
Abstract
The progranulin (PGRN) protein is tightly linked with TDP-43 proteinopathy in neurodegenerative diseases. However, how PGRN regulates TDP-43 proteinopathy remains unclear. In this study, we investigated the effect of PGRN loss on TDP-43 pathology in the TDP-43Q331K knock-in mice expressing an ALS-linked TDP-43 mutation at the endogenous level, and in the transgenic mice overexpressing human TDP-43 in neurons. We found that PGRN deficiency leads to mild glial activation and behavioral deficits in TDP-43Q331K mice without inducing typical TDP-43 pathology. RNA-seq analysis reveals upregulation of immune pathways and downregulation of myelination-related pathways in PGRN-deficient TDP-43Q331K mice. In addition, we observed myelination defects in human TDP-43 transgenic mice, but PGRN loss does not exacerbate TDP-43 pathology, myelination defects, and motor deficits in this mouse strain. Our studies demonstrated that PGRN deficiency exacerbates behavioral deficits and glial pathology caused by TDP-43 Q331K mutation but has minimal effect on TDP-43 pathology in mouse models.
Similar content being viewed by others


Introduction
Transactivation response (TAR) DNA-binding protein 43 (TDP-43), encoded by the Tardbp gene, is a DNA/RNA binding protein involved in multiple RNA-related processes1,2. In normal conditions, TDP-43 is predominantly localized in the nucleus but shuttles between the nucleus and cytoplasm to exert various functions, including regulation of RNA splicing, RNA trafficking, and RNA stability2,3. Due to its critical functions in RNA metabolism and other cellular processes, TDP-43 is essential for maintaining cellular health. TDP-43 proteinopathy, characterized by the accumulation of the hyper-phosphorylated, fragmented, and aggregated TDP-43 in the cytoplasm and depletion of TDP-43 from the nucleus, is a pathologic hallmark of many neurodegenerative diseases, including amyotrophic lateral sclerosis (ALS)4,5, frontotemporal lobar degeneration (FTLD)5,6,7, Alzheimer’s disease (AD)8,9,10, Huntington’s diseases (HD)11, and limbic-predominant age-related TDP-43 encephalopathy (LATE)12,13. However, molecular pathways leading to TDP-43 proteinopathy are still poorly understood.
Haploinsufficiency of progranulin (PGRN) protein caused by mutations in the granulin (GRN) gene is known to be a main cause of FTLD with TDP-43 proteinopathy14,15,16. In addition, the granulin (GRN) gene has also been identified as a major risk gene for LATE17,18, indicating an important role of PGRN in TDP-43 proteinopathy. However, little is known about how PGRN loss of function leads to TDP-43 proteinopathy. PGRN is an evolutionarily conserved glycoprotein comprised of 7.5 granulin repeats19,20,21, with critical roles in inflammation22. At the cellular level, PGRN is either secreted to the extracellular space or sorted into the lysosome23,24. Within the lysosome, PGRN is processed into granulin peptides, which are critical for proper lysosomal functions, especially during aging23,24. Since lysosomal dysfunction and neuroinflammation have both been linked to TDP-43 proteinopathy25,26,27, lysosomal dysfunction and enhanced neuroinflammation caused by PGRN loss may contribute to TDP-43 proteinopathy.
Despite the strong link between PGRN and TDP-43 proteinopathy in humans, minimal alteration in TDP-43 has been observed in PGRN-deficient mice28. To investigate the role of PGRN in TDP-43 proteinopathy, we utilize two TDP-43 mouse models. One is the TDP-43Q331K knock-in mouse line expressing TDP-43 Q331K mutant, which is associated with ALS, at endogenous levels. This mouse line does not develop cytoplasmic aggregation or nuclear loss of TDP-43 but shows slightly increased levels of TDP-43 in the nucleus29. Another one is the transgenic mouse line overexpressing the wild-type human TDP-43 in all neurons in the central nervous system, which shows accumulation of nuclear and cytoplasmic TDP-43 aggregates in neurons30. In both mouse models, we failed to detect significant effects of PGRN deficiency on TDP-43 protein levels, distribution, and solubility, which suggests that mouse models might not be an ideal system to study the role of PGRN in TDP-43 proteinopathy.
Results
PGRN deficiency does not induce TDP-43 pathology in TDP-43Q331K mice
Mutations in TDP-43 have been genetically linked to ALS and FTLD31,32,33. Mice expressing the ALS-linked TDP-43 mutant Q331K (TDP-43Q331K) at the endogenous TDP-43 loci do not display typical TDP-43 pathology, such as cytoplasmic aggregation and nuclear loss of TDP-43. Instead, they show increased TDP-43 levels due to the perturbed autoregulation caused by the Q331K mutation29. To investigate the role of PGRN in TDP-43 pathology, we crossed TDP-43Q331K mice with Grn−/− mice to assess how PGRN loss affects TDP-43 proteinopathy in this sensitized background. We first determined the effect of PGRN deficiency on TDP-43 homeostasis in the TDP-43Q331K mice. We failed to detect cytoplasmic TDP-43 accumulation in neurons in the cortex region of Grn−/−, TDP-43Q331K/Q331K, or TDP-43Q331K/Q331K Grn−/− mice, even at 16 months of age (Fig. 1A), suggesting PGRN loss does not lead to the cytoplasmic mislocalization of TDP-43 in these mice. Next, we measured the total TDP-43 levels and phosphorylated TDP-43 levels (pS409/410) using a validated pS409/410 antibody (Supplemental Fig. 1) in the 16.0-month-old mouse brain cortex. TDP-43 total levels, solubility, and phosphorylation state are not altered by PGRN loss in TDP-43Q331K/Q331K mice (Fig.1B, C). In addition, TDP-43 Q331K has little effect on PGRN protein levels (Fig.1B, D). Collectively, these results suggest that PGRN loss does not trigger any obvious TDP-43 pathology in the brain of TDP-43Q331K mice.

A Immunostaining of TDP-43 and NeuN in brain sections from 16-month-old mice WT, TDP-43Q331K/Q331K (Q331K), Grn−/−, TDP-43Q331K/Q331K Grn−/− (Q331K Grn−/−) mice. Representative images from the cortex were shown. Scale bar, 10 µm. B Analysis of TDP-43, phosphorylated TDP-43 (pS409/410), and PGRN levels in cortical lysates from 16-month-old mice of the indicated genotypes. C TDP-43 levels in RIPA- and urea-soluble fractions were quantified. Data are presented as mean ± SEM from 3 mice per group (n = 3). One-way ANOVA tests with Bonferroni’s multiple comparisons. D PGRN levels in RIPA soluble fractions were quantified and normalized to GAPDH. Data are presented as mean ± SEM from 3 mice per group (n = 3). E Total RNAs were extracted from the cortex of 10-month-old WT, TDP-43Q331K/Q331K, Grn−/−, TDP-43Q331K/Q331KGrn−/− male mice, and the RT-qPCR was performed to analyze the splicing changes in Sort1 exon 17b (left) and Mapt exons 2 and 3 (right). The relative mRNA levels of transcripts including or excluding exons 2 and 3 represent the inclusion of Mapt exons 2 and 3. Data are presented as mean ± SEM (n = 4 mice per genotype). p-values were determined using one-way ANOVA tests with Bonferroni’s multiple comparisons. F Expression levels of Tardbp in WT and Q331K mice. Total RNAs were extracted from the cortex of 10-month-old WT and Q331K male mice, and the RNA-seq was performed to analyze gene expression changes. Normalized read counts are shown. Data are presented as mean ± SEM (n = 5-6 mice per genotype). *p < 0.05, unpaired two-tailed Student's t-test.
PGRN ablation leads to upregulation of immune pathways and downregulation of myelination-related pathways in TDP-43Q331K mice
TDP-43Q331K has been shown to cause alterations in RNA splicing29. To determine whether PGRN loss affects TDP-43 function, we performed RT-qPCR to analyze changes in the splicing of known TDP-43 targets and RNA-seq analysis to identify gene expression alterations. A previous study has reported that the TDP-43 Q331K mutation leads to decreased inclusion of Sort1 exon 17b and increased inclusion of Mapt exons 2 and 329. However, we did not observe significant changes in Sort1 or Mapt splicing in RNA samples from the cortex of TDP-43Q331K/Q331K, Grn−/− or TDP-43Q331K/Q331kGrn−/− mice (Fig.1D). In addition, RNA-seq analysis revealed increased Tardbp expression in the cortex of 10-month-old TDP-43Q331K/Q331K mice, as demonstrated by increased normalized read counts in TDP-43Q331K/Q331K samples compared to the WT controls (Fig. 1F), confirming the dysregulation of Tardbp expression by Q331K mutation in these mice. However, no other significant gene expression changes were detected (Supplementary Data 1). In contrast, significant gene expression changes were identified in Grn−/− mouse brain, with 63 upregulated and 7 downregulated genes compared to WT controls (Fig. 2A), which are further exacerbated in TDP-43Q331K/Q331K Grn−/− mouse brain with 482 upregulated and 182 downregulated genes compared to WT (Fig. 2B–E). The expression changes of the DEGs ( | log2FC | ≥1.0) identified in TDP-43Q331K/Q331K Grn−/− mice versus WT mice were shown in the heatmap (Fig. 2F, G). We further compared the DEGs identified in the TDP-43Q331K/Q331K Grn−/− mice to those identified in the cortical samples from 5- and 20-month-old TDP-43Q331K/Q331K mice in the previous study29. When comparing DEGs with an FDR ≤ 0.05 and FC ≥ 1.5, only five upregulated genes (Car3, Dusp27, Lilra5, Prg4, and Tfap2d) overlap with those identified in 20-month-old TDP-43Q331K/Q331k mice. However, when using an FDR ≤ 0.05 (without an FC cutoff), more overlap is observed with DEGs from either the 5- or 20-month-old TDP-43Q331K/Q331k mice (Fig. 2H). The discrepancy in the gene expression alterations in the TDP-43Q331K discrepancy in our study versus previous studies29 could be attributed to several factors, such as differences in mouse housing environment or technical variations in RNA sequencing and analysis. The key methodology variations include the library preparation method and sequencing depth. Our study used rRNA depletion, which retains both mRNA and non-coding RNAs, potentially requiring deeper sequencing to achieve sufficient coverage of protein-coding genes. In contrast, the previous study employed poly(A) enrichment, which selectively targets mRNA and typically yields cleaner gene expression signals at lower sequencing depths. The prior study sequenced a library of 50 million 100 bp paired-end reads, providing higher read counts for enhanced statistical power in differential expression analysis. Our approach used 40 million 150 bp paired-end reads, which, while offering longer read lengths for alignment accuracy, resulted in fewer total reads. Moreover, differences in analytical pipelines (e.g., alignment tools, DEG-calling algorithms, or significance thresholds) may further contribute to the discrepancy.

Total RNAs were extracted from the cortex of 10-month-old WT, TDP-43Q331K/Q331K (Q331K), Grn−/−, and TDP-43Q331K/Q331KGrn−/− (Q331K Grn−/−) male mice, and the RNA-seq was performed to analyze gene expression changes (n = 5–6 mice per genotype). A Differentially expressed genes (DEGs) with FDR ≤ 0.05 and absolute log2(FC) ≥ 0.5 between WT and Grn−/− are plotted using Heatmapper. B DEGs identified in the TDP-43Q331K/Q331KGrn−/− compared with WT (FDR ≤ 0.05, absolute log2(FC) ≥ 0.5). C Heatmap showing the expression of the DEGs identified in the TDP-43Q331K/Q331KGrn−/− compared with TDP-43Q331K/Q331K (FDR ≤ 0.05, absolute log2(FC) ≥ 0.5). D Heatmap showing the expression of the DEGs identified in the TDP-43Q331K/Q331KGrn−/− compared with Grn−/− (FDR ≤ 0.05, absolute log2(FC) ≥ 0.5). E Venn diagrams showing the overlap of the DEGs identified in the indicated comparisons. F Heatmap illustrating the expression of the significantly upregulated genes (log2FC ≥ 1) identified in the TDP-43Q331K/Q331KGrn−/− mice compared with WT. G Heatmap illustrating the expression of the significantly downregulated genes (log2FC ≤ −1) identified in the TDP-43Q331K/Q331KGrn−/− mice compared with WT. H Venn diagrams showing the overlap of the DEGs identified in this study and the previous study by White et al. DEGs (FDR ≤ 0.05) identified in the TDP-43Q331K/Q331KGrn−/− mouse cortical samples were compared to those identified in the 5- and 20-month-old TDP-43Q331K/Q331K mouse cortical samples from the study of White et al.
Gene set enrichment analysis (GSEA) of all the genes revealed upregulation of immune-related pathways in Grn−/− mice compared with WT, as well as in TDP-43Q331K/Q331K Grn−/− mice compared with WT, TDP-43Q331K/Q331K, or Grn−/− (Fig. 3A–D), reflecting immune activation in the cortex in these mice. Notably, several pathways related to myelination, including axon ensheathment in the central nervous system and oligodendrocyte development and differentiation, were significantly downregulated in TDP-43Q331K/Q331K Grn−/− mice compared to WT or TDP-43Q331K/Q331K (Fig. 3B–E). These findings suggest that PGRN deficiency leads to myelination defects in TDP-43Q331K/Q331K mice. Taken together, PGRN ablation leads to the upregulation of immune pathways and downregulation of myelination pathways in TDP-43Q331K mice, reflecting immune activation and myelination defects caused by PGRN deficiency in TDP-43Q331K mice. Additionally, the axon ensheathment in the central nervous system is also significantly downregulated in TDP-43Q331K/Q331K Grn−/− mice compared to Grn−/− (Fig. 3D), indicating that the TDP-43 Q331K mutation might exacerbate myelination defects in the PGRN-deficient background.

A–D Pathway enrichment analysis was performed by GSEA using the gene sets of gene ontology biological process (GOBP). The significantly upregulated and downregulated gene sets (FDR q-value <0.1) identified in the indicated comparisons are listed. The gene sets with normalized enrichment score (NES) > 1.0 are upregulated, and those with NES < −1 are downregulated in the indicated comparisons. Myelination-related pathways were highlighted in orange. E Expression changes of the DEGs (FDR ≤ 0.05, absolute log2(FC) ≥ 0.5) identified in axon ensheathment in the central nervous system, oligodendrocyte development, and oligodendrocyte differentiation pathways in TDP-43Q331K/Q331K Grn−/− brain samples compared to WT based on GSEA results.
PGRN deficiency causes mild glial activation and behavioral deficits in TDP-43Q331K mice
Next, we examined glial activation in TDP-43Q331K/Q331K Grn−/− mice. A previous study demonstrated microglial activation in the cortex in TDP-43Q331K mouse brain, as evidenced by elevated levels of microglia marker IBA1 and decreased levels of the homeostatic microglia marker TMEM11934. While we similarly observed increased IBA1 levels in the cortex of TDP-43 mutant mice, no significant change was detected in the microglial activation marker CD68 (Fig. 4A, B). Consistent with the previous study34, we did not observe astrocyte activation in TDP-43Q331K/Q331K mice, as indicated by comparable GFAP levels between mutant and WT mice (Fig. 4A, B). As expected, PGRN deficiency leads to marked glial activation in the mouse brain, as demonstrated by significantly increased levels of IBA1, CD68, and GFAP in the cortex in both 10.0- and 16.0-month-old Grn−/− mice compared to age-matched WT (Fig. 4A, B). The glial activation was significantly higher in TDP-43Q331K/Q331K Grn−/− mice compared to TDP-43Q331K/Q331K mice, but only slightly increased relative to Grn−/− mice (Fig. 4A, B). PGRN deficiency is known to cause lipofuscin accumulation, an indicator for lysosomal dysfunction, in mouse cortex and thalamus35,36. To determine whether lysosomal defects are exacerbated in TDP-43 mutant mice under PGRN deficiency, we analyzed autofluorescent signals in the thalamus region of 10-month-old mice. As expected, Grn−/− mice exhibited significantly increased autofluorescence in the thalamus. However, the signals in TDP-43Q331K/Q331K Grn−/− mice were comparable to those in Grn−/− mice (Fig. 4C, D), indicating that TDP-43 Q331K may not exacerbate lysosomal defects caused by PGRN deficiency.

A, B Immunostaining of GFAP, CD68, and IBA1 in brain sections from 10 and 16-month-old WT, TDP-43Q331K/Q331K (Q331K), Grn−/−, and TDP-43Q331K/Q331KGrn−/− (Q331K Grn−/−) mice. Representative images from the cortex were shown. Scale bar, 100 µm. Quantification of GFAP, CD68, and IBA1 levels from 10.0-month-old mice sections was shown in (B). Data are presented as mean ± SEM from five mice per group (n = 5). p-values were determined using one-way ANOVA with Bonferroni’s multiple comparisons. *p < 0.05; **p < 0.01; ***p < 0.001;****p < 0.0001. C, D Lipofuscin accumulation in 10-month-old WT, Q331K, Grn−/−, and Q331K Grn−/− mice. Representative images for the autofluorescent signals from the thalamus were shown. Scale bar, 10 µm. Quantification of the autofluorescent signals was shown in (D). Data are presented as mean ± SEM from three mice per group (n = 3). p-values were determined using one-way ANOVA with Bonferroni’s multiple comparisons. ****p < 0.0001.
TDP-43Q331K mice display FTLD-like cognitive dysfunction, including executive and memory impairment, but do not develop significant motor impairments29. To determine the effect of PGRN on the motor function in TDP-43Q331K mice, we conducted open field tests to assess the locomotor activity of 10.0-month-old TDP-43Q331K/Q331K Grn−/− and TDP-43Q331K/+ Grn−/− mice. The total distance traveled by both TDP-43Q331K/Q331K and TDP-43Q331K/+ mice was similar to that of the WT mice, while PGRN deficiency in these mice led to a significant reduction (Fig. 5A, B), indicating that PGRN ablation impaired locomotor function in TDP-43Q331K mice. Additionally, both TDP-43Q331K/+ Grn−/− and TDP-43Q331K/Q331K Grn−/− mice spent less time in the center area of the open field compared to WT mice (Fig. 5C), suggesting that these mice may develop anxiety-like behavior. In balance beam tests, TDP-43Q331K/Q331K Grn−/− and TDP-43Q331K/+ Grn−/− mice spend more time crossing the beam compared with WT mice (Fig. 5D), indicating motor coordination impairments. For cognitive function assessment, novel object recognition tests and Y-maze tests were performed. TDP-43Q331K/Q331K Grn−/− and TDP-43Q331K/+ Grn−/− mice exhibit a reduced preference for novelty, similar to TDP-43Q331K/Q331K, TDP-43Q331K/+, and Grn−/− mice when compared to WT mice (Fig. 5E). However, TDP-43Q331K/Q331K Grn−/− and TDP-43Q331K/+ Grn−/− mice do not show any significant changes in alternation in the Y-maze tests (Fig. 5F, G), suggesting that spatial working memory is not influenced in these mice. In summary, PGRN deficiency causes mild glial activation and behavioral deficits in TDP-43Q331K mice.

A–C 10-month-old WT, TDP-43Q331K/+ (Q331K/ + ), TDP-43Q331K/Q331K (Q331K/Q331K), Grn−/−, TDP-43Q331K/+Grn−/− (Q331K/+ Grn−/−) and TDP-43Q331K/Q331KGrn−/− (Q331K/Q331K Grn−/−) male mice were subjected to the open field test. The total distance the mice traveled (A, B) and the percentage of the time spent in the center (time in center/total) (C) were quantified. Data are presented as means ± SEM (n = 9–17). One-way ANOVA tests. D Ten-month-old male mice of the indicated genotypes were subjected to balance beam tests. The average time across the beam was shown. Data are presented as means ± SEM (n = 8–19). P-values were determined using one-way ANOVA tests with Bonferroni’s multiple comparisons and were shown on top of the graphs. E 10-month-old male mice with indicated genotypes were subjected to novel object recognition tests. Novelty preference (exploration time of the novel object/exploration time of both objects) × 100%) was shown. Data are presented as means ± SEM (n = 9–17). One-way ANOVA tests with Bonferroni’s multiple comparisons. F, G 10-month-old male mice with indicated genotypes were subjected to Y-maze tests. Total arm entries (g) and alternation (%) (h) were shown. Data are presented as means ± SEM (n = 10–20). One-way ANOVA tests with Bonferroni’s multiple comparisons. *p < 0.05; **p < 0.01; ***p < 0.001;****p < 0.0001.
PGRN deficiency does not exacerbate TDP-43 pathology in human TDP-43 transgenic mice
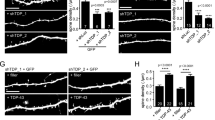
In addition to the TDP-43Q331K mouse model, we also utilized a mouse model overexpressing human wild-type TDP-4330 to determine the effect of PGRN loss on TDP-43 pathology. Heterozygous TDP-43 transgenic mice (hTDP-43Tg/+) were crossed with Grn−/− mice to generate hTDP-43Tg/+ Grn−/− and hTDP-43Tg/Tg Grn−/− mice. Due to early lethality, hTDP-43Tg/Tg Grn−/− mice, as well as hTDP-43Tg/Tg mice, were collected at 21 days. hTDP-43Tg/+ Grn−/− mice were collected at 6-month-old. Immunostaining with a rabbit anti-TDP-43 C-terminal domain antibody recognizing both human and mouse TDP-43 and a mouse anti-TDP-43 antibody that specifically recognizes human but not mouse TDP-43 showed no cytoplasmic TDP-43 accumulation in 21-day-old TDP-43Tg/Tg Grn−/− mice and 6-month-old TDP-43Tg/+ Grn−/− mice (Figs. 6A, 7A), suggesting PGRN loss does not lead to the cytoplasmic mislocalization of TDP-43 in hTDP-43Tg mice. In addition, total TDP-43 levels and human TDP-43 levels, as well as phosphorylated TDP-43 levels in RIPA-soluble and urea-insoluble fractions of the 21-day-old hTDP-43Tg/Tg Grn−/− and 6-month-old hTDP-43Tg/+ Grn−/−mouse brains, were analyzed. A significant increase in phosphorylated TDP-43 (pTDP-43) levels was detected in the urea fraction of 21-day-old TDP-43Tg/Tg and 6-month-old TDP-43Tg/+ mouse brains compared to age-matched WT controls, while PGRN loss did not significantly increase pTDP-43 levels in the brain lysates prepared from these mice (Figs. 6B, C, 7B, C). A significant increase in total TDP-43 levels was detected in TDP-43Tg/Tg mice compared to WT mice with the antibody against the N-terminal domain of TDP-43 but not with the antibody against the C-terminal domain of TDP-43 (Fig. 6B, C). This is likely caused by the C-terminal TDP-43 antibody having a higher affinity towards mouse TDP-43 compared to human TDP-43 due to the sequence difference towards the C-terminus of the protein. Nevertheless, loss of PGRN does not alter TDP-43 level or solubility in the 21-day-old hTDP-43Tg/Tg or 6-month-old hTDP-43Tg/+ mice (Figs. 6B, C, 7B, C). Additionally, the levels of PGRN in transgenic mice are largely unchanged (Fig. 6B, D). Taken together, our data support that PGRN loss does not exacerbate TDP-43 pathology in human TDP-43 transgenic mice.

A Immunostaining of TDP-43 and NeuN using rabbit anti-TDP-43 CTD antibodies and mouse anti-NeuN antibodies, respectively (left panel) or immunostaining of human TDP-43 (right panel) using mouse anti-human TDP-43 in brain sections from 21-day-old WT, hTDP-43Tg/Tg (Tg/Tg), Grn−/−, and hTDP-43Tg/Tg Grn−/− mice. Representative images from the cortex were shown. Scale bar, 10 µm. B Analysis of TDP-43 and phosphorylated TDP-43 (pS409/410) and PGRN levels in brain lysates from 21-day-old mice. Antibodies recognizing the C-terminal or N-terminal domain of TDP-43 were used to detect total TDP-43 levels. C TDP-43 and pTDP-43 levels in RIPA- and urea-soluble fractions were quantified. Data are presented as mean ± SEM from 3 mice per group (n = 3). One-way ANOVA tests with Bonferroni’s multiple comparisons. **p < 0.01. D PGRN levels in RIPA soluble fractions were quantified and normalized to GAPDH. Data are presented as mean ± SEM from 3 mice per group (n = 3).

A Immunostaining of TDP-43 and NeuN using rabbit anti-TDP-43 C-terminal domain (CTD) antibodies and mouse anti-NeuN antibodies in brain sections from 6-month-old WT, hTDP-43Tg/+(Tg/+), Grn−/−, and hTDP-43Tg/+ Grn−/− mice. Representative images from the cortex were shown. Scale bar, 10 µm. B Analysis of TDP-43 and phosphorylated TDP-43 (pS409/410) levels in brain lysates from 6-month-old mice. C TDP-43 and pTDP-43 levels in RIPA- and urea-soluble fractions were quantified. Data are presented as mean ± SEM from 3 mice per group (n = 3). One-way ANOVA tests with Bonferroni’s multiple comparisons. *p < 0.05.
PGRN deficiency does not exacerbate myelin and motor deficits in human TDP-43 transgenic mice
Our RNA-seq analysis from the TDP-43Q331K mouse model suggested that TDP-43 dysfunction might cause myelination defects. Interestingly, homozygous TDP-43 transgenic mice at the age of 21 days showed decreased levels of myelin proteins, including MAG, MBP, and PLP (Fig. 8A, B). PGRN deficiency does not cause a further decrease in myelin proteins (Fig. 8A, B). The previous study has shown that TDP-43 transgenic mice develop impaired motor function30. To determine the effect of PGRN loss on motor function in TDP-43 transgenic mice, we performed hindlimb clasping and footprint tests in 6-month-old mice. We found that hTDP-43Tg/+ mice displayed abnormal hindlimb reflexes and reduced hindlimb stride compared to the WT mice (Fig. 8C–E), indicating that these mice develop motor deficits as suggested previously. Grn−/− mice did not exhibit defects in these tests, while Grn−/− hTDP-43Tg/+ mice showed similar motor impairments as the hTDP-43Tg/+ mice (Fig. 8C–E). Taken together, PGRN deficiency does not exacerbate motor deficits in human TDP-43 transgenic mice.

A, B Western blot analysis of the myelin markers in the brain. The levels of MAG, MBP, and PLP in 21-day-old WT, hTDP-43Tg/Tg, Grn−/−, and hTDP-43Tg/Tg Grn−/− mice were analyzed by western blot. The levels were quantified by ImageJ and normalized to the loading control GAPDH. Data are presented as means ± SEM (n = 3–7). p-values were determined using one-way ANOVA tests with Bonferroni’s multiple comparisons. C, D The hindlimb clasping test was performed on 6-month-old WT, hTDP-43Tg/+(Tg/+), Grn−/−, and hTDP-43Tg/+ Grn−/− (Tg/+ Grn−/−) male and female mice. The hindlimb severity score was quantified. Data are presented as means ± SEM (n = 8–12). One-way ANOVA tests. E Six-month-old male and female mice were subjected to footprint tests. The hindlimb stride length was measured. Data are presented as means± SEM (n = 8–12). p-values were determined using one-way ANOVA tests with Bonferroni’s multiple comparisons. *p < 0.05; **p < 0.01; ***p < 0.001.
Discussion
PGRN haploinsufficiency caused by mutations in the GRN gene is a leading cause of FTLD with TDP-43 pathology14,15,16. Additionally, the GRN gene was identified as a key determinant of LATE, which is characterized by TDP-43 aggregation17,18. While the genetic studies point to an important role of PGRN in TDP-43 proteinopathy, it remains to be determined how PGRN regulates TDP-43 proteinopathy.
In mouse models, previous studies have shown that PGRN deficiency alone does not cause any obvious TDP-43 pathology28,37,38. Thus, it is worthwhile to investigate whether PGRN deficiency could lead to TDP-43 pathology in mouse models with disrupted TDP-43 protein homeostasis. In this study, we utilized two mouse models of TDP-43, the TDP-43Q331K knock-in and the human TDP-43 transgenic mice, to explore the role of PGRN in TDP-43 proteinopathy. TDP-43 knock-in mouse model harboring the ALS-associated mutation (Q331K) in the endogenous mouse Tardbp gene retains the endogenous TDP-43 gene structure29. This model does not develop typical TDP-43 pathology but exhibits perturbed TDP-43 homeostasis, which might allow us to investigate whether PGRN loss could induce TDP-43 aggregation in this sensitized background. In addition, TDP-43Q331K mice develop more pronounced FTD-like features29, potentially making them a more disease-related model for studying the role of PGRN deficiency in TDP-43 proteinopathy. However, PGRN loss does not seem to affect TDP-43 distribution, total levels and solubility as well as TDP-43’s function in splicing in TDP-43Q331K mouse brain (Fig. 1). In the second model, human TDP-43 transgenic mice, which overexpress wild-type human TDP-43 in neurons, develop TDP-43 aggregates in both the cytoplasm and nucleus of neurons in certain brain regions30. By using this model, we aim to determine whether PGRN deficiency could exacerbate TDP-43 pathology. Although phosphorylated TDP-43 was detected in the urea soluble fraction of the 21-day-old hTDP-43Tg/Tg and 6-month-old hTDP-43Tg/+ mouse brain lysates via western blot, TDP-43 distribution patterns were similar between transgenic mice and WT mice (Figs. 6, 7). Furthermore, PGRN loss does not significantly impact TDP-43 distribution, levels, or solubility (Figs. 6, 7). Taken together, our findings suggest that PGRN ablation has minimal effects on TDP-43 protein homeostasis in both mouse models used in this study. Future research using improved mouse models will be necessary to better understand how PGRN deficiency might regulate TDP-43 proteinopathy.
Studies have indicated that TDP-43 aggregates can be cleared by autophagy/lysosome pathways25,39. It is known that PGRN is essential for proper lysosomal function, and a deficiency in PGRN can lead to lysosomal defects, potentially disrupting TDP-43 protein homeostasis. Previous studies using mouse models and cell cultures have suggested that lysosomal dysfunction caused by PGRN deficiency may contribute to TDP-43 aggregation40,41. Additionally, PGRN is highly expressed in microglia, and its deficiency leads to microglial activation, which has been linked to TDP-43 pathology, although the mechanisms remain to be investigated42. Furthermore, PGRN-deficient mice also show myelination defects43. Myelin debris accumulates in PGRN-deficient microglia, likely due to the impaired lysosomal clearance, which exacerbates lysosomal defects by reducing the levels of lysosomal enzyme cathepsin D and further increases TDP-43 aggregation40. These findings support the hypothesis that both lysosomal dysfunction and microglia activation resulting from PGRN deficiency may contribute to TDP-43 proteinopathy. Although PGRN loss did not induce TDP-43 aggregation in TDP-43Q331K mice, our RNA-seq analysis revealed that PGRN deficiency leads to upregulation of immune pathways and downregulation of myelination pathways in the brains of TDP-43Q331K mice (Fig. 3), further supporting a role of PGRN in brain inflammation and myelination. Additionally, previous studies have implicated TDP-43 in myelination. In animal models, perturbation of TDP-43 protein homeostasis by either deletion or overexpression of TDP-43, or induction of cytoplasmic accumulation of truncated TDP-43 fragments in oligodendrocytes, has been shown to cause myelination defects44,45,46. Our findings that the TDP-43 Q331K downregulates myelination pathways in Grn−/− mice, as well as the observation that the transgenic mice with TDP-43 overexpression in the neurons exhibit reduced levels of myelin proteins in the brain, further suggest a role of TDP-43 in myelination (Figs. 2, 3, 8A, B).
As demonstrated previously, TDP-43Q331K mice display FTLD-like cognitive dysfunction but no significant motor impairment29. Consistently, the novel objection recognition test, open field test, and balance beam test suggested that the cognitive function, but not the motor function is impaired in TDP-43Q331K/+ and TDP-43Q331K/Q331K mice (Fig. 5). Previous studies have shown that Grn−/− or Grn+−/− mouse models develop cognitive dysfunction as well as anxiety or depressive-like behaviors28,38,47. Here, we found PGRN deficiency alone leads to both cognitive and motor dysfunction as well as increased open-field anxiety, but these deficits are not significantly worsened in Grn−/− TDP-43Q331K mice (Fig. 5). Human TDP-43 transgenic mice have been shown to develop motor deficits30, as evidenced by abnormal hindlimb reflexes and hindlimb footprints, which was also observed in this study (Fig 8). However, PGRN deficiency does not exacerbate the motor impairment in hTDP-43Tg mice (Fig. 8).
Taken together, our results support that PGRN deficiency or alteration of TDP-43 protein homeostasis leads to behavioral deficits and glial pathology in mice, and PGRN ablation exacerbates these phenotypes in TDP-43Q331K mice. Our results also show a critical role of both PGRN and TDP-43 in regulating the myelination process. However, PGRN deficiency does not have any obvious effect on TDP-43 protein homeostasis seen in human FTLD-GRN patients.
Methods
Antibodies and reagents
The following antibodies were used in this study: rabbit anti-IBA-1 (Wako, 01919741), goat anti-AIF-1/Iba1 (Novus Biologicals, NB100-1028), rat anti-CD68 (Bio-Rad, MCA1957), mouse anti-GFAP (Cell signaling, 3670S), rabbit anti-TDP43 (Proteintech Group, 12892-1-AP (C-terminal) and 10782-2-AP (N-terminal)), rabbit anti-phospho-TDP-43 (Ser409/410) (Proteintech group, 80007-1-RR), mouse anti-PLP (Millipore, MAB388), mouse anti-MBP (Millipore, SMI-99), rabbit anti-MAG (Proteintech Group, 14386-1-AP), sheep anti-mouse PGRN (R&D systems, AF2557), and mouse anti-GAPDH (Proteintech Group, 60004-1-Ig).
Mouse strains
Grn−/− mice48 and TDP-43Q331K mice29 in the C57BL/6 background and heterozygous transgenic mice overexpressing wild-type human TDP-4330 in the C57BL/6/SJL background (TDP-43Tg/+) were obtained from the Jackson laboratory. TDP-43Q331K/+ or TDP-43Q331K/Q331K Grn−/− mice were generated by crossing TDP-43Q331K/Q331K and Grn−/− mice. Human TDP-43Tg/+ Grn−/− or hTDP-43Tg/Tg Grn−/− mice were generated by crossing hTDP-43Tg/+ and Grn−/− mice. All the mice were housed in the Weill Hall animal facility at Cornell. All animal procedures have been approved by the Institutional Animal Care and Use Committee (IACUC) at Cornell. All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. The work under animal protocol 2017-0056 is approved by the Institutional Animal Care and Use Committee at Cornell University.
Behavioral test
Ten-month-old WT, TDP-43Q331K/+, TDP-43Q331K/Q331K, Grn−/−, TDP-43Q331K/+Grn−/− and TDP-43Q331K/Q331KGrn−/− male mice (8–20 mice/group) were subject to the following behavioral tests: (1) Open-field test: Mice were placed in a clean plastic chamber (45 × 45 × 45 cm) for 10 min. The total distance traveled was tracked by the Viewer III software (Biobserve, Bonn, Germany). The apparatus was thoroughly cleaned with 70% ethanol between trials. (2) Balance beam test: Beams that were 80 cm in length were used in the balance beam test for motor coordination. A bright light was used as an aversive stimulus at the start platform, and an escape box (20 × 20 x 20 cm) was placed at the end of the beam. The animals were trained for 3 consecutive trials on each beam for 3 consecutive days, and then were tested on the fourth day. The time taken to traverse the beam was recorded for each trial. The average time to cross the beam of the three trials was calculated. The maximum time cutoff was 60 s. (3) Y-Maze test: Spatial working memory performance was assessed by recording spontaneous alternation behavior in a Y-maze. Y-maze was made of light grey plastic and consisted of 3 arms at 120°. Each arm was 6 cm wide and 36 cm long and had 12.5 cm high walls. Each mouse was placed in the Y maze and allowed to move freely during an 8-min session. The series of arm entries was recorded visually, and arm entry was considered to be completed when the hind paws of the mouse were completely placed in the arm. The maze was cleaned with 70% ethanol after each mouse. Alternation was defined as successive entries into the 3 arms on overlapping triplet sets (e.g., ABC, BCA). The percentage of alternation was calculated as the ratio of actual to possible alternations (defined as the total number of arm entries minus two). (4) Novel object recognition test: Mice were first exposed to two identical objects for 10 min, followed by a 2 h retention interval, and then put back into the same chamber where a novel object was introduced and monitored for 3 min. Exploration, defined as any type of physical contact with an object (whisking, sniffing, rearing on, or touching the object), was recorded and analyzed using the tracing software Viewer III. The preference score (%) for the novel object was calculated as (exploration time of the novel object/exploration time of both objects) × 100%.
Six-month-old WT, hTDP-43Tg/+, Grn−/−, TDP-43Tg/+Grn−/− male and female mice (8-12 mice/group) were subject to the following behavioral tests: (1) Hindlimb clasping test: Mice were suspended by the base of the tail, and their behaviors were recorded for 30 s. Hindlimb clasping was rated from 0 to 3 based on severity49. (2) Footprint test: The footprint apparatus consists of a runway (60 cm in length) with a dark box at the end. Mice are trained to walk straight to the dark box on the white paper before the trial. The hind paws of the mice are painted blue. Then the mice are made to walk on the white paper. The footprint pattern is analyzed for stride length. Stride length is determined by measuring the distance between each step on the same side of the body. For all behavioral analyses, experimenters were blind to the genotypes of the mice.
Tissue lysate preparation
Mice were perfused with cold PBS, and tissues were dissected and snap-frozen with liquid nitrogen and kept at −80 °C. On the day of the experiment, frozen tissues were thawed and homogenized on ice with bead homogenizer (Moni International) in ice-cold RIPA buffer (150 mM NaCl, 50 mM Tris-HCl [pH 8.0], 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS) with 1 mM PMSF, and 1x protease inhibitors (Roche). After centrifugation at 14,000 × g for 15 min at 4 °C, supernatants were collected for analysis. The insoluble pellets were washed with RIPA buffer and extracted in 2× v/w of Urea buffer (7 M Urea, 2 M Thiourea, 4% CHAPS, 30 mM Tris, pH 8.5). After sonication, samples were centrifuged at 200,000 g at 24 °C for 1 h, and the supernatant was collected as the urea-soluble fraction.
Immunofluorescence staining, image acquisition, and analysis
For mouse brain section staining, mice were perfused with cold PBS, and tissues were post-fixed with 4% paraformaldehyde (PFA). After dehydration in 30% sucrose buffer, tissues were embedded in the O.C.T compound. Brain sections with an 18-µm-thickness were cut with a cryotome. Antigen retrieval was performed by microwaving in citrate buffer (pH 6.0) for 15 min. Tissue sections were blocked and permeabilized with 0.1% saponin in Odyssey blocking buffer before incubating with primary antibodies overnight at 4 °C. The next day, sections were washed 4 times with PBS, followed by incubation with secondary fluorescent antibodies and Hoechst at room temperature for 2 h. The sections were treated with the Autofluorescence Quencher (TRUE Black) to quench the autofluorescence. The slides were then mounted using a mounting medium (Vector Laboratories). Images were acquired on a CSU-X spinning disc confocal microscope (Intelligent Imaging Innovations) with an HQ2 CCD camera (Photometrics) using 40x or 63x objectives. Lower magnification images were captured by 10x or 20x objectives on a Leica DMi8 inverted microscope.
For the quantitative analysis of GFAP, CD68, and IBA1 levels in the brain sections, the fluorescence intensity was measured using ImageJ after a threshold application. To quantify the CD68 levels in microglia, IBA1-positive cells were selected, and CD68 signals within microglia were measured using the region of interest (ROI) tool in ImageJ. The mean from the three sections was used to be representative of each mouse. Data from ≥5 brains in each genotype were used for quantification.
Splicing analysis of the TDP-43 targets using RT-qPCR
Total RNAs were extracted from 10-month-old mouse cortex samples using TRIzol (Invitrogen). One microgram of total RNA was reverse transcribed to cDNA using SuperScript III Reverse Transcriptase (Invitrogen). Quantitative PCR was performed on a LightCyler 480 (Roche Applied Science), and the mRNA levels were measured using efficiency-adjusted ΔΔ-CT. The transcripts analyzed were normalized to β-actin. The primers used to detect the inclusion of Sort1 exon 17b and Mapt exon 2 and 3 were from the previous study29. The primer sequences for detecting Sort1 exon 17b inclusion were: 5′-AACCCCACAAAGCAGGACT-3′ and 5′-CTGCTACGACTGTGACAAGC-3′. The primers for Mapt exons 2 and 3 inclusion detection were: 5′-CATGGCTTAAAAGAGTCTCCCC-3′ and 5′-CCTGCTTCTTCGGCTGTAAT-3′ (for exon 2 and 3 included transcripts) and 5′-TTAAAAGCCGAAGAAGCAGGC-3' and 5'-CTGGAGGAGTCTTAGGGCTG-3′ (for exon 2 and 3 skipped transcripts). The primers for β-actin were: 5′-ACGAGGCCCAGAGCAAGAG-3′ and 5′-TCTCCAAGTCGTCCCAGTTG-3′.
RNA-seq analysis
Total RNA was extracted from the brain cortex of 10-month-old mice using Trizol (Thermo Scientific). RNA quality was checked using Nanodrop, gel electrophoresis, and Agilent Fragment Analyzer. RNAseq libraries were generated by the Cornell TREx Facility using the NEBNext Ultra II Directional RNA Library Prep Kit (New England Biolabs) using 700 ng input total RNA per sample. At least 40 M reads (2x150nt PE) were generated on a NovaSeq (Illumina). Reads were trimmed to remove low-quality and adaptor sequences with TrimGalore (a wrapper for cutadapt and fastQC), requiring a minimum trimmed length of 50 nt. Reads that passed quality control were aligned to the reference genome (mouse GRCm38/mm10)50 using STAR51, using ‘--quantMode GeneCounts’ to output counts per gene. SARTools52 and DESeq253 were used to generate normalized counts and perform differential gene expression analysis. Differentially expressed genes (DEGs) were identified by false discovery rates (FDR) or adjusted p-value ≤ 0.05 using multiple testing with Benjamini-Hochberg correction.
Heatmaps of the DEGs were generated by using Heatmapper54. An enrichment analysis using the hallmark gene sets or gene ontology biology process (GOBP) was performed using Gene Set Enrichment Analysis (GSEA)55. The normalized counts of all the expressed genes were uploaded to GSEA, and a standard GSEA was run with recommended default settings (1000 gene set permutations). The FDR was estimated to control the false-positive finding of a given normalized enrichment score (NES) by comparing the tails of the observed and null distributions derived from 1000 gene set permutations. The gene sets with absolute normalized enrichment score (NES) > 1.0, nominal p-value (NOM p-val) < 0.05, and false discovery rate q value (FDR q-val) <0.1 were considered as significantly enriched. The subset of genes that contributes most to the enrichment result was defined as the core enrichment genes.
Statistics analysis
Statistical analysis was performed using GraphPad Prism 10. Data are presented as mean ± SEM. Statistical significance was assessed by unpaired two-tailed Student’s t-test (for two-group comparison) and one-way ANOVA tests (for multiple comparisons), followed by Bonferroni’s multiple comparisons post hoc tests. P values less than or equal to 0.05 were considered statistically significant. The details about the sample sizes and number of replicates in each experiment are given in the respective sections of the methods and figure legends. *p < 0.05; **p < 0.01; ***p < 0.001.
Data availability
The data supporting the findings of this study are included in the supplemental material. A complete list of genes identified in RNA-seq is provided in Supplementary Data 1. Uncropped western blot images are provided in Supplementary Fig.2. Additional data are available from the corresponding author on request.
References
Ratti, A. & Buratti, E. Physiological functions and pathobiology of TDP-43 and FUS/TLS proteins. J. Neurochem. 138(Suppl 1), 95–111 (2016).
Birsa, N., Bentham, M. P. & Fratta, P. Cytoplasmic functions of TDP-43 and FUS and their role in ALS. Semin. cell developmental Biol. 99, 193–201 (2020).
Prasad, A., Bharathi, V., Sivalingam, V., Girdhar, A. & Patel, B. K. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 12, 25 (2019).
Suk, T. R. & Rousseaux, M. W. C. The role of TDP-43 mislocalization in amyotrophic lateral sclerosis. Mol. Neurodegener. 15, 45 (2020).
Neumann, M. et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Sci. (N.Y., N.Y.) 314, 130–133 (2006).
Foulds, P. et al. TDP-43 protein in plasma may index TDP-43 brain pathology in Alzheimer’s disease and frontotemporal lobar degeneration. Acta Neuropathol. 116, 141–146 (2008).
Van Deerlin, V. M. et al. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat. Genet 42, 234–239 (2010).
Uryu, K. et al. Concomitant TAR-DNA-binding protein 43 pathology is present in Alzheimer disease and corticobasal degeneration but not in other tauopathies. J. Neuropathol. Exp. Neurol. 67, 555–564 (2008).
Higashi, S. et al. Concurrence of TDP-43, tau and alpha-synuclein pathology in brains of Alzheimer’s disease and dementia with Lewy bodies. Brain Res 1184, 284–294 (2007).
Amador-Ortiz, C. et al. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann. Neurol. 61, 435–445 (2007).
Schwab, C., Arai, T., Hasegawa, M., Yu, S. & McGeer, P. L. Colocalization of transactivation-responsive DNA-binding protein 43 and huntingtin in inclusions of Huntington disease. J. Neuropathol. Exp. Neurol. 67, 1159–1165 (2008).
Nelson, P. T. et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain 142, 1503–1527 (2019).
Nag, S. & Schneider, J. A. Limbic-predominant age-related TDP43 encephalopathy (LATE) neuropathological change in neurodegenerative diseases. Nat. Rev. Neurol. 19, 525–541 (2023).
Gass, J. et al. Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum. Mol. Genet 15, 2988–3001 (2006).
Rademakers, R. & Hutton, M. The genetics of frontotemporal lobar degeneration. Curr. Neurol. Neurosci. Rep. 7, 434–442 (2007).
Mackenzie, I. R. & Rademakers, R. The molecular genetics and neuropathology of frontotemporal lobar degeneration: recent developments. Neurogenetics 8, 237–248 (2007).
Nelson, P. T. et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain: a J. Neurol. 142, 1503–1527 (2019).
Hokkanen, S. R. K. & Kero, M. Putative risk alleles for LATE-NC with hippocampal sclerosis in population-representative autopsy cohorts. 30, 364–372 https://doi.org/10.1111/bpa.12773 (2020).
Nicholson, A. M., Gass, J., Petrucelli, L. & Rademakers, R. Progranulin axis and recent developments in frontotemporal lobar degeneration. Alzheimers Res Ther. 4, 4 (2012).
Bateman, A. & Bennett, H. P. The granulin gene family: from cancer to dementia. Bioessays 31, 1245–1254 (2009).
Cenik, B., Sephton, C. F., Kutluk Cenik, B., Herz, J. & Yu, G. Progranulin: a proteolytically processed protein at the crossroads of inflammation and neurodegeneration. J. Biol. Chem. 287, 32298–32306 (2012).
Kao, A. W., McKay, A., Singh, P. P., Brunet, A. & Huang, E. J. Progranulin, lysosomal regulation and neurodegenerative disease. Nat. Rev. Neurosci. 18, 325–333 (2017).
Paushter, D. H., Du, H., Feng, T. & Hu, F. The lysosomal function of progranulin, a guardian against neurodegeneration. Acta Neuropathol. 136, 1–17 (2018).
Simon, M. J., Logan, T., DeVos, S. L. & Di Paolo, G. Lysosomal functions of progranulin and implications for treatment of frontotemporal dementia. Trends Cell Biol. https://doi.org/10.1016/j.tcb.2022.09.006 (2022).
Root, J., Merino, P., Nuckols, A., Johnson, M. & Kukar, T. Lysosome dysfunction as a cause of neurodegenerative diseases: Lessons from frontotemporal dementia and amyotrophic lateral sclerosis. Neurobiol. Dis. 154, 105360 (2021).
Tejwani, L. et al. Reduction of nemo-like kinase increases lysosome biogenesis and ameliorates TDP-43-related neurodegeneration. J. Clin. Invest. 133 https://doi.org/10.1172/JCI138207 (2023).
Bright, F., Chan, G., van Hummel, A., Ittner, L. M. & Ke, Y. D. TDP-43 and Inflammation: Implications for Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Int. J. Mol. Sci. 22 https://doi.org/10.3390/ijms22157781 (2021).
Petkau, T. L. et al. Synaptic dysfunction in progranulin-deficient mice. Neurobiol. Dis. 45, 711–722 (2012).
White, M. A. et al. TDP-43 gains function due to perturbed autoregulation in a Tardbp knock-in mouse model of ALS-FTD. Nat. Neurosci. 21, 552–563 (2018).
Wils, H. et al. TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc. Natl Acad. Sci. USA 107, 3858–3863 (2010).
Sreedharan, J. et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. (N.Y., N. Y.) 319, 1668–1672 (2008).
Rutherford, N. J. et al. Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet 4, e1000193 (2008).
Buratti, E. Functional Significance of TDP-43 Mutations in Disease. Adv. Genet 91, 1–53 (2015).
Lin, Z. et al. MRI-guided histology of TDP-43 knock-in mice implicates parvalbumin interneuron loss, impaired neurogenesis and aberrant neurodevelopment in amyotrophic lateral sclerosis-frontotemporal dementia. Brain Commun. 3, fcab114 (2021).
Root, J. et al. Granulins rescue inflammation, lysosome dysfunction, lipofuscin, and neuropathology in a mouse model of progranulin deficiency. Cell Rep. 43, 114985 (2024).
Logan, T. et al. Rescue of a lysosomal storage disorder caused by Grn loss of function with a brain penetrant progranulin biologic. Cell 184, 4651–4668.e4625 (2021).
Kayasuga, Y. et al. Alteration of behavioural phenotype in mice by targeted disruption of the progranulin gene. Behav. Brain Res 185, 110–118 (2007).
Yin, F. et al. Behavioral deficits and progressive neuropathology in progranulin-deficient mice: a mouse model of frontotemporal dementia. FASEB J. 24, 4639–4647 (2010).
Scotter, E. L., Chen, H. J. & Shaw, C. E. TDP-43 Proteinopathy and ALS: Insights into Disease Mechanisms and Therapeutic Targets. Neurotherapeutics 12, 352–363 (2015).
Wu, Y. et al. Microglial lysosome dysfunction contributes to white matter pathology and TDP-43 proteinopathy in GRN-associated FTD. Cell Rep. 36, 109581 (2021).
Sung, W. et al. Progranulin haploinsufficiency mediates cytoplasmic TDP-43 aggregation with lysosomal abnormalities in human microglia. J. Neuroinflamm. 21, 47 (2024).
Zhang, J. et al. Neurotoxic microglia promote TDP-43 proteinopathy in progranulin deficiency. Nature 588, 459–465 (2020).
Zhang, T. et al. Progranulin deficiency results in sex-dependent alterations in microglia in response to demyelination. Acta Neuropathol. 146, 97–119 (2023).
Yang, C. et al. Low-level overexpression of wild type TDP-43 causes late-onset, progressive neurodegeneration and paralysis in mice. PLoS One 17, e0255710 (2022).
Ho, W. Y. et al. TDP-43 mediates SREBF2-regulated gene expression required for oligodendrocyte myelination. J Cell Biol 220 https://doi.org/10.1083/jcb.201910213 (2021).
Zhu, L. et al. Pathologic TDP-43 downregulates myelin gene expression in the monkey brain. Brain Pathol. 34, e13277 (2024).
Filiano, A. J. et al. Dissociation of frontotemporal dementia-related deficits and neuroinflammation in progranulin haploinsufficient mice. J. Neurosci. 33, 5352–5361 (2013).
Yin, F. et al. Exaggerated inflammation, impaired host defense, and neuropathology in progranulin-deficient mice. J. Exp. Med 207, 117–128 (2010).
Miedel, C. J., Patton, J. M., Miedel, A. N., Miedel, E. S. & Levenson, J. M. Assessment of Spontaneous Alternation, Novel Object Recognition and Limb Clasping in Transgenic Mouse Models of Amyloid-beta and Tau Neuropathology. J. Vis. Exp. https://doi.org/10.3791/55523 (2017).
Zerbino, D. R. et al. Ensembl 2018. Nucleic Acids Res 46, D754–D761 (2018).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Varet, H., Brillet-Gueguen, L., Coppee, J. Y. & Dillies, M. A. SARTools: A DESeq2- and EdgeR-Based R Pipeline for Comprehensive Differential Analysis of RNA-Seq Data. PLoS One 11, e0157022 (2016).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Babicki, S. et al. Heatmapper: web-enabled heat mapping for all. Nucleic Acids Res 44, W147–153 (2016).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Acknowledgements
This work is supported by NINDS/NIA (R01 NS095954 & R21AG078741) to F.H., and the Weill Institute Fleming Postdoctoral Fellowship to C.Y. We thank Sara Down and Dr. Jeff Pleiss for their help with RNA splicing analysis. We thank Ms. Xiaochun Wu for her technical assistance and Dr. Chris Schaffer, Dr. Nozomi Nishimura, and Dr. Matthew David Isaacson for their assistance with behavioral tests.
Author information
Authors and Affiliations
Contributions
C.Y. performed the experiments, analyzed the data, and drafted the manuscript. F.T. helped with PGRN western blot and immunostaining analysis. F.H. supervised the project and edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, C., Feng, T. & Hu, F. Progranulin deficiency does not exacerbate TDP-43 pathology in TDP-43 transgenic mouse models. npj Dement. 1, 16 (2025). https://doi.org/10.1038/s44400-025-00020-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s44400-025-00020-4
