
Abstract
Circulating melanoma cells (CMCs) are responsible for the hematogenous spread of melanoma and, ultimately, metastasis. However, their study has been limited by the low abundance in patient blood and the heterogeneous expression of surface markers. The FDA-approved CellSearch platform enriches CD146-positive CMCs, whose number correlates with progression-free survival and overall survival. However, a single marker may not be sufficient to identify them all. The Parsortix system allows enrichment of CMCs based on their size and deformability, keeping them viable and suitable for downstream molecular analyses. In this study, we tested the strengths, weaknesses and potential convergences of both platforms to integrate the counting of CMCs with a protocol for their genetic analysis. Samples run on Parsortix were labeled with a customized melanoma antibody cocktail, which efficiently labeled and distinguished CMCs from endothelial cells/leukocytes. The capture rate of CellSearch and Parsortix was comparable for cell lines, but Parsortix had a higher capture rate in real-life samples. Moreover, double enrichment with both CellSearch and Parsortix succeeded in removing most of the leukocyte contamination, resulting in an almost pure CMC sample suitable for genetic analysis. In this regard, a proof-of-concept analysis of CMCs from a paradigmatic case of a metastatic uveal melanoma patient led to the identification of multiple genetic alterations. In particular, the GNAQ p.Q209L was identified as homozygous, while a deletion in BAP1 exon 9 was found hemizygous. Moreover, an isochromosome 8 and a homozygous deletion of the CDKN2A gene were detected. In conclusion, we have optimized an approach to successfully enrich and retrieve viable CMCs from metastatic melanoma patients. Moreover, this study provides proof-of-principle for the feasibility of a marker-agnostic CMC enrichment followed by CMC phenotypic identification and genetic analysis.Kindly check and confirm the processed contributed equally is correctly identify We confirm
Similar content being viewed by others

Introduction
Interest in exploring the potential of liquid biopsy as a companion tool for personalized medicine has been growing for more than a decade. It is becoming increasingly evident that it can be used to longitudinally monitor cancer progression in real time, detect early treatment resistance, identify minimal residual disease, and target the best treatment option1,2. Indeed, tissue biopsy, which remains the gold standard for cancer diagnosis, often does not reflect the spatial and temporal heterogeneity of the tumor itself3. If multiple clones coexist within a tumor, as it is now well known, their spatial and temporal distribution may affect their detection with important clinical implications4, whether they contain clinically relevant genetic information. In addition, performing multiple invasive biopsies to track tumor progression and resistance is clearly impractical5,6, as tissue biopsy carries significant risks when the tumor lesions are located in areas that are difficult to access5.
The main obstacles to the identification of reliable biomarkers useful to monitor response and resistance to therapy before clinical occurrence are the dynamic changes in the clonal composition of both tumors and metastases due to the selective pressure exerted by therapy, which promotes the growth of different subclones6,7,8. In the era of “personalized medicine”, much attention is being paid to finding approaches that can reveal the molecular landscape of the whole disease9,10. However, this kind of biomarkers is still missing for many tumors, including cutaneous (CM) and uveal (UM) melanoma. It is noteworthy that melanoma is a tumor with one of the highest levels of inter- and intra-tumor heterogeneity, from a spatial and temporal point of view4,11. Liquid biopsy has gained worldwide attention as a complementary tool to capture and characterize the tumor heterogeneity at the systemic level12,13, as it can be performed through minimally invasive blood sampling that contains the sum of tumor material released from each disease site5,6. Circulating melanoma cells (CMCs), which are responsible for the hematogenous spread of melanoma, and ultimately for metastasization, reflect tumor heterogeneity and could therefore be the key to identify the entire genetic/phenotypic pattern of the disease. Nonetheless, the heterogeneous expression of markers on the CMC surface, together with their low abundance (1–5 CMCs in 7.5 ml of blood), are the main hindrances hampering CMC isolation and limiting in-depth study of these cells in melanoma patients. On the other hand, despite the difficulty of analysis, cumulative evidence supports a role for CMC count in monitoring therapy response and predicting overall survival (OS), progression-free survival (PFS), and early progression14,15, even though the cut-off for melanoma patients is still under debate14,15,16,17,18. In addition to the undeniable potential of their count, studying CMCs is of paramount importance from a molecular point of view to assess in detail the genetic heterogeneity of melanoma. To this end, regardless of the type of downstream analysis, the first step is the enrichment of CMCs from the blood samples, which can be achieved using many different technologies exploiting different biological and physical characteristics of melanoma cells. Since its approval, the FDA-cleared CellSearch platform has become the gold standard for circulating tumor cell (CTC) count. As for CMC detection, it relies on the enrichment of CD146-positive cells, whose number has already been correlated with PFS and OS in various clinical settings14,15,16,17,18. However, because of the high phenotypic heterogeneity typical of CMCs, it is considerably difficult to identify all melanoma cells with a single marker19,20,21,22,23. This leads directly to the problem of identifying the most appropriate method for their isolation for downstream molecular analyses. For this reason, other platforms have been developed including the Parsortix system, which has recently received FDA approval for use in metastatic breast cancer1. This system allows the enrichment of CTCs according to their size and deformability, which are the main characteristics that distinguish CTCs from the other blood cells24. This size-based “filtration” mechanism enables the enrichment of intact and viable CTCs, which are in turn suitable for any type of downstream molecular analysis25.
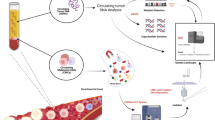
Considering all these aspects, we aimed to test the strengths, weaknesses and potential convergences of CellSearch and Parsortix platforms, in order to integrate CMC count with an efficient protocol for their genetic analysis. In this regard, we optimized and validated an approach to successfully enrich and recover viable CMCs from the blood of metastatic melanoma patients, which involved Parsortix size-based enrichment followed by customized phenotypic and genetic analysis. After careful testing of the Parsortix performance and the subsequent validation of the customized antibody cocktail on a series of spike-in samples, we tested the entire procedure in an exploratory analysis encompassing patients with CM or UM. We set up different scenarios where to analyze and combine the two enrichment platforms, as well as different downstream molecular strategies to achieve a satisfactory level of CMC genetic analysis (Fig. 1). One of the patients proved to be particularly interesting and represented a paradigmatic case for the full potential of the entire workflow, due to the large number of CMCs collected.

Workflow for CMC enrichment and genetic characterization. Blood from melanoma patients was processed through the CellSearch platform and/or the Parsortix system equipped with a customized antibody cocktail. DNA derived from harvested CMCs, superamplified or not, was analyzed with a customized NGS panel, ddPCR and MLPA to identify tumor genetic alterations. Paired cfDNA was used to assess/validate the genetic alterations identified in CMCs. The figure was created with BioRender (https://biorender.com/).
Methods
Enrichment, count, and phenotypic identification of melanoma cells
Cell lines
The human non-small cell lung cancer (NSCLC) H441 (kindly provided by Dr. Thomas Schlange within the Cancer ID consortium) and melanoma SK-MEL-28 (ATCC, Manassas, VA, USA) cell lines were grown in RPMI 1640 (Gibco, Life Technologies, Paisley, UK), while melanoma A375-MA2 (ATCC) cell line was cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) (Gibco, Life Technologies). All culture media were supplemented with 10% (vol/vol) Fetal Bovine Serum (FBS) (Life Technologies), 1 mM Hepes (Aurogene, Rome, Italy), 2–4 mM Ultraglutamine (LONZA, Basel, Switzerland), and, in the case of A375-MA2, 1 mM Sodium Pyruvate (LONZA). The human umbilical vein endothelial cells (HUVEC) (a kind gift from Dr Alice Biosa, Autifony, Padua, Italy) were grown in EBM-2 medium (LONZA), supplemented with EGM-2 SingleQuot Kit (LONZA).
Spiking of cell line cells into healthy donor blood
SK-MEL-28, A375-MA2, H441, and HUVEC cells were spiked into 5–10 ml of blood from healthy donors (HD) (range 10–1000 cells). Three 10-µl spots of cell suspension were double-checked by manual counting prior to spiking. Spiked samples were divided into two equal parts: the first was processed by Parsortix within 48 h, the second was transferred to a CellSave tube and processed by the CellSearch system within 96 h. When not stained in-cassette, SK-MEL-28 cells were pre-labeled prior to Parsortix run with CellTracker Orange CMRA Dye (Thermo Fisher Scientific, Waltham, MA, USA), according to the manufacturer’s instructions.
Optimization of antibody concentration for CMC identification
The antibody concentration was optimized for each antibody included in the cocktail using a live-cell immunofluorescence assay, and was finally assessed in the Parsortix system to ensure the most effective detection of melanoma cells. Cells were detached using Accutase (Euroclone, Milano, Italy) and adjusted to a final concentration of 105/ml. After a blocking step with 10% FBS/PBS (Euroclone) for 10 min, cells were incubated with specific antibodies at different concentrations for 30 min at RT, then washed and placed on a slide to be observed under a fluorescence inverted microscope (Nikon Eclipse Ti2, Nikon Corporation, Tokyo, Japan) to determine the optimal antibody concentration (Table 1).
Tumor cell enrichment and characterization
CellSearch system
Patient CMCs or spiked-in melanoma/endothelial cells were enriched from 7.5 ml peripheral blood through the CellSearch system, using the CellTracks Melanoma Cell Kit (Menarini Silicon Biosystems, Bologna, Italy) that relies on CD146/HMW-MAA (High Molecular Weight-Melanoma-Associated Antigen) for capture and detection, according to the manufacturer’s instructions. A semi-automated fluorescence-based microscope system (CellTracks Analyzer II, Menarini Silicon Biosystems) was used to identify CMCs/melanoma cells. An event was classified as a CMC/melanoma cell when its morphological features were consistent with those of a cell, and exhibited a CD146+, HMW-MAA+, DAPI+, CD34/CD45- phenotype26. Results were expressed as the total number of CMCs/melanoma cells per 7.5 ml of blood.
Parsortix system
Spiked-in melanoma-, NSCLC-, and endothelial cells or CMCs from patient blood were enriched using Parsortix separation protocol and a GEN3D6.5 Cell Separation Cassette (Angle plc, Guildford, Surrey, UK), in accordance with the manufacturer’s guidelines. In order to maximize cell harvesting, 1.8% bovine serum albumin (BSA) (Merck KGaA, Darmstadt, Germany) was added in the running buffer. After cell separation, pre-labeled cells (CellTracker Orange CMRA Dye) were directly counted within the cassette, while the unlabeled viable spiked cells or CMCs from patients were stained in cassette (program PX2_stain1) with our customized antibody cocktail. Finally, the enriched cells were harvested directly into a 16-well CultureWell Chambered Coverglass for Cell Culture (Thermo Fisher Scientific). The number of in-cassette captured or harvested cells was determined by manual counting under fluorescence guidance within the Parsortix cassette or the cell culture well, respectively.
Platform comparison
The capture capability of Parsortix was compared with that of the CellSearch, and expressed as the percentage of Parsortix vs. CellSearch captured cells (Capture Rate). The harvest rate was determined as the number of labeled cells harvested downstream divided by the number of captured cells.
Microscopy analysis
A Nikon Eclipse Ti2 inverted microscope (Nikon Corporation, Tokyo, Japan) equipped with a computer-controlled CCD camera was used for acquiring images from live-cell immunofluorescence coverglasses, Parsortix cassettes, and harvesting wells. Scanning and image acquisition were controlled by NIS Element AR software version 5.11.03 (Nikon BioImaging lab, Tokyo, Japan).
System testing with real-life samples
Patient selection and blood collection
Four blood samples belonging to 3 metastatic melanoma patients (stage III and IV CM, patient #1 and #2; stage IV UM, patient #3) were chosen to test the performance of the Parsortix system with real-life samples. Blood samples were collected at the baseline (T0), before the beginning of the first line immunotherapy for all patients, and also at the time of progression (P) for the uveal melanoma patient. Blood was collected in different tubes following this order: CellSave (Menarini Silicon Biosystems), EDTA, and Streck Cell-Free DNA BCT tubes (Streck, La Vista, NE, USA) for CellSearch, Parsortix, and cell-free DNA (cfDNA) analysis, respectively. The study was conducted according to the Declaration of Helsinki and the Good Clinical Practice, and approved by the Ethics Committee of the Veneto Institute of Oncology IOV-IRCCS (approval No. CESC-IOV 2018/36 on April 19, 2018).
CMC enrichment, phenotypic characterization, count, and harvest
CMCs were stained within the Parsortix system using the customized antibody cocktail, as described in Table 1. In addition to samples collected in EDTA or CellSave tubes and run on the Parsortix system (experiment #1 and #2), the content of the CellSearch cartridge of patient #3 at progression was subsequently run on Parsortix as a further enrichment step (experiment #3, Supplementary Fig. 1). After CMC count, each Parsortix enriched sample was harvested into one well of a 16-well CultureWell Chambered Coverglass for Cell Culture. After counting, the content of the well was transferred to a 0.2 ml tube and pelleted for subsequent analyses.
Molecular analyses on Parsortix-enriched CMCs derived from a paradigmatic uveal melanoma case
Sample lysis and whole genome amplification
CMCs from patient #3 collected at progression were either only lysed or also amplified using two different kits depending on the CMC status (vital or fixed) (Supplementary Fig. 1). More in detail, the samples obtained from Parsortix enrichment starting from blood collected in EDTA (experiment #1) were subjected to cell lysis using lysis buffer from REPLI-g Single Cell kit (Qiagen, Hilden, Germany); samples from CellSave preservative (experiment #2) were lysed with Ampli1 WGA kit lysis buffer (Menarini Silicon Biosystems). Each lysed sample was then divided into two parts. The first part was directly subjected to library preparation for NGS analysis (Exp#1–2_LYS, Supplementary Fig. 1), while the second was subjected to whole genome amplification (WGA) by means of the two different WGA kits followed by library preparation (Exp#1–2_WGA, Supplementary Fig. 1). The CMCs serially double enriched by CellSearch and Parsortix (experiment #3) were processed in the same way as in experiment #2. For each WGA experiment, a certified reference genomic DNA (Agilent Technologies, Santa Clara, CA, USA) and nuclease-free water were included as positive and negative controls, respectively. The quality of the WGA products was assessed using the Ampli1 WGA QC Kit (Menarini Silicon Biosystems), and WGA samples were quantified on a Qubit 4 Fluorometer (Thermo Fisher Scientific), following the manufacturer’s instructions. Size and integrity were assessed using the 4200 TapeStation system (Genomic DNA ScreenTape Assay, Agilent Technologies).
CfDNA extraction
The cfDNA was isolated from plasma obtained from Streck Cell-Free DNA BCT tubes, using the QIAamp Circulating Nucleic Acid Kit (Qiagen), and quantified on a Qubit 4 Fluorometer (Thermo Fisher Scientific), following the manufacturer’s instructions. Quality Control (QC) was performed using the 4200 TapeStation system (cfDNA ScreenTape Assay, Agilent Technologies). QC was passed if the percentage of cfDNA was > 82% (region table set from 50 to 700 bp15).
Next generation sequencing (NGS)
A hybridization capture-based target enrichment custom panel (SureSelect Cancer All-In-One custom panel, AIO, Agilent Technologies) was used for detecting single nucleotide variants (SNVs) and small deletions/insertions in 55 genes, and copy number variation (CNV) in 12 genes. The design covered hotspots for melanoma drivers, targetable mutations, and genes involved in pathways associated with treatment resistance, as described elsewhere15,27. Both lysed-only and WGA samples were subjected to enzymatic fragmentation before library preparation using the SureSelect Enzyme Fragmentation Kit (Agilent Technologies) following the manufacturer’s instructions. Sequencing libraries, from lysed-only, WGA-DNA, and cfDNA samples were prepared using the Magnis SureSelect XT HS Rev B Reagent Kit on the Magnis NGS Prep System (Agilent Technologies), according to the manufacturer’s instructions. QC was performed after library preparation with the 4200 TapeStation System (High Sensitivity D1000 ScreenTape Assay, Agilent Technologies) to ensure quality libraries. Library pools were sequenced using the 300 cycle NextSeq 500/550 Mid Output v2 kit on the NextSeq 550 (Illumina, San Diego, CA, USA). Alignment and variant calling were assessed using Alissa Align & Call (v 1.1.4–10), while interpretation and prioritization were assessed using Alissa Interpret software (v 5.3.4) (Agilent Technologies). Each variant was manually reviewed on Alissa Align & Call and Alissa Reporter (Agilent Technologies) in order to filter out false positives (i.e. indel calling in homopolymer regions28, single base calling near TTAA sites29, strand bias, pseudogene misalignments30). Variants found in the CMC samples subjected to different enrichment/WGA protocols were compared with those found in lysed-only CMC samples and in cfDNA as the custom NGS panel had previously been validated for cfDNA15.
Droplet digital PCR (ddPCR)
Specific SNVs and CNVs detected in CMC and cfDNA samples by NGS were assessed by ddPCR (BioRad Laboratories, Hercules, CA, USA). The entire list of commercial and customized probes is reported in Supplementary Table 1. For SNV analysis, positive (mutated DNA), negative (wild type cfDNA/DNA), and no-template (water) controls were included in each run. The reactions were performed in a 20 µl reaction mix containing 1× droplet PCR supermix, 250 nM of each probe, 450 nM of primers and 2–7 µL of cfDNA, which was quantified in parallel by ddPCR. Droplets were generated and analyzed using the QX600 ddPCR System (BioRad Laboratories). Data were acquired and analyzed using the QX Manager software version 2.1 (BioRad Laboratories). Samples were defined as positive when ≥ 3 FAM positive droplets were detected with no positive droplets in the negative control.
For CNV analysis, 3 probes, PTK2, TUSC331and PPARG31, were used to test chromosomal aberrations involving the chromosome regions 8q, 8p, and 3p, respectively, using as reference gene a probe located on chromosome 14 (TTC5 probe) as already reported32. For each sample, the reaction included 1 × ddPCR supermix, primers and probes at a final concentration of 900 nM and 250 nM, respectively, and 2–15 ng of cfDNA for a total volume of 20 µl. To set the cut-off for defining gain or loss from the cfDNA CNV analysis, 10 samples from healthy controls were used. The cut-off value was calculated as the mean copy number ± 1.96*SD (standard deviation), as already reported33.
Multiplex ligation-dependent probe amplification (MLPA)
The CNVs involving the region 9p21 were investigated using the SALSA MLPA Probemix ME024 9p21 CDKN2A/2B region kit (MRC Holland, Amsterdam, the Netherlands) containing 48 probes in the CDKN2A, CDKN2B and flanking genes. The MLPA assay was performed according to the manufacturer’s instructions, including 3 wild-type references and a no-template sample as negative control, starting from 60 ng of cfDNA (collected from patient #3 at progression) and DNA from Exp#3_WGA. Capillary electrophoresis was performed using the Applied Biosystems 3500xl Genetic Analyzer (Applied Biosystems, Foster City, CA). Coffalyser.Net (MRC Holland) data analysis software was used to analyze the raw data, to calculate the dosage quotients of MLPA probes that have been tested on DNA samples, and to assess the quality of raw MLPA data as well as the analysis itself.
Statistical analysis
Data were summarized by median, mean, range (min-max), and percentages using usual descriptive statistics. The cut-off value for the CNV ratios of loss and gain was based on the mean ± 1.96*SD of the normal (HD) cohort. The nonparametric Wilcoxon signed-rank test for paired data was used for comparison of data between groups (CellSearch vs. Parsortix captured cells). A p-value ≤ 0.05 was considered to be statistically significant. Statistical analysis was performed using GraphPad version 8.0 for Windows (GraphPad Software Inc., San Diego, CA, USA).
Results
CMC antibody cocktail optimization
The optimal antibody concentration for maximizing specificity and sensitivity in detecting melanoma cells was identified for each melanoma antigen by live-cell immunofluorescence, as reported in Table 1 (column 3) and shown in Fig. 2A. Subsequently, the ability of each antibody to properly detect melanoma cells in the Parsortix cassette was assessed (Fig. 2B, C), prior to adding each individual antibody into the cocktail. Finally, the whole customized melanoma antibody cocktail proved able to properly detect melanoma cells captured in the Parsortix cassette, and to successfully differentiate them from the endothelial cells (CD146+/CD34+) (Fig. 2D). Melanoma cells were identified as positive for any of the melanoma-specific markers present in the antibody cocktail, i.e., CD146, HMW-MAA, MART-1, GP100, while negative for CD45, CD16, and CD34, and with blue-stained nuclei. Representative images of stained melanoma cells captured using Parsortix are shown in Fig. 3A.

Representative images of live-cell immunofluorescence assays (A) and Parsortix experiments (B-D) of melanoma cells. (A-B) Melanoma cells (SK-MEL-28 or A375-MA2) were stained with antibodies against melanoma markers CD146/HMW-MAA/MART-1/GP100 (in green) and with DAPI for cell nuclei (in blue). (C) Bright-field images of cells captured in Parsortix cassette. (D) Endothelial cells (red arrows) can be discriminated from CMCs (white arrow) when positive for both CD146 (in green) and CD34 (in pink). Scale bar (lower right) represents 10 μm. Abbreviations: APC, allophycocyanin; FITC, fluorescein isothiocyanate; MEL, melanoma markers.
Performance of Parsortix system with customized antibody cocktail
The enrichment performance of the Parsortix system (Fig. 3A) was compared with that of the gold standard CellSearch (Fig. 3B), using both healthy donor blood spiked with cell lines or real-life samples from melanoma patients.
The overall capture of melanoma cells from spiked samples was comparable to that of the CellSearch (Fig. 4A; Table 2, column 4–5), although in several experiments Parsortix was able to recover a higher number of cells (from 14% to 67% more) (Fig. 4B; Table 2, column 6). When evaluating the harvest rate, defined as the percentage of harvested cells per total number of in-cassette captured cells, Parsortix demonstrated a mean/median harvest rate of 75%, in accordance with what already reported42,43 (Table 2, column 8; Fig. 4C). As recommended by the manufacturer and reported in previous studies24,44,45, the addition of BSA to the running buffer resulted in an increase in viable CTC harvesting by preventing cell attachment in the cassette. Indeed, the addition of 1.8% BSA was found to maximize the harvest rate, reaching 96% (mean of 87%; median of 90%) (Table 3, column 6; Fig. 4D). As Parsortix is an open platform, fully customizable, and suitable for transversal use, we evaluated the robustness of this optimized protocol and its applicability also with an epithelial cell line, together with the mesenchymal ones (melanoma cell lines). Therefore, a NSCLC cell line (H441) with putatively different intrinsic properties, such as cell adhesion, size, deformability, or mobility (which may influence the cell harvesting) was included. Notably, a high harvest rate was also achieved with this cell line, further validating the effectiveness of the protocol. As WBC contamination can impact downstream molecular analyses, the number of contaminant CD16+/CD45+ WBCs was determined for each spike-in experiment (Supplementary Table 2). According to previous studies11,46, the WBC background in the Parsortix harvested samples was notably lower (median of 2383, range 875–6760) when compared to that from CellSearch (median of 13802, range 3216–35108).

Representative images of captured melanoma cells identified by Parsortix and CellSearch systems. (A) Melanoma cells (A375-MA2) were enriched and stained with Parsortix system using melanoma markers (in green, CD146/MART-1/HMW-MAA/GP100), leukocyte and endothelial markers (in pink, CD45/CD16/CD34), and DAPI for cell nuclei (in blue). Scale bar (lower right) represents 10 μm. (B) Melanoma cells (A375-MA2) enriched and stained with CellSearch platform. Cells were immunomagnetically captured by a ferrofluid antibody against CD146, and were stained with HMW-MAA melanoma marker (MEL-PE), CD45/CD34 (APC) leukocyte and endothelial markers, respectively, and DAPI for cell nuclei. Abbreviations: APC, allophycocyanin; FITC, fluorescein isothiocyanate; MEL-FITC, melanoma markers; PE, phycoerythrin.
Enrichment and characterization of CMCs
The enrichment capability of the Parsortix system (together with the customized antibody cocktail) was tested on real-life samples, and compared with that of the CellSearch. Four blood samples (ps01-04, Table 4) from three metastatic melanoma patients were processed using both platforms. CMCs were detected in 3 out of 4 samples by both platforms, and a higher number of CMCs was enriched by Parsortix (Fig. 4E-F; Table 4 and Fig. 5). In addition, we defined the CMC purification from WBCs after double enrichment (CellSearch plus Parsortix, experiment #3, Supplementary Fig. 1) for ps04 sample. Indeed, a purification rate was determined and calculated as the ratio of the number of WBCs detected in experiments #1 and #3. Result revealed that the double enrichment process was able to purify CMCs from WBCs 220 times better than the single enrichment process (Supplementary Fig. 2; Supplementary Table 3). The case of the patient with stage IV uveal melanoma (ps03 and ps04) was particularly interesting due to the fast worsening of the disease (Supplementary data), as confirmed by an extraordinary high number of CMCs detected at progression (ps04, Table 4). It was therefore ideal to study the tumor genetic landscape of this patient.

Evaluation of the performance of the Parsortix system. Comparison of the captured cell line melanoma cells (A-B) or CMCs (E-F) by Parsortix vs. CellSearch in terms of number (A/E) or capture rate (B/F). Graphs C and D display the harvest rate of melanoma and NSCLC cells from the Parsortix cassette without (C) and with (D) the supplement of 1.8% BSA. The performance of CellSearch was set as 100% and indicated by the dotted line. Experiments #1 - #23 refer to spiking experiments of SK-MEL-28, A375-MA2, and H441 cells into healthy donor blood (A-D); experiments #01 - #04 refer to four blood samples belonging to 3 metastatic melanoma patients (E-F). The graphs were performed using GraphPad version 8.0 for Windows (GraphPad Software Inc., San Diego, CA, USA).

Representative images of CMCs from uveal and cutaneous melanoma patients captured with Parsortix system. CMCs derived from a uveal melanoma patient at the progression (A) and from a cutaneous melanoma patient at T0 (B). Cells were immunostained with melanoma markers (in green, CD146/MART-1/HMW-MAA/GP100), leukocyte/endothelial markers (in pink, CD45/CD16/CD34) and DAPI for cell nuclei (in blue). Scale bar (lower right) represents 10 μm. Abbreviations: APC, allophycocyanin; FITC, fluorescein isothiocyanate.
Whole genome amplification and sequencing QC
The high number of CMCs retrieved at progression from patient #3 allowed us to develop a workflow for CMC genetic analysis. Two samples, one consisting of DNA from lysed cells and the other of DNA from whole genome amplification, were obtained from the CMC harvested sample (experiment #1, #2, and #3, Supplementary Fig. 1). Each WGA sample (CMC-DNA amplified with Ampli1 WGA Kit or REPLI-g Single Cell Kit) passed the WGA quality control (4/4 PCR positive markers), and was therefore suitable for library preparation. Libraries from all WGA samples and Ampli1 lysates passed the quality control, while the library from REPLI-g Single Cell Kit lysate did not, and was therefore excluded from further analyses (Supplementary Fig. 1). The sequencing metrics reached different degrees of compliance as reported in Supplementary Table 4. Nevertheless, we were able to detect DNA variants and copy number variations belonging to melanoma cells in all sequenced samples.
Mutational and copy number profiling of CMCs and cfDNA
Mutational analysis was performed on CMC enriched samples from experiments #1, #2, #3 (Supplementary Fig. 1), and also on cfDNA available from both baseline and progression timepoints. The genetic alterations found in the different CMC samples were compared with those found in the cfDNA, and successfully validated (Table 5). NGS analysis revealed the GNAQ p.Q209L variant (confirmed by ddPCR, Table 5) and the not yet described BAP1 p.R238_V250 delinsM deletion in all samples. The allele frequency of the two variants at progression was very similar to each other in CMC and cfDNA sample (Table 5), suggesting that they were present in homozygosity or in a putative hemizygosity, respectively. Moreover, no loss of chromosome 3 and no change in BAP1 copy number was detected by either NGS or ddPCR analysis (Supplementary Table 5).
As shown in Table 5, the mutant allele fraction (MAF) of GNAQ and BAP1 from experiment #3 was the highest, indicating that the double enrichment (CellSearch plus Parsortix) removed most of the contaminating leukocytes and returned an almost completely pure CMC sample. Therefore, copy number changes were assessed on experiment #3 derivatives, first in the lysate sample (EXP#3_LYS) as it was free of potential bias introduced by the whole-genome amplification process, and then in the superamplified sample (EXP#3_WGA), for a double check (Table 6). The NGS analysis revealed an excess gain of 8q over 8p in both samples. This result was orthogonally validated by ddPCR analysis, which confirmed an unbalanced gain of 8q over 8p. Taken together, these imbalances are suggestive of the formation of an isochromosome, which is correlated with poor prognosis. 8q/8p copy number imbalances were also detected by NGS and/or ddPCR on cfDNA both at baseline and progression, which confirmed the isochromosomal status. Furthermore, a complete loss of the CDKN2A gene was detected by NGS in experiment #3 derivatives (lysate and amplified samples), and in cfDNA from progression. This finding was confirmed by MLPA analysis of the amplified sample of experiment #3 (EXP#3_WGA) and cfDNA at progression (cfDNA_P, Table 6). When comparing the WGA and lysed-only samples with cfDNA samples, we observed that some variant calls were only present in the amplified samples. By inspecting the.bam files on Alissa Align&Call, we realized that these calls were actually biases introduced during the whole genome amplification processes. In particular, the SNVs identified in Ampli1 WGA samples only were located near the restriction digestion TTAA sequences29, while the small indels identified at low frequency only in samples amplified with the REPLI-g Single Cell kit were located in homopolymeric regions28. Re-analysis of the FASTQ files with Alissa Reporter eliminated all calling errors in regions close to TTAA sequences, and part of the calling errors in homopolymeric regions. Therefore, this type of double check between WGA and lysed-only samples led to identify the areas that are most prone to change. Thus, such information could be applied to the next WGA samples as a roadmap for eliminating false calls.
Discussion
In this study, we developed a workflow that successfully enables the enrichment, detection, recovery, and molecular characterization of vital CMCs from metastatic melanoma patients. This approach was driven by the urgent need to isolate and analyze in detail the genetic and phenotypic profile of CMCs, and to gain significant knowledge in favor of their use as predictive/prognostic biomarker. Although CTC count was shown to provide prognostic insights in metastatic breast, prostate, and colorectal cancer7,10,12, CTCs (and CMCs) are not yet routinely used in clinical practice. Furthermore, the clinical relevance of CTC detection has been evaluated in several types of solid tumors, including melanoma47. In addition to their number, which was shown to be prognostic and predictive albeit in still small settings14,16,17,18, the molecular characterization of CMCs could be a valuable resource for better understanding tumor biology, in particular to uncover resistance mechanisms that can help guide the therapy choice in advanced melanoma patients15. CMCs are indeed the seeds responsible for the rise of distant metastases and, therefore, they are the perfect intermediate to be studied to uncover the genetic and phenotypic characteristics of future and/or ongoing metastatic disease.
The approach presented in this study is based on a marker-agnostic microfluidic-based technology, and takes advantage of an in-house customized melanoma antibody cocktail that comprises known major melanoma-associated antigens36,48. As already mentioned, the main barriers to a comprehensive analysis of CMCs are their heterogeneous expression of surface markers and their low abundance in blood48. Therefore, implementing methods to enrich and isolate CMCs is a mandatory step. The FDA-cleared CellSearch platform enriches CMCs using anti-CD146 antibody-labeled ferrofluids, but relying on a single melanoma marker for enrichment has some limitations that bias the number of CMCs that can be identified. Several studies have demonstrated that the presence of specific mesenchymal antigens, such as CD146, is related to an aggressive and higher metastatic phenotype2,49, somehow leading to infer that CellSearch analyzes the subpopulation with the higher potential to metastasize. However, due to the high heterogeneity of melanoma, including temporal heterogeneity, it cannot be excluded that a subclone capable of driving resistance may modulate, at earlier or temporally different stages of the disease, CD146 expression and therefore be excluded from capture and possible downstream analysis.
In this study we demonstrated that our customized melanoma antibody cocktail, combined with the Parsortix platform, is not only able to successfully identify melanoma cells, as reported very recently by Sementsov et al.50, but can also discriminate them from leukocytes and endothelial cells (Fig. 2C). As for the enrichment from melanoma cell lines, the performance of Parsortix was found to be comparable to the performance of CellSearch with a higher capture rate in several experiments (Table 2; Fig. 4A-B). Conversely, the scenario changed, at least in our very limited cohort, in the case of the enrichment of cells from melanoma patients, with a recovery rate of Parsortix exceeding that of CellSearch (Table 4; Fig. 4E-F). The observed variation in performance may be explained by the fact that, instead of using a single antibody, we took advantage of a custom antibody cocktail against four different melanoma antigens, which is likely to allow the detection of a greater number of melanoma cells, ultimately reflecting the heterogeneity of the metastatic disease.
The specific case of a patient with an exceptionally high number of CMCs was instrumental to test different sample processing strategies (enrichment/lysis/WGA) to obtain a satisfactory CMC genetic analysis. Indeed, we successfully identified SNVs and CNVs in genes typically involved in uveal melanoma in all the lysed and amplified samples analyzed by NGS. Furthermore, we showed that double enrichment with the combined use of CellSearch and Parsortix resulted in an almost completely pure sample of CMCs, as demonstrated by the allele frequency of GNAQ and BAP1 variants around 90%. Moreover, comparing the genetic profile of lysed and amplified samples, we could also rule out any bias introduced by the amplification process, thus establishing the boundaries and limits of the technique. Overall, we showed that CMCs can be analyzed either they are previously fixed and permeabilized by the CellSearch procedure, or viable and in good condition, with advantages in both cases. In the first case, a greater enrichment of cells can be achieved due to the use of the two platforms, but the fixed cells are less suitable for studies such as transcriptomics. In the second case, more heterogeneous, viable, cells can be obtained, therefore more suitable for any type of downstream molecular analysis. In our context, double purification was essential as no other tools were available for achieving a sufficient level of purity for downstream copy number analysis. The optimal solution would have been to use a micromanipulator downstream of the Parsortix system to enable single cell selection under fluorescence guidance, as discussed later. This approach would also make it possible to isolate cells belonging to different subclones depending on the antigens expressed. In addition, one platform (Parsortix or CellSearch) may be more appropriate than the other, or a combination of both may be required, depending on the extent of downstream analysis. For genetic analyses, the choice of platform is relatively flexible, as fixed and permeabilized cells are sufficient for most workflows. However, for transcriptomic studies, it is recommended to work with cells in the best possible condition, i.e. viable and non-permeabilized. This is particularly relevant when the aim is to proceed to cell culture after enrichment. Parsortix alone is more suited for these scenarios, as it preserves cell viability, also allowing greater flexibility in the choice of the super amplification kit, should one be required.
It is important to note that if cell count is not a priority, the staining step can be skipped within the Parsortix platform. On the other hand, when cell count is required and the aim is to generate data more comparable with the literature and general knowledge on the subject, CellSearch is often preferred due to its status of gold standard for CTC enumeration. Following this initial CellSearch enrichment, the sample can be further purified downstream using Parsortix, as demonstrated in the current work. However, this combined approach, while effective, inevitably leads to some degree of cell loss, which must always be taken into account.
Indeed, the effect of double enrichment should be evaluated according to the needs of the study. In some cases, if CMC count is sufficient, additional purification steps may not be necessary. Parsortix system may provide higher counts than CellSearch, but it must be remembered that the latter remains the gold standard51, as discussed above. Where detailed phenotypic characterization is required, Parsortix approach offers significant advantages, particularly in customizing antibody cocktails to target specific antigens, thereby improving CMC identification11,50. In these cases, additional purification may not be necessary if the primary goal is phenotypic assessment and/or cell harvest. On the other hand, if downstream genetic analysis is required, there is a trade-off between the scope of the analysis (e.g. SNV detection only, vs. CNV assessment and detection of variants at low MAF) and the number of cells available after double enrichment. Achieving higher purity through double enrichment may be particularly valuable, as reducing leukocyte contamination will improve the sensitivity and accuracy of variant detection51. Nonetheless, this option should only be chosen in specific contexts, where a sufficient number of cells is available and the downstream workflow is compatible with fixed and permeabilized cells (Supplementary Table 6).
Although the main objective of this work was to establish a complete workflow, from upstream CMC identification and recovery to downstream analysis, the substantial number of CMCs found in patient #3 allowed us to gather remarkable details regarding the genetics and evolution of this peculiar uveal melanoma case. Notably, we obtained a detailed molecular landscape of the tumor even in the absence of tumor tissue, and demonstrated the importance of merging CMC-derived information with data gathered from cfDNA in order to provide a complete and insightful tumor landscape in real time. The analysis highlighted both some peculiarities of uveal melanoma, such as its well-timed acquisition of different DNA alterations (each acquisition corresponds to an increase in aggressiveness and likelihood of disease progression), and a few particular differences from a classic uveal melanoma case. Indeed, NGS analysis revealed the presence of only two variants, according to the low mutation burden typical of this melanoma type52. The BAP1 inframe deletion of 36 base pairs in exon 9 results in the loss of 12 amino acids and the replacement with a methionine. This variant is classified as a VUS (Variant of Uncertain Significance) according to the AMP guidelines (https://franklin.genoox.com- Franklin by Genoox). It occurs in the ubiquitin carboxyl hydrolase (UCH) domain53of BAP1, a region reported to be more intolerant to missense changes and codon deletions than the central region of the protein54. This could be an indication in favor of a putative pathogenicity of the variant, which would be reinforced by the fact that all CMCs at progression carry this mutation and lack the wild-type allele (that is the second necessary hit for a tumor suppressor to be inactivated).
The MAF of the two variants (GNAQ p.Q209L and BAP1 p.R238_V250 delinsM) at progression was quite overlapping, in contrast to the condition at baseline. This suggests that they were present in homo- or hemi-zygosity at progression, and that the loss of the wild-type copy of BAP1 occurred later in the time course. However, due to the low MAF detected at baseline, the statement about mutation status, in terms of homo- or heterozygosity, is merely speculative. Inactivation of the tumor suppressor BAP1 is a key event for metastasization in uveal melanoma55, and the complete inactivation of BAP1 is usually associated with loss of chromosome 3 56. However, in our specific case there was no loss of chromosome 3 and no change in the number of copies of BAP1. This suggests that the apparent homozygosity of the p.R238_V250 delinsM variant was putatively hemizygosity with loss of the wild-type allele in a copy number neutral manner56, likely due to uniparental disomy or gene conversion process57,58. Another plausible scenario compatible with the MAFs observed52, would be that the BAP1 variant was already present in homo/hemizygosity at the baseline in a tumor subclone, which was then selected during disease progression due to the advantage conferred by the loss of the wild-type allele. This has already been observed by Karlsson et al., who found alterations in the BAP1 gene in 91% of the uveal melanoma metastases analyzed. In most cases, these were associated with a monosomy of chromosome 3, but in one case the BAP1 loss of heterozigosity occurred without copy number alterations56. Loss of wild-type GNAQ is also a rare event that has been found in very few cases of uveal melanoma52,59. Shain et al. reported 6 cases of metastatic uveal melanoma, in which GNAQ mutations were homozygous or hemizygous as a result of additional genetic changes that eliminated the wild-type allele. They found that loss of wild-type GNAQ tended to occur later in disease progression and suggested that GNAQ, unlike GNA11, requires a second hit to fully activate the pathway. Moreover, they also reported that a subclone with high levels of 8q and biallelic loss of CDKN2A had emerged from a primary melanoma, giving rise to liver metastases with even higher levels of 8q52. This picture mirrors the gene and chromosome rearrangement detected in the clinical case we analyzed (Supplementary Fig. 3), including the biallelic loss of CDKN2A seen at progression; this late event may be relevant in increasing the proliferative capacity and consequent aggressiveness of the disease56.
On the whole, our case seems to be related to the mechanism described by de Lange et al.4, as the temporally linear capacity to change progressively gives rise to different populations at different progression steps. The advantage of our setting is that we were able to detect these alterations in enriched CMC samples, with no need for tumor tissue. However, a major limitation of this study is the inability to compare the molecular profile of CMCs at baseline and at progression, due to the low number of CMCs available at baseline. At progression, the higher number of cells allowed us to gather significant molecular information through a bulk purification method (CellSearch plus Parsortix), even without a system for isolating individual cells. This approach, however, is not effective when only a small number of cells are present, highlighting the workflow primary limitation. To overcome this challenge, we thus took advantage of the cfDNA analysis for a comparison between the genetic status at the different time points, which is an excellent way of integrating information by gathering data from different liquid biopsy components60.
Conclusions
In conclusion, this study provides a proof-of-principle of the feasibility of enriching CMC starting from a marker-agnostic platform, with subsequent phenotypic identification by means of a customized antibody cocktail, and genetic analysis in different ways following specific workflows. The missing piece, which is also the main limitation of this study, would be the addition of a system capable of isolating single viable cells downstream of the workflow validated here, and prior to their analysis. This is a goal that should be pursued in the near future, as it is already feasible at least from fixed cells50,61, and should be incorporated into our workflow especially for samples from melanoma patients, where circulating tumor cells are in most cases very rare.
Finally, the case presented here is a good example of the complementary information that can be obtained from the combined analysis of cfDNA and CMCs, a system that provides maximum benefit in terms of information about the genetic landscape and evolution of the disease.
Data availability
The data are available from the corresponding authors on reasonable request. NGS raw data analysed in this study have been deposited in the NCBI Sequence Read Archive database under BioProject ID PRJNA1201193 and are available at the following link, https://www.ncbi.nlm.nih.gov/sra/PRJNA1201193.
References
Templeman, A. et al. Analytical performance of the FDA-cleared Parsortix® PC1 system. J. Circ. Biomark. 12, 26–33 (2023).
Gorges, K. et al. Intra-Patient Heterogeneity of Circulating Tumor Cells and Circulating Tumor DNA in Blood of Melanoma Patients. Cancers 11, (2019).
Lone, S. N. et al. Liquid biopsy: a step closer to transform diagnosis, prognosis and future of cancer treatments. Mol. Cancer. 21, 79 (2022).
de Lange, M. J., Nell, R. J. & van der Velden, P. A. Scientific and clinical implications of genetic and cellular heterogeneity in uveal melanoma. Mol. Biomed. 2, 25 (2021).
Pantel, K. & Alix-Panabières, C. Liquid biopsy: potential and challenges. Mol. Oncol. 10, 371–373 (2016).
Siravegna, G., Marsoni, S., Siena, S. & Bardelli, A. Integrating liquid biopsies into the management of cancer. Nat. Rev. Clin. Oncol. 14, 531–548 (2017).
Agashe, R. & Kurzrock, R. Circulating tumor cells: from the laboratory to the Cancer clinic. Cancers 12, 2361 (2020).
Kong, S. L. Complementary sequential Circulating tumor cell (CTC) and cell-Free tumor DNA (ctDNA) profiling reveals metastatic heterogeneity and genomic changes in lung Cancer and breast Cancer. Front. Oncol. 11, (2021).
Alix-Panabières, C. & Pantel, K. Liquid biopsy: from discovery to clinical application. Cancer Discov. 11, 858–873 (2021).
Cristofanilli, M. et al. Circulating tumor cells, disease progression, and survival in metastatic breast Cancer. N Engl. J. Med. 351, 781–791 (2004).
Aya-Bonilla, C. A. et al. Detection and prognostic role of heterogeneous populations of melanoma Circulating tumour cells. Br. J. Cancer. 122, 1059–1067 (2020).
Markou, A., Tzanikou, E. & Lianidou, E. The potential of liquid biopsy in the management of cancer patients. Semin Cancer Biol. 84, 69–79 (2022).
Russano, M. et al. Liquid biopsy and tumor heterogeneity in metastatic solid tumors: the potentiality of blood samples. J. Exp. Clin. Cancer Res. 39, 95 (2020).
Hall, C. S. et al. Circulating tumor cells in stage IV melanoma patients. J. Am. Coll. Surg. 227, 116–124 (2018).
Scaini, M. C. et al. A multiparameter liquid biopsy approach allows to track melanoma dynamics and identify early treatment resistance. Npj Precis Oncol. 8, 1–16 (2024).
Khoja, L. et al. Biomarker utility of Circulating tumor cells in metastatic cutaneous melanoma. J. Invest. Dermatol. 133, 1582–1590 (2013).
Khoja, L., Lorigan, P., Dive, C., Keilholz, U. & Fusi, A. Circulating tumour cells as tumour biomarkers in melanoma: detection methods and clinical relevance. Ann. Oncol. Off J. Eur. Soc. Med. Oncol. 26, 33–39 (2015).
Lucci, A. et al. Circulating tumor cells and early relapse in Node-positive melanoma. Clin. Cancer Res. Off J. Am. Assoc. Cancer Res. 26, 1886–1895 (2020).
Reid, A. L., Freeman, J. B., Millward, M., Ziman, M. & Gray, E. S. Detection of BRAF-V600E and V600K in melanoma Circulating tumour cells by droplet digital PCR. Clin. Biochem. (2015).
Aya-Bonilla, C. et al. Immunomagnetic-Enriched subpopulations of melanoma Circulating tumour cells (CTCs) exhibit distinct transcriptome profiles. Cancers 11, 157 (2019).
McEvoy, A. C. et al. Sensitive droplet digital PCR method for detection of TERT promoter mutations in cell free DNA from patients with metastatic melanoma. Oncotarget 8, 78890–78900 (2017).
Aya-Bonilla, C. A. et al. Isolation and detection of Circulating tumour cells from metastatic melanoma patients using a slanted spiral microfluidic device. Oncotarget 8, 67355–67368 (2017).
Hong, X. et al. Molecular signatures of Circulating melanoma cells for monitoring early response to immune checkpoint therapy. Proc. Natl. Acad. Sci. 115, 2467–2472 (2018).
Xu, L. et al. Optimization and evaluation of a novel size based Circulating tumor cell isolation system. PLOS ONE. 10, e0138032 (2015).
Miller, M. C., Robinson, P. S., Wagner, C. & O’Shannessy, D. J. The Parsortix™ cell separation System—A versatile liquid biopsy platform. Cytometry A. 93, 1234–1239 (2018).
Rao, C. et al. Circulating melanoma cells and survival in metastatic melanoma. Int. J. Oncol. 38, 755–760 (2011).
Catoni, C. et al. Investigating the retained inhibitory effect of Cobimetinib against p.P124L mutated MEK1: A combined liquid biopsy and in Silico approach. Cancers 14, 4153 (2022).
Buckley, A. R. et al. Pan-cancer analysis reveals technical artifacts in TCGA germline variant calls. BMC Genom. 18, 458 (2017).
Babayan, A. et al. Comparative study of whole genome amplification and next generation sequencing performance of single cancer cells. Oncotarget 8, 56066–56080 (2016).
Lim, J. Q., Lim, S. T. & Ong, C. K. Misaligned sequencing reads from the GNAQ-pseudogene locus May yield GNAQ artefact variants. Nat. Commun. 13, 458 (2022).
Versluis, M. et al. Digital PCR validates 8q dosage as prognostic tool in uveal melanoma. PloS One. 10, e0116371 (2015).
de Lange, M. J. et al. Heterogeneity revealed by integrated genomic analysis uncovers a molecular switch in malignant uveal melanoma. Oncotarget 6, 37824–37835 (2015).
Shoda, K. et al. Monitoring the HER2 copy number status in Circulating tumor DNA by droplet digital PCR in patients with gastric cancer. Gastric Cancer. 20, 126–135 (2017).
Freeman, J. B., Gray, E. S., Millward, M., Pearce, R. & Ziman, M. Evaluation of a multi-marker immunomagnetic enrichment assay for the quantification of Circulating melanoma cells. J. Transl Med. 10, 192 (2012).
Gray, E. S. et al. Circulating tumor DNA to monitor treatment response and detect acquired resistance in patients with metastatic melanoma. Oncotarget 6, 42008–42018 (2015).
Prieto, V. G. & Shea, C. R. Immunohistochemistry of melanocytic proliferations. Arch. Pathol. Lab. Med. 135, 853–859 (2011).
Kitago, M. et al. mRNA expression and BRAF mutation in Circulating melanoma cells isolated from peripheral blood with high molecular weight melanoma-associated antigen-specific monoclonal antibody beads. Clin. Chem. 55, 757–764 (2009).
Yang, J. et al. Melanoma chondroitin sulfate proteoglycan enhances FAK and ERK activation by distinct mechanisms. J. Cell. Biol. 165, 881–891 (2004).
Jungbluth, A. A. et al. Expression of melanocyte-associated markers gp-100 and Melan-A/MART-1 in angiomyolipomas. Virchows Arch. 434, 429–435 (1999).
Matsumoto, T. et al. Circulating endothelial/skeletal progenitor cells for bone regeneration and healing. Bone 43, 434–439 (2008).
Braakman, E., van de Winkel, J. G. J., van Krimpen, B. A., Jansze, M. & Bolhuis, R. L. H. CD16 on human Γδ T lymphocytes: expression, function, and specificity for mouse IgG isotypes. Cell. Immunol. 143, 97–107 (1992).
Neves, R. P. L. et al. Proficiency testing to assess technical performance for CTC-Processing and detection methods in CANCER-ID. Clin. Chem. 67, 631–641 (2021).
Smit, D. J. & Pantel, K. Circulating tumor cells as liquid biopsy markers in cancer patients. Mol. Aspects Med. 96, 101258 (2024).
Weidele, K. et al. Microfluidic enrichment, isolation and characterization of disseminated melanoma cells from lymph node samples. Int. J. Cancer. 145, 232–241 (2019).
Løppke, C. et al. Combined microfluidic enrichment and staining workflow for single-cell analysis of Circulating tumor cells in metastatic prostate cancer patients. Sci. Rep. 14, 17501 (2024).
Swennenhuis, J. F., van Dalum, G., Zeune, L. L. & Terstappen, L. W. M. M. Improving the CellSearch® system. Expert Rev. Mol. Diagn. 16, 1291–1305 (2016).
Pantel, K. & Alix-Panabières, C. Liquid biopsy and minimal residual disease - latest advances and implications for cure. Nat. Rev. Clin. Oncol. 16, 409–424 (2019).
Kamińska, P. et al. Liquid biopsy in melanoma: significance in diagnostics, prediction and treatment monitoring. Int. J. Mol. Sci. 22, 9714 (2021).
Tucci, M. et al. Dual-procedural separation of CTCs in cutaneous melanoma provides useful information for both molecular diagnosis and prognosis. Ther. Adv. Med. Oncol. 12, 1758835920905415 (2020).
Sementsov, M. et al. Mutation analysis in individual Circulating tumor cells depicts intratumor heterogeneity in melanoma. EMBO Mol. Med. 16, 1560–1578 (2024).
Alix-Panabières, C. & Pantel, K. Clinical applications of Circulating tumor cells and Circulating tumor DNA as liquid biopsy. Cancer Discov. 6, 479–491 (2016).
Shain, A. H. et al. The genetic evolution of metastatic uveal melanoma. Nat. Genet. 51, 1123–1130 (2019).
Louie, B. H. & Kurzrock, R. BAP1: not just a BRCA1-associated protein. Cancer Treat. Rev. 90, 102091 (2020).
Waters, A. J. et al. Saturation genome editing of BAP1 functionally classifies somatic and germline variants. Nat. Genet. 56, 1434–1445 (2024).
Harbour, J. W. et al. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science 330, 1410 (2010).
Karlsson, J. et al. Molecular profiling of driver events in metastatic uveal melanoma. Nat. Commun. 11, 1894 (2020).
Makishima, H. & Maciejewski, J. P. Pathogenesis and consequences of uniparental disomy in Cancer. Clin. Cancer Res. Off J. Am. Assoc. Cancer Res. 17, 3913–3923 (2011).
Takahashi, K. K. & Innan, H. Frequent somatic gene conversion as a mechanism for loss of heterozygosity in tumor suppressor genes. Genome Res. 32, 1017–1025 (2022).
Zhang, L. et al. The interplay of Circulating tumor DNA and chromatin modification, therapeutic resistance, and metastasis. Mol. Cancer. 18, 36 (2019).
Allen, T. A. The role of Circulating tumor cells as a liquid biopsy for cancer: advances, biology, technical challenges, and clinical relevance. Cancers 16, 1377 (2024).
Fernandez-Garcia, D. et al. Shallow WGS of individual CTCs identifies actionable targets for informing treatment decisions in metastatic breast cancer. Br. J. Cancer. 127, 1858–1864 (2022).
Acknowledgements
The CellSearch platform was gifted by the Association ASCOM, Padova (Italy).
Funding
This research was funded by the Italian Ministry of Health, Proposal code GR-2016-02361019, Liquid Biopsy in melanoma: investigating the prognostic/predictive potential of circulating melanoma cells (CMCs) and tumor DNA (ctDNA) - P.I. M.C. Scaini; 5 × 1000 Cancerplat-2, P23-Liquid biopsy in melanoma, P.I. M.C. Scaini; 5 × 1000 2016 Agon P.I. S. Mocellin; 5 × 1000 2019 BRIDGE. A.F.P. was supported by RETE-NET 2016-2361632, P.I. A. Rosato and Italian Ministry of Health Ricerca Corrente; I.G. was supported by 5 × 1000 2019 BRIDGE. This research received "Ricerca Corrente" funding from the Italian Ministry of Health to cover publication costs.
Author information
Authors and Affiliations
Contributions
C.P.: study design, data acquisition, validation, interpretation, image conceptualization, writing-original draft review and editing; A.F.P.: data acquisition and validation, writing-original draft; C.C.: study design, data acquisition and validation, interpretation, image conceptualization, writing-original draft review and editing; I.G.: data acquisition, writing-original draft; L.P.: sample contribution; S.P.: MLPA analysis; V.A.: sample contribution; V.S.: sample contribution; J.P.: study design and sample contribution; A.F.: sample contribution; A.F.: data acquisition; C.M.: data acquisition; P.D.F.: technical support; S.M.: funding acquisition, writing-review; V.C.S.: sample contribution, writing-review; A.R.: funding acquisition, supervision, writing-review, and editing; M.C.S.: funding acquisition, project administration, study design, conceptualization, data acquisition, validation, interpretation, supervision, writing-original draft review and editing, image conceptualization. All the authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study was conducted in accordance with the guidelines of the Declaration of Helsinki and was approved by the Ethics Committee of the ISTITUTO ONCOLOGICO VENETO (approval No. CESC-IOV 2018/36 on April 19, 2018). Informed consent was obtained from all subjects involved in the study.
Competing interests
M.C.S. received a travel grant from Agilent Technologies, all the other authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Poggiana, C., Piazza, A.F., Catoni, C. et al. A model workflow for microfluidic enrichment and genetic analysis of circulating melanoma cells. Sci Rep 15, 15329 (2025). https://doi.org/10.1038/s41598-025-99153-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-99153-y
