
Abstract
Amyotrophic lateral sclerosis (ALS) is a heritable disorder where rare variants with low-to-moderate penetrance are thought to dominate genetic risk. To identify such rare variants, we harmonized and analyzed exome data from 22 cohorts, totaling 17,919 individuals with ALS and 200,703 controls across discovery and replication phases. Rare variant analyses identified several new risk genes, with replication confirming association of YKT6 and supporting HTR3C, GBGT1 and KNTC1. We also provide strong, independent validation for genes with limited previous evidence: ARPP21, DNAJC7 and CFAP410. Notably, in ARPP21, we identified a new high-effect variant (p.P747L) and confirmed that p.P563L is an ALS-associated variant leading to an aggressive disease course. Beyond new discoveries, our analyses largely recapitulated the known genetic architecture of ALS, identifying risk variants in over 20% of cases and supporting a cumulative oligogenic risk model. These findings highlight new translational targets and show that rare variant analyses capture substantially more genetic risk than common variant genome-wide association studies.
Similar content being viewed by others

Main
ALS is a fatal neurodegenerative disease with a substantial genetic component1,2,3. Despite the discovery of several ALS genes, the genetic etiology remains elusive for most patients, with previous work indicating that a substantial portion of genetic risk for ALS is mediated by rare variants4. Identifying new ALS genes is key to furthering our understanding of the disease and may provide direct potential therapeutic targets, as evidenced by the recent approval of Tofersen for SOD1 mutation-positive ALS. The advent of next generation sequencing has opened the way for population-scale, genome-wide studies of both familial and sporadic ALS cases, which has already led to the identification of several ALS genes, including, among others, TBK1, NEK1 and KIF5A5,6,7,8,9. In continuing the search for ALS genes, sequencing data from tens of thousands of people are required to enable detection of ultrarare and low-to-modest impact variants. To this end, we assembled and harmonized data from 22 cohorts to generate the largest ALS exome sequencing dataset to date. This provided a discovery cohort encompassing 13,138 cases and 69,775 controls and an independent replication set of 4,781 cases and 130,928 controls. We processed and harmonized all samples uniformly, including realignment to the GRCh38 reference genome and joint variant calling, which we show to be crucial to eliminate structural biases. Through comprehensive single-variant and ultrarare variant (URV) burden analyses, we identified several new candidate variants, genes and genesets, thereby substantially expanding our understanding of the contribution of rare variants to ALS.
Results
Building a harmonized ALS exome dataset for rare variant analysis
To identify rare coding variants involved in ALS, we harmonized 18 whole-exome (WXS) and whole-genome (WGS) sequencing datasets into a discovery cohort totaling 94,545 people. All data were realigned uniformly to GRCh38 and called jointly using the functional equivalence pipeline10,11, substantially reducing technical variation (Supplementary Fig. 1a). Moreover, the distributions of exome-wide URV counts were aligned between ancestry-matched WGS (Project MinE) and WXS (UK Biobank) samples, indicating that sequencing technologies were comparable after joint processing and quality control (Supplementary Fig. 1b). Following strict filtering, the final dataset comprised 13,138 unrelated cases and 69,775 controls of predominantly European ancestry, with 5,207,138 variants (2,367,861 predicted moderate or high impact; Supplementary Figs. 2–4).
Rare single-variant analyses identify five new risk variants and largely recapitulate known rare variant architecture of ALS
We conducted single-variant analyses of 272,925 rare variants that fell within our testable minor allele frequency (MAF) range (5 × 10−5 < MAF < 0.05) while also satisfying variant effect prediction criteria of either moderate- (missense mutations, in-frame deletions and untranslated region (UTR) truncations) or high-impact (nonsense, splice acceptor/donor and frameshift mutations) annotations. For each variant, we used Firth’s logistic regression to test for an association between ALS status and minor allele count (MAC), adjusting for sex, ten principal components (PCs) and the total number of rare synonymous variants in each person12,13. The resulting test statistics showed no systematic inflation, indicating no residual confounding (λ1,000 = 1.01), and significant variants passed subsequent validation and sensitivity analyses (Extended Data Fig. 1a,b).
We identified 15 exome-wide significant variants across 11 distinct genes (P < 1.83 × 10−7; Fig. 1a, Table 1, Extended Data Table 1, Extended Data Figs. 1 and 2 and Supplementary Data 1), for all of which the minor allele was associated with increased ALS risk (Fig. 1c). Among the 15 associated variants, 10 were located in genes previously shown to be related to ALS: SOD1, CFAP410, NEK1, KIF5A, FUS and TBK1 (Fig. 1a and Extended Data Table 1). The remaining five have not been reported previously in ALS (Table 1). These encompass intermediate frequency variants with modest effect size, including HTR3C p.T186A (odds ratio (OR) = 3.41, P = 1.87 × 10−8) and YKT6 p.Y64C (OR = 2.84, P = 9.08 × 10−8) as well as rare variants with high effect size, including GBGT1 p.R152L (OR = 26.9, P = 1.68 × 10−10), CAPN2 p.I530V (OR = 25.3, P = 3.66 × 10−9), and KNTC1 p.W287R (OR = 27.7, P = 1.07 × 10−7).

a, y axis: exome-wide single variant associations estimated using Firth’s logistic regression with profile penalized likelihood CIs (−log10(P)); x axis: genomic coordinates (GRCh38). Dashed line: exome-wide significance threshold (P < 1.83 × 10−7). New variants are highlighted in orange. b, Rare single-variant analyses among ALS-linked genes curated by the ALS GCEP. y axis: single-variant associations estimated using Firth’s logistic regression with profile penalized likelihood CIs (−log10(P)); x axis: genomic coordinates (GRCh38). Variants are colored by the clinical validity classification as curated by the ALS GCEP. Lower dashed line: significance threshold across variants in ALS-linked genes (P < 3.20 × 10−5); upper dashed line: exome-wide significance threshold as presented in a. c, ORs (y axis) and 95% CIs (gray shaded area) plotted against the risk allele frequency in controls (x axis) for significant variants identified in either the exome-wide or GCEP analysis. For variants where the control risk allele frequency was 0, it was set to half the lowest nonzero risk allele frequency observed in the control group. P values are two-tailed and are presented uncorrected for multiple testing.
We also performed a targeted analysis of variants within 51 ALS-linked genes curated by the ALS Gene Curation Expert Panel (GCEP)14. To ensure the inclusion of the full set of GCEP-curated genes, we did not apply the per-supercohort call-rate filter for this analysis, allowing for the assessment of genes exhibiting subpar call-rates in certain subcohorts. This identified eight additional variants across six genes (P < 3.20 × 10−5; Fig. 1b,c, Extended Data Table 1 and Supplementary Data 2), including variants in genes that were not detected in the exome-wide analysis (ARPP21, ANXA11, UBQLN2 and TARDBP). For all identified variants, the minor allele was associated with increased ALS risk (Fig. 1c and Extended Data Table 1). We provide independent evidence for two rare variants in ARPP21 (p.P563L and p.P747L)—a gene that is currently considered as having limited evidence according to GCEP (p.P563L: OR = 44.8, P = 2.55 × 10−10; p.P747L: OR = 75.8, P = 1.45 × 10−6) (Fig. 1b,c). Of note, the ARPP21 p.P563L variant had subpar call-rates in some exome cohorts. However, even when restricting the analysis to cohorts meeting stringent call-rate thresholds, the association remained exome-wide significant with a similar odds ratio (P = 1.09 × 10−8, OR = 38.1; Extended Data Fig. 1c,d).
Principal component analysis (PCA) suggested a mixed pattern of geographical distribution for carriers of the identified variants (Supplementary Fig. 5). For some variants, we observed that carriers exhibited relatively tight clustering in PCA space. This was observed for both well-established ALS variants such as UBQLN2 p.P509S (Sweden) and SOD1 p.A5V (USA), as well for the new CAPN2 p.I540V variant (the Netherlands). Conversely, other variants were distributed more uniformly across patient populations (for example, YKT6 p.Y64C and ARPP21 p.P563L). In silico pathogenicity prediction tools also yielded varying annotations for both previously established and new ALS-associated variants (Supplementary Table 1). Nonetheless, we observed that YKT6 p.Y64C was consistently predicted as damaging by all predictors, and KNTC1 p.W287R was predicted as damaging by all but SIFT.
Ultrarare burden analyses identify new ALS-associated genes
To detect associations among URVs (five or fewer carriers), we performed burden tests using Firth’s logistic regression to evaluate their cumulative effects. URVs were aggregated across several functional units, including genes and protein domains. To enrich for potentially pathogenic variants, we used four filtering strategies based on two criteria: (1) variant frequency—either all URVs or singleton-only variants; (2) variant impact—either only high-impact variants or both high- and moderate-impact variants. Tests across these filtering strategies were combined using the ACAT omnibus test15. We observed no evidence of genomic inflation in any of the analyses performed (gene λ1,000 = 1.011, domain λ1,000 = 1.006; Extended Data Fig. 3a), and all presented genes passed subsequent sensitivity analyses (Extended Data Fig. 3e–g).
URV gene burden analyses across 17,324 protein-coding genes identified eight genes that reached exome-wide significance (P < 2.89 × 10−6) (Fig. 2a, Table 2, Extended Data Table 2, Extended Data Figs. 2 and 3, Supplementary Data 3 and 4 and Supplementary Fig. 6). Among these were four established ALS genes: SOD1 (P < 1 × 10−16), TBK1 (P < 1 × 10−16), NEK1 (P = 6.49 × 10−13) and TARDBP (P = 5.02 × 10−8) (Extended Data Table 2). Furthermore, we identified DNAJC7 (P = 8.77 × 10−8), which is currently classified as having limited evidence (ClinGen gene curation panel14), and here reaches exome-wide significance for the first time in an exome-wide discovery analysis. New candidate genes included TTC3 (P = 4.16 × 10−7), UNC13C (P = 2.80 × 10−7) and KIF4A (P = 1.62 × 10−6), in all of which higher URV burden increased risk of ALS (Table 2 and Extended Data Fig. 3b). A targeted analysis among the 51 ALS-linked genes classified by GCEP also revealed a significant association for OPTN (P = 1.56 × 10−5), which is classified by GCEP as a definitive ALS gene (Fig. 2b and Extended Data Table 2).

a, y axis: exome-wide gene-based URV associations (−log10(P)); x axis: genomic coordinates (GRCh38). Dashed line: exome-wide significance threshold (P < 2.9 × 10−6). New risk genes are highlighted in orange. b, URV burden analyses among ALS-linked genes curated by the ALS GCEP. y axis: gene-based URV associations (−log10(P)); x axis: genomic coordinates (GRCh38). Lower dashed line: significance threshold across ALS-linked genes (P < 1 × 10−3); upper dashed line: exome-wide significance threshold as presented in a. c, Domain-based URV analyses. y axis: domain associations (−log10(P)); x axis: genomic coordinates (GRCh38). Dashed line: exome-wide significance threshold (P < 7.68 × 10−7). d, Association P values for URV geneset burden analyses excluding exome-wide significant genes (y axis) versus including exome-wide significant genes (x axis). The dashed lines indicate the multiple testing threshold (P < 4.25 × 10−6). Association statistics were estimated using Firth’s logistic regression with profile penalized likelihood CIs. P values are from the ACAT omnibus test combining the four variant filtering strategies (Methods) and are two-tailed and uncorrected for multiple testing.
The URV domain analyses across 65,071 domains identified three partially overlapping domains in TBK1 (protein kinase, kinase-like and CCD1 domains), one domain in SOD1 (SOD_Cu/Zn_BS domain) and one domain in VCP (CDC48 domain 2-like domain) at exome-wide significance (P < 7.68 × 10−7; Fig. 2c, Extended Data Fig. 3c and Supplementary Data 5 and 6). Unlike SOD1 and TBK1, VCP did not reach significance in the whole-gene analysis (Pgene = 8.09 × 10−3), suggesting that the CDC48 domain 2-like region harbors the primary association signal with a markedly stronger effect (Pdomain = 2.16 × 10−7). This domain constitutes the second subdomain of the N-terminal domain, in which most known pathogenic mutations are concentrated16.
Across burden analyses, ORs were generally similar when including all URVs compared to including singletons only, with the notable exceptions of NEK1 and KIF4A, which showed markedly higher ORs in the singleton-only analyses (Extended Data Fig. 3b–d). The observed associations were driven primarily by moderate-impact variants: NEK1 and TBK1 were the only genes showing a significant signal when analyses were restricted to high-impact variants (Extended Data Fig. 3k–m), although some signal among high-impact variants was observed for DNAJC7 and OPTN. Single nucleotide variants (SNVs) were the primary drivers of the associations, with insertions/deletions (INDELs) contributing substantially to the association P values only for NEK1 and DNAJC7 (Extended Data Fig. 3h–j). For UNC13C, TTC3 and OPTN, we identified a small subset of people carrying two URVs, whereas for TBK1 and NEK1, we found people with both a URV and a more common (0.01 < MAF < 0.05) risk variant (p.V464A and p.R261H, respectively). No increased risk was observed in these cases, although this may be due to the low number of co-occurrences (Extended Data Fig. 4a).
Assessing geneset burden and variant co-occurrence
We performed URV geneset burden analyses across 11,777 Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG) and Reactome genesets from the Molecular Signatures Database (MSigDB v.7.5)17, using the same procedure as the single gene analyses (λ1,000 = 1.006, Supplementary Data 7 and 8). After excluding genesets driven solely by one highly significant gene, two genesets remained significant: ‘GOBP: regulation of mRNA splicing via spliceosome’ (GO:0048024, 96 genes, P = 2.97 × 10−7) and its parent term ‘GOBP: regulation of RNA splicing’ (GO:0043484, 142 genes, P = 3.50 × 10−6) (Fig. 2d and Extended Data Fig. 3d). As ‘regulation of mRNA splicing via spliceosome’ is a subset of ‘regulation of RNA splicing,’ we performed a conditional analysis to assess its independent contribution. This revealed that residual signal remains in ‘regulation of mRNA splicing via spliceosome’ (P = 0.0084), suggesting it captures a more specific association within this pathway. Among the 153 unique genes across these two genesets, 30 reached nominal significance (P < 0.05), with top genes including HSPA8, HABP4, NOVA2, HNRNPL and SNW1 (Supplementary Fig. 7a). We also performed a geneset analysis among the 51 ALS-linked genes curated by GCEP14. As expected, this showed that the ‘Definitive’ category was highly significant (P < 1 × 10−16) across allele frequency thresholds, whereas the ‘Limited’ category showed only modest enrichment (P = 0.0015), and no enrichment was seen among the other categories (Supplementary Fig. 7b,c).
We next examined whether carrying several variants among ‘Definitive’ ALS genes as classified by GCEP confers cumulative risk. We observed a clear dose–response relationship across low-frequency variants (MAF < 0.05): the OR increased progressively as people carried one (OR = 1.19, P = 2.11 × 10−15), two (OR = 1.35, P = 8.43 × 10−13), three (OR = 1.84, P = 2.78 × 10−8) or four (OR = 4.26, P = 5.35 × 10−5) qualifying variants (Extended Data Fig. 4c and Supplementary Data 9). This relationship persisted when burden was assessed at the gene level, where several variants within the same gene were counted as a single event (Extended Data Fig. 4d and Supplementary Data 9). Analyses restricted to rarer variants were underpowered due to the low number of people carrying several variants (Supplementary Data 9). We did not observe a similar dose–response relationship when we tested for an association with age at onset and survival (Supplementary Fig. 8).
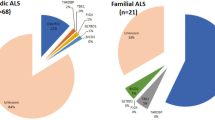
We next focused on co-occurrence among the specific risk variants identified in this study. Focusing on single variants in ‘Definitive’ GCEP genes, we found that 11.1% of cases carried one variant and 0.54% carried two, whereas the co-occurrence of three or more variants was not observed (Extended Data Table 3). When including variants in genes with ‘Limited’ evidence and new single variants identified in this study, the proportions increased to 14.5% for one, 1.1% for two and 0.0076% for three variants. The proportions increased further to 18.2%, 1.7% and 0.099%, respectively, when also including qualifying variants from the URV burden analyses. Finally, when C9orf72 repeat expansion status was also considered (available for 66% of cases), these totals rose to 23.5%, 3.12% and 0.22%, respectively, totaling 26.9% of cases. The observed co-occurrence rates did not deviate from those expected under an additive model using permutation analyses (P = 0.39). When examining specific variant pairs, we observed numerous instances of cases carrying several variants (Extended Data Fig. 5 and Supplementary Fig. 9). For example, 20% of C9orf72 repeat expansion carriers harbored additional risk variants. Furthermore, some pairs, including CFAP410 p.V58L × NEK1 p.R261H, showed trends suggestive of a synergistic effect (Extended Data Fig. 4e). To formally test whether any of these pairs showed nonadditive effects, we performed pairwise co-occurrence and interaction analyses. No pairs reached significance after correction for multiple testing (Extended Data Fig. 5 and Supplementary Fig. 9). This was consistent with our power calculations (Supplementary Fig. 10), which showed that the study was underpowered to detect all but the largest deviations from additivity for specific variant pairs, and then only for pairs including at least one low-frequency variant (0.01 < MAF < 0.05).
ARPP21 p.P563L is associated with earlier disease onset and shorter disease duration
To assess the impact of genetic variants on disease progression, we analyzed survival and age at onset across candidate genes and variants (Extended Data Fig. 6 and Supplementary Data 10). Consistent with previous reports, SOD1 p.A5V and p.D91A were associated significantly with a lower age at onset (p.A5V: B = −9.44, P = 5.61 × 10−4; p.D91A: B = −4.82, P = 1.11 × 10−5), with p.A5V linked to shorter survival and p.D91A to longer survival (p.A5V: hazard ratio (HR) = 13.0, P = 1.19 × 10−8; p.D91A: HR = 0.453, P = 1.48 × 10−7). Similarly, FUS p.R521C and p.P525L were associated with earlier onset (p.R521C: B = −16.2, P = 1.90 × 10−4; p.525L: B = −39.1, P = 1.53 × 10−10), with p.P525L specifically associated with shorter survival (HR = 41.75, P = 1.41 × 10−10). Notably, ARPP21 p.P563L was associated with a significantly lower age at onset (B = −12.7, P = 5.44 × 10−4) and shorter survival (HR = 5.96, ∆survival time = −19.5 months, P = 2.54 × 10−6), showing effect sizes comparable to SOD1 p.A5V (Extended Data Fig. 6a). Among URVs, SOD1 was associated with longer survival (HR = 0.45, P = 0.0022), whereas no significant associations were observed for other genes (Extended Data Fig. 6b).
Replication confirms YKT6 and supports HTR3C, GBGT1 and KNTC1 as ALS risk genes
For replication, we generated a cohort comprising 4,781 individuals with ALS and 130,928 controls after applying stringent quality control criteria identical to those used in the discovery set (Supplementary Figs. 11 and 12). Power analyses based on the (winner’s curse adjusted) effect sizes observed in the discovery dataset indicated that this provides between 32% and 91% statistical power for replication across candidate variants and genes (Supplementary Fig. 12).
Of the five new single variants identified in the discovery phase, all showed a consistent direction of effect in the replication cohort (Table 1; λ1,000 = 0.965). Moreover, all five reached exome-wide significance in a meta-analysis of the combined discovery and replication data, with all but CAPN2 p.I530V showing greater significance compared to the discovery phase alone (Table 1). Furthermore, YKT6 p.Y64C achieved replication-wide significance (P < 0.0063), correcting for the eight new associations from the discovery phase (five single variants and three URV genes). Among the three candidate URV genes, a consistent direction of effect was seen only for KIF4A (OR = 2.46, P = 0.26), and none reached replication-wide significance (Table 2; λ1,000 = 1.046).
Establishing independent evidence for ARPP21, DNAJC7 and CFAP410
Next, for the genes that were significant in our discovery analysis that are currently classified by GCEP with ‘Limited’ evidence (ARPP21, CFAP410 and DNAJC7), we aimed to confirm the independence of our findings.
For ARPP21, we identified two rare variants: p.P747L, which has not previously been reported in the scientific literature, and p.P563L, previously reported in UK and Spanish families as candidate variants18,19. To confirm independence for p.P563L, we excluded four potentially overlapping UK carriers (no Spanish carriers were identified). The association remained (OR = 28.3, P = 3.47 × 10−7; Extended Data Fig. 7a) and was further supported by our replication dataset, which had no potential overlap with previous studies (OR = 16.5, P = 3.29 × 10−3). A meta-analysis of these two independent datasets yielded a highly significant association (P = 4.31 × 10−9), confirming a strong, independent signal. We also validated the reported effects of age of onset and progression19 in our nonoverlapping cohort (Extended Data Fig. 6). Finally, ARPP21 carriers were observed across several cohorts beyond those from the UK and Spain, significantly expanding its known population distribution (Supplementary Fig. 5).
CFAP410 p.V58L was previously identified in two common variant genome-wide association studies (GWAS) (MAF = 0.013)4,20. To confirm independence, we excluded 8,372 cases and 4,159 controls that were duplicated or had second-degree or closer genetic relatedness to the original GWAS cohorts. The association remained highly significant after this exclusion (Pmeta = 1.34 × 10−14), with consistent ORs in both discovery (OR = 1.81, P = 1.32 × 10−10) and replication (OR = 1.61, P = 1.09 × 10−5) cohorts (Extended Data Fig. 7b).
For DNAJC7, implicated previously in a case–control study of ALS21, we re-evaluated the association after excluding overlapping cohorts (excluding 5,722 cases and 9,849 controls). In this reduced discovery dataset, there remained a robust association with a consistent odds ratio (ncases = 7,606, ncontrols = 59,926; OR = 2.56, P = 1.36 × 10−4; Extended Data Fig. 7c). This was further supported by our replication cohort, which had minimal overlap (190 cases) with the previous study (OR = 2.41, P = 2.82 × 10−3; Extended Data Fig. 7c). Meta-analysis across these two datasets confirmed a strong, independent signal (P = 2.96 × 10−6).
Discussion
This study represents one of the largest rare variant analysis of ALS until now, capturing nearly all of its known rare variant architecture. We expand upon known genetic factors contributing to ALS, demonstrating a substantially higher yield than common variant GWAS of comparable size4,20.
Our findings also provide a broader view of the genetic architecture of ALS. The variants we identified were mostly missense and spanned a spectrum of effect sizes, ranging from low-frequency variants with moderate effect sizes and URVs conferring large effects. At the far end of this spectrum, the latter category includes variants such as SOD1 p.A5V, which was absent from ~200,000 controls and conferred large effects (OR > 200), alongside other high impact variants such as those in ARPP21 (ORs > 40). Moreover, our data support an additive, oligogenic model22,23 wherein several rare variants cumulatively increase ALS risk without strong evidence of interaction. Although statistical power was limited, pairwise analyses of the best-powered variant combinations revealed no significant interactions, consistent with an additive model. Fully characterizing this oligogenic architecture will require studies extending beyond established ALS genes and, ultimately, beyond the exome.
Among the identified rare variants, YKT6 p.Y64C stood out due to its highly significant and consistent associations in both the discovery and replication cohorts. It was associated with a moderate increase in risk, with an OR comparable to that of established ALS variants SOD1 p.D91A and NEK1 p.R261H (Fig. 1c). YKT6 encodes a highly conserved SNARE protein that plays a key role in vesicular transport pathways, also implicated in GWAS20, and is critical for autophagosome-lysosome fusion24,25. The p.Y64C variant has been linked to a neurodevelopmental disorder in the homozygous state26, whereas we found it to be associated with ALS in the heterozygous state. The variant was shown to cause partial loss-of-function and impaired autophagy in Drosophila26, in line with it being consistently predicted as damaging by all in silico predictors we tested. YKT6 has also been implicated in the secretion of the MSP domain of the ALS-linked protein VAPB, suggesting that it may also play a role in extracellular signaling27,28. The identification of YKT6 thus highlights the central role of disrupted vesicle fusion and trafficking in ALS and specifically implicates downstream consequences including impaired autophagy and exocytosis. Beyond YKT6, three high-effect (OR > 20) missense variants in KNTC1, HTR3C and GBGT1 also represent strong candidates displaying consistent directions of effect across discovery and replication analyses, with meta-analysis achieving greater statistical significance than the discovery analysis alone. These genes implicate GTPase signaling, serotonergic function and glycosphingolipid metabolism respectively, all processes previous linked to ALS pathology29,30,31,32.
In addition to these new findings, a key contribution of our study is providing robust, independent evidence for several genes with limited previous evidence. In ARPP21, we identified two high-effect variants (p.P563L, p.P747L), with ORs comparable to those of highly penetrant variants such as FUS p.R521C and TARDBP p.N352S (Fig. 1c). Of these, p.P563L has been reported previously in two family studies18,19. Our study now firmly establishes not only the association of this variant with ALS, but also its previously reported effects on age of onset and survival. Moreover, this variant had been reported only in UK and Spanish families, whereas our study establishes its relevance in a broader population, identifying carriers across Dutch, US, Italian and Israeli cohorts. The second variant, p.P747L, has not previously been reported in the scientific literature. ARPP21, like TDP-43 and FUS, is an RNA-binding protein that localizes to stress granules under stress33. Our observed enrichment of URVs in splicing-related genes adds support to the relevance of this commonality, and suggests further insights remain to be discovered concerning the full depth of RNA processing dysfunction in ALS pathogenesis. For DNAJC7, which encodes a heat-shock protein implicated previously in an exome-wide burden analysis21, our study offers independent validation. Although the previous signal was driven by protein-truncating variants, our URV analysis identified a robust association that also included INDELs and missense variants. Crucially, this association was replicated across cohorts and remained after removal of cohorts overlapping with the previous study. Finally, in CFAP410 (also known as C21orf2), the low-frequency (MAF = 0.013) missense variant p.V58L was identified previously in two common variant GWASs4,20 and has been linked to primary cilia dysfunction34. We show that this is a robust and independent finding, as the association remained highly significant even after we excluded all participants who were duplicated or genetically related to the original GWAS cohorts.
Finally, despite inconclusive evidence from the replication analysis, UNC13C, KIF4A and CAPN2 remain candidates of interest for further study. UNC13C and KIF4A are paralogs of ALS genes UNC13A and KIF5A, respectively35,36,37,38, and are similarly involved in synaptic vesicle release and axonal transport (Supplementary Table 2 and Supplementary Data 11). CAPN2 is of interest due to previous literature supporting its role in ALS pathology and is being evaluated as a therapeutic target for antisense oligonucleotide (ASO) therapies39,40,41. Further investigation of these genes in independent datasets is warranted to fully elucidate their potential role in ALS.
Our findings have clear translational potential. ASO-based therapies such as Tofersen (SOD1) and Jacifusen (FUS)42 demonstrate the feasibility of gene-targeted treatment, and ongoing individualized approaches (for example, Silence ALS, n-Lorem) extend this to URVs. Our study increases the proportion of cases with an identifiable genetic risk factor from 11.6% (‘Definitive’ genes) to 15.6% with our validated and new single-variants, a figure that rises to 22.9% when C9orf72 repeat expansions are included. Although not all identified genes will be viable ASO targets, as evidenced by setbacks in trials targeting ATXN2 and C9orf72 (ref. 43), genes harboring variants with high ORs such as ARPP21 represent prime candidates to prioritize for future ASO-based studies.
Our study has limitations. First, we did not perform functional validation for the identified variants; therefore, the precise molecular mechanisms (for example, loss-of-function, gain-of-function or a combination thereof) remain to be determined. Second, by design, the exome analyses in this study precluded the investigation of noncoding variation. However, it is important to note that about three-quarters of the cases included in the discovery cohort and all replication cohorts underwent whole-genome sequencing from which exomes were derived in silico. In addition, the increasing availability of large-scale WGS data, particularly in the UK Biobank and All of Us, provides ample controls, meaning that much of what is needed to build large-scale WGS datasets for future analyses is already available44,45. Third, our analysis is restricted to germline variants and therefore does not address the potential role of somatic mutations, which accumulate in the central nervous system with aging and could therefore explain the late-onset of the disease46. Fourth, we acknowledge the importance of expanding beyond the predominantly European ancestry of participants included in this study. Finally, although our study captures most of the known ALS genetic architecture, a few genes remain undetected. These are either genes associated primarily with repeat expansions (ATXN2 and C9orf72) or genes harboring exceedingly rare variants (VAPB and PFN1).
To conclude, the assembly of the largest exome sequencing dataset for ALS to date, coupled with robust harmonization and replication, enabled the discovery of rare variant contributions to ALS. We show that rare variant analyses yield particularly high returns in ALS compared to common variant GWAS. The identification of several new genes, alongside the confirmation of genes with previous limited evidence, collectively provides a compelling set of potential new targets for translational ALS research.
Methods
Cohorts
This study was approved by the institutional review boards of all participating centers, written informed consent for research was obtained from each participant and the study was approved by the Medical Ethical Testing Committee NedMec and the Biobanks Testing Committee of UMC Utrecht. Cases were included in this study irrespective of their carrier status for variants in known ALS genes.
Discovery cohort
The discovery cohort included 15,862 participants with ALS and 78,683 controls, totaling 94,545 individuals, of which 21,102 were subjected to WGS and 73,443 to WXS. Case cohorts included the Project MinE ALS sequencing consortium (7,614 cases; 2,605 controls)47, the NYGC ALS Consortium (2,650 cases; 342 controls), the ALS Sequencing Consortium (2,851 cases)5, two cohorts from the FALS consortium (1,277 cases; phs001585), the National Institutes of Health (NIH) Exome Sequencing of FALS Project (194 cases; phs000101), two Australian cohorts described in ref. 48 (125 cases, 18 controls) and ref. 49 (568 cases), and a Chinese motor neurone disease (MND) cohort50 (583 cases, 182 controls). All cases were diagnosed with definite, probable or probable laboratory-supported ALS according to the revised El Escorial Criteria51. Control cohorts included 7,323 samples from the National Heart, Lung, and Blood Institute (NHLBI) TOPMed research program52, 49,981 samples from the UK Biobank44 and 18,232 samples across seven cohorts from dbGAP53.
Replication cohort
The replication cohort included 5,404 people with ALS and 133,823 controls, totaling 139,227 participants, all of whom were subjected to WGS. Cohorts include the Project MinE ALS sequencing consortium (1,510 cases; 169 controls), the NYGC ALS consortium (1,257 cases; 69 controls), ALS compute (1,870 cases; 1,820 controls; phs003184) and the UK Biobank (767 cases; 131,765 controls). During sample quality control, people who were duplicates or related up to the second degree to any participant in the discovery cohort were excluded.
Processing of sequencing data
Processing and annotation of sequencing data was performed as described previously54. All raw sequencing data were aligned to the GRCh38 reference genome using BWA-mem55 according to the functional equivalence pipeline described by Regier et al.11 (implementation can be found at https://github.com/maarten-k/realignment). Joint genotyping was performed using a uniform pipeline according to the GATK best practices (v.4.2.6.1)10. Genotype calls with a quality score < 20 were set to missing, variant calls supported by uninformative reads were excluded and multiallelic variants were split into biallelic variants. Male genotypes in nonpseudoautosomal regions on chromosome X were coded as 0 or 1 (according to 0 or 1 allele copies).
Variant annotation
Variants were annotated using snpEff56, dbscSNV57 and Ensembl Release v.105 gene models58. Variants were classified as high-impact when predicted by snpEff to have a high impact (including nonsense mutations, splice acceptor/donors and frameshift mutations) or predicted as potentially splice-altering by dbscSNV (‘ada’ or ‘rf’ score > 0.7). Variants were classified as having moderate impact when predicted as such by snpEff (including missense mutations, in-frame deletions and UTR truncations). For each gene, the impact of a variant was determined by its most severe consequence across protein-coding transcripts.
Sample quality control
Ancestry was estimated by projecting all samples on a reference ancestry space comprising samples from the 1000 Genomes project using the LASER software (v.2.04)59. We retained participants of predominantly European ancestry. We then excluded samples with low genotype call-rate (<0.9), discordant sex or deviating heterozygosity (inbreeding F < -0.1 or F > 0.1). These metrics were calculated in a set of autosomal variants meeting the following criteria: call-rate > 0.9 in each supercohort (discovery: WGS, WXSUKB, WXSother; replication: Project MinE, ALS compute, NYGC, UK Biobank), MAF > 0.01 and, for sex inference, heterozygosity, relatedness and PCA variants were also filtered based on Hardy–Weinberg equilibrium (HWE) (P < 0.0001; for nonpseudoautosomal regions on chromosome X, these were calculated among female participants only) and pruned if in linkage disequilibrium (LD) (r2 < 0.5, window size = 50, step = 5; furthermore, high LD regions were excluded before PCA60). We then excluded samples based on a high exome-wide number of SNVs, INDELs, singletons, high INDEL/SNV ratio or deviating Ti/Tv ratio (thresholds listed in Supplementary Fig. 2). Sample duplicates and relatives up to and including the second degree were identified using KING software61. An unrelated sample set was generated by first excluding samples with five or more relations, followed by iteratively excluding participants with the highest number of relations, resolving ties by prioritizing (in order) ALS over controls and WGS over WXS samples. Furthermore, in the replication cohort, samples that were duplicated or related up to the second degree to any sample in the discovery cohort were excluded. PCA was performed on the unrelated sample set using fastPCA as implemented in plink2 (ref. 62). In the discovery cohort, a distinct cluster was identified on the fourth and fifth PC consisting of an Amish population, which was excluded as the cluster contained only controls (Supplementary Fig. 2f).
Variant quality control
First, GATK variant quality score recalibration was applied to all variants using the training data and annotations as recommended by the GATK best practices10. Variants were excluded if they did not pass variant quality score recalibration, their genotyping rate was <0.9 in any of the supercohorts (discovery: WGS, WXSUKB, WXSother; replication: Project MinE, ALS compute, NYGC, UK Biobank) or if they did not pass the HWE test in controls (P < 0.0001). We then also excluded variants with subpar quality scores and variants located in regions showing signs of batch effects. Potential batch effects were identified by testing whether variant allele counts were associated with cohort membership within control subjects. Firth’s logistic regression with profile penalized likelihood CIs was used to perform these control–control analyses, adjusting for sex and four PCs12. This procedure was repeated for each cohort (that is, 1 = subject in respective cohort, 0 otherwise). In total, 16 cohorts were tested (including all WGS controls versus all WXS controls; cohorts with <100 controls were merged into one cohort) in the discovery cohort and four cohorts were tested in the replication cohort. The minimum P value across these analyses was used as a metric to identify variants associating with probable batch effects. The stringency of various standard variant quality control filters was then increased to eliminate variants exhibiting batch associated calling bias while maintaining maximal sensitivity for unbiased variant calls (Supplementary Fig. 4). Identical thresholds were used for SNVs and INDELs and we also excluded long insertions and deletions (>50 base pairs) and variants coding the reference allele in spanning deletions.
Single-variant analyses
Single-variant analyses were performed for all high and moderate impact variants with MAF < 0.05 and at least MAF > 5 × 10−5 (272,925 variants). For each variant, we tested for an association between ALS status and MAC using Firth’s logistic regression with profile penalized likelihood confidence intervals, which properly controls for type I error when testing rare variants in an unbalanced case–control setting12,63,64,65. We adjusted for sex, ten PCs and total number of rare synonymous variants in each participant. All tests were two-sided, and the Bonferroni correction was used to correct for multiple testing.
Candidate single-variant associations were screened for additional technical biases and excluded if (1) variant concordance <0.9 among 678 between-cohort duplicates included in the unfiltered dataset; (2) showed batch effects among case cohorts (Pcase–case < Pcase–control) based on the same procedure as used in the control–control analyses, where we tested for an association between cohort membership and MAC of the respective variant (that is, 1 = subject in respective cohort, 0 otherwise); (3) the minor allele was supported by one read in >25% of carriers; (4) significant heterogeneity (Phet < 0.001) between this study and a recent ALS common variant GWAS by van Rheenen et al.20, for variants that overlap between both studies.
A targeted analysis was conducted on variants within 51 ALS-linked genes curated by the ALS GCEP (accessed December 2024)14. Certain exons of known ALS genes had lower call rate within subcohorts of the dataset; to provide a more complete investigation of known ALS genes, we therefore did not apply the per-supercohort call-rate filter for this analysis.
URV burden analyses
URV burden analyses were performed using four filtering strategies based on two criteria: (1) variant frequency—either all URVs or singleton-only variants; (2) variant impact—either only high-impact variants (nonsense, splice acceptor/donor and frameshift mutations) or both high- and moderate-impact variants (missense mutations, in-frame deletions and UTR truncations). Burden analyses were performed by testing for an association between ALS status and the aggregate effect of minor alleles observed per sample per functional unit using Firth’s logistic regression with profile penalized likelihood confidence intervals12. Sex, ten PCs and the total number of qualifying synonymous variants in each participant were included as covariates. Tests were retained if there were at least ten carriers across the functional unit tested. Test-statistics across the four filtering strategies were combined using the Cauchy method (ACAT), which is designed to combine results from several statistical tests15. Candidate associations were screened for potential technical biases by assessing biases among case cohorts using the same procedure as used in the control–control analyses. Genes where Pcase–case < Pcase–control were flagged as potentially driven by technical variation.
Genes
Genes were defined using Ensembl gene models (release v.105), including only protein-coding genes without annotation errors.
Domains
Protein coordinates for Interpro domains, coiled coils, transmembrane helices, low complexity regions and cleavage sites were retrieved from Ensembl v.105 (http://dec2021.archive.ensembl.org/biomart/martview/)58. For each transcript, variants were annotated to domains by remapping both the domain coordinates and variant positions to coding sequence (CDS) relative coordinates using the mapToCDS method in RVAT13. Variants up to 12 base pairs from the CDS border (introns and UTRs) were mapped to the respective border. Domains that spanned more than 90% of the width of the transcript were excluded.
Genesets
To identify genesets or pathways associated with ALS, we performed geneset burden analysis on 13,347 GO, KEGG and Reactome genesets from the Molecular Signatures Database (MSigDB v.7.5)17. Genesets including fewer than 5 or more than 1,000 genes were excluded, resulting in a total of 11,777 tested genesets.
Variant co-occurrence analyses
We tested for a cumulative effect of carrying several risk variants among moderate- and high-impact variants in genes classified as ‘Definitive’ according to GCEP14. Participants were grouped into categories based on the number of variants carried: 0 (reference group), 1, 2‥. n variants. Both heterozygous and homozygous variants were treated as single events. We assessed the association between each variant count category and ALS status using Firth’s logistic regression with profile penalized likelihood CIs, with the 0-variant group as the reference category and adjusting for the same covariates used in the geneset burden analyses.
To identify nonrandom co-occurrence of variant pairs, we performed a permutation-based test within the case cohort. The set of variants tested included those defined above as well as C9orf72 (C9) repeat expansion status (available for n = 8,610 cases; 66%). For each pair, we generated an empirical null distribution by performing 100,000 permutations, shuffling the carrier status of one variant relative to the other. An empirical P value was then calculated by comparing the observed co-occurrence count to this null distribution. For each variant pair, only participants with nonmissing genotypes for both variants were included.
Power analyses for the co-occurrence analyses were performed through 10,000 simulations. For each variant pair, joint genotype counts were drawn from a multinomial distribution, with probabilities based on their allele frequencies and a given co-occurrence OR. We then tested then for a depletion or excess of co-occurrence using Fisher’s exact test comparing observed to expected counts under independence.
To test for statistical interactions among variant pairs, we used Firth’s logistic regression in the full case–control cohort. For each pair of variants, we fitted a model including their main effects and their interaction term, adjusting for the same covariates as used in the single-variant analyses.
Because C9 status was available for only a few controls, it was not included in these case–control interaction models.
Survival and age of onset analyses
Age at onset analyses (n = 10,557) were performed using linear regression, testing for an association between age at onset and either MAC (single-variant analyses) or the aggregate effect of minor alleles observed per sample per gene (URV burden analyses). Survival analyses (n = 7,194) were performed using a Cox proportional hazards model, testing for an association between right-censored survival time and either MAC (single-variant analyses) or the aggregate effect of minor alleles observed per sample per gene (URV burden analyses). Both age at onset and survival analyses were adjusted for sex, cohort, ten PCs and the total number of rare synonymous variants in each participant.
Replication analyses
Power analyses were performed through 10,000 simulations in which alleles were drawn from the binomial distribution with the probability set to the MAF of the respective variant. Simulated genotypes were tested for an association with the binary phenotype status using Firth’s logistic regression12. Power was calculated as the fraction of simulations with P values below the specified significance level. Effect sizes estimated in the discovery phase were corrected for winner’s curse bias using the parametric bootstrap approach implemented in the winnerscurse R package66. Power analyses for URV burden tests were performed in a similar manner, substituting MAF for the frequency of carrying at least one minor allele across the gene. These power estimates reflect an idealized scenario without covariate adjustment; in practice, necessary covariate inclusion may reduce power.
The processing of sequencing data and sample quality control were performed identically to the discovery stage; participants who were duplicates or related up to the second degree to any participant in the discovery cohort were excluded. Single-variant analyses were performed identically to those in the discovery analyses and included variants that were significant in the discovery analysis (all variants achieved per-supercohort call-rate >0.9, HWE P value > 0.0001). URV burden analyses were performed identically to those in the discovery analyses and included variants that passed strict quality control filters as applied in the discovery analysis. Meta-analyses were performed using Stouffer’s Z score method weighted by effective sample size, as implemented in METAL software67.
Gene annotation
Candidate genes were annotated with GO terms. The resulting list was summarized using the rrvgo R package68, where a matrix of pairwise semantic similarity scores was first calculated. The terms were subsequently clustered using the default similarity threshold of 0.7 and a representative term for each cluster was selected based on its uniqueness score.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Project MinE data are available here: https://www.projectmine.com/research/data-sharing/. dbGAP datasets used are available under the following accession numbers: ALS compute (phs003184); Alzheimer’s Disease Sequencing Project (ADSP) (phs000572); Autism Sequencing Consortium (ASC) (phs000298); Sweden-Schizophrenia Population-Based Case–Control Exome Sequencing (phs000473); Inflammatory Bowel Disease Exome Sequencing Study (phs001076); Myocardial Infarction Genetics Exome Sequencing Consortium: Ottawa Heart Study (phs000806); Myocardial Infarction Genetics Exome Sequencing Consortium: Malmo Diet and Cancer Study (phs001101); Myocardial Infarction Genetics Exome Sequencing Consortium: U. of Leicester (phs001000); Myocardial Infarction Genetics Exome Sequencing Consortium: Italian Atherosclerosis Thrombosis and Vascular Biology (phs000814); NHLBI GO-ESP: Women’s Health Initiative Exome Sequencing Project (WHI)—WHISP (phs000281); Building on GWAS for NHLBI diseases: the US CHARGE Consortium (CHARGE-S): CHS (phs000667); Building on GWAS for NHLBI Diseases: the US CHARGE Consortium (CHARGE-S): ARIC (phs000668); Building on GWAS for NHLBI diseases: the US CHARGE consortium (CHARGE-S): FHS (phs000651); NHLBI GO-ESP Family Studies: Idiopathic Bronchiectasis (phs000518); NHLBI GO-ESP: Family Studies (Hematological Cancers) (phs000632); NHLBI GO-ESP: Family Studies: (familial atrial fibrillation) (phs000362); NHLBI GO-ESP: Heart Cohorts Exome Sequencing Project (ARIC) (phs000398); NHLBI GO-ESP: Heart Cohorts Exome Sequencing Project (CHS) (phs000400); NHLBI GO-ESP: Heart Cohorts Exome Sequencing Project (FHS) (phs000401); NHLBI GO-ESP: Lung Cohorts Exome Sequencing Project (asthma) (phs000422); NHLBI GO-ESP: Lung Cohorts Exome Sequencing Project (COPDGene) (phs000296); GO-ESP: Family Studies (Thoracic aortic aneurysms leading to acute aortic dissections) (phs000347). NHLBI TOPMed: Genomic Activities such as Whole Genome Sequencing and Related Phenotypes in the Framingham Heart Study (phs000974); NHLBI TOPMed: Genetics of Cardiometabolic Health in the Amish (phs000956); NHLBI TOPMed: Genetic Epidemiology of COPD (COPDGene) (phs000951); NHLBI TOPMed: The Vanderbilt Atrial Fibrillation Registry (VU_AF) (phs001032); NHLBI TOPMed: Cleveland Clinic Atrial Fibrillation (CCAF) Study (phs001189); NHLBI TOPMed: Partners HealthCare Biobank (phs001024); NHLBI TOPMed—NHGRI CCDG: Massachusetts General Hospital (MGH) Atrial Fibrillation Study (phs001062); NHLBI TOPMed: Novel Risk Factors for the Development of Atrial Fibrillation in Women (phs001040); NHLBI TOPMed—NHGRI CCDG: The Vanderbilt AF Ablation Registry (phs000997); NHLBI TOPMed: Heart and Vascular Health Study (HVH) (phs000993); NHLBI TOPMed—NHGRI CCDG: Atherosclerosis Risk in Communities (ARIC) (phs001211); NHLBI TOPMed: The Genetics and Epidemiology of Asthma in Barbados (phs001143); NHLBI TOPMed: Women’s Health Initiative (WHI) (phs001237); NHLBI TOPMed: Whole Genome Sequencing of Venous Thromboembolism (WGS of VTE) (phs001402); NHLBI TOPMed: Trans-Omics for Precision Medicine (TOPMed) Whole Genome Sequencing Project: Cardiovascular Health Study (phs001368). All participants gave written informed consent, and all studies were approved by the institutional review boards of the respective participating centers.
Code availability
All raw sequencing data were aligned to the GRCh38 reference genome using BWA-mem (v.2.2.1) according to the pipeline described by Regier et al.11 (implementation is available on GitHub at https://github.com/maarten-k/realignment and via Zenodo at https://doi.org/10.5281/zenodo.10963076 (ref. 69)). Joint genotyping was performed using a uniform pipeline according to the GATK best practices (v.4.2.6.1). Handling and filtering of VCF files was performed using VCFtools (v.0.1.16), BCFtools (v.1.9) and PLINK (v.1.90b6.21). Ancestry was estimated using LASER (v.2.04). Variants were annotated using Ensembl (GRCh38.105), snpEff (v.5.1d) and dbNSFP (v.4.3a). Sample and variant quality control was performed using PLINK (v.1.90b6.21) and RVAT (v.0.2.0), whereas sample relatedness was inferred using KING (v.2.2.7). Meta-analyses were performed using METAL (v.2011-03-25). GO terms were summarized using the rrvgo R package (v.1.18.0). All downstream analyses were performed using custom R code (performed in R v.3.6.3) that we made available in the RVAT R package (v.0.2.0) (available on GitHub at https://github.com/kennalab/rvat and via Zenodo at https://doi.org/10.5281/zenodo.10973472 (ref. 70)). Other R packages used either as dependencies of RVAT or in other analyses and visualizations are ggplot2 (v.3.4.2), ggrepel (v.0.9.1), dplyr (v.1.0.7), readr (v.2.1.1), stringr (v.1.4.0), tidyr (v.1.1.4), magrittr (v.2.0.1), kinship2 (v.1.9.6), logistf (v.1.25.0), SKAT (v.2.2.5), SummarizedExperiment (v.1.16.1), S4Vectors (v.0.24.4), GenomicRanges (v.1.38.0), IRanges (v.2.20.2), libraDBI (v.1.1.3), RSQLite (v.2.3.1), survival (v.3.1.8), winnerscurse (v.0.1.1). Figures were generated using R v.4.2.3, using rvat (v.0.3.4), dplyr (v.1.1.4), ggplot (v.3.5.1), readr (v.2.15), ggrepel (v.0.9.5), colorblindr (v.0.1.0), stringr (v.1.5.1), tidyr (v.1.3.1) and magrittr (v.2.0.3).
References
Hardiman, O. et al. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primer 3, 17071 (2017).
van Es, M. A. et al. Amyotrophic lateral sclerosis. Lancet 390, 2084–2098 (2017).
Ryan, M., Heverin, M., McLaughlin, R. L. & Hardiman, O. Lifetime risk and heritability of amyotrophic lateral sclerosis. JAMA Neurol. 76, 1367–1374 (2019).
Van Rheenen, W. et al. Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat. Genet. 48, 1043–1048 (2016).
Cirulli, E. T. et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 347, 1436–1441 (2015).
Freischmidt, A. et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 18, 631–636 (2015).
Kenna, K. P. et al. NEK1 variants confer susceptibility to amyotrophic lateral sclerosis. Nat. Genet. 48, 1037–1042 (2016).
Brenner, D. et al. NEK1 mutations in familial amyotrophic lateral sclerosis. Brain 139, e28 (2016).
Nicolas, A. et al. Genome-wide analyses identify KIF5A as a novel ALS gene. Neuron 97, 1268–1283 (2018).
Van der Auwera, G. D. & O’Connor, B. D. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra (O’Reilly Media, Inc., 2020).
Regier, A. A. et al. Functional equivalence of genome sequencing analysis pipelines enables harmonized variant calling across human genetics projects. Nat. Commun. 9, 4038 (2018).
Firth, D. Bias reduction of maximum likelihood estimates. Biometrika 80, 27–38 (1993).
Hop, P. J. & Kenna, K. P. RVAT: Rare Variant Association Toolkit. GitHub https://github.com/kennalab/rvat (2026).
Dilliott, A. A. et al. Clinical testing panels for ALS: global distribution, consistency, and challenges. Amyotroph. Lateral Scler. Front. Degener. 24, 420–435 (2023).
Liu, Y. et al. ACAT: a fast and powerful p value combination method for rare-variant analysis in sequencing studies. Am. J. Hum. Genet. 104, 410–421 (2019).
Scarian, E. et al. The role of VCP mutations in the spectrum of amyotrophic lateral sclerosis—frontotemporal dementia. Front. Neurol. 13, 841394 (2022).
Liberzon, A. et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740 (2011).
Cooper-Knock, J. et al. Mutations in the glycosyltransferase domain of GLT8D1 are associated with familial amyotrophic lateral sclerosis. Cell Rep. 26, 2298–2306 (2019).
Dols-Icardo, O. et al. Identification of a pathogenic mutation in ARPP21 in patients with amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 96, 132–139 (2025).
van Rheenen, W. et al. Common and rare variant association analyses in amyotrophic lateral sclerosis identify 15 risk loci with distinct genetic architectures and neuron-specific biology. Nat. Genet. 53, 1636–1648 (2021).
Farhan, S. M. K. et al. Exome sequencing in amyotrophic lateral sclerosis implicates a novel gene, DNAJC7, encoding a heat-shock protein. Nat. Neurosci. 22, 1966–1974 (2019).
Van Blitterswijk, M. et al. Evidence for an oligogenic basis of amyotrophic lateral sclerosis. Hum. Mol. Genet. 21, 3776–3784 (2012).
Iacoangeli, A. et al. Oligogenic structure of amyotrophic lateral sclerosis has genetic testing, counselling and therapeutic implications. J. Neurol. Neurosurg. Psychiatry 96, 928–936 (2025).
Kriegenburg, F., Bas, L., Gao, J., Ungermann, C. & Kraft, C. The multi-functional SNARE protein Ykt6 in autophagosomal fusion processes. Cell Cycle 18, 639–651 (2019).
McGrath, K. et al. A conformational switch driven by phosphorylation regulates the activity of the evolutionarily conserved SNARE Ykt6. Proc. Natl Acad. Sci. USA 118, e2016730118 (2021).
Ma, M. et al. Homozygous missense variants in YKT6 result in loss of function and are associated with developmental delay, with or without severe infantile liver disease and risk for hepatocellular carcinoma. Genet. Med. 26, 101125 (2024).
Zein-Sabatto, H., Collawn, J., Chang, C. & Miller, M. A. An RNAi screen in C. elegans for genes that play a role in secretion and cleavage of VAPB MSP domain. Preprint at bioRxiv http://biorxiv.org/lookup/doi/10.1101/2021.01.02.425092 (2021).
Kamemura, K. et al. Secretion of endoplasmic reticulum protein VAPB/ALS8 requires topological inversion. Nat. Commun. 15, 8777 (2024).
Arnoux, A. et al. Evaluation of a 5-HT2B receptor agonist in a murine model of amyotrophic lateral sclerosis. Sci Rep. 11, 23582 (2021).
El Oussini, H. et al. Degeneration of serotonin neurons triggers spasticity in amyotrophic lateral sclerosis. Ann. Neurol. 82, 444–456 (2017).
Tracey, T. J., Kirk, S. E., Steyn, F. J. & Ngo, S. T. The role of lipids in the central nervous system and their pathological implications in amyotrophic lateral sclerosis. Semin. Cell Dev. Biol. 112, 69–81 (2021).
Mohassel, P. et al. Childhood amyotrophic lateral sclerosis caused by excess sphingolipid synthesis. Nat. Med. 27, 1197–1204 (2021).
Rehfeld, F. et al. The RNA-binding protein ARPP21 controls dendritic branching by functionally opposing the miRNA it hosts. Nat. Commun. 9, 1235 (2018).
De Decker, M. et al. C21ORF2 mutations point towards primary cilia dysfunction in amyotrophic lateral sclerosis. Brain 148, 803–816 (2025).
Willemse, S. W. et al. UNC13A in amyotrophic lateral sclerosis: from genetic association to therapeutic target. J. Neurol. Neurosurg. Psychiatry 94, 649–656 (2023).
Dittman, J. S. Unc13: a multifunctional synaptic marvel. Curr. Opin. Neurobiol. 57, 17–25 (2019).
Kalantari, S. et al. Expanding the KIF4A-associated phenotype. Am. J. Med. Genet. A. 185, 3728–3739 (2021).
Hirokawa, N., Noda, Y., Tanaka, Y. & Niwa, S. Kinesin superfamily motor proteins and intracellular transport. Nat. Rev. Mol. Cell Biol. 10, 682–696 (2009).
Yamashita, T. et al. A role for calpain-dependent cleavage of TDP-43 in amyotrophic lateral sclerosis pathology. Nat. Commun. 3, 1307 (2012).
Rao, M. V., Campbell, J., Palaniappan, A., Kumar, A. & Nixon, R. A. Calpastatin inhibits motor neuron death and increases survival of hSOD1G93A mice. J. Neurochem. 137, 253–265 (2016).
Cohen, J. et al. Next steps in development for AMX0114: an antisense oligonucleotide targeting calpain-2, a critical effector of axonal Degeneration. In MDA Clinical & Scientific Conference (Amylyx Pharmaceuticals, Inc., 2024); https://www.amylyx.com/document/next-steps-in-development-for-amx0114-an-antisense-oligonucleotide-targeting-calpain-2-a-critical-effector-of-axonal-degeneration.pdf
Korobeynikov, V. A., Lyashchenko, A. K., Blanco-Redondo, B., Jafar-Nejad, P. & Shneider, N. A. Antisense oligonucleotide silencing of FUS expression as a therapeutic approach in amyotrophic lateral sclerosis. Nat. Med. 28, 104–116 (2022).
van den Berg, L. H. et al. Safety, tolerability, and pharmacokinetics of antisense oligonucleotide BIIB078 in adults with C9orf72-associated amyotrophic lateral sclerosis: a phase 1, randomised, double blinded, placebo-controlled, multiple ascending dose study. Lancet Neurol. 23, 901–912 (2024).
Sudlow, C. et al. UK Biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 12, e1001779 (2015).
The All of Us Research Program Genomics Investigators. Genomic data in the All of Us Research Program. Nature 627, 340–346 (2024).
Miller, M. B. et al. Somatic genomic changes in single Alzheimer’s disease neurons. Nature 604, 714–722 (2022).
Project MinE ALS Sequencing Consortium. Project MinE: study design and pilot analyses of a large-scale whole-genome sequencing study in amyotrophic lateral sclerosis. Eur. J. Hum. Genet. 26, 1537–1546 (2018).
Garton, F. C. et al. Whole exome sequencing and DNA methylation analysis in a clinical amyotrophic lateral sclerosis cohort. Mol. Genet. Genomic Med. 5, 418–428 (2017).
McCann, E. P. et al. Evidence for polygenic and oligogenic basis of Australian sporadic amyotrophic lateral sclerosis. J. Med. Genet. 58, 87–95 (2021).
Gratten, J. et al. Whole-exome sequencing in amyotrophic lateral sclerosis suggests NEK1 is a risk gene in Chinese. Genome Med. 9, 97 (2017).
Brooks, B. R., Miller, R. G., Swash, M. & Munsat, T. L. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 1, 293–299 (2000).
Taliun, D. et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 590, 290–299 (2021).
Tryka, K. A. et al. NCBI’s database of genotypes and phenotypes: dbGaP. Nucleic Acids Res. 42, D975–D979 (2014).
Hop, P. J. et al. Systematic rare variant analyses identify RAB32 as a susceptibility gene for familial Parkinson’s disease. Nat. Genet. 56, 1371–1376 (2024).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w 1118; iso-2; iso-3. Fly (Austin) 6, 80–92 (2012).
Jian, X., Boerwinkle, E. & Liu, X. In silico prediction of splice-altering single nucleotide variants in the human genome. Nucleic Acids Res. 42, 13534–13544 (2014).
Zerbino, D. R. et al. Ensembl 2018. Nucleic Acids Res. 46, D754–D761 (2018).
Wang, C., Zhan, X., Liang, L., Abecasis, G. R. & Lin, X. Improved ancestry estimation for both genotyping and sequencing data using projection procrustes analysis and genotype Imputation. Am. J. Hum. Genet. 96, 926–937 (2015).
Anderson, C. A. et al. Data quality control in genetic case-control association studies. Nat. Protoc. 5, 1564–1573 (2010).
Manichaikul, A. et al. Robust relationship inference in genome-wide association studies. Bioinformatics 26, 2867–2873 (2010).
Galinsky, K. J. et al. Fast principal-component analysis reveals convergent evolution of ADH1B in Europe and East Asia. Am. J. Hum. Genet. 98, 456–472 (2016).
Heinze, G. & Schemper, M. A solution to the problem of separation in logistic regression. Stat. Med. 21, 2409–2419 (2002).
Ma, C., Blackwell, T., Boehnke, M., Scott, L. J. & GoT2D Investigators. Recommended joint and meta-analysis strategies for case-control association testing of single low-count variants: joint and meta-analysis of low-count variants. Genet. Epidemiol. 37, 539–550 (2013).
Mbatchou, J. et al. Computationally efficient whole-genome regression for quantitative and binary traits. Nat. Genet. 53, 1097–1103 (2021).
Forde, A., Hemani, G. & Ferguson, J. Review and further developments in statistical corrections for Winner’s Curse in genetic association studies. PLoS Genet. 19, e1010546 (2023).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Sayols, S. rrvgo: a Bioconductor package for interpreting lists of Gene Ontology terms. MicroPubl. Biol. https://doi.org/10.17912/micropub.biology.000811 (2023).
Kooyman, M. maarten-k/realignment: pipeline for exome and WGS(DF3) pipeline. Zenodo https://doi.org/10.5281/zenodo.10963075 (2024).
Hop, P. J. et al. KennaLab/rvat: v.2.0.9. Zenodo https://doi.org/10.5281/zenodo.10959178 (2025).
Acknowledgements
This research has been conducted using the UK Biobank Resource under application number 48361. This project has received funding from the European Research Council under the European Union’s Horizon 2020 research and innovation program (grant agreement no. 772376—EScORIAL. The collaboration project is cofunded by the PPP Allowance made available by Health–Holland, Top Sector Life Sciences and Health, to stimulate public–private partnerships. This study was supported by the ALS Foundation Netherlands. This work was sponsored by NWO–Domain Science for the use of supercomputer facilities. K.P.K. is supported by grants from the Dutch Research Council (grant no. ZonMW-VIDI 91719350) and the ALS Foundation Netherlands. Data from the New York Genome Center ALS consortium were used. All consortium members are listed in Supplementary Information. All New York Genome Center ALS Consortium activities are supported by the ALS Association (ALSA, grant no. 19-SI-459) and the Tow Foundation. R.L.M. receives support from the MND Association (grant no. 891-791). This publication has emanated from research conducted with the financial support of Taighde Éireann—Research Ireland, under grant no. 21/RC/10294_P2 at FutureNeuro Research Ireland Centre for Translational Brain Science. N.T. acknowledges the Italian Ministry of Health (grant no. RF-2021–12374238 ‘DRIVEALS’) and the Italian Ministry of University and Research (Dipartimenti di Eccellenza Program 2023–2027—Department of Pathophysiology and Transplantation, Università degli Studi di Milano). E.R. is supported by the G. Harry Sheppard Memorial Research Fund and the ALS Society of Canada. Project MinE Belgium was supported by a grant from IWT (no. 140935), the ALS Liga België, the National Lottery of Belgium and the KU Leuven Opening the Future Fund. N.A.B. gratefully acknowledges the use of the services and facilities of Koc University-KUTTAM and the generous support of Suna and Inan Kirac Foundation. National Health and Medical Research Council of Australia, grant no. 1176913. M.A.v.E. and V.S. serve on the board of the European Reference Network (ERN) on neuromuscular disease (EURO–NMD). This publication has been supported by the ERN–NMD. F.C.G. was supported by the Scott Sullivan Fellowship from MND and Me and MND Research Australia. P.M.A. is supported by the Knut and Alice Wallenberg Foundation, the Fort Knox Charity Foundation, The Olsson and Olsson Foundation and the Swedish Brain Foundation. A.A.K. is funded by The Motor Neurone Disease Association (grant no. 1122462), NIHR Maudsley Biomedical Research Centre, ALS Association Milton Safenowitz Research Fellowship (grant no. RE19765), the Darby Rimmer MND Foundation, LifeArc (grant no. RE23378), MRC (grant no. MR/Z505705/1) and the Dementia Consortium (grant no. 1819242). A.A.K is supported by the UK Dementia Research Institute through UK DRI Ltd, funded principally by the Medical Research Council. A.A.-C. is an NIHR Senior Investigator (grant no. NIHR202421) and a Visiting Professor at the Perron Institute for Neurological and Translational Science, Australia. A.A.-C. is supported through the UK MND Research Institute (funders MND Association of England, Wales and Northern Ireland, My Name’5 Doddie Foundation, MND Scotland, LifeArc, MRC and NIHR) and through the Alan Davidson Foundation and Darby Rimmer Foundation. This study represents independent research part funded by the National Institute for Health Research (NIHR) Biomedical Research Centre at South London and Maudsley NHS Foundation Trust and King’s College London. Samples used in this research were in part obtained from the UK National DNA Bank for MND Research, funded by the MND Association and the Wellcome Trust. We would like to thank people with MND and their families for their participation in this project. We acknowledge sample management undertaken by Biobanking Solutions funded by the Medical Research Council at the Centre for Integrated Genomic Medical Research, University of Manchester. This work was supported in part by the Intramural Research Program of the NIH, the National Institute on Aging (grant no. 1ZIAAG000933) and the National Institute of Neurological Disorders and Stroke (grant no. ZIANS003154). The contributions of the NIH authors were made as part of their official duties as NIH federal employees, are in compliance with agency policy requirements, and are considered Works of the United States Government. However, the findings and conclusions presented in this paper are those of the authors and do not necessarily reflect the views of NIH or the US Department of Health and Human Services.
Author information
Authors and Affiliations
Consortia
Contributions
Data processing and analysis were performed by P.J.H., M.K., B.J.K., K.P.K. and J.H.V. Sample ascertainment and data generation was carried out by P.J.H., M.K., B.J.K., R.A.J.Z., K.R.V.E., Y.W., C.H.v.D., E.B., W.v.R., P.B., J.J.F.A.v.V., A.A.K., A.I., J.C.-K., B.N.S., S.T., A.J.v.d.K., V.F., V.D., Y.L., Y.S., D.B.R., K.L.W., R.L.M., J.H., Y.H., C.-Y.C., E.T., H.R., E.A., E.J.N.G., M.A.v.E., R.J.P., S.M.K.F., F.C.G., A.F.M., P.A.M., R.D.H., D.F., L.Š., H.H., A.L.N., R.J.C., L.B., B.R., B.K., J.Z., T.S., J.S.M.P., M.G., M.P., P. Corcia, P.V., P. Couratier, M.W., M.C.K., R.P., I.P.B., M.d.C., N.A.B., C.I., P.M.A., L.Z., E.R., I.R.M., N.D., G.A.R., B.J.T., N.T., A.C., V.S., O.H., H.P., M.B.H., C.L.D., J.D.G., J.E.L., P.V.D., K.E.M., P.J.S., C.E.S., A.A.-C., L.H.V.d.B., K.P.K. and J.H.V. Writing of the paper was performed by P.J.H., K.P.K. and J.H.V. Study supervision was carried out by K.P.K. and J.H.V.
Corresponding authors
Ethics declarations
Competing interests
J.H., Y.H., C.-Y.C., E.A.T. and H.R. are current or previous employees of Biogen. P.M.A. has served on advisory boards for Biogen, Regeneron, uniQure, Orphazyme A/S and Mitsubishi Pharma (mostly paid to institution). A.A.K. received consulting fees from the UK National Endowment for Science, Technology and the Arts (NESTA). N.T. received compensation for consulting services from Amylyx Pharmaceutical, Biogen, Italfarmaco and Zambon Biotech SA. V.S. received compensation for consulting services and/or speaking activities from AveXis, Cytokinetics, Italfarmaco, Liquidweb S.r.l., Novartis Pharma AG, Amylyx Pharmaceuticals, Biogen and Zambon Biotech SA, receives or has received research support from the Italian Ministry of Health, AriSLA and E-Rare Joint Transnational Call and is on the Editorial Board of Amyotrophic Lateral Sclerosis and Frontotemporal Degeneration, European Neurology, American Journal of Neurodegenerative Diseases, Frontiers in Neurology and Exploration of Neuroprotective Therapy. P.V.D. has served in advisory boards for Biogen, CSL Behring, Alexion Pharmaceuticals, Ferrer, QurAlis, Cytokinetics, argenx, UCB, Muna Therapeutics, Alector, Augustine Therapeutics, VectorY, Zambon, Amylyx, Novartis, Prilenia, Verge Genomics, Sapreme Technologies, Trace Neuroscience and NRG Therapeutics (paid to institution). P.V.D. has received speaker fees from Biogen and Amylyx (paid to institution). P.V.D. is supported by the E. von Behring Chair for Neuromuscular and Neurodegenerative Disorders (paid to institution). A.A.-C. reports consultancies or advisory boards for Amylyx, Apellis, Biogen, Clene Therapeutics, Cytokinetics, GenieUs, GSK, Lilly, Mitsubishi Tanabe Pharma, Novartis, OrionPharma, Quralis, Sano Genetics, Sanofi, Voyager Therapeutics and Wave Pharmaceuticals and the following patents or patent applications: WO2024121173A1 and EP 25 306 172.5. J.H.V. reports to have sponsored research agreements with Biogen, Eli Lilly, Trace and Astra Zeneca. The other authors declare no competing interests.
Peer review
Peer review information
Nature Genetics thanks Rosa Rademakers, Caroline Rouaux and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Rare single variant quantile-quantile plot and post-hoc analyses.
a, Quantile-quantile (qq) plot of observed single variant association −log10 (P-values) versus expected −log10 (P-values) under the null model. The red dotted line indicates the exome-wide significance threshold (P = 1.83 × 10−7). λ indicates the observed genomic inflation factor. λ1000 indicates the genomic inflation factor for an equivalent study of 1,000 cases and 1,000 controls. b, Candidate single variant associations were screened for potential technical biases by assessing (i) variant concordance across 678 duplicates between cohorts present in the unfiltered dataset, (ii) biases among case cohorts using the same procedure as used in the control-control analyses, (iii) whether the minor allele was supported a low number of reads, and (iv) significant heterogeneity (Phet < 0.001) between this study and the most recent ALS common variant GWAS by van Rheenen et al.20 (for variants that overlap between both studies). c, Call-rates per cohort for the four variants in the targeted GCEP analysis that exhibited subpar call-rates. d, Forest plots comparing odds ratios (OR; center) with 95% confidence intervals (CI; error bars) for the variants shown in c. Results are displayed for analyses including all cohorts versus analyses restricted to cohorts meeting a call-rate threshold of 0.9. Association statistics were estimated using Firth’s logistic regression with profile penalized likelihood confidence intervals. P-values are two-tailed and presented uncorrected for multiple testing.
Extended Data Fig. 2 Mutation plots for genes identified in rare single variant or URV burden analyses.
The upper panel shows the coding sequence of the respective genes, with the y-axis showing the −log10(P-value) for single variants. The lower panel shows the whole-gene domains colored by the −log10(P-value). Only variants and genes supported by replication are displayed. For variants identified in the rare single variant analysis, all variants with MAF < 0.05 are displayed; for genes identified in the URV burden analysis URVs are displayed (≤5 carriers). Association statistics were estimated using Firth’s logistic regression with profile penalized likelihood confidence intervals. P-values for the gene-based and domain-based tests are from the ACAT omnibus test combining the four variant filtering strategies (see Methods). P-values shown are from two-tailed tests and are presented uncorrected for multiple testing.
Extended Data Fig. 3 Ultra-rare variant burden analyses.
a, Quantile-quantile (qq) plot of observed association −log10 (P-values) versus expected −log10 (P-values) under the null model for gene, domain and gene set analyses. b–d, Forest plots depicting odds-ratios (center) and 95% confidence intervals (error bars) of the exome-wide significant genes (b), domains (c) and gene sets (d). e–g, Significant associations were screened for technical biases arising from the inclusion of multiple case cohorts by testing for an association with cohort among ALS cases (e, gene-based analyses; f, domain-based analyses, g, gene-set analyses). h–j, Comparison of association P-values of ultra-rare gene burden analyses excluding INDELs (y-axis) compared to the main analysis including INDELs (x-axis) (h, gene-based analyses; i, domain-based analyses; j, gene-set analyses). k–m, Comparison of association P-values of ultra-rare gene burden analyses including high-impact variants only (y-axis) compared to including both moderate-impact and high-impact variants (x-axis) (k, gene-based analyses; l, domain-based analyses; m, gene-set analyses). Association statistics were estimated using Firth’s logistic regression with profile penalized likelihood confidence intervals. P-values for the gene-based, domain-based and gene set analyses are from the ACAT omnibus test combining the four variant filtering strategies (see Methods). P-values are two-tailed and are presented uncorrected for multiple testing.
Extended Data Fig. 4 Co-occurrence analyses.
a, b, Within-gene co-occurrence among genes identified in the ultra-rare variant burden analyses. The forest plots show the odds-ratio (OR; center) and 95% confidence intervals (CI; error bars), comparing the risk for individuals carrying a single qualifying variant to those carrying multiple variants. The panels depict genes in which co-occurrence of ultra-rare variants was observed (a), as well as genes in which co-occurrence of an ultra-rare variant with a more common variant from the single variant analyses was observed (b). c, d, Cumulative burden of carrying multiple high- or moderate-impact variants among Definitive ALS genes as curated by the GCEP. Shown are the odds-ratios (OR; center) and 95% confidence intervals (CI; error bars) (x-axis), stratified by the number of risk variants carried (y-axis). c, Analysis at the variant level, where each qualifying variant is counted individually. d, Analysis at the gene level, where carrying one or more qualifying variants within the same gene is counted as a single event. e, Forest plots showing the odds ratios (OR; center) and 95% confidence intervals (CI; error bars) for variant pairs. For each low-frequency variant (MAF 0.01-0.05), the odds-ratio is shown for carrying only the index variant (yellow, panel), only the target variant (green, y-axis), and carrying both variants (blue). The interaction odds-ratio (orange) indicates whether the variants act synergistically (OR > 1) or antagonistically (OR < 1). Association statistics are two-tailed and were estimated using Firth’s logistic regression with profile penalized likelihood confidence intervals. P-values are two-tailed and presented uncorrected for multiple testing.
Extended Data Fig. 5 Variant co-occurrence heatmap.
Heatmap displaying the observed co-occurrences in ALS patients among variant pairs. For genes identified in the URV burden analysis, an individual is considered a carrier if they had at least one qualifying variant in that gene. The lower triangle shows the log-transformed odds ratio, where red indicates a synergistic effect (greater co-occurrence than expected under an additive risk model), and blue indicates an antagonistic effect (lesser co-occurrence than expected under an additive risk model). The upper triangle shows the statistical significance from a permutation analysis (−log10(P-value)), with darker green indicating a more significant P-value. Pairs for which there was no co-occurrence in the full dataset (cases and controls) are greyed out. In addition to variants identified in this study, we included C9orf72 repeat expansion status in these co-occurrence analyses, which was available for 8,610 (66%) of cases. P-values are two-tailed and presented uncorrected for multiple testing.
Extended Data Fig. 6 Age at onset and survival analyses.
a, b, Age at onset analyses (left) and survival analyses (right) for significant variants (a) and significant genes among URV burden analyses (b). Estimates in years (center) and 95% confidence intervals (error bars) are shown for age at onset analyses in 10,557 participants (linear model), and log-transformed hazard ratios (center) and 95% confidence intervals (error bars) are shown for survival analyses in 7,194 participants (Cox proportional hazards model). Closed circles indicate variants or genes that reached significance corrected for the number of variants or genes tested.
Extended Data Fig. 7 Independent validation of genes with limited prior evidence.
Forest plots comparing odds ratios (OR; center) with 95% confidence intervals (CI; error bars) for main analyses versus sensitivity analyses that exclude potential overlap with prior studies. a, Plot comparing the ARPP21 p.P563L association from the full dataset to an analysis excluding four carriers who could potentially overlap with previous family studies. b, Plot comparing the CFAP410 p.V58L association in the full discovery (ncases = 11,763; ncontrols = 69,677) and replication (ncases = 4,781; ncontrols = 130,927) cohorts to a sensitivity analysis that excludes duplicated or genetically related individuals from the discovery (ncases = 4,474; ncontrols = 65,643) and replication (ncases = 3,698; ncontrol = 130,802) sets. c, Plot comparing the DNAJC7 URV association in the full discovery (ncases = 13,138; ncontrols = 69,775) and replication (ncases = 4,781; ncontrols = 130,928) cohorts to a sensitivity analysis that excludes individuals from cohorts that potentially overlap with the original study from the discovery (ncases = 7,606; ncontrols = 59,926) and replication (ncases = 4,591; ncontrols = 130,928) sets. Association statistics were estimated using Firth’s logistic regression with profile penalized likelihood confidence intervals.
Supplementary information
Supplementary Information (download PDF )
Supplementary Tables 1 and 2, Figs. 1–12 and Consortium lists.
Supplementary Data 1 (download CSV )
Summary statistics for the exome-wide rare single variant analyses.
Supplementary Data 2 (download XLSX )
Summary statistics for the rare single variant associations among ALS-linked genes curated by the ALS Gene Curation Expert Panel (GCEP). ‘GCEP sv’ includes the associations presented in the main text and Fig. 1. ‘GCEP sv (no MAF cutoff)’ includes all associations without the application of a lower MAF threshold.
Supplementary Data 3 (download CSV )
Summary statistics for the ultra-rare gene-based analyses: ACAT omnibus test across the four filtering strategies.
Supplementary Data 4 (download CSV )
Summary statistics for ultra-rare gene-based analyses: results for each of the four filtering strategies.
Supplementary Data 5 (download CSV )
Summary statistics for the ultra-rare domain-based analyses: ACAT omnibus test across the four filtering strategies.
Supplementary Data 6 (download CSV )
Summary statistics for the ultra-rare domain-based analyses: results for each of the four filtering strategies.
Supplementary Data 7 (download CSV )
Summary statistics for the ultra-rare gene set analyses: ACAT omnibus test across the four filtering strategies. Includes test-statistics both including and excluding significant single genes.
Supplementary Data 8 (download CSV )
Summary statistics for the ultra-rare gene set analyses: results for each of the four filtering strategies.
Supplementary Data 9 (download XLSX )
Summary statistics for co-occurrence analyses.
Supplementary Data 10 (download XLSX )
Summary statistics for age of onset and survival analyses.
Supplementary Data 11 (download XLSX )
Full list of Gene Ontology terms for candidate genes.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hop, P.J., Kooyman, M., Kenna, B.J. et al. Large-scale exome analyses reveal new rare variant contributions in amyotrophic lateral sclerosis. Nat Genet (2026). https://doi.org/10.1038/s41588-026-02535-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41588-026-02535-9
