
Abstract
Genetic and epigenetic variations contribute to the progression of glioma, but the mechanisms underlying these effects, particularly for enhancer-associated genetic variations in non-coding regions, still remain unclear. Here we performed high-throughput CRISPR interference screening to identify pro-tumour enhancers in glioma cells. By integrating genome-wide H3K27ac HiChIP data, we identified the target genes of these pro-tumour enhancers and revealed the essential role of enhancer connectomes in promoting glioma progression. Through systematic analysis of enhancers carrying glioma risk-associated single-nucleotide polymorphisms (SNPs), we found that these SNPs can promote glioma progression through the enhancer connectome. Using CRISPR–Cas9-mediated enhancer interference and SNP editing, we demonstrated that glioma-specific enhancer carrying the risk SNP rs2297440 regulates SOX18 expression by specifically recruiting transcription factor MEIS1 binding, thereby contributing to glioma progression. Our study sheds light on the molecular mechanisms underlying glioma susceptibility and provides potential therapeutic targets to treat glioma.
Similar content being viewed by others


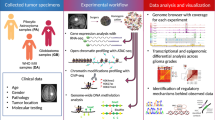
Main
Glioma is an aggressive type of brain cancer with a poor prognosis1,2,3,4. While moderate-to-high doses of ionizing radiation are established risk factors for glioma, some studies have noted an inverse association between a history of allergies and glioma risk5,6. However, few environmental risk factors have been firmly established for glioma6,7. The lack of environmental contributors has led researchers to focus on the genetic and epigenetic variations underlying glioma development8,9. Recently, genome-wide association studies (GWASs) revealed a correlation between single-nucleotide polymorphisms (SNPs) and glioma susceptibility and identified several risk SNPs associated with glioma occurrence10,11,12,13. Traditionally, neighbouring genes are assigned as the targeting gene for non-coding variants; however, this model is challenged by both experimental and computational evidence11,12,14,15. Identifying the target gene of these variants, especially for SNPs in non-coding regions, remains a challenge.
Long-range enhancer interactions play a key role in modulating gene expression by coordinating the activity of distal DNA elements located in non-coding regions15,16,17. In glioma, aberrant chromatin interactions have been implicated in the dysregulation of gene expression, contributing to tumour progression18,19. However, the precise mechanisms by which enhancer interactions regulate specific gene expression associated with tumour progression remain unclear. Furthermore, the role of genetic variants in modulating enhancer interactions and tumour progression is still poorly understood. Understanding the interplay between enhancer interactions and risk SNPs may provide insights into the molecular mechanisms underlying glioma development.
In this study, we applied an integrative approach combining enhancer CRISPR interference (CRISPRi) screening and HiChIP20 to systematically identify the pro-tumour enhancer connectome in glioma. Our analysis revealed that risk-associated SNPs located within enhancers can drive tumour progression through the enhancer connectome. This study underscores the functional significance of pro-tumour connectomes and pinpoints potential therapeutic target genes for glioma treatment.
Results
CRISPRi screening identifies pro-tumour enhancers in glioma
Enhancers play a crucial role in the development and progression of cancers by controlling the transcription of oncogenes14,16,21,22. To comprehensively identify enhancers involved in glioma development, we designed a pooled single guide RNA (sgRNA) library targeting 30,000+ H3K27ac peaks (with 1–3 sgRNAs per peak, totalling 90,049 sgRNAs) from chromatin immunoprecipitation sequencing (ChIP-seq) data obtained from glioma tissues and cell lines (Fig. 1a and Supplementary Table 1). We then performed a high-throughput CRISPRi screening in the human glioma cell line U25123 (Fig. 1b), with four independent replicates demonstrating remarkably high consistency across replicates (Extended Data Fig. 1a and Supplementary Table 2). We classified those enhancers with decreased sgRNA abundance as pro-tumour enhancers, as their disruption impaired tumour cell growth (Fig. 1c and Supplementary Table 3). Gene Ontology (GO) enrichment analysis showed that genes near pro-tumour enhancers were enriched for cell cycle and cell division regulation, mRNA splicing and translation-related pathways (Fig. 1d and Supplementary Table 4). Besides, our screening results revealed that pro-tumour enhancers not only localized to genic regions, but also resided in the intergenic regions (Fig. 1e and Extended Data Fig. 1b). For example, E34232 and E21248 are located in the promoter of a gene known to promote malignant cell progression24,25,26,27,28, while E12787 and E29716 are situated in the non-coding intergenic region (Fig. 1f). These results suggested that pro-tumour enhancers may influence oncogenesis by acting on oncogenes themselves or through long-range interactions with oncogenes. Understanding the mechanisms of how intergenic or non-coding region enhancers contribute to tumour progression requires the identification of their target genes.

a, The schematic workflow for identifying pro-tumour enhancers in glioma and designing sgRNAs targeting these enhancers. b, Diagram of CRISPRi-based functional enhancer screening. U251 cells expressing the dCas9-KRAB-MeCP2 are transduced by a pooled sgRNA library targeting enhancers. c, Identification of candidate pro-tumour enhancers in human glioma. The volcano plot shows each enhancer ranked by log2 fold change in sgRNA enrichment on day 21 relative to day 0 samples from CRISPRi screening. Statistical significance was determined by two-sided t-test. d, GO enrichment analysis for genes near pro-tumour enhancers. Statistical significance was determined by one-sided Fisher’s exact test. e, The proportion of enhancers located in the genic and intergenic regions. f, H3K27ac ChIP-seq signals of identified pro-tumour enhancers in normal brain and glioma. Orange shadows indicate genic regions, while green shadows highlight intergenic regions.
CRISPRi and HiChIP uncovers pro-tumour enhancer connectomes
We performed H3K27ac HiChIP on primary glioma tissues and cell lines to identify glioma-specific enhancer connectomes by comparing differential loops in glioma and normal brain (Fig. 2a, Extended Data Fig. 1c and Supplementary Table 5). Although most loops are shared between tumour and normal brain tissues, we identified a subset of glioma-specific loops (Fig. 2b). Our results revealed that binding motifs within glioma-specific loop anchors were enriched for transcription factors known to promote cell proliferation and DNA replication, including CREB1, MEIS1 and ETV129,30,31,32 (Fig. 2c), while normal brain-specific motifs were associated with genomic stability factors such as RUNX333,34,35 and E2F736,37 (Extended Data Fig. 1d). We then used glioma-specific loops to link pro-tumour enhancers to potential target genes, identifying 3,776 pro-tumour enhancer connectome-associated protein-coding genes (Fig. 2a,d). Among these, 1,998 genes were identified as pro-tumour hits in the CRISPRi screen, while the remaining 1,778 genes, although linked to pro-tumour enhancers via glioma-specific loops, were not identified as hits in the CRISPRi screen (Fig. 2a,d and Supplementary Table 6). For subsequent analysis, we focused on the 1,998 genes supported by both the glioma-specific loop and the CRISPRi screen results. GO enrichment analysis showed that these genes were enriched in cell division, DNA damage response and cycling (Fig. 2e), suggesting that the aberrant interaction between pro-tumour enhancers can contribute to tumourigenesis. For example, genes associated with glioma occurrence and development, such as MYC and VMP1, showed strong interactions with pro-tumour enhancers in glioma24,25,38,39 (Fig. 2f). Complementary validation using chromosome conformation capture (3C) assays40, confirmed direct physical interactions of E34235 –MYC and E15788–VMP1 (Extended Data Fig. 1e).

a, A schematic illustrating the identification of pro-tumour enhancer connectomes in glioma. Glioma-specific chromatin loops (red arcs) were used to link pro-tumour enhancers (red ovals) to potential target genes. b, The distribution of specific loops in glioma and normal brain tissue. c, Enrichment of glioma-specific loop motifs. Statistical significance was determined by one-sided Fisher’s exact test. d, A pie chart showing the distribution of the pro-tumour enhancer connectome-associated genes into functional pro-tumour hits identified by CRISPRi screen (type a) and those without functional hits in the screen (type b). e, GO enrichment analysis for genes identified in the intersection (in d). Statistical significance was determined by one-sided Fisher’s exact test. f, HiChIP contact maps illustrating interactions between pro-tumour enhancers and their target genes in glioma (patient 510) compared with normal brain tissue (middle frontal gyrus)52. Glioma-specific loops are highlighted with circles. Below each map, corresponding ChIP-seq signal tracks are presented. g, The interaction profile of the E13983 enhancer and its target genes. h, Validation of HiChIP target genes by RT-qPCR in CRISPRi U251 cells with targeting of E19383 enhancers. RT-qPCR measured mRNA levels of target genes in U251 cells with CRISPRi-mediated targeting of E19383 enhancers. Data represent means ± s.d. from three independent biological replicates. Statistical significance was determined by two-sided t-test. i, The effect of promoter CRISPRi of target genes on U251 cell proliferation. Data represent means ± s.d. from six independent biological replicates. Statistical significance was determined by two-sided t-test. NS, not significant.
To further validate the impact of the pro-tumour enhancer connectome on tumour development, we examined enhancer E19383, located within the intron of HS1BP3, a gene that encodes an HS1-binding protein not previously implicated in glioma41. H3K27ac HiChIP results showed that E19383 interacted with multiple genes (Fig. 2g). Using CRISPRi, we specifically silenced E19383 and observed a significant reduction in the transcript levels of six genes that physically interact with this enhancer (Fig. 2h). Among these genes, four of them (LAPTM4A, SDC1, PUM2 and RHOB) were also found in our CRISPRi screening (Extended Data Fig. 1f). Furthermore, CRISPR-mediated silencing of these four target genes significantly suppresses glioma cell proliferation (Fig. 2i). Survival analysis revealed that high expression levels of these four genes are associated with poorer prognosis in patients with glioma (Extended Data Fig. 1g). These findings collectively support that E19383 modulates glioma malignancy through enhancer–target gene interactions. Overall, our results suggest that targeting pro-tumour enhancers and their target genes may hold therapeutic potential for suppressing tumour growth in glioma.
Risk SNPs promote glioma progression via enhancer connectome
Previous GWASs identified several genetic variants associated with glioma risk3,11,12,42. Notably, we found that 85.18% of these SNPs were located in intergenic or intronic regions, suggesting that they may function as non-coding regulatory elements to regulate glioma development (Fig. 3a). This is reminiscent of the idea that non-coding regions can possess functional elements, such as enhancers, that regulate gene expression and influence disease susceptibility16,21,43,44. By integrating H3K27ac ChIP-seq data, we identified 12 SNPs that directly reside on enhancers, suggesting that these genetic variants may exert their function in an enhancer–promoter interaction manner (Fig. 3b). Through the luciferase reporter system, we found that these SNPs containing enhancers were all active in the U251 cell line, and 9 (75%) risk-associated alleles displayed more robust enhancer activity than their non-risk alleles (Fig. 3c). This suggests that these risk-associated alleles may increase enhancer activity and promote the expression of target genes that involved in glioma progression. Concurrently, our analysis of CRISPRi screening results identified nine enhancers with pro-tumour properties, five of which showed statistically significant fold changes (Fig. 3d). Using CRISPRi experiments, we validated the functional role of these enhancers in glioma cell proliferation (Extended Data Fig. 2a), and the results of the two most significant enhancers carrying risk SNPs (rs3851634 and rs2297440) are shown in Fig. 3e. Furthermore, we found that the frequencies of interactions between these two enhancers and their adjacent regions are elevated in glioma tissues compared with normal brain tissues (Extended Data Fig. 2b,c). Through HiChIP data, we identified 123 candidate target genes associated with these 12 enhancers carrying risk-associated SNPs (Supplementary Table 7), as exemplified by rs2297440 in Fig. 3f. Consistently, CRISPRi screening results showed that 30.10% of these candidate targets, including MYC, CASC11 (associated with rs55705857) and SOX18 (associated with rs2297440), promote glioma cell growth (Fig. 3g). These results suggest that the risk-associated SNP alleles may exert their oncogenic effects by affecting their target genes through the enhancer connectome, leading to aberrant activation of their target genes and subsequent promotion of glioma cell proliferation and tumourigenesis.

a, The genomic distribution of glioma-associated risk SNPs. b, Proportions of SNPs located in non-coding regions with or without H3K27ac modifications. c, Luciferase reporter assays reveal differential enhancer activity in glioma cells between risk and non-risk alleles for 12 risk SNP-associated enhancers. Top: the schematic of the luciferase reporter used to detect enhancer activity. Bottom: bar plots represent mean ± s.d. luciferase activity from three independent biological replicates. d, A scatter plot showing SNP-associated enhancers ranked by log2 fold change in sgRNA enrichment from CRISPRi screening. e, CRISPRi with rs2297440-associated enhancers significantly inhibits glioma cell proliferation. Data represent mean ± s.d. from three independent biological replicates. f, The interaction profile of an enhancer carrying risk-associated SNP rs2297440 and its target genes. g, Enrichment of target genes interacting with SNP-associated enhancers in glioma cells, as identified by CRISPRi screening. Statistical significance was determined by two-sided t-test. NS, not significant.
rs2297440-associated enhancer drives glioma cell growth
By comparing H3K27ac ChIP-seq data between glioma and normal brain, we identified the region containing rs2297440 as a glioma-specific enhancer (Fig. 4a), which interacts with over 30 candidate target genes (Supplementary Table 7) and displays increased activity compared with non-risk variants (Fig. 3c). To investigate rs2297440 function in glioma development, we used CRISPR–Cas9 technology to knock out the 1.3-kb enhancer region containing rs2297440 in U251 and LN229 cell lines (Fig. 4a and Extended Data Fig. 3a). Notably, the enhancer region is located within the intron of the RTEL1 gene. However, our results showed that knockout (KO) of the enhancer region did not affect RTEL1 expression (Fig. 4b and Extended Data Fig. 3b), suggesting that this enhancer regulates glioma cell expansion through the RTEL1-independent mechanism. Next, we performed MTS and colony formation assays in enhancer KO cells and found that the enhancer deletion significantly impeded cell growth and proliferation (Fig. 4c,d and Extended Data Fig. 3c). To further characterize the function of enhancers carrying rs2297440, we performed RNA-seq analysis in both U251 wild-type (WT) and enhancer KO cell lines and found that genes exhibiting significant expression changes in enhancer KO cells were related to cell migration and differentiation pathways (Extended Data Fig. 3d,e).

a, H3K27ac signal at the rs2297440-associated enhancer in normal brain, glioma tissues and glioma cell lines. b, The relative mRNA expression of RTEL1 in U251 cells after the rs2297440-associated enhancer KO. c, MTT assay showing the proliferation of enhancer KO (eKO) U251 cells. d, Colony formation assay in WT and eKO U251 cells. e, Scatter plots of differential expression of genes interacting with the rs2297440-associated enhancer between WT and eKO cells. f, The relative mRNA expression of SOX18 in U251 cells after enhancer KO. g, Top: genome architecture mapping contact matrix of the chr20: 62,500–64,200 kb genomic window, showing increased interaction between the rs2297440 locus and the SOX18 gene in glioma compared with normal brain tissues. Bottom: confirmation of 3C-PCR and Sanger sequencing detected contacting genomic fragments for SOX18 and rs2297440 locus. h, PCR–agarose gel electrophoresis of the ligation products of SOX18 TSS region and rs2297440 locus in U251, LN229 and U87 cells. This PCR–agarose gel electrophoresis was carried out once (n = 1). i, Relative connection frequency measured by 3C-qPCR in U251 WT cells and enhancer KO (eKO1 and eKO2) cell lines. Data represent mean ± s.d. of three independent experiments. Statistical significance was determined by two-sided t-test. NS, not significant.
SOX18 is a target of the rs2297440-associated enhancer
RNA-seq results showed that KO of the enhancer containing SNP rs2297440 led to a significant downregulation of multiple interacting genes, with SOX18 exhibiting the most significant decrease in expression (Fig. 4e). This finding was further validated by RT-qPCR (quantitative reverse transcription polymerase chain reaction) analysis (Fig. 4f). Besides, HiChIP analysis revealed that SOX18 is the target gene interacting with the rs2297440-resident enhancer (Fig. 3g), showing a strong interaction in glioma tissue compared with normal brain (Fig. 4g). Complementary validation using 3C-PCR confirmed direct physical interactions between the rs2297440-associated enhancer and SOX18 (Fig. 4g,h). Notably, enhancer KO abolished these chromatin contacts (Fig. 4i), confirming that SOX18 is the target gene of this regulatory element.
To validate the functional role of the rs2297440-associated enhancer and SOX18, we first conducted cell line experiments. Overexpression of SOX18 rescued the impaired cell growth and proliferation in enhancer KO cell lines (Fig. 5a,b and Extended Data Fig. 3f). In glioma stem cell (GSC) sphere models, enhancer KO reduced self-renewal capacity, and this defect was reversed by SOX18 overexpression (Fig. 5c,d and Extended Data Fig. 4a,b). Similarly, in the murine xenograft model, enhancer KO suppressed tumour formation, while SOX18 overexpression restored tumourigenicity (Fig. 5e,f).

a, MTT assay showing increased proliferation of enhancer KO (eKO) cells upon overexpression of SOX18. b, Colony formation assay demonstrating that SOX18 overexpression restores the colony-forming ability of eKO cells. c, GSC sphere models formed by WT, eKO and SOX18-overexpressing eKO cells. Scale bar, 100 μm. d, Quantification of GSC sphere formation in c. e, In vivo bioluminescence imaging of xenografts derived from WT, eKO and SOX18-overexpressing eKO cells at days 9, 18 and 27 post-implantation. f, Quantification of in vivo bioluminescence signals in e. Data represent mean photon flux ± s.d. from five independent mice per group. g, Cell proliferation assay in SOX18-KO U251 cells. h, Colony formation assay in SOX18-KO U251 cells. Unless otherwise specified, data are presented as mean ± s.d. from three independent biological replicates. Statistical significance was determined by two-sided t-test. NS, not significant.
To further investigate the functional role of SOX18, we used CRISPR–Cas9-mediated KO of SOX18 in glioma cells (Extended Data Fig. 4c,d), which resulted in significantly decreased cell growth and proliferation (Fig. 5g,h and Extended Data Fig. 4e). Collectively, these findings demonstrate that SOX18 is a critical downstream target of the rs2297440-associated enhancer, and its dysregulation contributes to the malignant phenotype of glioma cells.
rs2297440 modulates glioma cell growth through SOX18
To investigate the effect of rs2297440 (C/C) risk allele on glioma cell growth, we performed single-base editing of this SNP in U251 cells and obtained two identical non-risk allele (T/T) homozygous cell lines (Fig. 6a and Extended Data Fig. 5a). The non-risk allele (T/T) decreased the frequency of direct physical interaction between SOX18 and the rs2297440-associated enhancer (Fig. 6b). Subsequently, through MTS and colony formation assays, we demonstrated that the cell lines of (T/T) allele exhibited significantly reduced cell growth and proliferation capabilities (Fig. 6c,d and Extended Data Fig. 5b), supporting that the risk allele (C/C) increases glioma growth12. Consistent with the results of enhancer KO, we found that the rs2297440 mutation did not affect the expression of the RTEL1 (Extended Data Fig. 5c). RNA-seq analysis of the edited cell lines revealed that upregulated genes were enriched in biological processes related to cellular homeostasis and function, including translation, aerobic respiration, signal transduction and DNA repair (Extended Data Fig. 5d–f). We found that SOX18 expression was significantly downregulated in the mutant cell lines (Fig. 6e) and that overexpressing SOX18 in these cells rescued the cell proliferation and colony formation (Fig. 6f–h and Extended Data Fig. 5g, h). In addition, we demonstrated that the non-risk allele (T/T) exhibited significantly reduced GSC sphere formation, and this defect was rescued by SOX18 overexpression (Fig. 6i,j and Extended Data Fig. 5i–k). These results suggest that the rs2297440 risk allele promotes glioma progression by regulating SOX18 expression.

a, A schematic showing programmable editing of rs2297440 in U251 cell with a dsDNA donor. b, 3C-qPCR analysis showing the interaction frequency between the SOX18 TSS and the rs2297440 locus in WT and single-base edited mutant (MUT1 and MUT2) U251 cells. c, MTT assay showing the proliferation of WT cells and mutant cells over time. d, Quantification of colony formation in WT and mutant U251 cells. e, RT-qPCR analysis for SOX18 expression in U251 WT and mutant cells. f, The number of colonies formed by mutant cells with or without overexpression of SOX18. g,h, MTT assay assessing the proliferation of mutant U251 cells with or without SOX18 overexpression. i,j, GSC sphere formation assay evaluating the tumour-forming capacity of WT and mutant U251 cells, with and without SOX18 overexpression. Representative images of spheres (i). Scale bar, 100 μm. Bar plot quantifying the number of spheres (j). Data represent mean ± s.d. from three independent biological replicates. Statistical significance was determined by two-sided t-test. NS, not significant.
MEIS1 binds rs2297440-C allele to enhance SOX18 expression
Our results suggested that rs2297440 interacts with SOX18 through chromatin looping, which may involve the recruitment of transcription factors17,25,45,46. Sequence analysis of rs2297440 locus revealed putative transcription factor binding motifs, including MEIS1, PAX5 and SOX18 (Fig. 7a).

a, Motif analysis of rs2297440-associated enhancer, with relative scores obtained from the JASPAR database. b, ChIP–qPCR detection of MEIS1, SOX18 and PAX5 binding to rs2297440 locus between risk (WT) and non-risk (single-base editing) allele. IgG served as the negative control. c, SOX18 mRNA expression level in U251 cells after MEIS1 overexpression using dCAS9–CRISPRa. d, SOX18 mRNA expression level in U251 cells after MEIS1 overexpression using an exogenous overexpression plasmid. e, A schematic illustration of how rs2297440 allele modulates MEIS1 binding and regulates SOX18 expression and glioma progression. Data represent mean ± s.d. from three independent biological replicates. Statistical significance was determined by two-sided t-test. NS, not significant. TF, transcription factor.
Notably, transcription factor MEIS1 was found to be enriched in glioma-specific anchors (Fig. 2c). To examine if these transcription factors bind to the enhancer carrying rs2297440, we performed ChIP–qPCR and found that immunoprecipitation with the MEIS1 antibody resulted in a sevenfold enrichment of DNA fragments in glioma cells containing risk allele (C/C) compared with edited glioma cells containing the non-risk allele (T/T) (Fig. 7b). By contrast, immunoprecipitation with SOX18 or PAX5 antibody did not exhibit a differential binding signal (Fig. 7b), indicating that MEIS1 preferentially binds to the rs2297440 locus. We further investigated the impact of MEIS1 binding on SOX18 expression using a CRISPR activation (CRISPRa)–VP64 system and found that MEIS1 overexpression led to an approximately fivefold increase in SOX18 transcript levels (Fig. 7c). Consistently, MEIS1 overexpression vectors constructed in vitro also led to increased SOX18 expression (Fig. 7d). Taken together, our findings propose a model where the preferential binding of MEIS1 to the risk-associated allele rs2297440 facilitates enhancer interaction, leading to enhanced SOX18 expression and promoting glioma progression (Fig. 7e).
Discussion
Our study provides a valuable resource for understanding the role of enhancer connectome in glioma progression, shedding light on the relationships between non-coding genetic variants and their target genes. The integration of CRISPRi screening with HiChIP data allowed us to identify pro-tumour enhancer connectomes (Fig. 2), whose interactions are involved in essential cellular processes such as cell division, migration and DNA damage response. This finding highlights the potential of enhancer-targeted therapies in cancer treatment16,17,47,48,49,50.
The observation that many risk-associated SNPs are located in non-coding regions suggests these variants may exert their function through enhancer connectome. Consistent with a previous study that identified a specific SNP that correlates with increased risk of glioma25, our high-throughput approach revealed a series of non-coding SNPs associated with glioma development through enhancer interaction, providing a more comprehensive understanding of the genetic landscape underlying glioma susceptibility (Fig. 3g). We exemplified this by investigating the rs2297440 SNP locus. Previous studies highlighted rs2297440 as a critical genomic locus, where the C allele is associated with increased risk of glioma development, while the T allele may confer survival advantages36,51. In line with these findings, our study further elucidates that rs2297440 resides within a glioma-specific enhancer and regulates the expression of the target gene SOX18 through chromatin interactions, thereby promoting tumour proliferation. The functional characterization of the rs2297440-associated enhancer revealed that MEIS1, a transcription factor, preferentially binds to the risk allele to enhance SOX18 expression (Fig. 7e). These results suggest that rs2297440 risk allele contributes to glioma growth by enhancing the MEIS1–SOX18 axis.
Our study provides a systemic decoding of how non-coding genetic variants influence glioma progression through enhancer interactions. By mapping these interactions, we gain a deeper understanding of the regulatory mechanisms in glioma and identify potential genes as therapeutic targets. This strategy may also be applied in future studies of other cancers in the post-GWAS era.
Methods
The study was approved by the Ethics Institutional Review Boards of the First Affiliated Hospital of Sun Yat-sen University and complied with all relevant ethical regulations regarding human participants (2024SQXB001234 and [2024]341). Human glioma tissues were collected at the Department of Neurosurgery of the First Affiliated Hospital of Sun Yat-sen University with informed consent from the donors. Approximately 0.5–1 cm3 glioma tissues from each of five patients were used for HiChIP. Glioma cell lines LN229, U251 and U87 were used for H3K27ac HiChIP.
Cell culture
The glioma cells (LN229, U251 and U87) and HEK293T were cultured in Dulbecco’s modified Eagle medium (DMEM, Gibco) supplemented with 10% foetal bovine serum (AusGeneX) and 1× Pen Strep (ThermoFirsher), the medium was changed every 2 days and cells were passaged using trypsin–EDTA (0.25%) (Gibco).
Cell fixation
Primary tissues were mechanically disrupted, prepared as a single-cell suspension and cross-linked by 1% paraformaldehyde (Gibco, methanol-free). The glioma cell lines LN229, U251 and U87 were digested with trypsin before cross-linking reactions. Specifically, 1 × 107 cells were cross-linked for 10 min with rotation. Then, 125 mM glycine was added to quench the formaldehyde for 5 min and incubated on ice for 20 min to stop cross-linking completely. Cells were pelleted and washed with cold phosphate-buffered saline (PBS), pelleted again, snap-frozen in liquid nitrogen for 5 min and stored at −80 °C or immediately processed for the HiChIP protocol.
HiChIP library construction
The HiChIP protocol was performed as described with modifications20,53. In brief, 1 × 107 cross-linked cells were digested using 400 U MboI (NEB) in 37 °C with rotation (900 rpm) for 16 h. MboI (NEB) was heat-inactivated at 65 °C for 20 min. Then, 15 mM biotin14-d-ATP (Invitrogen) was added, and restriction-cut ends were ligated using 4,000 U T4 DNA ligase (NEB). For sonication, we kept the sonication constant at eight cycles for different amounts of cell starting material and preclearing with 60 μl Protein A beads (Gibco). The 300–700-bp genomic DNA was incubated with 7.5 μg H3K27ac antibody (Abcam) at 4 °C overnight with rotation. The next day, the complexes were captured by 60 μl Protein A magnetic beads and washed three times each with Low-Salt Wash Buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris–HCl pH 7.5 and 150 mM NaCl), High-Salt Wash Buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris–HCl pH 7.5 and 500 mM NaCl) and LiCl Wash Buffer (10 mM Tris–HCl pH 7.5, 250 mM LiCl, 1% NP-40, 1% sodium deoxycholate and 1 mM EDTA; make fresh). Washing was performed at room temperature on a magnet by adding 500 μl of a wash buffer, swishing the beads back and forth twice by moving the sample relative to the magnet, and then removing the supernatant. Beads were resuspended in DNA elution buffer. After elution, ChIP samples were incubated with 10 mg ml−1 Proteinase K (YEASEN) 4 h at 55 °C and 65 °C for 3 h. DNA was purified using AMPure XP Beads (Beckman). Streptavidin C1 beads (Invitrogen) were used to capture biotinylated DNA. QIAseq FX DNA Library Kits were used to generate the sequencing library. HiChIP libraries were size-selected to 300–700 bp using AMPure XP beads (Beckman) and subjected to 2× 150-bp paired-end sequencing on Novaseq. Two biological replicates were analysed for each experimental condition.
Quantitative chromosome conformation capture (3C-qPCR)
The 3C-qPCR protocol was performed as described with modifications40. In brief, 1 × 107 cross-linked cells were trypsinized and fixed by 1% formaldehyde in 10 ml of DMEM (Gibco) without foetal bovine serum for 10 min at room temperature. The reaction was quenched with ice-cold glycine. After centrifugation, the pellets were washed three times with 1× cold NEBuffer 3.1 and resuspended in a lysis buffer (10 mM NaCl; 10 mM Tris–HCl, pH 7.5; 0.2% NP-40; 1× protease inhibitor). Before digestion, we incubate the lysed cells in 0.1% of SDS (65 °C, 10 min) to eliminate proteins that are not cross-linked to DNA and open the chromatin for a better and more homogeneous digestion. The tubes were placed on ice immediately after opening the chromatin, and 43 μl of 10% Triton X-100 was added to the Hi-C-tube (423 µl total) to quench the SDS (1% Triton final), then mixed gently by pipetting up and down (avoiding bubbles). Then, 400 U MboI (NEB, R0147M) was used for genomic DNA digestion. The sample tube was incubated at 65 °C for 20 min to deactivate the endonuclease enzyme. The tubes were placed on ice until cooled to room temperature and 665 µl ligation mix (H2O, 242 μl; 5× ligation buffer (Invitrogen), 240 μl; 10% Triton X-100, 120 μl; 10 mg ml−1 bovine serum albumin, 12 μl; T4 DNA ligase (Invitrogen), 50 μl) was added. The samples were then incubated at 16 °C for 4 h in a ThermoMixer with intermittent shaking. After ligation, the nucleic acids were reverse cross-linked and purified by ethanol precipitation. The concentration of ligated DNA samples was measured by SYBR-qPCR, and data were normalized to GAPDH. Sequences for 3C-qPCR primers are listed in Supplementary Table 8.
ChIP-seq
A total of 1 × 107 cells were cross-linked in 1% formaldehyde (methanol free) and quenched as described above at a volume of 20 ml for every one million cells. Cells were cross-linked for 10 min at room temperature with rotation. Samples were incubated 20 min on ice and spun at 2,500g for 5 min. The supernatant was discarded, and pelleted nuclei were washed once with 500 μl prechilled cell lysis buffer (10 mM Tris–HCl pH 8.0, 10 mM NaCl, 0.2% NP-40, 1× protease inhibitors) and then rotated at 4 °C for 30 min. Samples were spun again at 2,500g for 5 min, supernatants were discarded and pelleted nuclei were washed twice with 500 μl of ice-cold ChIP lysis buffer. The volume of pelleted material was brought up to 1 ml in a nuclear lysis buffer (50 mM Tris–HCl pH 7.5, 10 mM EDTA, 1% SDS and 1× protease inhibitors) and incubated for 5 min at room temperature. Samples were then divided equally into three parts, sheared for ten cycles (30 s ‘ON’ and 30 s of ‘OFF’) and then spun down at 16,100g for 15 min. Supernatants were collected into new low-adsorption 1.5-ml Eppendorf tubes. Then, 60 μl of Protein A beads for every 107 cells were washed in ChIP dilution buffer and resuspended in 50 μl dilution buffer per tube (100 μl per ChIP). Protein A beads were added to samples, which were rotated 1 h at 4 °C for precleaning. Sample supernatants were then separated using a magnet and transferred into new tubes, and then 7.5 μg H3K27ac antibodies per 107 cells were incubated at 4 °C overnight with rotation. The next day, 60 μl Protein A beads were washed, added to samples and then rotated for 2 h at 4 °C. The beads were washed three times each with Low-Salt Wash Buffer, High-Salt Wash Buffer and LiCl Wash Buffer. ChIP-DNA was de-cross-linked with Proteinase K and purified using AMPure XP beads. According to the instruction document, DNA libraries were generated using an FX DNA Library Prep Kit (QIAGEN).
RT-qPCR
Total RNA was extracted from cells using TRIzol reagent (Life Technologies). cDNA synthesis was performed using a PrimerScript RT reagent kit with gDNA eraser (TaKaRa). Real-time PCR reactions were performed using Hieff qPCR SYBR Green Master Mix (YEASEN) and a Bio-Rad CFX Connect Real-Time system. PCR cycling conditions were as follows: 95 °C for 5 min, 40 cycles of 95 °C for 15 s, 60 °C for 15 s and 72 °C for 30 s. A melting curve of amplified DNA was subsequently acquired. The relative expression of target genes was determined using the comparative CT method and normalized to GAPDH. Sequences for RT-qPCR primers are listed in Supplementary Table 8.
ChIP–qPCR
ChIP–qPCR was performed in WT U251 cells (C/C) and SNP MUT (single-base edited mutant) cells (T/T) to investigate whether the two proteins could bind to the DNA sequence containing SNP rs2297440. A total of 7.5 μg antibody was used to assess the affinity of different mutant forms to the corresponding transcription factors, and the pull-down DNA content was measured by RT-qPCR. Sequences for ChIP–qPCR primers are listed in Supplementary Table 8.
CRISPRi and CRISPRa validation of functional enhancers and genes
For virus production, 5 × 106 HEK293T cells were plated per 10-cm dish (Corning). The following day, plasmid encoding lentivirus was co-transfected with plasmid dCas9-KRAB-MeCP2 (Addgene #122205) or lentiSAMv2 with 2A Blast (Addgene #75112), pMD2.G and psPAX2 (10 μg each) into the cells with Lipofectamine 3000 (Thermo Fisher) according to the manufacturer’s instructions. The supernatant was changed to fresh medium 12 h after transfection, and supernatant containing viral particles was collected 48 h after transfection and filtered with a 0.45-μm cell sieve. For lentivirus encoding individual sgRNAs, virus was concentrated tenfold using Lenti-X concentrator (Clontech) and stored at −80 °C.
To generate a glioma cell line expressing dCas9-KRAB-MeCP2 or dCas9-VP64-T2A-Blast, 2 × 106 glioma cells were plated per T25 flask. Twenty-four hours after plating, lentivirus harbouring the dCas9-KRAB-MeCP2 or dCas9-VP64-T2A-Blast was added with polybrene (4 µg ml−1). The medium was changed 12 h after infection and then again 48 h after infection with blasticidin (MedChemExpress LLC) at a concentration of 5 µg ml−1. Blasticidin-resistant cells were selected for 7 days, with the medium changed every other day.
As for sgRNA virus production, the sgRNA annealing program using a thermocycler was set as follows: 37 °C for 30 min, 95 °C for 5 min, followed by a ramp down at 0.1 °C s−1 from 95 °C to 25 °C. The annealed oligos can then be ligated into the plasmid (Adgene#52963, digested by BsmBI, NEB). The resulting sgRNA plasmid or a non-targeting control vector, along with pMD2.G and psPAX2 (10 μg each), were transfected into cells using Lipofectamine 3000 (Thermo Fisher) according to the manufacturer’s instructions. The next steps are same as those for dCas9-KRAB-MeCP2 virus generation.
To generate a glioma cells line expressing sgRNA, 2 × 106 glioma cells harbouring dCas9-KRAB-MeCP2 were plated per T25 flask. Twenty-four hours after plating, lentivirus harbouring the sgRNA or a non-targeting vector plasmid was added with polybrene (4 µg ml−1). The medium was changed 12 h after infection and then again 48 h after infection with 2 µg ml−1 puromycin (MCE) at a concentration of 5 µg ml−1. Puromycin-resistant cells were selected for 7 days, with the medium changed every other day.
CRISPRi screening
CRISPRi screening was performed as described54,55. As for enhancer sgRNA library design, the bed file containing H3K27ac peaks was called by MACS256,57 from 38 glioma samples (GSE145644) and 3 glioma cell lines (U251, LN229 and U87), and FlashFry58 was used for designing the library that targets enhancers in glioma. In total, 34,083 non-redundant enhancers were targeted by 90,049 sgRNAs. We selected no more than three sgRNAs from each H3K27ac region, with high Hsu2013 scores, high Doench2014 on-target scores and uniform protospacer adjacent motif distance distribution. All 90,049 sgRNAs (containing 539 control sgRNA) were ligated into lentiGuide-Puro to produce the sgRNA library lentivirus.
To establish glioma cell lines stably expressing dCas9-KRAB-MeCP2, U251 cells were infected by lentivirus and cultured with 4 μg ml−1 blasticidin over 1 week. A total of 4 × 107 cells were infected by sgRNA library lentivirus with a multiplicity of infection (MOI) of 0.3. In our study, we performed four independent biological replicates in the U251 cell line for the CRISPRi screen. After 24 h of infection, cells were cultured with 2 μg ml−1 puromycin to select cells stably expressing sgRNAs. After 7 days, 1 × 108 cells were taken as the day 0 group. The other cells were cultured for another 3 weeks; then 1 × 108 cells were collected as the day 21 group. Genome DNAs of day 0 and day 21 were extracted by the NucleoBond HMW DNA Kit. sgRNA libraries were amplified from all genome DNAs by TransStart FastPfu DNA Polymerase, using forward primers: TTGAAAGTATTT+ [8 bp barcode] + GCTTTATATATCTTGTGGAAAGGACGAAACACC, reverse primers: CCACTTTTTCAA+ [8 bp barcode] + CGGACTAGCCTTATTTTAACTTGC, 2.5 μg genome DNA per 50 μl PCR system. The run program was as follows: 98 °C 3 min; 30 cycles, 98 °C 15 s, 55 °C 15 s, 72 °C 15 s; 72 °C 1 min. Bands were cut and collected at 140 bp, and DNA was purified from agarose gel with NucleoSpin Gel and PCR Clean‑up kit. The products were used for sequencing, and sgRNAs were quantified from paired-end reads. To normalized counts unbiasedly, we computed the average of transcription terminate sgRNAs containing poly (~5–7T) as baseline, sgRNA counts were divided by the baseline and the primary screening results were analysed by calculating the fold change of sgRNA compared with the internal control at days 0 and 21.
The impact on specific enhancer regions was evaluated through individual analysis of each sgRNA. The t-test was performed across biological replicates to determine the statistical significance of changes in sgRNA abundance. Pro-tumour enhancers were defined as enhancers targeted by sgRNAs demonstrating significant depletion. Genes that overlap with pro-tumour enhancers were defined as genes near pro-tumour enhancers.
Luciferase reporter assays
Functional analysis of the risk SNP-containing H3K27ac region was performed through a luciferase reporter assay. Specifically, the genomic fragment encompassing the risk SNP locus within the H3K27ac-marked region was PCR-amplified from U251 cell genomic DNA and cloned into the pGL3-Promoter vector (Promega), positioned immediately upstream of the SV40 promoter. This candidate regulatory region was identified on the basis of U251-specific H3K27ac ChIP-seq signals. Site-directed mutagenesis was subsequently conducted using the Fast Mutagenesis System (TRANS) to generate both risk (mutant) and non-risk (WT) alleles at each SNP locus. The constructed reporter plasmids were co-transfected with the pRL-TK Renilla luciferase control vector (Promega) into U251 cells via Lipofectamine 3000 reagent (Invitrogen). After 48-h incubation, cells were collected for dual-luciferase measurement using the Dual-Luciferase Reporter Assay System (Promega). Firefly luciferase signals were normalized to Renilla luciferase internal controls for data standardization.
Western blot
Proteins were collected from U251 WT cells and SOX18-KO cells using RIPA buffer (Beyotime) supplemented with a protease inhibitor cocktail. Sample loading was based on the results of the bicinchoninic acid assay. Proteins were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis. Then, they were transferred to polyvinylidene difluoride membranes (Millipore), which were blocked and incubated with SOX18 antibody (Abcam, EPR2837(2), 1:1,000 dilution) or control GAPDH antibodies (Abcam, EPR16891, 1:500 dilution) overnight at 4 °C, followed by secondary antibodies (Abcam, ab288151, 1:2,000 dilution) for 1 h at room temperature. Bands were detected by Image Quant LAS 4000 with an Enhanced Chemiluminescence Kit (Thermo Pierce).
Generation of rs2297440-associated enhancer and SOX18 KOs
For enhancer and SOX18 KO, sgRNAs were designed using an online tool (CRISPick, http://broadinstitute.org/) (Supplementary Table 8). Oligos were synthesized and constructed with the CRISPR–Cas9 KO system following the manufacturer’s instructions. U251 cells were transfected with plasmids containing sgRNAs and plated as single cells in six-well plates. Then, 2 μg ml−1 puromycin was added 48 h after transfection to select stable lines. Medium-containing puromycin was changed every 2 days. Enhancer and SOX18 KO cells were verified by agarose gel electrophoresis and DNA sequencing, and KO efficiency was confirmed by RT-qPCR and western blotting.
Generation of rs2297440-mutated cell lines
First, the rs2297440 allele in the WT U251 cell line was identified by PCR and DNA sequencing. It was then determined that rs2297440 is a risk form (C/C) in WT U251 and LN229 cells. sgRNAs (Supplementary Table 8) for site-specific cleavage of genomic targets were cloned into the pSpCas9n(BB)-2A-Puro (PX462) V2.0 vector (Addgene, #62987). To generate independent single-cell-derived knock-in clones containing non-coding variants, 2.5 μg sgRNA vector, 3 μg double-stranded DNA (dsDNA) donor and the cell suspension were combined and transfected using the Lipofectamine 3000 system according to the manufacturer’s instructions. Then, 2 μg ml−1 puromycin was added for 7 days after transfection to enrich transfected cells, and the resulting clones were used for cell type identification. Genomic DNA was extracted and amplified by specific genotyping primers, and PCR products were sequenced.
Xenograft models and bioluminescence tumour tracking
Athymic nude mice (BALB/c nude background, male, 4 weeks old; Beijing Vital River Laboratory Animal Technology) were randomly divided into five animals per group. The mice were housed under a 12-h light/12-h dark cycle at a temperature of 18–24 °C (65–75 °F) with 40–60% humidity. Luciferase-expressing human glioma cell LN229 (3 × 105 cells suspended in 6 μl PBS) were implanted into the frontal lobes of nude mice using a stereotactic apparatus (68801, REWARD). Tumour growth was examined at 9, 18 and 27 days after implantation using bioluminescence imaging (IVIS spectrum in vivo imaging system, PerkinElmer). Animals were euthanized by cervical dislocation when they displayed any symptoms of continuous discomfort, such as severe hunchback posture, decreased activity, apathy, dragging legs or more than 20% weight loss. The Ethics Committee of Sun Yat-sen University Cancer Center and Animal Research Committee of Sun Yat-sen University Cancer Center approved this study.
Overexpression of SOX18 in KO, point-mutated glioma cells
The SOX18 gene sequence was obtained from the UCSC Genome Browser (http://genome.ucsc.edu/index.html), and primers were designed using the NCBI Primer-BLAST tool (https://www.ncbi.nlm.nih.gov/tools/primer-blast/index.cgi).The primers included appropriate restriction enzyme sites and protective bases, and were synthesized by Sangon Biotech. Sequences for primer are listed in Supplementary Table 8. The corresponding DNA fragments were synthesized, and the corresponding sequences were amplified with high-fidelity enzymes, then cloned into the pcDNA3.1 plasmid. U251 cells were transfected with plasmids containing sgRNAs and plated as single cells in six-well plates. Then, 2 μg ml−1 puromycin was added 48 h after transfection to select stable lines. The medium-containing puromycin was changed every 2–3 days. After collecting RNA from the cells, reverse transcription and RT-qPCR were performed to detect the expression of SOX18.
CRISPRa of MEIS1 in glioma
As for overexpression of MEIS1 in glioma cells in situ, three gRNAs for endogenous gene repression were selected to bind within −50 to +200 bp around the gene transcription start site (TSS), unless a different position was specified. Sequences for gRNAs are listed in Supplementary Table 8. The U251 cells stably expressing dCas9–VP64 were transfected with lentivirus expressing each gRNA; 48 h later, the medium was replaced with fresh medium with 2 μg ml−1 puromycin for 7 days. The medium-containing puromycin was changed every 2–3 days. After collecting RNA from the cells, reverse transcription and RT-qPCR were performed to detect the expression of MEIS1 and SOX18.
MTT and colony formation assays
For the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay, 2 × 103 cells were seeded in 96-well plates for 12 h (as day 0), 24 h (1 day), 48 h (2 days), 72 h (3 days) or 96 h (4 days). After cell culture, 20 μl of 5 mg ml−1 MTT (Sigma-Aldrich) was added to each well and incubated for 4 h at 37 °C. The plate was then gently shaken 30 s on a plate shaker at room temperature, and the optical density (OD) was measured at 490 nm. For colony formation assays, 500 cells were seeded into six-well plates for 10 days, stained with crystal violet and washed three times with PBS. The number of cell clones was determined using ImageJ59.
GSC sphere formation assays
A total of 0.5 × 104 U251 or LN229 cells were cultured in serum-free neural stem cell medium consisting of DMEM–F12 (Gibco) supplemented with 20 ng ml−1 recombinant human epidermal growth factor (MCE), 20 ng ml−1 recombinant human basic fibroblast growth factor (MCE), 1× B27 supplement (Gibco), 1× GlutaMAX (Gibco) and 1% penicillin–streptomycin (Gibco). Cells were maintained in ultra-low-attachment six-well flasks (Corning) at 37 °C in a humidified 5% CO2 incubator. After 7 days, the spheres with a diameter greater than 80 μm were collected and digested using StemPro Accutase (Thermo Fisher) for 5 min at 37 °C, followed by filtration through a 40-μm cell strainer to remove cell aggregates. Viable cells were counted using trypan blue exclusion and seeded at a density of 0.5 × 104 cells per well in six-well ultra-low attachment plates (Corning) with 200 μl of complete DMEM–F12 (Gibco) medium per well. Cells were cultured for 7 days, with half-medium changes every 2 days to replenish growth factors. GSC spheres with a diameter greater than 80 μm in each well were counted.
HiChIP data processing
HiChIP paired-end reads were aligned to the human genome (hg38) using the HiC-Pro pipeline60. The default settings were used to assign reads to MboI restriction fragments, filter out invalid interactions and generate binned interaction matrices. Valid read pairs of individual samples were merged for each cell line. The resulting interaction matrices were normalized using the Knight–Ruiz matrix balancing method. The normalized interaction maps were visualized using Juicebox (v2.20.00)61. Interaction loop calling was performed using hichipper62. We set a false discovery rate threshold of 0.1 and required that each loop be supported by at least two paired-end tags to ensure the reliability of the results. Diffloop (v1.3.4) was used to systematically assess changes in enhancer loops between glioma and normal brain samples63. Glioma-specific loops were defined as those exhibiting significant signal changes in glioma compared with normal brain, with a P value <0.1 and a log2(fold change) >1. The genomic coordinates for pro-tumour enhancers and glioma-specific loop anchors were intersected with bedtools64. For glioma-specific loops in which one anchor overlapped a pro-tumour enhancer, the gene located within 2 kb the either anchor was assigned as a putative target.
RNA-seq
Total RNA was extracted from cells using TRIzol reagent (Life Technologies), and DNA was removed with DNase I (Thermo Scientific). Total RNA (5 μg) was used for RNA-seq library preparation. Hisat2 (v2.2.1)65 was used to map reads to the hg38 genome. Samtools (v1.18) was used to process the alignment bam files. The number of reads mapped to genes was determined with htseq-count66. Genes with a mean count number <2.5 were filtered out. DESeq2 was used to analyse differentially expressed genes. Genes with expression log2(fold change) of ≥1 or ≤−1 and a P < 0.05 were considered to show significantly changed expression. DAVID (https://david.ncifcrf.gov/tools.jsp) was used for GO analysis.
Motif analysis
The JASPAR database67 was used to predict transcription factor binding to risk and non-risk alleles. DNA sequences 30 bp upstream and downstream of the rs2297440 variant were extracted, and the ‘SCAN’ option in JASPAR was used to predict transcription factor binding. Binding scores were compared between the T and C allele forms.
Statistics and reproducibility
Statistical analyses of the data were conducted as described in the respective figure legends. All results from representative experiments were collected at least three times independently. No statistical methods were used to predetermine sample sizes. No data were excluded from the analysis. Data distribution was assumed to be normal, but this was not formally tested. Data collection and analysis were not performed blind to the conditions of the experiments.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
HiChIP, RNA-seq and ChIP-seq data that support the findings of this study have been deposited in the Genome Sequence Archive for Human (https://ngdc.cncb.ac.cn/gsa-human/) under accession codes HRA006867 and HRA007675 and can be provided for scientific research only upon request owing to concerns about human patient privacy. Previously published data that were reanalysed here are available under accession codes GSE147672 (ref. 52) and GSE145644 (ref. 68). All other data supporting the findings of this study are available from the corresponding author on reasonable request. Unless stated otherwise, the experiments were not randomized. Source data are provided with this paper.
References
Panditharatna, E. et al. BAF complex maintains glioma stem cells in pediatric H3K27M glioma. Cancer Discov. 12, 2880–2905 (2022).
Chen, J. et al. circPTN sponges miR-145-5p/miR-330-5p to promote proliferation and stemness in glioma. J. Exp. Clin. Cancer Res. 38, 398 (2019).
Zhao, B. et al. Common genetic variation influencing human white matter microstructure. Science 372, eabf3736 (2021).
Chaligne, R. et al. Epigenetic encoding, heritability and plasticity of glioma transcriptional cell states. Nat. Genet. 53, 1469–1479 (2021).
Ostrom, Q. T. et al. The epidemiology of glioma in adults: a “state of the science” review. Neuro Oncol. 16, 896–913 (2014).
Ostrom, Q. T. et al. Risk factors for childhood and adult primary brain tumors. Neuro Oncol. 21, 1357–1375 (2019).
Weller, M. et al. Glioma. Nat. Rev. Dis. Primers 10, 33 (2024).
Verhaak, R. G. et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17, 98–110 (2010).
Neftel, C. et al. An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell 178, 835–849 (2019).
Alpen, K. et al. Australian genome-wide association study confirms higher female risk for adult glioma associated with variants in the region of CCDC26. Neuro Oncol. 25, 1355–1365 (2023).
Lindström, S. et al. Genome-wide analyses characterize shared heritability among cancers and identify novel cancer susceptibility regions. J. Natl Cancer Inst. 115, 712–732 (2023).
Melin, B. S. et al. Genome-wide association study of glioma subtypes identifies specific differences in genetic susceptibility to glioblastoma and non-glioblastoma tumors. Nat. Genet. 49, 789–794 (2017).
Saunders, C. N., Cornish, A. J., Kinnersley, B., Law, P. J. & Houlston, R. S. Searching for causal relationships of glioma: a phenome-wide Mendelian randomisation study. Br. J. Cancer 124, 447–454 (2021).
Ji, X. et al. 3D chromosome regulatory landscape of human pluripotent cells. Cell Stem Cell 18, 262–275 (2016).
van Steensel, B. & Furlong, E. E. M. The role of transcription in shaping the spatial organization of the genome. Nat. Rev. Mol. Cell Biol. 20, 327–337 (2019).
Schoenfelder, S. & Fraser, P. Long-range enhancer-promoter contacts in gene expression control. Nat. Rev. Genet. 20, 437–455 (2019).
Stadhouders, R., Filion, G. J. & Graf, T. Transcription factors and 3D genome conformation in cell-fate decisions. Nature 569, 345–354 (2019).
Wang, J. et al. Epigenomic landscape and 3D genome structure in pediatric high-grade glioma. Sci. Adv. 7, eabg4126 (2021).
Jessa, S. et al. K27M in canonical and noncanonical H3 variants occurs in distinct oligodendroglial cell lineages in brain midline gliomas. Nat. Genet. 54, 1865–1880 (2022).
Mumbach, M. R. et al. HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nat. Methods 13, 919–922 (2016).
Sabari, B. R. et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science 361, eaar3958 (2018).
Andersson, R. & Sandelin, A. Determinants of enhancer and promoter activities of regulatory elements. Nat. Rev. Genet. 21, 71–87 (2020).
Yeo, N. C. et al. An enhanced CRISPR repressor for targeted mammalian gene regulation. Nat. Methods 15, 611–616 (2018).
Gwynne, W. D. et al. Cancer-selective metabolic vulnerabilities in MYC-amplified medulloblastoma. Cancer Cell 40, 1488–1502.e1487 (2022).
Yanchus, C. et al. A noncoding single-nucleotide polymorphism at 8q24 drives IDH1-mutant glioma formation. Science 378, 68–78 (2022).
Ma, J., Zhou, W., Yuan, Y., Wang, B. & Meng, X. PSMD12 interacts with CDKN3 and facilitates pancreatic cancer progression. Cancer Gene Ther. 30, 1072–1083 (2023).
Wang, M. et al. Therapeutic blocking of VEGF binding to neuropilin-2 diminishes PD-L1 expression to activate antitumor immunity in prostate cancer. Sci. Transl. Med. 15, eade5855 (2023).
Polavaram, N. S. et al. Tumor- and osteoclast-derived NRP2 in prostate cancer bone metastases. Bone Res. 9, 24 (2021).
Rodón, L. et al. Active CREB1 promotes a malignant TGFβ2 autocrine loop in glioblastoma. Cancer Discov. 4, 1230–1241 (2014).
Yao, M., Gu, Y., Yang, Z., Zhong, K. & Chen, Z. MEIS1 and its potential as a cancer therapeutic target (Review). Int. J. Mol. Med. 48, 181 (2021).
Barbosa, K. et al. Transcriptional control of leukemogenesis by the chromatin reader SGF29. Blood 143, 697–712 (2023).
Qi, T. et al. Function and regulation of the PEA3 subfamily of ETS transcription factors in cancer. Am. J. Cancer Res. 10, 3083–3105 (2020).
Chuang, L. S. & Ito, Y. RUNX3 is multifunctional in carcinogenesis of multiple solid tumors. Oncogene 29, 2605–2615 (2010).
Krishnan, V. The RUNX family of proteins, DNA repair, and cancer. Cells 12, 1106 (2023).
Krishnan, V. et al. TGFβ promotes genomic instability after loss of RUNX3. Cancer Res. 78, 88–102 (2018).
Moreno, E. et al. E2F7 is a potent inhibitor of liver tumor growth in adult mice. Hepatology 73, 303–317 (2021).
Mitxelena, J. et al. An E2F7-dependent transcriptional program modulates DNA damage repair and genomic stability. Nucleic Acids Res. 46, 4546–4559 (2018).
Borgenvik, A. et al. Dormant SOX9-positive cells facilitate MYC-driven recurrence of medulloblastoma. Cancer Res. 82, 4586–4603 (2022).
Lin, W. et al. VMP1, a novel prognostic biomarker, contributes to glioma development by regulating autophagy. J. Neuroinflammation 18, 165 (2021).
Hagège, H. et al. Quantitative analysis of chromosome conformation capture assays (3C-qPCR). Nat. Protoc. 2, 1722–1733 (2007).
Takemoto, Y., Furuta, M., Sato, M., Kubo, M. & Hashimoto, Y. Isolation and characterization of a novel HS1 SH3 domain binding protein, HS1BP3. Int. Immunol. 11, 1957–1964 (1999).
Saunders, C. N. et al. Relationship between genetically determined telomere length and glioma risk. Neuro Oncol. 24, 171–181 (2022).
Goel, V. Y., Huseyin, M. K. & Hansen, A. S. Region capture Micro-C reveals coalescence of enhancers and promoters into nested microcompartments. Nat. Genet. 55, 1048–1056 (2023).
Javierre, B. M. et al. Lineage-specific genome architecture links enhancers and non-coding disease variants to target gene promoters. Cell 167, 1369–1384 (2016).
Qian, Y. et al. The prostate cancer risk variant rs55958994 regulates multiple gene expression through extreme long-range chromatin interaction to control tumor progression. Sci. Adv. 5, eaaw6710 (2019).
Yoshida, H. et al. The cis-regulatory atlas of the mouse immune system. Cell 176, 897–912 (2019).
Hung, K. L. et al. ecDNA hubs drive cooperative intermolecular oncogene expression. Nature 600, 731–736 (2021).
Hu, Y. et al. Lineage-specific 3D genome organization is assembled at multiple scales by IKAROS. Cell 186, 5269–5289 (2023).
Xu, J. et al. Subtype-specific 3D genome alteration in acute myeloid leukaemia. Nature 611, 387–398 (2022).
Mathur, R. et al. Glioblastoma evolution and heterogeneity from a 3D whole-tumor perspective. Cell 187, 446–463 (2024).
Liu, Y. et al. Polymorphisms of LIG4, BTBD2, HMGA2, and RTEL1 genes involved in the double-strand break repair pathway predict glioblastoma survival. J. Clin. Oncol. 28, 2467–2474 (2010).
Corces, M. R. et al. Single-cell epigenomic analyses implicate candidate causal variants at inherited risk loci for Alzheimer’s and Parkinson’s diseases. Nat. Genet. 52, 1158–1168 (2020).
Zelenka, T. & Spilianakis, C. HiChIP and Hi-C protocol optimized for primary murine T cells. Methods Protoc. 4, 49 (2021).
Joung, J. et al. Genome-scale CRISPR–Cas9 knockout and transcriptional activation screening. Nat. Protoc. 12, 828–863 (2017).
Diao, Y. et al. A tiling-deletion-based genetic screen for cis-regulatory element identification in mammalian cells. Nat. Methods 14, 629–635 (2017).
Feng, J., Liu, T., Qin, B., Zhang, Y. & Liu, X. S. Identifying ChIP-seq enrichment using MACS. Nat. Protoc. 7, 1728–1740 (2012).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137 (2008).
McKenna, A. & Shendure, J. FlashFry: a fast and flexible tool for large-scale CRISPR target design. BMC Biol. 16, 74 (2018).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259 (2015).
Rowley, M. J. & Corces, V. G. Minute-made data analysis: tools for rapid interrogation of Hi-C contacts. Mol. Cell 64, 9–11 (2016).
Lareau, C. A. & Aryee, M. J. hichipper: a preprocessing pipeline for calling DNA loops from HiChIP data. Nat. Methods 15, 155–156 (2018).
Lareau, C. A. & Aryee, M. J. diffloop: a computational framework for identifying and analyzing differential DNA loops from sequencing data. Bioinformatics 34, 672–674 (2018).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Anders, S., Pyl, P. T. & Huber, W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015).
Castro-Mondragon, J. A. et al. JASPAR 2022: the 9th release of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 50, D165–D173 (2022).
Xu, L. et al. Topography of transcriptionally active chromatin in glioblastoma. Sci. Adv. 7, eabd4676 (2021).
Acknowledgements
We thank all members of the Lu Lab for their scientific discussions and helpful comments. We also thank E. Lamar for revising our manuscript. The work was supported by the National Natural Science Foundation of China (grant no. 32130018), the National Key R&D Program of China (grant no. 2020YFA803700), the Postdoctoral Fellowship Program of CPSF (grant nos. GZC20233213 to J.B. and GZC20233231 to W.M.) and the China Postdoctoral Science Foundation (grant no. 2023M744084).
Author information
Authors and Affiliations
Contributions
J.B., N.Z. and W.L. designed the research; J.B., M.L., Y.S., Q.X., S.F., W.W., T.S., Y.Z., J.L., R.L., B.C., X.H., P.L., Z.Z., J.S., L.Z. and G.S. performed the experiments; J.B., M.L., W.M., N.Z. and W.L. analysed the data; and J.B., W.M., G.S., N.Z. and W.L. wrote the manuscript with input and approval from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Cell Biology thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Uncovers pro-tumor enhancer connectome in glioma by CRISPRi screening and H3K27ac HiChIP.
a. Comparison of CRISPRi screen replicates at different time points. The heatmap represents the Pearson correlation of normalized sgRNA counts between Day 0 (d0) and Day 21 (d21) in the CRISPRi screen. b. The classification of pro-tumor enhancers in glioma. c. Schematic outline of H3K27ac HiChIP experiments in glioma tissues and cell lines. d. Enrichment of normal brain-specific loop motifs. e. TOP: Confirmation of 3C-PCR detected contacting genomic fragments for MYC and E34235 locus; Bottom: Confirmation of 3C-PCR detected contacting genomic fragments for VMP1 and E15788 locus. f. Enrichment of target genes interacting with pro-tumor enhancer E19383 in glioma cells, as identified by CRISPRi screening. Data represent means ± SD from four independent biological replicates. Statistical significance was determined by two-sided t-test. ** P < 0.01, *** P < 0.001, **** P < 0.0001, N.S, not significant. g. Overall survival of cases stratified by E19383 target gene signature (LAPTM4A, SDC1, PUM2, and RHOB). Statistical analysis was performed using log-rank tests.
Extended Data Fig. 2 Profiles of enhancers associated with risk SNP.
a. CRISPR interference with risk SNP-associated enhancers significantly inhibits glioma cell proliferation. Data represent means ± SD from three independent biological replicates. Statistical significance was determined by two-sided t-test, ** P < 0.01, *** P < 0.001, N.S, not significant. b, c. H3K27ac HiChIP interaction maps at 5 Kb resolution, centered on the rs2297440 (b) and rs3851634 (c) loci, respectively, in glioma tissue, glioma cells, and normal brain.
Extended Data Fig. 3 Functional characterization of the rs2297440-containing enhancer in glioma cells.
a. CRISPR/Cas9-mediated enhancer knockout cells (KO1 and KO2) with deletions of 1317 bp sequences in U251 and LN229, respectively. b. Scatter plot of differential expression of genes between enhancer-KO and WT U251 cells. c. Colony-formation assay in WT and enhancer KO (eKO) U251 cells. d. Gene ontology (GO) analysis of down-regulated genes in enhancer-KO cell compared to WT U251 cell. e. Gene ontology (GO) analysis of up-regulated genes in in enhancer-KO cell compared to WT U251 cell. f. Colony formation assay in enhancer knockout U251 cells: Top, empty vector overexpression; Bottom, SOX18 overexpression.
Extended Data Fig. 4 SOX18 is a functional gene in glioma cells.
a, b. Glioma stem cell (GSC) sphere models grown in serum-free conditions to assess proliferation of U251 enhancer-KO cells overexpressing SOX18, Scale bar, 100 μm. Data represent means ± SD from three independent biological replicates. Statistical significance was determined by two-sided t-test, **** p < 0.0001. c. Schematic representation of the sgRNA design for SOX18 knockout (top) and DNA sequencing results confirming SOX18 knockout (bottom). d. Western blot analysis verifying SOX18 knockout, with GAPDH serving as a loading control. e. Colony formation assay results comparing wild-type and SOX18 knockout U251 cells.
Extended Data Fig. 5 mutation suppresses glioma progression through SOX18-dependent transcriptional regulation.
rs2297440a. DNA sequencing of rs2297440 in U251-WT and SNP edited cells. b. Colony formation assay in wild-type (WT) U251 cells containing the risk allele (C/C) and edited cells (MUT1 and MUT2) containing the non-risk allele (T/T). c. The relative mRNA expression of RTEL1 in U251 cells containing the risk allele and non-risk allele. d. Scatter plot showing differentially expressed genes between cells containing the risk allele and non-risk allele. e, f. Gene Ontology (GO) analysis of down-regulated genes (e) and up-regulated genes (f) in cells containing the non-risk allele compared to cells containing the risk allele. g. Detection of SOX18 expression in SNP mutated U251 cells with exogenous overexpression plasmid. h. Colony formation assay after overexpression of empty vector and SOX18 in cells containing the risk allele and non-risk allele. i. DNA sequencing of rs2297440 in LN229-WT and SNP mutated cells. j, k. Glioma stem cell sphere models (GSCs) grown in serum-free conditions to assess proliferation of rs2297440 mutant cells, with or without overexpressing SOX18. Data represent means ± SD from three independent biological replicates. Statistical significance was determined by two-sided t-test. *** P < 0.001, **** P < 0.0001, N.S, not significant, scale bar, 100 μm.
Supplementary information
Supplementary Tables (download XLSX )
Supplementary Tables 1–8.
Source data
Statistical Source Data for Figures and Extended Data Figures (download XLSX )
Statistical source data.
Unprocessed Blots for Extended Data Figures (download PDF )
Unprocessed western blots and/or gels.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Bi, J., Mo, W., Liu, M. et al. Systematic decoding of functional enhancer connectomes and risk variants in human glioma. Nat Cell Biol 27, 1838–1847 (2025). https://doi.org/10.1038/s41556-025-01737-3
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41556-025-01737-3
This article is cited by
-
Deciphering glioma susceptibility
Nature Cell Biology (2025)
