
Abstract
Macrophages are among the most abundant immune cells in solid tumors, yet how macrophage lineage and spatial organization shape antitumor immunity remains unclear. Here we uncovered a division of labor between tissue-resident CD206hi and CD206lo interstitial macrophage (IM) subsets and Ly6c2+Fn1+Vcan+ recruited macrophages (recMacs) in lung cancer. Using single-cell and spatial transcriptomics, we identified chemokine-expressing IM subsets with opposing functions. Cxcl13+CD206hi IMs, Cxcl9+CD206hi IMs and Cxcl10+CD206hi IMs positioned along bronchovascular regions drove tertiary lymphoid structure formation, lymphocyte recruitment and tumor control, whereas Ccl2+ IMs, localized within tumor regions, recruited protumorigenic Ly6c2+Fn1+Vcan+ recMacs. In addition, Ly6C+CD11b+ monocyte-derived dendritic cells (moDCs) functioned as immunosuppressive antigen-presenting cells in tumor-draining lymph nodes. During neoantigen vaccination, CCR5 blockade with maraviroc selectively inhibited antigen-bearing moDC migration, enhancing dendritic cell-mediated antitumor immunity. These findings showed how macrophage lineage and spatial compartmentalization govern tumor immunity and identified strategies to preserve protective IM functions, while disrupting macrophage-driven immunosuppression.
Similar content being viewed by others

Main
Macrophages are central regulators of tissue homeostasis and immune defense. In the lung, two tissue-resident populations coexist: CD11chiCD11bloCD64+SiglecF+ alveolar macrophages (AMs), which patrol the airspaces, and CD11c+CD11b+CD64+ interstitial macrophages (IMs), which reside within the interstitium1,2,3,4,5,6. AMs are tissue-specific macrophages, whereas CD11c+CD11b+CD64+ IMs are conserved across organs and species and display diverse transcriptional and functional states2,5,7,8,9,10,11,12. During inflammation and tumor growth, these resident macrophages are joined by Ly6C+CCR2+CD11b+ recruited macrophages (recMacs) derived from circulating Ly6C+ monocytes13,14,15. Within the tumor microenvironment (TME), recruited Ly6C+ monocytes differentiate either into CD206+ macrophages that acquire inflammatory, reparative or immunosuppressive states, or into Ly6C+MHCII+CD88+CD26lo monocyte-derived dendritic cells (moDCs)16. moDCs migrate to draining lymph nodes through CCR5, distinguishing them from classical dendritic cells (DCs) that rely on CCR7 (ref. 15).
Differentiating IMs from recMacs in vivo remains challenging because both populations share broad surface markers such as CD11c, CD11b, CD64, CD88, CD206 and MHCII, and because recMacs progressively downregulate Ly6C after tissue entry13,17,18,19,20; however, single-cell RNA sequencing (scRNA-seq) has uncovered clear transcriptional signatures that resolve these lineages and reveal far greater macrophage diversity than previously appreciated1. Although both AMs and many IM subsets express CD206 at high levels, CD206 (encoded by Mrc1) is neither macrophage-restricted nor predictive of tumor-promoting function, despite its widespread use as an ‘M2-like’ marker21,22,23. CD206hi AMs are heterogeneous, encompassing over a dozen distinct transcriptional programs such as reparative growth (IGF1+ and AREG+), antimicrobial defense (TLR2+, CXCL5+ and CCL18+), cholesterol metabolism (CYP51A1+ and INSIG1+) and metallothionein production (MT1X+MT2A+), with their proportions tightly regulated under homeostatic conditions1,3,11,24,25,26. Similarly, C1q+Itgam+C5ar1+ IMs and Ly6c2+Fn1+Vcan+ recMacs comprise multiple transcriptionally and functionally distinct populations including chemokine-specialized subsets such as Cxcl13+ and Ccl5+ IMs that are not captured by flow cytometry using canonical markers such as CD11b, Trem2 or CD169.
IMs can be subdivided into CD206hi IMs and CD206lo IMs with distinct gene expression programs and anatomical niches. CD206hi IMs coexpress Folr2, Cd163 and Mmp9 and exhibit variable levels of Cx3cr1, MhcII, Cd38, Siglec1, Lyve1, Clec10a and Timd4 (refs. 1,3,27). CD206lo IMs express Tmem119, Mmp12, Mmp13, Cd11c and Ccr2, and have high expression of Cx3cr1 and MhcII1,3,27,28,29. Many IM subsets produce chemokines, including Cxcl13, Cxcl9, Cxcl10, Ccl2 and Ccl24 (refs. 1,30), suggesting that IM heterogeneity may strongly influence immune outcomes. Such heterogeneity indicates the need for integrative transcriptional and spatial profiling to define macrophage subset function in vivo. Although no current genetic model selectively targets a single macrophage subset, with the exception of Ccl24Cre30, several systems allow interrogation of broader macrophage populations spanning multiple transcriptionally defined states.
Selective depletion of CD206hi IMs using Pf4CreCx3cr1DTR mice has shown that CD206hi IMs are required for tertiary lymphoid structure (TLS) formation and lymphocyte recruitment in the lung1. Because TLS abundance correlates with improved outcomes in lung cancer, here we investigated whether IM subsets differentially regulated tumor progression. Specifically, we asked whether Cxcl13+CD206hi IMs, Cxcl9+CD206hi IMs and Cxcl10+CD206hi IMs supported antitumor immunity, and whether Ccl2-expressing IMs promoted tumor growth by recruiting protumorigenic Ly6c2+Fn1+Vcan+ recMacs. Our findings revealed a spatially organized and functionally dichotomous IM network that governed the balance between tumor-suppressive and tumor-promoting macrophage programs.
Results
RecMacs display protumor transcriptional signatures
To identify macrophage subsets associated with tumor regression or progression, we flow-sorted extravascular CD64+CD11b+ mononuclear phagocytes, which include IMs and recMacs, from the lungs of wild-type (WT) C57BL/6 mice 16 days after intravenous injection of 4×105 B16F10 melanoma cells and analyzed them by scRNA-seq in two separate datasets (Fig. 1a and Extended Data Fig. 1a). Unbiased Uniform Manifold Approximation and Projection (UMAP) clustering of the scRNA-seq data resolved 23 immune cell populations defined by curated markers and differentially expressed genes (DEGs) (Fig. 1b and Extended Data Fig. 1b), including Ear1 and Cidec for AMs, Mafb, Folr2 and Nes for IMs, Cd14, Ly6c2 and Fn1 for recMacs, Xcr1 for DC1 and Ccr7 for migratory DCs11. Depletion of circulating monocytes enables clear separation of resident IMs as Pf4+C5ar1+Mafb+ cells and recMacs as Ly6c2+Fn1+Vcan+1, so we resolved AMs, IMs, recMacs and DCs based on these criteria (Fig. 1b and Extended Data Fig. 1b). Despite intravascular CD45 labeling, a small residual population of intravascular CD11b+ mononuclear phagocytes, consisting of Sell+ classical and Ace+ nonclassical monocytes was still detected in one of the scRNA-seq datasets (Extended Data Fig. 1b). C1q-expressing IMs were further characterized based on Mrc1 (CD206) expression (Fig. 1c,d). CD206hi IMs expressed Folr2, Cd163, Mmp9 and higher levels of Pf4 compared to CD206lo IMs, with a fraction of CD206hi IMs expressing Lyve1 (Fig. 1d), whereas CD206lo IMs expressed Ccr2, Mmp12, Mmp13 and Tmem119 (Fig. 1c,d and Extended Data Fig. 1c–i) Within the scRNA-seq cluster of Cd14+Csfr1+ recMacs, genes were enriched for Ly6c2, Vcan and Thbs1 with high levels of Ccr2 and Fn1 (Fig. 1c,e and Extended Data Fig. 1c–i).

a, Unbiased UMAP clustering of scRNA-seq data from flow-sorted extravascular CD64⁺CD11b⁺ mononuclear phagocytes isolated from the lungs of C57BL/6 WT mice on day 16 after intravenous injection of 4 × 105 B16F10 melanoma cells. Clusters include AMs, IMs, recruited macrophages (recMacs), dendritic cells 1 (DC1), dendritic cells 2 (DC2), inflammatory DCs (Inf DC2), migratory DCs (Mig DCs), pre-DCs, plasmacytoid DCs (pDCs), neutrophils, eosinophils, basophils, mast cells, CD8⁺ T cells, CD4⁺ T cells, regulatory T (Treg) cells, double-negative T (DN T) cells, γδ T cells, natural killer (NK) cells, NK progenitors, B cells, innate lymphoid cells (ILC2), melanoma cells and cycling cells. b, Key signature genes defining the unbiased clusters as in a. c, UMAP of Pf4+C5ar1+Mafb+ IMs and Ly6c2+Fn1+Vcan+ recMacs as in a. d, Feature plots showing the expression of C1qb and Pf4 in IMs (left), expression of Folr2, Cd163, Mmp9 and Lyve1 in CD206hi IMs (middle) and expression of Ccr2, Mmp12, Mmp13 and Tmem119 in CD206lo IMs (right). e, Feature plots showing expression for Fn1, Ly6c2, Vcan and Thbs1 in recMacs. f, Feature plots showing the expression of Cxcl13, Cxcl9 and Cxcl10 in CD206hi IMs (left) and Spp1, Vegfa, Arg1 and Cd274 in recMacs and Trem2 in recMacs and IMs (right).
We next assessed the expression of genes linked to anti- or protumorigenic programs. CD206hi IMs predominantly expressed antitumorigenic chemokines, including Cxcl13, Cxcl9 and Cxcl10, whereas CD206lo IMs and Fn1+Vcan+ recMacs expressed the protumorigenic genes Ccl2; all populations expressed Trem2 (Fig. 1f and Extended Data Fig. 1j)31. Fn1+Vcan+ recMacs were further enriched for canonical tumor-promoting transcripts, such as Spp1, Vegfa, Arg1 and Cd274 (which encodes PD-L1) (Fig. 1f and Extended Data Fig. 1h). Thus, IMs and recMacs displayed distinct gene expression profiles, with CD206hi IMs expressing chemokine transcripts associated with lymphocyte positioning (Cxcl13, Cxcl9 and Cxcl10), whereas Ccl2 expression was observed on both IMs and recMacs, with recMacs enriched for transcripts previously linked to tumor-promoting macrophage programs in lung cancer models32.
CD206hi IMs restrain tumor growth by enabling TLS formation
IMs differentiate into at least ten distinct chemokine-expressing (IMck) subsets, including IMck0 (nonspecific chemokine expression), IMck1 (Ccl2, Ccl7, Ccl12 and some Cxcl14), IMck2–IMck4 (Ccl3, Ccl4, Ccl5, Cxcl1, Cxcl2 and Cxcl3), IMck5 (Ccl8), IMck6 (Ccl6 and Ccl9), IMck7 (Cxcl9 and Cxcl10), IMck8 (Cxcl13) and IMck9 (Ccl24) which have protective roles in pulmonary inflammation and infection1. To determine the function of IMck7 (Cxcl9 and Cxcl10) and IMck8 (Cxcl13) in cancer, we used Pf4CreCx3cr1DTR mice, in which Pf4Cre activity is restricted to mature immune cells that transcriptionally express Pf4, including megakaryocytes, peritoneal macrophages and IMs, but only IMs coexpress Cx3cr1 and therefore selectively express the diphtheria toxin (DT) receptor (DTR)1. Following a single intravenous injection of DT, only high expressing Pf4⁺Cx3cr1⁺ IMs were targeted, resulting in preferential depletion of CD206hi IMs, including Cxcl13+ CD206hi IMs, Cxcl9+ and Cxcl10+ CD206hi IMs, as well as subsets of CD206hi IMs expressing Ccl6, Ccl8, Ccl9 and Ccl24 (ref. 1). CD206hi IMs, which also highly express FOLR2, were maximally depleted by day 7 post-DT injection and recovered by day 15 (Fig. 2a). Hematoxylin and eosin (H&E) staining, PECAM-1 (CD31) and SMA staining showed comparable microvessel density and tissue area in control Cx3cr1LSL-DTR mice (hereafter Cx3cr1DTR) and Pf4CreCx3cr1DTR mice at day 10 post-DT (Supplementary Fig. 1), indicating that lung morphology and vascular integrity were not affected by depletion of CD206hi IMs.

a, Representative flow cytometry plots showing the gating strategy for the identification of CD11b⁺CD64⁺CD206hi/loFOLR2+/− IMs (left) and time course analysis of the depletion of CD206hiFOLR2+ IMs at days 0, 7 and 15 (middle) and a scatter-plot showing the frequency of CD206hiFOLR2+ IM at day 0–15 (right) after intravenous injection of 700 ng DT in Pf4CreCx3cr1DTR mice. Two independent experiments, n = 2 mice per time point. b, Representative images of tumor-burdened lungs (left) and scatter-plot showing the number of surface metastases per lung (right) in Cx3cr1DTR (n = 5) and Pf4CreCx3cr1DTR (n = 5) mice at day 16 after intravenous injection of B16F10 melanoma cells. Shown is one of four independent experiments. Student’s two-tailed t-test with P < 0.0001. mets, metastases. c, Representative H&E-stained lung sections (left) and quantification of number of tumors per lung section (right) in AgerCreERT2KP mice reconstituted with Cx3cr1DTR (n = 5) and Pf4CreCx3cr1DTR (n = 5) BM. Scale bar, 1,000 µm. One of two independent experiments is shown. Student’s two-tailed t-test with P = 0.0008. d, Representative H&E-stained lung sections (left) and quantification of the number of tumors per section (right) in Cx3cr1DTR (n = 5) and Pf4CreCx3cr1DTR (n = 5) mice at day 16 post-intravenous injection of KPAR1.3 adenocarcinoma cells. Scale bar, 1,000 µm. One of four independent experiments is shown. Student’s two-tailed t-test with P < 0.0001. e, Representative immunohistochemistry images of B220 and CD3e antibodies staining (left) and quantification of B cell and T cell numbers (right) in the TME of Cx3cr1DTR (n = 5) and Pf4CreCx3cr1DTR (n = 5) mice at day 16 after intravenous injection with B16F10 melanoma cells. Scale bar, 100 µm. One of two independent experiments is shown. Student’s two-tailed t-test with P = 0.0027 (B cells) or P = 0.0006 (T cells). f, Representative immunohistochemistry images of B220 and CD3e antibodies staining (left) and quantification of B cell and T cell numbers (right) in the lungs of AgerCreERT2KP mice reconstituted with Cx3cr1DTR (n = 5) and Pf4CreCx3cr1DTR (n = 5) BM. Scale bar, 100 µm. One of two independent experiments is shown. Student’s two-tailed t-test with P = 0.0013 (B cells), P = 0.0008 (T cells). g, Representative immunohistochemistry images of B220 and CD3e antibodies staining (left) and quantification of histopathological scores (right) in the lungs of Cx3cr1DTR (n = 5) and Pf4creCx3cr1DTR (n = 5) mice at day 16 after intravenous injection of KPAR1.3 adenocarcinoma cells. Whole-lung images (stitched, scale bar 1,000 µm, top left) and magnified (×4 and ×10) views (scale bar, 100 µm, bottom left). One of three independent experiments is shown. Student’s two-tailed t-test with P = 0.0005. h, ELISA measurements of CXCL9, CXCL10 and CXCL13 in homogenized lungs from Cx3cr1DTR (n = 5) and Pf4CreCx3cr1DTR (n = 5) mice on day 16 after intravenous injection of KPAR1.3 adenocarcinoma. Two independent experiments. Student’s two-tailed t-test with P < 0.0001, P < 0.0001 and P = 0.0398. All data are represented as mean ± s.e.m. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
To assess the role of CD206hi IMs in tumor control, we intravenously injected melanoma (B16F10) or lung adenocarcinoma (KPAR1.3) cells into Cx3cr1DTR and Pf4CreCx3cr1DTR mice, followed by intravenous DT administration on days 3 and 7 post-tumor inoculation and quantified tumor burden at day 16. In addition, we used a primary lung adenocarcinoma model generated by crossing tamoxifen-inducible AgerCreERT2 mice, which drives Cre recombination selectively in alveolar type I epithelial cells, with KP mice, which are heterozygous for conditional oncogenic KrasG12D and carry a single floxed Trp53fl/+ allele, resulting in lung adenocarcinoma following Cre-mediated recombination (hereafter AgerCreERT2KP). To enable selective depletion of CD206hi IMs in the hematopoietic compartment, lethally irradiated AgerCreERT2KP mice were reconstituted with bone marrow (BM) from either Cx3cr1DTR (control) or Pf4CreCx3cr1DTR BM. Following 6 weeks of hematopoietic reconstitution, mice were placed on tamoxifen-containing chow for 30 days to induce Cre-mediated tumor initiation. Mice then received weekly intravenous injections of DT (500 ng per mouse) for 14 weeks, followed by tumor analysis in the lungs (Extended Data Fig. 2). In all three cancer models, DT administration in the Pf4CreCx3cr1DTR mice led to marked increased tumor burden (~3.7-fold in melanoma B16F10, 7.2-fold in lung adenocarcinoma KPAR1.3 and 2.3-fold in the spontaneous lung adenocarcinoma AgerCreERT2KP mice) relative to control Cx3cr1DTR mice (Fig. 2b–d and Supplementary Fig. 2a,b). Moreover, in the melanoma (B16F10) model, depletion of CD206hi IMs in Pf4CreCx3cr1DTR mice with a single intravenous DT dose administered on day 4 reproduced the phenotype observed with two DT doses administered on days 3 and 7 (Supplementary Fig. 2c), indicating that transient depletion of CD206hi IM after tumor seeding was sufficient to promote tumor growth.
We next used immunohistochemistry to analyze lymphocyte infiltration and TLS formation in melanoma (B16F10) and lung adenocarcinoma (KPAR1.3) transfer models, as well as in the AgerCreERT2KP spontaneous lung adenocarcinoma model. Control Cx3cr1DTR mice injected with B16F10 cells did not form visible TLS along the airways, similar with previous reports33, but had dense B220+ B cell and CD3+ T cell aggregates in the TME and peribronchial regions (Fig. 2e and Supplementary Fig. 2d). Following CD206hi IM depletion in Pf4CreCx3cr1DTR mice, B cell and T cell aggregates were markedly reduced (~80% reduction) by day 16 (Fig. 2e). In contrast, the KPAR1.3 lung adenocarcinoma transfer model and AgerCreERT2KP spontaneous mouse lung adenocarcinoma model, formed TLSs, which had compact, architecturally organized structures containing juxtaposed B220⁺ B cell clusters and CD3⁺ T cell zones (Fig. 2f,g). TLS were completely lost, along with the increased tumor burden, in the KPAR1.3 adenocarcinoma model and AgerCreERT2KP mice lung adenocarcinoma model following CD206hi IM depletion in Pf4CreCx3cr1DTR mice (Fig. 2c,d,f,g). ELISA of lung tissue from the lung adenocarcinoma KPAR1.3 model at day 16 post-tumor inoculation revealed substantial reduction in CXCL9, CXCL10 and CXCL13 protein levels (~2.7-fold, 2.6-fold and 1.5-fold, respectively) in CD206hi IM-deficient Pf4CreCx3cr1DTR mice compared to Cx3cr1DTR control mice (Fig. 2h). Together, these findings indicated that reduced lymphocyte recruitment and loss of TLS formation in CD206hi IM-depleted melanoma (B16F10) and lung adenocarcinoma (KPAR1.3) transfer models and spontaneous lung adenocarcinoma model (AgerCreERT2KP mice) were associated with accelerated tumor progression.
RecMacs and IMs establish spatial chemokine niches
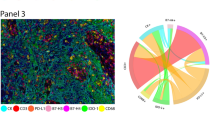
We next performed spatial transcriptomic profiling on tumor-bearing lungs collected at day 16 post-cancer cell inoculation from two WT mice bearing B16F10 melanoma lung metastases and two WT mice bearing KPAR1.3 lung adenocarcinoma tumors using the 10x Xenium platform and a targeted gene panel enriched for pan leukocytes, myeloid cells, chemokines and stromal transcripts (Supplementary Figs. 3 and 4). This subcellular-resolution analysis enabled direct spatial mapping of distinct macrophage populations, including Car4+ and Chil3+ AMs, Mafb+ and C1qc+ IMs, Fn1+ and Vcan+ recMacs, together with chemokine expression and major structural components of the TME (Fig. 3a). Structural components included Acta2⁺ stromal cells demarcating tumor boundaries, Tubb3⁺ neural structures, Lyve1⁺ lymphatic vessels, Pecam1⁺ blood vessels and Epcam⁺ epithelial cells (Fig. 3a and Extended Data Fig. 3). To validate spatial registration and chemokine detection, the Xenium section also included a lung-draining lymph node from one KPAR1.3 tumor-bearing mouse, which served as an internal control. As expected, Cxcl13 was confined to the B cell zone, whereas the the DC-associated chemokines Cxcl16, Ccl17 and Ccl22 were enriched in the T cell zone (Fig. 3a and Extended Data Fig. 3), confirming accurate chemokine localization and lymph node compartmentalization34,35. Within lung tumor sections, Xenium analysis revealed that tumor nodules were delineated by Acta2 mRNA expression, which encodes α-smooth muscle actin and marks activated stromal cells, whereas H&E staining independently confirmed that these Acta2⁺ stromal structures outlined tumor regions within the lung parenchyma (Fig. 3a,b). The TME from one KPAR1.3 tumor-bearing mouse was highly innervated (Tubb3 and Nes), whereas lymphatic (Lyve1 and Pdpn), vascular (Pecam1) and epithelial (Epcam) markers were comparatively sparse (Fig. 3a and Extended Data Fig. 3). Spatial mapping showed that AMs (annotated as Car4, Chil3 and Ear1 expressing cells) were largely excluded from tumor regions, whereas DCs (Zbtb46, Flt3 and Xcr1) were distributed throughout the lung, including the TME (Fig. 3a and Extended Data Fig. 3). RecMacs, defined by Fn1, Vcan, Plac8, Clec4n and Cd9, were abundant within the TME (Fig. 3a and Extended Data Fig. 3). Based on integrated scRNA-seq clustering and protein expression analyses, Mafb+C1qc+ IMs were annotated into two major subsets: a CD206hi IM subset characterized by high expression of Folr2, Cd163 and Mmp9, and a CD206lo IM subset characterized by high expression of Mmp12 (Figs. 1d and 2a and Supplementary Fig. 5)1,3. Spatial mapping revealed that CD206hi IMs (Folr2, Cd163 and Mmp9) localized preferentially adjacent to the bronchial airways and the visceral pleura, whereas CD206lo IMs (Mmp12) and Fn1+Vcan+ recMacs predominated in tumor-dense regions (Fig. 3a).

a, Xenium spatial transcriptomic analysis showing in situ gene expression of markers of structural cells (Acta2, Tubb3, Lyve1, Pecam1 and Epcam), AMs (Car4 and Chil3), recMacs (Fn1, Vcan and Plac8), IMs (Mafb, C1qc, Mmp12, Mmp9 and Cd163) and chemokines (Cxcl9, Cxcl13, Cxcl14 and Cxcl16) in the lungs of C57BL/6 WT mice at day 16 post-intravenous injection of 4 × 105 KPAR1.3 adenocarcinoma cells. White arrows, activated stromal structures within the tumor microenvironment. Scale bar, 1,000 µm. b, Representative H&E-stained section of the lung of C57BL/6 WT mice at day 16 post-intravenous injection of 4 × 105 KPAR1.3 adenocarcinoma cells, as in a. Scale bar, 1,000 µm. c, High-resolution imaging of spatial transcriptomics as in a showing expression of Folr2, CD163, Cxcl13 and Epcam. Red arrows highlight Cxcl13+CD206hi IMs. Scale bar, 100 µm. d,e, Spatial transcriptomics quantification of tumor associated macrophages in adenocarcinoma (n = 2) (d) or melanoma (n = 2) (e).
We next analyzed chemokine organization within the TME. Chemokine mapping revealed region-specific expression of Ccl3, Ccl4, Ccl6, Ccl7, Ccl8, Ccl9, Ccl17, Ccl22, Cxcl3, Cxcl9, Cxcl10, Cxcl13, Cxcl14 and Cxcl16 (Fig. 3a and Extended Data Fig. 3). Cxcl13 was enriched along bronchial airways coincident with Cd163⁺Folr2⁺ CD206hi IMs (Fig. 3a and Extended Data Figs. 4 and 5). In contrast, Cxcl14 localized to the outer tumor margins, whereas Cxcl16, a CXCR6 ligand associated with effector T cells and ILC2s, was concentrated in the tumor cores (Fig. 3a). High-resolution imaging indicated that CD163⁺Folr2⁺ CD206hi IMs lining Epcam⁺ bronchial epithelial airways coexpressed Cxcl13 in both the melanoma and adenocarcinoma models (Fig. 3c and Extended Data Fig. 5). Quantitative graph-based clustering of single-cell-resolved Xenium transcriptomic data was used to group cells based on transcriptional similarity, while Xenium Explorer visualization was used to validate the spatial localization of AMs, IMs and recMacs in the tumor-bearing lungs, as defined by marker-based annotation (Supplementary Figs. 3 and 4). Graph-based clustering of single-cell-resolved Xenium transcriptomic data indicated that Chil3+Ear1+Ccl6+ AMs and Cd163⁺Folr2⁺Pf4+ CD206hi IMs occupied airway- and vessel-associated niches, respectively and were less represented within the TME, whereas ~60–71% Vcan+Cx3cr1+Mafb+ recMacs and ~19–25% Mmp12+C5ar1+Mafb+CD206lo IMs of the total lung macrophages preferentially populated the tumor cores and margins (Fig. 3d,e). These data indicated a compartmentalized myeloid cell architecture that shaped the chemokine landscape and directed immune cell recruitment.
IM-derived CCL2 promotes tumor growth by recruiting recMacs
The chemokine CCL2 drives angiogenesis and monocyte recruitment16,36. The scRNA-seq analysis showed that Ccl2 was expressed by many mononuclear phagocyte populations, and Xenium spatial transcriptomics visualized Ccl2 expression throughout the TME in two WT mice bearing B16F10 melanoma lung metastases and two WT mice bearing KPAR1.3 lung adenocarcinoma (Fig. 4a,b and Extended Data Fig. 6). Furthermore, spatial transcriptomics localized expression of Ccl2 within the same tumor regions as those occupied by C1qb+ macrophages in B16F10 melanoma and KPAR1.3 adenocarcinoma-bearing lungs at day 16 post-tumor cell transfer (Fig. 4c and Extended Data Fig. 6). High-resolution imaging of Xenium transcriptomic data indicated that CD206lo IMs (Mmp12) and recMacs (Vcan) in the TME of B16F10 melanoma and KPAR1.3 adenocarcinoma-bearing lungs at day 16 coexpressed Ccl2 (Fig. 4d and Extended Data Fig. 7). To determine whether CD206hi and CD206lo IM-derived CCL2 specifically drove tumor progression, we lethally irradiated CD45.1 WT C57BL/6 mice and reconstituted them with CD45.2 WT or Ccl2−/− BM, an approach that preserves endothelial cell-derived CCL2 in the host, followed by injection with B16F10 melanoma cells 6 weeks after BM transfer. At 16 days post-B16F10 melanoma cell inoculation, BM chimeric Ccl2−/− mice showed an approximately 10.8-fold reduction in lung tumor burden and a 2.7-fold decrease in Ly6C+CD11b+ recMacs accumulation compared to BM chimeric WT mice (Fig. 4e and Supplementary Fig. 6a).

a, Xenium spatial transcriptomic analysis showing the expression of Ccl2, Il1b, Nos2, Arg1 and Ccl24 in the lungs of C57BL/6 WT mice at day 16 post-intravenous injection of 4 × 105 KPAR1.3 adenocarcinoma cells. b, UMAP visualization of Ccl2 expression in myeloid cells as in Fig. 1a. c, Spatial transcriptomics as in a showing the overlay of C1qb, Cd163 and Ccl2. Scale bar, 1,000 µm. d, High-resolution imaging of spatial transcriptomics as in a showing the expression of Vcan, Mmp12 and Ccl2 within the TME. Red arrows show Ccl2+ CD206lo IMs and Ccl2+ recMacs. Scale bar, 100 µm. e, Representative lung images (left), quantification of surface metastases (middle) and recMacs per lung (right) in mice reconstituted with CD45.2 WT (n = 5) or Ccl2−/− (n = 5) BM on day 16 post-intravenous injection with B16F10 melanoma cells. One of three independent experiments is shown. Statistical significance determined using Student’s two-tailed t-test P < 0.0001 (metastases), P = 0.0097 (recMacs). f, Histogram showing expression of CD45.1 in CD11b+CD64+ IMs, Ly6C+CD11b+ recMacs or CD11c+MHCII+CD64− DCs in C57BL/6 CD45.1 WT mice that were injected intraperitoneally with 25 mg kg−1 busulfan or lethally (900 rad) irradiated, reconstituted with WT CD45.2 BM and analyzed at 4 weeks after busulfan or 6 weeks after lethal irradiation. Three independent experiments with n = 3 mice per group. g, Representative lung images (left) and quantification of surface metastases (right) in irradiated or busulfan-conditioned C57BL/6 CD45.1 WT mice as in f reconstituted with WT (radiation, n = 4; busulfan, n = 5) or Ccl2−/− (radiation, n = 3; busulfan, n = 5) BM at day 16 post-intravenous injection of B16F10 melanoma cells. One of two independent experiments is shown. Statistical significance was determined via one-way analysis of variance (ANOVA) followed by Bonferroni’s multiple comparisons test with P = 0.0012 (radiation WT versus radiation Ccl2−/−), P = 0.001 (radiation Ccl2−/− versus busulfan WT) and P = 0.0034 (radiation Ccl2−/− versus busulfan Ccl2−/−). All data are represented as mean ± s.e.m. **P < 0.01; ****P < 0.0001.
Because lethal irradiation resulted in complete replacement of the host myeloid compartment by BM donor-derived cells, irradiated mice reconstituted with Ccl2−/− BM lacked CCL2 expression across all mononuclear phagocyte populations, including IMs, recMacs and DCs. To selectively preserve CCL2 expression by long-lived, tissue-resident IMs, while eliminating CCL2 production from short-lived circulating myeloid populations such as recMacs and DCs, we used conditioning with busulfan, which depletes hematopoietic progenitors and circulating myeloid cells while sparing long-lived IMs. CD45.1 WT recipients that were either treated intraperitoneally with 25 mg kg−1 busulfan or lethally irradiated were reconstituted with either CD45.2 WT or CD45.2 Ccl2−/− BM. Four weeks after Ccl2−/− BM transfer in the busulfan-treated mice, flow cytometric analysis of lung tissue using CD45.1 (host) and CD45.2 (donor) congenic markers indicated that CD11b⁺CD64⁺ IMs were predominantly CD45.1 WT host-derived and retained CCL2 expression, whereas Ly6C⁺CD11b⁺ recMacs and CD11c⁺MHCII⁺CD88⁻ DCs were CD45.2 Ccl2−/− donor-derived and therefore lacked CCL2 expression (Fig. 4f). In contrast, irradiation resulted in BM donor-derived replacement of all mononuclear phagocyte populations (Fig. 4f). Consistent with this reconstitution pattern, CCL2-producing IMs were detected in busulfan-treated BM chimeric mice but were absent in irradiated BM chimeric mice reconstituted with Ccl2−/− BM (Fig. 4f).
When irradiated mice and busulfan-treated mice were intravenously injected with B16F10 melanoma cells at 6 weeks or 4 weeks post-conditioning, respectively, and lung metastases were quantified at day 16 post-tumor inoculation, irradiated BM chimeric mice reconstituted with Ccl2−/− BM exhibited a 7.0-fold reduction in lung metastatic burden compared to busulfan-treated BM chimeric mice reconstituted with Ccl2−/− BM (Fig. 4e,g). In contrast, busulfan-treated Ccl2−/− BM chimeric mice, which retained CCL2-producing IMs but lacked CCL2 production by recMacs and DCs, exhibited metastatic tumor growth comparable to busulfan-treated WT BM chimeric mice and irradiated WT BM chimeric mice (Fig. 4g and Supplementary Fig. 6b). Together, these data demonstrated that IM-derived CCL2, rather than endothelial, recMac or DC derived CCL2, was required for recMac recruitment and the progression of B16F10 melanoma lung metastasis model.
MoDCs suppress vaccine-induced antitumor immunity
Ly6C+CD11b+ recMacs migrate to the draining lymph nodes, where they differentiate into moDCs that function as antigen-presenting cells and induce antigen-specific regulatory T cells that limit DC-driven type 2 inflammation and cytotoxic T cell responses against tumors15,16,37. moDCs depend on CCR5-CCL5 signaling, rather than on CCR7, for entry into the lymph node15. UMAP analysis of the scRNA-seq dataset from the extravascular CD64+CD11b+ mononuclear phagocytes as above indicated expression of Ccr5 in Ly6c2⁺ recMacs and Ccl5 expression in Ccr7⁺ migratory DCs (Fig. 5a). Spatial transcriptomic analysis of lung-draining lymph nodes from mice injected with KPAR1.3 adenocarcinoma cells further indicated that Ccl5 expression was localized in the Cd3+ T cell zone, where migratory DCs reside (Fig. 5b). To test whether CCR5-dependent moDC migration elicited an immunosuppressive function in cancer, we set up competitive mixed BM chimera to selectively disrupt CCR5 expression in monocyte-derived cells while preserving CCR5 expression in other lineages37. To set up competitive BM chimeras that could test the functional contribution of CCR5⁺ moDCs, independent of effects on conventional DCs, lethally irradiated CD45.2 C57BL/6 WT recipient mice were reconstituted with mixed whole BM consisting of 80% Ccr2−/− to 20% WT BM or 80% Ccr2−/− to 20% Ccr5−/− BM. In this competitive setting, CCR2-sufficient BM cells outcompete CCR2-deficent cells in the reconstitution of CCR2-dependent cell types, resulting in the complete repopulation of lymph node monocytes by Ccr5−/–derived BM cells (or WT BM cells in the control mice)37. In contrast, DCs, which do not depend on CCR2 for tissue or lymph node migration, are reconstituted according to the original BM input ratio, thereby maintaining normal functional DC representation across groups37. Because DCs do not require CCR2 or CCR5 to enter the lymph nodes37, this experimental setup could test the role of CCR5⁺ moDCs, and implicitly Ly6C+CD11b+ recMacs, in the B16F10 melanoma and KPAR1.3 lung adenocarcinoma models. Six weeks after BM reconstitution, mice were intravenously injected with B16F10 melanoma or KPAR1.3 lung adenocarcinoma cells, and lung metastases were quantified at day 16 post-tumor inoculation. Chimeric mice reconstituted with 80% Ccr2−/− to 20% Ccr5−/− BM, which lacked CCR5 expression in monocyte-derived cells, had a 6.8-fold reduction in lung metastatic melanoma burden and a 9.3-fold reduction in lung adenocarcinoma burden compared to 80% Ccr2−/− to 20% WT control chimeric mice (Fig. 5c,d and Supplementary Fig. 7a,b). These data demonstrated that CCR5 expression in monocyte-derived cells was required to support lung tumor growth in both the B16F10 melanoma and KPAR1.3 adenocarcinoma models.

a, UMAP visualization of Ccr5, Ccl5 and Ccr7 expression in myeloid cells as in Fig. 1a. b, Xenium spatial transcriptomics expression of Cd19, Cd3e and Ccl5 in the lung-draining lymph nodes from C57BL/6 WT mice on day 16 post-intravenous injection of 4 × 105 KPAR1.3 adenocarcinoma cells. Scale bar, 100 µm. c, Representative lung images (left) and quantification of surface metastases (right) in lethally irradiated C57BL/6 WT mice reconstituted with Ccr2−/−:WT (80:20) or Ccr2−/−:Ccr5−/− (80:20) BM on day 16 post-intravenous injection with B16F10 melanoma cells. One of two independent experiments with n = 5 mice per group is shown. Statistical significance was determined via a Student’s two-tailed t-test with P < 0.0001. d, Representative H&E staining (left) and quantification of the number of tumors per section (right) in the lungs of Ccr2−/−:WT (80:20) (n = 5) and Ccr2−/−:Ccr5−/− (80:20) (n = 4) mixed BM chimeras as in c on day 16 post-intravenous injection with KPAR adenocarcinoma cells. Two independent experiments were conducted. Statistical significance was determined via a Student’s two-tailed t-test with P < 0.0001. e, Representative flow cytometry plots showing the gating strategy for the identification of antigen (Ag)+ Ly6C⁺ moDCs and CD26⁺ DCs (top, left to right) and quantification of the frequency of Ag+ monocytes (bottom right) in the lung-draining lymph nodes of WT mice treated with or without 500 μg maraviroc 3 h before intranasal administration of 5 μg OVA Ag + 50 μg poly(I:C) (top and middle) or nondraining distal skin lymph nodes from mice receiving 5 μg OVA Ag + 50 μg poly(I:C) without maraviroc (bottom) and analyzed 24 h post-antigen administration. Three independent experiments were conducted with n = 4 per group. Statistical significance was determined via a Student’s two-tailed t-test with P < 0.0001. f, Schematics showing the experimental flow (top), representative lung images (middle) and number of surface metastases per lung (bottom) in WT mice treated with PBS (n = 5), neoantigen (NeoAg) peptides (n = 5), NeoAg peptides + poly(I:C) (n = 4) or NeoAg peptides + poly(I:C) + maraviroc (n = 5) on day 14 and 7 before intravenous injection with B16F10 melanoma cells and analyzed on day 16 post-tumor injection. Two independent experiments were conducted. Statistical significance was determined via one-way ANOVA followed by Bonferroni’s multiple comparison test with P = 0.0035 (NeoAg + poly(I:C) versus NeoAg + poly(I:C) + maraviroc). All data are represented as mean ± s.e.m. **P < 0.01; ***P < 0.001; ****P < 0.0001.
To define the functional contribution of antigen-bearing Ly6C⁺ moDCs15, we transiently blocked CCR5-dependent moDC migration using the small-molecule CCR5 antagonist maraviroc during DC-based vaccination. To measure directly the effects of maraviroc on the migration of antigen-bearing Ly6C⁺ moDCs to the lung-draining lymph nodes, WT C57BL/6 mice were injected intraperitoneally with maraviroc or vehicle, followed 3 h later by intranasal administration of fluorescently labeled ovalbumin (OVA)+poly(I:C). At 24 h after antigen delivery, both antigen-bearing CD26⁺ DCs and Ly6C⁺ moDCs were detected in the lung-draining lymph nodes (Fig. 5e); however, mice treated with maraviroc exhibited an approximately 76% reduction in antigen-bearing Ly6C⁺ moDCs compared to vehicle-treated mice (Fig. 5e)15, indicating impaired moDC migration without affecting DC migration. To determine the duration of CCR5 blockade, maraviroc was administered 3, 24 or 48 h before intranasal OVA+poly(I:C) delivery. Inhibition of antigen-bearing Ly6C⁺ moDC migration was observed only when maraviroc was administered 3 h before antigen delivery, but not at 24 or 48 h (Extended Data Fig. 8), indicating that CCR5 blockade was transient. This temporal restriction is consistent with the reported short half-life of maraviroc (~16 h)38.
Based on these kinetics, we next examined whether blocking migration of antigen-bearing Ly6C⁺ moDC during the priming phase of DC-based cancer vaccination altered antitumor immunity. Mice were injected intraperitoneally with maraviroc or vehicle and vaccinated 3 h later with melanoma neoantigen peptides+poly(I:C). Control groups received neoantigen peptides without poly(I:C) or were left unvaccinated. Mice were then challenged intravenously with B16F10 melanoma cells, and lung metastases were quantified at day 16 post-tumor inoculation. Mice that received neoantigen peptides+ poly(I:C) together with maraviroc exhibited a 7.5-fold reduction in metastatic burden compared to unvaccinated mice and a 3.1-fold reduction compared to mice vaccinated with neoantigen peptides+poly(I:C) without maraviroc (Fig. 5f and Supplementary Fig. 7c). These data indicated that Ly6C⁺ moDCs in the draining lymph node acted as immunosuppressive antigen-presenting cells during early vaccination and that transient CCR5 blockade during this defined window enhanced the efficacy of DC-based neoantigen vaccination.
Discussion
Here we defined a spatially organized division of labor between tissue-resident IMs and recMacs in lung cancer. Our data indicated that tissue-resident macrophage function was best predicted by anatomical niche, with distinct chemokine programs organizing either protective immunity or tumor-promoting circuits. In three complementary lung cancer models, melanoma or lung adenocarcinoma cell transfer models and AgerCreERT2KP spontaneous lung adenocarcinoma, CD206hi IMs were positioned along bronchovascular and pleural regions and acted as local producers of CXCL13, CXCL9 and CXCL10, supporting lymphocyte recruitment and the formation of TLS required for effective tumor control1. Removal of CD206hi IMs dismantled this immunologic scaffold and accelerated tumor growth.
Spatial transcriptomic analysis indicated that AMs were largely excluded from tumor regions, whereas Fn1+Vcan+ recMacs accumulated directly within tumor-dense regions and adopted a different transcriptional state (expression of Vegfa, Spp1, Arg1, Cd274 and Ccl2) and reinforced tumor progression, consistent with previous descriptions of monocyte-derived macrophages in tumor regions16,36,39. Dissection of hematopoietic versus nonhematopoietic sources identified CD206hi and CD206lo IM-derived CCL2 as the dominant source that drove the continued recruitment of Ly6C+CD11b+ recMacs into the tumor. Loss of IM-derived CCL2 sharply curtailed Ly6C+CD11b+ recMac influx and limited tumor growth, suggesting it represents a nonredundant pathway. These data refined prevailing models emphasizing stromal, endothelial or cancer cell expression of CCL2 (ref. 16) and identifies tissue-resident Ccl2+ IMs as key contributors to the suppressive myeloid environment. Because both CD206hi and CD206lo IM subsets produced Ccl2, it is possible that both IM subsets contributed to this protumor loop, likely with distinct timing and context.
Our data further linked recMac accumulation to immunosuppressive antigen presentation in tumor-draining lymph nodes. Ly6C+ recMacs differentiated into moDCs that migrated to lymph nodes through CCR5-dependent trafficking and suppressed adaptive immunity, distinguishing them from conventional DCs, which rely on CCR7-dependent migration to elicit adaptive immune activation15,37,40. Using competitive mixed BM chimeras to selectively disrupt CCR5 in monocyte-derived cells, while maintaining normal DC representation, we found that loss of CCR5 in the monocyte lineage reduced metastatic burden in both melanoma and lung adenocarcinoma models. These findings supported an in vivo requirement for CCR5-dependent monocyte-derived cells in sustaining lung tumor growth and aligned with previous work showing that monocyte-derived antigen-presenting cells can restrain effective T cell priming15,37.
We also asked whether migration of antigen-bearing moDCs to lung-draining lymph nodes, and their associated suppressive effects, could be transiently disrupted during neoantigen vaccination designed to enhance DC cross-priming using an established adjuvant poly(I:C) in conjunction with tumor-specific peptides41,42. Brief inhibition of CCR5 with maraviroc before a DC-based neoantigen vaccination selectively reduced migration of antigen-bearing Ly6C+ moDCs to lung-draining lymph nodes without impairing migration of antigen-bearing DCs. This short intervention enhanced vaccine efficacy and reduced metastatic burden, indicating that early monocyte-derived antigen presentation constrained protective immunity and that temporary disruption of this pathway favored DC-mediated priming. This approach differed fundamentally from previous studies using prolonged systemic CCR5 blockade in patients with cancer38,43,44, which broadly affected multiple CCR5-expressing immune cell populations beyond monocytes, including effector T cells, tissue-resident macrophages and neutrophils, encompassing cell types with both pro- and antitumor functions. Last, although recruited monocytes in our cancer models predominantly functioned as immunosuppressive cells, scRNA-seq also identified inflammatory monocyte clusters that, in specific contexts such as fungal, viral and bacterial infections, outcompete immunosuppressive programs and support type 2 and type 1 T cell priming45,46,47,48,49,50.
In summary, macrophage function in vivo is best understood by integrating spatial context with transcriptional state rather than relying on surface markers alone. New experimental models will be required to selectively interrogate individual chemokine-defined IM subsets, as current genetic tools do not resolve these populations with sufficient precision. Macrophage programs, including those of IMs and monocytes, are strongly conserved between mice and humans1,11, underscoring the translational reach of these findings. Together, these findings establish spatial organization as a central determinant of macrophage function in tumors and suggest that durable immunotherapeutic benefit will require selectively disrupting suppressive monocyte-driven pathways while preserving resident macrophage programs that coordinate protective immune architecture.
Methods
Mice
C57BL/6 Ly5.1 (CD45.1) and Ly5.2 (CD45.2) WT mice were purchased from Charles River/NCI. Pf4Cre (C57BL/6-Tg (Pf4-icre) Q3Rsko/J), Cx3cr1DTR (B6N.129P2-Cx3cr1tm3(Hbegf)Litt/J), Ccl2−/− (B6.129S4-Ccl2tm1Rol/J), Ccr5−/− (B6.129P2-Ccr5tm1Kuz/J) and Ccr2−/− (B6.129S4-Ccr2tm1Ifc/J) mice were obtained from The Jackson Laboratory. Agertm2.1(cre/ERT2)Blh/2J mice (AgercreERT2) were crossed with B6.129-Krastm4Tyj Trp53tm1Brn/J (KP mice) (Extended Data Fig. 2). All mice were bred in-house, genotyped before studies and used at 6–12 weeks of age. Both male and female mice were used for the experiments. Experiments were performed on age-matched cohorts. Pf4CreCx3cr1DTR mice were compared to Cx3cr1DTR littermate controls in BM chimera studies. Mice were housed under specific-pathogen-free conditions at Dartmouth Hitchcock Medical Center. Facilities were maintained at an ambient temperature of 24 °C with a 12-h light–dark cycle. Mice had access to food and water ad libitum. All procedures followed protocol no. 00002229 approved by the Dartmouth College Institutional Animal Care and Use Committee. For lung tumor studies, mice were monitored daily using a standardized clinical scoring system assessing body weight, activity, grooming and respiration. Animals reaching predefined humane end points, defined as a total clinical score ≥6 or any individual score of 3, were killed. In addition, day 16 after intravenous injection of 4 × 105 B16F10 or KPAR1.3 was designated as the experimental ethical end point, as approved by the institutional animal ethics committee.
Bone marrow chimeras
Six-week-old Ly5.1 (CD45.1) WT mice were lethally irradiated with a single 900 rad dose. We used 25 mg kg−1 busulfan instead of radiation, to retain host IMs. Following irradiation, mice received 5 × 106 donor BM cells intravenously from the following genotypes: Ccr2−/−:WT (80:20 ratio), Ccr2−/−:Ccr5−/− (80:20 ratio) or pure BM chimeras from Pf4CreCx3cr1DTR,Cx3cr1DTR, Ccl2−/− or WT donors. Chimerism was verified using congenic markers before experimental use.
Eukaryotic cell lines and cancer models
Melanoma (B16F10-ATCC CRL-6475) were cultured according to ATCC and the adenocarcinoma (KPAR1.3) cell line was kindly provided by J. Downward. Cells were maintained in DMEM (ATCC 30-2002) supplemented with 10% heat-inactivated fetal bovine serum, 1% L-glutamine and 1% penicillin–streptomycin. Cells were collected using trypsin-EDTA, washed with PBS and HBSS, and 4 × 105 cells were injected intravenously into mice via the tail vein. On day 16, lungs were perfused with PBS, inflated with 0.5% agarose, and fixed overnight in 10% neutral-buffered formalin (NBF) at 4 °C. Tumor metastases were counted the following day. For the B16F10 model, tumors were categorized into four size groups and counted across all lobes using a blinded approach. In the adenocarcinoma models, tumors were quantified with H&E-stained sections and plotted as tumor number per section. Pf4CreCx3cr1DTR were given 700 ng intravenous DT at day 0 for time course analysis; and days 3 and 7 (or single dose at day 4) for cancer models.
Spontaneous genetic tumor model of lung adenocarcinoma
Agertm2.1(cre/ERT2)Blh/2J mice (JAX:036942) were crossed with B6.129-Krastm4Tyj Trp53tm1Brn/J (KP; LSL-K-ras G12D; p53LoxP mice) (JAX:032435). Ager(cre/ERT2)+/−Kras+/−Trp53+/− were selected for the BM chimera host. Mice were divided into two groups and reconstituted with BM from Cx3cr1DTR and Pf4CreCx3cr1DTR donors, followed by a 6-week recovery period for immune cell reconstitution. Chimeric mice were then fed tamoxifen chow for 30 days to activate the AgerCre allele. After tamoxifen chow, both groups received 500 ng DT per mouse by intravenous injection once per week for up to 14 weeks, until tissue collection.
ELISA chemokine protein analysis
Mouse IP-10 (CXCL10), CXCL13 and CXCL9 levels were quantified using Invitrogen ELISA kits following the manufacturer’s instructions.
Flow cytometry
Lungs were perfused with PBS, minced and digested in 1 ml solution of 2.5 mg ml−1 collagenase D and 400 μg ml−1 Liberase in RPMI at 37 °C for 30 min. Digestion was stopped with 100 μl of 100 mM EDTA. The cell suspensions were filtered through a 70-μm filter and centrifuged at 300g for 5 min. Samples were stained with monoclonal antibodies and isotype controls from BioLegend or BD bioscience, including Alexa Fluor 488-conjugated CD206, CD4, FITC-conjugated CD26; PE-conjugated CD206, CD45.1 and CD88; PerCP-Cy5.5–conjugated CD64, XCR1, Ly6C and CD8; PE-Cy7–conjugated CD11c; BUV395-conjugated CD11b; APC-conjugated CD26, FOLR2 and CD19; APC-Cy7–conjugated Ly6C and CD45; BV421-conjugated Ly6G and SiglecF; and BV510-conjugated MHCII and CD45.2. The viability dye 4,6-diamidino-2-phenylindole (DAPI) was added immediately before sample acquisition on a BD Symphony A3 analyzer. Data were analyzed using FlowJo software. For extravascular leukocyte analysis, mice were injected intravenously with 5 μl anti-CD45 in 200 μl PBS 5 min before killing to exclude intravascular cells.
CCR5 inhibitor study
WT mice were injected intraperitoneally with 500 μg Maraviroc (Cayman no. 14641) 3 h before intranasal delivery of 5 μg OVA-Alexa Fluor 488 and 50 μg poly(I:C). After 24 h, mice were collected for the analysis of antigen-bearing cell migration to the draining lymph node. For immunotherapy, mice received an intranasal immunization with 50 μl containing 20 μg Pmel17, 20 μg Trp2 and 50 μg polyIC. The same immunization was repeated on day 7. On day 14, mice were injected intravenously with 4 × 105 B16F10 cells. On day 30, lungs were collected for tumor quantification.
Microscopy
Lungs were perfused with PBS (Corning), inflated with 10% NBF (Sigma-Aldrich) and paraffin-embedded. Then, 5-μm sections were stained with H&E and imaged by ×4, ×10, ×20 and ×40 objective lenses using a Keyence BZ-X800 microscope. For histopathological scoring, infiltrates were assessed based on severity, with a scale from 0 (no infiltrates) to 4 (severe infiltrates with complete perivascular or peribronchial thicker than ten cells). Each lobe was scored separately, and the average histopathology score was reported. Immunohistochemistry: 5-μm sections were stained with rat anti-mouse/human B220 (BioLegend no. 103226) and rabbit anti-mouse CD3e (Cell Signaling no. 99940).
Xenium sample preparation and data acquisition
Sample processing
Four lung sections were prepared with tumor-bearing WT C57BL/6 mice (two intravenously injected with B16F10 melanoma and two with KPAR1.3 adenocarcinoma); lungs were collected on day 16. For 10x Genomics Xenium spatial transcriptomics, mice were perfused with 10% NBF to remove circulating blood. The lungs were then inflated with NBF to preserve tissue architecture and fixed by submersion in 10% NBF for 12 h at room temperature. After fixation, lung tissues were processed for paraffin embedding and formalin-fixed paraffin-embedded (FFPE) blocks were prepared. Then, 5-μm sections were placed onto Xenium slides in the Pathology Shared Resource at Dartmouth (RRID:SCR_023479) according to 10x Genomics protocol CG000580). Slides were then transferred to the Genomics and Molecular Biology Shared Resource (RRID:SCR_021293) and processed following the manufacturer’s instructions for FFPE tissue sections (protocol CG000581) followed by probe hybridization, ligation and amplification (protocol CG000582). Slides were run on a Xenium Analyzer instrument running Xenium instrument software v.2.0.1.0 and On-Board Analysis software v.2.0.0.10 to produce the output data bundle used for downstream analysis. Following the Xenium run, slides were H&E-stained on a Sakura Tissue-Tek Prism stainer and whole slide imaging conducted at ×40 magnification using an Aperio GT450 instrument (Leica). Xenium spatial transcriptomics analysis was then processed and performed using Xenium Explorer 3 (10x Genomics), with cell population identification conducted using R v.4.2.
Data preparation
Graph-based clustering results and DEGs from the 10x Xenium pipeline were utilized for cell-type identification based on known marker combinations, enabling the identification of IM clusters. Spatial localization of IM subsets was explored using the selection tool in Xenium Explorer 3, retrieving relevant cell IDs. Additional analyses were conducted in R v.4.2 using the Seurat Xenium pipeline.
Single-cell RNA sequencing data and references
The scRNA-seq data were obtained from mouse pulmonary cells 16 days post-intravenous injection of 400,000 B16F10 melanoma cells n = 6 B16F10 samples were loaded on separate lanes, each loaded sample contains three pooled lungs (Fig. 1) GSE225667. A second set (Extended Data Fig. 1), n = 3 pulmonary B16F10 samples, contained three pooled lungs per sample. To differentiate intravascular and extravascular leukocytes, mice were injected intravenously with APC-Cy7-conjugated anti-CD45 antibody 5 min before collection. Lung single-cell suspensions were enriched using CD11b Miltenyi beads. Extravascular monocyte-macrophage populations, CD64+CD11b+ cells, were sorted using the FACS Aria Fusion (BD Biosciences). Approximately 30,000 cells per sample were loaded on the Chromium Next GEM Single Cell 3′ Platform (10x Genomics) and sequenced on an Illumina NextSeq 500/550 with an average depth of approximately 50,000 reads per cell.
Data preparation
Raw sequencing reads were demultiplexed and mapped to the GRCm38 genome using CellRanger v.6.1. Data processing and analysis were performed in R v.4.2 and Python v.3.6, with Seurat v.4.3 used for data integration and visualization. Cell type identification followed methods detailed in ref. 1, wherein IMs and recMacs were distinguished based on characteristic marker genes and clustering profiles.
Statistics and reproducibility
All measurements were taken from distinct samples and the number of individuals in each experiment or analysis is clearly indicated either in the text or in figure legends. Significance was evaluated using a two-tailed Student’s t-test and ordinary one-way ANOVA. Data distribution for the transgenic mouse experiment was assumed to be normal, but this was not formally tested. In the selection of experimental cohorts of mice, randomization was not the dominant driver of the process. Littermate controls were assigned appropriately to match mice that were genetically altered, so that controls were tested side by side with those bearing a different genotype. Experimental analysis was carried out so that for any given length of a protocol, all experimental cohorts were dealt with simultaneously; no one whole group was processed first before the next, but the cohorts were evenly distributed throughout the procedure. Data collection and analysis were performed blind to the genotypes of the mice. The investigators were blinded to allocations during experiments and outcome assessments. No animals or data points were excluded from the study.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
10x Xenium spatial transcriptomic data are available in the Gene Expression Omnibus under accession codes GSE313081. 10x scRNA-seq processed data are available in the Gene Expression Omnibus under accession codes GSE225664, GSE225667 and GSE313092 and are accessible for online visualization at UCSC Cell Browser at https://cells-test.gi.ucsc.edu/?ds=macrophage-atlas+mTumor (GSE225667) and https://cells-test.gi.ucsc.edu/?ds=mouse-lung-b16 (GSE313092). Source data are provided with this paper.
References
Li, X. et al. Coordinated chemokine expression defines macrophage subsets across tissues. Nat. Immunol. 25, 1110–1122 (2024).
Aegerter, H., Lambrecht, B. N. & Jakubzick, C. V. Biology of lung macrophages in health and disease. Immunity 55, 1564–1580 (2022).
Moore, P. K. et al. Single-cell RNA sequencing reveals unique monocyte-derived interstitial macrophage subsets during lipopolysaccharide-induced acute lung inflammation. Am. J. Physiol. Lung Cell. Mol. Physiol. https://doi.org/10.1152/ajplung.00223.2022 (2023).
Dick, S. A. et al. Three tissue resident macrophage subsets coexist across organs with conserved origins and life cycles. Sci. Immunol. 7, eabf7777 (2022).
Yeung, S. T. Nerve- and airway-associated interstitial macrophages mitigate SARS-CoV-2 pathogenesis via type I interferon signaling. Immunity 58, 1327–1342.e5 (2025).
Verwaerde, S. et al. Innate type 2 lymphocytes trigger an inflammatory switch in alveolar macrophages. Immunity 59, 60–78.e9 (2026).
Kim, S. H. et al. IL-10 sensing by lung interstitial macrophages prevents bacterial dysbiosis-driven pulmonary inflammation and maintains immune homeostasis. Immunity 58, 1306–1326 e1307 (2025).
Kumar, R. et al. Monocytes and interstitial macrophages contribute to hypoxic pulmonary hypertension. J. Clin. Invest. 135, e176865 (2025).
King, E. M. et al. Gpnmb and Spp1 mark a conserved macrophage injury response masking fibrosis-specific programming in the lung. JCI Insight 9, e182700 (2024).
Legrand, C. Lung interstitial macrophages can present soluble antigens and induce Foxp3+ regulatory T cells. Am. J. Respir. Cell Mol. Biol. 70, 446–456 (2024).
Li, X. & Jakubzick, C. V. The heterogeneity, parallels, and divergence of alveolar macrophages in humans and mice. Am. J. Respir. Cell Mol. Biol. 72, 335–337 (2025).
Dagnachew, Y. M. et al. Collagen deposition in lung parenchyma driven by depletion of interstitial Lyve-1+ macrophages prevents cigarette smoke-induced emphysema and loss of airway function. Front. Immunol. 15, 1493395 (2024).
Jakubzick, C. et al. Minimal differentiation of classical monocytes as they survey steady-state tissues and transport antigen to lymph nodes. Immunity 39, 599–610 (2013).
Jakubzick, C. V., Randolph, G. J. & Henson, P. M. Monocyte differentiation and antigen-presenting functions. Nat. Rev. Immunol. 17, 349–362 (2017).
Rawat, K. et al. CCL5-producing migratory dendritic cells guide CCR5+ monocytes into the draining lymph nodes. J. Exp. Med. 220, e20222129 (2023).
Qian, B. Z. et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 475, 222–225 (2011).
Tamoutounour, S. et al. CD64 distinguishes macrophages from dendritic cells in the gut and reveals the Th1-inducing role of mesenteric lymph node macrophages during colitis. Eur. J. Immunol. 42, 3150–3166 (2012).
Langlet, C. et al. CD64 expression distinguishes monocyte-derived and conventional dendritic cells and reveals their distinct role during intramuscular immunization. J. Immunol. 188, 1751–1760 (2012).
Bain, C. C. et al. Resident and pro-inflammatory macrophages in the colon represent alternative context-dependent fates of the same Ly6Chi monocyte precursors. Mucosal Immunol. 6, 498–510 (2013).
Yona, S. et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38, 79–91 (2013).
Desch, A. N. et al. Flow cytometric analysis of mononuclear phagocytes in non-diseased human lung and lung-draining lymph nodes. Am. J. Respir. Crit. Care Med. https://doi.org/10.1164/rccm.201507-1376oc (2015).
Ray, A. et al. Targeting CD206+ macrophages disrupts the establishment of a key antitumor immune axis. J. Exp. Med. 222, e20240957 (2025).
Nalio Ramos, R. et al. Tissue-resident FOLR2+ macrophages associate with CD8+ T cell infiltration in human breast cancer. Cell 185, 1189–1207.e25 (2022).
Li, X. et al. ScRNA-seq expression of IFI27 and APOC2 identifies four alveolar macrophage superclusters in healthy BALF. Life Sci. Alliance 5, e202201458 (2022).
Mould, K. J. et al. Airspace macrophages and monocytes exist in transcriptionally distinct subsets in healthy adults. Am. J. Respir. Crit. Care Med. 203, 946–956 (2021).
Peng, W. et al. Endothelial-driven TGFβ signaling supports lung interstitial macrophage development from monocytes. Sci. Immunol. 10, eadr4977 (2025).
Gibbings, S. L. et al. Three unique interstitial macrophages in the murine lung at steady state. Am. J. Respir. Cell Mol. Biol. 57, 66–76 (2017).
Lim, H. Y. et al. Hyaluronan receptor LYVE-1-expressing macrophages maintain arterial tone through hyaluronan-mediated regulation of smooth muscle cell collagen. Immunity 49, 326–341.e7 (2018).
Vanneste, D. et al. MafB-restricted local monocyte proliferation precedes lung interstitial macrophage differentiation. Nat. Immunol. 24, 827–840 (2023).
Lee, S. H. et al. Dermis resident macrophages orchestrate localized ILC2 eosinophil circuitries to promote non-healing cutaneous leishmaniasis. Nat. Commun. 14, 7852 (2023).
Park, M. D. et al. TREM2 macrophages drive NK cell paucity and dysfunction in lung cancer. Nat. Immunol. 24, 792–801 (2023).
Loyher, P. L. et al. Macrophages of distinct origins contribute to tumor development in the lung. J. Exp. Med. 215, 2536–2553 (2018).
Petroni, G., Pillozzi, S. & Antonuzzo, L. Exploiting tertiary lymphoid structures to stimulate antitumor immunity and improve immunotherapy efficacy. Cancer Res. 84, 1199–1209 (2024).
Vella, J. L. et al. Dendritic cells maintain anti-tumor immunity by positioning CD8 skin-resident memory T cells. Life Sci. Alliance 4, e202101056 (2021).
Beaty, S. R., Rose, C. E. & Sung, S. S. Diverse and potent chemokine production by lung CD11bhigh dendritic cells in homeostasis and in allergic lung inflammation. J. Immunol. 178, 1882–1895 (2007).
Kitamura, T. et al. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J. Exp. Med. 212, 1043–1059 (2015).
Tewari, A., Prabagar, M. G., Gibbings, S. L., Rawat, K. & Jakubzick, C. V. L. N. Monocytes limit DC-poly I:C induced cytotoxic T cell response via IL-10 and induction of suppressor CD4 T cells. Front. Immunol. 12, 763379 (2021).
Abel, S., Back, D. J. & Vourvahis, M. Maraviroc: pharmacokinetics and drug interactions. Antivir. Ther. 14, 607–618 (2009).
Nasrollahzadeh, E., Razi, S., Keshavarz-Fathi, M., Mazzone, M. & Rezaei, N. Pro-tumorigenic functions of macrophages at the primary, invasive and metastatic tumor site. Cancer Immunol. Immunother. 69, 1673–1697 (2020).
Perry, J. A. et al. PD-L1-PD-1 interactions limit effector regulatory T cell populations at homeostasis and during infection. Nat. Immunol. 23, 743–756 (2022).
Ott, P. A. et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 547, 217–221 (2017).
Desch, A. N. et al. Dendritic cell subsets require cis-activation for cytotoxic CD8 T-cell induction. Nat. Commun. 5, 4674 (2014).
Haag, G. M. et al. Pembrolizumab and maraviroc in refractory mismatch repair proficient/microsatellite-stable metastatic colorectal cancer: the PICCASSO phase I trial. Eur. J. Cancer 167, 112–122 (2022).
Jiao, X. et al. Recent advances targeting CCR5 for cancer and its role in immuno-oncology. Cancer Res. 79, 4801–4807 (2019).
Misharin, A. V. et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J. Exp. Med. 214, 2387–2404 (2017).
Wuthrich, M., Ersland, K., Sullivan, T., Galles, K. & Klein, B. S. Fungi subvert vaccine T cell priming at the respiratory mucosa by preventing chemokine-induced influx of inflammatory monocytes. Immunity 36, 680–692 (2012).
Serbina, N. V., Hohl, T. M., Cherny, M. & Pamer, E. G. Selective expansion of the monocytic lineage directed by bacterial infection. J. Immunol. 183, 1900–1910 (2009).
Larson, S. R. Ly6C+ monocyte efferocytosis and cross-presentation of cell-associated antigens. Cell Death Differ. 23, 997–1003 (2016).
De Koker, S. et al. Inflammatory monocytes regulate Th1 oriented immunity to CpG adjuvanted protein vaccines through production of IL-12. Sci. Rep. 7, 5986 (2017).
Plantinga, M. et al. Conventional and monocyte-derived CD11b+ dendritic cells initiate and maintain T helper 2 cell-mediated immunity to house dust mite allergen. Immunity 38, 322–335 (2013).
Acknowledgements
This work was funded by National Institutes of Health (NIH) grant R35 HL155458 (C.V.J.); National Cancer Institute Cancer Center Support Grant 5P30CA023108 (F.W.K.); NIH S10 1S10OD030242 (F.W.K.); NIH NIGMS P20GM130454 (F.W.K.); and NIH S10 S10OD025235 (F.W.K.). We thank S. M. Palisoul of the Department of Pathology at Dartmouth Hitchcock Medical Center for performing H&E staining of lung samples, and S. Leach for assistance with Xenium data deposition.
Author information
Authors and Affiliations
Contributions
S.G and C.V.J. conceived and designed the experiments, interpreted results and wrote the paper. S.G., X.L., K.R., A.D., F.W.K., C.S.R. and C.V.J. developed the methodology. S.G., X.L., S.K., A.D., R.H., F.W.K. and C.V.J. conducted experiments and analyzed data; C.V.J. provided supervision, obtained regulatory compliance and funding; S.G. and C.V.J. wrote the paper with intellectual and editing input from all the authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Immunology thanks the anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available. Primary Handling Editor: Ioana Staicu, in collaboration with the Nature Immunology team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Second set: scRNA-seq of pulmonary melanoma identifies gene signatures in CD206hi IMs, CD206lo IMs and recMacs.
(a) Unbiased UMAP clustering of scRNA-seq data from flow-sorted extravascular CD64⁺CD11b⁺ mononuclear phagocytes isolated from the lungs of WT C57BL/6 mice 16 days after intravenous injection of 4×105 B16F10 melanoma cells. Alveolar macrophages (AMs), interstitial macrophages (CD206hi IMs and CD206lo IMs); recruited macrophages (recMacs), classical and nonclassical monocytes, dendritic cells (DC1), DC2 (CD301+ and CD301−), migratory DCs (Mig DCs), neutrophils, T cells, B cells, fibroblasts, endothelial cells and cycling cells. (b) Top 3 DEGs defining the UMAP clusters as in a (c-h) Violin plots comparing gene expression in CD206hi IMs (dark blue) and CD206lo IMs (light blue) and recMacs (pink). (c) Violin plot shows the expression of C1qb as a key IM signature gene. Violin plots depict expression of (d) Mrc1, Pf4 Folr2, Cd163, and Mmp9 for CD206hi IM, (e) H2-Ab1, Tmem119, and Mmp12 for CD206lo IM, (f) Ccr2, Ly6c2, Vcan, and Thbs1 for recMacs, (g) Cxcl13, and Cxcl10 for IMs, and (h) Arg1, Vegfa and Cd274 by recMacs, and Trem2 for IMs and recMacs. (i) Dot plot shows the expression of curated genes in 16 immune cell populations (as in a). (j) Feature plot showing the expression of Ccl2 (as in a).
Extended Data Fig. 2 A spontaneous genetic model of lung adenocarcinoma.
Agertm2.1(Cre/ERT2)Blh/2 J mice were crossed with B6.129-Krastm4Tyj Trp53tm1Brn/J (KP; LSL-K-ras G12D; p53LoxP mice). Ager(Cre/ERT2)+/−Kras+/−Trp53+/− were selected for BM chimera recipients. Mice were divided into two groups and reconstituted with BM from either Cx3cr1DTR control or Pf4CreCx3cr1DTR mice. Six weeks after BM transplant and immune cell reconstitution, chimeric mice were fed tamoxifen chow for 30 days to activate AgerCre allele. After 30 days on tamoxifen chow, mice were returned to regular chow and received intravenous injections of diphtheria toxin (DT; 500 ng per mouse) once weekly for 14 weeks, after which mice were harvested for tumor analysis.
Extended Data Fig. 3 Spatial transcriptomics reveals abundant recMacs and IMs with distinct chemokine programs in lung adenocarcinoma.
(a) Xenium spatial expression analysis of the lungs of two naïve WT C57BL/6 mice showing the expression of Acta2 (alpha-smooth muscle actin) (b) Spatial expression analysis of the lungs of WT C57BL/6 mice 16 days after intravenous injection of 4×105 KPAR1.3 adenocarcinoma cells shows various genes in different compartments including: nerves (Nes), lymphatic vessels (Pdpn), alveolar macrophages (Ear1), monocyte/macrophage markers (Cd14), recruited macrophages (Clec4n, Cd9), CD206hi IMs (Folr2), B cells (Cd19), T cells (CD3e), chemokines (Ccl3, Ccl4, Ccl6, Ccl7, Ccl8, Ccl9, Ccl17, Ccl22, Cxcl10, Cxcl3). Macrophages (Mafb) and dendritic cells (Zbtb46, Flt3, Xcr1). Scale bar, 1000 µm.
Extended Data Fig. 4 Xenium spatial transcriptomic mapping of macrophages and chemokine co-localization.
(a) Xenium H&E-stained section of four lungs of WT C57BL/6 mice 16 days after intravenous injection of 4×105 KPAR1.3 adenocarcinoma (n = 2) and B16F10 melanoma cells (n = 2). Scale bar, 1000 µm. (b) Stromal cell spatial map (same samples as in a). ImageDimPlot highlighting endothelial, epithelial and tumor cell populations. Cell type identities were assigned by matching graph-based clusters to known markers (Supplementary Figs 3-4), illustrating the anatomical distribution of airway/bronchial epithelial structures and vascular endothelium in each sample. Scale bar, 1000 µm. (c) Spatial localization (same samples as in a) of chemokine transcripts, spatial molecule plot showing per-molecule transcript coordinates for Cxcl9, Cxcl10, Cxcl13, and Ccl2 detected in each sample. Molecules are overlaid on the cell segmentation map. Spatial distributions highlight chemokine-producing regions within each tumor, allowing direct comparison of inflammatory niches between KPAR and B16F10 models. Scale bar, 1000 µm. (d) Myeloid cell spatial map (same samples as in a). ImageDimPlot visualizing four major macrophage and monocyte-derived populations: AMs; CD206hi IMs; CD206lo IMs; recMacs. Cluster-to-cell type assignments were performed using sample-specific marker-enriched clusters defined a priori (Supplementary Figs 3-4). Plots show the spatial localization of each myeloid subset within the TME, revealing region-specific enrichment patterns across KPAR and B16F10 samples. Scale bar, 1000 µm. (e) Spatial distances (same samples as in a) between myeloid cell subsets and tumor or bronchovascular structures. Violin plots show the distribution of nearest-neighbor distances from individual myeloid cells AMs, CD206hi IMs, CD206lo IMs, and recMacs to the closest tumor cell. Distances were calculated in two-dimensional space as the Euclidean distance from each myeloid cell centroid to its nearest target cell centroid. Values are plotted as log1p-transformed distances. Violin plots represent the full distribution of single-cell distances. Box plots indicate the median (center line), the 25th and 75th percentiles (box), and whiskers extending to the minimum and maximum values; outliers are shown as individual points.
Extended Data Fig. 5 Spatial transcriptomic visualization of Cxcl13 expression in CD206hi IMs in lung adenocarcinoma and melanoma.
(a) Spatial RNA expression of Cd163, Folr2, and Cxcl13 in lungs of WT C57BL/6 mice 16 days after intravenous injection of 4×105 KPAR1.3 adenocarcinoma (n = 2) and B16F10 melanoma cells (n = 2). Scale bar, 1000 µm. (b) High-resolution imaging of spatial transcriptomics (as in a) showing that Folr2⁺ CD163⁺ (purple and blue) CD206hi IMs coexpress Cxcl13 (orange), located close to Epcam⁺ (green) bronchial airways. Scale bar, 100 µm (c) High-resolution imaging of one adenocarcinoma (as in a) spatial expression analysis showing Folr2⁺ (blue) CD206hi IMs lining the bronchial airways coexpress Cxcl13 (orange), while Epcam⁺ (green) bronchial epithelial cells and Cd3⁺ T cells (pink) do not. Note: Other cell types in the lungs besides CD206hi IMs also express Cxcl13 (as in a). Scale bar, 50 µm.
Extended Data Fig. 6 Ccl2 expression in the TME.
Xenium spatial transcriptomics overlay of C1qb (blue), Cd163 (red) and Ccl2 (yellow) visualized in four tumor samples: lungs of WT C57BL/6 mice 16 days after intravenous injection of 4×105 KPAR1.3 adenocarcinoma (n = 2) and B16F10 melanoma cells (n = 2). Scale bar, 1000 µm.
Extended Data Fig. 7 Spatial transcriptomic visualization of Ccl2 expression in IMs and recMacs in lung TME of adenocarcinoma and melanoma.
(a) Spatial expression analysis of Vcan, Mmp12 and Ccl2 in lungs of WT C57BL/6 mice 16 days after intravenous injection of 4×105 KPAR1.3 adenocarcinoma (n = 2) and B16F10 melanoma cells (n = 2). Scale bar, 1000 µm. (b) High-resolution imaging of spatial transcriptomics (as in a) showing that cellular expression of Vcan (green), Mmp12 (blue), and Ccl2 (orange). Scale bar, 100 µm Note: Endothelial cells, CD206hi IMs and DCs can also express Ccl2, data not shown.
Extended Data Fig. 8 Maraviroc transiently inhibits CCR5-dependent moDC migration.
(a) Experimental design, WT mice were treated intraperitoneal with 500 μg of a CCR5 antagonist, maraviroc, 3, 24, or 48 hours prior to intranasal delivery of 5 μg OVA-Alexa Fluor 488 and 50 μg poly(I:C). Twenty-four hours after antigen intranasal delivery, the lung-draining lymph nodes were harvested, (b) Live cells were first plotted as CD11c versus CD11b, gated CD11c + CD11b+ myeloid cells (left plots) were then plotted as Ly6C versus Alexa Fluor 488 to gate on fluorescently labeled migratory antigen-bearing cells in the lung-draining lymph node (middle plots), antigen-bearing cells were then plotted as Ly6C versus CD26 to identify antigen-bearing Ly6C⁺CD26lo moDCs, as well as antigen-bearing Ly6C−CD26⁺ DCs (right plots). Scatter plot analysis of antigen-bearing Ly6C⁺ moDC migration following Maraviroc treatment at different time points, 3, 24, or 48 hours prior to antigen delivery. Two independent experiments were conducted, n = 3 per group. Data represented as mean ± s.e.m. Statistical significance was determined via one-way ANOVA followed by Bonferroni’s multiple comparison with p < 0.0001. ****p < 0.0001, ns=nonsignificant.
Supplementary information
Source data
Source Data Fig. 1 (download TXT )
Source data link for scRNA-seq deposited as GSE225667.
Source Data Fig. 2 (download XLSX )
Flow data points, tumor count values, statistical source data.
Source Data Fig. 3 (download XLSX )
Source data link for Xenium data deposited as GSE313081 and statistical source data.
Source Data Fig. 4 (download XLSX )
Source data link for deposited Xenium and scRNA-seq, tumor counts, flow data points, statistical source files.
Source Data Fig. 5 (download XLSX )
Source data link for deposited scRNA-seq, tumor counts, flow data points, statistical source files.
Source Data Extended Data Fig.1 (download TXT )
Source data link for scRNA-seq deposited as GSE313092.
Source Data Extended Data Figs. 3–7 (download TXT )
Source data link for Xenium data deposited as GSE313081.
Source Data Extended Data Fig. 4 (download XLSX )
Source data link for Xenium data deposited as GSE313081 and statistical source data.
Source Data Extended Data Fig. 8 (download XLSX )
Flow cytometry data points and statistical source data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ghosh, S., Li, X., Rawat, K. et al. Chemokine-defined macrophage niches establish spatial organization of tumor immunity. Nat Immunol 27, 715–724 (2026). https://doi.org/10.1038/s41590-026-02445-2
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41590-026-02445-2
