
Abstract
Directed evolution methods face trade-offs between the control of discrete approaches and the throughput of modern continuous systems. Here, we engineered a method called lytic selection and evolution (LySE) for near-continuous evolution of bacterial gene clusters while maintaining discrete checkpoints. We developed a hypermutagenic T7 DNA polymerase variant fused to a dual adenine-cytosine deaminase to install all possible transition mutations at similar frequencies. By relieving pressure from maintaining genome fidelity, we obtained mutation rates of 3.82 × 10−5 substitutions per base. For biocontainment, the T7 DNA polymerase was encoded on an accessory plasmid, while the target gene cluster was encoded on a T7 DNA polymerase-lacking T7 phagemid. Alternating cycles of lysis and transduction enable selective replication and mutagenesis of target genes, while off-target genomic mutations are discarded. LySE evolved a 25-fold increase in tetA-encoded tigecycline resistance in 5 cycles, and a 50.9% increase in endpoint biomass of a bacterial strain that uses the polyethylene terephthalate monomer, ethylene glycol, as its sole carbon source. Our method balances speed and control for directed bacterial evolution.
Similar content being viewed by others

Main
Directed evolution enables discovery of new protein variants and is an indispensable tool for bioengineering1. Classical directed evolution platforms based on discrete cycles of mutagenesis, selection and amplification have produced remarkable successes across diverse applications2. However, these approaches are time consuming to iterate, difficult to scale up and limited in their ability to explore the full sequence space of a gene of interest (GOI)3. To overcome these limitations, continuous evolution systems, such as phage-assisted continuous evolution (PACE)4 and orthogonal replication5,6,7,8, have been developed to selectively mutagenize target GOIs inside cells, enabling evolutionary cycles to run without manually initiated steps9. Although these platforms enable unprecedented speed, they sacrifice control over evolutionary trajectories. PACE requires coupling of phage replication to production of protein pIII, which restricts the flexibility of selection strategies10. With basal mutation rates, orthogonal replication still risks accumulating genomic off-target mutations that make non-GOI genes contribute to the phenotype, which complicates the attribution of phenotypic effects to specific genetic changes11. ‘Cheater mutations’ can bypass selection pressure without altering the GOI, or disable the selection system itself, especially in biosensor-based selection12. These result in a complicated troubleshooting process when evolutionary runs fail, limiting the accessibility of these techniques to experienced researchers10,13.
The enduring popularity of classical directed evolution methods, despite newer continuous alternatives, testifies to the value of controllability in protein engineering workflows13,14,15,16,17. Classical approaches, and the manually performed phage-assisted non-continuous evolution (PANCE)18, offer discrete checkpoints between evolutionary rounds, enabling researchers to analyse intermediate results, adjust selection parameters and prevent the accumulation of off-target mutations in the host genome. An ideal evolution system would therefore combine this controllability with the speed and throughput advantages of continuous evolution platforms. Here we developed lytic selection and evolution (LySE), a robust T7 phage-based system that bridges classical discrete and continuous directed evolution paradigms. LySE uses a T7 phagemid containing the gene cluster of interest (GCOI), which is maintained as a stable plasmid during the cell cycle but packaged into the phage capsid upon infection19. The system selectively replicates and mutagenizes the GCOI phagemid while carrying it through alternating cycles of lysis and transduction in Escherichia coli. The process is facilitated by simple mixing of phage lysates and cell cultures and monitored by changes in cell density, improving controllability and accessibility to new users. Each LySE cycle completely eliminates the E. coli culture after selection, effectively removing all off-target mutations in the host genome. We engineered a hypermutagenic T7 DNA polymerase (T7 DNAP) that achieves 3.82 × 10−5 substitutions per base—160,000× higher than the genomic mutation rate of the host E. coli. The mutational spectrum was assessed by next-generation sequencing (NGS) and we found an even incorporation of all transition (purine-to-purine and pyrimidine-to-pyrimidine) mutations. Selection can be done by coupling GCOI to cellular fitness or with high-throughput screening methods, such as cell sorting20. Most importantly, LySE enables selection of slow-manifesting metabolic functions by coupling large gene cluster expression to host fitness, with a capacity of up to 39 kilobases (kb). Such metabolic pathways are unsuitable for evolution through PACE and they are also too large for in vitro diversification. We show this by evolving a pathway that allows the host bacterium to use and grow on the polyethylene terephthalate (PET) monomer, ethylene glycol (EG), as its sole carbon source. LySE combines continuous evolution with discrete evolution cycles, enabling directed evolution of large gene clusters while maintaining stringent control over mutational trajectories.
Results
Evolution through a lytic cycle
Bacteriophage T7 is a lytic phage that infects E. coli, replicates its 40-kb genome and lyses the host cell within 17 minutes, releasing approximately 180 progeny phages21,22 (Extended Data Fig. 1a). We exploited the ability of the T7 phage for rapid multiplication and distribution of large genetic material to develop a system for near-continuous, accelerated evolution of large GCOIs. First, we engineered a T7 phage variant lacking the T7 DNAP, by in vitro assembly of the complete phage genome except gene gp5, and introducing it into a cell-free extract from E. coli (Supplementary Data 1 and Supplementary Table 1). This process, known as ‘rebooting’, allowed the cell-free system to express the phage genes, assemble a new capsid and produce infectious phage particles23. This phage, T7∆DNAP, efficiently propagates only in hosts that carry an accessory plasmid (AP) expressing the T7 DNAP under a T7 promoter (Extended Data Fig. 1b–d, Supplementary Data 2 and Supplementary Table 2). The absence of phage propagation without AP shows the strict biocontainment of the system.
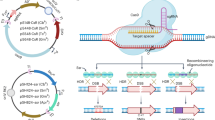
Phage T7 can replicate, package and transduce phagemids: circular plasmids containing the T7 origin of replication (T7 ori), a T7 packaging signal (pac site), a T7 terminal repeat and a host origin of choice24. Importantly, the phagemid is replicated by the host replication machinery during the cell cycle and by the phage replication machinery during phage infection. We constructed phagemids of different sizes with a p15a host origin and showed that they were packaged and transduced with similar efficiency by phage T7∆DNAP in cells containing the AP (Fig. 1a, Extended Data Fig. 1e,f and Supplementary Data 3).

a, Controlled replication and phage packaging. T7 DNAP expression from the AP is tightly regulated under a T7 promoter during the cell cycle. Upon T7 phage infection, the T7 RNA polymerase (T7 RNAP) induces expression of error-prone T7 DNAP, which replicates the phagemid (PM) that carries the GCOI, for subsequent packaging and transduction. b, Schematic of the LySE workflow. At a high MOI, the system enters a lytic phase where the GCOI, carried on a phagemid, undergoes error-prone replication by error-prone T7 DNAP expressed from the AP. Mutated variants are packaged into T7 phage particles and released. With high error rates, it is possible to have multiple mutations in one phagemid or different mutations on separate phagemids. Uncontrolled lytic phage propagation is avoided as the error-prone T7 DNAP severely compromises replication fidelity and makes phage lysis dependent on a high MOI. During subsequent transduction at a low MOI, these mutated phagemids are introduced into new host cells, enabling fitness-based selection through cell growth. Faded cells represent cells that carry GCOI mutants of reduced fitness, which become depleted from the pool. The evolved gene cluster pool is then cycled back to the lytic phase by reintroducing phage T7∆DNAP. c, Temporal representation of a typical LySE campaign, enabling day-scale evolution cycles through exploitation of the short life cycle of the T7 phage. d, LySE is a hybrid system between discrete and continuous evolution. Transduction replaces transformation to enable seamless transition between mutagenesis and selection, a characteristic of continuous evolution. Cheater mutations are a problem in orthogonal replication systems because the GOI replicates with the host. LySE creates discrete checkpoints to prevent cheater accumulation by refreshing the host in each cycle. Star symbols on plasmids indicate mutations. host ori, E. coli host origin of replication. SP, selection plasmid; O-DNAP, orthogonal DNA polymerase. Figure created in BioRender; Fredens, J. https://biorender.com/lmo9gzz (2026).
When the AP is equipped with an error-prone T7 DNAP variant, this system will enable cyclic evolution, where the phagemid undergoes error-prone replication and packaging during a lytic phase (high multiplicity of infection (MOI), the phage:cell ratio), followed by transduction to fresh hosts for growth and selection during a cellular phase (low MOI) (Fig. 1b). Uncontrolled lytic phage propagation is avoided as the error-prone T7 DNAP severely compromises replication fidelity and makes phage lysis dependent on a high MOI25. During the cellular phase, T7 DNAP expression will cease and host DNA polymerase III maintains the phagemid with high fidelity. The selection of improved GCOI variants is achieved by linking phagemid-encoded functions to host cell fitness during the cellular phase. The short lysis time and large burst size of T7 facilitate quick turnover of large genetic pools, thus reducing the time for directed evolution from days to hours (Fig. 1c). The workflow requires only simple manual mixing of phage lysates and cell cultures using standard laboratory tools, making LySE readily portable and accessible to most laboratories regardless of experience. This fundamental simplicity also enables straightforward scaling and automation for high-throughput campaigns. Using T7 as an efficient gene shuttle between cells, LySE creates a hybrid system between continuous and discrete evolution by seamlessly transitioning between mutagenesis and selection while preserving discrete checkpoints (Fig. 1d).
Engineering hypermutagenic T7 DNAPs
We engineered and tested several error-prone T7 DNAP variants to drive targeted mutagenesis of phagemid-encoded GCOIs (Supplementary Table 3). By targeting error-prone replication to the phagemid and not the host genome, the GCOI can undergo accelerated evolution at hypermutagenic rates that would be toxic to the host. Our design strategy incorporated complementary error-inducing mechanisms for additive effects (Fig. 2a).

a, AlphaFold3-predicted67 structure of variant v8, highlighting key engineered modifications: v1 (S399T), thumb domain relaxation affecting template minor groove interactions; v2 (T523R), fingers domain modification proximal to active site; v4 (D5A, E7A, Y64C and F120L), exonuclease domain inactivation; v7–9, fusion with TadA-8e adenosine deaminase or TadDE general deaminase enabling concurrent DNA deamination during replication. b, Mutational frequencies of engineered T7 DNAP variants measured by a fluctuation assay using a phagemid-encoded chloramphenicol resistance gene containing an internal stop codon. Wild-type T7 DNAP (WT), mutant T7 DNAPs (v1–v4), T7 DNAP v2.4 fused to TadA-8e (v7), T7 DNAP v4 fused to TadA-8e (v8) and T7 DNAP v4 fused to TadDE (v9) are shown. Data are shown as mean ± s.d. of n = 3 independent experiments. c, Snapshots from molecular dynamics simulations of T7 DNAP (PDB: 1T7P): wild type (top) and T523R variant mutated in silico (bottom) with A–G nucleotide mismatch. Hydrogens are not shown. Distances are measured in Ångström. d, Mutational frequencies of T7 DNAP variants determined through Illumina NGS using a 39-kb BAC phagemid. Mismatches were corrected for sequencing errors by subtracting wild-type mismatches. Data points represent mismatches across 39 kb binned into 20 equal-width bins. e, Substitution frequencies and ratios of A:T → G:C and C:G → T:A transitions for v8 and v9 variants derived from NGS data. Data points represent substitution frequencies for the respective transition types across 39 kb binned into 20 equal-width bins (Illumina sequencing, mean coverage >14,000× per base pair). Centre line represents the mean. f, Mutational spectra heat maps of LySE v8 and v9 showing the frequency of each substitution type calculated as a percentage of the total number of mutations observed. Rows represent the reference base; columns represent the mutated base. The sum of all values in the matrix equals 100%. g, Distribution of LySE v8 and v9 substitution frequencies across a 39-kb BAC phagemid (mean coverage = 14,098× per base pair). Diagram in g created in BioRender; Fredens, J. https://biorender.com/jomazxk (2026).
Mutation rates were quantified by fluctuation analysis using a chloramphenicol resistance gene (CmR) containing a premature stop codon (Q38TAG)26,27. After one generation of phagemid replication and transduction, we measured the frequency of cells acquiring chloramphenicol resistance through point mutations converting the TAG to sense codons. We calculated apparent mutation rates in substitutions per base pair per generation (s.p.b.) by correcting for multiple phagemid copies per phage particle.
We began with a previously characterized error-prone variant, T7 DNAP v1, containing mutations in both the thumb domain (S399T) and exonuclease domain (Y64C and F120L)25. S399T has been proposed to increase error rates by disturbing the nucleic acid binding cleft, while Y64C and F120L probably reduce proofreading activity. This variant showed a 10.5-fold increased error rate compared with wild-type T7 DNAP (Fig. 2b), in line with previous reports.
To further enhance mutagenesis, we used homology-guided design based on fidelity-reduced E. coli DNA polymerase I variants28. We identified several potential mutations in the fingers domain of T7 DNAP (L479N, H506Y, T523R and P560H), some of which are positioned near the polymerase active site. Through systematic experimental testing, we determined T523R significantly increased error rates and, when combined with v1 (v2.4: Y64C, F120L, S399T and T523R), achieved error rates 70-fold higher than wild type (Extended Data Fig. 2a). Molecular dynamics simulations of the T7 DNAP crystal structure29 revealed that T523R significantly alters nucleotide positioning in the active site (Fig. 2c and Supplementary Data 4 and 5). An incoming triphosphate nucleotide is pulled deeper into the active site in the presence of the arginine substitution. Thus, we propose that the arginine stabilizes incorrect base pairing.
Complete inactivation of the exonuclease function through D5A and E7A mutations further reduced replication fidelity30. Incorporating D5A and E7A into T7 DNAP v2.4 generated variant v4 with a 1.7-fold increased error rate (Fig. 2b). In parallel, we explored error-prone replication by fusing T7 DNAP to DNA deaminases for concurrent deamination during replication, analogous to previous work on RNA polymerase–deaminase fusions31,32. We fused the adenosine deaminase TadA-8e to the amino terminus of wild-type T7 DNAP with linkers of varying lengths33 (v5.1–5.5; Supplementary Table 3). A 24-residue linker (v5.3) resulted in the highest error rate of 1.84 × 10−5, comparable to the rate of v2.4 (Extended Data Fig. 2b). The cytosine deaminase PmCDA1 fusion (v6.1) proved to be less effective for mutagenesis compared with TadA-8e (v5.3).
Finally, we combined the two complementary approaches by fusing TadA-8e to the most error-prone T7 DNAP variants with mutations in the thumb, fingers and exonuclease domain (v2.4) and exonuclease inactivation (v4), creating v7 and v8, respectively. Unexpectedly, v7 showed a reduced error rate, indicating potential incompatibility between the error-prone mechanisms. T7 DNAP v8, however, increased error rates by 1.78-fold compared with v4 (Fig. 2b), achieving an error rate of 3.82 × 10−5 s.p.b.—160,000× higher than the determined genomic mutation rate in E. coli (2.39 ± 1.10 × 10−10 s.p.b.). Variant v8 had a negligible impact on host growth kinetics (Extended Data Fig. 3a), showing tight control of error-prone replication. Given that v8 uses an adenosine deaminase, which catalyses adenosine-to-inosine deamination, resulting in A:T → G:C transitions during DNA replication, we anticipated imbalances in the mutational spectrum and replaced TadA-8e with a recently engineered dual adenine-cytosine deaminase, TadDE, to create v9. TadDE is capable of performing both adenosine and cytidine deamination34, facilitating A:T → G:C and C:G → T:A transitions, respectively (Fig. 2a). T7 DNAP v9 had a slightly lower but comparable error rate as v8 at 2.45 × 10−5 s.p.b. (Fig. 2b and Supplementary Data 6).
To validate fluctuation analysis error rates and to determine the mutational spectrum, we performed Illumina NGS on bacterial artificial chromosome (BAC) phagemids mutated by the engineered T7 DNAP variants. We first assembled a 39-kb T7 phagemid with a BAC origin by homologous recombination in Saccharomyces cerevisiae (Supplementary Data 7). After performing one generation of LySE with this BAC phagemid, nanopore sequencing of transductants confirmed that the intact vector was successfully transferred into fresh host cells (Supplementary Data 8). We then performed NGS to analyse the mutational profiles. To correct for sequencing errors, the observed fraction of mismatches at each nucleotide position in samples with the wild-type T7 DNAP was subtracted from the fraction of mutations for engineered T7 DNAP variants. NGS-determined error rates were slightly higher, but comparable to fluctuation analysis error rates, at 7.21 × 10−5 s.p.b. and 5.66 × 10−5 s.p.b. for v8 and v9, respectively (Fig. 2d).
Analysis of the mutational spectra revealed that DNAP v4 showed an intrinsic bias for C → T transitions compared with A:T → G:C transitions (Extended Data Fig. 4a,b). Increasing A:T → G:C transitions by TadA-8e fusion (v8) complements this bias to achieve a balanced A:T → G:C to C:G → T:A transitions ratio of 1.00:0.97 (Fig. 2e). In contrast, installing TadDE in v9 reduced adenosine deamination activity, reverting to a biased ratio of 1.00:1.61 (Fig. 2e,f). Both v8 and v9 showed clear bias towards transition mutations (67.9–69.2%) over transversion mutations (Fig. 2f). The 2 variants showed a uniform distribution of mutations and base transitions throughout the entire 39-kb BAC phagemid, while maintaining the biologically relevant preference for transitions over transversions35 (Fig. 2g and Extended Data Fig. 4c). Taken together, LySE presents a notable improvement over existing continuous evolution tools by enabling the potential evolution of large gene clusters up to 40 kb, equivalent to the size of the complete T7 genome19.
Multiplicity tuning for controlled lysis
Next we assessed how our error-prone T7 DNAP variants affect T7 phage replication. E. coli cultures harbouring T7 DNAP variants were infected with T7ΔDNAP at varying MOIs while we monitored cell density over time (Fig. 3a). Wild-type T7 DNAP caused efficient cell lysis independent of the initial MOI, due to rapid phage propagation21. However, as the error rate of replication increased (v2.4–v9), the resulting lysis became highly MOI dependent. In cultures with T7 DNAP v9, the cell density increased during infection at a low MOI, while it decreased at a high MOI (Fig. 3b). These findings show that lysis by T7 phage—a strictly lytic phage in nature—can be tuned by MOI under error-prone replication.

a, Change in optical density of E. coli cultures expressing T7 DNAP wild-type and variants (v2.4–v9) when infected with T7ΔDNAP over 3 hours at varying MOIs (0.01–10). Data are shown as mean ± s.d. of n = 3 independent experiments. b, Lysis kinetics of E. coli cultures expressing wild-type (dashed lines) or hypermutagenic (v9; solid lines) T7 DNAP at different MOIs (0–10). c, Schematic representation of proposed multiplicity tuning mechanism: after infection (step I), wild-type T7 DNAP enables efficient phage replication at a low MOI (step IIa), while error-prone variant v9 requires a higher MOI due to increased phage inactivation during replication (step IIb). d, Quantification of phagemid packaging efficiency across T7 DNAP variants, showing maintained library diversity despite reduced transduction rates in error-prone variants. Packaging efficiency is reported as CFU ml−1 phage lysate. Data are shown as mean ± s.d. of n = 3 independent experiments. e, Representative 20-hour time course of a complete LySE cycle, showing distinct phases of bacterial growth and phage-mediated lysis initiated by simple mixing of phage lysates and cell cultures. Diagrams in c and e created in BioRender; Fredens, J. https://biorender.com/8aqhp97 (2026).
During faithful replication with wild-type T7 DNAP, the phage propagates and lyses the cells exponentially, independent of MOIs (Fig. 3c). Conversely, error-prone variants such as v9 compromise replication fidelity, resulting in unsustainable propagation and MOI-dependent lysis25. The reduction of functional T7 ΔDNAP helper phages increases the proportion of phagemids in the mutant pool (Extended Data Fig. 5). We verified that this compromised propagation still permits substantial phagemid packaging and transduction even with the highly error-prone DNAP v9 (Fig. 3d and Extended Data Fig. 3b). The substantial phagemid packaging efficiency provides a respectable effective library size, which can be further tuned by increasing the volume of phagemids transduced and selected for, particularly if a higher diversity is required to kick-start a challenging LySE campaign (Table 1). Given that the increased mutation rate of v8 was accompanied by declining phagemid packaging efficiency (Fig. 3d and Extended Data Fig. 3b), while v9 maintained relatively high packaging rates, we proceeded with DNAP v9, which provided an optimal balance between mutation rates and library size.
The ability to tune the degree of culture lysis by adjusting T7 phage multiplicity enables controlled and distinct phases of the LySE cycle. Addition of phage T7∆DNAP at a high MOI prompts mutagenesis of the phagemids and phages, followed by packaging into virions, released through efficient cell lysis (Fig. 3e). The resulting lysate consists of virions that either contain mutated phagemids or mutated phage T7∆DNAP genomes. When this is used to infect a fresh cell culture at a low MOI, phagemid libraries get transduced to fresh cells without widespread lysis by the mutated phages. This library of transductants can be grown under selective conditions to enrich for improved GCOI performance before initiating another round of lysis. These distinct LySE phases allow adjustment of growth and selection duration, facilitating evolution of slow-manifesting phenotypes, such as low-rate metabolic pathways36 or slow-folding protein complexes37.
Accelerated evolution of genes and gene clusters
To show the utility of LySE for accelerated evolution, we first evolved the tetracycline resistance gene tetA to confer resistance to tigecycline. We expressed tetA from a phagemid and subjected it to five generations of LySE with progressively increasing tigecycline concentrations (Fig. 4a and Supplementary Data 9). For comparison, we performed adaptive laboratory evolution (ALE), where the same host cell with phagemid was continuously passaged to allow evolution at the basal mutation rate under identical selection conditions38. Following five generations of evolution by LySE (E5 LySE), we obtained cells tolerating 2.5 μg ml−1 of tigecycline—a 25-fold increase over the wild-type tolerance of 0.1 μg ml−1 (Fig. 4b). In contrast, cells evolved by ALE tolerated only up to 1 μg ml−1 (E5 ALE) (Fig. 4c). Furthermore, when the E5 ALE-evolved phagemid was transferred to fresh cells (E5T ALE), the acquired resistance was lost. This indicates that the evolved resistance was caused by genomic mutations rather than mutations in the target phagemid, which shows that ALE is highly sensitive to off-target and cheater mutations11. LySE overcomes these issues by refreshing the host in each cycle, thus only allowing evolution through on-target mutations of the phagemid. We transferred a LySE-evolved phagemid (E5_2 LySE) to fresh cells and observed that it maintained acquired resistance (E5_2T LySE) (Fig. 4b).

a, Comparative workflows of LySE versus ALE. LySE confines mutations to the phagemid through phage-mediated cycling, whereas ALE allows accumulation of genome-wide mutations during serial passaging. Star symbols on plasmids indicate mutations. b, Tigecycline resistance profiles of an evolved pool after five generations of LySE (E5 LySE), compared with the culture before evolution (E1 LySE). A representative single clone from E5 is also shown immediately after evolution (E5_2 LySE) and after transformation to a fresh host (E5_2T LySE). c, The same phagemid and tetA evolved by ALE. Resistance profile of the pool after five passages (E5 ALE). The acquired resistance was almost entirely lost after transferring the phagemid to a fresh host cell (E5T ALE), indicating genome-dependent rather than phagemid-encoded adaptation. d, Structure of TetA and identified mutations in the evolved pool by Sanger sequencing of 32 clones. e, Metabolic pathway for EG assimilation in E. coli. f, Linearized representation of the phagemid with the EG assimilation pathway for evolution. Identified mutations from eight clones are labelled in red, with corresponding clone numbers (EGA1–8). g, Comparative workflows for semi-relaxing evolution of the EG assimilation pathway using LySE versus ALE. Both approaches used progressive selection from 1 g l−1 glucose + 8 g l−1 EG (E1) to 0 g l−1 glucose + 12 g l−1 EG (E5). The LySE protocol included cell recovery in ampicillin and kanamycin to maintain phagemid and AP, followed by washing and selection in minimal medium. Similarly, the ALE protocol comprised initial growth, washing, minimal medium selection, antibiotic recovery to maintain phagemid and AP, and final washing before the next selection cycle. h, Growth curves of E. coli bearing 4 isolated evolved phagemids transformed into fresh hosts (EGA3, EGA5, EGA7 and EGA8) and wild-type phagemid in M9 media with 10 g l−1 EG and no glucose. For panel g and h, data are shown as mean ± s.d. of n = 3 independent experiments. Diagrams in a, f and g created in BioRender; Fredens, J. https://biorender.com/zx2vu3i (2026).
From evolved clones of E5 LySE, we identified 4 point mutations in tetA (Fig. 4d, Extended Data Fig. 6a,b and Supplementary Data 10) and a mutation in the promoter that increased tetA expression approximately 200-fold (Extended Data Fig. 6c). This demonstrates the ability of LySE to simultaneously evolve both regulatory and coding regions, which is particularly useful for optimizing complex phenotypes39.
We next explored the capability to evolve a complex process—a multigene cluster constituting a complete metabolic pathway for assimilation of EG, a monomer derived from PET degradation. This pathway was chosen due to its substantial implications for plastic recycling and its potential contribution towards developing a sustainable circular economy for plastic waste management36,40.
We constructed the pathway starting with EG reduction to glycolaldehyde using Gluconobacter oxydans alcohol dehydrogenase (Gox0313), selected for its superior performance and use of only NAD+ as cofactor41 (Fig. 4e). Glycolaldehyde is further reduced to glycolate and then to glyoxylate by endogenous aldehyde dehydrogenase (aldA) and glycolate oxidase complex (glcDEF) from E. coli. The resulting glyoxylate enters the glycerate pathway for biomass and energy production42 (Fig. 4e). We cloned all 5 genes into a T7 phagemid, creating a 9,715 bp plasmid (Fig. 4f and Supplementary Data 11), and subjected the pathway to 5 generations of evolution by LySE or ALE under the same selection regime. We implemented a semi-relaxing selection protocol with gradual glucose withdrawal from the culture medium, resulting in improved normalized growth rates for LySE-evolved cells compared with ALE-evolved cells (Fig. 4g and Extended Data Figs. 7 and 8). After 5 generations, the ALE-evolved cell culture showed a 1.7-fold increase in growth compared with wild-type cells, whereas LySE-evolved cell growth increased by 2.8-fold (Extended Data Fig. 9). Sequencing of 32 ALE clones revealed that none of the phagemids had accumulated any mutations, illustrating how ALE confers systemic adaptation that may be caused by genomic mutations rather than mutations in the GCOI. In contrast, sequencing of clones evolved by LySE revealed mutations in the T7 ori–pac site, the p15a ori, near the aldA promoter and nonsynonymous mutations in three of the five genes (Fig. 4f, Extended Data Fig. 9, and Supplementary Tables 4 and 5). Five clones showed accelerated growth on EG compared with wild-type cells: one clone acquired a mutation in the T7 ori–pac site, which may have benefitted phagemid replication or transduction (Extended Data Fig. 10). All four remaining clones acquired mutations in the genes glcd, glcf and gox0313. When introducing each of the four mutations individually into the wild-type phagemid in a new host cell, the accelerated growth was fully restored, showing that each mutation was the sole cause for improved growth on EG (Fig. 4h). Two clones featured a Y94F substitution in Gox0313 (EGA3) and I312F substitution in glcD (EGA7), with improved endpoint biomass of 50.9% and 46.1%, respectively, after just 5 generations (Fig. 4h, Extended Data Fig. 10 and Supplementary Data 12). These experiments demonstrate LySE as a powerful directed evolution platform that enables rapid, targeted genetic optimization across both single genes and complex gene clusters.
Discussion
Here we developed LySE, a robust T7 phage-based system that bridges the fundamental trade-off between controllability and speed in directed evolution. By leveraging T7 as an efficient gene shuttle between cells, LySE transfers DNA of up to 40 kb without transformation losses while performing hypermutagenesis on target gene clusters. The system preserves discrete checkpoints for mutagenesis and selection yet enables seamless transitions between phases, combining continuous evolution with controlled discrete cycles.
Unlike previous phage-based continuous evolution methods such as PACE4 and T7AE43 that insert GOIs directly into the phage genome—thereby limiting the capacity to up to 8 kb (refs. 10,44)—our approach uses a T7 phagemid design using T7 ori–pac signals to direct packaging of the GCOI into the phage capsid. This strategy leverages the full capacity of the T7 capsid to potentially enable evolution of 40-kb constructs, of which we have demonstrated transduction of a 29-kb GCOI and evolution of a 7-kb GCOI in this work (Figs. 2g and 4f). This substantially exceeds the capabilities of existing phage-assisted and in vivo continuous evolution methods4,28,32,45. The expanded capacity of LySE substantially broadens the potential applications for near-continuous evolution. It enables work with anabolic pathways for small molecule synthesis46, catabolic pathways for waste assimilation36,41,42 (Fig. 4e–h) or carbon capture47, and evolution of protein complexes48—many of which exceed 10 kb when including regulatory elements. This versatility eliminates size constraints that have previously limited directed evolution approaches.
Orthogonal replication systems have gained traction for handling larger constructs of up to a 16.5-kb replicon7. However, continuous evolution systems, where host cells are not refreshed, remain vulnerable to accumulation of genomic off-target mutations11. These can be hitchhiking mutations that complicate phenotypic attribution or cheater mutations that bypass the applied selection pressure, especially in biosensor-based evolution. These vulnerabilities stem from the fundamental mechanism of orthogonal replication, where the GOI replicates with the host, allowing off-target mutations to be carried over to subsequent generations. Similarly, P1 phage-based methods, such as Inducible Directed Evolution (IDE), despite accommodating large inserts of up to 36 kb (ref. 49), are known to transfer flanking genomic regions after integration—a property that, while useful for genome engineering50, presents challenges for precise directed evolution. LySE overcomes these limitations through its lytic cycle. T7 phage-mediated cell lysis completely eliminates the E. coli culture after each selection cycle, effectively removing all off-target mutations. This process mirrors the discrete checkpoints of classical directed evolution but operates with the speed and throughput of continuous systems. It is worth noting that in LySE, PACE and orthogonal replication, mutations can accumulate throughout the phagemid or plasmid under evolution, including outside the GOIs. We observed this during evolution of the EG degradation pathway, where one of five clones with accelerated growth displayed a mutation in the T7 ori–pac site of the phagemid, which may have benefitted replication or transduction (Fig. 4f). The remaining four clones with accelerated growth had acquired causational mutations in the EG pathway genes. Thus, the risk of beneficial mutations outside the GCOI is limited and can be mitigated through stringent selection pressure, particularly when evolving enzyme specificity for synthetic metabolism51.
Furthermore, as an orthogonal DNA polymerase, T7 DNAP can be engineered to achieve extremely high error rates without affecting cell viability5. Our implementation of controlled error-prone replication and multiplicity tuning redirects pressure away from maintaining T7 genome fidelity, enabling us to exceed standard T7 phage error thresholds25 (Fig. 2a), achieving an estimated 3.82 × 10−5 s.p.b. A potential strength of LySE lies in its modularity and compatibility with other diversification methods that operate on plasmids. LySE can be combined with EvolvR, which uses a CRISPR-guided DNA polymerase for targeted mutagenesis, to further increase error rates in specific regions of the phagemid15,28. As LySE refreshes the host in every cycle, no genomic mutations can accumulate, opening up the possibility to further increase error rates using global mutagens as used for PACE52, mutator strains like XL-1 red53 or even chemical mutagens and UV irradiation54. If further diversity is required to kick-start a LySE campaign, in vitro diversification methods such as error-prone PCR55 and DNA shuffling56 can be used to generate an initial library.
Unlike most other phage-assisted evolution methods, LySE uses intracellular selection rather than viral fitness coupling. This enables direct coupling of GCOI function to cellular fitness, which is more appropriate for applications ultimately deployed in cellular contexts, such as strain or therapeutic cell engineering57, while also enabling selections based on physical cell properties through techniques like cell sorting20. Using the T7 bacteriophage as a gene delivery vector, LySE can switch hosts between generations, allowing selection schemes to change within a single evolution campaign without genetic modifications. In growth-coupled metabolic engineering, this facilitates rapid switching between different auxotrophic biosensor strains, each offering distinct selection stringency levels suited to specific target genes or clusters58. Although LySE may have a longer generation time than PACE and its derivatives4,18,59,60,61,62, it accommodates slow-manifesting phenotypes, which ultimately bottlenecks the selection time.
The LySE workflow (lysis, transduction, recovery and selection) requires only mixing phage lysates with cell cultures, making it accessible to users regardless of experience or access to equipment. Although manually performed here, the process is amenable to automation via liquid-handling systems for increased throughput similar to other evolution systems61,63,64. Our results establish a generalizable method for accelerated evolution of whole gene clusters, providing researchers with a robust tool for engineering large metabolic pathways and protein complexes without the complications of off-target mutations or mutations that circumvent selection.
Methods
General methods
All E. coli and phage strains created in this study are listed in Supplementary Table 1, plasmids in Supplementary Table 2 and all oligonucleotides in Supplementary Table 6. Phage T7∆DNAP is available upon request and relevant plasmids are available from Addgene. Phage T7 was obtained from ATCC (BAA-1025-B2). Cloning of all plasmids was carried out using MDS42 cells (E-6265-05K; Scarab Genomics). Plasmids were constructed with NEBuilder HiFi DNA Assembly (New England Biolabs) unless otherwise stated. Native E. coli and T7 genes were amplified by PCR directly from genomic DNA. Other genes were synthesized as gBlocks Gene Fragments (Integrated DNA Technologies), unless otherwise stated. PCR reactions were performed using PrimeSTAR GXL DNA Polymerase (Takara) for cloning and using Rapid Taq DNA Polymerase (Vazyme) for genotyping. All oligonucleotides were synthesized by Integrated DNA Technologies. Sanger sequencing was performed by 1st BASE. Nanopore sequencing was performed by Plasmidsaurus. Sample preparation for sequencing was done according to each company’s protocol.
Culture media
E. coli was grown in LB medium or on LB agar (Bio Basic Asia Pacific) with added kanamycin (50 μg ml−1), ampicillin (100 μg ml−1), streptomycin (100 μg ml−1), chloramphenicol (20 μg ml−1), tetracycline (10 μg ml−1) or hygromycin (200 μg ml−1) where appropriate. EG selection and growth experiments were carried out without antibiotics in standard M9 minimal media (50 mM Na2HPO4, 20 mM KH2PO4, 1 mM NaCl, 20 mM NH4Cl, 2 mM MgSO4, 100 μM CaCl2, 134 μM EDTA, 13 μM FeCl3·6H2O, 6.2 μM ZnCl2, 0.76 μM CuCl2·2H2O, 0.42 μM CoCl2·2H2O, 1.62 μM H3BO3 and 0.081 μM MnCl2·4H2O). Carbon sources were used as indicated in the text.
Electrocompetent cell preparation
E. coli cells were inoculated in 10 ml LB and grown overnight at 37 °C with shaking at 225 rpm, with appropriate antibiotics if applicable. The next morning, cells were diluted 1:50-fold in the same media to a final volume of 500 ml, grown to an optical density at 600 nm (OD600) of 0.5 and incubated on ice for 20 minutes. The cells were spun down at 4,000g for 10 minutes at 4 °C. The supernatant was discarded and the cells were resuspended in 50 ml of chilled sterile dH2O. The cells were washed once again with sterile dH2O followed by 16% (w/v) chilled glycerol. The cells were then resuspended in 1 ml of chilled 16% glycerol and used immediately for transformation by electroporation or they were flash frozen and stored at −80 °C.
Scarless gene deletion by homologous recombination
Gene deletions were performed as previously described50. We created plasmid pPBG01 (number 253997; Addgene) (Supplementary Data 13) for arabinose-inducible λ-Red recombination by cloning genes gam, beta and exo from E. coli C321.∆A.opt (87359; Addgene) into a CloDF13 vector (69669; Addgene).
E. coli MG1655rpsLK43R was used for all gene deletions. It was created by PCR amplification of rpsLK43R from E. coli DH10b, which we electroporated into competent and induced MG1655 cells harbouring pPBG01. After recovery, the cells were plated on LB agar with streptomycin to select for successful recombination. We further created a double-selection cassette of negative selection gene rpsL (from MG1655) and positive selection gene hygR (104405; Addgene) combined by overhang extension PCR. For gene deletions, we amplified the rpsL-hygR cassette by PCR using primers containing homology to the regions flanking the target gene to be deleted. We electroporated the PCR product into competent, induced MG1655rpsLK43R cells harbouring pPBG01 and selected for genomic integration with hygromycin. Successful gene deletions were verified by genotyping. To achieve scarless gene deletion, the rpsL-hygR cassette was subsequently removed by a second round of recombineering by electroporating 10 μg of a 90-base oligonucleotide covering the genomic homology regions targeting the lagging strand. Loss of the rpsL-hygR cassette was selected for with streptomycin and verified by genotyping and Sanger sequencing.
Phage plaque assay
Phage from clarified phage lysate (or from rebooting in cell-free TXTL extract, see later) were serially diluted tenfold in LB medium. Overnight cultures of E. coli host strains were prepared, diluted 4-fold in LB and incubated with shaking for 1 hour at 37 °C. For each 100-mm petri dish, 100 µl cells were mixed with 1 µl of serially diluted phage and incubated for 5 minutes at 37 °C. The phage-cell mixtures were then mixed with 4 ml pre-warmed molten 0.35% top LB agar and immediately poured uniformly onto 100-mm petri dishes containing 15 ml of solidified 1.5% bottom LB agar. The top agar was left to solidify for 1 hour and then incubated for 4 hours at 37 °C for plaque formation.
Phage titre determination
Overnight cultures of E. coli were diluted and embedded in top agar as mentioned earlier, but without phage. Plates were dried for 1 hour. Each phage was serially diluted, and 5 µl was spotted onto the top agar and left to dry for 15 minutes. The plates were incubated at 37 °C, facing down, for 4 hours. The number of plaques was counted at each dilution to determine plaque-forming units (PFUs). The MOI was determined by calculating the ratio of PFU of phage added to the colony-forming units (CFUs) of cells used for phage infection. We empirically determined that phage lysates are, on average, approximately 1 × 1013 PFU ml−1, and diluted overnight cell cultures are approximately 1 × 1011 CFU ml−1.
T7ΔDNAP phage assembly and rebooting in TXTL
T7 genomic DNA excluding the T7 DNAP gene, gp5, was PCR amplified in 5 fragments of 10.0 kb (F1), 4.4 kb (F2), 3.5 kb (F4), 10.0 kb (F5) and 10.0 kb (F6), with 25–30 bp overlapping sequences. Fragment F3 containing the trxA gene was amplified from the E. coli genome, with overlapping sequences with F2 and F4 to replace T7 DNAP. Purified PCR fragments were mixed, including 300 ng of F3 and 100 ng of each of the other fragments. They were then assembled by Gibson Assembly using NEBuilder HiFi DNA Assembly (New England Biolabs). The assembly mix, together with 50 ng of T7 genomic DNA, was mixed using the myTXTL Linear DNA Expression Kit (Arbor Biosciences), according to the manufacturer’s protocol, and incubated overnight at 37 °C to generate a mixture of rebooted wild-type T7 and T7ΔDNAP phage. To select for the mutant phage, a plaque assay was performed by infecting MG1655ΔtrxA containing pSJ55 (253998; Addgene), which expresses wild-type T7 DNAP. Wild-type T7 not expressing trxA would not propagate in this strain. Only T7ΔDNAP trxA would be able to propagate in this strain and form plaques. We confirmed successful generation of T7ΔDNAP trxA by genotyping and Sanger sequencing the genomic region from which T7 DNAP was deleted (GenBank:PZ151113) (Supplementary Data 1 and Supplementary Table 1).
Phage infection kinetic assay
Infection kinetics were carried out in 96-well plates using an BioTek Synergy HTX Microplate Reader (Agilent). Overnight cultures of E. coli host strains were prepared, diluted 1:1 in LB medium and 100 μl was added to each well. In each well, 20 µl of different serial phage dilutions were added to tune MOI. Each condition was replicated in three different wells. A lid was added to the 96-well plates to reduce evaporation during acquisition. The microplate reader was set to 37 °C with continuous orbital shaking at 300 rpm. OD600 was measured every 3 minutes and monitored for at least 2 hours.
Yeast assembly
Large phagemids were assembled by transformation-associated recombination in yeast65 as previously described66. S. cerevisiae BY4741 (MATα his3Δ1 leu2Δ0 met15Δ0 ura3Δ0) was obtained from ATCC (201388). We modified the cell wall digestion step slightly: we only used 1 µl zymolyase and we measured the OD660 every 10 minutes after 30 minutes of zymolyase digestion. Digested cells were resuspended by swirling and inverting the tubes.
Phagemid assembly was assessed by genotyping the resulting yeast colonies, amplifying 350–1,000 bp across the intersections of assembled fragments. Positive yeast colonies were cultured, phagemids were isolated with the Zymoprep Yeast Plasmid Miniprep Kit (Zymo Research) and electroporated into E. coli MDS42 under selection. Individual colonies were genotyped again with the same primers, and phagemids were isolated from positive clones using the Monarch Plasmid Miniprep Kit and verified by nanopore sequencing.
General LySE strain preparation
All LySE experiments were conducted with E. coli MG1655. Cells were first transformed with APs containing different T7 DNAP variants by electroporation as described earlier and selected with ampicillin. The protein sequences of the T7 DNAP variants are given in Supplementary Table 3. Cells containing the AP were transformed with the phagemid by electroporation and selected on media containing both ampicillin and kanamycin. Genotyping was performed at each step to confirm successful transformation.
LySE cycle
To propagate the T7ΔDNAP phage, MG1655 with pSJ55 was grown overnight in LB medium with ampicillin, then diluted with equal volumes of LB the next day. T7ΔDNAP phage lysates were mixed with the diluted cells at a volume ratio of 1:1,000 (approximate MOI = 0.1) and incubated at 37 °C with shaking for 2 hours until there was no further reduction in OD600. The phage lysates were then washed once with equal volumes of chloroform to remove residual cells and debris.
The LySE cycle begins with an overnight culture of MG1655 cells containing AP and phagemid. Overnight culture cells were diluted with equal volumes of LB the next day. To 100 µl of diluted cells, 20 µl of phage was added (approximate MOI = 10; high MOI) and incubated at 37 °C with shaking for 2 hours until there was no further reduction in OD600. The phage lysates were washed once with equal volumes of chloroform. Next, to 1 ml overnight culture of MG1655 cells containing only AP diluted with equal volumes of LB, we mixed 10 µl phage lysates containing phagemids and incubated at 37 °C with shaking for 1 hour for complete transduction of phagemids (approximate MOI = 1; low MOI). Phagemid packaging efficiency was determined by serial dilution of the transduced cells, followed by spot plating on LB agar with kanamycin to determine the CFU of transduced phagemid. To continue the LySE cycle, transduced cells were diluted in selection media and grown to confluency. The exact protocol for selection and culture recovery is specific to each evolution experiment but should include addition of ampicillin to maintain the AP. The next LySE cycle is continued by adding phage at high MOI to lyse cultured cells.
Fluctuation assay
We performed Luria–Delbrück fluctuation analysis to quantify the mutation rate per generation of LySE for each T7 DNAP variant. We cloned pSJ51, a phagemid constitutively expressing a chloramphenicol resistance gene (CmR) containing a premature stop codon (Q38TAG). MG1655 containing pSJ51 and an AP encoding the T7 DNAP variant to be tested was grown overnight, diluted and lysed as per standard LySE protocols. After one generation of phagemid replication and transduction, transduced cells were serially diluted tenfold and spotted on LB agar with kanamycin to quantify total transduction CFU and on LB agar with chloramphenicol to quantify stop-codon reversion CFU, respectively. After overnight incubation at 37 °C, CFUs from 3 independent replicates were counted. The apparent mutation rate, µ (substitutions per base pair per generation, s.p.b.) was calculated as µ = m / (R × C). Mutation frequency (m) was calculated based on the ratio of cells grown on chloramphenicol to that grown on kanamycin. R is the number of distinct mutation sites that make the resistance gene effective, that is, yielding a sense codon. For TAG, 8/9 possible single base substitutions yield a sense codon. Averaged over the three positions in the codon, R = (8/9) × 3 = 8/3. C is the gene copy number. As T7 packages concatemeric phagemid DNA via a head-full mechanism, multiple CmR copies are expected per phage, while only a single reverted TAG is sufficient to confer chloramphenicol resistance. We therefore determined C by taking the fraction of phagemid to T7 genome size, so C = 39,937/3,469 = 11.5.
Molecular dynamics simulation
We used the crystal structure published by ref. 29 (PDB:1T7P) as the basis for our molecular dynamics simulations of the wild-type and T523R mutant T7 DNAP. 1T7P contains a growing DNA strand terminated with a dideoxy cytosine nucleotide, and the incoming nucleotide is dideoxyguanosine triphosphate (ddGTP). To represent the real biomolecules as closely as possible, we manually added 3′-hydroxyl groups to the chain-terminating nucleotide of the growing DNA strand and ddGTP. Wild-type and mutant T523R T7 DNAP variants were created in silico, and subsets of these containing DNA substitutions of the leading cytosine nucleotide on the template strand were implemented to study the effect of T523R on base mispairing. The structure is dissolved in water under physiological conditions using the solution builder on CHARMM-GUI. We placed each protein structure in a 130 Å × 130 Å × 130 Å simulation box with water containing 215/216 sodium ions and 183 chloride ions (150 mM) to balance protein charges at a pH of 7.0. For each condition, we ran a 5,000-step steepest descent energy minimization. This was followed by a number of particles, volume and temperature (NVT) equilibration step with a simulation time of 125 ps (125,000 steps) at 303.15 K. The subsequent number of particles, pressure and temperature (NPT) production simulation is 10 ns (5,000,000 steps) at 303.15 K. All simulations used the CHARMM36m force field and were run using CUDA-supported GROMACS (v.2023.3) on high-performance computing facilities (National University of Singapore HPC).
Genomic fluctuation assay
E. coli MG1655 was grown from glycerol stocks overnight in LB. To determine total CFU, the cells were serially diluted tenfold and spotted on LB agar. To determine frequency of rifampicin resistance, 2 ml of cells were spun down and the cell pellet was plated onto selective LB agar with 50 µg ml−1 rifampicin. After overnight incubation at 37 °C, CFUs from 3 independent replicates were counted. Mutation frequency was calculated based on the ratio of cells grown on rifampicin to that grown on LB. To calculate substitutions per base pair (s.p.b.), the mutation rate was normalized by the number of mutations in the rpoB gene that impart rifampicin resistance (77 known point mutations, divide observed mutation rate by 77/3)31. We determined E. coli genomic mutation rates to be 2.39 ± 1.10 × 10−10 s.p.b., comparable to those previously reported16,18.
Illumina NGS and data analysis
We cloned pSJ77 (GenBank:PZ151114), a 39-kb BAC phagemid by yeast assembly of fragments from pBeloBAC11 (60342; Addgene), pRS316 (110533; Addgene) and yeast genomic DNA. E. coli MG1655 containing pSJ77 and an AP encoding a T7 DNAP variant was grown overnight, diluted and lysed as per standard LySE protocols. After one generation of phagemid replication and transduction into MG1655 with no plasmids, transduced cells were diluted tenfold in LB with kanamycin and recovered overnight. The recovered mutated BAC-phagemid library was purified using the ZymoPURE Plasmid Miniprep Kit (Zymo Research). Illumina NGS of the BAC-phagemid library was performed by Bio Basic Asia Pacific. NGS library preparations were constructed following the manufacturer’s protocol. For each sample, 200 μg DNA was randomly fragmented by Covaris to an average size of 300–350 bp. The fragments were treated with End Prep Enzyme Mix for end repairing, 5′ phosphorylation and 3′ adenylation to add adaptors to both ends. Size selection of adaptor-ligated DNA was then performed using DNA Cleanup beads. Each sample was then amplified by PCR for eight cycles using P5 and P7 primers, with both primers carrying sequences that can anneal to the flow cell to perform bridge PCR, and the P7 primer carrying a six-base index allowing multiplexing. The PCR products were cleaned up and validated using an Agilent 2100 Bioanalyzer. The qualified libraries were sequenced paired-end (PE150) on the Illumina NovaSeq system. Fastp (v.0.23.0) was used for quality control and preprocessing, including removal of adaptor sequences, PCR primers, reads with more than 14 N bases and reads with less than 40% bases above a Phred quality score of 20 (Q20). The cleaned data were then mapped to the reference genome using the Sentieon pipeline (v.202112.02). A custom python script using the pysam (v.0.23.0) module was used to align the NGS reads with Q score ≥30 to the reference sequence and count the nucleotide positions from which the experimental sample deviates from the reference sequence. To correct for sequencing errors, the observed fraction of mismatches at each nucleotide position mutated by the wild-type T7 DNAP was subtracted from the fraction of mutations for engineered T7 DNAP variants. Corrected mismatch rates and A:T → G:C and C:G → T:A mutation rates were binned into 20 equal-width bins and then calculated and plotted. For mutational rates spread across 39 kb, corrected mutation rates were binned every 100 bp and then plotted. Overall mutational spectra, and for every 10,000 bp, were calculated by taking the fraction of each of 12 mutation types relative to the overall mutation rate for each T7DNAP variant. We yielded an average of >14,000 reads per position for each of the sequenced samples.
LySE evolution of tetA
We cloned pSJ78 (254000; Addgene), a phagemid constitutively expressing the tetA tetracycline efflux gene. E. coli MG1655 containing pSJ78 and pSJ139 (T7 DNAP v9; 254001; Addgene) was grown overnight, diluted and lysed as per standard LySE protocols. After one generation of phagemid replication and transduction into MG1655 with pSJ139, transduced cells were diluted 10-fold with LB with ampicillin, kanamycin and 0.1 μg ml−1 tigecycline. The cells were incubated overnight at 37 °C with shaking at 225 rpm. The next day, the cells were diluted with equal volumes of LB and T7ΔDNAP phages were added to lyse the culture, starting another round of LySE. Evolution cycles were repeated another four times for a total of five evolution cycles, using the best growing cultures from the previous cycle as the starting material for the next cycle. Tigecycline concentrations were incrementally increased from LySE E1 to E5, with concentrations 0.1 μg ml−1, 0.25 μg ml−1, 0.5 μg ml−1, 0.75 μg ml−1 and 1 μg ml−1, respectively. After the 5th LySE cycle, the best growing culture was streaked on LB agar with kanamycin and 1 μg ml−1 tigecycline. Individual colonies were picked and inoculated separately in 1 ml LB with kanamycin in 24-well plates and grown overnight. A total of 32 evolved clones were spotted on LB agar with increasing concentrations of tigecycline to test resistance. The 32 clones were PCR amplified for the tetA cassette using PrimeSTAR GXL DNA Polymerase and the amplicon was sequenced by Sanger sequencing.
For ALE, E. coli MG1655 containing pSJ78 and pSJ55 (wild-type T7 DNAP) was grown overnight in LB with kanamycin and ampicillin and then diluted 10-fold with LB with kanamycin, ampicillin and 0.1 μg ml−1 tigecycline. The cells were incubated overnight at 37 °C with shaking at 225 rpm. The next day, the cells were diluted another tenfold to continue ALE. A total of five passages were performed, each time with increasing concentrations of tigecycline identical to the LySE schedule. Cells at the fifth passage were spotted on LB agar with increasing concentrations of tigecycline to test resistance. The cells were also lysed by addition of T7ΔDNAP, the lysate washed with chloroform and phagemids transduced to fresh MG1655 with no plasmids. The transduced cells were diluted tenfold with LB with kanamycin and then grown overnight. The recovered E5T ALE cells were then spotted on LB agar with tigecycline to test resistance after transduction.
Quantitative real-time PCR
Total RNA was first isolated from the E. coli cells. Cells were grown overnight and diluted 1:50 with LB and appropriate antibiotics and cultured until OD600 = 0.5. Then, 500 µl of cells were transferred into an Eppendorf tube, spun down and the pellet dried by dabbing on a paper towel. The pellet was resuspended in 100 µl of Tris-EDTA buffer pH 8.5 with 15 mg ml−1 lysozyme, vortexed for 10 seconds and incubated at room temperature for 5 minutes with shaking. To the mixture, 400 µl of TRK Lysis Buffer (Omega Biotek) with 4 µl 2-mercaptoethanol was added and total RNA was extracted immediately using the E.Z.N.A RNA Isolation Kit (Omega Biotek). Purified RNA was quantified with NanoDrop (Thermo Fisher Scientific). One microgram of RNA was converted to cDNA using the GoScript Reverse Transcriptase (Promega). Quantitative real-time PCR (qPCR) was performed with GoTaq qPCR (Promega) using the CFX Opus 96 Real-Time PCR System (Bio-Rad). Fold changes were normalized to 16S rRNA and are based on relative expression values calculated using the \({2}^{-\varDelta \varDelta {C}_{{\rm{T}}}}\) method.
LySE evolution of EG assimilation pathway
The gox0313 gene (UniProt: Q5FU50) was synthesized by GentleGen. We cloned pAN29 (253996; Addgene), a phagemid containing metabolic pathway genes for complete assimilation of EG. E. coli MG1655 containing pAN29 and pSJ139 (T7 DNAP v9) was grown overnight, diluted and lysed as per standard LySE protocols. After 1 generation of phagemid replication and transduction into MG1655 with pSJ139, transduced cells were diluted 10-fold with LB with ampicillin and kanamycin and incubated overnight at 37 °C with shaking at 225 rpm. The next day, 5 ml of recovered cells were pelleted by centrifugation at 3,900g for 3 minutes, washed 3× with M9 medium and then resuspended in M9 medium with EG, with or without glucose at concentrations as indicated in the text until the OD600 was approximately 0.2. No antibiotics were used for selection. To each well in a 24-well plate, 1 ml of the cell suspension was added and cultured overnight at 37 °C with shaking at 300 rpm in a Thermo-Shaker PST-60HL-4 Microplate Reader (BioSan). Every hour, the OD600 was measured to monitor cell growth. Evolution cycles were repeated another four times for a total of five evolution cycles, using the best growing cultures from the previous cycle as the starting material for the next cycle. After the 5th LySE cycle, the best growing culture was streaked on M9 minimal agar plates supplemented with 2 g l−1 of glucose and 10 g l−1 of EG. Individual colonies were picked and inoculated separately in 1 ml LB with kanamycin in 24-well plates and grown overnight. The following day, the cultures were washed with M9 media at 3,900g for 1 minute. Subsequently, the cells were inoculated in M9 medium supplemented with 10 g l−1 EG in 12 ml culture tubes for 6 hours to reach OD600 = 0.1. For each culture, 200 µl was transferred into a sterile 96-well microplate as triplicates and incubated overnight with continuous orbital shaking at 600 rpm. Growth rates were determined by measuring the OD600 at 12 hours, 18 hours, 24 hours, 36 hours, 42 hours and 48 hours post-inoculation using the microplate reader. Eight clones were PCR amplified for the phagemid using PrimeSTAR GXL DNA Polymerase and the amplicon was sequenced by nanopore sequencing.
For ALE, 5 ml E. coli MG1655 containing pAN29 was grown overnight in LB with kanamycin, washed 3× with M9 medium, and then resuspended in M9 medium with EG, with or without glucose at concentrations as indicated in the text until the OD600 was approximately 0.2. To each well in a 24-well plate, 1 ml of the cell suspension was added and cultured overnight. The next day, 1 μl of cells was added to 10 ml of LB with kanamycin and grown overnight, before 5 ml was taken and washed following the same washing and resuspension procedure as described previously for the next round of ALE. Evolution cycles were repeated another four times for a total of five evolution cycles, using the best growing cultures from the previous cycle as the starting material for the next cycle.
Statistical analysis
Significance was determined by two-tailed unpaired two-sample t-tests performed with Microsoft Excel Analysis ToolPak v.16.97.2.
Software
SnapGene 8.2.2 was used for plasmid design. GraphPad Prism 11.0.0 was used to generate all graphs. Illustrations were created in Adobe Illustrator 30.2.1 and BioRender.com.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All raw sequencing data generated in this study are available in the NCBI Sequence Read Archive under BioProject accession PRJNA1443325. Relevant nucleotide sequences are found in Supplementary Tables and GenBank (PZ151113 and PZ151114), and key protein sequences are provided in Supplementary Tables. Essential plasmids are available from Addgene (numbers 253996–254001). Phage T7∆DNAP is available upon request.
Code availability
The code used in this study is available on GitHub at https://github.com/shujian-ong/LySE.
References
Packer, M. S. & Liu, D. R. Methods for the directed evolution of proteins. Nat. Rev. Genet. 16, 379–394 (2015).
Chen, K. & Arnold, F. H. Engineering new catalytic activities in enzymes. Nat. Catal. 3, 203–213 (2020).
Arnold, F. H. Design by directed evolution. Acc. Chem. Res. 31, 125–131 (1998).
Esvelt, K. M., Carlson, J. C. & Liu, D. R. A system for the continuous directed evolution of biomolecules. Nature 472, 499–503 (2011).
Diercks, C. S. et al. An orthogonal T7 replisome for continuous hypermutation and accelerated evolution in E. coli. Science 389, 618–622 (2025).
Ravikumar, A., Arrieta, A. & Liu, C. C. An orthogonal DNA replication system in yeast. Nat. Chem. Biol. 10, 175–177 (2014).
Tian, R. et al. Establishing a synthetic orthogonal replication system enables accelerated evolution in E. coli. Science 383, 421–426 (2024).
Tian, R. et al. Engineered bacterial orthogonal DNA replication system for continuous evolution. Nat. Chem. Biol. 19, 1504–1512 (2023).
Molina, R. S. et al. In vivo hypermutation and continuous evolution. Nat. Rev. Methods Prim. 2, 37 (2022).
Miller, S. M., Wang, T. & Liu, D. R. Phage-assisted continuous and non-continuous evolution. Nat. Protoc. 15, 4101–4127 (2020).
Buskirk, S. W., Peace, R. E. & Lang, G. I. Hitchhiking and epistasis give rise to cohort dynamics in adapting populations. Proc. Natl Acad. Sci. USA 114, 8330–8335 (2017).
Roth, T. B., Woolston, B. M., Stephanopoulos, G. & Liu, D. R. Phage-assisted evolution of Bacillus methanolicus methanol dehydrogenase 2. ACS Synth. Biol. 8, 796–806 (2019).
Witte, I. P. et al. Programmable gene insertion in human cells with a laboratory-evolved CRISPR-associated transposase. Science 388, eadt5199 (2025).
Lee, J. K. et al. Directed evolution of CRISPR-Cas9 to increase its specificity. Nat. Commun. 9, 3048 (2018).
Ma, E. et al. Directed evolution expands CRISPR-Cas12a genome-editing capacity. Nucleic Acids Res. https://doi.org/10.1093/nar/gkaf649 (2025).
Oh, J. et al. Engineering a membrane protein chaperone to ameliorate the proteotoxicity of mutant huntingtin. Nat. Commun. 16, 737 (2025).
Studer, S. et al. Evolution of a highly active and enantiospecific metalloenzyme from short peptides. Science 362, 1285–1288 (2018).
Suzuki, T. et al. Crystal structures reveal an elusive functional domain of pyrrolysyl-tRNA synthetase. Nat. Chem. Biol. 13, 1261–1266 (2017).
Yosef, I., Goren, M. G., Globus, R., Molshanski-Mor, S. & Qimron, U. Extending the host range of bacteriophage particles for DNA transduction. Mol. Cell 66, 721–728.e723 (2017).
Wellner, A. et al. Rapid generation of potent antibodies by autonomous hypermutation in yeast. Nat. Chem. Biol. 17, 1057–1064 (2021).
Nguyen, H. M. & Kang, C. Lysis delay and burst shrinkage of coliphage T7 by deletion of terminator Tφ reversed by deletion of early genes. J. Virol. 88, 2107–2115 (2014).
Pryor, J. M., Potapov, V., Bilotti, K., Pokhrel, N. & Lohman, G. J. S. Rapid 40 kb genome construction from 52 parts through data-optimized assembly design. ACS Synth. Biol. 11, 2036–2042 (2022).
Levrier, A. et al. PHEIGES: all-cell-free phage synthesis and selection from engineered genomes. Nat. Commun. 15, 2223 (2024).
Chung, Y. B. & Hinkle, D. C. Bacteriophage T7 DNA packaging. I. Plasmids containing a T7 replication origin and the T7 concatemer junction are packaged into transducing particles during phage infection. J. Mol. Biol. 216, 911–926 (1990).
Söte, S., Kleine, S., Schlicke, M. & Brakmann, S. Directed evolution of an error-prone T7 DNA polymerase that attenuates viral replication. ChemBioChem 12, 1551–1558 (2011).
Foster, P. L. Methods for determining spontaneous mutation rates. Methods Enzymol. 409, 195–213 (2006).
Hall, B. M., Ma, C. X., Liang, P. & Singh, K. K. Fluctuation analysis CalculatOR: a web tool for the determination of mutation rate using Luria-Delbruck fluctuation analysis. Bioinformatics 25, 1564–1565 (2009).
Halperin, S. O. et al. CRISPR-guided DNA polymerases enable diversification of all nucleotides in a tunable window. Nature 560, 248–252 (2018).
Doublié, S., Tabor, S., Long, A. M., Richardson, C. C. & Ellenberger, T. Crystal structure of a bacteriophage T7 DNA replication complex at 2.2 Å resolution. Nature 391, 251–258 (1998).
Patel, S. S., Wong, I. & Johnson, K. A. Pre-steady-state kinetic analysis of processive DNA replication including complete characterization of an exonuclease-deficient mutant. Biochemistry 30, 511–525 (1991).
Cravens, A., Jamil, O. K., Kong, D., Sockolosky, J. T. & Smolke, C. D. Polymerase-guided base editing enables in vivo mutagenesis and rapid protein engineering. Nat. Commun. 12, 1579 (2021).
Moore, C. L., Papa, L. J. III. & Shoulders, M. D. A processive protein chimera introduces mutations across defined DNA regions in vivo. J. Am. Chem. Soc. 140, 11560–11564 (2018).
Richter, M. F. et al. Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat. Biotechnol. 38, 883–891 (2020).
Neugebauer, M. E. et al. Evolution of an adenine base editor into a small, efficient cytosine base editor with low off-target activity. Nat. Biotechnol. 41, 673–685 (2023).
Stoltzfus, A. & McCandlish, D. M. Mutational biases influence parallel adaptation. Mol. Biol. Evol. 34, 2163–2172 (2017).
Schada von Borzyskowski, L. et al. Implementation of the β-hydroxyaspartate cycle increases growth performance of Pseudomonas putida on the PET monomer ethylene glycol. Metab. Eng. 76, 97–109 (2023).
Bandyopadhyay, B., Mondal, T., Unger, R. & Horovitz, A. Contact order is a determinant for the dependence of GFP folding on the chaperonin GroEL. Biophys. J. 116, 42–48 (2019).
Dragosits, M. & Mattanovich, D. Adaptive laboratory evolution—principles and applications for biotechnology. Microb. Cell Fact. 12 https://doi.org/10.1186/1475-2859-12-64 (2013).
Liew, F. E. et al. Carbon-negative production of acetone and isopropanol by gas fermentation at industrial pilot scale. Nat. Biotechnol. 40, 335–344 (2022).
Tiso, T. et al. The metabolic potential of plastics as biotechnological carbon sources—review and targets for the future. Metab. Eng. 71, 77–98 (2022).
Zhang, X., Zhang, B., Lin, J. & Wei, D. Oxidation of ethylene glycol to glycolaldehyde using a highly selective alcohol dehydrogenase from Gluconobacter oxydans. J. Mol. Catal. B: Enzym. 112, 69–75 (2015).
Chi, J. et al. Engineering Escherichia coli for utilization of PET degraded ethylene glycol as sole feedstock. Biotechnol. Biofuels Bioprod. 17, 121 (2024).
Goicoechea Serrano, E., Blázquez-Bondia, C. & Jaramillo, A. T7 phage-assisted evolution of riboswitches using error-prone replication and dual selection. Sci. Rep. 14, 2377 (2024).
Johnston, C. W., Badran, A. H. & Collins, J. J. Continuous bioactivity-dependent evolution of an antibiotic biosynthetic pathway. Nat. Commun. 11, 4202 (2020).
Yi, X., Khey, J., Kazlauskas, R. J. & Travisano, M. Plasmid hypermutation using a targeted artificial DNA replisome. Sci. Adv. 7, eabg8712 (2021).
Zhang, J. et al. A microbial supply chain for production of the anti-cancer drug vinblastine. Nature 609, 341–347 (2022).
Luo, S. et al. Construction and modular implementation of the THETA cycle for synthetic CO2 fixation. Nat. Catal. 6, 1228–1240 (2023).
Aigner, H. et al. Plant RuBisCo assembly in E. coli with five chloroplast chaperones including BSD2. Science 358, 1272–1278 (2017).
Al’Abri, I. S., Haller, D. J., Li, Z. & Crook, N. Inducible directed evolution of complex phenotypes in bacteria. Nucleic Acids Res. 50, e58 (2022).
Thomason, L. C., Costantino, N. & Court, D. L. E. coli genome manipulation by P1 transduction. Curr. Protoc. Mol. Biol. Chapter 1, 1.17.11–11.17.18 (2007).
He, H. et al. Adaptive laboratory evolution recruits the promiscuity of succinate semialdehyde dehydrogenase to repair different metabolic deficiencies. Nat. Commun. 15, 8898 (2024).
Badran, A. H. & Liu, D. R. Development of potent in vivo mutagenesis plasmids with broad mutational spectra. Nat. Commun. 6, 8425 (2015).
Muteeb, G. & Sen, R. Random mutagenesis using a mutator strain. Methods Mol. Biol. 634, 411–419 (2010).
Cupples, C. G. & Miller, J. H. A set of lacZ mutations in Escherichia coli that allow rapid detection of each of the six base substitutions. Proc. Natl Acad. Sci. USA 86, 5345–5349 (1989).
Wilson, D. S. & Keefe, A. D. Random mutagenesis by PCR. Curr. Protoc. Mol. Biol. Chapter 8, Unit 8.3 (2001).
Stemmer, W. P. DNA shuffling by random fragmentation and reassembly: in vitro recombination for molecular evolution. Proc. Natl Acad. Sci. USA 91, 10747–10751 (1994).
Rix, G. et al. Continuous evolution of user-defined genes at 1 million times the genomic mutation rate. Science 386, eadm9073 (2024).
Schulz-Mirbach, H., Dronsella, B. & Erb, T. J. Escherichia coli selection strains for growth-coupled metabolic engineering. Trends Biotechnol. https://doi.org/10.1016/j.tibtech.2025.06.015 (2026).
Brodel, A. K., Jaramillo, A. & Isalan, M. Engineering orthogonal dual transcription factors for multi-input synthetic promoters. Nat. Commun. 7, 13858 (2016).
Brodel, A. K., Rodrigues, R., Jaramillo, A. & Isalan, M. Accelerated evolution of a minimal 63-amino acid dual transcription factor. Sci. Adv. 6, eaba2728 (2020).
DeBenedictis, E. A. et al. Systematic molecular evolution enables robust biomolecule discovery. Nat. Methods 19, 55–64 (2022).
Zinkus-Boltz, J., DeValk, C. & Dickinson, B. C. A phage-assisted continuous selection approach for deep mutational scanning of protein–protein interactions. ACS Chem. Biol. 14, 2757–2767 (2019).
Huang, T. P. et al. High-throughput continuous evolution of compact Cas9 variants targeting single-nucleotide-pyrimidine PAMs. Nat. Biotechnol. 41, 96–107 (2023).
Zhong, Z. et al. Automated continuous evolution of proteins in vivo. ACS Synth. Biol. 9, 1270–1276 (2020).
Kouprina, N., Noskov, V. N. & Larionov, V. Selective isolation of large chromosomal regions by transformation-associated recombination cloning for structural and functional analysis of mammalian genomes. Methods Mol. Biol. 349, 85–101 (2006).
Robertson, W. E. et al. Creating custom synthetic genomes in Escherichia coli with REXER and GENESIS. Nat. Protoc. 16, 2345–2380 (2021).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
Acknowledgements
We thank L. F. H. Funke for helpful comments on the paper, and B. Xue for helpful discussions on molecular dynamics simulations. This work was supported by the National University of Singapore Presidential Young Professorship (NUHSRO/2021/071/Startup/06) and the National Research Foundation Fellowship (NRF-NRFF14-2022-0009) to J.F., while S.O. was supported by the National University of Singapore President’s Graduate Fellowship, Integrative Science and Engineering Programme. This work was also supported by the National University of Singapore (NUHSRO/2024/064/NUSMed/05/SynCTI2.0) and the National Research Foundation, Singapore (NRF-MSG-2023-0003).
Author information
Authors and Affiliations
Contributions
S.O. and J.F. conceived and designed the project. P.G. established T7 phagemid packaging and transduction. S.O., J.F. and F.W. designed the error-prone T7 DNAP variants. S.O., P.G., A.N., F.W. and S.L. performed cloning and strain engineering. S.O. and P.G. performed fluctuation analysis. S.O. performed experimental characterizations and NGS analysis. T.V.E. and C.O.M. performed molecular dynamics simulations. S.O., A.N., S.L. and F.W. performed accelerated evolution experiments. W.S.Y. supervised C.O.M., while J.F. supervised the project. S.O. and J.F. wrote the paper with input from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests. The National University of Singapore has filed patent application PCT/SG2025/050734 in Singapore covering LySE.
Peer review
Peer review information
Nature Microbiology thanks Maryam Ali, Jumi Shin, B. Thuronyi and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 T7 phagemid packaging during lysis.
a. Schematic of the T7 lytic cycle. T7 bacteriophage attaches to specific lipopolysaccharides (LPS) on the outer membrane of the E. coli host and ejects its DNA. Early genes like T7 DNA polymerase (T7 DNAP) and T7 RNA polymerase (T7 RNAP) are expressed first. T7 DNAP replicates the T7 genome in a bidirectional manner, producing concatemers. Meanwhile, T7 RNAP transcribes phage genes, which are then translated by host ribosomes to produce phage structural proteins. Towards the end of the lytic cycle, new phage particles are assembled, the T7 genome is packaged, and the cell lyses to release 180 progeny phages. The whole process takes 17 min. b. Biocontainment of phage T7∆DNAP. Phage T7∆DNAP is engineered to lack T7 DNAP, and the accessory plasmid (AP) carries T7 DNAP under the control of a T7 promoter. Upon infection, phage T7∆DNAP expresses T7 RNAP that induces expression of T7 DNAP, leading to phage genome replication, packaging, and production of phage progeny. Phage T7∆DNAP is biocontained and propagates only in E. coli cells harbouring AP. c. Plaque assay of T7ΔDNAP infecting E. coli cells with or without AP carrying T7 DNAP. For cells expressing T7 DNAP, the bacteria lawn was completed cleared upon infection with T7∆DNAP of 5 × 104 PFU or higher. No plaques were observed on cells not expressing T7 DNAP. d. Lysis kinetics of T7ΔDNAP infecting E. coli cells with or without AP carrying T7 DNAP. MOI = 0.1. e. Transduction of a phagemid into new host cells by phage T7∆DNAP. In triplicates, E. coli cells containing the AP and phagemid were lysed by addition of phage T7∆DNAP. The lysate was washed with chloroform, transduced 1:100 into fresh E. coli cells and directly spotted with dilution on LB agar with 50 µg/mL kanamycin to select for cells that received the phagemid. Three independent replicates are shown. f. Quantification of packaging efficiency for phagemids of sizes 3.5 kb, 5.9 kb, and 9.7 kb with wild-type T7 DNAP by selection with kanamycin after transduction. Data shown as mean ± SD of three independent experiments, each with three technical replicates (n = 9).
Extended Data Fig. 2 Optimization of hypermutagenic T7 DNA polymerase variants.
a. Mutational frequencies of combinatorial mutations on the fingers domain (v2) of engineered T7 DNAP variants measured by a fluctuation assay using a phagemid-encoded chloramphenicol resistance gene containing an internal stop codon, with genotypes listed on the right. Mutations L479N and P560H did not increase mutation rates when added to T523R, and were therefore left out from future variants. b. Mutational frequencies of mutant T7 DNAP v3 (v1 with exonuclease inactivation), and wild-type T7 DNAP fused to TadA-8e or PmCDA1 with different linker lengths. Data shown as mean ± SD of n = 3 independent experiments. Significance was tested via two-tailed unpaired two-sample t-test. ns: p > 0.05.
Extended Data Fig. 3 Transduction efficiency and growth rates for T7 DNAP variants.
a. Growth kinetics of E. coli strains harboring no T7 DNAP (Empty), wild-type T7 DNAP (wt), or the hypermutagenic T7 DNAP variant v8. b. Quantification of phagemid packaging efficiency across wild-type (wt) and engineered (v1-9) T7 DNAP variants by selection with kanamycin after transduction. Data shown as mean ± SD of n = 3 independent experiments.
Extended Data Fig. 4 Mutational rates and spectra of LySE T7 DNAP variants.
a. Mutational frequencies of engineered T7 DNAP variants (v1-v9) determined by Illumina NGS. Colours indicate specific transition and transversion types. ‘n.d.’ indicates not detected. b. Mutational spectra heatmaps displaying the frequency of each substitution type calculated as a percentage of the total number of mutations observed. Rows represent the reference base; columns represent the mutated base. The sum of all values in the matrix equals 100%. c. Distribution of Mutational spectra across a 39-kb BAC-phagemid. Top: Schematic of the DNA construct including T7 ori/pac, antibiotic selection markers (KanR/CmR), bacterial artificial chromosome (BAC), Yeast artificial chromosome (YAC), and yeast chromosomal DNA elements. Bottom: Mutational spectra calculated for consecutive 10,000 bp windows for variants v8 and v9.
Extended Data Fig. 5 Comparing proportion of phagemids in lysate pools generated by wild-type T7 DNAP and v9 variant.
Left: Count of plaque forming units (PFU) per mL of lysate to measure the amount of functional phage T7∆DNAP in E. coli cells harbouring the AP. Middle: Count of colony forming units (CFU) per mL of lysate to measure the amount of phagemids transduced into recipient E. coli hosts. Right: Ratio of phagemid CFU to functional phage T7∆DNAP PFU (%). The error-prone T7DNAP v9 reduced both T7∆DNAP PFU and phagemid CFU, but reduced T7∆DNAP PFU to a greater extent, thereby increasing the proportion of phagemids in the lysate pools. Data shown as mean ± SD of n = 3 independent experiments.
Extended Data Fig. 6 tetA mutants obtained from LySE evolution for tigecycline resistance.
a. Screening of 32 clones from LySE E5 evolution of tetA gene for tigecycline resistance. b. Sanger sequencing of the tetA gene in the 32 evolved clones. Convergence of sequences in the promoter region (pCAT) as well as in the coding sequence (V145A, G283S). c. Normalized tetA expression levels in E5 LySE compared to E1 pools quantified by RT-qPCR. Data shown as mean ± SD of n = 3 independent experiments.
Extended Data Fig. 7 LySE evolution of ethylene glycol (EG) assimilation pathway and selection with EG as the sole carbon source.
Growth rate analysis of E. coli MG1655 containing the EG assimilation pathway phagemid following four rounds (E1 to E4) of LySE selection with EG as the sole carbon source. Initial LySE evolution showed no improvements, likely due to combined mutagenesis and carbon restriction stresses causing poor growth and loss of potential mutants. We therefore implemented a semi-relaxing selection protocol with initial glucose supplementation followed by gradual glucose withdrawal (Extended Data Fig. 11). Blue lines (MG1655 + pAN29) represent the growth curves of cells containing the EG assimilation pathway phagemid. Red lines (Control) show the negative control which is E. coli MG1655 without the phagemid. Cultures were grown in M9 minimal medium with 10 g/L EG. Data shown as mean ± SD of n = 3 independent experiments. The best replicate from each round was picked for subsequent rounds of LySE.
Extended Data Fig. 8 LySE evolution of EG assimilation pathway with semi-relaxing selection.
Growth rate analysis of E. coli MG1655 containing the EG assimilation pathway phagemid following five rounds (E1 to E5) of LySE versus adaptive laboratory evolution (ALE) following a semi-relaxing selection workflow. Both approaches employed progressive selection from 1 g/L glucose + 8 g/L EG (E1) to 0 g/L glucose + 12 g/L EG (E5). Data shown as mean ± SD of n = 3 independent experiments.
Extended Data Fig. 9 Characterization of phagemid pools obtained from LySE evolution versus ALE evolution for EG assimilation.
a. Growth curves of E. coli strains containing EG assimilation phagemids evolved by five generations of LySE (LySE E5), evolved by five generations of ALE (ALE E5), and the unevolved E. coli strain containing the wild-type EG assimilation phagemid (WT). We observed improved growth for both evolved strains compared to the wild type, and a greater improvement for the LySE evolved strain. Data shown as mean ± SD of n = 3 independent experiments. b. Nanopore sequencing of the EG assimilation phagemid in 32 ALE-evolved clones detected no mutations, indicating that improved growth is likely attributed to genomic mutations. c. Nanopore sequencing of the EG assimilation phagemid in 28 LySE-evolved clones detected mutations throughout the phagemids.
Extended Data Fig. 10 Characterization of eight isolated phagemids evolved by LySE for EG assimilation.
a. Growth curves of E. coli bearing eight isolated evolved phagemids (EGA1-8) after five generations of LySE, and wild-type phagemid (WT) in M9 media with 10 g/L EG and no glucose. b. Endpoint biomass (t = 48 h) of the growth curves. Five clones exhibited accelerated growth on EG compared to the wild type (Starting from best: EGA5, EGA3, EGA7, EGA8, EGA4). Data are the mean ± standard deviation from three (n = 3) independent biological replicates. Significance was tested via two-tailed unpaired two-sample t-test between each respective mutant and the wild-type variant. EGA3: p = 2.29 × 10−5; EGA4: p = 8.21 × 10−3; EGA5: p = 2.53 × 10−5; EGA7: p = 3.26 × 10−4; EGA8: p = 1.35 × 10−4. * p < 0.01; ** p < 0.001; *** p < 0.0001.
Supplementary information
Supplementary Information (download PDF )
Supplementary Data 1–13.
Supplementary Tables 1–6 (download XLSX )
Strains, plasmids, T7 DNAP variants, sequencing, characterization and oligonucleotides.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ong, S., Ghode, P., Narenderan, A. et al. Bridging continuous and discrete evolution through a controllable, hypermutagenic phage-bacteria system. Nat Microbiol (2026). https://doi.org/10.1038/s41564-026-02346-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41564-026-02346-y
