
Abstract
Metabolic dysfunction–associated steatotic liver disease (MASLD) is a globally increasing metabolic disorder associated with serious health complications. The molecular mechanisms linking stress-response proteins to hepatic lipogenesis in MASLD remain poorly understood. Here, we identified GADD45β as a key suppressor of de novo lipogenesis through SIRT1 stabilization. In both methionine-choline-deficient (MCD) diet-fed mice and palmitic acid (PA)-treated hepatocytes, GADD45β deficiency exacerbated lipid accumulation and upregulated lipogenic genes (SREBP1, FASN, ACC). Mechanistically, GADD45β directly bound to SIRT1 and inhibited its ubiquitination, thereby prolonging SIRT1 protein stability. Enhanced SIRT1 stability increased AMPK phosphorylation, which suppressed SREBP1-mediated transcription of lipogenic targets. Crucially, hepatic overexpression of GADD45β reversed PA-induced steatosis in vitro. Our study uncovered a GADD45β/SIRT1-/AMPK axis as a central regulator of hepatic lipogenesis, proposing GADD45β as a therapeutic target for MASLD.
Similar content being viewed by others


Introduction
The increasing prevalence of metabolic dysfunction–associated steatotic liver disease (MASLD) is a significant global health concern. In accordance with recent consensus recommendations, the condition previously termed nonalcoholic fatty liver disease (NAFLD) is now defined as MASLD, and nonalcoholic steatohepatitis (NASH) is now termed metabolic dysfunction–associated steatohepatitis (MASH). Over the last decade, MASLD has become the predominant cause of chronic liver disease worldwide and now affects more than 25% of the global population1. MASLD is often linked to obesity, insulin resistance, and metabolic syndrome2. It is characterized by excessive lipid accumulation in hepatocytes, which can progress from simple steatosis to more severe forms, such as steatohepatitis and cirrhosis. The mechanisms underlying hepatic lipid metabolism are critical for understanding the pathophysiology of MASLD, yet they remain inadequately explored. De novo lipogenesis plays a crucial role in the development of MASLD3. This process converts excess glucose or fructose into fatty acids and triglycerides4 and is important for maintaining metabolic homeostasis; however, its increased activation may cause hepatic steatosis5. Therefore, inhibition of lipogenesis represents a potential therapeutic strategy for lipid metabolism-related diseases. Previous studies have focused primarily on signaling pathways involved in lipid regulation, such as sirtuin 1 (SIRT1) and AMP-activated protein kinase (AMPK), whereas the specific contributions of growth arrest and DNA damage-inducible 45β (GADD45β) in this context remain poorly characterized6,7.
GADD45β, a member of the GADD45 family, participates in cellular stress responses, DNA repair, and apoptosis8. Recent studies have highlighted its potential involvement in glucose and lipid metabolism, suggesting that GADD45β may be an important regulator in the pathogenesis of MASLD9. However, whether GADD45β contributes to lipogenesis and thereby affects MASLD remains unclear. A significant gap persists regarding the mechanistic role of GADD45β in hepatic lipid metabolism, particularly its interactions with established pathways such as SIRT1/AMPK/sterol regulatory element-binding protein 1 (SREBP1) signaling, which are crucial for maintaining metabolic homeostasis10,11.
In this study, we provided evidence that SIRT1 was regulated by GADD45β and demonstrated an important association between them in MASLD. GADD45β interacted with SIRT1 and attenuated its ubiquitin–proteasome degradation, thereby enhancing its stability. GADD45β deficiency decreased the activity of the SIRT1 target AMPK, resulting in lipogenesis exacerbation. Our findings highlight a critical role of GADD45β in maintaining SIRT1 stability and limiting lipogenesis, providing new insights and potential therapeutic strategies for MASLD.
Materials and methods
Reagents and antibodies
Cycloheximide (CHX, Cat# M4879) was purchased from AbMole Bioscience, Inc. MG132 (Cat# S2619) was purchased from Selleck Chemicals. Palmitic acid (PA, Cat# S-A9165-5G) was purchased from Sigma‒Aldrich. Compound C (CC, Cat# HY-13418) was purchased from Med ChemExpress. Antibodies against FASN (Cat# ARG55898, 1:1000), HSP90 (Cat# ARG55781, 1:1000), Flag Tag (Cat# ARG62342, 1:1000) and β-Actin (Cat# ARG65683, 1:1000) were purchased from Arigo. Antibodies against ubiquitin (Cat# 3936, 1:400), AMPK (Cat# 2532 S, 1:1000), and p-AMPK (Cat# 2531 S, 1:1000) were purchased from Cell Signaling Technology. Antibodies against SIRT1 (Cat# ab189494, 1:1000), SREBP1 (Cat# ab28481, 1:1000), and ACC1 (Cat# ab45174, 1:1000) were purchased from Abcam. Antibody against GADD45β (Cat#sc377311, 1:1000) was purchased from Santa Cruz. HRP-conjugated secondary antibodies against mouse (Servicebio, GB23301) or rabbit (Servicebio, GB23303) were used.
Animal treatment
All animal experiments were approved by the Animal Research Ethics Committee of Shanghai Sixth People’s Hospital and conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The study followed all institutional and national guidelines for animal research and complied with the ARRIVE guidelines to ensure ethical and reproducible practices12.
Eight-week-old C57BL/6J mice weighing 23–28 g were purchased from Shanghai Sippe-Bk Lab Animal Co., Ltd. (Shanghai, China). Animals were maintained in a specific pathogen-free facility under a 12 h light/dark cycle. After one week of acclimatization, mice were randomly assigned to experimental groups. The methionine-choline-deficient (MCD) diet (A02082002B; Research Diets, USA) was administered for 4 weeks to induce MASH, and age-matched mice on a standard chow diet were used as controls.
To knock down hepatic GADD45β, an adeno-associated virus (AAV8; OBiO Technology, Shanghai, China) carrying a short hairpin RNA targeting GADD45β (pscAAV-U6-shGADD45β-CMV-EGFP-tWPA) was constructed (AAV-shGADD45β). The target sequence was 5’-GCGACAATGACATTGACATCG-3’. Control mice received an AAV-shNC construct (pscAAV-U6-shNC-CMV-EGFP-tWPA). Viruses (2 × 1011 PFU per mouse) were purified using cesium chloride gradient centrifugation, dialyzed in PBS containing 10% glycerol, and administered via tail vein injection 14 days before euthanasia. At the study endpoint, after 12 h of fasting, mice were euthanized by intraperitoneal injection of 3% sodium pentobarbital (60 mg/kg). Liver tissues were rapidly dissected, snap-frozen in liquid nitrogen, and stored at − 80 °C for subsequent RNA and protein analysis.
All animal procedures were performed in adherence to the 3R principles (Replacement, Reduction, and Refinement). Sample size was determined based on prior pilot studies and ethical guidelines, with a minimum of eight mice per group considered sufficient to achieve statistical power while minimizing animal use. Although investigators were aware of group allocation during procedures and outcome assessment, all interventions and data collection followed standardized protocols to minimize bias. Humane endpoints were predefined, and any animals showing signs of severe distress or irreversible suffering were promptly euthanized to ensure animal welfare.
Biochemical assays
Blood samples collected from the mice were left at room temperature for 30 min and then centrifuged at 5000 rcf for 5 min at 4 °C. The supernatant was centrifuged again at 10,000 rcf for 5 min at 4 °C. Serum was carefully transferred into clean 1.5 mL EP tubes and stored at -20 °C until use. Serum levels of aspartate aminotransferase (AST), alanine aminotransferase (ALT), triglycerides (TG), and gamma-glutamyltransferase (γ-GT) were measured using a fully automated biochemical analyzer (Rayto Technologies, China).
Hematoxylin and eosin (H&E) and Oil Red O staining
For H&E staining, paraffin-embedded liver tissues were sectioned at 4 μm thickness. Slides were deparaffinized in xylene for 5 min (twice), rehydrated through a graded ethanol series (100%, 95%, 85%, 75%), and rinsed in distilled water. Sections were stained with hematoxylin for 7 min, rinsed, counterstained with eosin for 30 s, differentiated in acid ethanol for 30 s, washed in distilled water, dehydrated through graded alcohols, cleared in xylene, and mounted with neutral resin.
For Oil Red O staining, frozen liver Sect. (7 μm) were fixed in 4% paraformaldehyde for 10 min, washed with distilled water, and incubated with freshly prepared Oil Red O working solution (Sigma-Aldrich; 0.5 g Oil Red O dissolved in 100 mL isopropanol, diluted 3:2 with distilled water, filtered before use) for 8–10 min. Sections were differentiated in 75% ethanol, rinsed with PBS, counterstained with hematoxylin for 90 s, and mounted using glycerol gelatin. Slides were examined under an Olympus light microscope.
Cell culture and treatment
HepG2 cells were cultured in DMEM/high glucose (Cat# SH30243.01; HyClone) supplemented with 10% fetal bovine serum (Cat#04-001-1ACS; Biological Industries) and 1% penicillin‒streptomycin (Cat#C0222; Beyotime, China) in a 5% CO2 incubator at 37 °C. Cells were treated with fatty acid-free bovine serum albumin or 400 µM PA for 24 h. Intracellular lipid droplets were stained with Oil Red O and visualized under a light microscope (Olympus, Tokyo, Japan).
Cell transfection
pSLenti-GADD45β-3xFlag (GADD45β), control pSLenti-MCS-3xFlag (vector), pLenti-U6-siGADD45β-CMV (siGADD45β:GCATACTCCTTCCACGTTA), and control pSLenti-U6-shNC2-CMV (siNC: TTCTCCGAACGTGTCACGT) were constructed by OBiO Technology (Shanghai, China). Transfections were performed using Lipofectamine® LTX & PLUS™ Reagent (Cat# A12621; Invitrogen, NY, USA) according to the manufacturer’s instructions. Briefly, when HepG2 cells reached 70–80% confluence, solutions A (2.5 µg plasmid DNA, 2.5 µL PLUS reagent, and 125 µL Opti-MEM) and solution B (5 µL LTX reagent and 125 µL Opti-MEM) were prepared separately, combined, and incubated for 5 min before being added dropwise to the cell culture medium. Medium was replaced after 6–8 h. Cells were collected 48 h later for RNA and protein extraction.
In addition, two lentivirus (LV) constructs, LentiCRISPRv2-SIRT1-sgRNA (LV-shSIRT1) and a blank vector (LV-shNC), were purchased from Fenghbio Biotechnology Co., Ltd. (Hunan, China). HepG2 cells at 80% confluence were transduced with these constructs at a multiplicity of infection of 50. After 24 h, the medium was replaced with fresh medium containing FBS, and puromycin (30 µg/mL; Cat# HY-K1057; MedChemExpress) was applied to select successfully transfected cells. The culture was continued for 2 days, after which cells were harvested and lysed for Western blotting.
Western blot analysis
Total protein extracts from liver tissues and HepG2 cells were prepared using RIPA buffer containing 50 mM Tris-HCl, 150 mM NaCl, 5 mM MgCl2, 2 mM EDTA, 1 mM NaF, 1% NP40, and 0.1% SDS. Equal amounts of protein were denatured in loading buffer, separated by 10–12% SDS‒PAGE, and transferred onto polyvinylidene fluoride membranes. Membranes were blocked with 5% nonfat milk for 1 h at room temperature and then incubated with primary antibodies overnight at 4 °C. After five washes with PBS containing Tween-20, membranes were incubated with HRP-conjugated secondary antibodies for 1 h. Protein bands were visualized using an enhanced chemiluminescence system.
Coimmunoprecipitation (Co-IP)
Cells were washed with PBS and lysed on ice for 30 min with IP lysis solution (Beyotime) supplemented with protease and phosphatase inhibitors (Thermo Fisher Scientific). After centrifugation, equal amounts of lysates were incubated overnight at 4 °C with the corresponding antibodies or control IgG, followed by incubation with 20 µl of A/G agarose beads (Cat# 88802; Thermo Fisher Scientific) for 4 h. Immunocomplexes were pelleted, washed three times with lysis buffer, and eluted with SDS loading buffer (Cat# BL502A; Biosharp Life Sciences, Hefei, Anhui, China). Samples were then analyzed by immunoblotting with the indicated antibodies.
Quantitative real-time polymerase chain reaction (qRT‒PCR)
Total RNA was extracted from cells or liver tissues using TRIzol reagent and reverse transcribed into cDNA with PrimeScript™ RT Master Mix (Cat# R433-01; Vazyme) according to the manufacturer’s instructions. qRT‒PCR was performed with SYBR Premix Ex Taq™ (Cat#Q312-02; Vazyme), and results were analyzed using LightCycler 480 software (Roche Diagnostics GmbH, Mannheim, Germany). Relative gene expression was quantified using the 2−ΔΔCt method, with GAPDH as the internal control. Primer sequences are listed in Table S1.
Statistics
Statistical analyses were performed using GraphPad InStat Software (San Diego, CA, USA). Data are presented as the means ± SEM. For animal and cellular experiments, comparisons between two groups were made using a two-tailed unpaired Student’s t-test. Western blot and morphological images are representative of at least three experiments. The normality of quantitative data was assessed using the Kolmogorov‒Smirnov test. For multiple group comparisons, one-way ANOVA was applied. Statistical significance was expressed as *P < 0.05, **P < 0.01, and ***P < 0.001.
Results
Hepatic GADD45β expression is decreased in MASLD
By accessing the GEO database, two MASLD datasets, GSE48452 and GSE33814, were analyzed, comprising 37 healthy controls, 39 MASLD patients, and 31 MASH patients. Notably, GADD45β was consistently downregulated at the mRNA level in both MASLD and MASH groups (Fig. 1A,B). To further validate this finding, GADD45β mRNA and protein levels were examined in mouse liver samples. Consistent with the human data, GADD45β expression was substantially reduced in the livers of MCD diet-fed mice compared with controls (Fig. 1C,D).

Hepatic GADD45β expression was decreased in MASLD. (A, B) GADD45β mRNA expression levels in two different MASLD GEO datasets. (C) GADD45β mRNA expression levels in the livers of control diet-fed (CON) and methionine- and choline-deficient (MCD) diet-fed mice (n = 6). (D) GADD45β protein expression levels in the livers of control diet-fed mice (CON) and methionine- and choline-deficient diet-fed mice (MCD) (n = 6). nsP > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001.
Hepatic knockdown of GADD45β exacerbates steatohepatitis in MCD mice
To investigate the role of hepatic GADD45β in steatohepatitis, C57BL/6J mice fed an MCD diet were injected with adenoviral shRNA targeting GADD45β via the tail vein, which effectively reduced hepatic GADD45β expression (Fig. 2A,B). Although body weight remained unchanged, liver weight, liver index, and hepatic TG content were significantly increased in the knockdown group compared with controls (Fig. 2C–E). Consistently, H&E and Oil Red O staining revealed markedly aggravated hepatic steatosis in GADD45β-deficient mice (Fig. 2F,G). Interestingly, while serum TG, TC, ALT, and AST levels showed no significant differences between groups (Fig. 2H–K), γGT levels were significantly elevated in the GADD45β-knockdown group (Fig. 2L), suggesting worsened liver injury. Collectively, these findings demonstrate that hepatic GADD45β knockdown aggravates steatohepatitis in MCD-fed mice.

Hepatic knockdown of GADD45β aggravated steatohepatitis in MCD diet-fed mice. (A) Schematic Diagram of Animal Experimental Procedures (B) GADD45β expression in liver tissues of GADD45β hepatic knockdown mice compared with the control group (n = 6). (C–E) Liver weight, liver weight/body weight ratio (liver index), and liver TG levels of each group (n = 6). (F, G) Representative images of H&E and Oil Red O staining of liver sections from each group (n = 5); scale bar, 100 μm. (H–L) Serum TG, TC, ALT, AST and γGT levels in each group (n = 5). *P < 0.05, **P < 0.01, ***P < 0.001.
Hepatic knockdown of GADD45β enhances hepatic lipogenenic gene expression in MCD diet-fed mice
To investigate how GADD45β knockdown aggravates hepatic steatosis, we compared the expression of lipogenesis-related genes in the livers of GADD45β-knockdown and control mice fed an MCD diet. MCD feeding alone significantly upregulated hepatic SREBP1 mRNA expression, and liver-specific knockdown of GADD45β further increased its expression. Liver-specific knockdown of GADD45β also significantly elevated the mRNA levels of additional lipogenic genes, FASN and ACC (Fig. 3A). However, the hepatic mRNA levels of genes related to fatty acid oxidation, fatty acid transport, and lipolysis were unaffected (Fig. 3A). Western blot analysis confirmed increased hepatic protein levels of SREBP1, FASN, and ACC1 in GADD45β-deficient mice, consistent with the mRNA changes (Fig. 3B). These results indicate that liver-specific knockdown of GADD45β enhances hepatic lipogenesis in MCD diet-fed mice.

Hepatic knockdown of GADD45β increased hepatic lipogenic gene expression in MCD diet-fed mice. (A) Quantitative PCR analysis of the mRNA expression levels of lipid metabolism-related genes in the liver tissue of different groups (n = 6). (B) Western blot of ACC1, FASN, nuclear SREBP1 (SREBP1-N) and cytoplasmic SREBP1 (SREBP1-P) in the liver tissues of different groups (n = 6). *P < 0.05, **P < 0.01, ***P < 0.001.
GADD45β inhibits PA-induced steatosis and lipogenesis in hepatocytes
To confirm the function of GADD45β in hepatocytes, HepG2 cells were transfected with a GADD45β plasmid. Western blot analysis verified a significant increase in GADD45β protein levels after transfection (Fig. 4F). Cells were then treated with 400 µM PA for 24 h to induce steatosis. Oil Red O staining revealed that GADD45β overexpression significantly reduced intracellular lipid accumulation. Consistently, PA stimulation significantly elevated intracellular TG content, which was blocked by GADD45β overexpression (Fig. 4A–C). These results indicate that GADD45β upregulation protects hepatocytes from PA-induced steatosis.

GADD45β inhibited palmitic acid-induced steatosis and lipogenesis in hepatocytes. (A) Oil Red O staining and area quantification of GADD45β-overexpressing or GADD45β-knockdown HepG2 cells after treatment with PA. (B–E) Lipid drops, intracellular TG levels, and relative quantification of GADD45β-overexpressing or GADD45β-knockdown HepG2 cells after treatment with PA. (F–H) Protein and mRNA expression levels of FASN, ACC1, and SREBP1 in PA-induced GADD45β-overexpressing HepG2 cells (n = 3). (I–K) Protein and mRNA expression levels of FASN, ACC1, and SREBP1 in PA-induced GADD45β-knockdown HepG2 cells (n = 3).
The effect of GADD45β on lipogenesis was further examined. qRT-PCR and Western blot analyses demonstrated that GADD45β overexpression significantly suppressed PA-induced expression of lipogenic regulators, including SREBP1, ACC1, and FASN (Fig. 4F‒H). Conversely, knockdown of GADD45β enhanced the expression of these genes after PA treatment (Fig. 4A,I–K), which was accompanied by increased lipid accumulation and aggravated steatosis in hepatocytes (Fig. 4A,D,E). These findings collectively suggest that GADD45β exerts a protective effect against hepatic steatosis by inhibiting lipogenesis.
GADD45β mediates lipogenesis via the SIRT1/AMPK/SREBP1 pathway in hepatocytes
To determine the molecular mechanism by which GADD45β regulates hepatic lipogenesis, we first examined its effects on the SIRT1/AMPK pathway and downstream SREBP1. In vivo, hepatic knockdown of GADD45β in MCD-fed mice markedly reduced SIRT1 expression and the p-AMPK/AMPK ratio as shown by both western blotting and immunohistochemistry (Fig. 5A–C). Consistently, in PA-treated HepG2 cells, GADD45β silencing decreased SIRT1 and p-AMPK/AMPK expression and increased SREBP1 levels, whereas GADD45β overexpression enhanced SIRT1/AMPK signaling and suppressed SREBP1 expression (Fig. 5D,E). These data indicate that GADD45β positively regulates the SIRT1/AMPK pathway and thereby inhibits SREBP1-mediated lipogenesis.

GADD45β mediated lipogenesis via the SIRT1/AMPK/SREBP1 pathway in hepatocytes. (A) Western blot analysis of p-AMPK, AMPK, and SIRT1 expression in liver tissues from GADD45β-knockdown and control mice (n = 6). (B, C) Representative IHC staining and relative quantification of p-AMPK and SIRT1 in liver sections from the indicated individuals. (n = 6). (D) Western blot of p-AMPK, AMPK, and SIRT1 expression in GADD45β-knockdown HepG2 cells treated with PA (n = 6). (E) Western blot of p-AMPK, AMPK, and SIRT1 expression in GADD45β-overexpressing HepG2 cells treated with PA (n = 6). (F) Western blot analysis of SIRT1 expression in PA-induced HepG2 cells infected with LV-shSIRT1 or LV-shControl. (G) SIRT1, p-AMPK/AMPK, and SREBP-1 expression in PA-induced SIRT1 knockdown and GADD45β-overexpressing HepG2 cells (n = 6). (H) Western blot of p-AMPK/AMPK expression in PA-induced HepG2 cells treated with multiple doses of Compound C. (I) p-AMPK/AMPK and SREBP-1 expression in PA-induced GADD45β-overexpressing and Compound C-treated HepG2 cells (n = 6).
To further confirm whether SIRT1 functions as an upstream regulator of AMPK, rescue experiments were performed. Lentiviral knockdown of SIRT1 in PA-stimulated HepG2 cells abrogated the increase in p-AMPK/AMPK and the suppression of SREBP1 conferred by GADD45β overexpression (Fig. 5F,G). These results identify SIRT1 as a key upstream mediator of the p-AMPK/AMPK axis.
Finally, to evaluate whether AMPK is essential for the protective role of GADD45β, HepG2 cells were treated with the AMPK inhibitor CC. Inhibition of AMPK abolished the effects of GADD45β overexpression, as indicated by decreased p-AMPK and restored SREBP1 expression (Fig. 5H,I). Together, these findings demonstrate that GADD45β suppresses lipogenesis in hepatocytes through the SIRT1/AMPK/SREBP1 signaling pathway.
GADD45β directly interacts with SIRT1
Although GADD45β overexpression increased SIRT1 protein levels, it did not affect SIRT1 mRNA expression as determined by qRT-PCR (Fig. 6A). We therefore hypothesized that GADD45β may physically associate with SIRT1. Co-IP assays in PA-induced HepG2 cells confirmed this interaction, as GADD45β was pulled down with SIRT1 and reciprocally, SIRT1 was detected in anti-GADD45β immunoprecipitates (Fig. 6B). These findings indicate that GADD45β directly interacts with SIRT1.

GADD45β directly interacted with SIRT1. (A) GADD45β and SIRT1 mRNA expression in HepG2 cells after the overexpression of GADD45β. (B) Coimmunoprecipitation (co-IP) of GADD45β with SIRT1 proteins in HepG2 cells. The co-IP and western blot data shown are representative of at least 3 independent experiments with consistent results.
GADD45β upregulates SIRT1 levels by inhibiting ubiquitin‒proteasome degradation
To exclude the influence of protein synthesis on SIRT1 levels, and because SIRT1 can be regulated through ubiquitination, we further examined whether GADD45β affects SIRT1 stability. HepG2 cells transfected with GADD45β plasmids were treated with CHX, a protein synthesis inhibitor, and SIRT1 protein levels were measured. As expected, GADD45β significantly prolonged the half-life of SIRT1, which was decreased by CHX (Fig. 7A). Conversely, GADD45β knockdown decreased SIRT1 stability in PA-induced HepG2 cells (Fig. 7B), suggesting that GADD45β maintains SIRT1 levels by inhibiting its degradation rather than promoting protein synthesis.

GADD45β facilitatef SIRT1 stability by inhibiting its ubiquitin‒proteasome degradation. (A) Western blots of PA-induced GADD45β-overexpressing HeG2 cells treated with cycloheximide (CHX; 20 μg/ml) for the indicated time periods and semiquantification of SIRT1 levels. (B) Western blots of PA-induced GADD45β-knockdown HeG2 cells treated with cycloheximide (CHX; 20 μg/ml) for the indicated time periods and semiquantification of SIRT1 levels. (C) Coimmunoprecipitation (co-IP) of SIRT1 with ubiquitin proteins in PA-induced GADD45β-overexpressing HepG2 cells after treatment with 10 µM MG132 for 8 h (n = 3). The Co-IP and western blot data shown are representative of 3 independent experiments with consistent results.
The K48-linked ubiquitin‒proteasome system (UPS) is a major intracellular protein degradation mechanism. As shown in Fig. 7C, in the condition of exposing to PA, treatment HepG2 cells with the proteasome inhibitor MG132, GADD45β overexpression significantly increased SIRT1 protein expression levels compare with the control group, implicating that the UPS pathway may involve in this process. Furthermore the ubiquitin immunoprecipitation further revealed that PA exposure mediates SIRT1 ubiquitination, whereas GADD45β overexpression reverses these effects. All of these results indicate that GADD45β increases SIRT1 levels by inhibiting its ubiquitin‒proteasome-mediated degradation.
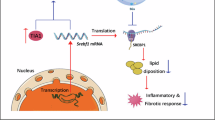
Based on these results, we propose a mechanistic model: reduced GADD45β expression in hepatocytes of MASH patients and MCD-fed mice diminishes its interaction with SIRT1, leading to enhanced ubiquitin–proteasome-mediated degradation of SIRT1. The resulting loss of SIRT1 impairs AMPK phosphorylation, thereby augmenting SREBP1-mediated de novo fatty acid synthesis. This cascade increases lipogenesis and exacerbates hepatic steatosis (Fig. 8).

Working model of GADD45β in the regulation of hepatic steatosis. GADD45β expression is decreased in hepatocytes fed an MCD diet. A decrease in GADD45β does not interact with Sirtuin 1 (SIRT1) and is subsequently degraded by ubiquitination. This results in increased lipogenesis and thereby synergistically aggravates fatty acid accumulation and hepatic steatosis.
Discussion
MASLD has emerged as a significant global health concern due to its complex pathogenesis, which involves insulin resistance, oxidative stress, and inflammatory pathways. With the increasing prevalence of obesity and metabolic syndrome, MASLD is recognized not only as a hepatic disorder but also as a major risk factor for cardiovascular diseases, type 2 diabetes, and other metabolic disorders. This underscores the urgent need to understand its mechanisms and develop effective therapeutic strategies13,14. In this study, we identified GADD45β as a novel factor contributing to the development and progression of MASLD. GADD45β is an acidic stress-responsive protein involved in DNA repair, cell cycle regulation, apoptosis, inflammation, and stress responses through protein‒protein interactions15. Several studies have also implicated GADD45β in lipid metabolism. Dong et al. have demonstrated that GADD45β was downregulated in multiple clinical datasets and in a murine MASLD model, where its expression mitigated lipid accumulation and insulin resistance in high-fat diet (HFD)-induced disease6. Fuhrmeister et al. have reported that under fasting conditions, GADD45β binds FABP1, promoting its cytoplasmic retention, thereby inhibiting fatty acid uptake and enhancing lipid metabolism16. Kim et al. have revealed that GADD45β suppresses hepatic gluconeogenesis by stabilizing and activating forkhead box protein O1 (FoxO1), while GADD45β knockout increased expression of lipogenesis-related genes. Furthermore, GADD45β has been identified as an inducible coactivator of the constitutive androstane receptor (CAR), promoting rapid liver growth. Cai et al. have also reported that CAR-mediated metabolic regulation depends on GADD45β, as GADD45β knockout blunted the weight reduction and insulin sensitivity induced by the CAR agonist TCPOBOP in HFD-fed mice, mechanistically linked to lipogenesis inhibition in hepatocytes17. However, further mechanistic studies remain limited. In the present study, we observed that GADD45β mRNA and protein levels progressively decreased in human and mouse MASLD livers as the disease advanced from MASLD to MASH. Using a liver-specific GADD45β knockdown model under an MCD diet, we found that steatosis was aggravated compared with control diet-fed mice. This was accompanied by increased hepatic expression of lipogenic genes, including SREBP1, FASN, and ACC, consistent with previous findings. In vitro, GADD45β overexpression in HepG2 cells significantly suppressed fatty acid-induced expression of these genes, whereas GADD45β knockdown enhanced their expression and lipid accumulation. These data indicate that GADD45β is involved in hepatic lipid metabolism and fat deposition in MASLD.
To clarify how GADD45β influences hepatic lipid metabolism, we investigated its relationship with AMPK, a heterotrimeric kinase that regulates cellular energy balance through lipid metabolism. AMPK plays a key role in promoting hepatic fatty acid oxidation, inhibiting cholesterol and TG synthesis, and repressing lipogenesis18. Among its downstream targets, SREBP-1c is a key transcription factor for lipogenesis in the liver19. AMPK reduces lipogenic gene expression by phosphorylating SREBP-1c at Ser372, thereby preventing its proteolytic activation20. Furthermore, AMPK may suppress SREBP-1c expression through mTOR and LXRa21. Once activated, SREBP1 translocates to the nucleus and stimulates the transcription of lipogenic genes such as ACC, FASN, and stearoyl-CoA desaturase (SCD1)22,23,24. Although AMPK is known to strongly influence lipogenic gene expression, it has not been established whether GADD45β regulates lipid metabolism through this pathway. Our study revealed that protein levels associated with the AMPK/SREBP1 axis were significantly altered in vitro and in vivo under MASLD conditions, and these changes were further modulated by GADD45β. Moreover, treatment with CC, an ATP-competitive inhibitor of AMPK25, abolished GADD45β-induced activation of AMPK and its inhibitory effect on lipogenesis. These results indicate that GADD45β may ameliorate lipid metabolism dysfunction via the AMPK/SREBP1 pathway in MASLD.
Even though GADD45β affects the AMPK signaling pathway, AMPK activity is regulated primarily by phosphorylation, and GADD45β does not possess phosphatase activity. In addition, as a scaffold protein, GADD45β does not directly regulate transcription, prompting us to identify downstream factors potentially affected by GADD45β. We found that SIRT1, a known GADD45β-binding partner, mediated its effects on the AMPK/SREBP1 pathway. This is a key finding of our study in understanding how GADD45β regulates lipogenesis in MASLD. SIRT1 is a class III histone deacetylase that requires nicotinamide adenine (NAD+) to deacetylate histone and non-histone proteins involved in diverse metabolic processes and stress responses26,27. It is widely expressed in multiple tissues and has been implicated in metabolic and age-related diseases28. SIRT1 regulates adipocyte differentiation, hepatic lipid metabolism, systemic inflammation, nutrient sensing, circadian rhythms, and particularly lipogenesis29. Often described as a “master metabolic regulator”, it maintains lipid homeostasis by modulating several metabolic proteins30,31. Previous studies have demonstrated that SIRT1 inhibits lipogenic enzymes such as SREBP-1c, thereby alleviating hepatic lipid dysregulation32,33,34, and can also suppress ACC and FASN in MASLD35. Importantly, AMPK, the natural regulator of SREBP1, is activated indirectly by SIRT1 through deacetylation of its upstream kinase36. Thus, SIRT1 amplifies AMPK activity to reinforce lipid homeostasis37,38. These findings align with clinical evidence showing that SIRT1 activators can alleviate fatty liver in MASLD patients, highlighting its pivotal role in lipid-related disease through the AMPK pathway39,40. In this study, disruption of SIRT1 expression revealed that the impact of GADD45β on hepatic lipid synthesis was mediated through the SIRT1/AMPK signaling pathway. We further confirmed the interaction between GADD45β and SIRT1 in both animal and cell models of steatosis. Consistent with previous reports, the MCD diet significantly downregulated hepatic SIRT1 expression. In GADD45β liver-specific knockdown mice, SIRT1 expression was reduced, and lipogenic genes were significantly increased. Conversely, GADD45β overexpression in HepG2 cells reversed the PA-induced suppression of SIRT1 and upregulation of lipogenic genes. Together, these results suggest that GADD45β regulates hepatic steatosis by stabilizing SIRT1 expression and subsequently modulating downstream lipogenic genes.
Another novel finding of our study is that GADD45β was involved in the ubiquitination and stabilization of SIRT1. We found that GADD45β not only interacted with SIRT1 but also modulated its protein stability. Previous studies have shown that SIRT1 is regulated through ubiquitin‒proteasome degradation and that control of its ubiquitination is crucial for maintaining stability. Ubiquitination is a key signal for protein degradation, with ubiquitin ligases catalyzing the covalent attachment of ubiquitin to substrates and extending polyubiquitin chains. In MASLD, SIRT1 acts as a substrate of E3 ligases, undergoing K48-linked ubiquitination and degradation, which exacerbates hepatic steatosis41. Proteomic studies have identified multiple E3 ligases that target SIRT1, including MDM2 (under oxidative stress-induced cell senescence)42, SMURF2 (to inhibit proliferation and tumor formation)43, CUL4 (to promote autophagy)44, and COP1 (to exacerbate lipid toxicity)45. Our results provide the first evidence that GADD45β regulates SIRT1 ubiquitination. GADD45β attenuated the PA-induced SIRT1 ubiquitination, thereby preserving SIRT1 protein expression and preventing proteasomal degradation. This novel mechanism highlights GADD45β as a stabilizer of SIRT1, extending its role beyond transcriptional and metabolic regulation. Although our study revealed that GADD45β bound to SIRT1 and influenced its ubiquitination, GADD45β itself is not a ubiquitin ligase. Previous studies suggest that the association of adaptor proteins can provide additional layers of regulation to ubiquitin ligase activity46. Ubiquitin ligases often function as part of complexes that include substrate receptors or adaptor proteins, which guide substrate recognition and catalyze ubiquitination and subsequent degradation47. Therefore, we speculate that GADD45β may act as an adaptor protein, facilitating ubiquitin ligase-mediated regulation of SIRT1. Among known E3 ligases, MDM2, SMURF2, COP1, and CUL4B have been implicated in SIRT1 turnover under different stress and metabolic conditions, including oxidative stress, lipotoxicity, and tumorigenesis43,48,49,50. These findings suggest that similar mechanisms may operate in hepatocytes, and that GADD45β could modulate SIRT1 stability by interacting with one or more of these ligase complexes. Proteomic screening and targeted co-IP assays are necessary to determine which ligase mediates this effect in MASLD. Additionally, the specific ubiquitination site on SIRT1 that is affected by GADD45β warrants further exploration.
Notably, the GADD45 family consists of three isoforms, GADD45α, GADD45β, and GADD45γ, that are structurally homologous and share partially overlapping functions in stress responses51. GADD45α activates AMPK to enhance fatty acid oxidation and energy expenditure while suppressing lipogenesis, protecting against acetaminophen- and diet-induced liver injury52. Its downregulation under ER stress or fibrosis promotes steatosis and inflammation53. GADD45β also suppresses hepatic lipogenesis by regulating the SIRT1/AMPK/SREBP1 axis, as shown in our study. Similarly, GADD45γ is expressed in the liver and induced by stress, but its role in hepatic lipid metabolism remains undefined. Thus, GADD45α and GADD45β may exert related but mechanistically distinct effects in hepatic lipid metabolism.
Evidence also suggests that GADD45β regulates multiple metabolic and pathological processes in the liver. It promotes gluconeogenesis through TET1–PGC-1α demethylation, contributing to hyperglycemia in obesity and diabetes54. Stabilization by HSP72 improves insulin resistance and reduces triglyceride accumulation in MASLD55. GADD45β also inhibits liver fibrosis by attenuating hepatocyte injury, inflammatory responses, and fibrogenic signaling pathways56. Metformin protects against acetaminophen-induced hepatotoxicity via GADD45β-dependent JNK inhibition57. Since metformin improves insulin sensitivity and steatosis in MASLD58, these findings support a protective role of GADD45β in limiting inflammation, fibrosis, and progression to MASH.
This study has several limitations. First, palmitate-treated HepG2 cells model lipotoxic stress rather than classical de novo lipogenesis, and the relevance of our findings to glucose-mediated de novo lipogenesis requires further clarification. Second, the MCD model does not robustly activate de novo lipogenesis, and GADD45β may influence lipid accumulation via alternative pathways, such as fatty acid uptake or oxidation, which were not functionally explored. Third, genetic rescue experiments targeting SREBP1 or ACC are necessary to establish the causal role of this pathway in mediating the GADD45β knockdown phenotype, but could not be performed due to current resource constraints. Fourth, while GADD45β expression was analyzed at the mRNA level using human GEO datasets, direct validation in clinical liver tissues at the protein level was not feasible, limiting translational strength. Lastly, as HepG2 cells are hepatocarcinoma-derived, their regulatory fidelity may not fully reflect that of normal hepatocytes. Future studies using human liver tissues, primary hepatocytes, or organoid models are warranted to validate the GADD45β/SIRT1 axis under more physiologically relevant conditions.
In conclusion, this study underscores the important role of GADD45β in modulating hepatic lipogenesis through its interaction with SIRT1. By stabilizing SIRT1 and preventing its ubiquitin–proteasome degradation, GADD45β enhances SIRT1 activity and activates the SIRT1/AMPK signaling pathway, thereby suppressing lipogenesis. These findings provide novel insights into the molecular mechanisms of hepatic lipid metabolism and identify GADD45β as a potential therapeutic target for the management of MAFLD. Given the increasing global burden of metabolic liver disorders, further exploration of GADD45β function may support the development of targeted interventions to mitigate disease progression.
Data availability
RNA sequencing data were obtained from the GEO database (GSE33814, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi? acc = GSE33814 and GSE48452, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi? acc = GSE48452). All other data generated or analyzed during this study are included in the published manuscript and the Supplementary information files.
References
Friedman, S. L., Neuschwander-Tetri, B. A., Rinella, M. & Sanyal, A. J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 24, 908–922. https://doi.org/10.1038/s41591-018-0104-9 (2018).
Eslam, M. et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 73, 202–209. https://doi.org/10.1016/j.jhep.2020.03.039 (2020).
Buzzetti, E., Pinzani, M. & Tsochatzis, E. A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 65, 1038–1048. https://doi.org/10.1016/j.metabol.2015.12.012 (2016).
Ter Horst, K. W. & Serlie, M. J. Fructose consumption, lipogenesis, and non-alcoholic fatty liver disease. Nutrients 9, 981. https://doi.org/10.3390/nu9090981 (2017).
Knebel, B. et al. Fatty liver due to increased de novo lipogenesis: Alterations in the hepatic peroxisomal proteome. Front. Cell. Dev. Biol. 7, 248. https://doi.org/10.3389/fcell.2019.00248 (2019).
Dong, Y. et al. GADD45beta stabilized by direct interaction with HSP72 ameliorates insulin resistance and lipid accumulation. Pharmacol. Res. 173, 105879. https://doi.org/10.1016/j.phrs.2021.105879 (2021).
Wilkins, T., Tadkod, A., Hepburn, I. & Schade, R. R. Nonalcoholic fatty liver disease: Diagnosis and management. Am. Fam Physician. 88, 35–42 (2013).
Yu, Y. et al. GADD45beta mediates p53 protein degradation via Src/PP2A/MDM2 pathway upon arsenite treatment. Cell. Death Dis. 4, e637. https://doi.org/10.1038/cddis.2013.162 (2013).
Kim, H. et al. GADD45beta regulates hepatic gluconeogenesis via modulating the protein stability of FoxO1. Biomedicines 9, 50. https://doi.org/10.3390/biomedicines9010050 (2021).
Yang, Y. et al. Regulation of yak longissimus lumborum energy metabolism and tenderness by the AMPK/SIRT1 signaling pathways during postmortem storage. PLoS One. 17, e0277410. https://doi.org/10.1371/journal.pone.0277410 (2022).
Dong, H. W., Zhang, L. F. & Bao, S. L. AMPK regulates energy metabolism through the SIRT1 signaling pathway to improve myocardial hypertrophy. Eur. Rev. Med. Pharmacol. Sci. 22, 2757–2766. https://doi.org/10.26355/eurrev_201805_14973 (2018).
Percie du Sert, N. et al. The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. PLoS Biol. 18, e3000410. https://doi.org/10.1371/journal.pbio.3000410 (2020).
Manne, V., Handa, P. & Kowdley, K. V. Pathophysiology of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Clin. Liver Dis. 22, 23–37. https://doi.org/10.1016/j.cld.2017.08.007 (2018).
Fulop, P. & Paragh, G. [Patomechanisms of hepatic steatosis]. Orv Hetil. 151, 323–329. https://doi.org/10.1556/OH.2010.28816 (2010).
Moskalev, A. A. et al. Gadd45 proteins: Relevance to aging, longevity and age-related pathologies. Ageing Res. Rev. 11, 51–66. https://doi.org/10.1016/j.arr.2011.09.003 (2012).
Fuhrmeister, J. et al. Fasting-induced liver GADD45beta restrains hepatic fatty acid uptake and improves metabolic health. EMBO Mol. Med. 8, 654–669. https://doi.org/10.15252/emmm.201505801 (2016).
Cai, X., Feng, Y., Xu, M., Yu, C. & Xie, W. Gadd45b is required in part for the anti-obesity effect of constitutive androstane receptor (CAR). Acta Pharm. Sin B. 11, 434–441. https://doi.org/10.1016/j.apsb.2020.08.015 (2021).
Winder, W. W. & Hardie, D. G. AMP-activated protein kinase, a metabolic master switch: Possible roles in type 2 diabetes. Am. J. Physiol. 277, E1–E10. https://doi.org/10.1152/ajpendo.1999.277.1.E1 (1999).
Smith, B. K. et al. Treatment of nonalcoholic fatty liver disease: Role of AMPK. Am. J. Physiol. Endocrinol. Metab. 311, E730–E740. https://doi.org/10.1152/ajpendo.00225.2016 (2016).
Li, Y. et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell. Metab. 13, 376–388. https://doi.org/10.1016/j.cmet.2011.03.009 (2011).
Zhou, Y. et al. LXRa participates in the mTOR/S6K1/SREBP-1c signaling pathway during sodium palmitate-induced lipogenesis in HepG2 cells. Nutr. Metab. (Lond). 15, 31. https://doi.org/10.1186/s12986-018-0268-9 (2018).
Nagle, C. A., Klett, E. L. & Coleman, R. A. Hepatic triacylglycerol accumulation and insulin resistance. J. Lipid Res. 50 Suppl, 74–79. https://doi.org/10.1194/jlr.R800053-JLR200 (2009).
Edwards, P. A., Tabor, D., Kast, H. R. & Venkateswaran, A. Regulation of gene expression by SREBP and SCAP. Biochim. Biophys. Acta. 1529, 103–113. https://doi.org/10.1016/s1388-1981(00)00140-2 (2000).
Sekiya, M. et al. SREBP-1-independent regulation of lipogenic gene expression in adipocytes. J. Lipid Res. 48, 1581–1591. https://doi.org/10.1194/jlr.M700033-JLR200 (2007).
Calderin, E. P. et al. Exercise-induced specialized proresolving mediators stimulate AMPK phosphorylation to promote mitochondrial respiration in macrophages. Mol. Metab. 66, 101637. https://doi.org/10.1016/j.molmet.2022.101637 (2022).
Canto, C. & Auwerx, J. Targeting sirtuin 1 to improve metabolism: All you need is NAD(+)? Pharmacol. Rev. 64, 166–187. https://doi.org/10.1124/pr.110.003905 (2012).
Rahman, S. & Islam, R. Mammalian Sirt1: Insights on its biological functions. Cell. Commun. Signal. 9, 11. https://doi.org/10.1186/1478-811X-9-11 (2011).
Elibol, B. & Kilic, U. High levels of SIRT1 expression as a protective mechanism against disease-related conditions. Front. Endocrinol. (Lausanne). 9, 614. https://doi.org/10.3389/fendo.2018.00614 (2018).
Schug, T. T. & Li, X. Sirtuin 1 in lipid metabolism and obesity. Ann. Med. 43, 198–211. https://doi.org/10.3109/07853890.2010.547211 (2011).
Canto, C. & Auwerx, J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 20, 98–105. https://doi.org/10.1097/MOL.0b013e328328d0a4 (2009).
Banks, A. S. et al. SirT1 gain of function increases energy efficiency and prevents diabetes in mice. Cell. Metab. 8, 333–341. https://doi.org/10.1016/j.cmet.2008.08.014 (2008).
Wang, R. H., Li, C. & Deng, C. X. Liver steatosis and increased ChREBP expression in mice carrying a liver specific SIRT1 null mutation under a normal feeding condition. Int. J. Biol. Sci. 6, 682–690. https://doi.org/10.7150/ijbs.6.682 (2010).
Ponugoti, B. et al. SIRT1 deacetylates and inhibits SREBP-1 C activity in regulation of hepatic lipid metabolism. J. Biol. Chem. 285, 33959–33970. https://doi.org/10.1074/jbc.M110.122978 (2010).
Noriega, L. G. et al. CREB and ChREBP oppositely regulate SIRT1 expression in response to energy availability. EMBO Rep. 12, 1069–1076. https://doi.org/10.1038/embor.2011.151 (2011).
Andrade, J. M. et al. Resveratrol attenuates hepatic steatosis in high-fat fed mice by decreasing lipogenesis and inflammation. Nutrition 30, 915–919. https://doi.org/10.1016/j.nut.2013.11.016 (2014).
Hou, X. et al. SIRT1 regulates hepatocyte lipid metabolism through activating AMP-activated protein kinase. J. Biol. Chem. 283, 20015–20026. https://doi.org/10.1074/jbc.M802187200 (2008).
Price, N. L. et al. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell. Metab. 15, 675–690. https://doi.org/10.1016/j.cmet.2012.04.003 (2012).
Ford, R. J., Desjardins, E. M. & Steinberg, G. R. Are SIRT1 activators another indirect method to increase AMPK for beneficial effects on aging and the metabolic syndrome? EBioMedicine 19, 16–17. https://doi.org/10.1016/j.ebiom.2017.04.027 (2017).
Ajmo, J. M., Liang, X., Rogers, C. Q., Pennock, B. & You, M. Resveratrol alleviates alcoholic fatty liver in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 295, G833–842. https://doi.org/10.1152/ajpgi.90358.2008 (2008).
Heeboll, S. et al. Placebo-controlled, randomised clinical trial: High-dose Resveratrol treatment for non-alcoholic fatty liver disease. Scand. J. Gastroenterol. 51, 456–464. https://doi.org/10.3109/00365521.2015.1107620 (2016).
Liu, P. Y. et al. E3 ubiquitin ligase Grail promotes hepatic steatosis through Sirt1 Inhibition. Cell. Death Dis. 12, 323. https://doi.org/10.1038/s41419-021-03608-9 (2021).
Yu, Y. et al. Oxidative stress impairs the Nur77-Sirt1 axis resulting in a decline in organism homeostasis during aging. Aging Cell. 22, e13812. https://doi.org/10.1111/acel.13812 (2023).
Yu, L. et al. Ubiquitination-mediated degradation of SIRT1 by SMURF2 suppresses CRC cell proliferation and tumorigenesis. Oncogene 39, 4450–4464. https://doi.org/10.1038/s41388-020-1298-0 (2020).
Leng, S. et al. SIRT1 coordinates with the CRL4B complex to regulate pancreatic cancer stem cells to promote tumorigenesis. Cell. Death Differ. 28, 3329–3343. https://doi.org/10.1038/s41418-021-00821-z (2021).
Ren, X., Chen, N., Chen, Y., Liu, W. & Hu, Y. TRB3 stimulates SIRT1 degradation and induces insulin resistance by lipotoxicity via COP1. Exp. Cell. Res. 382, 111428. https://doi.org/10.1016/j.yexcr.2019.05.009 (2019).
Cruz Walma, D. A., Chen, Z., Bullock, A. N. & Yamada, K. M. Ubiquitin ligases: Guardians of mammalian development. Nat. Rev. Mol. Cell. Biol. 23, 350–367. https://doi.org/10.1038/s41580-021-00448-5 (2022).
Steklov, M. et al. Mutations in LZTR1 drive human disease by dysregulating RAS ubiquitination. Science 362, 1177–1182. https://doi.org/10.1126/science.aap7607 (2018).
Park, J. J. et al. MDM2-dependent Sirt1 degradation is a prerequisite for Sirt6-mediated cell death in head and neck cancers. Exp. Mol. Med. 53, 422–431 (2021).
Ren, X., Chen, N., Chen, Y., Liu, W. & Hu, Y. TRB3 stimulates SIRT1 degradation and induces insulin resistance by lipotoxicity via COP1. Exp. Cell Res. 382, 111428 (2019).
Leng, S. et al. SIRT1 coordinates with the CRL4B complex to regulate pancreatic cancer stem cells to promote tumorigenesis. Cell. Death Differ. 28, 3329–3343 (2021).
Palomer, X. et al. GADD45A: With or without you. Med. Res. Rev. 44, 1375–1403 (2024).
Li, C. et al. GADD45α alleviates acetaminophen-induced hepatotoxicity by promoting AMPK activation. Cell. Mol. Life Sci. 76, 129–145 (2019).
Tanaka, N. et al. Growth arrest and DNA damage-inducible 45α protects against nonalcoholic steatohepatitis induced by methionine-and choline-deficient diet. Biochim. Biophys. Acta (BBA)-Mol. Basis Disease. 1863, 3170–3182 (2017).
Wu, L. et al. Hepatic Gadd45β promotes hyperglycemia and glucose intolerance through DNA demethylation of PGC-1α. J. Exp. Med. 218, e20201475 (2021).
Dong, Y. et al. GADD45β stabilized by direct interaction with HSP72 ameliorates insulin resistance and lipid accumulation. Pharmacol. Res. 173, 105879 (2021).
Wu, C. et al. Contradictory role of Gadd45β in liver diseases. J. Cell. Mol. Med. 28, e70267 (2024).
Cai, L. et al. AMPK dependent protective effects of Metformin on tumor necrosis factor-induced apoptotic liver injury. Biochem. Biophys. Res. Commun. 465, 381–386 (2015).
Perazza, F. et al. Metformin and the liver: Unlocking the full therapeutic potential. Metabolites 14, 186 (2024).
Funding
This work was supported by the Shanghai Sixth People’s Hospital Clinic Research Project (ynhg202103) to Yuanyuan Xiao; Foundation of Shanghai University of Medicine and Health Sciences (SSF-23-14-001) to Chaoyu Zhu; Fundamental Research Funds for the Central Universities (24X010301321) to Qianqian Wang; Institutional Project of Shanghai Sixth People’s Hospital (ynhglg202405) to Xinyi Wang; Shanghai Municipal Science and Technology Commission Project (20ZR1442500) to Li Wei. All the funding sources were not involved in the study design; collection, analysis, and interpretation of data; writing of the manuscript; and the decision to submit the article for publication.
Author information
Authors and Affiliations
Contributions
Yuanyuan Xiao, Renjie Wang, and Li Wei conceptualized the study. Yuanyuan Xiao, Renjie Wang, Jun Yin, and Li Wei designed the methodology. Yuanyuan Xiao, Renjie Wang, and Chaoyu Zhu performed validation. Yuanyuan Xiao, Renjie Wang, Chaoyu Zhu, Qianqian Wang, Xinyi Wang, Wenjing Song, Shouxia Li, and Fusong Jianga conducted the investigation and data collection. Yuanyuan Xiao and Renjie Wang drafted the manuscript. Jun Yin and Li Wei revised the manuscript. Yuanyuan Xiao and Renjie Wang prepared the figures and tables. Jun Yin and Li Wei supervised the project. Li Wei provided resources and administered the project. Yuanyuan Xiao, Chaoyu Zhu, Qianqian Wang, Xinyi Wang, and Li Wei acquired funding.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xiao, Y., Wang, R., Zhu, C. et al. GADD45β inhibits hepatic lipogenesis through the AMPK/SREBP1 pathway via reducing the ubiquitination-mediated degradation of SIRT1. Sci Rep 15, 39026 (2025). https://doi.org/10.1038/s41598-025-24864-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-24864-1
Keywords
This article is cited by
-
Dysregulation of the AMPK-SREBP1-FASN axis in MASLD: driving a vicious cycle of lipotoxicity and metabolic-immune crosstalk
Lipids in Health and Disease (2026)
