
Abstract
This study presents significant advances in understanding the complex interplay between inorganic scaling and asphaltene behavior in oil–water systems through innovative experimental and analytical approaches. We aim to elucidate non-linear, concentration-dependent scaling phenomena, molecular-level asphaltene transformations during scaling, and the coupled impacts on interfacial properties. To that end, we conducted carefully designed static incompatibility tests using formation waters (FWs) containing Ba2+ or Sr2+ and modified seawater (SW) with controlled sulfate (4 × Na2SO4), quantified precipitates and characterized them by energy dispersive X-ray spectroscopy (EDS)/X-ray diffraction (XRD), tracked molecular signatures by Fourier transform infrared spectroscopy (FTIR), and monitored polarity indices and interfacial tension (IFT). The results show that sulfate concentration thresholds govern scaling; at 4 × Na2SO4, Sr2+ systems yield 85.43% gypsum with 14.57% halite, whereas Ba2+ systems form 75.08% gypsum and 24.92% barite, confirming barium’s stronger scaling tendency. Polarity indices shift from 0.1296 (crude oil) to 0.1515 (a non-scaling Sr2+ condition) and then decrease to 0.1388 under scaling, with more pronounced effects in Ba2+ systems; EDS indicates sequestration of oxygen and sulfur heteroatoms within scale matrices. Consistently, IFT rises from 27.33 to 39.04 mN/m in Sr2+ systems and from 33.41 to 42.73 mN/m in Ba2+ systems under scaling. These findings provide a quantitative framework for predicting and managing coupled scaling–asphaltene interactions in carbonate reservoirs and enable optimized water-injection strategies that jointly control scale formation and enhance oil recovery, improving on conventional approaches that treat these challenges separately.
Similar content being viewed by others


Introduction
Enhanced oil recovery (EOR) techniques have become crucial for extending the life of aging reservoirs and improving production efficiency, particularly as primary recovery methods decline1. One of the most common and cost-effective methods employed in secondary oil recovery is water flooding, with seawater (SW) injection being a widely used practice2. SW offers operational advantages due to its accessibility, low cost, and relatively simple injection system requirements. However, despite its benefits, SW injection can result in scale formation, permeability decline, and operational inefficiencies, especially when incompatibility arises between the injected SW and the formation water (FW)3. This incompatibility often results in the formation of mineral scales, which can negatively impact reservoir performance by blocking pore spaces, reducing permeability, and causing equipment failures4,5. The formation of scale during SW injection is primarily due to the interaction between ions present in the SW and the FW. The presence of barium, calcium, and strontium ions in FW, when combined with sulfate ions in SW, can lead to the precipitation of insoluble minerals, such as calcium sulfate (CaSO4) and barium sulfate (BaSO4)6. While numerous divalent cations exist in reservoir brines, Ba2+ and Sr2+ were selected as representative cases due to: (1) their contrasting solubility products with sulfate (Ksp(BaSO4) = 1.1 × 10−10 vs Ksp(SrSO4) = 3.4 × 10−7), (2) their prevalence in problematic scaling scenarios, and (3) their differential impacts on asphaltene behavior documented in preliminary studies7.
The mixing of SW and FW, which contains high concentrations of calcium, barium, and strontium ions, can lead to the precipitation of various scales, such as CaSO4, BaSO4, and strontium sulfate (SrSO4). These scale formations can significantly reduce the permeability of the reservoir rock, leading to production losses and operational difficulties5. A study by Merdhah et al. (2008) investigated the permeability reduction caused by scale deposition in sandstone cores. The results demonstrated that the extent of permeability damage was strongly influenced by the concentration of ions and temperature, with higher temperatures promoting the precipitation of CaSO4 and SrSO4 and reducing BaSO4 precipitation. The study also found that the mixing of SW and FW at elevated temperatures and pressures led to significant scale formation, which was confirmed through scanning electron microscopy (SEM) analysis. The solubility of the scales was found to decrease with rising temperature for calcium and strontium sulfate, while barium sulfate showed increased solubility at higher temperatures8. The experimental study by Abbasi et al. (2020) examined mixed-salt precipitation during smart water injection in carbonate formations. The study aims to explore the impact of increasing sulfate ion concentrations in injection water and its potential to form sulfate scales upon mixing with FW. The results show that increasing sulfate concentrations, particularly in smart water, significantly enhances the precipitation of sulfate scales like SrSO4 and calcium carbonate (CaCO3). This phenomenon introduces a significant challenge in field operations, as it can potentially block pores and reduce permeability. The study also highlights that smart water’s composition, especially the sulfate ion concentration, plays a key role in both wettability alteration and scale formation9. Mohammadi and Riahi (2020) scrutinized the effects of water incompatibility during waterflooding in carbonate reservoirs, focusing on scale formation caused by mixing injected brine with FW. Key findings suggest that sulfate ions are the primary contributors to scale formation when mixed with cation-rich FW, particularly causing the precipitation of CaSO4 and BaSO4. The study also highlights the importance of ionic composition in the formation of scale and the effectiveness of scale inhibitors in mitigating scale risks10.
Al-Samhan et al. (2020) evaluated the scale deposition and scale tendency of effluent water mixed with seawater for injection in oilfields. The study focuses on the challenges posed by scale formation during water injection. The researchers examined effluent water samples from various oil fields in Kuwait and mixed them with seawater in different ratios to assess compatibility and scale formation risks. The results showed that mixing effluent water from the North Kuwait field with seawater in a 1:1 ratio produced scale deposits, predominantly CaSO4 and silicon oxide, but with minimal BaSO4 precipitation. The study used X-ray diffraction (XRD), energy-dispersive spectroscopy (EDS), and SEM to characterize the precipitates. The findings revealed that the risk of scale formation, particularly BaSO4, can be minimized by adjusting the seawater/effluent ratio11. Razavirad et al. (2024) studied the compatibility between formation water and candidate injection water, focusing on the effects of temperature and pressure on scale formation in oil reservoirs. Their findings revealed that temperature significantly impacts the concentration of sulfate in seawater, where an increase in temperature led to a decrease in sulfate concentration due to the precipitation of CaSO4. However, in smart water mixtures, sulfate concentrations increased with temperature, likely due to the transformation of sulfite into sulfate. The study also examined pressure effects, finding minimal impact on sulfate and calcium solubility under varied pressures at ambient temperatures. Notably, while SrSO4 precipitation did not occur, calcium carbonate formation was observed in mixtures with higher smart water ratios12. Hussein et al. (2024) conducted an experimental study to examine the factors influencing BaSO4 scale formation in water injection operations. The study focused on the impact of injection rate and temperature on scale formation under controlled laboratory conditions. The results revealed that higher injection rates and elevated temperatures significantly increased the formation of BaSO4 scale. Specifically, scale formation increased from 15.2 to 22.3 mg/L with an increase in injection rate from 1 to 5 mL/min at 25 °C. Likewise, at a constant injection rate of 3 mL/min, scale formation rose from 18.1 mg/L at 25 °C to 26.5 mg/L at 75 °C. SEM and XRD analyses indicate a systematic shift in scale habit with thermal and hydrodynamic conditions. At lower temperatures and slower injection rates, crystals adopt dendritic or needle-like morphologies, whereas higher temperatures and higher rates favor more compact, near-spherical crystals. This distinction is operationally important, because dendritic networks, with higher specific surface area and asperity-driven interlocking, more readily bridge pore throats, adhere to mineral surfaces, and intensify permeability loss and wettability perturbation. In contrast, spherical crystals are more easily entrained and transported, tend to migrate and redeposit deeper in the formation or in surface facilities, and present lower interfacial area per unit mass. Recognizing and managing this habit evolution helps predict the localization of damage, such as near-wellbore plugging versus deeper migration, and it aligns with our FTIR and EDS evidence that rougher dendritic surfaces more effectively sequester polar asphaltene fragments13.
The study of asphaltenes dates back to the 1970s, when Yen proposed a dense aromatic cluster model, which extended aromatic rings into large sheets with side chains14. In recent years, substantial progress has been made in the study of asphaltene precipitation and its prevention. Asphaltene precipitation is a significant challenge in oil production, particularly in EOR methods. Asphaltene, a complex fraction of crude oil, can precipitate and deposit within the porous media during water injection, causing permeability reduction and altering wettability, thus affecting production efficiency15. Previous research has identified several factors influencing asphaltene instability, such as temperature, pressure, and oil composition. However, the role of brine, especially emulsified brine, on asphaltene precipitation and deposition has received limited attention. The impact of brine on asphaltene precipitation is largely driven by the concentration and type of salts, which can significantly alter the interfacial tension (IFT) between water and oil phases16,17. Tokali et al. (2016) focused on the effect of salinity on asphaltene stability and emulsification, demonstrating that emulsions could significantly affect asphaltene instability in certain crude oils18. Studies by Shojaati et al. (2017) have demonstrated that salts like MgCl2 enhance asphaltene precipitation, with the concentration threshold for optimal effects observed between 25,000 and 40,000 ppm. Similarly, the dual effect of salts, salting in and salting out, further complicates this relationship19. Additionally, the type of brine, particularly the presence of divalent cations like Ca2+ and Mg2+, plays a crucial role in modifying asphaltene stability. For instance, the studies by Alizadeh and Soulgani (2021) revealed that different cation types and concentrations influence the surface excess concentration of asphaltenes, thereby affecting their precipitation behavior. These findings underscore the complex interplay between water, brine composition, and asphaltene behavior in reservoir conditions20. The study by Doryani et al. (2018) explored the effects of various connate water types on asphaltene behavior in porous media. They found that different cations in connate water, particularly divalent cations like Ca2+, play a significant role in asphaltene precipitation due to their affinity for asphaltene molecules. The study also revealed that higher salinity and brine concentration could enhance the precipitation and deposition of asphaltenes, with brines containing divalent ions exhibiting a more pronounced effect compared to monovalent ions. Furthermore, the researchers highlighted the role of water saturation levels, where higher connate water saturation led to increased asphaltene precipitation21. Mokhtari et al. (2022) focused on investigating the destabilization of asphaltenes in crude oil during interactions with aqueous phases, specifically brine solutions of varying salinities. The research explored the influence of brine salinity, ion types, and contact time on asphaltene precipitation, an essential phenomenon for waterflooding in oil reservoirs. At higher brine salinities, ions like sulfate and magnesium are more likely to act as nuclei, promoting asphaltene precipitation. Conversely, at lower salinities, the brine-oil interface’s polarity leads to adsorption of asphaltenes, which can destabilize their structure in the bulk oil. The findings also highlight the role of naphthenic acids (NAs) in modulating the extent of asphaltene destabilization, with higher salinity reducing the impact of NAs by restricting their ability to affect asphaltene precipitation22. Mahdavi and Dehaghani (2024) investigated the behavior of water-in-oil emulsions in the context of smart water flooding, particularly focusing on the role of asphaltene and clay particles in the emulsion formation process. It highlights how ion-tuned water, which is injected during EOR processes, interacts with crude oil and clay, affecting the stability of emulsions. The research explores the effects of different ions such as calcium, magnesium, and sulfate in the aqueous phase and their influence on the IFT and asphaltene precipitation onset point (AOP). The results indicate that the presence of clay, especially kaolinite, enhances the tendency of polar asphaltene components to migrate to the interface, altering the viscosity and rheological properties of the emulsions23.
To better highlight the novelty of this work, a comparative summary of the most relevant previous studies on scaling and asphaltene behavior is provided in Table 1. This table contrasts their focus, findings, and limitations with the present study, emphasizing how our integrated framework bridges existing gaps.
While previous studies have established the fundamental interactions between sulfate-rich waters and Ba2+/Sr2+ ions in scale formation (e.g., barite and gypsum precipitation)6,24, and some have examined scaling effects on asphaltene behavior and IFT10,17, critical knowledge gaps remain in understanding the complex, coupled physicochemical processes under realistic reservoir conditions (see Table 1). In contrast to previous studies that mainly investigated single-ion effects or treated scaling and asphaltene behavior separately, the present work introduces three distinctive contributions. First, it systematically explores the coupled impacts of Mg2+, Ca2+, and SO42−concentrations on both scaling dynamics and asphaltene transformations. Second, it integrates multi-scale analytical techniques (FTIR, EDS, XRD) to directly correlate mineral precipitation with molecular-level asphaltene alterations. Third, it identifies a critical sulfate threshold (4 × Na2SO4) at which barite-dominated scaling triggers abrupt changes in asphaltene polarity and interfacial tension. Together, these contributions provide novel mechanistic insights and practical guidelines for designing water injection strategies beyond the scope of existing research. Beyond filling existing knowledge gaps, the present work establishes a broader framework for linking mineral precipitation with molecular changes in crude oil systems. By bridging laboratory-scale observations with reservoir-scale implications, this study provides a new perspective on how water chemistry can be engineered to balance scale control and asphaltene stability. The insights gained here not only refine the mechanistic understanding of oil–brine interactions but also offer a practical roadmap for designing injection strategies that are more predictive, integrative, and field-relevant than conventional approaches.
Our integrated experimental framework provides new predictive capabilities for managing the dual challenges of mineral scaling and asphaltene instability in carbonate reservoirs. The findings enable more precise optimization of injection water chemistry to simultaneously control scale formation and enhance oil recovery, thereby advancing beyond current practices that typically address these phenomena separately.
Experimental section
This section presents a detailed description of the materials, equipment, and experimental procedures used throughout the study, from the preparation of brine solutions to the analysis of scale formation and its effects on asphaltene behavior. To replicate realistic reservoir conditions and simulate subsequent seawater injection, synthetic formation water (FW) and seawater (SW) solutions were carefully prepared in the laboratory using deionized water and high-purity salts. The ionic compositions of these solutions were designed to closely match the actual concentrations observed in reservoir and seawater samples. The experiments were conducted to evaluate the type and quantity of precipitated scales resulting from SW–FW incompatibility and to assess the impact of these precipitates on the molecular structure and interfacial behavior of asphaltenes.
Materials
Brine preparation
To prepare synthetic FW and SW solutions, high-purity salts (≥ 99%) were purchased from Merck (Germany). The ionic composition of the formation water was modeled after the brine from the Azadegan oil field located in southern Iran (Ahvaz region). Two types of formation water were designed for this study: one containing strontium ions (Sr2+), reflecting the actual composition of the reservoir brine, and a second synthetic variant in which strontium chloride (SrCl2) was replaced by barium chloride (BaCl2) at an equivalent molar concentration. Although barium ions are not naturally present in Azadegan formation water, they were included in this study due to their prevalence in other carbonate reservoirs worldwide and their well-documented strong scaling tendency with sulfate ions. This allows comparative analysis of scaling behaviors between common divalent cations (Sr2+ and Ba2+) under identical experimental conditions. The detailed compositions of the two FW types, containing either SrCl2 or BaCl2, and the synthetic SW are presented in Table 2.
For a more comprehensive investigation, the prepared SW solution was diluted in four different ratios (1:2, 1:5, 1:10, and 1:20) using deionized water to study the effect of salinity variation on scale formation and interfacial properties. These diluted versions are referred to as modified SW solutions throughout the experiments25.
To assess the role of specific scaling ions on asphaltene behavior and IFT, three salts including magnesium chloride (MgCl2), calcium chloride (CaCl2), and sodium sulfate (Na2SO4) were also examined. Each of these salts was added to the seawater solution at three concentration levels: 1.5 × , 2 × , and 4 × of their standard values. This generated an additional nine modified brine solutions. In total, fourteen distinct fluids were prepared and used in various experiments, as summarized in Table 3.
Oil sample preparation
To systematically investigate the influence of inorganic ions and scale formation on asphaltene behavior while eliminating confounding factors present in whole crude oil, we utilized a synthetic oil model prepared by dissolving 5000 ppm of extracted Azadegan asphaltenes in toluene. This approach follows established practices in fundamental asphaltene studies and provides three key advantages: (1) elimination of matrix interference from resins and waxes that could obscure asphaltene-specific interactions, (2) enhanced experimental reproducibility through precise control of asphaltene concentration, and (3) unambiguous attribution of observed effects to scaling-asphaltene dynamics. While natural crude oil offers greater ecological relevance, the synthetic model, which preserves the critical functional groups of Azadegan asphaltenes as confirmed by different analyses, enables the parametric precision required for mechanistic understanding, as demonstrated in similar fundamental studies26.
To synthesize the model oil, asphaltenes were first extracted from Azadegan crude oil using the standard IP-143 procedure, which will be described in detail in the following section. The extracted asphaltene was then dried and mixed with high-purity toluene to create a stable synthetic oil solution. Based on previous research findings, a concentration of 5000 ppm was selected for the mixture, meaning that 0.5 g of dried asphaltene powder was dissolved in 100 g of toluene. This synthetic oil was used in all interfacial tension and phase behavior tests involving different brine and scale-forming conditions27.
The reagents used for asphaltene extraction and synthetic oil preparation, as well as their physical properties, are summarized in Table 4.
To isolate asphaltene-specific mechanisms and ensure reproducible, mechanism-focused comparisons across multi-ion brines, we used a toluene-based model oil (5000 ppm asphaltene). Representativeness was checked by comparing FTIR spectra of whole-crude asphaltenes and a dried film from the model oil; see Supplementary Sect. S2 (Fig. S2; Table S1).
Asphaltene extraction and synthetic oil preparation
We extracted asphaltenes from crude oil following the IP-143 standard using a series of controlled solvent-based separation steps. The procedure was designed to isolate the asphaltene fraction from other heavy components such as waxes and resins, which were removed using n-heptane. The required equipment included a round-bottom flask, funnel, Whatman filter paper with 40 µm mesh size, and a reflux condenser.
In the first step, 10 mL of crude oil obtained from the Azadegan oil field was transferred into a round-bottom flask, followed by the addition of 200 mL of n-heptane, equivalent to 20 times the oil volume. The mixture was stirred using a magnetic stirrer for 10–15 min to initiate asphaltene precipitation. The flask was then kept in a dark environment at room temperature for 24 h to allow complete sedimentation of asphaltenes. After this resting period, the contents were filtered through Whatman filter paper (40 μm) placed in a funnel. The filter retained the solid residue, composed primarily of waxes and asphaltenes, while the clarified solution passed through. To ensure full removal of waxes, approximately 50 mL of fresh n-heptane was added to a clean flask containing the filter paper, and the setup was placed under a reflux condenser. Refluxing continued for at least 1 h, or until the dripping n-heptane became completely colorless, confirming the elimination of waxes and leaving only purified asphaltenes on the filter. Next, the filter paper was transferred into the original flask and treated with 50 mL of pure toluene. A second refluxing process was conducted for 1 h, during which the condenser was rotated every 15 min to ensure uniform exposure of the filter to toluene. This continued until the toluene extract was entirely colorless, indicating full desorption of asphaltenes from the filter. The resulting toluene–asphaltene solution was then poured into a glass beaker and left under a fume hood until all the toluene evaporated, leaving behind a dry layer of asphaltenes. The dry residue was gently scraped off, ground into a fine powder, and stored in an airtight container for subsequent use.
In summary, the model synthetic oil was prepared by dissolving 0.5 g of dried asphaltene powder in 100 g of high-purity toluene, yielding a solution with a concentration of 5000 ppm. This synthetic oil was used as the standard hydrocarbon phase in all experiments involving interfacial tension and asphaltene stability conducted in this study.
Experimental methods and procedures
This section outlines the experimental techniques, instruments, and materials used to evaluate scale formation, interfacial tension changes, and asphaltene behavior under simulated reservoir conditions. All experimental tests, including precipitate separation from SW-FW mixtures (for mass quantification) and interfacial tension measurements, were conducted in triplicate to ensure reproducibility. Mean values are reported with error bars representing standard deviation in all relevant figures. In this study, we divided the experimental methodology into six main categories (See supplementary file).
Scale separation test
In the scale-separation experiment, we used a vacuum pump (Model VE215, China) and Whatman 0.22 µm membrane filters (UK) to isolate and quantify scale formed from water incompatibility. First, two types of synthetic formation water, one containing SrCl2 and the other BaCl2, were prepared. In parallel, fourteen seawater-based brines were formulated, including both diluted samples and those modified by varying concentrations of specific ions. Each of the two formation waters was mixed with all fourteen seawater samples, yielding a total of 28 unique combinations. These mixtures were transferred into separate test tubes and left to rest at room temperature to allow natural precipitation of scales. Once the resting period was complete, the mixtures were filtered through the 0.22 µm membrane under vacuum conditions to separate the solid precipitates. The resulting filtrates, which were free of visible scale, were stored for subsequent analyses. Meanwhile, the collected precipitates were dried and weighed using a precision balance to quantify the extent of scaling under different conditions.
X-ray diffraction (XRD) analysis
We used XRD to determine the crystalline structure and composition of the precipitated scales. This technique, based on Bragg’s law, allows for the identification of crystalline phases by analyzing their unique diffraction patterns. The experiments were conducted using an X’Pert MPD X-ray diffractometer, manufactured by Philips (Netherlands). The instrument was equipped with a Cu X-ray tube operating at a wavelength (λ) of 1.54056 Å, with a step size of 0.02°. The system operated at a voltage of 40 kV and a current of 40 mA, providing high-resolution data suitable for accurate characterization of mineral precipitates28.
Static bottle test
We conducted static bottle tests to simulate emulsion formation and evaluate how scale-forming brines influence asphaltene behavior under reservoir-like conditions. For this purpose, test tubes were filled with equal volumes (1:1) of synthetic oil, prepared by dissolving 5000 ppm asphaltene in high-purity toluene, and the various brine mixtures obtained from earlier steps. The filled tubes were then placed in a laboratory oven set at 70 ℃ and incubated for a period of 3 to 4 weeks to mimic subsurface thermal conditions and promote potential emulsion development. At the end of the incubation period, the emulsion-free fluid phases were carefully separated and stored. These samples were subsequently used for further characterization and analysis using FTIR spectroscopy, XRD, and IFT measurements.
Fourier transform infrared spectroscopy (FTIR) analysis
We collected FTIR spectra in transmission mode using KBr supports (pellets/windows). For each sample, a fixed drop volume of the solution/suspension was drop-cast onto KBr of identical diameter and dried under the same conditions prior to measurement (typical range 4000–650 cm−1, resolution 4 cm−1, ≥ 32 co-added scans). A clean KBr background was recorded before each series. Spectra were baseline-corrected, atmospheric H2O/CO2 compensated, and band areas were integrated over standard windows, e.g. aliphatic C–H stretching (2954, 2925, 2872, 2853 cm−1), aromatic C = C (~ 1600 cm−1), carbonyl C = O (∼1710–1680 cm−1), and sulfoxide S = O (∼1035–1015 cm−1).
Because drop-cast films on KBr do not provide a rigorously defined optical path length (l), absolute Beer–Lambert quantitation is not applied. Instead, we use normalized band-area indices and ratios acquired under identical deposition (same drop volume and drying protocol), which yield robust semi-quantitative trends while minimizing sensitivity to l and matrix-dependent molar absorptivities (ε). Representative indices include AC=O/A1600, AS=O/A1600, and CH2/CH3 from the 2925/2954 cm−1 bands. These metrics allow us to track relative changes in functional-group content of model-oil and whole-crude asphaltenes during oil–brine interactions and scale formation (see supplementary file)29.
Energy dispersive X-ray spectroscopy (EDS)
The elemental composition of the scale deposits formed during brine interactions was determined using EDS performed on a Zeiss Sigma 300-HV device (Germany). This analytical technique operates on the principle that incident X-ray photons eject inner-shell electrons from sample atoms, creating vacancies that are subsequently filled by higher-energy electrons. The resulting energy difference is emitted as characteristic X-rays, which are element-specific and enable precise identification in accordance with Moseley’s law. The EDS data provided critical insights into the elemental constituents of the scales, facilitating mechanistic analysis of precipitation behavior30.
Interfacial tension (IFT) measurement
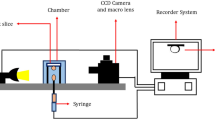
We measured IFT between the synthetic oil and each brine using the pendant drop method, a well-established technique for quantifying the interfacial tension between two immiscible phases, such as liquid–liquid or liquid–gas systems. The IFT between synthetic oil and brine solutions was measured using the pendant drop method, a well-established technique for evaluating the interfacial tension between two immiscible phases, such as liquid–liquid or liquid–gas systems. In this method, a droplet of the lighter fluid (in this case, oil) is suspended from the tip of a fine needle and immersed in a denser medium (the brine). The shape of the stabilized droplet is governed by the balance between interfacial tension and gravitational forces. By analyzing the droplet’s curvature profile captured in high-resolution images or video frames, the interfacial tension is determined through the Young–Laplace equation, which relates pressure difference across the interface to surface curvature and interfacial tension. The schematic diagram of this device is illustrated in Fig. 1.

The schematic diagram of measuring interfacial tension setup.
Results and discussion
This section presents the analysis and interpretation of the experimental results obtained throughout the study. It begins with a detailed presentation of the results from the scale separation test, XRD, FTIR, EDS, and IFT measurements. Following this, a comprehensive comparison and analysis of the FTIR and EDS outcomes will be provided to examine the impact of ions and precipitates on the molecular properties of asphaltene and the interfacial properties between oil and brine. These analyses will contribute to a deeper understanding of the chemical and physical processes occurring in SW and FW systems, particularly focusing on the effects of various precipitates and their interactions with asphaltene and oil phase characteristics.
Precipitate formation analysis from SW-FW interactions
A systematic investigation of inorganic scale deposition from SW-FW interactions was conducted using 46 mL borosilicate test tubes containing 1:1 mixtures of FW (with 9 mM Sr2+ or Ba2+) and 14 distinct SW formulations (including dilutions and ion-enriched variants). The mixtures were maintained at reservoir temperature (70 °C) for 72 h to enable complete precipitation, followed by vacuum filtration through 0.22 μm Whatman membranes (VE215 pump, 25 inHg) and drying at 105 °C for 24 h before triplicate gravimetric measurements (± 0.1 mg precision). As shown in Fig. 2, comparative analysis revealed Ba2+ systems generated 2.2–2.8 × more precipitate than Sr2+ in base conditions, with 4 × Na2SO4 SW causing the most dramatic increases (+ 728% for Sr2+ vs + 151% for Ba2+). Table 5 further demonstrates these trends through consolidated precipitation data [experimental error for precipitate measurements: ± 0.0001 g], highlighting a decrease in the Ba/Sr scaling ratio from 2.81 in base SW to 0.85 under high-sulfate conditions. This pattern indicates that Sr2+ is more sensitive to sulfate enrichment than Ba2+, despite Ba2+’s higher overall scaling tendency.

Absolute precipitate mass (g) for Sr2+ and Ba2+ in variably modified seawater: dilutions and selective salt enrichments, with %Δ vs base concentrations.
The comparative analysis revealed three interconnected scaling mechanisms with significant implications for reservoir management. First, concentration-dependent scaling was observed in both Sr2+ and Ba2+ systems, where precipitate mass increased dramatically with higher salt concentrations, particularly Na2SO4, which produced 0.1449 g (Sr2+) and 0.1237 g (Ba2+) at 4 × concentration. This trend stems from sulfate-driven supersaturation, where elevated [SO42−] exceeds the solubility limits of both cations (Ksp(BaSO4) = 1.1 × 10−10 vs. Ksp(SrSO4) = 3.4 × 10−7), accelerating nucleation kinetics31,32. Second, cation-specific behavior emerged: Ba2+ systems generated 2.2–2.8 × more precipitate in base conditions, yet Sr2+ exhibited greater relative sensitivity to sulfate (+ 728% vs. Ba2+’s + 151%). The inversion of the Ba/Sr ratio (0.85 at 4 × Na2SO4) underscores Sr2+’s unique sulfate affinity, likely due to its intermediate solubility promoting rapid crystal growth at threshold concentrations33,34. Third, mineralogical controls (XRD, Sect. "XRD analysis") dictated practical outcomes: Ba2+ formed dense barite (BaSO4) resistant to membrane filtration, while Sr2+ produced finer SrSO4 crystals prone to penetration, alongside co-precipitated CaSO4 in both systems35.
Mechanistically, sulfate dominance (Fig. 2) arose from strong ionic pairing, while divalent cation effects showed Ca2+ induced 1.9 × more scaling than Mg2+ in Ba2+ systems, reflecting competitive hydration energies. Dilution mitigated scaling asymmetrically (− 29.9% for Ba2+ vs. − 8.6% for Sr2+ at 1:20 SW), suggesting Sr2+’s persistence at low concentrations. Practical insights include: (1) Ba2+ reservoirs demand stringent sulfate control (< 2 × baseline) to avoid pore-clogging barite, (2) Sr2+ systems require monitoring even at moderate sulfate due to rapid celestine nucleation, and (3) filtration efficiency must adapt to scale morphology, barite’s compactness reduces membrane throughput, whereas SrSO4’s fines increase post-treatment risks. These findings collectively refine scale prediction models by quantifying threshold concentrations, cation-specific kinetics, and mineral-dependent recovery factors36.
To proceed with XRD analysis, a minimum precipitate mass of 0.1 g was required to ensure reliable characterization of the crystalline phases. Based on the results, only the samples derived from the mixtures of seawater enriched with 4 × concentration of sodium sulfate and formation waters containing either strontium or barium ions produced sufficient precipitate for XRD testing. Consequently, these two specific precipitates, one from FW-Sr2+ with 4 × Na2SO4 SW and the other from FW-Ba2+ with 4 × Na2SO4 SW were selected for XRD analysis to determine the mineralogical nature of the scale formed under the most severe scaling conditions.
XRD analysis
We used XRD to identify the mineralogical composition and the relative abundance of crystalline phases formed as scale during brine incompatibility. The XRD output consists of intensity values plotted against the 2θ angle, which represents the angle between the incident and diffracted X-ray beams. This 2θ value is experimentally measurable and reflects the diffraction conditions unique to each crystalline plane in the sample. The intensity of each diffraction peak correlates with the number of diffracted X-rays from a specific atomic plane; therefore, higher intensities generally correspond to more dominant crystal structures. Typically, peak intensities decrease at higher diffraction angles due to lower scattering from higher-index planes. In this study, the objective of XRD testing was to determine both the types and relative quantities of precipitated scales. To quantify the results, the area under each diffraction peak was calculated to estimate the overall percentage of crystalline content. The relative proportions of different scales were then determined by comparing the peak intensities, with the strongest peak used as a reference. This approach enabled the calculation of the percentage contribution of each mineral phase within the precipitate37.
Mineralogical composition and phase distribution of Sr2+/Ba2+ scales
We performed XRD on precipitates generated by mixing FW (containing Sr2+ or Ba2+) with SW with 4 × Na2SO4. The diffractograms (Fig. 3) reveal distinct crystalline phases, with quantitative phase abundances calculated via peak intensity normalization (Table 6).

XRD spectra of precipitates from (a) Sr2+-FW + 4 × Na2SO4 SW and (b) Ba2+-FW + 4 × Na2SO4 SW. Peaks marked for gypsum (●), halite (■), and barite (▲).
The XRD results in Fig. 3 and Table 6 reveal that gypsum (CaSO4) dominates the precipitated mineral scales in both Sr2+ and Ba2+ systems (85.43% in Sr2+-FW and 75.08% in Ba2+-FW). This predominance stems from the rapid reaction between calcium ions in the formation water (FW) and excess sulfate from modified seawater (SW), driven by the low solubility product (Ksp) of CaSO4 and its favorable precipitation kinetics. While Ba2+ exhibits a strong affinity for sulfate, forming barite (BaSO4, 24.92%), the high background concentration of Ca2+ in FW and SW outcompetes Ba2+, leading to gypsum as the primary scale. This aligns with previous observations, where gypsum is frequently reported due to ubiquitous calcium and sulfate interactions38.
The differential scaling behavior between gypsum (CaSO4) and barite (BaSO4) presents critical operational implications. while gypsum can be readily mitigated through conventional acidizing or mechanical removal methods, barite’s chemical inertness necessitates expensive inhibitors or chelating agents, with even minor barite precipitation (24.92%) posing severe permeability impairment risks due to its irreversible deposition39. This distinction stems from fundamental precipitation dynamics: despite Ba2+'s greater thermodynamic affinity for sulfate (lower Ksp), CaSO4's kinetic precipitation advantage under typical reservoir conditions ensures gypsum dominance in mixed-cation systems unless sulfate concentrations surpass critical thresholds for barite nucleation. Consequently, optimal waterflooding strategies must be tailored to the specific cation composition: Ba2+-rich reservoirs require strict sulfate control to prevent persistent barite scaling, while Sr2+-containing formations can tolerate higher sulfate concentrations due to gypsum’s more manageable remediation profile. This underscores the need for customized injection water formulations based on thorough FW characterization.
FTIR analysis of asphaltene interactions and functional group alterations in oil–brine systems
In this section, the FTIR technique was employed to investigate the functional groups present in the asphaltene components of the oil phase after prolonged contact with brine solutions. As shown in Fig. 4, a 1:1 volume ratio of synthetic oil and various brine mixtures (comprising FW and SW) was prepared in test tubes and incubated at reservoir temperature (70 °C) for a period of 1 month to simulate in-situ reservoir conditions. After the aging period, the bulk oil phase was carefully separated from the emulsion, dried under a fume hood, and the resulting asphaltene-rich residues were collected for FTIR analysis. This analysis aimed to identify the key functional groups responsible for asphaltene behavior and to assess any molecular-level changes in response to scaling and ion interactions. The FTIR spectra provided insight into the nature and intensity of bonds such as C = O, C = C, O–H, and S = O, which are known to influence emulsion stability and interfacial behavior in oil–brine systems.

Schematic illustration of sample preparation for FTIR analysis: synthetic oil mixed with brine (FW + SW) at a 1:1 ratio.
The FTIR spectra of all various asphaltene samples revealed consistent and representative absorption peaks located at 3400, 3040, 2962, 2921, 2851, 1700, 1600, 1450, 1375, 1262, 1116, 1030, 864, 801, 754, and 727 cm−1. The interpretation of these peaks is summarized in Table 7, which assigns each wavenumber range to its most probable functional group. Notably, the wide absorption band around 3400 cm−1 corresponds to O–H and N–H stretching vibrations, indicating the presence of hydroxyl groups (from phenols, alcohols, and carboxylic acids) and amides within the asphaltene structure. The aromatic C–H stretch at 3040 cm−1 confirms the aromatic nature of asphaltene, while strong CH3 and CH2 stretches at 2962 and 2921 cm−1 reflect the aliphatic side chains. Carbonyl (C = O) functionality was detected around 1700 cm−1, suggesting the presence of ketones, aldehydes, or carboxylic acids. The peaks at 1600 cm−1 (aromatic C = C), 1450–1375 cm−1 (aliphatic CH bending), and 1262 cm−1 (C–O stretching in acids) further confirm the complexity and multifunctionality of asphaltene molecules. Peaks below 1116 cm−1, especially at 1030 cm−1 are indicative of sulfoxide groups (S = O), suggesting oxidative aging or interaction with sulfate ions.
Quantitative comparisons among the samples were enabled by calculating several structural indices based on peak area integrations, as outlined in Table 8. These indices include aromaticity, aliphaticity, polarity, sulfoxide index, carbonyl index, and aliphatic chain length. The total integrated peak area (ΣA) was used as the normalization factor for each calculation. For instance, the polarity index was derived from A3400/ΣA, while aromatic and aliphatic indices were calculated using designated aromatic and aliphatic peaks, respectively. These indices facilitated the assessment of structural modifications in asphaltenes after exposure to different brine chemistries40.
In this section, to investigate the molecular structure of asphaltene and its interactions with inorganic ions and scale precipitates, nine different asphaltene samples extracted from oil under various conditions were subjected to FTIR analysis. These samples were prepared to simulate contact between oil and synthetic waters containing strontium or barium ions, as well as seawater with or without elevated sodium sulfate concentrations. The main objective was to identify structural changes in functional groups and chemical bonds affecting the stability of oil–water emulsions and the precipitation behavior of scales. The FTIR results for these nine samples were analyzed to assess the influence of divalent ions and their associated scales on asphaltene chemistry. The experimental design included both precipitation and non-precipitation conditions in the presence of Sr2+- or Ba2+-based FWs combined with seawater or seawater enriched with 4 × sodium sulfate. This dataset enables a detailed evaluation of the effects of divalent ions on asphaltene structure, as well as the influence of mineral scaling on the polar, aromatic, aliphatic, and heteroatomic functional characteristics of asphaltenes. Table 9 presents the full list of the asphaltene samples analyzed in this study, along with a brief description of their preparation conditions, providing a framework for the detailed comparison and FTIR-based index calculations that follow in the subsequent sections41.
FTIR analysis of asphaltene extracted from base crude oil
As previously noted, the asphaltene content in the crude oil was extracted using the IP-143 standard method and subjected to FTIR spectroscopy to identify its functional groups. The resulting spectrum, shown in Fig. 5, serves as the baseline molecular fingerprint for untreated asphaltene. The FTIR spectrum reveals several prominent absorption bands characteristic of asphaltene structures. A broad and strong peak centered around 3400 cm−1 corresponds to O–H and N–H stretching vibrations, indicating the presence of hydroxyl, phenolic, and amide functionalities. These groups reflect the polar nature of asphaltenes and their potential for hydrogen bonding.

FTIR spectrum of asphaltene extracted from base crude oil.
Intense bands at 2921 cm−1 and 2851 cm−1 are assigned to asymmetric and symmetric C–H stretching vibrations in aliphatic CH2 and CH3 groups, suggesting the presence of long alkyl chains. Additionally, the peak at 3040 cm−1 is attributed to aromatic C–H stretching, confirming the polyaromatic character of the sample. Absorptions near 1600 cm−1 and 1450–1375 cm−1 represent aromatic C = C stretching and C–H bending in aliphatic chains, respectively. A clear absorption at 1700 cm−1 suggests the presence of C = O stretching in carbonyl-containing groups such as ketones, aldehydes, and carboxylic acids. Minor peaks at 1030 cm−1 and 1116 cm−1 are attributed to S = O stretching in sulfoxides and C–N stretching in amines, indicating the presence of heteroatoms. The fingerprint region also shows bands near 864, 801, 754, and 727 cm−1, which correspond to aromatic C–H out-of-plane bending and C–H vibrations in long-chain alkanes.
This initial FTIR analysis serves as a baseline for comparison with subsequent spectra obtained from crude oil samples that were contacted with FWs containing divalent cations (Sr2+ and Ba2+), as well as various synthetic SW solutions, including both standard SW and SW enriched with four times the concentration of Na2SO4. The comparative analysis includes conditions both with and without the formation of mineral scales. This comparative approach enables the identification of any structural or chemical alterations in asphaltene molecules resulting from brine interaction and scale precipitation phenomena.
FTIR analysis of crude oil asphaltenes in contact with Sr2+-based FW and SW: With and without scale formation
To assess the structural and functional changes in asphaltene due to interaction with brine components and scale formation, FTIR spectroscopy was conducted on three different samples: (1) native crude oil asphaltene, (2) crude oil exposed to Sr2+-rich FW and SW with scale formation, and (3) crude oil exposed to the same brine system without scale formation. In both treated cases, the bulk oil phase was sampled after exposure, and toluene was used to extract the asphaltenes, followed by solvent evaporation under a fume hood. The resulting solid was then analyzed using FTIR. Figure 6 presents an overlay of the three spectra (crude oil, Sr2+ + SW with scale, Sr2+ + SW without scale), enabling direct comparison of functional group variations among the samples. This comparative analysis highlights the influence of scaling conditions on the interfacial chemistry and structural features of the asphaltenes.

Comparative FTIR spectra of asphaltenes: crude oil vs. Sr2+-based FW + SW system with and without scale formation.
According to the absorption bands identified in the FTIR spectra and the corresponding functional group assignments summarized in Table 6, notable structural changes in asphaltene were observed under varying experimental conditions. These changes were quantitatively analyzed using specific functional indices, such as S = O, C = O, polarity, aromaticity, and aliphaticity, as presented in Table 10, and visually compared across conditions in Fig. 7. The FTIR analysis reveals distinct variations in molecular composition when crude oil is exposed to SW and strontium-containing FW under conditions that either allow or prevent mineral scale formation.

Comparison of aliphatic, aromatic, polar, carbonyl, and sulfoxide indices of asphaltene in crude oil, Sr2+-based FW + SW system (with and without scale formation).
As illustrated in Fig. 7, the comparative analysis of asphaltene characteristics under different conditions reveals several important trends. The formation of mineral scale clearly results in a reduction in the relative abundance of polar and carbonyl functional groups, as indicated by the decline in their corresponding indices. This suggests that these polar moieties may be preferentially incorporated into the scale matrix or emulsified phase. Furthermore, asphaltene molecules in the bulk oil phase exhibit a stronger aliphatic character following scale precipitation, contrasting with the enhanced polarity observed when scale does not form. Notably, the presence of divalent ions such as Sr2+ promotes an increase in asphaltene polarity only in the absence of scale formation; however, once scaling occurs, this effect is diminished or even reversed. This highlights the complex interplay between ionic composition and scale dynamics in governing the molecular structure and interfacial behavior of asphaltenes.
Furthermore, exposure of crude oil to Sr2+-based FW and standard SW (in the absence of mineral scale formation) led to a noticeable increase in polar functional groups. Specifically, the sulfoxide index (S = O) rose from 0.0308 (crude oil) to 0.0359, while the carbonyl index (C = O) increased from 0.0026 to 0.0444. These shifts indicate strong interactions between asphaltene molecules and polar species present in the injected brine. In line with this trend, the polarity index also increased from 0.1296 to 0.1515, suggesting an accumulation of polar moieties in the bulk oil phase. Conversely, the aliphatic index decreased from 0.6110 to 0.4318, implying that aliphatic chains may have migrated toward the interface or aqueous phase. This redistribution facilitates the relative enrichment of polar functionalities in the oil phase, potentially increasing the polarity and interfacial activity of asphaltenes.
Following mineral scale formation, both polar indices declined: the S = O index dropped to 0.0409, and the C = O index to 0.0330. This suggests that some polar components may have become incorporated into the precipitated scale or the emulsion phase. Supporting this, the polarity index also decreased to 0.1388, reinforcing the idea that polar groups were partially removed from the oil phase upon scale precipitation. Interestingly, the aromatic index increased slightly from 0.1458 to 0.1509, indicating the relative retention or enrichment of aromatic structures, which are generally more stable. Moreover, the aliphatic index rose to 0.4644, suggesting a lower tendency of aliphatic groups to partition out of the bulk phase, potentially due to interface saturation or steric hindrance caused by the precipitated scale.
This analysis highlights the dual role of divalent cations (e.g., Sr2+) and scale formation in influencing the molecular structure and partitioning behavior of asphaltenes. FTIR data show that both chemical interactions (ion exchange, solvation) and physical processes (precipitation, emulsification) significantly modulate the distribution of functional groups within asphaltene molecules. These insights have important implications for understanding emulsion stability, wettability alterations, and the design of effective strategies for EOR in carbonate reservoirs.
FTIR analysis of crude oil asphaltenes in contact with Sr2+-based FW and SW containing 4 × Na2SO4: With and without scale formation
To investigate the molecular changes in asphaltenes under high-sulfate conditions, FTIR spectroscopy was performed on two asphaltene samples: one extracted from crude oil in contact with Sr2+-based FW and SW enriched with four times the concentration of Na2SO4 under scale-forming conditions, and another obtained from the same brine system under non-scale-forming conditions. In both cases, the bulk oil phase was sampled, and after solvent evaporation, the dried asphaltenes were analyzed using FTIR. Figure 8 presents a comparative FTIR overlay of these two treated samples along with the baseline crude oil spectrum, allowing direct evaluation of structural variations in functional groups due to sulfate-induced scaling. Table 11 summarizes the numerical indices of key functional groups, including S = O (sulfoxide), C = O (carbonyl), polarity, aromaticity, and aliphaticity, for all three conditions including crude oil, post-scaling, and non-scaling exposure. Furthermore, a comparative bar chart of these indices is presented in Fig. 9 to enhance visual interpretation.

Comparative FTIR spectra of asphaltenes extracted from crude oil, and crude oil exposed to Sr2+-based FW and 4 × Na2SO4 SW under scale and non-scale conditions.

Comparison of FTIR-derived indices (S = O, C = O, polarity, aromaticity, aliphaticity) for asphaltenes in crude oil and after exposure to Sr2+-based FW and SW with 4 × Na2SO4 (with and without scale formation).
As shown in Table 11, the FTIR-derived indices provide critical insights into how asphaltene molecular structure is altered by interaction with brines containing Sr2+ and highly concentrated Na2SO4, both under scale and non-scale conditions. Upon exposure of crude oil to Sr2+-based FW and SW containing four times the concentration of sodium sulfate (4 × Na2SO4), significant molecular transformations in the asphaltene structure were observed, particularly in relation to polar and hydrocarbon indices derived from FTIR analysis. In the absence of scale formation, the sulfoxide index (S = O) increased markedly from 0.0308 in the untreated crude oil to 0.0470, indicating a higher presence or retention of oxidized sulfur-containing groups within the asphaltene matrix. Similarly, the carbonyl index (C = O) rose from 0.0026 to 0.0323, reflecting a greater abundance of ketones, aldehydes, or carboxylic acids. However, despite these increases in functional polar groups, the overall polarity index declined from 0.1296 to 0.1035. This apparent contradiction suggests that while polar functionalities formed or accumulated in asphaltenes, the most hydrophilic fractions may have partitioned into the aqueous or interfacial phase, thereby reducing their net representation in the bulk oil. Concurrently, the aromatic index exhibited a notable increase from 0.1458 to 0.1700, likely due to the selective enrichment of stable aromatic cores as aliphatic chains migrated outward. The aliphatic index dropped significantly from 0.6110 to 0.5024, confirming that aliphatic structures were more susceptible to interfacial migration or solubilization in the brine phase when scale was not present.
In contrast, under conditions where mineral scale formation occurred, the changes in asphaltene structure followed a slightly different pattern. The S = O index increased to 0.0417, higher than the crude baseline but lower than the non-scale condition, suggesting partial sequestration of sulfur-bearing functionalities within the precipitated scale or at the emulsion interface. The C = O index similarly rose to 0.0372, which again exceeds the crude reference but falls short of the value observed without scale, indicating that mineral scaling likely restricts the retention of these oxygenated functionalities in the oil phase. The polarity index showed a modest decline to 0.1239, supporting the notion that polar compounds may be increasingly incorporated into scale matrices or emulsified regions rather than remaining dissolved in the bulk oil. The aromatic index rose slightly to 0.1570, consistent with the trend of relative aromatic enrichment, likely driven by the preferential migration of aliphatic species out of the oil phase. Correspondingly, the aliphatic index declined to 0.4638, suggesting that scale presence further facilitates the selective exclusion of aliphatic components from the crude matrix, possibly due to steric hindrance or interfacial crowding introduced by scale particles.
Furthermore, as illustrated in Fig. 9, the compositional trends of asphaltene samples subjected to Sr2+-based FW and 4 × Na2SO4-enriched SW, under both scale and non-scale conditions, reveal several notable molecular transformations. The presence of mineral scale clearly dampens the expression of polar functional groups, such as sulfoxides (S = O) and carbonyls (C = O), compared to the corresponding non-scale environment. This is likely due to the partial sequestration of these functionalities within the mineral precipitate or at oil–water interfaces. Aromatic content is found to be enriched in both treated samples, with the highest aromatic index observed under non-scaling conditions. This enrichment suggests a preferential retention of chemically stable aromatic cores in the oil phase, as less stable aliphatic structures are displaced. Aliphaticity, while most preserved in the untreated crude oil, declines under both brine treatment scenarios, particularly in the presence of scale, indicating the outward migration of aliphatic chains toward emulsified or aqueous regions.
The quantitative trends presented in Table 11 support this visual interpretation. Injection of SW enriched with 4 × sodium sulfate increases oxidative functionalities in asphaltenes, with more pronounced effects when mineral scale formation is absent. Under these non-scaling conditions, the asphaltene matrix retains a higher content of S = O and C = O groups, indicating strong interactions between asphaltene molecules and oxidizing agents in the brine. In contrast, when scale forms, polar groups are partially removed from the bulk oil, either by entrapment within the precipitated matrix or through redistribution at the interface, resulting in reduced polarity indices. Meanwhile, aromatic systems persist and slightly concentrate, and the sharp decline in the aliphatic index confirms selective depletion of these moieties from the crude phase.
FTIR analysis of asphaltenes in crude oil after exposure to Ba2+-based FW and SW: With and without scale formation
To assess asphaltene structural variations under Ba2+-rich brine conditions, two treated oil samples, one with and one without scale formation, were analyzed. The asphaltenes were extracted from the bulk oil phase using the IP-143 method after toluene evaporation. Figure 10 presents a comparative FTIR spectrum of the two treated samples along with untreated crude oil, enabling direct visual comparison of functional group variations. Figure 11 shows the relative distribution of key functional groups derived from peak area integration, while Table 12 summarizes the corresponding functional indices (S = O, C = O, polarity, aromaticity, aliphaticity) for all three scenarios.

Comparative FTIR spectra of asphaltenes in crude oil and in samples treated with Ba2+-based FW and SW, under scale and non-scale conditions.

Comparison of FTIR-derived indices for asphaltenes in crude oil and after treatment with Ba2+-based FW and SW, with and without scale formation.
According to the data summarized in Table 12 and visually presented in Fig. 11, significant molecular changes in asphaltene structure occur upon exposure to Ba2+-based FW and SW under both scale and non-scale conditions. When the crude oil is contacted with high-salinity brine without inducing mineral scale formation, a notable increase is observed in the sulfoxide (S = O) index, rising from 0.0308 in crude to 0.0477, and the carbonyl (C = O) index, which elevates sharply to 0.0421. These enhancements suggest intensified interaction between asphaltenes and oxidized sulfur and oxygen species in the ionic medium. Interestingly, despite this enrichment of polar functionalities, the polarity index drops from 0.1296 to 0.0558. This counterintuitive observation is likely due to the migration of highly polar moieties into the aqueous or interfacial phases, leading to a relative depletion of polar groups in the bulk oil. Concurrently, the aromatic index rises significantly to 0.164, implying that chemically stable aromatic cores remain concentrated in the oil phase as other components are partitioned out. Meanwhile, the aliphatic index decreases from 0.6110 to 0.5376, supporting the notion that linear aliphatic chains preferentially migrate out of the bulk oil and into surrounding water-rich regions or interfaces.
In the presence of mineral scale, induced by Ba2+–SW interactions, the functional group distribution shifts again. The S = O index increases to 0.0383 and the C = O index to 0.0518, values that are still elevated relative to crude oil but lower than under non-scaling conditions. This suggests that part of these oxidized functionalities becomes sequestered within the scale precipitate or adsorbed into the interfacial emulsion matrix. The polarity index in this case rises to 0.1274, slightly below that of crude but significantly higher than in the non-scale scenario, indicating a partial retention of polar groups in the bulk or their entrapment in loosely bound interfacial zones. Moreover, the aromatic index shows a moderate rise to 0.1538, confirming the trend of aromatic enrichment across all treatments. A more substantial decline in the aliphatic index to 0.4641 further supports the conclusion that scale formation facilitates the removal or exclusion of aliphatic moieties, possibly through steric hindrance or interfacial destabilization caused by barium sulfate crystals.
Overall, this analysis reveals that S = O and C = O groups are maximally retained in non-scaling environments, while scale formation limits their persistence in the oil phase. The polarity index exhibits its lowest value when no scale forms, suggesting migration of polar compounds to surrounding phases. In contrast, aromatic structures are consistently enriched in the bulk oil due to their inherent stability, whereas aliphatic content shows the greatest loss in scale-forming systems.
It should be noted that the Ba2+ brines displayed higher oxygen contents by EDS while showing lower FTIR S = O indices. This outcome is not contradictory. EDS quantifies total elemental oxygen from all phases present within the electron-beam interaction volume, including inorganic O-bearing species such as interfacial BaSO4 microcrystals, Ba-carbonates/hydroxides, and residual/brine-bound water. In contrast, the FTIR S = O index specifically reflects organic sulfoxide/sulfonyl vibrations within the asphaltene-rich interfacial film and does not capture sulfate S–O modes from mineral barite. The pronounced propensity of Ba2+ to precipitate sulfate as insoluble barite therefore enriches the surface in inorganic oxygen (elevating EDS-O) without increasing the organic S = O signature. Additionally, the two techniques probe different length scales (localized μm-scale EDS vs. bulk thin-film FTIR), so spatially heterogeneous mineral aggregates can amplify the EDS oxygen signal while exerting a comparatively minor effect on the averaged FTIR spectrum.
FTIR analysis of crude oil asphaltenes in contact with Ba2+-Based FW and SW enriched with 4 × Na2SO4: Effects of scale and non-scale conditions
In this section, the structural transformations of asphaltenes in crude oil upon exposure to Ba2+-based FW and synthetic SW enriched with four times the standard concentration of sodium sulfate (4 × Na2SO4) were investigated under both scale-forming and non-scale-forming conditions. As in previous analyses, samples were obtained from the bulk oil phase after contact with the brine, followed by toluene evaporation and asphaltene isolation using the IP-143 method. Figure 12 presents an overlay comparison of the FTIR spectra for untreated crude oil, and the two Ba2+-treated systems, one under scaling conditions and the other without visible scale formation, highlighting the differences in functional group distributions resulting from scale-related interactions. Quantitative indices derived from FTIR data are listed in Table 13, while a graphical comparison of the functional group indices is shown in Fig. 13.

Overlay comparison of FTIR spectra for asphaltenes in crude oil, and after exposure to Ba2+-based FW and SW with 4 × Na2SO4, under both scale and non-scale conditions.

Comparison of FTIR-derived functional group indices (S = O, C = O, polar, aromatic, aliphatic) for asphaltenes in crude oil and after brine treatment with Ba2+ and 4 × Na2SO4, with and without scale formation.
Upon injection of the high-sulfate brine into the crude oil system, the sulfoxide index (S = O) exhibited a noticeable increase from 0.0308 to 0.0472, while the carbonyl index (C = O) rose from 0.0026 to 0.0334. These shifts clearly reflect the enhanced incorporation or stabilization of oxidized sulfur- and oxygen-bearing groups in the asphaltene molecules, indicating stronger interactions between asphaltenes and the brine components. However, this chemical enrichment was accompanied by a pronounced drop in the polarity index from 0.1296 to 0.086. This paradoxical trend can be attributed to the migration of highly polar compounds from the oil bulk phase to either the aqueous domain or the oil–water interface. Such migration reduces the detectable polarity within the bulk oil but simultaneously contributes to emulsion development and interfacial tension modification. The aromatic index increased significantly to 0.1794, suggesting that the relatively stable aromatic cores are retained in the oil phase while less stable, more reactive groups, including aliphatic chains, are expelled. Supporting this, the aliphatic index decreased from 0.6110 to 0.5069, which implies that long hydrocarbon chains were more susceptible to migration into the emulsion or aqueous phase, especially under elevated ionic strength conditions induced by the concentrated sulfate solution.
When scale formation was allowed to occur under the same high-salinity conditions, the behavior of functional groups and molecular fractions was notably altered. The S = O index reached 0.0336, still higher than crude oil but lower than the non-scaling condition, suggesting partial retention of oxidized sulfur functionalities, possibly due to entrapment within the barium sulfate precipitate or associated interfacial domains. Interestingly, the C = O index increased more markedly to 0.0583, indicating that carbonyl-containing structures remain prominently in the bulk oil even after scale precipitation. This may be due to selective affinity of carboxylic or ketonic groups for oil-phase retention in the presence of Ba2+ or their relative exclusion from crystal surfaces. One of the most remarkable shifts was observed in the polarity index, which surged to 0.2402, the highest among all tested conditions. This sharp rise implies that a substantial fraction of polar compounds, rather than fully migrating to the aqueous or emulsion phases, remain within the oil, likely due to entrapment in interfacial microdomains created by mineral scaling. The aromatic index in this case also increased to 0.1666, consistent with prior trends showing that aromatic systems are preferentially retained in the bulk phase due to their structural stability. In contrast, the aliphatic index decreased steeply to 0.3816, representing the lowest observed value. This suggests that linear hydrocarbon chains are most prone to exclusion from the oil phase under scale-forming conditions, potentially due to steric hindrance, interfacial disruptions, or chemical incompatibility with precipitated scale structures. The main findings from the FTIR analysis section are summarized as follows:
-
Oxidized functional groups, particularly sulfoxides (S = O) and carbonyls (C = O), generally increased after exposure to high-sulfate brines. The highest enrichment occurred under non-scaling conditions. When mineral scale formed, especially barium sulfate, these polar groups were partially removed or incorporated into the scale matrix.
-
The polarity index showed a significant decline in non-scaling conditions, indicating that polar compounds migrated from the oil phase into the aqueous or interfacial regions. In contrast, under scaling conditions, a considerable portion of these polar species remained in the oil phase or were trapped within the oil–scale interface, resulting in a sharp increase in the polarity index.
-
Aromatic compounds tended to accumulate in the bulk oil phase due to their inherent chemical stability. Meanwhile, aliphatic chains were progressively excluded, particularly in samples exposed to barium-based scale. This exclusion is likely due to steric hindrance or disruption at the oil–water interface caused by scale formation.
-
These findings highlight the intricate relationship between divalent ion-rich brine chemistry, scale precipitation, and the structural response of asphaltene molecules. Understanding these mechanisms is essential for predicting emulsion behavior, managing flow assurance challenges, and designing effective EOR operations in carbonate reservoirs.
Elemental composition analysis using energy dispersive x-ray spectroscopy (EDS)
As previously noted, EDS is a powerful analytical technique used to determine the elemental composition of solid materials. In this section, EDS was employed to evaluate the elemental distribution within asphaltenes extracted from the bulk oil phase. The primary objective was to investigate potential changes in elemental composition, particularly the heteroatoms nitrogen (N), sulfur (S), and oxygen (O) in the asphaltene structure following emulsification with formation water and synthetic SW. This analysis provides insight into the extent of chemical modification and elemental partitioning in the oil phase as a result of brine-oil interactions and the presence of mineral scale.
EDS analysis of asphaltene extracted from crude oil
As previously discussed, the asphaltene fraction from crude oil was extracted using the IP-143 method and subsequently analyzed by EDS. The resulting spectrum is shown in Fig. 14, while the quantitative elemental composition is summarized in Table 14.

EDS spectrum of asphaltene extracted from crude oil.
The EDS spectrum and elemental analysis indicate that the crude oil asphaltene is predominantly composed of C, with a weight percentage of 89.69%, confirming its hydrocarbon-rich molecular structure. This high carbon content is a typical characteristic of asphaltenes, which consist mainly of polycyclic aromatic hydrocarbons with alkyl side chains. The presence of heteroatoms, particularly S at 6.32%, O at 3.26%, and N at 0.42%, is also notable. These elements are integral to asphaltene functionality, contributing to their polarity, reactivity, and tendency to participate in intermolecular interactions such as hydrogen bonding and metal complexation. In particular, sulfur is typically found in thiophenic or sulfoxide structures, which contribute to the oxidation potential and aggregation behavior of asphaltenes. Oxygen may exist in the form of carboxyl, hydroxyl, or carbonyl groups, thereby enhancing the overall polarity and acidity of the molecules. Although present in lower concentrations, nitrogen commonly occurs in pyrrolic or pyridinic forms and plays a significant role in the coordination of metal ions, further influencing the stability and interfacial behavior of asphaltenes.
In terms of metals, trace amounts of vanadium (V, 0.03%) and nickel (Ni, 0.01%) were detected. These are commonly found in crude oil as metalloporphyrins and are known to influence asphaltene aggregation and catalytic fouling in refining processes. Other metals such as Fe, Cr, and Al were below detectable limits in this particular sample, as reflected by their absence in Table 18. Minor levels of silicon (Si, 0.27%) may originate from siloxane contaminants or formation rock interaction and are not uncommon in asphaltenic systems. This baseline elemental profile of crude oil asphaltene serves as a reference point for comparing the effects of brine interaction and mineral scaling, which will be further examined through subsequent EDS analyses under varying reservoir water compositions and scale-forming conditions.
EDS analysis of asphaltene in crude oil after exposure to Sr2+-based FW and SW: With and without scale formation
In this section, the elemental composition of asphaltenes extracted from crude oil samples contacted with Sr2+-based FW and standard SW was analyzed using EDS. Two scenarios were examined: (1) in the presence of mineral scale, and (2) under non-scaling conditions. After asphaltene extraction and toluene evaporation, the dried samples were subjected to EDS analysis. The resulting spectra are shown in Fig. 15a and b, while Table 15 summarizes the corresponding elemental compositions in terms of weight percentage.

EDS spectrum of asphaltenes extracted from crude oil after contact with Sr2+-based FW and SW (a) with scale formation, (b) without scale formation.
The elemental analysis of asphaltenes extracted from crude oil after contact with Sr2+-based FW and SW, under both scale-forming and non-scaling conditions, reveals distinct variations in heteroatom content and trace element distribution (Fig. 15; Table 15).
Carbon remained the dominant element in both samples, consistent with the hydrocarbon-rich backbone of asphaltenes. A slightly higher carbon content was observed in the scale-forming condition (89.22%) compared to the non-scaling scenario (88.86%), suggesting better preservation of the hydrocarbon framework due to the protective effect of scale deposition, which may limit structural fragmentation. Sulfur, typically present in thiophenic and sulfoxide forms, exhibited comparable levels in both cases: 5.17% with scale and 5.15% without, indicating that scale formation has a negligible impact on the retention of sulfur-containing moieties.
In contrast, oxygen content exhibited a pronounced difference between the two conditions. The non-scale sample had a significantly higher oxygen content (6.24%) compared to the scale-forming sample (2.33%). This reduction suggests that oxygenated functional groups (e.g., hydroxyl, carbonyl, and carboxyl) are either removed, incorporated into the scale matrix, or redistributed into the emulsion phase during scale formation, resulting in their depletion from the oil-borne asphaltenes. Nitrogen content showed an opposite trend, with a markedly higher value in the scale-forming sample (0.98%) compared to the non-scaling condition (0.04%). This may be attributed to the preferential retention or stabilization of pyrrolic and pyridinic nitrogen species in the oil phase under scaling conditions, potentially through metal coordination or interactions at the oil–scale interface. Silicon was detected in both samples but was notably higher in the non-scale sample (0.79%) than in the scale-forming sample (0.34%), possibly due to the presence of siliceous particles or additives that remain in the oil phase when no precipitation occurs.
Trace metal distributions further underscore the influence of scaling. Nickel was detected only in the scale-forming sample (0.11%), likely reflecting complexation with nitrogen or sulfur sites in the asphaltene matrix stabilized by scale. Vanadium was present in both samples, with a slightly higher concentration in the non-scale condition (0.08% versus 0.04%). Iron, chromium, and aluminum were detected exclusively in the non-scale sample at low levels (≤ 0.05%), suggesting that these elements may be incorporated into the scale precipitate or otherwise removed from the oil phase during scale formation.
In summary, the EDS results confirm that scale formation significantly alters the elemental composition of asphaltenes. Oxygenated polar groups are retained at higher levels when scale does not form, potentially enhancing oil phase polarity and emulsion stability. Conversely, nitrogen-containing species become more prominent in the presence of scale, likely due to stabilization or complexation effects at the oil–scale interface.
EDS analysis of crude oil asphaltenes in contact with Sr2+-based FW and SW enriched with 4 × Na2SO4 under scale-forming and non-scale conditions
To investigate the impact of high-sulfate SW and Sr2+-rich FW on the elemental composition of asphaltenes, two sets of EDS analyses were conducted. The first sample corresponds to the scale-forming condition (Fig. 16a and Table 16), while the second sample represents the non-scaling environment (Fig. 16b and Table 16).

EDS spectra of asphaltenes extracted from crude oil in contact with Sr2+-based FW and 4 × Na2SO4-enriched SW: (a) under scale-forming conditions, (b) under non-scaling conditions.
The elemental analysis of asphaltenes extracted after contact with Sr2+-based FW and 4 × Na2SO4-enriched SW, under scale-forming and non-scaling conditions, highlights significant differences that reflect strong chemical interactions at the oil–brine interface (Fig. 16; Table 16).
Carbon remained the dominant element in both cases, slightly higher under scaling (88.64%) than without scale (88.16%), suggesting that the hydrocarbon backbone is better preserved when scale forms, likely due to reduced emulsification and interfacial exchange. In contrast, sulfur content was notably lower with scale (5.78%) compared to the non-scale condition (7.21%). This reduction indicates possible sequestration of sulfur-containing functional groups (e.g., thiophenic and sulfoxide) into the scale matrix or their migration to the aqueous phase during precipitation, highlighting the role of scale in modulating asphaltene polarity and reactivity.
Nitrogen content was slightly higher under scale-forming conditions (0.95%), implying stabilization of nitrogenous moieties through interactions with Sr2+ ions or surface complexation with scale particles, which could hinder their migration into the emulsion phase. Silicon and nickel showed greater retention without scale, reflecting possible accumulation of siliceous impurities and organometallic complexes in the oil phase when precipitation is absent. The exclusive presence of vanadium under scaling conditions and the detection of iron and chromium only without scale suggest a selective redistribution of trace metals driven by scale formation.
Overall, these observations indicate that scale formation not only alters the overall heteroatom balance in asphaltenes but also influences the distribution of metal species. The preferential retention or removal of polar and metallic components during scaling processes directly affects the interfacial properties and stability of asphaltene-rich oil systems, providing mechanistic insights into wettability alteration and emulsion behavior in carbonate reservoirs.
Although sodium and potassium were not varied independently in this study, their influence on sulfate-scale kinetics warrants clarification. Monovalent ions modify the effective driving force for precipitation by changing solution non-ideality (through ionic-strength effects) and by forming weak ion pairs with sulfate that transiently reduce the freely available sulfate and divalent cations. These factors can lengthen or shorten induction times and adjust growth rates depending on salinity and composition. Specific-ion behavior also plays a role: compared with sodium, potassium is less strongly hydrated and can more efficiently screen surface charge at mineral–solution interfaces, which can modestly facilitate primary nucleation of barite under otherwise similar conditions. Under our salinity window, and given the overwhelming thermodynamic preference for barite over celestite, these kinetic modulations are secondary to the contrast between barium- and strontium-bearing systems. Accordingly, the sulfate threshold reported here is best interpreted in terms of sulfate activity rather than nominal concentration, and some field-to-field variability in induction times is expected as a function of the background Na+/K+ balance. A targeted parametric study that varies Na+/K+ at fixed ionic strength would be a valuable extension.
Elemental analysis of crude oil asphaltenes in the presence of Ba2+-based FW and SW: Comparison between scale and non-scale conditions
To investigate how barium-rich FW and SW affect the elemental composition of asphaltenes, two crude oil samples were analyzed. One sample was exposed to a system where mineral scale formed, and the other to a system without scale precipitation. After solvent evaporation, the dried asphaltenes from both samples were subjected to EDS analysis. The resulting spectra are presented in Fig. 17a (with scale) and Fig. 17b (without scale), and the corresponding elemental compositions are summarized in Table 17.

EDS spectra of asphaltenes extracted from crude oil in contact with Ba2+-based FW and SW: (a) under scale-forming conditions, (b) under non-scaling conditions.
Carbon remained the predominant element in both samples, reflecting the hydrocarbon-rich nature of asphaltenes (Fig. 17; Table 17). Its slightly higher content under scale-forming conditions (87.27%) compared to the non-scale scenario (87.05%) suggests that in the presence of Ba2+-induced scale, the carbon backbone is better conserved, likely due to reduced molecular fragmentation and lower interfacial exchange. Interestingly, sulfur content was marginally higher under scaling conditions (6.50%) than in the non-scale case (6.42%). This contrasts with the Sr2+ system, where sulfur tended to decrease in the presence of scale. The slight sulfur increase here suggests that sulfur-containing groups such as thiophenes or sulfoxides may become partially trapped within the denser and chemically more inert barite (BaSO4) scale, rather than being released or removed.
Oxygen content was higher in the scale-forming condition (5.69%) compared to non-scale (4.72%), indicating that Ba2+-based scale formation may facilitate the retention of oxygenated polar functionalities (e.g., hydroxyl or carbonyl groups) within the oil phase, possibly through strong coordination with barium or by preventing their migration into the aqueous phase. This behavior differs from the Sr2+ case, where scale formation generally led to a decline in oxygen content, highlighting the distinct chemical affinity and trapping capacity of Ba2+. Nitrogen content was slightly higher in the non-scale sample (0.38%) than under scale-forming conditions (0.31%). This suggests that in the absence of scale, nitrogenous moieties such as pyrroles and pyridines remain more mobile and persist in the oil phase. When scale is present, these groups might be partially immobilized through interactions with Ba2+ or become incorporated into interfacial complexes. Silicon content was significantly higher in the non-scale condition (1.21%) than in the scale-forming sample (0.20%), implying that silicate or clay-derived particles are more stably retained in the oil phase without mineral precipitation. Among trace metals, nickel was only detected in the non-scale sample (0.07%), indicating its possible loss via co-precipitation when scale forms. Iron and aluminum were more abundant in the absence of scale (Fe: 0.11%, Al: 0.03%), while vanadium and chromium were absent in both conditions.
Overall, these findings underscore that Ba2+-induced scale formation alters the distribution of heteroatoms and metals in asphaltenes more profoundly than Sr2+, due to the higher density and lower solubility of barite. The retention of sulfur and oxygen functionalities suggests stronger interactions or entrapment effects in Ba2+ systems, which can significantly impact asphaltene stability, interfacial properties, and ultimately emulsion behavior during water injection processes in carbonate reservoirs.
Elemental analysis of crude oil asphaltenes in the presence of Ba2+-based FW and SW with 4 × Na2SO4: Comparison between scale and non-scale conditions
The elemental composition of asphaltenes exposed to Ba2+-based FW and SW containing 4 × Na2SO4 was investigated using EDS under both scale-forming and non-scaling conditions (Fig. 18; Table 18). Carbon remained the predominant element, with higher content under scale-forming conditions (88.90%) compared to the non-scale case (86.06%). This suggests that the hydrocarbon backbone is more strongly preserved when barium sulfate (barite) scale is present, likely due to the formation of a protective interfacial barrier that restricts molecular exchange and fragmentation. Sulfur content was lower with scale (5.11%) than without (6.44%), implying that sulfur-bearing groups, such as thiophenic and sulfoxide functionalities, may be sequestered within the precipitated scale or partitioned into the emulsion phase. Interestingly, this trend is similar to what was observed in the Sr2+ system, reinforcing the role of scaling in limiting sulfur retention in the oil phase. Oxygen content showed an opposite trend: higher in the non-scaling condition (5.99%) than with scale (5.26%). This indicates that polar oxygenated groups (e.g., hydroxyl, carboxyl, and carbonyl functionalities) are more retained in the oil phase in the absence of scale, possibly because scaling reduces available interfacial sites for these groups to remain solubilized. Nitrogen levels were also higher without scale (0.79% vs. 0.48%), suggesting that nitrogen-containing heterocycles (pyrroles, pyridines) are more stable and remain in the oil phase when no barite forms. Silicon was more pronounced without scale (0.15% vs. 0.05%), indicating stronger retention of silicate or mineral particles in the oil phase when no precipitation occurs, similar to what was observed in Sr2+ tests. Trace metals, including nickel and vanadium, were present only in the non-scaling scenario, supporting the hypothesis that scale formation either traps these metals within the solid matrix or promotes their migration into the aqueous phase. Iron, chromium, and aluminum appeared in both conditions at trace levels, with slightly higher concentrations in the absence of scale.

EDS spectra of asphaltenes extracted from crude oil in contact with Ba2+-based FW and SW containing 4 × Na2SO4: (a) under scale-forming conditions, (b) under non-scaling conditions.
Overall, these observations emphasize the strong influence of barite scale formation on heteroatom and metal distributions within asphaltenes. Compared to the Sr2+ system, Ba2+ not only enhances carbon preservation but also exhibits a stronger tendency to exclude polar and metallic species from the oil phase through selective trapping or precipitation. This has significant implications for interfacial stability, emulsion behavior, and wettability alteration during enhanced oil recovery operations in carbonate reservoirs.
Correlation between EDS and FTIR analyses: Heteroatom behavior under SW and FW interactions
One of the core objectives of this work was to determine how SW injection and the accompanying inorganic scaling alter the molecular architecture of asphaltenes. To that end, we correlate functional-group indices from FTIR with bulk elemental data from EDS across scale-forming and non-scale-forming conditions. For transparency and readability, the complete set of FTIR-derived indices (S = O, C = O, Polarity, Aromatic, Aliphatic) for all FW–SW pairings are compiled in Table 18, and the corresponding EDS elemental compositions (C, S, O, N and trace elements) are reported in Table 19. To decouple FW versus SW effects, asphaltene samples were contacted with Sr2+- or Ba2+-based FW and subsequently exposed to either base SW or high-sulfate SW (4 × Na2SO4). FTIR tracks oxidized and polar functionalities (notably S = O and C = O) and the composite polarity index (Table 19), while EDS quantifies retention or loss of heteroatoms (N, S, O) in the bulk oil phase (Table 20).
The results reveal a strong correlation between the presence of scale and the retention or migration of heteroatoms. In general, under non-scaling conditions, the EDS data indicate elevated oxygen and sulfur contents, which are consistent with higher FTIR-derived sulfoxide and carbonyl indices. This suggests that in the absence of scale, polar groups remain stabilized in the oil phase, possibly due to emulsification or lack of mineral entrapment. For example, in the Sr2+-based system with 4 × Na2SO4 SW (no scale), O and S contents reached 4.91% and 7.21%, respectively, aligning with elevated S = O and C = O peaks in FTIR spectra.
Conversely, in scale-forming conditions, EDS showed a drop in O and S concentrations, sometimes by more than 2% absolute, despite the presence of sulfur and oxygen functionalities in the FTIR profile. This discrepancy indicates that while oxidized groups still exist in the molecular fingerprint, part of them may be sequestered into mineral precipitates or emulsified layers rather than being retained in the bulk oil phase. The oxygen content, in particular, exhibited a sharper decline in Ba2+-based systems with scaling, pointing to the stronger impact of barite (BaSO4) formation on asphaltene polarity suppression.
Nitrogen content showed a more complex pattern. In certain scale-forming cases (e.g., Sr2+ + SW), nitrogen increased in EDS results despite a decrease in FTIR polarity index. This may reflect the persistence of stable nitrogenous moieties (pyrrolic/pyridinic) in the bulk oil, even as other polar species migrated or precipitated. Alternatively, it could suggest preferential retention of N-bearing sites on the asphaltene core due to chelation or weak coordination with divalent cations. These findings highlight that:
-
Oxygen and sulfur heteroatoms are more vulnerable to removal from the oil phase during scaling, likely via physical incorporation into mineral matrices or redistribution to the oil–water interface.
-
Nitrogen groups, while less abundant, may exhibit relative stability or become more detectable in scale-containing environments.
-
The combined use of EDS and FTIR provides complementary insights: EDS quantifies elemental loss or retention, while FTIR captures bond-level transformations and functional behavior.
Therefore, this integrative analysis confirms that scale formation induced by SW, FW incompatibility not only alters the physicochemical behavior of the asphaltenes but also drives selective heteroatom migration and transformation, which has direct implications for interfacial tension, emulsion stability, and ultimately oil recovery efficiency.
Interfacial tension (IFT) measurement between synthetic oil and brine combinations
To investigate the influence of brine composition and inorganic scaling on the interfacial properties of crude oil systems, pendant drop IFT measurements were carried out between synthetic oil and eight different brine solutions. These solutions were composed of combinations of FW containing either Ba2+ or Sr2+ ions, and SW in its standard or sulfate-enriched form (4 × Na2SO4). The tests were conducted under both scale-forming and non-scale-forming conditions to isolate the individual and combined effects of ionic composition and precipitate presence on IFT behavior. Table 21 presents the complete set of IFT values [experimental error: ± 0.1 mN/m] for all eight FW–SW combinations, and Fig. 19 provides a comprehensive visual comparison.

Comparison of IFT values for different FW–SW brine systems with and without scale formation.
As shown in Fig. 19, the following trends were identified:
-
Effect of cation type (Ba2+ vs. Sr2+): In both scale and non-scale scenarios, brines containing Ba2+ consistently exhibited higher IFT values compared to their Sr2+ counterparts. For instance, in the non-scale case, IFT increased from 27.33 mN/m (SW + Sr2+) to 33.41 mN/m (SW + Ba2+), and from 26.72 to 32.65 mN/m in the sulfate-enriched brines. This supports the interpretation that barium ions reduce interfacial compatibility, potentially due to stronger interactions with asphaltene molecules or their polar components.
-
Effect of sulfate concentration: As clearly evident in Table 18 and Fig. 19, increasing the Na2SO4 concentration in the SW from standard to 4 × resulted in minor variations in IFT, especially under non-scaling conditions. For example, in the Sr2+ system, IFT changed slightly from 27.33 to 26.72 mN/m. Thus, sulfate enrichment alone does not have a major impact on IFT, unless it leads to scale formation.
-
Effect of scale formation: The presence of scale significantly increased the IFT values in all cases. For example, in the Sr2+ system, IFT rose from 27.33 to 39.04 mN/m, while in the Ba2⁺ system it increased from 33.41 to 41.51 mN/m. This increase is attributed to the partial sequestration of polar asphaltene groups within the scale matrix or interfacial emulsions, which reduces the stabilizing interactions at the oil–water boundary.
-
Synergistic effect of Ba2+ and scale: The highest IFT value (42.73 mN/m) was observed in the 4 × Na2SO4 + Ba2+ system under scale-forming conditions, indicating a synergistic effect between barium ions and sulfate-induced scaling on destabilizing the interface.
-
Relative to Sr2+, Ba2+ exhibits weaker hydration and higher polarizability, which facilitates specific, inner-sphere complexation with asphaltene heteroatoms (e.g., sulfoxide/carbonyl and basic nitrogen sites). Such cation-bridging promotes aggregation of asphaltene sub-fractions and depletes amphiphilic fragments from the oil–water interface. Under sulfate-rich conditions, the very low solubility of BaSO4 accelerates co-precipitation/entrapment of O- and S-bearing moieties into barite-rich solids, consistent with the reduced S = O/C = O FTIR indices and lower O and S detected by EDS in the bulk oil for Ba-systems. The combined outcome is a thinner, less elastic interfacial film and consistently higher IFT in Ba2+ brines, whereas Sr2+ with stronger hydration, weaker specific complexation, and more soluble sulfate preserves more surface-active species at the interface.
The findings from this study underline that barium ions are more disruptive to interfacial equilibrium compared to strontium, making their presence a greater challenge in EOR operations. Additionally, scale formation plays a dominant role in increasing IFT, which may negatively affect emulsion stability and hinder oil–water separation. While sulfate enrichment alone does not significantly alter IFT, its combination with Ba2+ becomes particularly impactful due to the high tendency for precipitation and scale formation. These conclusions are in agreement with previous interpretations derived from FTIR and EDS analyses, which also demonstrated a reduction in interfacial polarity and the retention or redistribution of polar heteroatoms under scale-forming conditions.
Conclusions
While previous studies have examined single-ion effects on scaling or asphaltene behavior independently, the combined impact of multi-ion interactions (Mg2+/Ca2+/SO42−) on coupled scaling-asphaltene phenomena remains unclear. This study specifically addresses: (1) whether a critical sulfate threshold exists beyond which scaling dominantly controls asphaltene interfacial activity, and (2) how barium vs. strontium ions differentially modify this threshold. The following conclusions can be drawn from the experimental findings:
-
The degree of incompatibility between FW and SW is highly dependent on the ionic content, especially the presence of divalent cations such as Ba2+ and Sr2+. While varying the concentrations of MgCl2 and CaCl2 in seawater had a negligible effect on the amount of precipitate formed, a substantial increase in scaling was observed when the concentration of Na2SO4 was increased fourfold. This spike in precipitation occurred regardless of whether the FW contained barium or strontium ions, indicating the critical role of sulfate in enhancing scale formation, particularly in the presence of divalent cations.
-
The incompatibility between 4 × Na2SO4-enriched SW and Sr2+-rich FW resulted in a scale primarily composed of calcium sulfate (75.3%) and sodium chloride (12.99%). In contrast, the precipitate formed with Ba2+-rich FW included a significant fraction of barium sulfate (17.09%) along with calcium sulfate (51.27%). These results highlight the higher tendency of barium to form insoluble scales compared to strontium, consistent with known solubility trends.
-
Dilution of SW was found to moderately reduce scale formation, although not completely preventing it. Furthermore, polarity index assessments revealed that the injection of standard SW into Sr2+-containing FW increased the polarity of the bulk oil phase, whereas injecting sulfate-enriched SW caused a decrease, likely due to the migration of polar asphaltene components into the emulsion phase. This behavior was reversed in the Ba2+-based system, where polarity consistently decreased with both standard and high-sulfate SW, indicating unfavorable interfacial interactions and a diminished capacity to stabilize emulsions.
-
Interfacial tension (IFT) measurements confirmed that brine composition and scale formation significantly affect the oil–water interfacial behavior. IFT values were consistently higher in systems containing barium ions than those with strontium, regardless of sulfate concentration. Notably, the combination of Ba2+-based FW and 4 × Na2SO4-enriched SW produced the highest IFT values, especially under scale-forming conditions, pointing to a synergistic disruption of interfacial compatibility. In contrast, sulfate enrichment had little effect on IFT when used with Sr2+-rich FW, and no significant difference was observed between 1 × and 4 × sulfate concentrations in terms of IFT.
-
The presence of inorganic precipitates in oil–brine systems resulted in increased IFT across all brine types. This increase is attributed to the partial sequestration of amphiphilic and polar molecules, particularly those associated with asphaltenes within the mineral scale or at oil–water interfaces, thereby diminishing interfacial cohesion.
-
The sulfate-threshold framework generalizes beyond Azadegan to Ba2+-rich reservoirs (e.g., North Sea), where mixing SW with Ba2+-bearing formation brines readily drives barite supersaturation. Operationally, the allowable SW fraction is governed primarily by the injected sulfate budget; deploying sulfate-reduced SW (e.g., via nanofiltration/SRU (sulfate-removal units)) and applying our threshold calculation provides quantitative limits for SW blend ratios across startup, breakthrough, and WAG (water-alternating-gas injection) scenarios, while balancing scale control against desired wettability effects in carbonate EOR (see supplementary Sect. S3 for more details).
-
Acute and chronic toxicity is associated with dissolved Ba2+, which is systemically absorbed and regulated in discharges and drinking water (U.S. EPA MCL = 2 mg L−1; WHO guideline = 1.3 mg L−1) [EPA, WHO]. By contrast, barium precipitated as barite (BaSO4) under sulfate-rich conditions is extremely sparingly soluble (K_sp ≈ 10−10 at 25 °C) and has negligible oral bioavailability (clinically used as an inert radiographic contrast agent), so its acute toxicity is minimal compared with dissolved barium. High-sulfate injection therefore scavenges Ba2+ into barite, typically lowering dissolved barium in produced water; the trade-off is greater barite-rich scale/solids that must be captured and disposed. A further implication is that radium (Ra, mainly Ra-226/Ra-228) can co-precipitate/isomorphically substitute into the BaSO4 lattice due to similar ionic radii, decreasing dissolved Ra but concentrating NORM (naturally occurring radioactive material) in the solid phase, which requires compliant handling and disposal NORM. Operationally, we recommend: (i) routine monitoring of dissolved Ba2+ (especially around start-up, acidizing, and chelant treatments that can re-mobilize Ba/Ra from scales), and (ii) solids management (hydrocyclones/filtration and segregated handling of barite-rich fines) under NORM protocols appropriate to local regulation.
Overall, this study highlights the critical impact of water chemistry on oil–brine interfacial behavior, scale formation potential, and asphaltene stability. These findings have direct implications for EOR, particularly in carbonate reservoirs, where controlling SW sulfate concentration, especially in barium-rich formations, is vital to mitigate scale-related damage and maintain interfacial stability. The use of strontium-containing systems appears less problematic compared to barium-based ones, both in terms of scale formation and emulsion stability. Moreover, the integration of FTIR and EDS analyses offers a predictive tool for assessing scale-induced wettability alteration, supporting the design of smart water injection strategies in EOR operations.
Recommendations
Based on the findings of this study and in light of the observed effects of ionic composition, scale formation, and interfacial alterations on crude oil systems, the following recommendations are proposed for future research and field-scale implementation:
-
Advanced molecular characterization: It is recommended to perform NMR spectroscopy to further investigate the behavior of heteroatoms (particularly nitrogen and hydrogen) within asphaltene molecules before and after exposure to incompatible brine systems. This will help elucidate subtle changes in molecular structure and hydrogen-bonding environments that are not detectable through FTIR alone.
-
Selective salt impact assessment: While this study focused on Na2SO4-induced scaling, further experiments should be conducted to isolate and quantify the impact of magnesium chloride (MgCl2) and calcium chloride (CaCl2) at varying concentrations on both scale formation and asphaltene stability. Understanding their individual effects can provide greater insight into the competitive interactions in multivalent brine systems.
-
Mitigation strategies for scale deposition: Field implementation of seawater injection should be accompanied by pre-injection compatibility screening and potential dilution or softening of seawater, particularly in barium-rich reservoirs. Moreover, the identification and testing of effective scale inhibitors or chelating agents is essential, especially for managing CaSO4 and BaSO4 precipitates under high-sulfate conditions.
-
Customized brine formulations: Engineering ion-tuned water formulations that minimize the availability of scaling ions while optimizing wettability alteration and interfacial properties is highly recommended. Tailoring brines to the specific cation profile of the formation water can help reduce risks of precipitation and improve oil recovery efficiency.
-
Interfacial behavior and asphaltene interaction analysis: Further studies should examine the interfacial tension and functional group evolution of asphaltenes in brine–oil systems using both static and dynamic (flow-based) conditions to better simulate reservoir flow environments. The integration of FTIR and interfacial rheology would offer a more comprehensive view of emulsion behavior and asphaltene aggregation mechanisms.
-
Comparative evaluation with distilled water: A control series using distilled water should be systematically compared with ion-rich brine systems to distinguish between effects caused by salinity and those resulting from specific ionic interactions. This baseline comparison will clarify the chemical nature of asphaltene–brine interactions.
-
Impact of individual ions on functional groups: Dedicated studies focusing on the effect of increasing Mg2+ and Ca2+ concentrations on specific functional groups of asphaltenes (e.g., carboxyl, hydroxyl, sulfoxide) using spectroscopic methods are essential for molecular-level understanding of destabilization pathways.
-
While Na+/K+ were held constant here to isolate divalent–sulfate chemistry, background monovalent ions can shift the effective driving force for sulfate precipitation and subtly modify nucleation and growth. We recommend a targeted study that varies Na+/K+ at fixed ionic strength to quantify these specific-ion effects across relevant salinity windows.
By addressing these areas, future work can contribute to the design of more compatible and efficient water injection schemes, minimize operational issues related to scaling and emulsion stability, and enhance the understanding of oil–brine interfacial dynamics under complex reservoir conditions.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Saeedi Dehaghani, A. H. & Ghalamizade Elyaderani, S. M. Application of ion-engineered Persian Gulf seawater in EOR: Effects of different ions on interfacial tension, contact angle, zeta potential, and oil recovery. Pet. Sci. https://doi.org/10.1007/s12182-020-00541-y (2021).
Dehaghani, A. H. S. & Daneshfar, R. Experimental investigation of the sequence injection effect of sea water and smart water into an offshore carbonate reservoir for enhanced oil recovery. Sci. Rep. 14, 4595. https://doi.org/10.1038/s41598-024-55440-8 (2024).
Mahmoud, M., Elkatatny, S. & Abdelgawad, K. Z. Using high- and low-salinity seawater injection to maintain the oil reservoir pressure without damage. J. Petrol. Explor. Prod. Technol. 7, 589–596. https://doi.org/10.1007/s13202-016-0279-x (2017).
Moghadasi, J., Müller-Steinhagen, H., Jamialahmadi, M., Sharif, A. Scale deposits in porous media and their removal by edta injection (accessed 10 May 2025). https://dc.engconfintl.org/heatexchanger2007/10/
Khodabakhshi, M. J. & Bijani, M. Predicting scale deposition in oil reservoirs using machine learning optimization algorithms. Results Eng. 22, 102263. https://doi.org/10.1016/j.rineng.2024.102263 (2024).
Merdhah, A. B. B. & Yassin, A. A. M. Scale formation in oil reservoir during water injection at high-salinity formation water. J. Appl. Sci. 7, 3198–3207. https://doi.org/10.3923/jas.2007.3198.3207 (2007).
Saeedi Dehaghani, A. & Alizadeh, S. Investigation of the effect of sulfate sediment particles on water-oil emulsion formation when mixing smart water-formation water. J. Pet. Res. 29, 64–78 (2020).
Merdhah, A. B., Yassin, M. & Azam, A. Study of scale formation due to incompatible water. J. Teknol. 49, 9–26 (2008).
Abbasi, P., Abbasi, S. & Moghadasi, J. Experimental investigation of mixed-salt precipitation during smart water injection in the carbonate formation. J. Mol. Liq. 299, 112131. https://doi.org/10.1016/j.molliq.2019.112131 (2020).
Mohammadi, M. & Riahi, S. Experimental investigation of water incompatibility and rock/fluid and fluid/fluid interactions in the absence and presence of scale inhibitors. SPE J. 25, 2615–2631. https://doi.org/10.2118/201117-PA (2020).
Al-Samhan, M. et al. Evaluating scale deposition and scale tendency of effluent water mix with seawater for compatible injection water. J. Pet. Explor. Prod. Technol. 10, 2105–2111. https://doi.org/10.1007/s13202-020-00849-w (2020).
Razavirad, F., Heidari, S. & Shahrabadi, A. Evaluation of compatibility between formation and Injection water into the reservoir rock. Colloids Surf. A 690, 133787. https://doi.org/10.1016/j.colsurfa.2024.133787 (2024).
Hussein, M. F., Mubarak, M. F., Al-Sirhani, A. M. & Hosny, R. Examining the factors that impact the formation of barite scale in water injection operations: Experimental study and quantification of scale formation. Discov. Appl. Sci. 6, 519. https://doi.org/10.1007/s42452-024-06176-7 (2024).
Yen, T. F. Asphaltenes. In Structures and Dynamics of Asphaltenes (eds Mullins, O. C. & Sheu, E. Y.) 1–20 (Springer US, Boston, MA, 1998). https://doi.org/10.1007/978-1-4899-1615-0_1.
Yazdani, B., Dehaghani, A. H. S. & Karami, S. Fundamental study of asphaltene cracking and desorption from rock surface due to microwave radiation: A molecular insight into catalytic effect of minerals and asphaltene chemistry. Geoenergy Sci. Eng. 227, 211946. https://doi.org/10.1016/j.geoen.2023.211946 (2023).
Ameri, A., Esmaeilzadeh, F. & Mowla, D. Effect of brine on asphaltene precipitation at high pressures in oil reservoirs. Pet. Chem. 59, 57–65. https://doi.org/10.1134/S0965544119010031 (2019).
Shahsavani, B., Riazi, M. & Malayeri, M. R. Asphaltene instability in the presence of emulsified aqueous phase. Fuel 305, 121528. https://doi.org/10.1134/S0965544119010031 (2021).
Tavakkoli, M. et al. Effect of emulsified water on asphaltene instability in crude oils. Energy Fuels 30, 3676–3686. https://doi.org/10.1021/acs.energyfuels.5b02180 (2016).
Shojaati, F., Mousavi, S. H., Riazi, M., Torabi, F. & Osat, M. Investigating the effect of salinity on the behavior of asphaltene precipitation in the presence of emulsified water. Ind. Eng. Chem. Res. 56, 14362–14368. https://doi.org/10.1021/acs.iecr.7b03331 (2017).
Alizadeh, S. & Soulgani, B. S. Experimental investigation of the brine effect on asphaltene precipitation and deposition during water injection in porous media using static and dynamic systems. Energy Fuels 35, 11141–11153. https://doi.org/10.1021/acs.energyfuels.1c00090 (2021).
Doryani, H., Malayeri, M. R. & Riazi, M. Precipitation and deposition of asphaltene in porous media: Impact of various connate water types. J. Mol. Liq. 258, 124–132. https://doi.org/10.1016/j.molliq.2018.02.124 (2018).
Mokhtari, R., Hosseini, A., Fatemi, M., Andersen, S. I. & Ayatollahi, S. Asphaltene destabilization in the presence of an aqueous phase: The effects of salinity, ion type, and contact time. J. Petrol. Sci. Eng. 208, 109757. https://doi.org/10.1016/j.petrol.2021.109757 (2022).
Mahdavi, M. S. & Dehaghani, A. H. S. Experimental study of the simultaneous effect of ion-tuned water and clay particles on the behavior of water-in-oil emulsion; new insight into asphaltene molecular structure (2024). https://www.researchsquare.com/article/rs-5073626/latest (Accessed 7 May 2025). https://doi.org/10.1016/j.petrol.2021.109757
Almulhim, N., Alotaibi, F., Rafie, M. & Alrufail, T. Sulfate-based scales: Why sulfate ion concentrations matter. In International Petroleum Technology Conference, IPTC, 2024 D031S118R005. https://onepetro.org/IPTCONF/proceedings-abstract/24IPTC/3-24IPTC/542512 (accessed 18 May 2025). https://doi.org/10.2523/IPTC-23383-MS.
Behnammotlagh, M. A., Hashemi, R., Taheri Rizi, Z., Mohammadtaheri, M. & Mohammadi, M. Experimental study of the effect of the combined monoethylene glycol with NaCl/CaCl2 salts on sour gas hydrate inhibition with low-concentration hydrogen sulfide. J. Chem. Eng. Data 67, 1250–1258. https://doi.org/10.1021/acs.jced.1c00888 (2022).
Hamidian, R., Lashkarbolooki, M. & Hezave, A. Z. Interfacial tension and contact angle of asphaltenic and resinous model oil in the presence of binary salts mixtures. Sci. Rep. 14, 18018. https://doi.org/10.1038/s41598-024-68740-w (2024).
Behnam Motlagh, M. A. et al. Comprehensive review of the impact of thermodynamic inhibitors and the predictive power of machine learning models on hydrate formation pressure and temperature. J. Chem. Eng. Data https://doi.org/10.1021/acs.jced.5c00025 (2025).
Kimura, K. et al. Application of anomalous x-ray scattering method to liquid electrolytes used in a battery: Local structural analysis around a dilute metallic ion. Anal. Chem. 92, 9956–9962. https://doi.org/10.1021/acs.analchem.0c01525 (2020).
Undavalli, V. K., Ling, C. & Khandelwal, B. Impact of alternative fuels and properties on elastomer compatibility. In: Aviation Fuels 113–132 (Elsevier, 2021). https://www.sciencedirect.com/science/article/pii/B9780128183144000017 (accessed 16 May 2025). https://doi.org/10.1016/B978-0-12-818314-4.00001-7
Hodoroaba, V. -D. Energy-dispersive X-ray spectroscopy (EDS). In Characterization of Nanoparticles 397–417 (Elsevier, 2020). https://doi.org/10.1016/B978-0-12-814182-3.00021-3
Yousuf, M. A., Shaik, F. & Nageswara, L. R. Studies on scale deposition in oil industries & their control. Int. J. Innov. Res. Sci. Technol. 3, 152 (2017).
Ghasemian, J. & Riahi, S. Effects of salinity and ionic composition of smart water on mineral scaling in carbonate reservoirs during water flooding. Pet. Explor. Dev. 48, 421–429. https://doi.org/10.1016/S1876-3804(21)60033-2 (2021).
Wu, J. et al. Improved oil recovery from carbonate reservoirs by tuning injection seawater composition. Energies 15, 6405. https://doi.org/10.3390/en15176405 (2022).
Valadbeygian, V., Hajipour, M. & Behnood, M. Static and dynamic evaluation of formation damage due to barium sulfate scale during water injection in carbonate reservoirs. J. Pet. Explor. Prod. Technol. 13, 1819–1831. https://doi.org/10.1007/s13202-023-01652-z (2023).
Al-Shalabi, E. W. & Sepehrnoori, K. A comprehensive review of low salinity/engineered water injections and their applications in sandstone and carbonate rocks. J. Pet. Sci. Eng. 139, 137–161. https://doi.org/10.1016/j.petrol.2015.11.027 (2016).
Saeedi-Dehaghani, A. H. & Behnam-Motlagh, M. A. Experimental investigation of foam stability under various salinity levels, oil types, and surfactant conditions: Effect of natural polymer lignin. Sci. Iran. 45, 456. https://doi.org/10.24200/sci.2025.66165.9885 (2025).
Grangeon, S. et al. Mineralogical and geochemical composition of a cementitious grout and its evolution during interaction with water. Npj Mater. Degrad. 8, 75. https://doi.org/10.1038/s41529-024-00488-0 (2024).
Amiri, G., Jahangiri, H. R. & Jalalpoor, M. Evaluating water injection compatibility and scale-formation reduction in a middle east reservoir using direct pore-scale visualization and NaOH treatment. Sci. Rep. 15, 12925. https://doi.org/10.1038/s41598-024-80965-3 (2025).
Zayed, A. M. et al. From non-conventional agricultural waste into sustainable and eco-friendly activated carbon through specified thermo-chemical protocol. Appl. Nanosci. 14, 21–32. https://doi.org/10.1007/s13204-023-02939-7 (2024).
Tanhaei, H. & Dehaghani, A. H. S. Independent effects of microwave irradiation and acidic solutions on asphaltene content and upgrading of heavy crude oil. J. Mol. Liquids 431, 127743. https://doi.org/10.1016/j.molliq.2025.127743 (2025).
Tanhaei, H., Hossein, A. & Dehaghani, S. Improvement of the quality of heavy crude oil and reducing the concentration of asphaltene hydrocarbons using microwave radiation during acidizing. Sci. Rep. 15, 7434. https://doi.org/10.1038/s41598-025-91932-x (2025).
Funding
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Author information
Authors and Affiliations
Contributions
Mohammad Amin Behnam Motlagh: Conceptualization, Formal analysis, Investigation, Software, Writing—original draft, Writing—review & editing. Amir Hossein Saeedi Dehaghani: Data curation, Project administration, Supervision, Validation, Writing—review and editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Behnam Motlagh, M., Saeedi Dehaghani, A. Influence of inorganic scale formation on asphaltene behavior during water injection. Sci Rep 15, 41240 (2025). https://doi.org/10.1038/s41598-025-25144-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-25144-8
