
Abstract
Preeclampsia (PE) is a pregnancy-specific complication and there remains no effective treatment. Given the limitations on medication use during pregnancy, exploring natural, safe, and effective drugs for PE is worthwhile. We investigate the causal relationship between ferroptosis, inflammation, and PE, and determine the protective effects of quercetin (QCT), a representative compound that is classified as a flavanol, against endothelial dysfunction. Then, the target of QCT is predicted and verified. The prophylactic addition of a low dose of QCT rescues endothelial dysfunction, aiding in endothelial repair. Furthermore, QCT alleviates PE-like maternal manifestations and endothelial dysfunction in the placenta of the selective reduced uteroplacental perfusion (sRUPP) rat model through binding to the epidermal growth factor receptor (EGFR). The potential applications of QCT are expanded, offering the possibility of further development as a safe and effective preventive molecule for PE.

Similar content being viewed by others
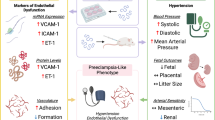


Introduction
Preeclampsia (PE) is a complication unique to pregnancy, marked by new-onset hypertension (systolic ≥ 140 mmHg and/or diastolic ≥ 90 mmHg) after 20 weeks of gestation. It’s accompanied by proteinuria (> 300 mg/24 h or positive random urine protein) or various degrees of end-organ dysfunction, significantly impacting both maternal and child health1. Statistics show that globally, PE accounts for at least 46,000 maternal and 500,000 perinatal deaths annually, with its incidence and mortality rates increasing each year2. As previously reported, women with a history of PE are at an increased risk of long-term coronary heart disease, heart failure, stroke, and cardiovascular disease-related death later in life3, and their offspring are also at an elevated risk of hypertension and cardiovascular disease4. At present, the only effective treatment for PE is the termination of the pregnancy. Additionally, the exact etiology and pathogenesis of PE remain somewhat unclear, but the “two-stage” theory is widely attached by numerous scholars as the pathogenesis of PE5. In the first stage, various risk factors lead to placental ischemia/hypoxia, releasing a diverse range of placental factors. In the second stage, these factors enter the maternal blood circulation and promote systemic inflammatory responses and endothelial dysfunction, resulting in a series of clinical manifestations. Notably, inflammation and endothelial dysfunction in the placenta have been proposed as two essential hallmarks of PE6,7.
Ferroptosis is a form of cell death dependent on reactive oxygen species (ROS) and is characterized by two main biochemical features: lipid peroxidation and iron accumulation8. Recent evidence has increasingly linked ferroptosis to several diseases, such as liver ischemia/reperfusion injury, diabetes, acute kidney injury, and stroke, and in the context of the female reproductive system, our previous research has established a link between ferroptosis and PE9,10. Moreover, as reported in a previous study11,12, ferroptotic cell death is capable of releasing its intracellular components as pro-inflammatory damage-associated molecular patterns (DAMPs), and the releasing and accumulation of DAMPs can trigger tissue inflammation, resulting in the development of various inflammatory diseases (e.g., PE12). Therefore, it is reasonable to hypothesize that ferroptosis may be involved in the development of PE as an upstream of inflammation. Considering these findings, targeting ferroptosis and/or inflammation could be effective in preventing or treating PE. Given the constraints on medication use during pregnancy, it is valuable to explore natural, safe, and effective dietary-derived drugs that have anti-ferroptosis or combined anti-ferroptosis and anti-inflammatory properties.
Over the past few years, substantial advancements have been made in researching dietary phytochemicals, such as natural vitamins and food polyphenols. Among these polyphenols, flavonoids stand out. Quercetin (QCT) is the most well-known, widely distributed, abundantly found, and extensively studied compound in nature given the classification of flavonols. It is found in a range of foods, including peppers, tomatoes, onions, broccoli, berries, cherries, apples, and pears, as well as teas and red wine13. In addition, QCT exhibits a variety of biological effects, including but not limited to antioxidant, anti-inflammatory, anti-cancer, and anti-ischemic, etc14. Furthermore, it has been designated as a ferroptosis inhibitor for its ability to prevent typical morphological changes in ferroptotic cells15. However a high concentration of QCT could also induce ferroptosis in several types of human cancer cell lines16. Additionally, studies have verified QCT’s safety, showing no toxic effects even with relatively high daily consumption during pregnancy17,18. Taking appropriate doses of QCT as a dietary supplement during pregnancy has been suggested to confer health benefits19. Moreover, QCT has shown beneficial effects on embryonic, fetal, and placental development, without causing teratogenic or abortifacient effects20,21. Thus, QCT emerges as a potentially safe and effective drug for protecting against PE. However, it is worth noting that, at present, there is a lack of relevant research on this subject.
In this study, we first investigated the effects of ferroptosis on inflammation and endothelial dysfunction. Subsequently, we shifted our attention to QCT, aiming to understand if/how QCT might inhibit ferroptosis and/or inflammation, with the ultimate goal of alleviating endothelial dysfunction to protect against PE.
Results
Ferroptosis, inflammatory imbalance, and endothelial dysfunction are present in patients with PE
To assess the changes in ferroptosis-related markers in the placenta and investigate whether there is an imbalance in inflammatory and angiogenic factors in the plasma, we collected placental and plasma samples from 34 normal pregnant women and 72 patients with preeclampsia (PE), respectively (Fig. 1A). Western blot analysis revealed changes in ferroptosis-related proteins in the placenta of PE patients, with increased ACSL4 expression and decreased GPX4 and SLC7A11 expressions (Fig. 1B–B3). Meanwhile, lipid peroxidation and iron colorimetric assays indicated elevated MDA and Fe2+ concentrations, and reduced GSH concentration and GSH-Px activity in the placenta of PE patients (Fig. 1C–F). In addition, clinical correlation analysis showed that mean arterial pressure (MAP) positively correlated with MDA (r = 0.825, P < 0.001) and Fe2+ (r = 0.839, P < 0.001) concentrations, and negatively correlated with GSH concentration (r = −0.828, P < 0.001) and GSH-Px activity (r = −0.767, P < 0.001) (Fig. 1G–J). Furthermore, we also observed elevated levels of pro-inflammatory factors IL-6 and TNF-α and reduced levels of anti-inflammatory factor IL-10 in the plasma of PE patients (Fig. 1K–M). Then, the ELISA results for angiogenesis-related factors indicated an increase in anti-angiogenic factor sFlt-1 and a decrease in pro-angiogenic factor PLGF in PE (Fig. 1N, O). These findings suggest the presence of ferroptosis, inflammatory imbalance, and endothelial dysfunction in PE patients.

A Schematic representation of tissue sources and detection indicators. B–B3 Representative Western blot images of ACSL4, GPX4, and SLC7A11 (B) and bar charts of quantitative analysis of relative expressions of ACSL4 (B1) (n = 15 per group), GPX4 (B2) (n = 14 per group) and SLC7A11 (B3) (n = 15 per group). C–J Detection of MDA concentration (C), GSH concentration (D), GSH-Px activity (E), and Fe2+ concentration (F) and correlation analysis was implemented between mean arterial pressure (MAP) and MDA concentration (G), GSH concentration (H), GSH-Px activity (I) and Fe2+ concentration (J) in the placenta tissues of patients, respectively (n = 34 per group). K–M The levels of IL-6 (K), IL-10 (L), and TNF-α (M) in the plasma of control and PE patients (n = 72 per group). N–O The levels of sFlt-1 (N) and PLGF (O) in the plasma of control and PE patients (n = 72 per group). The error bars in (B1–B3) represent means ± SD, in (C–F, K–O) represent the median and 25–75% quartiles. *P < 0.05; **P < 0.01; ***P < 0.001.
Ferroptosis inducer Erastin induces PE-like phenotype and is accompanied by inflammatory imbalance and endothelial dysfunction in rats
To explore the causal relationship between ferroptosis, inflammation, endothelial dysfunction, and PE, we administered Erastin intraperitoneally to pregnant rats. We then monitored their blood pressure and collected various samples (kidney, fetus, urine, placenta, and plasma) for detailed analysis (Fig. 2A). The results showed that Erastin-induced rats exhibited increased blood pressure, with significant elevations in both systolic (SBP) and diastolic blood pressure (DBP) on GD 17.5 (Fig. 2B–B2). Additionally, Erastin markedly increased 24-h proteinuria levels (Fig. 2C). HE and PAS staining revealed morphological damage in the glomerulus caused by Erastin, such as a narrower Bowman’s capsule (Supplementary Fig. 1A, B). Subsequently, Erastin-induced pregnant rats exhibited a lower embryo survival rate, fetal growth restriction (FGR), and reductions in placental diameter and weight compared to the Sham group (Fig. 2D–F and Supplementary Fig. 1C–G). Western blot analysis of ferroptosis markers (ACSL4, SLC7A11, and GPX4) and lipid peroxidation assays (measuring MDA, GSH, and GSH-Px) collectively demonstrate that Erastin induces ferroptosis in rat placentas (Fig. 2G–J). These data strongly suggest that induced ferroptosis can result in PE-like manifestations in pregnant rats.

A Schematic illustration of the establishment of an Erastin-induced pregnant rat model. B–B2 The systolic and diastolic pressure from Sham and Erastin groups (B), the systolic blood pressure (B1), and the diastolic blood pressure (B2) on GD17.5 (n = 8 in Sham, n = 9 in Erastin). C The 24-h proteinuria from Sham and Erastin groups (n = 7 in Sham, n = 5 in Erastin). D–F Placental diameter (D) (n = 40 in Sham, n = 63 in Erastin), HE staining of placental sections (E), and placental weight (F) (n = 40 in Sham, n = 63 in Erastin). G–G3 Representative Western blot images of ACSL4, GPX4, and SLC7A11 (G) and bar charts of quantitative analysis of relative expressions of ACSL4 (G1) (n = 18 per group), GPX4 (G2) (n = 12 per group) and SLC7A11 (G3) (n = 18 per group). H–J Detection of MDA concentration (H), GSH concentration (I), and GSH-Px activity (J) in the placenta of rats (n = 5 in Sham, n = 6 in Erastin). K–M The levels of IL-6 (K), IL-10 (L), and TNF-α (M) in the plasma of rats (n = 8 in Sham, n = 6 in Erastin). N The levels of sFlt-1 in plasma of rats (n = 8 in Sham, n = 6 in Erastin). O–P Representative images of Caveolin 1 immunofluorescence staining of labyrinth zone of Sham and Erastin rats (O), and the bar charts of Caveolin 1 (+) vessel density (P) (n = 3 per group). Scale bars = 100 μm in (O). The error bars in (B1–D, F, and H–N) represent the median, and 25–75% quartiles, in (G1–G3, P) represent means ± SD. *P < 0.05; **P < 0.01; ***P < 0.001.
Next, the detection of inflammatory, as well as angiogenic factors in plasma was performed. The results of ELISA manifested that Erastin induced increased levels of IL-6 and TNF-α, and decreased IL-10, indicating an inflammatory imbalance (Fig. 2K–M). Additionally, ELISA identified an angiogenic imbalance, evidenced by elevated sFlt-1 levels (Fig. 2N). Furthermore, immunofluorescence of Caveolin 1 (Caveolin 1-positive vessels in the placental labyrinth assess placental vascularization and respond to placental vascular endothelial damage22,23) on the cross-sections of placenta displayed that Erastin led to significantly reduced vascular density, indicated that Erastin could result in endothelial damage (Fig. 2O, P).
QCT inhibits ferroptosis, inflammation, and endothelial dysfunction in vitro
To investigate whether QCT, a small-molecular drug with both anti-inflammatory and anti-ferroptosis properties, can protect against Erastin-induced ferroptosis and associated disruptions in cell viability. Cells were exposed to 10–80 μM Erastin and 1-5 μM QCT at various time points. The results showed that Erastin had time- and dose-dependent effects on cell viability (Fig. 3A). After 24 h, 30 μM Erastin led to about a 50% reduction in cell viability, whereas 1 μM QCT at 48 h had a negligible effect on cell viability (Fig. 3A, B). In subsequent rescue experiments, we observed that medium (3 μM) to high (5 μM) concentrations of QCT acted as a therapeutic agent against Erastin-induced reduction in cell viability, whereas low (1 μM) to medium concentrations of QCT exhibited a preventive effect (Fig. 3C, D). Therefore, we opted for a concentration and duration combination of 1 μM QCT for 48 h (for prevention) and 30 μM Erastin for 24 h for subsequent experiments.

A CCK-8 shows the effect of Erastin on cell viability. B CCK-8 shows the effect of QCT on cell viability. C CCK-8 shows the effect of Erastin 24 h and QCT 48 h on cell viability (n = 3 per group). D CCK-8 shows the effect of QCT 24 h, Erastin, and QCT 24 h on cell viability (n = 3 per group). E, F Representative images of PI staining of HUVEC from the Control, Erastin, and Erastin + QCT groups (E) and the bar charts of the ratio of PI-positive cells (F) (n = 3 per group). G Representative western blot images and bar charts of quantitative analysis of relative expressions of ACSL4 (n = 4 per group), GPX4 (n = 5 per group), and SLC7A11 (n = 3 per group) from the aforementioned three groups. H The relative mRNA expression of IL-6, IL-10, and TNF-α in HUVEC from the aforementioned three groups (n = 3 per group). I–J Representative fluorescent detection of DCFH-DA from the aforementioned three groups (I) and the bar charts of ROS mean fluorescent intensity (J) (n = 3 per group). K–L Representative fluorescent detection of JC-1 from the aforementioned three groups (E) and the bar charts of the ratio of red/green fluorescent intensity (F) (n = 3 per group). Scale bars = 100 μm in (E), (I), and (K). The error bars in the figure represent means ± SD. *P < 0.05; **P < 0.01; ***P < 0.001.
First, the PI staining was performed to evaluate the cell death, and the results displayed that QCT effectively reversed cell death induced by Erastin (Fig. 3E, F). Then, the Western blot was used to confirm whether this cell death was ferroptosis, and the result showed that QCT effectively reversed the changes in ferroptotic proteins (ACSL4, GPX4, and SLC7A11) induced by Erastin (Fig. 3G). The qPCR analysis showed that QCT effectively ameliorated the changes in inflammatory indicators (IL-6, TNF-α, and IL-10) induced by Erastin (Fig. 3H). In addition, ROS production and changes in mitochondrial membrane potential are associated with ferroptosis, so we examined the levels of intracellular ROS, as well as mitochondrial membrane potential changes. The fluorescent detection of DCFH-DA revealed the reversal of Erastin-induced increases in intracellular ROS levels after pre-processing with QCT (Fig. 3I, J) and JC-1 staining demonstrated a clear recovery of mitochondrial membrane potential alterations induced by Erastin following QCT pre-treatment (Fig. 3K, L).
Furthermore, the barrier function of endothelial cells and damage repair were assessed. The FITC-dextran assay demonstrated that the addition of QCT significantly attenuated the increased endothelial cell permeability caused by Erastin (Fig. 4A–A2). Then, the immunofluorescence of tight junction protein ZO-1 exhibited that QCT mitigated Erastin’s disruption of cell junctions (Fig. 4B, C). Moreover, QCT effectively facilitated the repair of endothelial damage (Fig. 4D–D2). Collectively, these findings suggest that QCT can counteract Erastin-induced ferroptosis, inflammation, and endothelial dysfunction and promote the repair of endothelial damage.

A–A2 The FITC-dextran assay of HUVEC. Schematic illustration of the FITC-dextran assay (A), a standard curve of FITC (A1), and the bar charts of 1 h FITC-dextran permeability (A2) from the Control, Erastin, and Erastin + QCT groups (n = 4 per group). B, C Representative images of ZO-1 immunofluorescence staining of HUVEC from the aforementioned three groups (B), and the bar charts of ZO-1 mean fluorescent intensity (C) (n = 3 per group). D–D2 Representative images of the scratch experiment of HUVEC (D) and the bar charts showing 24 h (D1) (n = 3 per group) and 48 h (D2) (n = 3 per group) relative wound closure from the aforementioned three groups. Scale bars = 100 μm in (B) and (D). The error bars in the figure represent means ± SD. **P < 0.01; ***P < 0.001.
QCT regulates excess ferroptosis-induced endothelial intracellular ROS production and endothelial damage by binding to EGFR
To identify the potential targets of QCT, the online website “Swiss Target Prediction” was used to perform the prediction of targets of QCT based on its chemical structure, and a total of 100 targets were obtained. The classification of predicted targets showed that 28.0% were kinases, 26.0% enzymes, and 11.0% lyases (Supplementary Fig. 2). Due to the extensive range of QCT targets, we focused on identifying key targets with potentially crucial roles or central significance. The Matthews correlation coefficient (MCC) algorithm of the Cytohubba plug-in in Cytoscape software was employed to pinpoint QCT’s top target proteins. The top three target proteins identified were SRC, AKT1, and EGFR, based on their high core content (Fig. 5A). Molecular docking of QCT was then performed with each of these three proteins. Among the three proteins, EGFR exhibited the highest total score and C-score. Notably, QCT formed a bond within the docking pocket of EGFR, in contrast to its external bonding with SRC and AKT1. This indicates that EGFR is the most probable target of QCT (Fig. 5B–B3). Additionally, Bioinformatics analysis of the three largest sample size datasets in the Gene Expression Omnibus (GEO) database showed that only EGFR, among SRC, AKT1, and EGFR, was differentially expressed in the placenta of PE patients (Fig. 5C–E). Further molecular docking showed that QCT formed hydrogen bonds with amino acid residues in the EGFR pocket at six sites: Aspartic acid (ASP)-800, Cystine (CYS)-797, Methionine (MET)-793, Glutamine (GLN)-791, Threonine (THR)-790, and THR-854 (Fig. 5F). The docking process was repeated four times, yielding 35 individual values. The docking affinity was calculated as −6.3 ± 0.75 kcal/mol, with a pKi of −1.36 ± 0.55 μM. Furthermore, we performed the cellular thermal shift assay (CETSA) at a concentration of 60 µM of QCT by its predicted Ki value (60.351 µM), and the results indicated that EGFR expression decreased with rising temperature in both control and QCT groups, with lower expression observed in the control group (Fig. 5G, H). Additionally, concentrations of QCT above 60 µM can be used for its antioxidant and anti-aging research24,25, and our CCK-8 results showed that 60 µM QCT had no significant effect on cell viability (Supplementary Fig. 3). Next, we performed CETSA with 1 μM QCT. The results showed that EGFR levels decreased with increasing temperature between 45 °C and 57 °C in both the control and QCT groups, with a statistically significant difference at 57 °C between the two groups (Supplementary Fig. 4). This suggests that 1 μM QCT also binds to EGFR.

A PPI network of top ten targets of QCT. B–B3 The score of molecular docking of QCT with SRC, AKT1, and EGFR (B), and the representative images of molecular docking of QCT with SRC (B1), AKT1 (B2), and EGFR (B3). C–E Volcano plot of GSE114691 (C), GSE25906 (D), and GSE10588 (E) of differentially expressed genes (P value < 0.01) in placenta between control and PE patients. F Representative images of molecular docking of QCT with EGFR. G, H Representative Western blot images of cellular thermal shift assay (G) and the line graph of quantitative analysis of relative expressions of EGFR from the Control and QCT groups (H) (n = 6 per group of each temperature). The error bars in the figure represent means ± SD. ns no significance; **P < 0.01.
Subsequently, the expression of EGFR in HUVECs was down-regulated to confirm that QCT’s biological effects are mediated through binding to EGFR (Supplementary Fig. 5). Fluorescent probe results showed that QCT failed to reduce intracellular ROS production or enhance mitochondrial membrane potential, but still diminished cell death under conditions of EGFR down-regulation (Fig. 6A–C and Supplementary Fig. 6A–C). As for endothelial function assessment, the immunofluorescence of ZO-1 and wound healing assay results displayed that following EGFR down-regulation, QCT could not alleviate Erastin-induced disruption of cell junctions or promote endothelial damage repair (Fig. 6D–F and Supplementary Fig. 6D, E). The above results clearly demonstrated that QCT’s actions are indeed mediated through its binding to EGFR.

A–F The bar charts of the ratio of PI-positive cells (A), ROS mean fluorescent intensity (B), the ratio of red/green fluorescent intensity (C), ZO-1 mean fluorescent intensity (D), 24 h (E) and 48 h (F) relative wound closure from the si-NC, si-NC + Erastin, si-NC + Erastin + QCT, si-EGFR, siEGFR + Erastin and si-EGFR + Erastin + QCT groups (n = 6 per group). G–G5 Representative western blot images of EGFR, pEGFR, AKT, pAKT, ERK, pERK and GPX4 (G) and bar charts of quantitative analysis of relative expressions of EGFR (G1), pEGFR/EGFR (G2) from the si-NC, si-NC + Erastin and si-NC + Erastin + QCT groups (n = 3 per group), and relative expressions of pAKT/AKT (G3), pERK/ERK (G4) and GPX4 (G5) from the si-NC, si-NC + Erastin, si-NC + Erastin + QCT, si-EGFR, siEGFR + Erastin and si-EGFR + Erastin + QCT groups (n = 3 per group). The error bars in the figure represent means ± SD. ns no significance; *P < 0.05; **P < 0.01; ***P < 0.001; #P < 0.05; ##P < 0.01; ###P < 0.001.
In general, AKT was regulated by EGFR and/or pEGFR. Thus, we also determined the changes in EGFR/AKT signaling. The Western blot results indicated that QCT markedly reduced the elevated EGFR, as well as the ratio of pEGFR/EGFR caused by Erastin. Then, QCT could suppress the Erastin-induced elevation in the phosphorylated AKT (pAKT) to AKT ratio and attenuated the reduction in GPX4 expression, which was not exerted by QCT after down-regulation of EGFR. However, neither Erastin nor QCT has an effect on the ratio of phosphorylated ERK (pERK) to ERK (Fig. 6G–G5).
QCT mitigates PE in sRUPP rats by reducing ferroptosis, alleviating inflammation, and preventing endothelial dysfunction
Having shown that QCT could improve endothelial dysfunction caused by Erastin in vitro, our next question was whether QCT could exert a protective role in a PE-like animal model. Therefore, we chose the sRUPP rat model, a well-established animal model of PE that directly mimics insufficient placental perfusion, a primary characteristic of PE26,27. QCT was administered prophylactically intraperitoneally to pregnant rats, and ferrostatin-1 (Fer-1), a selective ferroptosis inhibitor, served as a positive control (Fig. 7A). The results indicated that both Fer-1 and QCT significantly reduced the elevated blood pressure observed in the sRUPP model (Fig. 7B–B2). Meanwhile, Fer-1 and QCT effectively reversed the increased 24-h proteinuria and morphological damage in the glomerulus (Fig. 7C–E). However, neither Fer-1 nor QCT could reverse the reduced embryo survival rate, FGR (i.e., reduced crown-rump length and fetal weight), and placental abnormalities (e.g., decreased placental diameter and weight) (Supplementary Fig. 7).

A The schematic illustration that ferroptosis is blocked in a pregnant rat model of sRUPP. B–B2 The systolic and diastolic pressure from Sham, sRUPP, sRUPP + Fer-1 and sRUPP + QCT groups (B), the systolic blood pressure (B1) and the diastolic blood pressure (B2) on GD17.5 (n = 8 in Sham, n = 10 in sRUPP, n = 6 in sRUPP + Fer-1, n = 8 in sRUPP + QCT). C The 24-h proteinuria from the aforementioned four groups (n = 7 in Sham, n = 7 in sRUPP, n = 6 in sRUPP + Fer-1, n = 8 in sRUPP + QCT). D, E Representative images of glomeruli stained with HE and PAS (D) and the open capillary areas from the aforementioned four groups (E) (n = 74 in Sham, n = 52 in sRUPP, n = 24 in sRUPP + Fer-1, n = 51 in sRUPP + QCT). Scale bars = 100 μm in (D). The error bars in (B) represent means ± SD, in (B1–C, E) represent the median and 25–75% quartiles. **P < 0.01; ***P < 0.001.
Western blot analysis demonstrated that both Fer-1 and QCT could reverse the increase in ACSL4 and the decrease in GPX4 and SLC7A11 (Fig. 8A–A3). Lipid peroxidation results indicated that QCT reversed the elevated MDA levels and restored the decreased GSH concentration and GSH-Px activity in the placenta. Interestingly, Fer-1 did not show improvements in GSH-Px activity (Fig. 8B–D). Additionally, the ELISA results demonstrated that both Fer-1 and QCT significantly alleviated the inflammatory and angiogenic imbalance. This was evident from the reversal of increased IL-6 and TNF-α levels, decreased IL-10 levels, and correction of elevated sFlt-1 (Fig. 8E–H). The immunofluorescence results of Caveolin 1 on placental cross-sections revealed a restoration of vascular density in the presence of QCT in the sRUPP rat model, a result not observed with Fer-1 (Fig. 8I, J). Finally, immunohistochemistry analysis of EGFR and pEGFR in placental tissues showed a marked increase in both expression and phosphorylation of EGFR in the sRUPP group compared to the Sham group (Supplementary Fig. 8). This increase was effectively reversed by QCT, but not by Fer-1, indicating that QCT’s effects are mediated through the inhibition of EGFR expression and phosphorylation.

A–A3 Representative Western blot images of ACSL4, GPX4, and SLC7A11 (A) and bar charts of quantitative analysis of relative expressions of ACSL4 (A1) (n = 8 per group), GPX4 (A2) (n = 9 per group) and SLC7A11 (A3) (n = 11 per group) from the Sham, sRUPP, sRUPP + Fer-1 and sRUPP + QCT groups. B–D The levels of MDA concentration (B), GSH concentration (C), and GSH-Px activity (D) in the placenta of the aforementioned four groups (n = 7 in Sham, n = 7 in sRUPP, n = 5 in sRUPP + Fer-1, n = 5 in sRUPP + QCT). E–G Detection of IL-6 (E) (n = 7 in Sham, n = 9 in sRUPP, n = 6 in sRUPP + Fer-1, n = 8 in sRUPP + QCT), IL-10 (F) (n = 7 in Sham, n = 10 in sRUPP, n = 6 in sRUPP + Fer-1, n = 8 in sRUPP + QCT), and TNF-α (G) (n = 7 in Sham, n = 9 in sRUPP, n = 6 in sRUPP + Fer-1, n = 8 in sRUPP + QCT) in plasma of the aforementioned four groups. H The levels of sFlt-1 in plasma of the aforementioned four groups (n = 8 in Sham, n = 9 in sRUPP, n = 6 in sRUPP + Fer-1, n = 8 in sRUPP + QCT). I–J Representative images of Caveolin 1 immunofluorescence staining of labyrinth zone of the aforementioned four groups rats (I), and the bar charts of Caveolin 1 (+) vessel density (J) (n = 3 per group). Scale bars = 100 μm in (J). The error bars in (A1–A3, J) represent means ± SD, in (B–H) represent median and 25–75% quartiles. ns no significance; *P < 0.05; **P < 0.01; ***P < 0.001.
Discussion
PE stands as a prominent cause of maternal and perinatal morbidity and mortality, with no effective treatment currently available. Recently, there has been growing interest in exploring small-molecular drugs, derived from dietary sources, for disease prevention and treatment. QCT, a representative compound that is classified as a flavanol, possesses diverse biological effects. However, the precise mechanisms by which QCT ameliorates endothelial dysfunction in PE are not yet fully understood. In this study, we discovered that QCT can alleviate endothelial dysfunction by inhibiting ferroptosis and inflammation via its interaction with EGFR.
Firstly, we indicated that ferroptosis, inflammation, and endothelial dysfunction were present in patients with PE (Fig. 1). The causal relationship between them was further substantiated when PE-like phenotypes, such as elevated blood pressure, increased 24-h proteinuria, kidney injury, FGR, and abnormal placental development, were observed in pregnant rats following intraperitoneal administration of the ferroptosis inducer Erastin (Fig. 2B–F and Supplementary Fig. 1). The injection of Erastin in pregnant rats also revealed that ferroptosis could induce the occurrence of inflammation and endothelial damage (Figs. 2K–M, O, and P). Despite the precise etiology of PE remaining unclear at present, increasing evidence suggests an association between PE and an imbalance in inflammation, contributing to endothelial dysfunction. This hypothesis is widely accepted in the pathogenesis of PE28. Consequently, our follow-up studies primarily focus on investigating whether QCT can attenuate endothelial dysfunction.
While in vitro experiments demonstrated QCT’s therapeutic and preventive effects on Erastin-induced HUVEC (Fig. 3C, D), prevention often takes precedence over treatment in clinical settings. Additionally, a medium dose of QCT did not yield better functional improvement. Hence, we chose a prophylactic low dose (1 μM) of QCT to counteract Erastin-induced endothelial dysfunction. For endothelium, it has multiple functions, including, but not limited to, hemostasis, fibrinolysis, regulation of vascular tone, inflammatory cascades, and mediation of osmolality29,30. In PE, endothelial dysfunction primarily manifests as barrier disruption and impaired vasodilatory capacity31,32. Even though PE patients experience elevated blood pressure, their overall blood and plasma volumes are reduced33,34, and in such hypovolemic conditions, body fluids are distributed more into the tissue interstitium than into the vasculature, indicating capillary leakage35. Therefore, we assessed endothelial barrier function, indicated by monolayer HUVEC permeability and cell-tight junction integrity. The results displayed that QCT could mitigate the damage of the endothelial barrier caused by Erastin (Fig. 4A–C). Furthermore, QCT addition reduced Erastin-induced cell death (e.g., ferroptosis), inflammation, intracellular ROS production, and mitochondrial membrane potential changes, and facilitated the repair of endothelial damage (Figs. 3E–L and 4D–D2).
The subsequent step was identifying the target through which QCT counters Erastin-induced endothelial dysfunction. Based on target prediction, bioinformatics analysis, molecular docking, and CETSA, we identified EGFR as the target of QCT (Fig. 5). Activation of EGFR triggers various downstream signaling cascades, such as MAPK, PI3K/AKT/mTOR, JAK-STAT, and PLCγ/PKC36,37,38. It is worth noting that a previous study has already reported that QCT exhibits strong affinity and stability towards EGFR and AKT1, two key targets of QCT, and inhibits the EGFR/AKT pathway. This inhibition results in the downregulation of phosphorylation of EGFR, AKT, and ERK39. Interestingly, our experiments yielded similar results, showing that QCT suppresses the upregulated pEGFR/EGFR and pAKT/AKT induced by Erastin. However, QCT did not affect the pERK/ERK ratio (Fig. 6G–G4). This inconsistency may be due to the diversities of cell types or different concentrations of QCT. Additionally, research by Poursaitidis et al. suggested a role for GSH-Px, particularly GPX4, in modulating sensitivity to ferroptosis. Activated EGFR determines cellular sensitivity to ferroptosis by stimulating MAPK signaling and reducing the expression of GPX440. Ferroptotic cell death can exacerbate inflammation through DAMP release, like activating NF-κB via its receptor for advanced glycation end-products (RAGE)41. Our results indicate that QCT, by binding to EGFR, inhibits EGFR’s expression and phosphorylation, counters the decrease in GPX4, and alleviates inflammatory imbalance (Fig. 6G and Supplementary Fig. 8). However, it is worth noting that QCT, as a functionally diverse molecule, affects multiple intracellular targets42. Furthermore, our study predicted a total of 100 targets for QCT (Supplementary Table 7), with EGFR being just one potentially significant or central target. Therefore, it is unsurprising that QCT continues to play a role in reducing cell death even after EGFR down-regulation (Fig. 6A and Supplementary Fig. 6A), possibly via other targets.
Since QCT could enhance the endothelial function in vitro, we wonder if it can exert a protective effect against PE in vivo. Therefore, we selected the sRUPP rat model that specifically targets placental ischemia/hypoxia. This model circumvents the main limitations of the reduced uterine-perfusion pressure (RUPP) model: hindlimb ischemic paraplegia and hypertension due to toxemia from ischemia/hypoxia in non-placental tissues26. We then administered QCT prophylactically via intraperitoneal injection to pregnant rats. As there were no specifically suitable positive control drugs for protective roles in PE, we chose the ferroptosis inhibitor Fer-1 as our option. Fer-1 has been reported in previous studies to not only inhibit ferroptosis but also rescue inflammatory43. Additionally, in earlier research, we utilized Fer-1 and confirmed its ability to alleviate PE manifestations in a rat model10. In this study, QCT similarly to Fer-1, played a rescue role in mitigating maternal PE-like symptoms (Fig. 7). However, neither QCT nor Fer-1 improved FGR or placental dysplasia in the sRUPP model (Supplementary Fig. 7), likely because the surgery-induced ischemia/hypoxia had a more significant effect on fetal and placental development than ferroptosis and inflammation. Notably, QCT showed a more significant effect than Fer-1 in improving GSH-Px activity and placental vascular density (Fig. 8D, I, and J). This may be due to QCT’s multiple targets beyond its anti-ferroptosis and anti-inflammatory capabilities. It is noteworthy that while multiple drug concentrations may be designed in other studies, for our study, minimizing the drug dose while achieving its effect was considered for safety reasons, given that PE is a pregnancy-related disease, and medication use during pregnancy is restricted. Hence, we chose only a single concentration of QCT (2 mg/kg) based on a prior study44, a relatively low dose compared to the commonly used range of 10–100 mg/kg45,46, for our in vivo experiments.
In summary, this study provides comprehensive evidence that QCT can alleviate endothelial dysfunction in PE by inhibiting ferroptosis and inflammation through specific binding to EGFR. The potential applications of QCT have been expanded, offering the possibility of further development as a safe and effective preventive molecule for PE. However, it should be noted that we did not intervene with QCT in Erastin-induced PE-like rats, and in addition, we did not further explore the molecular mechanism by which QCT exerts its effects after binding to EGFR, which is also the shortcoming of our study and deserves further investigation and discussion.
Materials and methods
Human tissue collection
A total of 144 blood samples were collected (Control = 72, PE = 72) and plasma was extracted and stored at −80 °C until processing. Placentas were obtained from normal pregnancies (n = 34) and patients with PE (n = 34), and all of them had undergone elective or emergency cesarean deliveries from Jan 2021 to Aug 2022 at the Department of Gynecology and Obstetrics, the First Affiliated Hospital of Jinan University (Guangzhou, China). The PE diagnosis was based on the criteria issued by the International Society for the Study of Hypertension in Pregnancy (ISSHP) in 201847. The inclusion and exclusion criteria are shown in Supplementary Table 1. The clinical data of all subjects was listed in Supplementary Table 2. This study was conducted in accordance with the Declaration of Helsinki. Signed informed consent was obtained from all the study participants. All ethical regulations relevant to human research participants were followed. All processes involving human tissues in this study were approved by the Ethics Committee of the First Affiliated Hospital of Jinan University (approval number: KY-2021-092).
Animal model
Sprague–Dawley rats (6–8 weeks, 180–230 g) obtained from Guangdong Vital River Laboratory Animal Technology Co., Ltd. (SCXK 2022-0174, Guangdong, China) were individually housed at 24 °C with a 12:12 h light-dark cycle, with ad libitum access to food and water. Pairs of females and males were matched at a 1:1 ratio, designating the presence of a vaginal plug as gestation day (GD) 0.5. A ferroptosis-induced rat model was established by intraperitoneal injection of Erastin (GC16630-50mg, GLPBIO, USA), randomly assigning rats to Sham (n = 8) and Erastin (n = 9) groups. On GD 12.5, Erastin (10 mg/kg/day) or saline (Sham) was administered intraperitoneally.
The selective reduced uteroplacental perfusion (sRUPP) model of preeclampsia (PE) was established as previously described in ref. 48. Briefly, rats on GD13.5 were anesthetized with 2% pentobarbital sodium, and a midabdominal incision was made. The ligations were placed on both bilateral uterine and ovarian arteries in the sRUPP model, while the Sham group placed no ligations. Rats were randomly assigned to Sham (n = 8), sRUPP (n = 10), sRUPP + Fer-1 (n = 6), and sRUPP + QCT (n = 8) groups. On GD 7.5, Ferrostatin-1 (Fer-1, GC10380-25mg, GLPBIO, USA) and Quercetin (QCT, GN10266-5g, GLPBIO, USA) were intraperitoneally injected at 2 μmol/kg/day and 2 mg/kg/day, respectively, while Sham and sRUPP groups received equivalent saline injections. 24-h urine samples were collected from GD 16.5 to GD 17.5, and on GD 17.5, pregnant rats were euthanized by pentobarbital sodium overdose for placental tissues, peripheral blood, kidney, and fetuses. Blood pressure was measured every other day from GD 1.5 to GD 17.5 using an automated computerized tail-cuff system (Softron BP2010-A, BP-2010 Series, Softron Biotechnology Ltd., China). We have complied with all relevant ethical regulations for animal use. All animal experiments adhered to the Ethical Committee for Animal Experimentation, Jinan University (approval number: IACUC-20221104-01).
Cell culture and cell viability assay
The human umbilical vein endothelial cell (HUVEC) (CRL-1730) was purchased from Shanghai Kanglang Biological Technology Co., Ltd. (Shanghai, China). Cells of passage between P3 and P10 were used for all experiments, and cells for the same experiment originated from the same number of passages. HUVECs were maintained in 10% fetal bovine serum (FBS) (10270-106, Gibco, USA) at 37 °C and 5% CO2. The small interfering RNA (si-RNA) was purchased from OBiO Technology Co., Ltd. (Shanghai, China) and the sequences are listed in Supplementary Table 3. All cells were cultured in 96-well plates (2.5 × 104 cells/ml) and different concentrations of Erastin and QCT were at 0 h, 24 h, 36 h, and 48 h after plating, CCK-8 reagent (10 µl) was added to the wells, followed by a 2-h incubation at 37 °C. Absorbance values were measured at 450 nm using the Bio-Rad Model 450 microplate reader (Bio-Rad, CA, USA). Each experiment was conducted independently at least three times.
Histological analysis
Rat tissues were serially sectioned at 4 μm thicknesses and then mounted on slides for further study. Hematoxylin and eosin (HE) staining was conducted according to routine protocols49. Tissue sections underwent deparaffinization in xylene and a graded ethanol series, followed by rehydration. Then, Harris hematoxylin staining was applied to visualize the nuclei, after which the sections were rinsed and treated with an alkaline solution to produce blue nuclear staining. Differentiation with a weak acid alcohol solution removed excess hematoxylin, and eosin counterstaining was then used to impart a pink hue to the cytoplasm. The stained sections were subsequently dehydrated through an ethanol series, cleared with xylene, and mounted using a resinous medium for microscopic analysis. Glycogen staining was conducted using a periodic acid-schiff (PAS) kit (395B, Sigma-Aldrich, USA) following the manufacturer’s instructions. Immunofluorescence and immunohistochemistry staining were carried out as previously described in ref. 50. For immunofluorescence staining, tissues or cells were first fixed with 4% paraformaldehyde and then permeabilized using 0.1% Triton X-100 in PBS to facilitate antibody penetration into cells. This was followed by a blocking step with 10% normal goat serum (NGS) in PBS for 1 h at room temperature to minimize non-specific binding. The samples were then incubated with the primary antibody against the protein of interest at 4 °C overnight. After thorough washing, the sections were treated with a secondary antibody conjugated to a fluorophore for 1–2 h at room temperature. Finally, the sections were mounted using an anti-fade reagent with DAPI to counterstain the nuclei before being examined under a fluorescence microscope. In the immunohistochemistry process, tissue sections underwent dewaxing with xylene and a gradient ethanol series. To unveil concealed antigens, antigen retrieval was executed. A 3% hydrogen peroxide solution was utilized to inactivate endogenous peroxidase, thereby reducing background staining. Following incubation with the primary antibody recognizing the antigen of interest and subsequent PBS washing, a biotinylated secondary antibody was introduced, which was then complexed with streptavidin-horseradish peroxidase (HRP). The presence of the antigen was visualized by developing the signal with a chromogen-like diaminobenzidine (DAB), resulting in a brown color at the antigenic sites. The sections were subsequently hematoxylin counterstained, dehydrated through graded ethanol, cleared with xylene, and mounted for microscopic analysis. At least five random images from three samples per group were analyzed. The details of the antibodies are listed in Supplementary Table 4.
Western blot
Proteins extracted from placental tissues and HUVECs were analyzed with a minimum of three replicates, following previously described procedures51. Briefly, Proteins, loaded equally into the sample buffer, were denatured by boiling for 5–10 min prior to separation via SDS-PAGE. The resolved proteins were transferred to a PVDF membrane. Following a 1-h blocking step with 5% non-fat milk in TBST at room temperature, the membrane was probed with a primary antibody directed against the protein of interest overnight at 4 °C. The membrane was then washed and incubated with an HRP-conjugated secondary antibody for 1 h. Finally, the protein bands were detected using an ECL substrate. The details of the antibodies are listed in Supplementary Table 5.
Lipid peroxidation assay and enzyme-linked immunosorbent assay (ELISA) analysis
Tissue lipid peroxidation markers, including malondialdehyde (MDA), glutathione (GSH), glutathione peroxidase (GSH-Px), and Fe2+ contents, were detected following the respective manufacturer’s protocols.
Intracellular ROS levels were quantitatively measured using a 2’,7’-dichlorodihydrofluorescein diacetate (DCFH-DA) red or green fluorescent probe, as previously reported52. Cells were incubated with the ROS indicator, DCFH-DA, a membrane-permeable compound that becomes fluorescent upon intracellular esterase-mediated hydrolysis, yielding DCFH. This transformation occurs within the cell, and subsequent ROS-mediated oxidation of DCFH results in the formation of brightly fluorescent DCF, whose intensity correlates with ROS concentration. Mean fluorescence intensity was utilized for statistical analysis. Cell death was assessed using propidium iodide (PI) red fluorescent probe to quantify the proportion of PI-positive cells, as previously reported53. PI is a fluorescent nucleic acid stain that penetrates cells with damaged membranes, resulting in red fluorescence emission. This property enables the distinction between viable, apoptotic, and necrotic cells, relying on the cell membrane’s integrity and the dye’s DNA interaction. The JC-1 Mitochondrial Membrane Potential Assay Kit was employed to examine mitochondrial membrane potential following the manufacturer’s instructions.
Whole blood samples from humans and rats were collected. Plasma substances were measured using ultraviolet (UV) spectrophotometry with detection kits, following the manufacturer’s protocols (Mbbiology, China). The details of the kits are listed in Supplementary Table 6.
Wound healing assay
A total of 5 × 105 HUVECs were seeded in one well of 6-well plates and grown to reach confluent monolayers, with or without si-RNA transfection or drug treatment. Then, scratches were created by using a 200 μl pipette tip. The images of migrated cells were recorded at 0 h, 24 h, and 48 h. The percentage of wound closure was analyzed. The experiment was conducted independently at least three times.
Fluorescein isothiocyanate (FITC)-dextran assay
HUVECs were cultured into monolayer cells in the upper chamber of transwells with a pore size of 0.4 μm. The serum-free medium was used to starve and culture for 3 h so that HUVECs grow synchronously and are in the stationary phase. Then pretreated HUVECs with QCT for 2 h and treated with Erastin for 1 h. The upper chamber of each well was added with FITC-labeled dextran (70 kDa) at a final concentration of 10 mg/ml. After incubation for 1 h, an equal amount of medium from the upper and lower chambers was aspirated from each well and added to 96-well plates, respectively. The standard curve was set up with a concentration gradient of 0 mg/ml, 2 mg/ml, 4 mg/ml, 6 mg/ml, 8 mg/ml, 10 mg/ml, 12 mg/ml. The absorbance at 490 nm was measured and the concentration of FITC-labeled dextran was calculated from the standard curve for the upper and lower chambers in each group before calculating the penetration rate. The experiment was conducted independently at least three times.
Target prediction and molecular docking
The SMILES notation for QCT (PubChem CID: 5280343) obtained from PubChem (https://pubchem.ncbi.nlm.nih.gov/) was utilized to depict its chemical structure. The Swiss Target Prediction online platform (http://www.swisstargetprediction.ch/) was employed to predict potential targets for QCT. Detailed information on all predicted targets is provided in Supplementary Table 7.
Protein-protein interaction (PPI) network analysis was conducted using Cytoscape V3.10.1 software. The structures of Tyrosine-protein kinase SRC (SRC) (PDB: 1O43), Serine/threonine-protein kinase AKT (AKT1) (PDB: 1UNR), and epidermal growth factor receptor (EGFR) (PDB: 8A2D) were obtained from the RCSB Protein Data Bank (PDB) (http://www.rcsb.org/). Before molecular docking, Chem3D V21.0.0 was employed to preprocess QCT for energy minimization. The open-source PyMOL (https://pymol.org) was used to remove non-standard amino acids, excess ligands, crystal water, and impurity chains from the primary structure of EGFR. For molecular docking, the Surflex-Dock Geom (SFXC) docking model of SYBYL-X V2.1.1 was utilized to dock QCT to the binding site of the target proteins. The total score and C-score were recorded for a comprehensive evaluation of the docking effect. Bioinformatics analysis of differentially expressed proteins was performed using the “limma” package. The subsequent molecular docking of QCT with EGFR followed the procedure described previously in ref. 54. Molecular docking calculations were performed four times.
The cellular thermal shift assay (CETSA)
The CETSA was performed according to the protocols as previously described in ref. 55. The cell suspension was initially divided into two groups. The QCT groups were exposed to 60 μM QCT (with a predicted Ki of 60.351 μM) or 1 μM QCT (concentration used in cellular experiments), while the control group received an isovolumetric Dimethyl sulfoxide (DMSO) solution, both incubated at room temperature for 3 h. Following this, cells from each group were diluted with phosphate-buffered saline (PBS) and divided into aliquots. The cells were then subjected to temperature variations (37 °C, 41 °C, 45 °C, 49 °C, 53 °C, and 57 °C) for 3 min, followed by a 3 min cooling period at room temperature. Subsequently, the cells underwent repeated freezing and thawing in liquid nitrogen to lyse them. The resulting lysates were centrifuged at 4 °C, 12,000 rpm for 30 min for Western blot experiments. This process was repeated in at least three independent experiments.
RNA isolation and quantitative polymerase chain reaction (qPCR)
Total RNA was extracted from HUVECs, and real-time quantitative PCR (qPCR) procedures were conducted following established protocols as previously described in refs. 56,57. RNA was isolated from cells or tissues using an appropriate kit, adhering to the provider’s guidelines. Samples with a 260/280 ratio within the range of 1.8 to 2.0, as measured by a spectrophotometer, were selected for RNA integrity and concentration assessment. From 1 µg of this RNA, cDNA was reverse-transcribed using oligo (dT) primers and a reverse transcription kit, following the manufacturer’s recommendations. Subsequently, qPCR was conducted with a SYBR Green master mix and gene-specific primers in a 20 µL reaction volume, which included the cDNA template. The qPCR was executed on a qPCR instrument, and the relative gene expression data were processed using the 2−ΔΔCt method for analysis. The relative expression of messenger RNA (mRNA) was normalized to GAPDH. The results presented are representative of three independent experiments. The specific primer sequences used can be found in Supplementary Table 8.
Statistics and reproducibility
Analysis of statistics was performed by using the SPSS V22.0 (SPSS, Chicago, IL, USA). The construction of statistical charts was performed by using the GraphPad Prism V8.0.2.263 software (GraphPad Software, CA, USA). The normally distributed continuous variables were analyzed by T-test and the skewed variables were analyzed by Mann–Whitney U-test. The correlation between the two variables was analyzed by using Spearman correlation. All values were presented as mean ± standard deviation (SD) or median (interquartile range). P value < 0.05 was considered statistically significant.
Compliance with ethics requirements
We have obtained informed consent from all participants, and all processes involving human tissues in this study were approved by the Ethics Committee of the First Affiliated Hospital of Jinan University (approval number: KY-2021-092). All animal experiments were conducted in accordance with the procedures of the Ethical Committee for Animal Experimentation, Jinan University (approval number: IACUC-20221104-01).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Publicly available data sets (GSE11469158, GSE2590659, and GSE1058860) were analyzed in this study and the data are available via https://www.ncbi.nlm.nih.gov/ geo/. The source data underlying the graphs and charts presented in the main figures are available in the file named Supplementary Data 1.
References
Wu, P., Green, M. & Myers, J. E. Hypertensive disorders of pregnancy. BMJ 381, e071653 (2023).
Magee, L. A., Nicolaides, K. H. & von Dadelszen, P. Preeclampsia. N. Engl. J. Med. 386, 1817–1832 (2022).
Wu, P et al. Preeclampsia and future cardiovascular health: a systematic review and meta-analysis. Circ. Cardiovasc. Qual. Outcomes. https://doi.org/10.1161/CIRCOUTCOMES.116.003497 (2017).
Karatza, A. A. & Dimitriou, G. Preeclampsia emerging as a novel risk factor for cardiovascular disease in the offspring. Curr. Pediatr. Rev. 16, 194–199 (2020).
Bisson, C. et al. Preeclampsia pathophysiology and adverse outcomes during pregnancy and postpartum. Front. Med. 10, 1144170 (2023).
Du, M. et al. Serum inflammatory markers and preeclampsia in type 1 diabetes: a prospective study. Diab. Care 36, 2054–2061 (2013).
Lawlor, D. A. et al. Cardiovascular biomarkers and vascular function during childhood in the offspring of mothers with hypertensive disorders of pregnancy: findings from the avon longitudinal study of parents and children. Eur. Heart J. 33, 335–345 (2012).
Tang, D., Chen, X., Kang, R. & Kroemer, G. Ferroptosis: molecular mechanisms and health implications. Cell Res. 31, 107–125 (2021).
Zheng, J. & Conrad, M. The metabolic underpinnings of ferroptosis. Cell Metab. 32, 920–937 (2020).
Yang, X. et al. Ferritin light chain deficiency-induced ferroptosis is involved in preeclampsia pathophysiology by disturbing uterine spiral artery remodelling. Redox Biol. 58, 102555 (2022).
Yu, Y. et al. Ferroptosis: a cell death connecting oxidative stress, inflammation and cardiovascular diseases. Cell Death Discov. 7, 193 (2021).
Murthi, P, Pinar, AA, Dimitriadis, E & Samuel, CS. Inflammasomes—a molecular link for altered immunoregulation and inflammation mediated vascular dysfunction in preeclampsia. Int. J. Mol. Sci. https://doi.org/10.3390/ijms21041406 (2020).
Popiolek-Kalisz, J. & Fornal, E. The effects of quercetin supplementation on blood pressure—meta-analysis. Curr. Probl. Cardiol. 47, 101350 (2022).
Di Petrillo, A., Orrù, G., Fais, A. & Fantini, M. C. Quercetin and its derivates as antiviral potentials: a comprehensive review. Phytother. Res. 36, 266–278 (2022).
Wang, Y. et al. Quercetin alleviates acute kidney injury by inhibiting ferroptosis. J. Adv. Res. 28, 231–243 (2021).
Wang, Z.-X. et al. Quercetin induces p53-independent cancer cell death through lysosome activation by the transcription factor EB and reactive oxygen species-dependent ferroptosis. Br. J. Pharmacol. 178, 1133–1148 (2021).
Willhite, C. C. Teratogenic potential of quercetin in the rat. Food Chem. Toxicol. 20, 75–79 (1982).
Wu, Z. et al. Maternal quercetin administration during gestation and lactation decrease endoplasmic reticulum stress and related inflammation in the adult offspring of obese female rats. Eur. J. Nutr. 53, 1669–1683 (2014).
Elfarra, J. et al. Natural killer cells mediate pathophysiology in response to reduced uterine perfusion pressure. Clin. Sci. 131, 2753–2762 (2017).
Yang, D., Wang, T., Long, M. & Li, P. Quercetin: its main pharmacological activity and potential application in clinical medicine. Oxid. Med. Cell Longev. 2020, 8825387 (2020).
Zhang, M. et al. Neurodevelopmental toxicity induced by maternal PM2.5 exposure and protective effects of quercetin and Vitamin C. Chemosphere 213, 182–196 (2018).
Zhao, Y. et al. Vascular endothelium deploys caveolin-1 to regulate oligodendrogenesis after chronic cerebral ischemia in mice. Nat. Commun. 13, 6813 (2022).
Courtney, JA, Wilson, RL, Cnota, J & Jones, HN. Conditional mutation of Hand1 in the mouse placenta disrupts placental vascular development resulting in fetal loss in both early and late pregnancy. Int. J. Mol. Sci. https://doi.org/10.3390/ijms22179532 (2021).
Bai, L. et al. CETSA-MS-based target profiling of anti-aging natural compound quercetin. Eur. J. Med. Chem. 267, 116203 (2024).
Kook, D. et al. The protective effect of quercetin against oxidative stress in the human RPE in vitro. Invest. Ophthalmol. Vis. Sci. 49, 1712–1720 (2008).
Morton, J. S. et al. Characterisation of the selective reduced uteroplacental perfusion (sRUPP) model of preeclampsia. Sci. Rep. 9, 9565 (2019).
Gatford, K. L., Andraweera, P. H., Roberts, C. T. & Care, A. S. Animal models of preeclampsia: causes, consequences, and interventions. Hypertension 75, 1363–1381 (2020).
Sankaralingam, S, Arenas, IA, Lalu, MM & Davidge, ST. Preeclampsia: current understanding of the molecular basis of vascular dysfunction. Expert Rev. Mol. Med. 8, 1–20 (2006).
Deanfield, J. E., Halcox, J. P. & Rabelink, T. J. Endothelial function and dysfunction: testing and clinical relevance. Circulation 115, 1285–1295 (2007).
Marti, C. N. et al. Endothelial dysfunction, arterial stiffness, and heart failure. J. Am. Coll. Cardiol. 60, 1455–1469 (2012).
Chambers, J. C. et al. Association of maternal endothelial dysfunction with preeclampsia. JAMA 285, 1607–1612 (2001).
Opichka, MA, Rappelt, MW, Gutterman, DD, Grobe, JL & McIntosh, JJ. Vascular dysfunction in preeclampsia. Cells. https://doi.org/10.3390/cells10113055 (2021).
Silver, H. M., Seebeck, M. & Carlson, R. Comparison of total blood volume in normal, preeclamptic, and nonproteinuric gestational hypertensive pregnancy by simultaneous measurement of red blood cell and plasma volumes. Am. J. Obstet. Gynecol. 179, 87–93 (1998).
Salas, S. P., Marshall, G., Gutiérrez, B. L. & Rosso, P. Time course of maternal plasma volume and hormonal changes in women with preeclampsia or fetal growth restriction. Hypertension 47, 203–208 (2006).
Anthony, J. & Schoeman, L. K. Fluid management in pre-eclampsia. Obstet. Med. 6, 100–104 (2013).
Herbst, R. S. Review of epidermal growth factor receptor biology. Int J. Radiat. Oncol. Biol. Phys. 59, 21–26 (2004).
Jones, S. & Rappoport, J. Z. Interdependent epidermal growth factor receptor signalling and trafficking. Int J. Biochem. Cell Biol. 51, 23–28 (2014).
Wee, P & Wang, Z Epidermal growth factor receptor cell proliferation signaling pathways. Cancers. https://doi.org/10.3390/cancers9050052 (2017).
Zhang, Z., Wu, H., Zhang, Y., Shen, C. & Zhou, F. Dietary antioxidant quercetin overcomes the acquired resistance of Sorafenib in Sorafenib-resistant hepatocellular carcinoma cells through epidermal growth factor receptor signaling inactivation. Naunyn Schmiedebergs Arch. Pharmacol. 397, 559–574 (2024).
Poursaitidis, I. et al. Oncogene-selective sensitivity to synchronous cell death following modulation of the amino acid nutrient cystine. Cell Rep. 18, 2547–2556 (2017).
Gong, T, Liu, L, Jiang, W & Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. https://doi.org/10.1038/s41577-019-0215-7 (2020).
Wang, D., Sun-Waterhouse, D., Li, F., Xin, L. & Li, D. MicroRNAs as molecular targets of quercetin and its derivatives underlying their biological effects: a preclinical strategy. Crit. Rev. Food Sci. Nutr. 59, 2189–2201 (2019).
Shou, Y., Yang, L., Yang, Y. & Xu, J. Inhibition of keratinocyte ferroptosis suppresses psoriatic inflammation. Cell Death Dis. 12, 1009 (2021).
Yang, S., Song, L., Shi, X., Zhao, N. & Ma, Y. Ameliorative effects of pre-eclampsia by quercetin supplement to aspirin in a rat model induced by L-NAME. Biomed. Pharmacother. 116, 108969 (2019).
Wang, Y. et al. Quercetin reduces neural tissue damage and promotes astrocyte activation after spinal cord injury in rats. J. Cell Biochem. 119, 2298–2306 (2018).
Shahzad, H., Giribabu, N., Sekaran, M. & Salleh, N. Quercetin Induces dose-dependent differential morphological and proliferative changes in rat uteri in the presence and in the absence of estrogen. J. Med. Food 18, 1307–1316 (2015).
Brown, M. A. et al. The hypertensive disorders of pregnancy: ISSHP classification, diagnosis & management recommendations for international practice. Pregnancy Hypertens. 13, 291–310 (2018).
Shi, M. et al. Comparison of different modified operations in the reduced uteroplacental perfusion pressure rat model of preeclampsia. J. Reprod. Immunol. 156, 103815 (2023).
Wang, J. et al. Alpha-2-macroglobulin is involved in the occurrence of early-onset pre-eclampsia via its negative impact on uterine spiral artery remodeling and placental angiogenesis. BMC Med. 21, 90 (2023).
Yang, Q. et al. MNSFβ regulates placental development by conjugating IGF2BP2 to enhance trophoblast cell invasiveness. Cell Prolif. 54, e13145 (2021).
Luo, X. et al. The role of inactivated NF-κB in premature ovarian failure. Am. J. Pathol. 192, 468–483 (2022).
Ko, H. S. et al. Oncostatin M stimulates cell migration and proliferation by down-regulating E-cadherin in HTR8/SVneo cell line through STAT3 activation. Reprod. Biol. Endocrinol. 11, 93 (2013). RB&E.
Jung, EJ et al. p53 Enhances Artemisia annua L. polyphenols-induced cell death through upregulation of p53-dependent targets and cleavage of PARP1 and Lamin A/C in HCT116 colorectal cancer cells. Int. J. Mol. Sci. https://doi.org/10.3390/ijms21239315 (2020).
Wang, G et al. EGFR/MAPK signaling pathway acts as a potential therapeutic target for sulforaphane-rescued heart tube malformation induced by various concentrations of PhIP exposure. Phytomedicine. https://doi.org/10.1016/j.phymed.2023.155270 (2023).
Jafari, R. et al. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat. Protoc. 9, 2100–2122 (2014).
He, M-Y et al. Nrf2 signalling and autophagy are involved in diabetes mellitus-induced defects in the development of mouse placenta. Open Biol. https://doi.org/10.1098/rsob.160064 (2016).
Li, Y.-X. et al. Gestational diabetes mellitus in women increased the risk of neonatal infection via inflammation and autophagy in the placenta. Medicine 99, e22152 (2020).
Awamleh, Z., Gloor, G. B. & Han, V. K. M. Placental microRNAs in pregnancies with early onset intrauterine growth restriction and preeclampsia: potential impact on gene expression and pathophysiology. BMC Med. Genomics 12, 91 (2019).
Tsai, S. et al. Transcriptional profiling of human placentas from pregnancies complicated by preeclampsia reveals disregulation of sialic acid acetylesterase and immune signalling pathways. Placenta 32, 175–182 (2011).
Sitras, V. et al. Differential placental gene expression in severe preeclampsia. Placenta 30, 424–433 (2009).
Acknowledgements
The authors sincerely thank all participants involved in this study. In addition, the authors would like to express their sincere gratitude to the Medical Experimental Center of the School of Basic Medicine and Public Health, Jinan University, for providing some of the experimental equipment for this study. This study was supported by NSFC grants (no. 32170825 and 82371692), Guangdong Natural Science Foundation (2023A1515010424 and 2023A1515012741), the Science and Technology Planning Project of Guangzhou (no. 202201020007), the Clinical Frontier Technology Program of the First Affiliated Hospital of Jinan University, China (no. JNU1AF-CFTP-2022-a01209), the Fundamental Research Funds for the Central Universities (21624111), and the City-University Joint Sponsorship (Dengfeng Hospital) Basic Research Project, China (2023A03J1009).
Author information
Authors and Affiliations
Contributions
Meiting Shi: conceptualization, methodology, formal analysis, investigation, and writing—original draft. Lu Sun: validation, methodology, formal analysis, investigation, and resources. Jiachun Wei: methodology, formal analysis, investigation, and resources. Yao Shen: methodology, formal analysis, validation, and resources. Jian Wang: validation, investigation, and resources. Ping Zhang: methodology, validation, and resources. Xiaofeng Yang: investigation and resources. Yuzhen Ding: investigation and resources. Wanchang Yin: resources. Xinyao Lu: resources. Xuesong Yang: conceptualization, supervision, project administration, and funding acquisition. Guang Wang: conceptualization, writing—review and editing, supervision, project administration, and funding acquisition. Ruiman Li: conceptualization, writing—review and editing, supervision, project administration, and funding acquisition.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Yoko Hirata and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Christopher Hine and Dario Ummarino.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Shi, M., Sun, L., Wei, J. et al. Quercetin alleviates endothelial dysfunction in preeclampsia by inhibiting ferroptosis and inflammation through EGFR binding. Commun Biol 8, 90 (2025). https://doi.org/10.1038/s42003-025-07547-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-07547-5
