) and cloned into the pCold TF vector. All recombinant proteins were expressed in Escherichia coli strain Rosetta (DE3) cells and induced with isopropyl-β-D-thiogalactoside. ZnCl2 was added during expression of the Ehd3, SDG724, SDG725, AtEHD3, and SDG7 proteins.
To purify GST-tagged proteins, cells were harvested and suspended in phosphate-buffered saline (1× PBS) containing 10% (v/v) glycerol, 0.1% (v/v) Triton X-100, 2 mM 1,4-dithiothreitol (DTT), and 0.1 mM ZnCl2, pH 7.4. After sonicating and centrifuging the lysed cells, the supernatant was incubated with Glutathione Sepharose 4 Fast Flow (Cytiva, Uppsala, Sweden) at 4 °C for 2 h. Subsequently, the supernatants were discarded and the beads were washed three times with 1× PBS containing 10% (v/v) glycerol, 2 mM DTT, and 0.1 mM ZnCl2, pH 7.4. The GST-tagged proteins were eluted with an elution buffer (150 mM NaCl, 20 mM Tris-HCl, and 20 mM reduced glutathione, pH 7.4). The flow-through was concentrated and substituted with the buffer containing 150 mM NaCl, 20 mM Tris-HCl, 2 mM DTT, and 0.1 mM ZnCl2, pH 7.4, using an Amicon 10-KDa cutoff (Millipore, Burlington, MA, USA).
To purify the proteins cloned in the pSUMO vector, cells were harvested and suspended in the binding buffer (500 mM NaCl, 20 mM Tris-HCl, and 25 mM imidazole, pH 8.0). The lysed cells, processed using a high-pressure disruptor, were centrifuged, and the supernatant was loaded onto a Ni-NTA column (GE Healthcare). The proteins were gradually eluted using an elution buffer (500 mM NaCl, 20 mM Tris-HCl, and 500 mM imidazole, pH 8.0). Subsequently, the eluted proteins were incubated with ULP1 protease and then diluted for 3 h in buffer containing 500 mM NaCl and 20 mM Tris-HCl, pH 8.0. The diluted proteins were then loaded onto a Ni-NTA column, and the flow-through was further purified using a Superdex 75 column (GE Healthcare) equilibrated with buffer containing 100 mM NaCl, 20 mM Tris-HCl, and 1 mM Tris-(2-carboxyethyl)-phosphine hydrochloride (TCEP), pH 8.0. Finally, the proteins were concentrated using an Amicon 10-KDa cutoff (Millipore).
To purify the protein expressed in the pCold TF vector, cells were harvested and suspended in the binding buffer (20 mM Tris-HCl, 500 mM NaCl, 25 mM Imidazole, 5% (v/v) Glycerol, pH 8.0). After sonicating and centrifuging the lysed cells, the supernatant was incubated with cOmplete His-Tag Purification Resin (Roche) at 4 °C for 2 h. Subsequently, the supernatants were discarded and the beads were washed three times with binding buffer. The His-tagged proteins were eluted with the elution buffer (20 mM Tris-HCl, 500 mM NaCl, 500 mM Imidazole, 5% v/v Glycerol, pH 8.0). The flow-through was concentrated using an Amicon 10-KDa cutoff (Millipore).
Crystallization and structure determination
The purified Ehd3 protein (417–547 aa) was incubated with H3(1–12) K4me1 histone peptide (Supplementary Data 1) at a molar ratio of 1:1.2 at 4 °C for 1 h. Crystallization was performed by a Gryphon crystallization robot system with a sitting-drop vapor diffusion method. Specifically, 0.2 μl Ehd3 protein (10 mg/ml) and peptide mixture was combined with an equal volume of a crystallization kit solution (Hampton, Aliso Viejo, CA, USA) and incubated at 18 °C. The optimization procedure was performed using the hanging-drop vapor diffusion method at 18 °C. X-ray data for the crystals were collected at the B18U1 beamline of the Shanghai Synchrotron Radiation Facility and were processed using HKL300075. The crystal structure was solved by the autosol and refined by the phenix.refine programs from Phenix76. The 2Fo–Fc and Fo–Fc electron density maps and unbiased omission map were regularly calculated and used as guides for the building of the H3K4me1 peptide, Zn2+ ions, and water molecules in COOT77. The data collection and refinement statistics are summarized in Supplementary Data 3. All molecular graphics were generated using PyMOL (https://pymol.org/).
Structure prediction by AlphaFold
AlphaFold version 2.1.032 was used to predict the structures of Ehd3 tandem PHD finger homologs, i.e., A0A2K3D188 (117–217 aa) in Chlamydomonas reinhardtii, A0A2K1L966 (364–464 aa) in Physcomitrium patens, A0A1D6LDK6 (401–498 aa) in Zea mays, A0A3B6RKR3 (432–532 aa) in Triticum aestivum, Q1JPM3 (354–450 aa) in Arabidopsis thaliana, A0A162A0W2 (424–519 aa) in Daucus carota, A0A3R7Q333 (793–889 aa) in Penaeus vannamei, A0A433TT05 (688–784 aa) in Elysia chlorotica, O14686 (1379–1475 aa) in Homo sapiens, and Q8BRH4 (914–1010 aa) in Mus musculus. AlphaFold version 3.055 was used to predict the structures of SRI domain-containing homologs, i.e., A0A2K3DEA3 (1–1870 aa) in Chlamydomonas reinhardtii, A0A2K1LAV4 (1–1709 aa) and A0A2K1L6H4 (1–1814 aa) in Physcomitrium patens, SK05G17270.mRNA1 (1–1295 aa) in Selaginella kraussiana, Ceric.12G094400.1 (1–1600 aa) in Ceratopteris richardii, F4JLS1 (1–1628 aa) in Arabidopsis thaliana, and Q336Y0 (1–1832 aa) in Oryza sativa. The protein codes correspond to the UniProt entries (https://www.uniprot.org), except for SK05G17270.mRNA1 and Ceric.12G094400.1, which were obtained from https://sk.ccgg.fun/blast.
Peptide microarray
The MODified™ Histone Peptide Array (Active Motif, Carlsbad, CA, USA) was blocked with TTBS–milk buffer (20 mM Tris-HCl, 150 mM NaCl, 0.1% (v/v) Triton X-100, and 5% (w/v) non-fat dried milk, pH 7.4) at room temperature for 2 h. After washing three times with TTBS buffer (20 mM Tris-HCl, 150 mM NaCl, and 0.1% (v/v) Triton X-100, pH 7.4), the peptide array was incubated overnight at 4 °C with 500 nM GST-tagged full-length Ehd3 protein in TTBS–milk buffer. Subsequently, the peptide array was washed three times with TTBS buffer and incubated with the Mouse anti-GST antibody (M20007, Abmart, Shanghai, China, 1:2000) in the TTBS–milk buffer at room temperature for 1 h. After washing three times with TTBS buffer, the peptide array was incubated with the Goat anti-Mouse IgG HRP antibody (M21001, Abmart, 1:5000) in the TTBS–milk buffer at room temperature for 1 h. The peptide array was washed three times with TBS buffer (20 mM Tris-HCl and 150 mM NaCl, pH 7.4) and imaged using a luminescence imaging workstation (Tanon, Shanghai, China). The results were analyzed using Array Analyze software (Active Motif).
Peptide pulldown
Biotinylated histone peptides (2 μg) (H3(1–21), H3(1–21)K4me1, H3(1–21)K4me2, H3(1–21)K4me3, H3(1–21)K9me2, H3(1–21)K9ac, H3(1–21)K14ac, H3(21–41)K27me3, H3(16–34)K27ac, and H3(21–41)K36me3) (Supplementary Data 1) were incubated with 20 μl prewashed streptavidin magnetic beads (NEB, Ipswich, MA, USA) at 4 °C for 2 h in 500 μl binding buffer (150 mM NaCl, 20 mM Tris-HCl, and 0.1% (v/v) Nonidet P-40). After washing three times, the peptide-bound beads were incubated with 2 μg GST-tagged full-length or truncated Ehd3 proteins at 4 °C for 2 h in 500 μl binding buffer. The beads were then washed three times with the binding buffer and boiled in 1× SDS loading buffer (50 mM Tris-HCl, 2% (w/v) sodium dodecyl sulfate, 0.01% (w/v) bromophenol blue, 2 mM DTT, and 2 mM β-Mercaptoethanol, pH 6.8). The resulting products were subjected to SDS-PAGE and detected using the Mouse anti-GST antibody (M20007, Abmart, 1:2000).
Semi-in vivo pulldown
Total nuclear proteins were extracted from wild-type NiP using a buffer containing 50 mM Tris-HCl, 5 mM MgCl2, 60 mM NaCl, 60 mM KCl, 2 mM DTT, and proteinase inhibitor cocktail (Roche), pH 7.5. The nuclear protein supernatant was incubated at 4 °C for 2 h with beads precoated with GST, GST-OsLDL3 SRI, GST-SDG724, or GST-SDG725 CW-SET proteins. After washing three times, the samples were analyzed by SDS-PAGE and western blot using Rabbit anti-Ser2P RNAPII (ab5095, Abcam, 1:2000) and Rabbit anti-Ser5P RNAPII (ab5131, Abcam, 1:2000) antibodies.
Isothermal titration calorimetry (ITC)
The ITC experiments were conducted using an iTC200 MicroCalorimeter (Malvern Panalytical, Malvern, UK) at the National Center for Protein Science Shanghai. The interactions involving the full-length Ehd3 (100 μM), wild-type or mutated Ehd3 truncations (417–547 aa, 100 μM), A0A2K3D188 (173–217 aa, 100 μM), A0A2K1L966 (364–473 aa, 100 μM), Q1JPM3 (342–474 aa, 100 μM), Q8BRH4 (338–468 aa, 50 μM), and Q8BRH4 (911–1043 aa, 50 μM) were analyzed in the presence of the peptides H3(1–12)K4me0, H3(1–12)K4me1, H3(1–12)K4me2, or H3(1–12)K4me3 (1 mM), as well as OsLDL3 (1744–1828 aa, 100 μM) with the peptides NP CTD, S2P CTD, or S5P CTD (1 mM) (Supplementary Data 1). All reactions were performed at 25 °C in buffer containing 100 mM NaCl, 20 mM Tris-HCl, and 1 mM TCEP, pH 8.0. The titration data were fitted using Origin version 7.0 software (Supplementary Data 2).
Immunoprecipitation followed by mass spectrometry (IP-MS)
Transgenic seedlings (2 g) were ground in liquid nitrogen and homogenized in lysis buffer (150 mM NaCl, 50 mM Tris-HCl, 5 mM MgCl2, 10% (v/v) glycerol, 0.1% (v/v) Nonidet P-40, 5 mM DTT, 0.1 mM PMSF, and proteinase inhibitor cocktail (Roche), pH 7.6). After sonication and centrifugation, the supernatant was incubated with the Rabbit anti-GFP antibody (ab290, Abcam, 1:100) at 4 °C for 3 h. The mixture was then incubated with protein A magnetic beads (Thermo Fisher Scientific, Waltham, MA, USA) at 4 °C for 3 h. Subsequently, the beads were washed with the same buffer and subjected to SDS-PAGE. The protein bands were then excised and subjected to in-gel digestion with trypsin. The resulting peptides were collected and analyzed using a nanoflow EASY-nLC 1200 system (Thermo Fisher Scientific) coupled with an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific). Raw data analysis was performed using Proteome Discoverer version 2.4 (Thermo Fisher Scientific) and an in-house Mascot server version 2.7 (Matrix Science, London, UK). The rice protein database was obtained from RGAP (http://rice.uga.edu). Data were searched using the following parameters: trypsin/P as the enzyme; allowing up to two missed cleavage sites; 10 ppm mass tolerance for MS and 0.05 Da for MS/MS fragment ions; carbamidomethylation on cysteine as fixed modifications; and protein N-terminal acetylation, oxidation on methionine, and N-terminal pyroQ as variable modifications. The incorporated Percolator in Proteome Discoverer was used for validation, accepting only the hits with false discovery rate ≤ 0.01 for the following analysis.
GST pulldown
The GST, GST-SDG724, or GST-AtEHD3 beads-coated protein was incubated with the full-length Ehd3 or His-SDG7 proteins at 4 °C for 2 h in buffer containing 75 mM NaCl, 75 mM KCl, 50 mM HEPES, 5% (v/v) glycerol, 8 mM DTT, and 2 mM MgCl2, pH 8.0. After washing three times, the beads were boiled in 1× SDS loading buffer, subjected to SDS-PAGE, stained with Coomassie Brilliant Blue, and analyzed by western blot using Mouse anti-His antibody (M20001, Abmart, 1:2000).
Yeast two-hybrid
The Ehd3, SDG724, Arabidopsis AtEHD3, or SDG7 cDNA sequence was cloned into the pGADT7 and pGBKT7 vectors (Clontech, Kyoto, Japan), respectively, using specific primers (Supplementary Data 10). The resulting constructs were transformed into the Y2HGold yeast strain following the manufacturer’s instructions (WeidiBio, Shanghai, China). The interaction was screened on a synthetic defined medium lacking leucine, tryptophan, and histidine (SD/−L/−W/−H), or on medium additionally lacking adenine (SD/−L/−W/−H/−A).
Co-immunoprecipitation (Co-IP)
Rice protoplast isolation and transformation were performed as described previously78. The Ehd3 and SDG724 cDNA sequences were cloned into the pRTVnMyc and pRTVnHA vectors72, respectively. After transformation of the constructs, the protoplasts were incubated at 28 °C for 20 h. The protoplasts were subsequently harvested in buffer containing 20 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 2 mM DTT, 0.5% (v/v) Triton X-100, and proteinase inhibitor cocktail (Roche), pH 8.0. The harvested protoplasts were sonicated at 4 °C. After centrifugation, the supernatant was incubated with the agarose-conjugated Mouse anti-Myc antibody (M20012, Abmart, 1:100) at 4 °C for 2 h. The immunoprecipitants were washed three times, subjected to SDS-PAGE, and detected with the Mouse anti-Myc (M20002, Abmart, 1:2000) and Rabbit anti-HA (ab9110, Abcam, 1:2000) antibodies.
Bimolecular fluorescence complementation (BiFC)
The Ehd3 and SDG724 cDNA sequences were cloned into the pRTVnVC and pRTVnVN vectors using the primers listed in Supplementary Data 1072, respectively. After transformation of the constructs, the protoplasts were incubated at 28 °C for 20 h. Fluorescence in the protoplasts was observed using an LSM 710 confocal laser scanning microscope (Carl Zeiss, Jena, Germany; https://www.zeiss.com).
Histone methyltransferase assay
Either 500 nM recombinant Ehd3, GST-SDG724, GST-SDG724 mixed with Ehd3, or 100 nM SDG725 (CW-SET domain, 1240–1898 aa) were incubated with 1 μM unmodified (31467, Active Motif) or H3K4me1-modified (31585, Active Motif) mononucleosomes in histone methyltransferase buffer (50 mM Tris-HCl, 0.02% (v/v) Triton X-100, 1 mM TCEP, and 160 μM SAM, pH 8.5) at room temperature for 15 h. Reactions were stopped by adding 1× SDS loading buffer, then were subjected to SDS-PAGE and detected by western blot using the Rabbit anti-H3K36me2 (ab9049, Abcam, 1:2000) and Rabbit anti-H3K36me3 (ab9050, Abcam, 1:2000) antibodies.
RNA extraction, library construction and sequencing
Fourteen-day-old Oryza sativa and Arabidopsis thaliana seedlings (100 mg) grown on MS medium, 7-day-old Physcomitrium patens grown on BCD medium, and Chlamydomonas reinhardtii cultured in TAP medium to a density of 4–8 × 106 cells/ml were used for mRNA extraction. The mRNA was extracted using the RNAprep Pure Plant Kit (Tiangen Biotech, Beijing, China). Three independent biological replicates of RNA-seq libraries were prepared using the KAPA Stranded mRNA-seq Kit (Kapa Biosystems, Wilmington, MA, USA) and sequenced on an Illumina NovaSeq 6000 instrument by NEO BIO (Shanghai, China). Raw reads were cleaned with Cutadapt version 3.579 to remove low-quality bases and sequencing adapters. Trimmed reads were aligned to the reference genomes of different species using HISAT2 version 2.1.080. Samtools version 1.1781 was used to remove reads with low mapping quality (MAPQ < 20), and then featureCounts version 2.0.182 was used to quantify the number of reads mapped to exons. RPKM (reads per kilobase per million reads) was used to characterize the transcription level of RNA. For rice RNA-seq, the DESeq2 version 1.36.083 package for R (version 4.2.1) was used to screen DEGs (|fold of change| ≥ 1.5 and Benjamini–Hochberg adjusted p-value ≤ 0.05, by two-sided Wald test). Gene ontology (GO) functional enrichment analysis for specific gene sets was conducted through the online platform CARMO84 with a modified one-sided Fisher’s exact test (EASE score)85, which was applied to identify significantly enriched GO terms.
The reference genomes used for rice and Arabidopsis were MSU7 (http://rice.uga.edu/) and TAIR10 (https://www.arabidopsis.org/), respectively. The reference genomes for other species were obtained from Ensembl (https://www.ensembl.org/). The genomes correspond to specific species: R64-1-1 for Saccharomyces cerevisiae, BDGP6.32 for Drosophila melanogaster, GRCm39 for Mus musculus, GRCh38.p14 for Homo sapiens, Chlamydomonas_reinhardtii_v5.5 for Chlamydomonas reinhardtii, and Phypa_V3 for Physcomitrium patens.
Chromatin immunoprecipitation (ChIP)
Fourteen-day-old Oryza sativa and Arabidopsis thaliana seedlings (2 g) grown on MS medium, and 7-day-old Physcomitrium patens grown on BCD medium, were fixed in cross-linking buffer (0.4 M sucrose, 10 mM Tris-HCl, 1 mM PMSF, 1 mM EDTA, and 1% (v/v) formaldehyde, pH 8.0). Chlamydomonas reinhardtii grown in 1 L TAP medium to a density of 4–8 × 106 cells/ml were harvested and fixed in cross-linking buffer (20 mM HEPES-KOH, 80 mM KCl, and 0.4% (v/v) formaldehyde, pH 7.6). The ground seedlings or cells were resuspended in isolation buffer (0.25 M sucrose, 15 mM PIPES, 5 mM MgCl2, 60 mM KCl, 15 mM NaCl, 1 mM CaCl2, 0.9% (v/v) Triton X-100, 1 mM PMSF, and proteinase inhibitor cocktail (Roche), pH 6.8). Chromatin was sonicated in lysis buffer (50 mM HEPES, 150 mM NaCl, 1 mM EDTA, 1% (w/v) SDS, 0.1% (w/v) sodium deoxycholate, 1% (v/v) Triton X-100, and proteinase inhibitor cocktail (Roche), pH 7.5) using a Bioruptor™ Plus sonication device (Diagenode, Liege, Belgium). After centrifugation, the supernatant was incubated with antibodies including Rabbit anti-H3K4me1 (ab8895, Abcam, 1:200), Rabbit anti-H3K4me2 (07-030, Millipore, 1:200), Rabbit anti-H3K4me3 (07-473, Millipore, 1:200), Rabbit anti-H3K36me1 (ab9048, Abcam, 1:200), Rabbit anti-H3K36me2 (ab9049, Abcam, 1:200), Rabbit anti-H3K36me3 (ab9050, Abcam, 1:200), Rabbit anti-H3 (ab1791, Abcam, 1:200), or Rabbit anti-GFP (ab290, Abcam, 1:200) overnight at 4 °C. The prewashed protein A magnetic beads (Thermo Fisher Scientific) were added to the mixture and incubated at 4 °C for 2 h. The immunoprecipitates were washed sequentially: once with low-salt buffer (150 mM NaCl, 20 mM Tris-HCl, 0.1% (w/v) SDS, 1% (v/v) Triton X-100, and 2 mM EDTA, pH 8.0), once with high-salt buffer (500 mM NaCl, 20 mM Tris-HCl, 0.1% (w/v) SDS, 1% (v/v) Triton X-100, and 2 mM EDTA, pH 8.0), once with LiCl buffer (10 mM Tris-HCl, 0.25 M LiCl, 1% (w/v) sodium deoxycholate, 1% (v/v) Nonidet P-40, and 1 mM EDTA, pH 8.0), and twice with Tris-EDTA buffer (10 mM Tris-HCl and 1 mM EDTA, pH 8.0). The immunoprecipitates were eluted with elution buffer (0.5% (w/v) SDS and 0.1 M NaHCO3), followed by reverse crosslinking overnight at 65 °C. Subsequently, the samples were treated with RNase A and proteinase K. DNA extraction and ChIP-seq library construction were conducted as described previously86.
ChIP sequencing (ChIP-seq) analysis
DNA fragments for ChIP assays were prepared for sequencing on an Illumina NovaSeq 6000 instrument by NEO BIO (Shanghai, China). Raw reads trimmed by Cutadapt version 3.579 were aligned to genomes using Bowtie2 version 2.4.587. Reads with low-quality mapping (MAPQ < 20) and duplicate reads were filtered out using Samtools version 1.1781. Macs2 version 2.2.7.1/Macs3 version 3.0.0b388 (q-value ≤ 0.05) and MAnorm version 1.3.089 (with a Bayesian model for significance testing) were used for peak-calling and differential peaks detection for histone modifications, respectively. SICER version 1.190 was used for peak-calling for Ehd3- and SDG724-enriched peaks (W200-G600-FDR0.05), which were subtracted by the corresponding NiP control. The ChIPpeakAnno version 3.30.191 package for R version 4.2.1 was used to annotate peaks to genes. BEDtools version 2.31.092 was used to perform data format conversion. deepTools version 3.5.393 was used to calculate the normalized signals across the whole genome or on the selected genes. Genomic track files, generated by the bamCoverage tool of deepTools version 3.5.393 (with the parameter ‘--normalizeUsing RPKM’), were visualized in the genome browser IGV version 2.11.194. Metagene plots and heatmaps were plotted using the computeMatrix and plotHeatmap tools in deepTools version 3.5.393. Each ChIP-seq sample included at least two independent biological replicates.
Cleavage under targets and tagmentation (CUT&Tag) and data analysis
Fourteen-day-old Oryza sativa seedlings (2 g) grown on MS medium were ground in liquid nitrogen and resuspended in ChIP isolation buffer containing 0.1% (v/v) formaldehyde. Cross-linking was quenched by adding 2.5 M Glycine. The extracted nuclei were used for CUT&Tag assay with the Hyperactive Universal CUT&Tag Assay Kit for Illumina Pro (Vazyme, Nanjing, China), according to the manufacturer’s protocol. Briefly, nuclei were bound to Concanavalin A (ConA) beads and incubated overnight at 4 °C with a Rabbit anti-HA (AT1747, Engibody, Shanghai, China, 1:50) antibody. After washing, samples were incubated with a Goat anti-rabbit IgG H&L (ab6702, Abcam, 1:50) antibody for 1 h at room temperature, followed by incubation with Tn5 transposase for 1 h at room temperature. Target chromatin was fragmented at 37 °C for 1 h, and reverse cross-linking was performed at 55 °C for 3 h. DNA was purified, and libraries were constructed and amplified. Sequencing was carried out on an Illumina NovaSeq 6000 instrument by NEO BIO (Shanghai, China). The raw reads from CUT&Tag libraries were processed using a bioinformatic pipeline largely consistent with that applied to the ChIP-seq data described above. Due to the inherent affinity of the transposome for exposed DNA95, an additional correction step was implemented: genomic regions corresponding to NiP peaks (identified by SICER version 1.190 with parameters W200-G200-E100) were excluded from the coverage tracks via the --blackListFileName option in bamCoverage (deepTools version 3.5.393). Each CUT&Tag sample included two independent biological replicates.
Statistics and reproducibility
For rice heading date analysis, 20 individual plants grown in Shanghai or Sanya were randomly selected, with no data excluded. Statistical analyses were conducted using GraphPad Prism (version 9.5.0). For multigroup comparisons in rice heading date analysis, one-sided Ordinary one-way ANOVA test followed two-sided Tukey’s Honestly Significant Difference test was applied. An adjusted p-value < 0.01 was considered statistically significant. No statistical method was used to predetermine sample size. For RNA-seq, ChIP-seq and CUT&Tag data, the independent biological replicates for each sample were analyzed to be consistent and no data were excluded. But not all the independent biological replicates of ChIP-seq or CUT&Tag data were shown in the figures. Statistical analyses were performed using R (version 4.2.1), including two-sided correlation tests and one-sided Fisher’s exact tests. The experiments were not randomized, and investigators were not blinded to allocation during experiments and outcome assessment.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data supporting the findings of this work are available within the main text, supplementary materials, the listed Protein Data Bank (PDB) accession, the listed National Center for Biotechnology Information (NCBI) GEO accessions, or the listed ProteomeXchange accession. The atomic coordinates of the Ehd3-bound H3K4me1 peptide structure in this work have been deposited in the PDB database under accession code 9VNO. The RNA-seq, ChIP-seq (anti-H3K4me1/me2/me3, anti-H3K36me1/me2/me3, anti-H3, anti-HA, and anti-GFP), and CUT&Tag data for Chlamydomonas reinhardtii, Physcomitrium patens, and Oryza sativa in this work have been deposited in NCBI database under accession number GSE266911, GSE311864, and GSE311865. The proteomic datasets in this work were deposited in the ProteomeXchange database under accession number PXD068723. The ChIP-seq data for Oryza sativa used in correlation analyses were obtained from the NCBI database (accession numbers GSE79033, GSE126436, and GSE109616). The Homo sapiens ChIP-seq data used in correlation analyses were obtained from the NCBI database (accession numbers GSE175752, GSE179461, GSE107599, and GSE200770). The RNA-seq and ChIP-seq data for H3K4 and H3K36 methylations were obtained from the NCBI database (accession numbers GSE73407 and GSE242874 for Saccharomyces cerevisiae; GSE47281, GSE55555, GSE47285, GSE118785, and SRR1197331 for Drosophila melanogaster; GSE95781 and GSE118785 for Mus musculus; GSE51176, GSE169207, and GSE174252 for Homo sapiens; GSE258772 and GSE188493 for Arabidopsis thaliana; GSE128434 for Eutrema salsugineum, Populus trichocarpa, Phaseolus vulgaris, Asparagus officinalis, Brachypodium distachyon, Setaria viridis, Sorghum bicolor, and Zea mays). Source data are provided with this paper.
References
Flavahan, W. A., Gaskell, E. & Bernstein, B. E. Epigenetic plasticity and the hallmarks of cancer. Science 357, eaal2380 (2017).
Lloyd, J. P. B. & Lister, R. Epigenome plasticity in plants. Nat. Rev. Genet. 23, 55–68 (2022).
Musselman, C. A., Lalonde, M. E., Côté, J. & Kutateladze, T. G. Perceiving the epigenetic landscape through histone readers. Nat. Struct. Mol. Biol. 19, 1218–1227 (2012).
Strahl, B. D. & Allis, C. D. The language of covalent histone modifications. Nature 403, 41–45 (2000).
Black, J. C., Van Rechem, C. & Whetstine, J. R. Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol. Cell 48, 491–507 (2012).
Soares, L. M. et al. Determinants of histone H3K4 methylation patterns. Mol. Cell 68, 773–785.e6 (2017).
Mikkelsen, T. S. et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448, 553–560 (2007).
Barski, A. et al. High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837 (2007).
Liu, Y. et al. H3K4me2 functions as a repressive epigenetic mark in plants. Epigenet. Chromatin 12, 40 (2019).
Creyghton, M. P. et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 107, 21931–21936 (2010).
Zentner, G. E., Tesar, P. J. & Scacheri, P. C. Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Res. 21, 1273–1283 (2011).
Rada-Iglesias, A. et al. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470, 279–283 (2011).
Oka, R. et al. Genome-wide mapping of transcriptional enhancer candidates using DNA and chromatin features in maize. Genome Biol. 18, 137 (2017).
Lukauskas, S. et al. Decoding chromatin states by proteomic profiling of nucleosome readers. Nature 627, 671–679 (2024).
Liu, Y. C. & Huang, Y. Uncovering the mechanistic basis for specific recognition of monomethylated H3K4 by the CW domain of Arabidopsis histone methyltransferase SDG8. J. Biol. Chem. 293, 6470–6481 (2018).
Niu, Q. F. et al. A histone H3K4me1-specific binding protein is required for siRNA accumulation and DNA methylation at a subset of loci targeted by RNA-directed DNA methylation. Nat. Commun. 12, 3367 (2021).
Ramakrishnan, S. et al. Counteracting H3K4 methylation modulators Set1 and Jhd2 co-regulate chromatin dynamics and gene transcription. Nat. Commun. 7, 11949 (2016).
Rickels, R. et al. Histone H3K4 monomethylation catalyzed by Trr and mammalian COMPASS-like proteins at enhancers is dispensable for development and viability. Nat. Genet. 49, 1647–1653 (2017).
Hu, D. Q. et al. The MLL3/MLL4 branches of the COMPASS family function as major histone H3K4 monomethylases at enhancers. Mol. Cell. Biol. 33, 4745–4754 (2013).
Ieranò, C. et al. In PD-1+human colon cancer cells NIVOLUMAB promotes survival and could protect tumor cells from conventional therapies. J. Immunother. Cancer 10, e004032 (2022).
Liu, B. et al. The H3K4 demethylase JMJ1 is required for proper timing of flowering in Brachypodium distachyon. Plant Cell 36, 2729–2745 (2024).
Lu, Z. et al. The prevalence, evolution and chromatin signatures of plant regulatory elements. Nat. Plants 5, 1250–1259 (2019).
Matsubara, K. et al. Ehd3, encoding a plant homeodomain finger-containing protein, is a critical promoter of rice flowering. Plant J. 66, 603–612 (2011).
Zeng, L. et al. Mechanism and regulation of acetylated histone binding by the tandem PHD finger of DPF3b. Nature 466, 258–262 (2010).
Local, A. et al. Identification of H3K4me1-associated proteins at mammalian enhancers. Nat. Genet. 50, 73–82 (2018).
Li, H. T. et al. Molecular basis for site-specific read-out of histone H3K4me3 by the BPTF PHD finger of NURF. Nature 442, 91–95 (2006).
Wen, H. et al. Recognition of histone H3K4 trimethylation by the plant homeodomain of PHF2 modulates histone demethylation. J. Biol. Chem. 285, 9322–9326 (2010).
Taverna, S. D. et al. Yng1 PHD finger binding to H3 trimethylated at K4 promotes NuA3 HAT activity at K14 of H3 and transcription at a subset of targeted ORFs. Mol. Cell 24, 785–796 (2006).
Peña, P. V. et al. Molecular mechanism of histone H3K4me3 recognition by plant homeodomain of ING2. Nature 442, 100–103 (2006).
Choi, S. C. et al. Trithorax group protein Oryza sativa Trithorax1 controls flowering time in rice via interaction with early heading date3. Plant Physiol. 164, 1326–1337 (2014).
Sun, C. H. et al. The histone methyltransferase SDG724 mediates H3K36me2/3 deposition at MADS50 and RFT1 and promotes flowering in rice. Plant Cell 24, 3235–3247 (2012).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Fang, Y. et al. Functional characterization of open chromatin in bidirectional promoters of rice. Sci. Rep. 6, 32088 (2016).
Zheng, D. Y. et al. Salt-Responsive Genes are Differentially Regulated at the Chromatin Levels Between Seedlings and Roots in Rice. Plant Cell Physiol. 60, 1790–1803 (2019).
Van Rechem, C. et al. Collective regulation of chromatin modifications predicts replication timing during cell cycle. Cell Rep. 37, 109799 (2021).
Kim, H. J., Moon, S. J., Hong, S., Won, H. H. & Kim, J. H. DBC1 is a key positive regulator of enhancer epigenomic writers KMT2D and p300. Nucleic Acids Res. 50, 7873–7888 (2022).
Peeters, J. G. C. et al. Transcriptional and epigenetic profiling of nutrient-deprived cells to identify novel regulators of autophagy. Autophagy 15, 98–112 (2019).
Pal, D. et al. H4K16ac activates the transcription of transposable elements and contributes to their cis-regulatory function. Nat. Struct. Mol. Biol. 30, 935–947 (2023).
Weinberg, D. N. et al. The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape. Nature 573, 281–286 (2019).
Dixon, G. et al. QSER1 protects DNA methylation valleys from de novo methylation. Science 372, eabd0875 (2021).
Zhong, Z. H. et al. Histone chaperone ASF1 mediates H3.3-H4 deposition in Arabidopsis. Nat. Commun. 13, 6970 (2022).
Swaffer, M. P. et al. RNA polymerase II dynamics and mRNA stability feedback scale mRNA amounts with cell size. Cell 186, 5254–5268.e26 (2023).
Andrews, F. H. et al. Multivalent chromatin engagement and inter-domain crosstalk regulate MORC3 ATPase. Cell Rep. 16, 3195–3207 (2016).
Sanchez, R. & Zhou, M. M. The PHD finger: a versatile epigenome reader. Trends Biochem. Sci. 36, 364–372 (2011).
Rajakumara, E. et al. PHD finger recognition of unmodified histone H3R2 links UHRF1 to regulation of euchromatic gene expression. Mol. Cell 43, 275–284 (2011).
Ramón-Maiques, S. et al. The plant homeodomain finger of RAG2 recognizes histone H3 methylated at both lysine-4 and arginine-2. Proc. Natl. Acad. Sci. USA 104, 18993–18998 (2007).
Vermeulen, M. et al. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell 131, 58–69 (2007).
Qian, S. M. et al. Dual recognition of H3K4me3 and H3K27me3 by a plant histone reader SHL. Nat. Commun. 9, 2425 (2018).
Qiu, Y. et al. Combinatorial readout of unmodified H3R2 and acetylated H3K14 by the tandem PHD finger of MOZ reveals a regulatory mechanism for HOXA9 transcription. Genes Dev. 26, 1376–1391 (2012).
Xiong, X. Z. et al. Selective recognition of Histone crotonylation by double PHD fingers of MOZ and DPF2. Nat. Chem. Biol. 12, 1111–1118 (2016).
Oya, S., Takahashi, M., Takashima, K., Kakutani, T. & Inagaki, S. Transcription-coupled and epigenome-encoded mechanisms direct H3K4 methylation. Nat. Commun. 13, 4521 (2022).
Kizer, K. O. et al. A novel domain in Set2 mediates RNA polymerase II interaction and couples histone H3 K36 methylation with transcript elongation. Mol. Cell Biol. 25, 3305–3316 (2005).
Li, M. et al. Solution structure of the Set2-Rpb1 interacting domain of human Set2 and its interaction with the hyperphosphorylated C-terminal domain of Rpb1. Proc. Natl. Acad. Sci. USA 102, 17636–17641 (2005).
Mori, S. et al. Cotranscriptional demethylation induces global loss of H3K4me2 from active genes in Arabidopsis. EMBO J. 42, e113798 (2023).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
Carrozza, M. J. et al. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell 123, 581–592 (2005).
Keogh, M. C. et al. Cotranscriptional Set2 methylation of histone H3 lysine 36 recruits a repressive Rpd3 complex. Cell 123, 593–605 (2005).
Li, B. et al. Histone H3 lysine 36 dimethylation (H3K36me2) is sufficient to recruit the Rpd3s histone deacetylase complex and to repress spurious transcription. J. Biol. Chem. 284, 7970–7976 (2009).
Neri, F. et al. Intragenic DNA methylation prevents spurious transcription initiation. Nature 543, 72–77 (2017).
Guan, H. et al. Diverse modes of H3K36me3-guided nucleosomal deacetylation by Rpd3S. Nature 620, 669–675 (2023).
Baubec, T. et al. Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature 520, 243–247 (2015).
Zhu, J. K. Abiotic stress signaling and responses in plants. Cell 167, 313–324 (2016).
Ueda, M. & Seki, M. Histone modifications form epigenetic regulatory networks to regulate abiotic stress response. Plant Physiol. 182, 15–26 (2020).
Lämke, J. & Bäurle, I. Epigenetic and chromatin-based mechanisms in environmental stress adaptation and stress memory in plants. Genome Biol. 18, 124 (2017).
Asensi-Fabado, M. A., Amtmann, A. & Perrella, G. Plant responses to abiotic stress: The chromatin context of transcriptional regulation. Biochim. Biophys. Acta Gene Regul. Mech. 1860, 106–122 (2017).
Liu, H. P., Able, A. J. & Able, J. A. Priming crops for the future: rewiring stress memory. Trends Plant Sci. 27, 699–716 (2022).
Chen, K. et al. H3K36 methyltransferase SDG708 enhances drought tolerance by promoting abscisic acid biosynthesis in rice. N. Phytologist 230, 1967–1984 (2021).
Berr, A. et al. Arabidopsis histone methyltransferase SET DOMAIN GROUP8 mediates induction of the jasmonate/ethylene pathway genes in plant defense response to necrotrophic fungi. Plant Physiol. 154, 1403–1414 (2010).
Xu, J. et al. Natural antisense transcripts are significantly involved in regulation of drought stress in maize. Nucleic Acids Res. 45, 5126–5141 (2017).
Zhang, C. M., Wu-Scharf, D., Jeong, B. & Cerutti, H. A WD40-repeat containing protein, similar to a fungal co-repressor, is required for transcriptional gene silencing in Chlamydomonas. Plant J. 31, 25–36 (2002).
Nishiyama, T., Hiwatashi, Y., Sakakibara, I., Kato, M. & Hasebe, M. Tagged mutagenesis and gene-trap in the moss, Physcomitrella patens by shuttle mutagenesis. DNA Res 7, 9–17 (2000).
He, F., Zhang, F., Sun, W., Ning, Y. & Wang, G. L. A versatile vector toolkit for functional analysis of rice genes. Rice 11, 27 (2018).
Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549 (2018).
Wang, B. et al. Structural insights into partner selection for MYB and bHLH transcription factor complexes. Nat. Plants 8, 1108–1117 (2022).
Minor, W., Cymborowski, M., Otwinowski, Z. & Chruszcz, M. HKL-3000: the integration of data reduction and structure solution–from diffraction images to an initial model in minutes. Acta Crystallogr. D. Biol. Crystallogr. 62, 859–866 (2006).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr. 66, 213–221 (2010).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D. Biol. Crystallogr. 60, 2126–2132 (2004).
Du, K. et al. OsChz1 acts as a histone chaperone in modulating chromatin organization and genome function in rice. Nat. Commun. 11, 5717 (2020).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10–12 (2011).
Kim, D., Landmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Wang, J. W., Qi, M. F., Liu, J. & Zhang, Y. J. CARMO: a comprehensive annotation platform for functional exploration of rice multi-omics data. Plant J. 83, 359–374 (2015).
Huang da, W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009).
Saleh, A., Alvarez-Venegas, R. & Avramova, Z. An efficient chromatin immunoprecipitation (ChIP) protocol for studying histone modifications in Arabidopsis plants. Nat. Protoc. 3, 1018–1025 (2008).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Shao, Z., Zhang, Y., Yuan, G. C., Orkin, S. H. & Waxman, D. J. MAnorm: a robust model for quantitative comparison of ChIP-Seq data sets. Genome Biol. 13, R16 (2012).
Zang, C. et al. A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics 25, 1952–1958 (2009).
Zhu, L. J. et al. ChIPpeakAnno: a Bioconductor package to annotate ChIP-seq and ChIP-chip data. BMC Bioinforma. 11, 237 (2010).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Ramirez, F. et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016).
Thorvaldsdottir, H., Robinson, J. T. & Mesirov, J. P. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 14, 178–192 (2013).
Kaya-Okur, H. S. et al. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun. 10, 1930 (2019).
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2024YFE0105100 to A.D.), the National Natural Science Foundation of China (NSFC31930017 to A.D., NSFC31800207 to B.L., NSFC32100453 to K.D., and NSFC32400467 to J.Wu), and the Open Research Fund of State Key Laboratory of Crop Gene Exploration and Utilization in Southwest China, Sichuan Agricultural University (SKL-KF202412 to J.Wu). We thank the Shanghai Synchrotron Radiation Facility for providing technical support and assistance with crystallization data collection; the staff members of the Large-scale Protein Preparation System (https://cstr.cn/31129.02.NFPS.LSPS) at the National Facility for Protein Science in Shanghai for providing technical support and assistance in data collection and analysis; and Robert McKenzie, PhD, from Liwen Bianji (Edanz) (www.liwenbianji.cn) for editing a draft of this manuscript.
Author information
Authors and Affiliations
Contributions
A.D. conceived and designed the research. A.D., B.L., and J.G. supervised the experiments and interpreted the data; J.Wu, K.D., Y.L., C.H., Q.X., X.Z., Z.W., L.Z., F.L., and J.G. collected and analyzed the data; J.Wang, W.X., X.L., and X.W. analyzed the bioinformatic data; J.Wu, J.Wang, K.D., Y.L., W.-H.S., J.G., B.L., and A.D. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jiamu Du, Soichi Inagaki and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wu, J., Wang, J., Du, K. et al. H3K4me1 directs H3K36me2 and H3K36me3 deposition in land plants. Nat Commun 17, 2831 (2026). https://doi.org/10.1038/s41467-026-69632-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-69632-5
