
Abstract
The genetics of β-cell function (BCF) offer valuable insights into the aetiology of type 2 diabetes (T2D)1,2. Previous studies have expanded the catalogue of BCF genetic associations through candidate gene studies3,4,5,6,7, large-scale genome-wide association studies (GWAS) of fasting BCF8,9 or functional islet studies on T2D risk variants10,11,12,13,14. Nonetheless, GWAS focused on BCF traits derived from oral glucose tolerance test (OGTT) data have been limited in sample size15,16 and have often overlooked the potential for related traits to capture distinct genetic features of insulin-producing β-cells17,18. We reasoned that investigating the genetic basis of multiple BCF estimates could provide a broader understanding of β-cell physiology. Here, we aggregate GWAS data of eight OGTT-based BCF traits from ~26,000 individuals of European descent, identifying 55 independent genetic associations at 44 loci. By examining the effects of BCF genetic signals on related phenotypes, we uncover diverse disease mechanisms whereby genetic regulation of BCF may influence T2D risk. Integrating BCF-GWAS data with pancreatic islet transcriptomic and epigenomic datasets reveals 92 candidate effector genes. Gene silencing in β-cell models highlights ACSL1 and FAM46C as key regulators of insulin secretion. Overall, our findings yield insights into the biology of insulin release and the molecular processes linking BCF to T2D risk, shedding light on the heterogeneity of T2D pathophysiology.
Similar content being viewed by others


Main
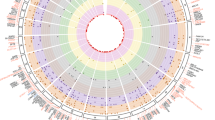
We conducted a meta-analysis of nine GWAS involving up to 26,356 non-diabetic individuals of European descent. We assessed eight BCF measures: (1) BCF insulin sensitivity glucose tolerance test for acute insulin response (BIGTT-AIR), (2) corrected insulin response (CIR), (3) first-phase Stumvoll (Stumvoll), (4) insulinogenic index (xinsdG30), (5) modified insulinogenic index (xinsG30), (6) disposition index for modified insulinogenic index and Matsuda (DI), (7) disposition index for BIGTT-AIR and BIGTT-SI (DIBIG) and the HOMA-β index (HOMA-β)19,20,21,22,23,24,25 (Fig. 1 and Supplementary Tables 1 and 2). After supplementing GWAS discovery with approximate conditional analyses, we identified 55 independent genetic signals at 44 GWAS loci, each detected in at least one BCF trait (Fig. 1b, Extended Data Fig. 1 and Supplementary Tables 3 and 4). Following Bonferroni correction for the eight BCF traits, 31 loci remained significant (Supplementary Table 3).

a, Overview of the signal discovery strategy. We mapped genetic effects on BCF indices using oral glucose-stimulated and steady-state fasting measurements from ~26,000 non-diabetic individuals. In-house BCF groupings are highlighted on the right. b. Multi-trait Manhattan plot of single-variant genome-wide association meta-analyses for eight BCF estimates. From inner to outer tracks: HOMA-β, DI, DIBIG, BIGTT-AIR, CIR, Stumvoll, xinsdG30 and xinsG30. Genome-wide significant variants (P ≤ 5 × 10−8) are coloured in dark blue and previously unreported loci are in red. Loci meeting Bonferroni-correction thresholds are detailed in Supplementary Table 3. Y axis, association P-values on the –log10 scale; x axis, genomic position in hg19.
Next, we investigated the extent to which GWAS loci were specific to a single index or a recognizable group of BCF traits. Hierarchical clustering of genetic (rg) and phenotypic (rp) correlations clustered BCF indices into three main groups (Extended Data Fig. 2a,b). This is consistent with our expectations, as the traits are grouped according to whether the BCF indices are adjusted for insulin sensitivity or derived from dynamic (OGTT) or steady-state measurements. The groups include insulin secretion measurements during an OGTT (mean rg = 0.90, rp = 0.85), disposition indexes (rg = 0.93, rp = 0.78) and a steady-state BCF surrogate. We observed a significant negative phenotype correlation between DI and HOMA-β, which was unexpected. HOMA-β summarizes the feedback loop between β-cells and the liver that balances the hepatic glucose output and insulin secretion in the fasting state26. Previous work has reported that progressive β-cell failure in impaired glucose tolerance is captured by disposition indexes but not by HOMA-β27. This indicates that HOMA-β may inadequately reflect glucose-responsive β-cell activity28, limiting its utility in predicting the role of β-cell dysfunction in T2D progression29.
We observed that 48% of all GWAS loci were shared across all glucose-stimulated BCF-based indices (18 out of 44) or together with HOMA-β (3 out of 44). Nonetheless, 52% of them (23 out of 44) were confined to selective BCF groups or single traits (Extended Data Fig. 2c,d). Equivalent sample sizes across BCF indices indicate that differences in signal discovery between traits are not due to statistical power. Accordingly, heterogeneity tests for the effect sizes revealed high heterogeneity within BCF groups at selective signals (Supplementary Table 5). For example, GWAS associations were identified at the GIPR locus for xinsG30, CIR, Stumvoll, xinsdG30 and DI. Heterogeneity tests at rs2238691 found significant differences in the effect sizes between the two insulinogenic indexes (xinsG30 and xinsdG30) and CIR (CIR vs xinsG30, P = 1.36 × 10−8 and CIR vs xinsdG30, P = 4.37 × 10−9) and Stumvoll (Stumvoll vs xinsG30, P = 3.36 × 10−13 and Stumvoll vs xinsdG30 P = 1.15 × 10−6), respectively. Collectively, these results provide evidence that despite high genetic and phenotypic correlations, different estimates for BCF probably encapsulate distinct genetic profiles.
We compared BCF-GWAS with and without body mass index (BMI) adjustment to assess whether genetic effects influencing β-cell secretory function might be confounded by obesity. We found substantial concordance in effect sizes regardless of BMI adjustment (Extended Data Fig. 3 and Supplementary Table 6), despite a sizable number of shared genetic loci between BCF and BMI (Supplementary Table 7). This suggests that BCF associations are unlikely to be steered by the effects of obesity-related insulin resistance30,31.
To characterize the distinct biological mechanisms underlying genetic effects on glucose-responsive BCF, we examined their relationship with other traits involved in glucose homeostasis. We identified 14 unknown loci, eight of which had been previously associated with metabolic or anthropometric traits (for example, fasting glucose, T2D or BMI), albeit lacking prior evidence of association with BCF. Similarly, we observed a high genetic correlation between BCF and glycaemic traits as well as with other diabetes-related phenotypes (Fig. 2a). We then investigated the effects of 42 non-palindromic BCF loci on glucose metabolism and other complex traits (Fig. 2b). Colocalization evidence (posterior probability (PP) > 0.6 for at least one BCF trait) was observed between BCF associations and variants influencing T2D (25 out of 42) and/or fasting glucose (24 out of 42) (Fig. 2b, Extended Data Fig. 4a and Supplementary Table 8) across numerous loci. Interestingly, only three BCF loci show colocalization with BMI–GWAS associations, also suggesting a minor influence of BMI on BCF-GWAS signals. We further characterized the allelic effects of BCF associations on T2D risk (Fig. 2B). We observed a subset of GWAS signals in which the allele associated with decreased BCF also increased T2D risk but lowered BMI. These findings align with previous efforts identifying a similar genetic signature (in which increased adiposity is coupled with lower cardiometabolic risk) for some of these loci (for example, ARAP1, IGF2BP2, TCF7L2)32. Conversely, allelic effects of the insulin-sensitivity LOC646736; MIR5702 (IRS1) signal (Extended Data Fig. 4b,c) were associated with both reduced BCF and T2D risk but increased BMI. This locus was reported to uncouple adiposity from cardiometabolic risk by promoting a more favourable fat distribution32,33,34. Effects on BCF could be explained by an indirect influence of insulin sensitivity or a simultaneous effect on IRS1 signalling in β-cells. We then examined causal links between BCF on T2D risk using Mendelian randomization, providing evidence of a causal effect of increased BCF on lower T2D risk (inverse-variance weighted P = 8.08 × 10−20 to 1.82 × 10−6). Interestingly, this was not observed for HOMA-β (Supplementary Table 9). Altogether, our analysis delineated key genetic determinants of BCF that modulate T2D risk through different molecular mechanisms.

a, Heatmap of genetic correlations (Z-score) between each BCF trait and other complex traits, as calculated by LDSC regression implemented in LDHub. Asterisks indicate significant correlations after Bonferroni multiple-test correction (P ≤ 0.05 / (8 BCF traits × 9 complex trait categories)). Rows are grouped using hierarchical clustering with Euclidean distance. b, Effect sizes (Z-scores) for multiple pancreatic islet-relevant traits among 42 BCF loci. Loci are named as follows: gene | chromosome:position [effect allele]. WHR, waist-to-hip ratio; IFC, insulin fold change; ISI, Stumvoll insulin sensitivity; T1D, type 1 diabetes; IVGTT, intravenous glucose tolerance test; MRI, magnetic resonance imaging; Kidney function, estimated glomerular filtration rate based on serum creatinine; TG, triglycerides. Variant effect sizes were aligned to a negative BIGTT-AIR effect. The sentinel variant at each locus corresponds to the lead variant or a proxy (LD r2 > 0.7), excluding non-palindromic alleles and with available data among all BCF, BMI and T2D traits. **Genome-wide significant (P ≤ 5 × 10−8), *Bonferroni-corrected P-value (P ≤ 0.05 / 42 variants), | Nominal significance (P ≤ 0.05). Red, yellow and blue dots on the left indicate moderate evidence of colocalization at a given BCF locus between at least one BCF trait and T2D, fasting glucose or BMI, respectively.
We next identified the most relevant tissues and cell types in which genetic signals exert their effects on BCF. We examined the enrichment of BCF heritability among enhancer and promoter epigenomic annotations in human pancreatic islets13 as well as in other tissues and cell types35. All BCF traits except HOMA-β showed the strongest heritability enrichments in islet non-coding elements (Fig. 3a, Supplementary Fig. 4 and Supplementary Table 10). Likewise, we found significant BCF-GWAS enrichment in genes expressed in mouse pancreatic tissues36 as well as in intestine and brain tissues and in human endocrine cells37 (Fig. 3b,c). Notably, we observed an attenuated pattern of enrichments for HOMA-β across tissue and cell-type-specific annotations. These findings reinforce the notion that steady-state measurements are not sensitive enough to capture the glucose-responsive function of pancreatic islet cells. The adequacy of HOMA-β has been previously discussed38,39 and is also reflected in both the small genetic overlap with other BCF traits (Extended Data Fig. 2c) and the relatively low number of genetic findings in a previous meta-analysis of HOMA-β in ~95,000 individuals8. Interestingly, we observed heritability enrichments in peripheral tissues (for example, omental fat pad, small intestine, cerebellum) for insulin-sensitivity-corrected BCF measures. This further indicates that disposition indexes might capture effects from glucose metabolism in addition to BCF40. Collectively, these findings highlight pancreatic islets as the most relevant tissue of action, together with the influence of other tissues and cell types.

a, Heatmap of heritability (h2) enrichments for BCF and other complex traits and diseases, calculated using S-LDSC, across cis-regulatory transcriptional annotations from multiple tissues and cell types. Self rep.: self-reported. b,c, Heatmap showing the association of BCF genetic signals with cell-type-specific expression in relevant cells from Tabula Muris36 (b) and pancreatic islet cells from human donors37 (c). Z-scores displayed on the heatmap were calculated using CELLECT-LDSC. **Bonferroni-corrected P ≤ 0.05 / (8 BCF traits × 7 complex trait categories) in b and P ≤ 0.05 / (8 BCF traits × 14 pancreatic cell types) in c. *Nominal significance. Rows are grouped by hierarchical clustering with Euclidean distance in a–c. d, Scheme illustrating the prioritization of effector transcripts for BCF traits based on epigenomic annotations and enhancer-gene assignments in human pancreatic islets, and effects on human islet gene expression and alternative splicing through TWAS and colocalization approaches (adapted from a previous publication65). e, Enrichments of BCF candidate target genes within transcription factor (TF) co-expression networks from ARCHS4, calculated using enrichR. Barplot representation shows enrichR combined scores, with bars coloured according to the Benjamini–Hochberg-adjusted P value. *Significant enrichments at a false discovery rate of 5%. f, Illustrative examples of candidate BCF protein-coding genes contributing to the enrichment of annotations that are central for islet function and identity, and diabetes, as calculated using enrichR. The number of BCF genes contributing to each functional annotation is indicated in brackets. Adjusted P values using the Benjamini–Hochberg method are provided.
To identify molecular effectors influencing the β-cell secretory function, we integrated BCF-GWAS data with pancreatic islet epigenomic and chromatin interaction maps, as well as genetic effects on islet expression and splicing activity (Fig. 3d). We created credible sets of variants with 99% causal PP (Supplementary Table 11) and overlaid BCF fine-mapped variants on non-coding elements, connecting them to their target genes using human islet promoter capture Hi-C (pcHi-C) chromatin maps13 (Fig. 3d, left panel). From these data, we identified 72 candidate effector genes (Supplementary Table 12). For example, pcHi-C data linked enhancers harbouring BCF-GWAS variants in the CLEC14AA/LINC00639 and SLC39A11 loci to somatostatin receptor genes SSTR1 and SSTR2, respectively (Extended Data Fig. 5, Supplementary Table 12). Somatostatin inhibits insulin and glucagon secretion by activating G-protein-coupled somatostatin receptors. Although SSTR2 has a functionally dominant role in both α-cells and β-cells, selective effects on glucagon secretion have been reported via SSTR1-agonists41.
We next harnessed transcriptome-wide association studies (TWAS), which integrate transcriptomic reference panels42 with GWAS summary statistics to identify genes whose expression or splicing usage is associated with BCF traits (Fig. 3d, right panel). TWAS predicts gene expression or splicing activity within a GWAS cohort, uncovering associations between gene expression (eTWAS) or splicing junction usage (sTWAS) and GWAS traits. By reducing multiple comparison penalties, TWAS aids in identifying genetic associations that may not have reached GWAS significance43. We found 21 genes (15 eTWAS and six sTWAS) whose islet transcriptome activity is associated with BCF traits, supported by colocalization between islet expression or splicing quantitative trait loci (eQTLs and sQTLs, respectively) and BCF-GWAS signals (Supplementary Figs. 5 and 6 and Supplementary Tables 13 and 14). Among these genes, five loci harboured significant eTWAS associations but showed suggestive significance in our BCF meta-analyses. This includes UBE2E2 (rs35352848, xinsdG30 P = 2.60 × 10−7) and FAM234A (also known as ITFG3; rs56038902, Stumvoll P = 9.54 × 10−7) (Supplementary Fig. 5). Allelic effects increasing ubiquitin-conjugating enzyme E2E2 (UBE2E2) expression reduce BCF and are associated with higher T2D risk (Extended Data Fig. 6a–d and Supplementary Table 14). This supports the long-standing notion that β-cell homeostasis relies on the ubiquitin–proteasome system44,45. eTWAS also revealed associations between ITFG3 expression and BCF (Supplementary Fig. 5). The non-coding rs56038902-T allele, which increases islet ITFG3 expression, is also associated with lower CIR and Stumvoll measures (Extended Data Fig. 6e–h) but higher T2D risk and glucose levels46 (Supplementary Table 15). Consistent with this, exome-sequencing analysis identified significant associations between ITFG3 loss-of-function variants and lower blood glucose levels and a 36% reduction in T2D risk, respectively47. Additionally, genetic effects on splicing variation supplied eight target genes, three of which were unique to splicing regulation (Supplementary Fig. 6, Extended Data Fig. 7 and Supplementary Table 14). Overall, we identified 92 candidate molecular effectors (82 protein-coding genes and six long non-coding RNAs), 31 of which lack functional islet evidence such as that provided by previous large-scale transcriptomic and epigenomic studies (Supplementary Table 16). Protein-coding genes were enriched in islet-specific co-expression networks (Fig. 3e and Supplementary Table 17), known regulators of BCF (for example, ADCY5, KCNJ11, TCF7L2) and genes implicated in monogenic (for example, HNF1A, PDX1, ABCC8) and common (for example, IGF2BP2, C2CD4A/4B, PAM) forms of diabetes (Fig. 3f and Supplementary Table 17). Our findings provide a comprehensive list of candidate genes relevant to insulin release regulation and diabetes pathophysiology.
We next explored the mechanisms by which candidate genes at two distinct loci might influence insulin release. Disposition indexes identified the ACSL1 locus. Integrating fine-mapped DI and DIBIG associations with epigenomic annotations localized rs4862423 (DI PP = 0.23 and DIBIG PP = 0.13, linkage disequilibrium (LD) with ACSL1-lead rs10022124r2 = 0.85) and rs72695654 (DI PP = 0.00040, LD with ACSL1-lead rs10022124r2 = 0.12) to islet enhancers connected through chromatin interactions13 to the ACSL1 gene, encoding the long-chain fatty acyl-CoA synthetase 1 (Fig. 4a and Supplementary Table 12). Although TWAS did not report a significant association between ACSL1 expression and BCF, a previous large-scale human islet eQTL mapping study found strong colocalization between ACSL1 eQTLs and T2D risk through rs58730668(ref. 48), which was also prioritized through DI fine-mapping (DI PP = 0.0004, LD with ACSL1-lead rs10022124r2 = 0.12). We observed that the DI rs58730668-C allele is associated with higher ACSL1 expression, lower T2D risk and higher BCF (Supplementary Table 15). Additionally, DI and DIBIG rs4862423-T and rs72695654-T alleles, located in islet enhancers, were also associated with decreased BCF, increased glycaemic levels49 and T2D risk50 (Fig. 4b and Supplementary Table 15). ACSL1 showed robust colocalization between disposition indexes and fasting glucose (DI PP.H4 = 0.998, DIBIG PP.H4 = 0.998) and with T2D via other BCF traits (for example, BIGTT-AIR PP.H4 = 0.85) (Supplementary Table 8). We then examined the adjacent CENPU gene as a potential molecular effector in addition to ACSL1. Despite the close proximity of the enhancer containing rs4862423 to the CENPU promoter, colocalization of molecular QTLs or TWAS data from this study or others did not support a role for CENPU in BCF or diabetes-related traits. Single-cell RNA sequencing (RNA-seq) datasets specifically highlighted ACSL1 as relevant in human pancreatic endocrine cells (Extended Data Fig. 8). Based on the convergence of islet 3D-chromatin interactions, eQTLs and single-cell RNA-seq data, ACSL1 was nominated as a strong molecular mediator of this GWAS association. Earlier work demonstrated that silencing two ACSL1 paralogs impairs insulin release by altering the fatty acid composition of insulin granules and mitochondrial function51. We thus silenced ACSL1 mRNA using small interfering RNA (siRNA) in EndoC-βH1 and INS-1 832/13 cell lines (Extended Data Fig. 9a,b), resulting in impaired insulin release in response to stimulation by glucose, pyruvate and KCl (Fig. 4c–g). Acyl-CoAs have long been promoted as important metabolic coupling factors regulating fuel-mediated insulin secretion52. Hence, the abrogated formation of acyl-CoA upon ACSL1 knockdown was expected to inhibit insulin release, as we observed. Thus, the human genetic evidence presented here supports this widely held53, though only partially proven, mechanism of insulin release regulation54. We also examined the adjacent gene Cenpu by silencing it in INS-1 832/13 cells, which resulted in a significant decrease in insulin release and total insulin content, along with abnormal cell morphology (Extended Data Fig. 9e,f). However, Cenpu gene silencing did not affect fractional insulin release when adjusted for insulin content, unlike Acsl1 (Extended Data Fig. 9c,d,g,h). Given that CENPU encodes for centromere protein U, which holds a crucial role in cell proliferation, we presume that CENPU modulates growth or differentiation in the immortalized β-cell line rather than stimulus-secretion coupling. Collectively, cellular, genetic and epigenomic evidence supports the role of ACSL1 as the most likely molecular mediator of this BCF association, offering the potential for diabetes-modifying strategies.

a, Regional signal plot at the ACSL1 locus, showing P values on a −log10 scale (y axis) in hg19 locations (x axis) from DIBIG single-variant GWAS meta-analyses. Variants are coloured by their LD correlation (r2) with the lead variant (rs10022124). Below, epigenomic datasets are shown, including ATAC-seq and ChIP-seq data in human pancreatic islets. Enhancer-gene assignments from pcHi-C data are represented as pink arcs; those inferred using pcHi-C data are in purple. Fine-mapped DI and DIBIG signals located in islet active enhancers connected to ACSL1 and CENPU, or identified as T2D-colocalizing islet ACSL1 eQTLs, are highlighted. b, Forest plot of the strongest functionally prioritized signal at the ACSL1 locus (rs4862423, DIBIG). For each of the BCF traits, glycaemic levels and T2D risk, the square represents the β estimate with 95% CI error bars. The square size reflects precision. The effect allele and variant RSID are provided at the top. Summary statistics data were generated in this study or obtained from previous publications49,50 and FinnGen (release v.8). OR, odds ratio. c,d, Insulin secretion in human EndoC-β H1 cells following ACSL1 siRNA silencing in response to high glucose (c) and KCl stimuli (d). e–g, Insulin secretion in INS-1 832/13 cells upon Acsl1 gene silencing in response to high glucose (e), KCl (f) and pyruvate (g). Bar plots show the means of three (in c and d) and four (in e–g) independent replicates; error bars, s.e.m. Statistical significance was assessed using one-way ANOVA followed by Tukey’s post hoc test of each condition against the negative control group (scrambled siRNA).
At the BIGTT-AIR TENT5C-DT locus (also known as LOC100996263), TWAS identified associations between FAM46C expression (encoding a member of the nucleotidyltransferase gene superfamily) and several BCF indices (Supplementary Fig. 5). Fine-mapping of BIGTT-AIR signals prioritized rs1975283 (PP = 0.028, LD with GWAS-lead rs320370r2 = 0.83), which also leads eTWAS associations between BCF traits and FAM46C expression. This variant resides within an islet enhancer connected to FAM46C through chromatin-based enhancer-gene assignments (Extended Data Fig. 10a). Strong colocalization between BCF traits and T2D was detected at the FAM46C locus (Supplementary Table 8). The rs1975283-A allele was associated with increased FAM46C islet expression, negative effects on BCF, higher T2D risk (odds ratio, 1.03, P = 3.51 × 10−8) and elevated blood glucose levels (β = 0.01, P = 2.29 × 10−18)55,56 (Extended Data Fig. 10b–d and Supplementary Table 13). This epigenomic and transcriptomic evidence supports FAM46C as a strong candidate effector for this locus. The polyadenylation activity of FAM46C has been linked to enhanced expression of endoplasmic reticulum-targeted proteins57, concordant with the role of FAM46 proteins in RNA stabilization58. To delve into the effects of FAM46C on BCF, we silenced FAM46C mRNA in EndoC-βH1 and INS-1 832/13 cells (Extended Data Fig. 10e,h). This resulted in a significant inhibition of insulin release in response to all the secretagogues tested (Extended Data Fig. 10f,g,i–n). Consistently, the International Mouse Phenotyping Consortium reported mild glucose intolerance for female heterozygous Tent5c knock-out mice (area under glucose-response curve, wild-type 11,408.5 ± 2,532.2 vs heterozygous 13,763.2 ± 2,011.7 min × mg dl−1, P = 0.04)59. FAM46C mRNA levels in 188 human islet samples showed significant positive correlations with glucagon (r = 0.65, P = 6.06 × 10−24) and IAPP (r = 0.68, P = 1.14 × 10−26) expression60, alongside the co-expression of genes relevant for insulin exocytosis (SYT13, SYT16, STXBP4), proinsulin processing (PCSK1, CPE) and β-cell differentiation (NEUROD1)83 (Supplementary Table 18). These results underscore the important role of FAM46C in BCF and glucose homeostasis. However, further research is needed to fully elucidate its precise role in the insulin secretory function.
Altogether, this study offers a more comprehensive perspective of the genetic architecture of oral glucose-stimulated insulin release. Our data show that the eight BCF indices provide both shared and specific contributions to the genetics of β-cell secretory function. We report intricate patterns of shared genetic effects on BCF, T2D risk, BMI and glucose metabolism-relevant traits. Our results expand the repertoire of effector genes potentially influencing T2D risk through insulin secretory pathways, highlighting ACSL1 and FAM46C as additional BCF regulators. These findings advance the understanding of glucose-stimulated β-cell response and offer promising avenues for drug discovery in diabetes.
Methods
Ethics statement
All contributing studies included in this analysis were approved by relevant ethical committees and participants provided informed consent. Specific ethics statements for each study are provided in Supplementary Table 2.
Statistics and reproducibility
To investigate the genetic architecture of the BCF, we conducted multiple analyses, including single-variant association studies across nine cohorts assembled in eight meta-analyses for each of the BCF traits. We examined the independence of the eight BCF traits through genetic and phenotypic correlations, heterogeneity tests of the effect sizes and pleiotropic effects using colocalization, Mendelian randomization and by assessing BCF allelic effects in other relevant complex traits. To identify molecular effectors for BCF-GWAS loci, we carried out heritability enrichment analyses across tissue-specific chromatin annotations and cell-type gene expression data. We prioritized candidate target genes using epigenomic data and genetic effects on transcriptome activity. Finally, we assessed the role of selected candidate target genes in insulin secretion using gene silencing in human and rat cell lines. Detailed descriptions of all analyses are provided below.
BCF estimates
This study examined the genetic basis of eight BCF estimates derived from a 75 g OGTT. Plasma glucose and serum insulin data at 0, 30 and 120 min during the OGTT were used to calculate the BCF indices, which include BIGTT-AIR (BIGTT-AIR0–30–120)19, CIR21, Stumvoll21, xinsdG30 (ref. 22), xinsG30 (ref. 24), DI23 and DIBIG19. We also incorporated HOMA-β25, derived from steady-state measures of fasting glucose and insulin. For mathematical formulations, see Supplementary Table 1. Even though multiple indices, such as disposition indexes, have emerged to model the complex hyperbolic relationship between β-function and insulin resistance, there is no consensus on the most appropriate estimate40,61,62,63. We selected DI, which is widely accepted in the literature, and DIBIG, developed by our lab.
We categorized these eight BCF measures into three groups: insulin secretion estimates during an OGTT, insulin secretion estimates during an OGTT adjusted for insulin sensitivity (that is, disposition indexes) and a surrogate BCF index based on steady-state fasting glucose and insulin measurements.
Study-specific analyses
BCF measures were analysed alongside genetic data from 26,356 individuals of Northern European descent, recruited across nine cohorts. Participants were genotyped using various genome-wide single-nucleotide polymorphism (SNP) arrays (Supplementary Table 2). We implemented a quality control to each cohort for genotype data that excluded samples if the call rate was <95%, heterozygosity was greater than the median plus three times the interquartile range, if there was discordance between genetically inferred and self-reported sex, duplicates and outliers identified using PCA projection of the study samples onto 1 KG reference samples. Variants were excluded if the variant call rate was <98%, there was a departure from Hardy–Weinberg equilibrium of P < 10−5 (or <10−4 if the data were genotyped in batches; also by inspecting Hardy–Weinberg P value distribution to minimize peaks of low P values) and monomorphic markers. Variants were imputed using the Michigan Imputation Server (https://imputationserver.sph.umich.edu)64 with the HRC reference panel (GRCh37 genome build, HRC r1.1 2016 release)65 and Eagle (v.2.3) for haplotype phasing. For the PPP-Botnia Study, genotype imputation was performed using a workflow linked to the THL Biobank’s SISu v.3 WGS imputation reference panel (https://doi.org/10.17504/protocols.io.nmndc5e).
For each study, we used linear models to test genetic associations with BCF indices using inverse normal rank-transformed values adjusted by BMI, age, age2 and sex. No sex-specific analyses were performed, as the aim of this study was to map the genetic architecture of glucose-stimulated insulin secretory function independently of sex. Covariates used in BCF calculations, such as BMI and sex in BIGTT-AIR and DIBIG, were not included in the adjustments. To account for population structure and relatedness, association testing was also adjusted by genetic principal components calculated with PLINK66 (Supplementary Table 2) and kinship using a genetic relationship matrix (Balding–Nicols model) computed with vcf2kinship (https://github.com/zhanxw/rvtests/blob/master/vcfUtils/vcf2kinship.cpp). Before calculating the principal components, data pruning was performed with PLINK (v.1.9) using the settings (–indep 200 20 2–maf 0.01–thin 0.1)66. Additional association testing was conducted with linear models after removing related individuals (PI_HAT > 0.125) and adjusting for the same covariates as the linear mixed models (Supplementary Table 2). Individuals with known diabetes or those meeting the World Health Organization’s 2006 criteria for diabetes (fasting glucose of >7 mmol l−1 and 2 h glucose of ≥11.1 mmol l−1) were excluded before association testing. The Danish-based cohorts and PPP-Botnia Study were analysed using the RVTEST (v.20171009) software67, the Botnia Family Study was analysed using EPACTS (v.3.3.0)68 and METSIM was analysed using GTCA69. Complementary GWAS analyses were conducted without BMI adjustment.
Meta-analysis
Summary statistics from nine GWAS were combined using a fixed-effects meta-analysis with METAL (v.2011-03-25)70, using standard error-based weights for effect sizes. Variants with multiallelic sites, a minor allele frequency (MAF) of <0.001 (<0.01 for the GDM offspring cohort) and imputation quality of <0.6 were excluded. Genomic control was applied to standard errors to address between-study structure. METAL used Cochran's Q-test to assess heterogeneity, excluding variants with heterogeneity of >75% and n × MAF > 43. Genomic inflation was estimated using variants with MAF × n > 50, MAF > 1% and MAF > 5% (Supplementary Figs. 1–3). Consistent results were observed across all MAF filters. GWAS genomic loci were defined using the IRanges package in R (v.3.6.1) as non-overlapping 500 kb windows (merged otherwise) with a GWAS-significant variant (P ≤ 5 × 10−8). For multiple-test correction across eight BCF traits, significance was set at P ≤ 6.25 × 10−9. BCF-GWAS loci were named based on the nearest gene.
Detection of independent BCF signals
Conditional analysis to identify independent signals at each genome-wide significant BCF was performed using genome-wide complex trait analysis (GCTA (v.1.91.2 beta)71, GCTA-COJO). The Inter99 cohort (Supplementary Table 2) served as the reference dataset to model LD structure, excluding SNPs with low imputation quality (MINIMAC R2 < 0.6) and individuals with relatedness (PI_HAT > 0.125). We examined genome-wide significant variants within a ~250 kb range centred on the lead BCF signal. Signals with P < 10−5 were considered independent. Conditional analysis was iterated until no additional independent variants were found, with reciprocal conditional analysis confirming each additional signal. The final set of independent BCF variants was determined by clumping all independent lead variants across the eight BCF estimates using the R package TwoSampleMR with an R2 cut-off of 0.05, using individuals of European descent as the reference dataset.
Assessing differences between various estimates of BCF
Phenotype correlations between the eight BCF estimates were calculated at the study-specific level using Pearson correlation with rank-based inverse normal transformed values, adjusting for sex, age and BMI. Diabetic individuals were removed from this analysis. Phenotype correlations were aggregated in a meta-analysis conducted in R (v.3.4.2) using the meta package (v.4.9-2)72, applying a fixed-effect model based on Fisher’s Z-transformation of the correlation coefficients.
LD score (LDSC) regression was used to calculate pairwise genetic correlations between the eight BCF estimates based on the proportion of shared genetic variance73,74 (https://github.com/hillfung/rg-matrix). Following the developers’ guidelines, calculations were based on GWAS data without genomic control, using association estimates derived from linear models.
For clustering BCF estimates based on genetic and phenotypic correlations, we used complete hierarchical clustering with Euclidean distance, as implemented in the R (v.3.6.1) stats package.
The heterogeneity of the effect sizes between BCF estimates was assessed using estimated standardized differences:
Here, t1 and t2 represent the two traits being compared, ‘effect’ is the effect size, ‘stderror’ is the standard error of the effect size and ρ is the genetic correlation between the two traits, which was calculated considering all variants and using Pearson correlation as implemented in the R (v.4.0.2) stats package.
Known BCF-GWAS loci
We classified a BCF locus as ‘previously known’ if it met any of the following criteria: (1) association with any of the eight BCF estimates listed in the National Human Genome Research Institute–European Bioinformatics Institute (NHGRI-EBI) GWAS Catalog75 (Supplementary Table 7), (2) association reported with any of the eight BCF traits through T2D candidate gene studies or (3) identification as a T2D genetic association linked to a β-cell dysfunction genetic cluster or a fasting proinsulin genetic signal. For criterion (3), islet functional evidence was required to confirm known BCF associations.
The NHGRI-EBI GWAS Catalog was accessed on 30 January 2023 and lifted from GRCh38 to GRCh37 using LiftOver (https://genome.ucsc.edu/cgi-bin/hgLiftOver). We investigated two sets of variants: those with a P value of <5 × 10−8 or with a P value of <5 × 10−6 in LD with BCF lead variants (R2 > 0.6 or R2 > 0.1, respectively). Proxies were identified using the LDlinkR R (v.4.1.0) package and European individuals as the reference dataset76. We also checked LD (R2 > 0.1) using LDLINK (https://ldlink.nih.gov) between BCF lead variants of this study and those from known GWAS of insulin secretory function or related traits that account for islet functional evidence. Details on the different datasets used are provided in Supplementary Table 19. BCF loci not previously reported in GWAS datasets for BMI, T2D or glycaemic-related traits were further investigated using the T2D Knowledge Portal (https://t2d.hugeamp.org) and the MR-Base platform (https://gwas.mrcieu.ac.uk/phewas)77,78.
BCF heritability and the relationship with other traits
Genetic correlations with relevant traits were calculated using LDHub (http://ldsc.broadinstitute.org). Metabolic-related traits were investigated together with brain-related traits owing to the neuroendocrine origin of β-cells. We also excluded traits with negative heritability, those that are duplicated and traits connected to the following categories: (1) education, (2) cancer, (3) smoking and alcohol, (4) personality, (5) reproductive system, (6) skin and nails, (7) uric acid, (8) metal, (9) medication, supplements and consumption, (10) eyes and vision, (11) certain infectious and parasitic diseases, (12) other, (13) aging, (14) bone, (15) autoimmune and inflammatory, (16) sleeping, (17) lung system and (18) physical activity.
Only GWAS summary statistics data from individuals of European descent were included. LDHub leverages LDSC regression; therefore, we used the same GWAS summary statistics used for genetic correlation calculations. Multiple testing correction was applied based on eight main categories of complex traits (P < 0.05 in seven categories: P = 0.00714).
Heterogeneity tests between BMI-adjusted and BMI-unadjusted models were assessed as described in equation 1.
For the study-specific heatmap, we selected the strongest non-palindromic association for each BCF locus that was significant for most traits and had GWAS data available for BCF, T2D and BMI (LD R2 > 0.7 with BCF variant). Effect sizes were aligned to the BCF-lowering allele. Proxies could not be identified for two loci with palindromic sequences: LOC100996263 and PROX1-AS1. The allelic effects of the final set of 42 qualifying BCF signals were examined using GWAS datasets for T2D, BMI and other relevant traits (Supplementary Table 20).
GWAS summary statistics for body fat percentage were calculated for individuals of European ancestry from UK Biobank using REGENIE (v.3.0)79. Body fat percentage was rank-based inverse normal transformed per sex. The following covariates were used for association testing: age, age2, PC1-10, sex, chip and centre.
GWAS summary statistics for body incretins were calculated as follows: blood samples for measurement of GIP and GLP-1 were drawn into EDTA tubes and immediately put on ice. Radioimmunological determinations of total plasma GIP and GLP-1 concentrations were performed as previously described80,81,82,83. The analytical detection limit was 1 pmol l−1 and intra-assay and inter-assay coefficients of variation were <6% and <15%, respectively. The incremental areas under the curves (iAUCs) of plasma GIP and GLP-1 concentrations were calculated using the trapezoid rule, from 0 to 30 min (iAUC 0–30 min) and 0 to 120 min (iAUC 0–120 min) during an OGTT. Participants were genotyped by the Illumina Infinium HumanCoreExome Beadchip platform. Genotypes were called using the Genotyping module (v.1.9.4) of GenomeStudio software (v.2011.1; Illumina). Closely related individuals, individuals with an extreme inbreeding coefficient, individuals with mislabelled sex, individuals with a call rate of <95%, duplicates and individuals identified as ethnic outliers as well as genetic markers with a call rate of <98%, MAF < 0.01 and a Hardy–Weinberg equilibrium P value of <1 × 10−5 were removed during quality control. Imputation was performed based on the 2,184 haplotypes available from the 1000 Genomes Project Phase 1, version 3 (ref. 84) using IMPUTE85. Associations between genetic variants and plasma incretin hormone levels were studied using a linear mixed model (EMMAX) implemented in the EPACTS software package86 using inverse-normalized residuals of the trait adjusted for age, sex and BMI in each cohort separately. Individuals with missing information were excluded from the analysis.
Colocalization analysis with T2D, fasting glucose and BMI
Colocalization was conducted using the coloc package (v.5.2.2) in R (v.4.3.1) with default settings. We analysed the fraction of genetic variants with MAF > 0.001 within a 500 kb window around each BCF lead variant. For T2D colocalization analysis, we harnessed a large-scale European-based T2D meta-analysis55. Genetic variants were aligned with those in our BCF-GWAS meta-analysis. The same approach was applied to BMI87 and BMI-adjusted fasting glucose49 colocalization analyses.
Mendelian randomization
Mendelian randomization was conducted using the TwoSampleMR package in R (v.4.3.1; 2023-06-16)78,88 with summary statistics from a recent large-scale European-based T2D meta-analysis55. T2D GWAS data were harmonized to align with BCF-GWAS summary statistics. Steiger filtering was applied to select BCF genetic variants that explained more variance (r2) in BCF than in T2D.
Fine-mapping of variants associated with BCF estimates
We fine-mapped 500 kb genomic regions centred on each BCF-independent signal, including all variants with LD r2 ≥ 0.1 with the lead SNP. LD calculations were based on individuals of European descent from the Genomes Project (http://www.1000genomes.org)84. Summary statistics from the conditional analysis were used to fine-map independent signals identified in subsequent conditional analyses. Credible sets were built using a Bayesian approach89. For each variant, the approximate Bayes’ factor was calculated based on effect size and standard error. PPs were then computed, variants were ranked in descending order of approximate Bayes’ factor and cumulative PPs were generated. Genetic signals were included in the credible set until cumulative PP exceeded 99%.
Heritability enrichments on chromatin annotations
To estimate the enrichment for BCF heritability in functional genomic regions, we used stratified LDSC regression (S-LDSC)90. For the sake of comparison, in addition to our BCF-GWAS data, we also included additional traits connected to pancreatic islet cell biology as well as a broad range of complex traits and diseases (https://alkesgroup.broadinstitute.org/LDSCORE/all_sumstats). S-LDSC was performed across epigenomic annotations comprising enhancer and promoter elements identified in human pancreatic islets but also across a wide range of tissues and cell types. For pancreatic islets, we selected active enhancer and active or bivalent promoter states from a pancreatic islet regulome published elsewhere13. For other tissues and cell types, we relied on chromHMM states generated by EpiMap (https://personal.broadinstitute.org/cboix/epimap/ChromHMM/observed_aux_18_hg19/CALLS)35. We overlaid each set of enhancer and promoter annotations identified in each of the selected tissues with 1000 Genomes Phase 3 variants, as provided by S-LDSC authors. We next generated binary annotations and custom LDSC files. For each trait, we estimated heritability enrichments relative to the annotations included in the baseline LD model (v.2.2). We computed two-sided P values for per-SNP heritability Z-scores. Enrichment P values and per-SNP heritability P values were corrected for multiple testing using the Benjamini–Hochberg procedure (α = 0.01).
Cell-type gene expression enrichments using CELLECT
Cell-type-specific gene expression enrichments were calculated using the CELL-type Expression-specific integration for Complex Traits (CELLECT)-LDSC91, based on BCF-GWAS summary statistics and single-cell RNA-seq data from human donors37 and the Tabula Muris36. For the Tabula Muris dataset, the following tissues were excluded: lung, mammary, heart, thymus, trachea, bladder, skin, spleen, tongue and marrow.
Target gene identification using epigenomic and chromatin interaction datasets
We leveraged 99% credible sets for all our BCF sentinel SNPs and overlaid them with islet regulome annotations13. Fine-mapped variants located in islet promoter elements were directly connected to their target genes through transcription start site overlap. For fine-mapped variants within distal regulatory elements, we used previously identified enhancer-gene assignments13 to establish links to their target genes.
Target gene identification using transcriptomic datasets
Candidate genes for BCF traits were also identified by (1) TWAS, (2) colocalization between eQTLs or sQTLs and BCF associations and (3) the overlap between fine-mapped eQTLs and sQTLs and fine-mapped BCF variants. For approach (3), we overlaid BCF credible set variants with fine-mapped eQTL and sQTL variants42. For approach (1), we conducted TWAS as in FUSION43. We used the FUSION_assoc_test.R script to test associations between imputed gene expression (or splicing activity) and each BCF trait, respectively. We advocated for the FUSION software because of its ability to test expression or splicing trait associations using pre-computed SNP weights for gene expression or splicing activity (trained in a panel of 399 human islet samples with genotype and transcriptome data available42) and BCF-GWAS summary statistics data. This avoids using individual-level data to impute gene expression or splicing junction activity in our BCF-GWAS cohorts. We filtered the BCF-GWAS summary statistics data by the genomic coverage of the panel used to train per-gene predictive models of genetic effects on islet gene expression and splicing junction usage (~6.5 common genetic variants). Multiple-testing correction for the TWAS P values was applied using Bonferroni correction, considering 2,851 and 5,804 features for expression and alternative splicing, respectively. For approach (2), we also relied on the FUSION software that allowed us to implement colocalization analysis while performing TWAS. This approach looks for evidence of a shared causal genetic association between each BCF trait and islet gene expression or splicing activity. FUSION uses the coloc software to compute approximate colocalization statistics based on the marginal FUSION weights. We used the median sample size across all variants tested for each BCF study (–GWASN) and set–coloc_P to 0.00001. Note that the TWAS results are unfitted to distinguish causal relationships from linkage92. Consequently, we discarded TWAS signals with low colocalization evidence and are hereby confounded by linkage (coloc PP4 < 0.6). Finally, TWAS associations were linked to BCF-GWAS loci if the TWAS lead variant was in LD (r2 ≥ 0.1, using the genotypes of individuals with European descent from Phase 3 of the 1000 Genomes Project) or within 500 kb from any of the BCF sentinel SNPs.
To visualize colocalization between islet eQTLs and BCF associations at selective GWAS loci or TWAS associations, we used LocusCompareR (v.1.0.0) in R (v.3.6.1). If the LD (r2) correlation information for the lead QTL or TWAS association was not available in pre-calculated LD datasets, we used PLINK66 (v.1.9) with the settings –ld-window-kb 1000–ld-window 99999–ld-window-r2 0, and genotypes from 399 human islet samples used in the TWAS analysis as reference dataset. eQTL P values for the ITFG3 eTWAS association were computed here using genotype and RNA-seq data from 399 human islet samples42, as this signal was missed in the original eQTL mapping analysis. We applied the same eQTL mapping guidelines as in the original study. eQTLs for ITFG3 have also been identified in other islet eQTL mapping studies, including rs56314769 (ref. 12) and rs6600191(ref. 48), which are in LD with the lead ITFG3 eTWAS association (rs56038902r2 = 0.6 and r2 = 0.8, respectively).
We observed that pcHi-C assignments supported three and five candidate genes identified by eTWAS and sTWAS, respectively. Limited overlap between BCF candidate genes identified through eTWAS and pcHi-C approaches could be partially explained by the limited discovery power of eQTL mapping on GWAS-relevant traits, as recently discussed93. Likewise, the ability of pcHi-C assignments to identify BCF target genes from credible set variants is hampered by several factors, including stringent significance thresholds for chromatin loop calling, distance to the target gene, enhancer discovery in the disease-relevant cell state and the resolution of genetic fine-mapping to prioritize causal regulatory variants. Therefore, using complementary functional approaches could surmount the intrinsic technical limitations of each individual strategy.
Visualization of regional association plots
Regional association plots for BCF susceptibility regions were created using LocusZoom94 (v.1.4) in R (v.3.6.1).
Visualization of splicing events
IGV95 was used to visualize islet splice junctions identified elsewhere42 as arcs.
Assessing the impact of non-coding fine-mapped variants on transcription factor binding
We used motifbreakR96 to predict the impact of BCF fine-mapped variants located in islet regulatory elements on transcription factor binding. Human motif information was sourced from the HOCOMOCO and Jaspar databases via MotifDb. The parameters used where filterp = TRUE, threshold = 5 × 10−5, method = ‘ic’. We repeated the calculations using nucleotide background frequencies estimated in islet enhancers (A = 0.270182, C = 0.2290216, G = 0.2297711, T = 0.2710253). Results were filtered for ‘strong’ effects with a score greater than 0.8 for any of the alleles tested (pctRef > 0.8 | pctAlt > 0.8).
Enrichment of functional annotations among candidate target genes
We used the R interface of enrichR97 (last accessed on 13 June 2023) to identify relevant functional annotations enriched by the protein-coding molecular effector transcripts nominated using islet epigenomic and transcriptomic datasets.
Cell culture
The EndoC-βH1 cell line (MTA from Endocells Sarl) is derived from female human foetal pancreatic buds implanted into SCID mice, maturing into pancreatic tissue and eventually giving rise to insulinoma cells after differentiation. The sex was not considered in the choice of the cell line sources. EndoC-βH1 cells were cultured as previously described98 on Matrigel-fibronectin-coated culture vessels (Matrigel at 100 μg ml−1 and fibronectin at 2 μg ml−1, both from Sigma-Aldrich) in DMEM supplemented with 5.6 mM glucose, 2% BSA fraction V (Roche Diagnostics), 10 mM nicotinamide (Merck Millipore), 50 μM 2-mercaptoethanol, 5.5 μg ml−1 transferrin and 6.7 ng ml−1 sodium selenite (Sigma-Aldrich). The INS-1 832/13 cell line is male and derived from the rat insulinoma cell line INS-1, following stable transfection with a plasmid containing the human proinsulin gene. One of the authors (H.M.) is a co-creator of the INS-1 832/13 cell line and has unrestricted access to it99. INS-1 832/13 cells were cultured in RPMI-1640 containing 11.1 mM glucose, 10% fetal bovine serum, 10 mM HEPES, 2 mM glutamine, 1 mM sodium pyruvate and 50 μM β-mercaptoethanol as previously described99. Both cell lines were cultured at 37 °C with 5% CO2. INS-1 832/13 cells were previously authenticated through PCR analysis of Ins1 and Pdx1 mRNAs. EndoC-βH1 cells were also previously authenticated through PCR analysis of insulin and β-cell transcription factors PDX1, PAX6, FOXA2, NEUROD1, MAFB and NKX6.1.
Gene silencing
Gene silencing of ACSL1, FAM46C and CENPU expression by RNA interference was performed in EndoC-βH1 and INS-1 832/13 insulin-producing cells. Lipofectamine RNAiMAX Reagent and 10 nM siRNAs specific for human ACSL1 and FAM46C, rat Acsl1, Fam46c and Cenpu, as well as a scrambled siRNA as negative control were used. After 72 h of transfection, gene expression and β-cell secretory activity were examined by quantitative PCR (qPCR) and insulin secretion assays, respectively.
Human ACSL1: Thermo Fisher Scientific; s129379 and s129380. Rat Acsl1: Thermo Fisher Scientific; s4994 and s4995. Human FAM46C: Thermo Fisher Scientific; s29596 and s29597. Rat Fam46c: Thermo Fisher Scientific; s160621 and s221157. Rat Cenpu (Thermo Fisher Scientific; s157528). Scrambled siRNA as a negative control for rat, custom select siRNA 50- GAGACCCUAUCCGUGAUUAUU-30. Scrambled siRNA as a negative control for human, Silencer Select Negative Control No. 1 siRNA (Thermo Fisher, 4390843).
mRNA expression analysis
Total RNA was extracted using the RNeasy Mini Kit (QIAGENiagen) and cDNA was generated with the RevertAid First-Strand cDNA synthesis kit. For qPCR, 10 ng of cDNA per well (in duplicates) was used with human ACSL1 and FAM46C, or rat Acsl1, Fam46c and Cenpu Taqman gene expression assays (Thermo Fisher Scientific; code: Rn01488229_m1, Rn01534138_m1, Rn01488384_m1, Hs00960561_m1, Hs01933465_s1). qPCR assays were performed using the Applied Biosystems QuantStudio7 Flex Real-Time PCR System (Thermo Fisher). Relative gene abundance was calculated using the ΔΔCt method with β-actin as the housekeeping reference gene. Gene expression values were reported as fold changes relative to the control.
Measurement of insulin secretion in EndoC-βH1 cells
The day before the experiment, EndoC-βH1 cells were switched to an overnight starvation medium containing 2.8 mM of glucose. On the experiment day, the starvation medium was replaced with HEPES-buffered Krebs–Ringer Buffer (115 mM NaCl, 24 mM NaHCO3, 5 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 10 mM HEPES, 0.2% BSA pH 7.4) containing 1 mM of glucose for 1 h. The cells were then stimulated in KRB with either 1 mM or 20 mM glucose, or 35 mM KCl for an additional 1 h. Insulin secretion was measured using a human insulin ELISA (Mercodia) according to the manufacturer’s instructions.
Measurement of insulin secretion in INS-11 832/13 cells
INS-1 832/13 cells were starved for 2 h in a secretion assay buffer (114 mM NaCl, 1.2 mM KH2PO4, 4.7 mM KCl, 1.16 mM MgSO4, 2.5 mM CaCl2, 100 mM HEPES, 25 mM NaHCO3, 0.2% BSA pH 7.2) containing 2.8 mM glucose. Following starvation, the cells were stimulated for 1 h with secretion assay buffer containing either 2.8 mM or 16.7 mM glucose, 10 mM pyruvate or 35 mM KCl at 37 °C. Insulin concentration in the supernatants was measured by a high-range rat insulin ELISA (Mercodia, cat. no. 10-1145-01).
Normalization and statistical analyses
Insulin values for both INS-1 832/13 cells and EndoC-βH1 cells were normalized to the total protein and insulin content. Protein was extracted using RIPA buffer and measured with the BCA Protein Assay Kit. Total insulin content was measured in the same cell lysates to normalize insulin secretion to insulin content, which was then expressed as a percentage of secreted insulin. Analyses were conducted using GraphPad Prism software (v.9) and data are presented as means ± s.e.m. Statistical significance was assessed by Student’s t-test for single comparison and/or one-way ANOVA followed by Tukey’s post-hoc test for multiple comparisons. P values of <0.05 were considered statistically significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
We have made all data related to this study available through Source Data Files and Supplementary Materials wherever possible. Additional resources and relevant datasets can be found as follows. The GWAS summary statistics for the main GWAS model (linear mixed model with outcome variable = genetic variant; predictor variable = inverse normal rank-transformed values; covariates = BMI (excluding BIGTT-AIR and DI), age, age2, sex, principal components and genetic relationship matrix) for eight BCF traits are available on the T2D knowledge portal downloads page (type2diabetesgenetics.org). Summary statistics data from human islet eQTL and sQTLs can be accessed at https://zenodo.org/records/6546807. From the same islet eQTL and sQTL mapping study, RNA-seq and genotyped SNP array data are available at the European Genome-Phenome Archive (EGA) (EGAS00001006440). Processed data files for human islet pcHi-C interactions, epigenomic annotations and enhancer–promoter assignments are available at https://www.crg.eu/en/programmes-groups/ferrer-lab#datasets. Source data are provided with this paper.
References
Defronzo, R. A. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitis. Diabetes 58, 773–795 (2009).
Bonnefond, A. & Froguel, P. Rare and common genetic events in type 2 diabetes: What should biologists know? Cell Metab. 21, 357–368 (2015).
Rose, C. S. et al. A −30G>A polymorphism of the β-cell-specific glucokinase promoter associates with hyperglycemia in the general population of whites. Diabetes 54, 3026–3031 (2005).
Ingelsson, E. et al. Detailed physiologic characterization reveals diverse mechanisms for novel genetic loci regulating glucose and insulin metabolism in humans. Diabetes 59, 1266–1275 (2010).
Grarup, N. et al. Studies of association of variants near the HHEX, CDKN2A/B, and IGF2BP2 genes with type 2 diabetes and impaired insulin release in 10,705 Danish subjects. Diabetes 56, 3105–3111 (2007).
Boesgaard, T. W. et al. Variants at DGKB/TMEM195, ADRA2A, GLIS3 and C2CD4B loci are associated with reduced glucose-stimulated beta cell function in middle-aged Danish people. Diabetologia 53, 1647–1655 (2010).
Udler, M. S. et al. Type 2 diabetes genetic loci informed by multi-trait associations point to disease mechanisms and subtypes: a soft clustering analysis. PLoS Med. 15, e1002654 (2018).
Dupuis, J. et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat. Genet. 42, 105–116 (2010).
Broadaway, K. A. et al. Loci for insulin processing and secretion provide insight into type 2 diabetes risk. Am. J. Hum. Genet. 110, 284–299 (2023).
Gaulton, K. J. et al. A map of open chromatin in human pancreatic islets. Nat. Genet. 42, 255–259 (2010).
Pasquali, L. et al. Pancreatic islet enhancer clusters enriched in type 2 diabetes risk-associated variants. Nat. Genet. 46, 136–143 (2014).
Viñuela, A. et al. Genetic variant effects on gene expression in human pancreatic islets and their implications for T2D. Nat. Commun. 11, 4912 (2020).
Miguel-Escalada, I. et al. Human pancreatic islet three-dimensional chromatin architecture provides insights into the genetics of type 2 diabetes. Nat. Genet. 51, 1137–1148 (2019).
Mahajan, A. et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat. Genet. 50, 1505–1513 (2018).
Prokopenko, I. et al. A central role for GRB10 in regulation of islet function in man. PLoS Genet. 10, e1004235 (2014).
Huyghe, J. R. et al. Exome array analysis identifies novel loci and low-frequency variants for insulin processing and secretion. Nat. Genet. 45, 197–201 (2013).
Gjesing, A. P. et al. Genetic and phenotypic correlations between surrogate measures of insulin release obtained from OGTT data. Diabetologia 58, 1006–1012 (2015).
Williamson, A. et al. Genome-wide association study and functional characterization identifies candidate genes for insulin-stimulated glucose uptake. Nat. Genet. 55, 973–983 (2023).
Hansen, T. et al. The BIGTT test: a novel test for simultaneous measurement of pancreatic-cell function, insulin sensitivity, and glucose tolerance. Diabetes Care 30, 257–262 (2007).
Sluiter, W. J., Erkelens, D. W., Reitsma, W. D. & Doorenbos, H. Glucose tolerance and insulin release, a mathematical approach I. Assay of the beta-cell response after oral glucose loading. Diabetes 25, 241–244 (1976).
Stumvoll, M. et al. Use of the oral glucose tolerance test to assess insulin release and insulin sensitivity. Diabetes Care 23, 295–301 (2000).
Seltzer, H. S., Allen, E. W., Herron, A. L. & Brennan, M. T. Insulin secretion in response to glycemic stimulus: relation of delayed initial release to carbohydrate intolerance in mild diabetes mellitus. J. Clin. Invest. 46, 323–335 (1967).
Matsuda, M. & DeFronzo, R. A. Insulin sensitivity indices obtained from oral glucose tolerance testing: comparison with the euglycemic glucose clamp. Diabetes Care 22, 1462–1470 (1999).
Kadowaki, T., Miyake, Y. & Hagura, R. Risk factors for worsening to diabetes in subjects with impaired glucose tolerance. Diabetologia 1, 44–49 (1984).
Matthews, D. R. et al. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 28, 412–419 (1985).
Turner, R. C., Holman, R. R., Matthews, D., Hockaday, T. D. R. & Peto, J. Insulin deficiency and insulin resistance interaction in diabetes: estimation of their relative contribution by feedback analysis from basal plasma insulin and glucose concentrations. Metabolism 28, 1086–1096 (1979).
Festa, A., Williams, K., Hanley, A. J. G. & Haffner, S. M. Beta-cell dysfunction in subjects with impaired glucose tolerance and early type 2 diabetes: comparison of surrogate markers with first-phase insulin secretion from an intravenous glucose tolerance test. Diabetes 57, 1638–1644 (2008).
Cersosimo, E., Solis-Herrera, C., Trautmann, M., Malloy, J. & Triplitt, C. Assessment of pancreatic β-cell function: review of methods and clinical applications. Curr. Diabetes Rev. 10, 2–42 (2014).
Sung, K. C., Reaven, G. M. & Kim, S. H. Utility of homeostasis model assessment of β-cell function in predicting diabetes in 12,924 healthy Koreans. Diabetes Care 33, 200–202 (2010).
Kahn, S. E. et al. Obesity, body fat distribution, insulin sensitivity and islet β-cell function as explanations for metabolic diversity. J. Nutr. 131, 354S–360S (2001).
Kahn, S. E., Hull, R. L. & Utzschneider, K. M. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 444, 840–846 (2006).
Huang, L. O. et al. Genome-wide discovery of genetic loci that uncouple excess adiposity from its comorbidities. Nat. Metab. 3, 228–243 (2021).
J, R. et al. Genetic variant near IRS1 is associated with type 2 diabetes, insulin resistance and hyperinsulinemia. Nat. Genet. 41, 1110–1115 (2009).
Kilpeläinen, T. O. et al. Genetic variation near IRS1 associates with reduced adiposity and an impaired metabolic profile. Nat. Genet. 43, 753–760 (2011).
Boix, C. A., James, B. T., Park, Y. P., Meuleman, W. & Kellis, M. Regulatory genomic circuitry of human disease loci by integrative epigenomics. Nature 590, 300–307 (2021).
Schaum, N. et al. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 562, 367–372 (2018).
Baron, M. et al. A single-cell transcriptomic map of the human and mouse pancreas reveals inter- and intra-cell population structure. Cell Syst. 3, 346–360.e4 (2016).
Boyko, E. J. & Jensen, C. C. Do we know what homeostasis model assessment measures? If not, does it matter? Diabetes Care 30, 2725–2728 (2007).
Wallace, T. M., Levy, J. C. & Matthews, D. R. Use and abuse of HOMA modeling. Diabetes Care 27, 1487–1495 (2004).
Mari, A., Ahrén, B. & Pacini, G. Assessment of insulin secretion in relation to insulin resistance. Curr. Opin. Clin. Nutr. Metab. Care 8, 529–533 (2005).
Kailey, B. et al. SSTR2 is the functionally dominant somatostatin receptor in human pancreatic β- and α-cells. Am. J. Physiol. Endocrinol. Metab. 303, 1107–1116 (2012).
Atla, G. et al. Genetic regulation of RNA splicing in human pancreatic islets. Genome Biol. 23, 196 (2022).
Gusev, A. et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 48, 245–252 (2016).
López-Avalos, M. D. et al. Evidence for a role of the ubiquitin–proteasome pathway in pancreatic islets. Diabetes 55, 1223–1231 (2006).
Kawaguchi, M., Minami, K., Nagashima, K. & Seino, S. Essential role of ubiquitin–proteasome system in normal regulation of insulin secretion. J. Biol. Chem. 281, 13015–13020 (2006).
Zhao, W. et al. Identification of new susceptibility loci for type 2 diabetes and shared etiological pathways with coronary heart disease. Nat. Genet. 49, 1450–1457 (2017).
Backman, J. D. et al. Exome sequencing and analysis of 454,787 UK Biobank participants. Nature 599, 628–634 (2021).
Alonso, L. et al. TIGER: the gene expression regulatory variation landscape of human pancreatic islets. Cell Rep. 37, 109807 (2021).
Chen, J. et al. The trans-ancestral genomic architecture of glycemic traits. Nat. Genet. 53, 840–860 (2021).
Suzuki, K. et al. Genetic drivers of heterogeneity in type 2 diabetes pathophysiology. Nature 627, 347–357 (2024).
Ansari, I. Ul. H. et al. Characterization of Acyl-CoA synthetase isoforms in pancreatic beta cells: gene silencing shows participation of ACSL3 and ACSL4 in insulin secretion. Arch. Biochem. Biophys. 618, 32–43 (2017).
Prentki, M. et al. Malonyl-CoA and long chain acyl-CoA esters as metabolic coupling factors in nutrient-induced insulin secretion. J. Biol. Chem. 267, 5802–5810 (1992).
Prentki, M., Matschinsky, F. M. & Madiraju, S. R. M. Metabolic signaling in fuel-induced insulin secretion. Cell Metab. 18, 162–185 (2013).
Mulder, H. et al. Overexpression of a modified human malonyl-CoA decarboxylase blocks the glucose-induced increase in malonyl-CoA level but has no impact on insulin secretion in INS-1-derived (832/13) β-cells. J. Biol. Chem. 276, 6479–6484 (2001).
Mahajan, A. et al. Multi-ancestry genetic study of type 2 diabetes highlights the power of diverse populations for discovery and translation. Nat. Genet. 54, 560–572 (2022).
Forgetta, V. et al. An effector index to predict target genes at GWAS loci. Hum. Genet. 141, 1431–1447 (2022).
Mroczek, S. et al. The non-canonical poly(A) polymerase FAM46C acts as an onco-suppressor in multiple myeloma. Nat. Commun. 8, 619 (2017).
Warkocki, Z., Liudkovska, V., Gewartowska, O., Mroczek, S. & Dziembowski, A. Terminal nucleotidyl transferases (TENTs) in mammalian RNA metabolism. Philos. Trans. R. Soc. Lond. B Biol. Sci. 373, 20180162 (2018).
Groza, T. et al. The International Mouse Phenotyping Consortium: comprehensive knockout phenotyping underpinning the study of human disease. Nucleic Acids Res. 51, D1038–D1045 (2023).
Asplund, O. et al. Islet Gene View—a tool to facilitate islet research. Life Sci. Alliance 5, e202201376 (2022).
Pacini, G. & Mari, A. Methods for clinical assessment of insulin sensitivity and β-cell function. Best. Pr. Res Clin. Endocrinol. Metab. 17, 305–322 (2003).
Albareda, M., Rodríguez-Espinosa, J., Murugo, M., de Leiva, A. & Corcoy, R. Assessment of insulin sensitivity and beta-cell function from measurements in the fasting state and during an oral glucose tolerance test. Diabetologia 43, 1507–1511 (2000).
Retnakaran, R. et al. Hyperbolic relationship between insulin secretion and sensitivity on oral glucose tolerance test. Obesity 16, 1901–1907 (2008).
Das, S. et al. Next-generation genotype imputation service and methods. Nat. Genet. 48, 1284–1287 (2016).
Loh, P. R. et al. Reference-based phasing using the Haplotype Reference Consortium panel. Nat. Genet. 48, 1443–1448 (2016).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015).
Zhan, X., Hu, Y., Li, B., Abecasis, G. R. & Liu, D. J. RVTESTS: an efficient and comprehensive tool for rare variant association analysis using sequence data. Bioinformatics 32, 1423–1426 (2016).
Chen, W. et al. A rare variant analysis framework using public genotype summary counts to prioritize disease-predisposition genes. Nat. Commun. 13, 2592 (2022).
Yang, J., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82 (2011).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Yang, J. et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat. Genet. 44, 369–375 (2012).
Balduzzi, S., Rücker, G. & Schwarzer, G. How to perform a meta-analysis with R: a practical tutorial. Evid. Based Ment. Health 22, 153–160 (2019).
Bulik-sullivan, B. K. et al. LD score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
Bulik-Sullivan, B. et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet. 47, 1236–1241 (2015).
Sollis, E. et al. The NHGRI-EBI GWAS Catalog: knowledgebase and deposition resource. Nucleic Acids Res. 51, D977–D985 (2023).
Machiela, M. J. & Chanock, S. J. LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics 31, 3555–3557 (2015).
Hemani, G. et al. The MR-Base platform supports systematic causal inference across the human phenome. eLife https://doi.org/10.7554/eLife.34408 (2018).
Hemani, G. et al. The MR-base platform supports systematic causal inference across the human phenome. eLife 7, e34408 (2018).
Mbatchou, J. et al. Computationally efficient whole-genome regression for quantitative and binary traits. Nat. Genet. 53, 1097–1103 (2021).
Ørskov, C., Rabenhøj, L., Wettergren, A., Kofod, H. & Holst, J. J. Tissue and plasma concentrations of amidated and glycine-extended glucagon-like peptide I in humans. Diabetes 43, 535–539 (1994).
Krarup, T. & Holst, J. J. The heterogeneity of gastric inhibitory polypeptide in porcine and human gastrointestinal mucosa evaluated with five different antisera. Regul. Pept. 9, 35–46 (1984).
Krarup, T. et al. Diminished immunoreactive gastric inhibitory polypeptide response to a meal in newly diagnosed type I (insulin-dependent) diabetics. J. Clin. Endocrinol. Metab. 56, 1306–1312 (1983).
Hoist, J. J. Molecular heterogeneity of glucagon in normal subjects and in patients with glucagon-producing tumours. Diabetologia 24, 359–365 (1983).
Auton, A. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Howie, B. N., Donnelly, P. & Marchini, J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 5, e1000529 (2009).
Kang, H. M. et al. Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 42, 348–354 (2010).
Pulit, S. L. et al. Meta-analysis of genome-wide association studies for body fat distribution in 694 649 individuals of European ancestry. Hum. Mol. Genet. 28, 166–174 (2019).
Hemani, G., Tilling, K. & Davey Smith, G. Orienting the causal relationship between imprecisely measured traits using GWAS summary data. PLoS Genet. 13, e1007081 (2017).
Wakefield, J. A Bayesian measure of the probability of false discovery in genetic epidemiology studies. Am. J. Hum. Genet. 81, 208–227 (2007).
Finucane, H. K. et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 47, 1228–1235 (2015).
Timshel, P. N., Thompson, J. J. & Pers, T. H. Genetic mapping of etiologic brain cell types for obesity. eLife 9, e55851 (2020).
Yao, D. W., O’Connor, L. J., Price, A. L. & Gusev, A. Quantifying genetic effects on disease mediated by assayed gene expression levels. Nat. Genet. 52, 626–633 (2020).
Mostafavi, H., Spence, J. P., Naqvi, S. & Pritchard, J. K. Systematic differences in discovery of genetic effects on gene expression and complex traits. Nat. Genet. 55, 1866–1875 (2023).
Pruim, R. J. et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 26, 2336–2337 (2010).
Robinson, J. T. et al. Integrative Genomics Viewer. Nat. Biotechnol. 29, 24–26 (2011).
Coetzee, S. G., Coetzee, G. A. & Hazelett, D. J. motifbreakR: an R/Bioconductor package for predicting variant effects at transcription factor binding sites. Bioinformatics 31, 3847–3849 (2015).
Kuleshov, M. V. et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97 (2016).
Andersson, L. E. et al. Characterization of stimulus-secretion coupling in the human pancreatic EndoC-βH1 beta cell line. PLoS ONE 10, e0120879 (2015).
Hohmeier, H. E. et al. Isolation of INS-1-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin secretion. Diabetes 49, 424–430 (2000).
Acknowledgements
A.L.M. was supported by a research grant from the Danish Diabetes Academy, which is funded by the Novo Nordisk Foundation, grant number NNF17SA0031406. T.H. was supported by a research grant from the Novo Nordisk Foundation NNF18OC0034532. The Novo Nordisk Foundation Center for Basic Metabolic Research is also supported by an unrestricted grant from the Novo Nordisk Foundation (NNF18CC0034900). S.B.G was supported by a Juan de la Cierva postdoctoral fellowship (MINECO; FJCI-2017-32090). J.F. was supported by Instituto de Salud Carlos III (PMP21/00067), Wellcome Trust (WT101033), European Research Council Advanced Grant (789055), Spanish Ministry of Science and Innovation (RTI2018-095666-B-I00, PID2021-122522OB-I00) and Departament de Recerca i Universitats de la Generalitat de Catalunya (SGR2021-01371). Work in the Centre for Genomic Regulation was supported by the CERCA Programme, Generalitat de Catalunya and Centro de Excelencia Severo Ochoa (CEX2020-001049) and support from the Spanish Ministry of Science and Innovation to the EMBL partnership. M.G.M. was supported by a research grant from the Danish Cardiovascular Academy, which is funded by the Novo Nordisk Foundation, grant number NNF20SA0067242 and the Danish Heart Foundation. T.S.A. was supported by the Novo Nordisk Foundation NNF18OC0052457. The ADDITION-PRO study was funded by an unrestricted grant from the European Foundation for the Study of Diabetes/Pfizer for Research into Cardiovascular Disease Risk Reduction in Patients with Diabetes (74550801), by the Danish Council for Strategic Research and by internal research and equipment funds from Steno Diabetes Center together with Danish Medical Research Council, the Danish Heart Foundation, Danish Health Insurance Foundation, the Foundation of 1870, Fanny Miranda and Willy Rottbøll Rokbølls Foundation, D.M.Sci Torben Geils Foundation, the grant of 22 July 1959 and Director E. Danielsen and Wife’s Foundation. ADIGEN was supported by grants from The Danish Medical Research Council, The Danish National Research Foundation and The Velux Foundation. The Botnia Family Study and the PPP-Botnia Study have been financially supported by grants from Folkhälsan Research Foundation, the Sigrid Juselius Foundation, The Academy of Finland (grant nos. 263401, 267882, 312063, 336822, 312072 and 336826), University of Helsinki, Nordic Center of Excellence in Disease Genetics, EU (EXGENESIS, MOSAIC FP7-600914), Ollqvist Foundation, Swedish Cultural Foundation in Finland, Finnish Diabetes Research Foundation, Foundation for Life and Health in Finland, Finnish Medical Society, State Research Funding via the Helsinki University Hospital, Perklén Foundation, Närpes Health Care Foundation and Ahokas Foundation. The PPP-Botnia Study was genotyped by the Finngen project and the Botnia Family Study by Regeneron Pharmaceuticals. The study sponsors had no role in the analysis and interpretation of the data, writing of the manuscript or decision to submit the manuscript for publication. GDM offspring was supported by Copenhagen University, the P. Carl Petersens Foundation, the Danish Medical Research Council, the Copenhagen University Hospital Foundation, the Danish Diabetes Association, Lægeforeningens Forskningsfond/MC and JK Moltums Fond. Health08 was funded by Timber Merchant Vilhelm Bang’s Foundation, The Danish Heart Foundation and The Health Insurance Foundation (Helsefonden). Inter99 is supported economically by The Danish Medical Research Council, The Danish Centre for Evaluation and Health Technology Assessment, Novo Nordisk, Copenhagen County, The Danish Heart Foundation, The Danish Pharmaceutical Association, Augustinus Foundation, Ib Henriksen Foundation and Becket Foundation. Metabolic Syndrome in Men (METSIM) was supported by the Academy of Finland (contract no. 124243), the Finnish Heart Foundation, the Finnish Diabetes Foundation, Tekes (contract 1510/31/06), the Commission of the European Community (HEALTH-F2-2007 201681) and the US National Institutes of Health grants DK093757, DK072193, DK062370 and ZIA-HG000024. Analyses of UK Biobank data were performed under application no. 1251 (fat percentage GWAS) and no. 32683 (the rest).
Author information
Authors and Affiliations
Contributions
A.L.M., S.B.G., J.F. and T.H. conceived, coordinated and supervised the study. A.L.M. centralized the core of the genetic analysis, including meta-analysis, fine-mapping and genetic correlations. N.G. oversaw the research strategy for the analysis and formulation of BCF estimates. M.G.M. conducted a fat percentage GWAS on UK Biobank data. R.J.F.L. coordinated GWAS analysis on fat percentage. A.E.J. performed a GWAS on incretin levels. A.M.J. and S.H. conducted single-cell expression analysis using CELLECT under the supervision of T.H.P. S.B.G. conducted heritability enrichment analysis in non-coding annotations and in silico variant-to-function analysis, including integration of epigenomic datasets and TWAS analysis. G.A. contributed to the primary analysis of islet sQTLs. I.M. performed chromatin analysis. S.G. and L.R.C. conducted functional analysis for ACSL1 and FAM46C, respectively. M.F. and H.M. supervised the functional analysis on ACSL1 and FAM46C. A.L.M., R.P., G.H., J.V., V.A., O.D., A.L., T.S.A., T.D., O.P., T.I.A.S., A.A., D.W., P.D., T.D.C., E.M., T.T., L.H. and M.L. performed genotyping, phenotyping and association analysis for each study cohort. A.L.M., S.B.G., J.F. and T.H. wrote the manuscript. All authors reviewed, edited and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
G.A. became a full-time employee of Novo Nordisk Ltd. and N.G. became a full-time employee at Novo Nordisk A/S while this manuscript was being drafted. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Metabolism thanks Xianyong Yin, Philippe Froguel and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Yanina-Yasmin Pesch, in collaboration with the Nature Metabolism team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Genome-wide association meta-analyses for eight β-cell function (BCF) estimates.
Manhattan plots (a) and q-q plots (b) using summary statistics of eight single-variant BCF GWAS meta-analyses in up to 26,000 individuals. (a) For each variant tested, association P-values on -log10 scale (y-axis) are plotted against their genomic locations in hg19 (x-axis). The black horizontal line denotes genome-wide significance, P-value ≤ 5x10−8. (b) Observed -log10 (P-values) are compared against expected -log10 (P-values) according to a uniform distribution. Significant loci after Bonferroni multiple-test correction can be found in Supplementary Table 3.
Extended Data Fig. 2 Genetic and phenotypic sharing of β-cell function (BCF) traits.
(a) Genetic (top left) and phenotypic correlations (bottom right) between eight BCF estimates. Phenotypic correlations were calculated using Pearson correlation, and genetic correlations using LDSC. *Significant correlations after Bonferroni multiple test correction, P-value ≤ 0.05 / (8 estimates * 2 correlations tests). (b) Hierarchical clustering using Euclidean distance of genetic and phenotypic correlations among the eight BCF estimates. Colouring highlights each identified subgroup. (c) Venn diagram of shared loci between three subgroups of BCF estimates. Loci are named by the nearest gene. Colour indicates BCF sub-grouping from panel (b), (i) Disposition indexes: DI and DIBIG, (ii) 30’ and 120’ OGTT measurements: BIGTT-AIR, CIR, Stumvoll, xinsdG30, and xinsG30, (iii) fasting values: HOMA-β. (d) Venn diagram of BCF-GWAS loci. Each independent BCF lead association is named according to the nearest gene.
Extended Data Fig. 3 BMI influence on β-cell function (BCF) loci.
BMI influence on BCF-loci for (a) xinsG30, (b) xinsdG30, (c) CIR, (d) Stumvoll, (e) DI, and (f) HOMA-β. Each panel includes Miami plots showing -log10 association P-values with and without BMI-adjustment on the y-axis. Chromosome and genome locations in hg19 are provided on the x-axis. Variants that reached genome-wide significance (P-value ≤ 5x10-8) (indicated by orange dots) were tested for heterogeneity (two-sided P-values) of the effect sizes (right panel). Significant heterogeneous variants are coloured red. The right panel compares effect sizes (Z-scores) without and with BMI-adjustment (x and y-axis, respectively). The -log10 P-values from the heterogeneity test are used as the colouring scheme, with significant heterogeneous loci labelled by the nearest gene. The significance threshold was Bonferroni-adjusted for the number of independent loci (P-value < 0.05 / 55).
Extended Data Fig. 4 Pleiotropic genetic effects between β-cell function (BCF) loci and other relevant traits and diseases.
(a) Heatmap representations show colocalization posterior probabilities from COLOC between each BCF trait and T2D, FG and BMI for 44 BCF loci. Heatmap colours indicate the strength of a shared causal variant hypothesis H4 between each combination of traits being examined. Grouping of BCF loci on the left follows Fig. 2b. (b-c) Insulin sensitivity effects on BCF were assessed by testing (two-sided P-values) for heterogeneity in effect sizes (as Z-scores) among genome-wide significant variants between (b) DI (x-axis) and xinsG30 (y-axis), and (c) DIBIG (x-axis) and BIGTT-AIR (y-axis). The colour legend indicates the -log10 (P-value) of the heterogeneity test estimate. Significant heterogeneous loci are labelled by the nearest gene. The significance threshold was adjusted by the number of independent loci using Bonferroni correction (P-value < 0.05 / 55).
Extended Data Fig. 5 Epigenomic datasets and chromatin interaction maps in human pancreatic islets connect non-coding β-cell function (BCF) genetic associations with (a) SSTR1 and (b) SSTR2 genes.
Regional signal plots show P-values calculated from single-variant BCF GWAS meta-analyses on a -log10 scale (y-axis) across the hg19 genome build (x-axis). Variants are coloured according to their LD correlation (r2) with the lead association. Epigenomic datasets in human pancreatic islets, including chromatin accessibility, histone modifications and TF binding profiles, are shown along with enhancer-gene assignments from pcHi-C. Fine-mapped enhancer variants connected to each molecular effector gene are highlighted with circles coloured according to their LD (r2) with the lead association in the locuszoom plot.
Extended Data Fig. 6 eTWAS associations between β-cell function (BCF) indices and UBE2E2 and ITFG3 islet gene expression.
Forest plots of the lead eTWAS associations for UBE2E2 (a) and ITFG3 (f). For each of the eight BCF traits and T2D, the square represents the β-estimate, and the error bars indicate the 95% confidence intervals. Square size is based on precision, and the effect allele and variant rsid are provided at the top. Summary statistics data were generated here or obtained from Mahajan, A. et al.55. Regional signal plots depict single-variant BCF GWAS meta-analyses P-values (-log10 scale, y-axis) and hg19 locations (x-axis) at the (b) UBE2E2 and (e) ITFG3 loci. LocusCompare scatter plots compare P-values from single-variant BCF GWAS meta-analyses and (c) UBE2E2 and (g) ITFG3 pancreatic islet eQTLs42 on the -log10 scale, respectively. The eTWAS P-value and the colocalization posterior probability (CPP) are provided at the top. Variants in regional signal plots and LocusCompare plots are coloured based on their LD correlation (r2) with the lead eTWAS association. Boxplots in panels (d) and (h) represent COMBAT normalized islet gene expression for each genotype of the lead eTWAS association for UBE2E2 and ITFG3, respectively. The data are derived from 399 human islet samples42. Boxplots show first (lower) quartile, median, and third (upper) quartile, and whiskers indicate 1.5 x interquartile range. Chromatin state profiles in panel (e) highlight the enhancer harbouring the ITFG3-eTWAS rs56038902 signal, which is proximal to the ITFG3 promoter.
Extended Data Fig. 7 sTWAS associations between β-cell function (BCF) traits and islet mRNA splicing of G6PC2 and GRB10 genes.
Forest plots of the lead sTWAS associations at G6PC2 (a) and GRB10 (e). For each of the eight BCF traits and glycaemic traits, the square shows the β-estimate, and the error bars indicate the 95% confidence intervals. Summary statistics data were generated in this study or obtained from large-scale transancestral meta-analyses (European-based estimates from Chen, J. et al.49). Square sizes are based on precision. LocusCompare scatter plots compare the -log10 P-values for sQTLs of (b-c) G6PC2 and (g) GRB10 from Atla, A. et al.42, on the y-axis, with the -log10 P-values from BCF single-variant GWAS meta-analyses, on the x-axis. Variants are coloured based on the LD correlation (r2) with the lead sTWAS association. The sTWAS P-value and the colocalization posterior probability (CPP) are provided at the top. In panels (d) and (h), boxplots show normalized, batch-corrected junction PSI values stratified by the genotype of the lead sTWAS association. The data are derived from 399 human islet samples42. Boxplots show the first (lower) quartile, median, and third (upper) quartile, with whiskers extending to 1.5 times the interquartile range. IGV was used for the visualization of splice junctions depicted at the top of the boxplots, with arrow indicating the splice junction selected from sTWAS analysis. Regional signal plot at panel (f) shows single-variant BCF-GWAS meta-analyses P-values (-log10 scale, y-axis) and hg19 locations (x-axis) for the GRB10 locus.
Extended Data Fig. 8 Human pancreatic islet cell type-specific gene expression of selected β-cell function (BCF) candidate genes.
(a) The heatmap displays raw count matrices for selected genes from human pancreatic cells, as reported by Baron, M. et al.37, measured by single-cell RNA-seq. The data underwent log1p normalization with a scale factor of 10000 and were averaged across each available cell type.
Extended Data Fig. 9 Examining the effects of ACSL1 and CENPU downregulation on insulin release.
(a) ACSL1 mRNA expression levels assessed by qPCR after 72h transfection with scramble (control) and ACSL1 siRNA1-2 in human EndoC-βH1 cells. (b) Acsl1 mRNA expression levels after 72h transfection with scramble (control) and Acsl1 siRNA1-2 in INS-1 832/13 rat insulinoma cells, measured by qPCR. (c) Measurement of insulin content relative to total protein level in INS-1 832/13 cells. (d) Insulin secretion levels normalized by total insulin content in response to high glucose (16.7 mM) in INS-1 832/13 cells. (e) Cenpu mRNA expression levels assessed by qPCR after 72h transfection with scramble (control) and Cenpu siRNA1 in INS-1 832/13 cells. (f) Insulin secretion levels in response to high glucose (16.7 mM) concentration in INS-1 832/13 cells. (g) Measurement of total insulin content in INS-1 832/13 cells. (h) Insulin secretion normalization for total insulin content in response to high glucose (16.7 mM) concentration. Bar plots in panels (a-h) depict mean +/- SE from three (a) and four (b-h) independent replicates. Two-sided Student’s t-test was used for panels (c,e,g). One-way analysis of variance (ANOVA) followed by Tukey post-hoc test was used for panels (a,b,d,f,h). Panels (i) and (j) show morphology changes in INS-1 832/13 cells after transfection with scramble and Cenpu siRNA1, respectively. Images from a basic optical microscope, where scale bars are not available.
Extended Data Fig. 10 β-cell function (BCF) associations influence FAM46C pancreatic islet gene expression and T2D related traits.
(a) ATAC-seq and ChIP-seq data for histone modifications and islet-specific transcription factors are presented. An enhancer-gene assignment, inferred from pcHi-C data (coloured in purple), connects the islet enhancer containing rs1975283 to the FAM46C promoter. The rs1975283 variant, fine-mapped by BIGTT-AIR GWAS data and identified as a FAM46C islet eQTL, leads the eTWAS association between FAM46C islet expression and multiple BCF traits. (b) Forest plot of the lead rs1975283FAM46C eTWAS association. For each of the eight BCF traits, as well as glycaemic levels and T2D risk, the square represents the β-estimate, and the error bars indicate the 95% confidence intervals. Summary statistics data were generated in this study or obtained from Chen, J. et al.49 (European-based estimates) and Mahajan, A. et al.55. Square sizes are based on precision. (c) LocusCompare scatter plot compares FAM46C islet eQTL42 (x-axis) and single-variant BIGTT-AIR GWAS meta-analyses (y-axis) P-values on the -log10 scale. The eTWAS P-value and the colocalization posterior probability (CPP) are provided at the top. Variants are coloured based on their LD correlation with the lead eTWAS association (rs1975283). (d) Boxplot of COMBAT normalized expression values for the FAM46C gene stratified by the genotype of the lead rs1975283 eTWAS association. The data are derived from 399 human islet samples42. Boxplots show first (lower) quartile, median, and third (upper) quartile, and whiskers indicate 1.5 x interquartile range. (e) FAM46C gene expression in human EndoC-βH1 cells assessed by qPCR 72h after transfection with scramble (control) and FAM46C siRNA1-2. (f-g) Insulin secretion levels in response to (f) high glucose (16.7 mM) and (g) KCl in EndoC-βH1 cells. (h) Fam46c gene expression in rat INS-1 832/13 cells assessed by qPCR 72h after transfection with scramble (control) and Fam46c siRNA1-2. (i-k) Insulin secretion in response to (i) high glucose, (j) KCl, and (k) pyruvate in INS-1 832/13 cells. (l-n) Insulin secretion levels, normalized to total insulin content in response to (l) high glucose, (m) KCl, and (n) pyruvate in INS-1 832/13 cells. Bar plots in panels (e-n) represent mean +/- SE. Panels (e-g) include data from three independent replicates, while panels (h-n) include data from four independent replicates for Fam46c siRNA1, and three replicates for Fam46c siRNA2. One-way analysis of variance (ANOVA) followed by Tukey’s post-hoc test was used for statistical analysis in all panels (e-n).
Supplementary information
Supplementary Information
Supplementary Tables 1–18. Supplementary Figs. 1–6. Supplementary Methods
Supplementary Tables
All supplementary tables
Source data
Source Data Fig. 2
Plotted values
Source Data Fig. 3
Plotted values
Source Data Fig. 4
Plotted values
Source Data Extended Data Fig. 4
Plotted values
Source Data Extended Data Fig. 6
Plotted values
Source Data Extended Data Fig. 7
Plotted values
Source Data Extended Data Fig. 8
Plotted values
Source Data Extended Data Fig. 9
Plotted values
Source Data Extended Data Fig. 10
Plotted values
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Madsen, A.L., Bonàs-Guarch, S., Gheibi, S. et al. Genetic architecture of oral glucose-stimulated insulin release provides biological insights into type 2 diabetes aetiology. Nat Metab 6, 1897–1912 (2024). https://doi.org/10.1038/s42255-024-01140-6
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s42255-024-01140-6
This article is cited by
-
Functional ADAM12 variants modulate proteolytic activity and influence metabolic traits
Scientific Reports (2026)
-
Maternal and fetal genetic predispositions to insulin deficiency and resistance affect fetal growth through distinct pathways
Diabetologia (2026)
-
The role of the beta cell in type 2 diabetes: new findings from the last 5 years
Diabetologia (2025)
-
Single-cell mRNA-regulation analysis reveals cell type-specific mechanisms of type 2 diabetes
Nature Communications (2025)
-
Genetics brings new insight to β-cell function
Nature Metabolism (2024)
