
Abstract
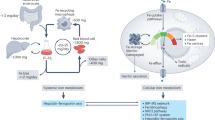
Iron transporters are essential for numerous iron-dependent biological processes. Among them, SLC22A17 plays a key role in lipocalin-2 (LCN2)-mediated iron transport and is implicated in several human diseases. However, its precise molecular and physiological functions remain poorly understood. In this study, we demonstrate that Slc22a17 is critical for postnatal neurogenesis through its regulation of iron homeostasis in the hippocampus. Conditional knockout of Slc22a17 in the murine brain results in early postnatal mortality, severe growth retardation, excessive neural stem cell (NSC) apoptosis, and cognitive impairments, all driven by oxidative stress caused by iron overload. Mechanistically, using TurboID-based proximity labeling and immunoprecipitation, we identify an interaction between Slc22a17 and p62, which modulates Nrf2 activity. Loss of Slc22a17 activates the Nrf2/HO-1 pathway, paradoxically enhancing iron release while impairing iron efflux. This imbalance triggers the production of iron-catalyzed reactive oxygen species (ROS), leading to oxidative stress. Together, our findings highlight Slc22a17 as a potential therapeutic target for neurological disorders associated with iron dysregulation.
Similar content being viewed by others


Introduction
The generation of functional neurons from neural stem cells (NSCs) is a crucial step in mammalian brain development, occurring primarily during the embryonic and early postnatal stages1,2. This neurogenic process is most active in two specific regions: the subventricular zone adjacent to the lateral ventricles and the sub-granular zone of the dentate gyrus3,4. During these stages, mammals experience an increased demand for iron to support various developmental processes, including neurogenesis, synaptogenesis, and the initiation of myelination5. Despite its importance, the relationship between iron metabolism and neurogenesis has received limited attention6. In contrast, excess iron accelerates the production of reactive oxygen species (ROS), triggering a cascade that results in cellular oxidative stress, which can lead to apoptosis or ferroptosis7,8,9. The harmful effects of ROS on cellular integrity not only compromise the self-renewal capacity of NSCs but also disrupt neurogenesis, long-term potentiation in the hippocampus, and, ultimately, cognitive function in mice4,10,11.
Among iron-homeostasis proteins, SLC22A17 was initially identified as a cell-surface receptor for lipocalin-2 (LCN2), also referred to as 24p3R, NGALR, or LCN2R12,13. Upon binding to SLC22A17, both apo-LCN2 (iron-free) and holo-LCN2 (iron-loaded) are internalized, facilitating iron efflux or influx, respectively12. SLC22A17 is highly expressed in the mammalian brain, particularly in neurons and oligodendrocytes14,15,16. Clinically, SLC22A17 has been implicated in the pathogenesis of several cancers, including glioma, as well as other diseases17,18,19,20. Despite extensive studies on its ligand LCN2, the physiological and molecular roles of SLC22A17 in the brain and the mechanisms underlying its function remain poorly understood.
Disruptions in iron metabolism promote cell death and contribute to human diseases by increasing the production of ROS9. In response to oxidative stress, nuclear factor erythroid 2-related factor 2 (Nrf2) provides well-established protective and neuroprotective effects by mitigating oxidative damage and mitochondrial dysfunction21,22. Within this critical ROS-response pathway, Kelch-like ECH-associated protein 1 (Keap1) regulates Nrf2 activity through ubiquitination and proteasome-dependent degradation23, maintaining oxidative balance. Sequestome 1 (SQSTM1/p62) regulates the Nrf2-Keap1 signaling pathway by binding to Keap1, thereby disrupting Nrf2 ubiquitination and degradation24. Among the key targets of Nrf2, heme oxygenase-1 (Hmox1, HO-1) is induced by oxidative peroxides and catalyzes the degradation of heme to carbon monoxide, free iron, and biliverdin25,26. Biliverdin is subsequently reduced to bilirubin, which exhibits antioxidant properties27,28. On the other hand, HO-1 can also generate reactive iron, which can become toxic if it accumulates excessively27,29. Under disease conditions, overexpression of HO-1 may release pro-oxidative heme and hemoproteins, further exacerbating oxidative stress30,31. Elevated HO-1 levels are commonly observed in age-related neurodegenerative diseases, such as Alzheimer’s and Parkinson’s diseases32,33.
In this study, we characterized the unique physiological and molecular roles of Slc22a17 in iron homeostasis and hippocampal neurogenesis. We observed that whole-brain knockout of Slc22a17 in mice resulted in significant body weight loss, severe impairment of hippocampal NSC development, and early postnatal mortality. We demonstrated that Slc22a17, localized in the endoplasmic reticulum, regulates iron export in NSCs through an endosomal recycling pathway, thereby maintaining iron homeostasis. Furthermore, we found that Slc22a17 interacts with p62, and its loss activates the Nrf2/HO-1 pathway, leading to intracellular ROS accumulation. This accumulation results from the dual effects of increased iron release and impaired iron efflux. Notably, deletion of Nrf2 or pharmacological inhibition of HO-1 alleviated ROS-induced deficits in hippocampal neurogenesis.
Results
Slc22a17 conditional knockout (cKO) mice exhibit postnatal lethality and impaired neuronal development
Slc22a17 is one of the most highly expressed iron transporters in the hippocampus, second only to ferritin, as revealed by our transcriptomic analysis of hippocampal tissues (Fig. 1A; Supplementary Data 1). We first performed immunoblotting of Slc22a17 in 12 mouse organs and tissues, which revealed its high expression levels in the brain (Fig. S1A). To investigate its distribution, we used immunofluorescence staining on mouse brain sections. This analysis showed that Slc22a17 is present across various brain regions, including the hippocampus (Fig. S1B). These findings align with previous studies20 and underscore the functional importance of Slc22a17 in hippocampal processes. We next quantified Slc22a17 levels in the hippocampus at different developmental stages. Slc22a17 expression was low in neonatal mice (postnatal day 0, P0) but increased during development, paralleling the expression of the presynaptic marker Synapsin I (Fig. S1C). To further investigate, we assessed iron levels in fluorescence-activated cell sorting (FACS) -isolated neural progenitor cells (NPCs) at multiple time points, starting from embryonic day 14 (E14). Our analysis revealed a progressive increase in iron content from E14, peaking at birth, followed by a sharp decline between P7 and P14. This iron accumulation pattern was inversely correlated with Slc22a17 expression in NPCs during these developmental stages (Fig. 1B). These observations suggest a potential role of Slc22a17 in early neurogenesis and its involvement in iron homeostasis within NSCs.

A Expression of iron-related genes in the hippocampus (n = 3). B The expression pattern of Slc22a17 in the neural stem cells (NSCs) during brain development negatively correlated with intracellular iron contents. Blue line indicates Slc22a17 expression and gray line indicates iron contents (n = 3). C Survival curves of the WT (Slc22a17 flox/flox) and cKO (Slc22a17 flox/flox; Nestin-Cre) mice. Most cKO mice died by P21 (WT, n = 36; cKO, n = 72). D Representative photos showing that cKO mice and brain tissues are smaller than WT at P14. E Body weights of WT and cKO mice (WT, n = 24, 39, 53, 5 at P0, P7, P14, and P21 respectively; cKO, n = 8, 21, 36, 4). F Representative photos showing immunostaining of Nestin in the dentate gyrus (DG) of WT and cKO mice at P14. Scale bar, 50 μm. G Representative photos showing immunostaining of Neurod1 and Dcx in the DG of WT and cKO mice at P14. Scale bar, 50 μm. H Decreased number of Nestin+, Neurod1+, and Dcx+ cells in the DG of cKO mice (n = 8). I Left: Representative photos showing Golgi-staining results of the hippocampus of WT and cKO mice at P14. Right: Representative photos showing that the dendrite numbers were reduced in the hippocampus of cKO; Thy1-EGFP mice compared with WT; Thy1-EGFP mice. Scale bar, 100 μm. J, K Representative photos and statistical analysis showing reduced primary basal dendrite numbers and dendritic spine numbers in cKO hippocampus at P14 (n = 24 cells from 3 mice/group). Left: Scale bar, 100 μm; Right: Scale bar, 5 μm. L Western blotting results showing synaptic components changes in the hippocampus of WT and cKO mice at P14. Syn, Synapsin I. M Left: Representative images of neuronal maker Tuj1 staining in vitro. Scale bar, 50 μm. Right: Statistical analysis of neurite length (WT, n = 18 cells from 3 mice; cKO, n = 14 cells from 3 mice). Statistical significance was determined by using Student’s t-test, two-sided. Data are presented as mean ± SEM. Source data are provided as a Source Data file.
We generated conditional knockout mice by crossing Slc22a17 flox/flox mice with Nestin-Cre mice (Slc22a17 flox/flox; Nestin-Cre, cKO). In these cKO mice, Slc22a17 mRNA was nearly undetectable in the hippocampus and cortex (Fig. S1D), and they exhibited marked postnatal lethality. Notably, 80% of the cKO mice died by postnatal day 21 (P21), whereas none of the wild-type (Slc22a17 flox/flox, WT) littermates succumbed (Fig. 1C). The cKO mice also displayed significantly reduced body weight compared to WT, Nestin-Cre, and Het (Slc22a17 flox/+; Nestin-Cre) mice (Fig. 1D and Fig. S1E). This body weight discrepancy became more pronounced by P7 and persisted until P21 (Fig. 1E). At P14, cKO mice exhibited a significantly increased brain-to-body mass ratio, indicating a disproportionate reduction in body weight relative to brain weight (Fig. S1F). In contrast, no significant difference was observed in other cell-type-specific knockout mice (Fig. S1G).
To assess the effect of Slc22a17 deletion on postnatal neurogenesis, we examined the hippocampal structure of WT and cKO littermates at P14. The cKO mice exhibited nearly a 40% reduction in the number of Nestin+ NSCs (Fig. 1F, H) and a 20% decrease in the dentate gyrus (DG) volume compared to WT (Fig. S1H). Similarly, the number of immature neurons, as indicated by Dcx and Neurod1 markers, was significantly reduced in the cKO mice (Fig. 1G, H). Our data indicate that deletion of Slc22a17 disrupts hippocampal neurogenesis, particularly affecting postnatal hippocampal development.
Next, we examined neuronal morphology in the cKO hippocampus at P14 by crossing WT and cKO mice with Thy1-EGFP mice16. We found that Slc22a17 deletion reduced dendrite complexity and spine density in cKO GFP+ granule cells (Fig. 1I–K). Similar results were obtained using Golgi-Cox staining (Fig. 1I and Fig. S1I). Quantification of proteins involved in the pre- and postsynaptic compartments, including Synapsin I (Syn), NMDAR2A, GluR1, and neuronal nuclear protein NeuN, revealed significant decreases in all these proteins in cKO mice compared to WT littermates (Fig. 1L). In vitro culture of primary hippocampal neurons confirmed that Slc22a17 cKO neurons exhibited significantly reduced total neurite length (Fig. 1M) and lower Synapsin levels (Fig. S1J). Additionally, we observed an increase in the relative fluorescence intensity of the astrocytic marker GFAP, but no change in the number of Iba1+ microglia (Fig. S1K, L), which may indicate the presence of reactive astrocytes triggered by synaptic loss34. Together, these findings suggest that Slc22a17 cKO severely impairs neuronal development in the hippocampus.
Neural stem cell (NSC) proliferation and differentiation are impaired in the hippocampus of Slc22a17 cKO mice
A previous study demonstrated that Slc22a17 is expressed in Sox2+ and Nestin+ NSCs in mice and cultured neurospheres35, although its precise functions remain unclear. We first quantified the number of Pax6+ NSCs, Sox2+ NSCs, and Ki67+ proliferating cells and found a reduction in proliferating NSCs in the cKO DG at P14 (Fig. S2A, B). To further assess the impact of Slc22a17 deletion on NSC proliferation, we injected BrdU intraperitoneally into WT and cKO mice 2 h before sacrifice at P14 to label proliferating cells. Quantifications showed that the number of BrdU+/Ki67+ and BrdU+/Sox2+ cells was significantly lower in the cKO DG compared with WT, indicating a reduced proliferation rate of NSCs in Slc22a17 cKO mice (Fig. 2A, B, and Fig. S2C). Next, we conducted clonal neurosphere culture assays and confirmed the deletion of Slc22a17 in cKO neurospheres using real-time qPCR and immunofluorescence (Fig. S2D, E). The number of primary and secondary neurospheres derived from cKO hippocampus revealed a significant reduction, as quantified per 1000 plated cells. Moreover, the diameter of primary and secondary neurospheres was also significantly decreased (Fig. 2C–E). In line with this, the number of BrdU+/Ki67+ and BrdU+/Sox2+ cells was also significantly decreased in cKO neurospheres (Fig. 2F, G). These results collectively suggest that Slc22a17 is essential for NSC proliferation.

A Representative image showing Ki67 (red)/BrdU (green) and Sox2 (red)/BrdU (green) co-immunostaining in the DG. Arrows indicate the double-labeled cells. Scale bar, 50 μm. B Decreased number of Ki67+/BrdU+ and Sox2+/BrdU+ cells in the DG of cKO mice compared with WT mice (WT, n = 12, 9 for Ki67, Sox2, respectively; cKO, n = 12, 6). C Representative images of the cultured primary and secondary neurospheres from WT and cKO brains. Scale bar, 100 μm. D Quantifications of primary and secondary neurospheres formed per 1000 plated cells derived from WT and cKO hippocampus (n = 8). E Quantifications of the diameter of cultured primary and secondary neurospheres (WT, n = 111 primary and 128 secondary neurospheres; cKO, n = 91 primary and 130 secondary neurospheres from 8 mice). F Representative images of secondary neurospheres stained for Ki67 (red)/BrdU (green) and Sox2 (red)/BrdU (green) co-immunostaining. Scale bar, 50 μm. G Quantifications of Ki67+/BrdU+ and Sox2+/BrdU+ cells in the secondary neuropheres derived from cKO mice (WT, n = 19, 18 neurospheres for Ki67, Sox2, respectively; cKO, n = 17, 26). H BrdU is intraperitoneally injected at P14, and the hippocampal neurogenesis is detected at P21. I Left: Representative images of NeuN (red)/BrdU (green) co-immunostaining in the DG. Arrows indicate the double-labeled cells. Scale bar, 50 μm. Right: Decreased number of NeuN+/BrdU+ cells in the DG of cKO mice (n = 19). J Quantification of neuronal differentiation from WT and cKO neurospheres after 7 days of induction. The percentage of MAP2⁺ neurons relative to total DAPI⁺ cells was calculated (n = 6). K Left: Representative images for Tuj1 (green) and MAP2 (red) staining in neurons differentiated from neurospheres after 7 and 14 days of culture (DIV7 and 14). Scale bar, 100 μm. Right: Quantification of neurite length for neurons differentiated from WT and cKO neurospheres (Tuj1: WT n = 6, 16 neurons at DIV7 and 14, respectively, cKO n = 17, 17; MAP2: WT n = 19, 16, cKO n = 27, 22). Statistical significance was determined by using Student’s t-test, two-sided. Data are presented as mean ± SEM. Source data are provided as a Source Data file.
We then analyzed NSC differentiation 7 days after BrdU injection at P1436. The number of NeuN+/BrdU+ cells in the DG of Slc22a17 cKO mice was significantly lower than in WT mice, indicating that Slc22a17 deletion disrupted NSC differentiation in the hippocampus (Fig. 2H, I and Fig. S2F). Furthermore, we cultured neurospheres in neuronal differentiation medium supplemented with fibronectin for 7 or 14 days. Immunofluorescence staining revealed that the proportion of neurons differentiated from cKO neurospheres was reduced by more than half compared to WT after 7 days of culture (Fig. 2J). Additionally, cKO neurosphere-derived neurons exhibited neurites that were, on average, 50% shorter than those of WT counterparts at both time points (Fig. 2K). Together, these findings suggest that Slc22a17 regulates hippocampal neuron development by modulating NSC proliferation and differentiation.
Slc22a17 deficiency impairs hippocampal adult neurogenesis and spatial learning and memory
After observing postnatal lethality and impaired NSC development in Slc22a17 cKO mice during the neonatal stage, we next sought to determine whether similar effects would occur in adult hippocampal neurogenesis. To investigate this, we generated inducible conditional knockout (iKO) mice by crossing Slc22a17 flox/flox mice with tamoxifen-inducible Nestin-CreERT2 transgenic mice. Administration of intraperitoneal tamoxifen efficiently induced Cre-mediated recombination at LoxP sites in Nestin+ cells. To assess changes in NSCs in the DG, we used a Rosa26R-CAG::tdTomato Cre-reporter, which labels NSCs and their progeny, including proliferating cells (Ki67+), stem-like cells (Sox2+), immature neurons (Dcx+), and mature neurons (NeuN+) with tdTomato fluorescence (Fig. 3A). qPCR analysis confirmed a significant reduction in Slc22a17 mRNA expression in iKO hippocampus and neural progenitor cells (NPCs) isolated from iKO mice (Fig. S3A). We compared the number of tdTomato+ cells in the hippocampus of WT and iKO mice at 7, 14, and 28 days post-injection (dpi). In WT hippocampus, the number of tdTomato+ cells increased rapidly after 7 dpi, whereas in iKO hippocampus, this increase was slower (Fig. 3B, C). Immunofluorescence staining of brain sections revealed a significant reduction in the density of Ki67+, Sox2+, and Dcx+ cells (Fig. 3D, E). Moreover, the relative proportion of these cells among BrdU+ cells was also significantly decreased in the DG of iKO mice compared with WT (Fig. 3F, G). These results indicate that deletion of Slc22a17 impairs adult hippocampal neurogenesis.

A Co-localization of tdTomato (red) with Ki67, Sox2, Dcx, and NeuN (green) in representing cells at different developmental stages. Scale bar, 50 μm. B, C Representative images and quantifications of tdTomato+ cells at 1 week, 2 weeks, and 4 weeks in Slc22a17 +/+, Nestin-CreERT2 (WT), and Slc22a17 flox/flox, Nestin-CreERT2 (iKO) mice post tamoxifen treatment (WT, n = 8, 7, 10 at 1, 2, and 4 weeks respectively; iKO, n = 7, 6, 6). Scale bar, 50 μm. D Up: Scheme of TAM/BrdU protocol: 5 days of TAM followed by BrdU injection. Bottom: Immunostaining of Ki67, Sox2, and Dcx in the DG 4 weeks post TAM. Scale bar, 50 μm. E Quantifications of Ki67+, Sox2+, and Dcx+ cells in the DG (WT, n = 9, 19, 23 for Ki67, Sox2 and Dcx respectively; iKO, n = 9, 12, 17). F Representative photos showing co-immunostaining of Ki67 (red)/BrdU (green), Sox2 (red)/BrdU (green), and Dcx (red)/BrdU (green) in the DG 4 weeks post the first TAM treatment. Arrows indicate the double-labeled cells. Scale bar, 50 μm. G Quantification of the proportion of Ki67+/BrdU+, Sox2+/BrdU+, and Dcx+/BrdU+ cells in the total BrdU+ cells (WT, n = 10, 8, 4 for Ki67, Sox2 and Dcx respectively; iKO, n = 9, 6, 9). H Analysis of Morris water maze (MWM) test showing the latency to reach the platform during the 6-day training. iKO mice spent more time reaching the platform compared with WT mice (WT, n = 31; iKO, n = 18). I Track the probe trial on day 7. J Decreased time spent in the target quadrant of iKO mice at probe trials on day 7 (WT, n = 31; iKO, n = 18). K Reduced number of platform crossings of iKO mice at probe trials on day 7 (WT, n = 31; iKO, n = 18). L Similar freezing behavior was monitored during the training session and impaired contextual memory in iKO mice (n = 5). Statistical significance in H was determined by using two-way ANOVA. Statistical significance in other panels was determined by using Student’s t-test, two-sided. Data are presented as mean ± SEM. Source data are provided as a Source Data file.
Impaired adult neurogenesis has been linked to deficits in learning and memory37. To assess whether learning and memory were affected in iKO mice, we performed behavioral tests in 8-week-old animals. The Morris Water Maze (MWM) test, widely used to evaluate spatial learning and memory38, was employed to assess cognitive function in these mice. We recorded the latency of WT and iKO mice in finding the hidden platform during the 6-day training period, followed by probe trials on the 7th day after removing the platform. iKO mice displayed a significant deficit in spatial memory acquisition compared to their WT littermates. This was evidenced by a longer latency to locate the platform during training (Fig. 3H and Fig. S3B), more erratic movement paths (Fig. 3I), reduced time spent in the target quadrant after the platform was removed (Fig. 3J), and fewer platform crossings (Fig. 3K). To further assess hippocampal function, we conducted a contextual fear conditioning task. Although there was no difference in freezing behavior between iKO and WT mice during the training phase, iKO mice exhibited a significant decrease in freezing percentage when re-exposed to the conditioning context 24 h later (Fig. 3L and Fig. S3C). Collectively, these results indicate that impaired adult hippocampal neurogenesis in iKO mice leads to deficits in hippocampal-dependent brain functions.
Given the neuronal expression pattern of Slc22a17 illustrated in whole-brain immunofluorescence staining (Fig. S1B), we next examined the effects of Slc22a17 knockout in specific mature neuron populations. We crossed Slc22a17 flox/flox mice with transgenic lines expressing CaMKIIα-Cre (for glutamatergic neurons) and VGAT-Cre (for GABAergic inhibitory neurons)39. Slc22a17 flox/flox, CaMKIIα-Cre mice exhibited longer latency in the MWM and swam longer distances to find the platform compared to WT controls (Fig. S3D–F). In contrast, Slc22a17 flox/flox, VGAT-Cre mice showed no significant differences in behavior compared to WT mice (Fig. S3G–I). These findings suggest that Slc22a17 may play a functional role in glutamatergic neurons.
Slc22a17 deficiency induces iron accumulation and oxidative damage
We performed RNA sequencing (RNA-seq) analysis of primary cultured neurospheres from control and cKO mice, and revealed a set of 209 differentially expressed genes (DEGs) under Slc22a17 deletion (Supplementary Data 2). Up-regulated DEGs were enriched in iron-responsive cues and neuron apoptotic process in cKO neurospheres, while down-regulated DEGs were enriched for cell cycle, neuron migration, and neuron differentiation (Fig. S4A). Since Slc22a17 has been reported to mediate iron trafficking12, we measured the concentration of several divalent metal ions in the brain, liver, and spleen of cKO and WT mice at P14 using inductively coupled plasma-mass spectrometry (ICP-MS). The results revealed that iron concentrations were elevated in the whole brain (Fig. 4A) and hippocampus (Fig. 4B) of cKO mice, whereas zinc, copper, and manganese levels remained unchanged (Fig. S4B), indicating that Slc22a17 specifically mediates iron transport. In line with these findings, non-heme iron concentrations were also markedly increased in the brains of cKO mice (Fig. 4C), further supporting a selective role of Slc22a17 in regulating brain iron homeostasis. In the liver and spleen—two primary storage organs for iron—only iron concentrations were significantly higher in cKO mice compared to WT mice (Fig. S4C, D)40, potentially due to brain-specific Slc22a17 deletion indirectly causing tissue iron accumulation via central-peripheral regulation. We further observed increased hepatic hepcidin expression and elevated serum hepcidin levels in cKO mice (Fig. S4E). These changes in hepatic hepcidin could impair iron export, leading to increased tissue iron levels, which may contribute to the elevated liver iron content, or alternatively, may result from the increased liver iron levels. We also measured metal ion levels in neurospheres and observed a significant increase in iron concentration in cKO neurospheres, while the culture medium showed no change (Fig. 4D and Fig. S4F). To confirm this, we utilized Perl’s staining to visualize hippocampal iron accumulation in cKO mice and employed the iron-sensitive probes FerroOrange and Calcein to demonstrate elevated cellular iron levels in cKO neurospheres (Fig. 4E, F), where the green fluorescence of Calcein is quenched by iron, allowing for the detection of increased cellular iron. These findings indicate that Slc22a17 deficiency leads to severe iron overload.

A Brain iron, zinc, and copper levels in WT and cKO mice by inductively coupled plasma-mass spectroscopy (ICP-MS) (n = 4). B Iron concentrations in the hippocampus and cortex (WT, n = 11, 11 for hippocampus, cortex respectively; cKO, n = 10, 9). C Brain non-heme iron levels, measured by the standard chromogen method (n = 6). D Iron, zinc, and copper in WT and cKO neurospheres (WT, n = 6, 8, 8 for Fe, Zn, and Cu, respectively; cKO, n = 7, 8, 8). E Perls’ staining in the hippocampus. Left: Scale bar, 50 μm; Right: Scale bar, 20 μm. F Left: FerroOrange and Calcein labeling in neurospheres. Scale bar, 20 μm. Right: Quantifications of FerroOrange and Calcein fluorescence intensity (FerroOrange, n = 14; Calcein, n = 5 neurospheres from 5 mice/group). G Flow cytometry showing elevated ROS in cKO neurospheres, rescued by DFO or NAC (WT, n = 10; cKO, n = 11; cKO-DFO, n = 4; cKO-NAC, n = 4). H Immunostaining for 8-OHdG recognizing DNA damage and quantifications (WT, n = 11; cKO, n = 9; cKO-DFO, n = 6; cKO-NAC, n = 6 neurospheres from 3 mice/group. Scale bar, 20 μm. I Malondialdehyde (MDA) levels in hippocampus (n = 4). J Annexin V/PI staining showing increased NSCs apoptosis in cKO NSCs (n = 5). K Apoptosis of cKO neurospheres rescued by DFO (n = 6). L Western blot of apoptosis-related proteins in WT and cKO hippocampus at P14. Target proteins were normalized to their respective loading controls. M Left: IF staining of cleaved Caspase3 in the DG. Scale bar, 50 μm. Right: Quantification of Casp3+ cells per DG (n = 4). N Left: JC-1 assay showing reduced mitochondrial membrane potential in cKO neurospheres. Scale bar, 20 μm. Right: Quantifications of red (J-aggregates) to green (monomers) ratio (n = 30 neurospheres from 3 mice/group). O TEM images of hippocampal mitochondria showing swelling and cristae disorganization in cKO. Red arrows indicate abnormal mitochondrial cristae. Scale bar, 200 nm. Statistical significance in I was determined by using Chi-square test, Bonferroni correction. Statistical significance in F, G, J, N was determined by using one-way ANOVA and Scheffe’s multiple comparisons test. Statistical significance in other panels was determined by using Student’s t-test, two-sided. Data are presented as mean ± SEM. Source data are provided as a Source Data file.
Excessive iron accumulation promotes oxidative stress by generating ROS, which can cause DNA damage, cell cycle arrest, and cell death41. To investigate this, we evaluated ROS levels and found that intracellular ROS was significantly higher in cKO neurospheres compared to WT. This ROS increase was abolished following 24 h of exposure to the iron chelator deferoxamine (DFO) or the antioxidant N-acetylcysteine (NAC) (Fig. 4G). To investigate DNA damage associated with oxidative stress, we used 8-hydroxy-2’-deoxyguanosine (8-OHdG), a marker of oxidized guanine lesions. As expected, elevated 8-OHdG intensity was detected in cKO neurospheres, which was reduced upon treatment with the iron chelator DFO or the NAC (Fig. 4H). Consistent with this, the malondialdehyde (MDA) level, a marker of lipid peroxidation, was also significantly higher in cKO neurospheres (Fig. 4I).
Excessive ROS levels often trigger apoptotic cell death. To assess apoptosis, we performed flow cytometry, western blotting, and immunofluorescence analysis. Flow cytometry revealed a significant increase in the proportion of apoptotic cells in cKO neurospheres, an effect that was reversed by DFO treatment (Fig. 4J, K). Western blot analysis showed elevated levels of pro-apoptotic proteins, including cleaved PARP, cytochrome c, cleaved Caspase-9, and cleaved Caspase-3 in cKO neurospheres (Fig. 4L). Immunofluorescence staining for cleaved caspase-3 further confirmed neuronal apoptosis in the cKO hippocampus (Fig. 4M). The release of cytochrome c and activation of caspases suggested mitochondrial dysfunction, which was further supported by JC-1 staining and electron microscopy. These techniques revealed severe mitochondrial damage in cKO neurospheres, including mitochondrial depolarization and swelling (Fig. 4N, O). To determine whether elevated iron exacerbated mitochondrial damage, we added ferric ammonium citrate (FAC), a soluble iron supplement, to increase intracellular iron levels. This treatment further aggravated mitochondrial damage in cKO neurospheres (Fig. 4N). Notably, the oxidative damage caused by Slc22a17 deficiency appears to be independent of ferroptosis, as the expression of ferroptosis-related drivers and suppressors remained unchanged between WT and cKO groups at both the RNA and protein levels (Fig. S4 G–I). Together, these findings demonstrate that Slc22a17 deficiency leads to iron overload-induced oxidative stress, resulting in ROS-dependent cell death.
Slc22a17 facilitates cellular iron efflux in response to iron overload
To investigate the effects of aberrant iron accumulation in the cKO hippocampus and neurospheres, we examined the expression of key proteins involved in iron uptake and storage. We observed decreased levels of transferrin receptor (TfR) and its regulator IREB2, as well as increased levels of Ferritin-H and Ferritin-L, which are indicative of iron overload in the cKO hippocampus (Fig. 5A). Based on these findings, we hypothesized that Slc22a17 may facilitate iron efflux in neural cells in response to elevated intracellular iron levels. Interestingly, we did not observe compensatory upregulation of ferroportin (Fpn, Slc40a1), but rather a reduction in Fpn protein levels in the hippocampus, suggesting that Fpn-mediated iron export may be impaired in the absence of Slc22a17. To test this hypothesis, we performed iron release experiments. Primary neurons were pre-incubated with FAC, cultured in fresh medium, and then analyzed for a decrease in intracellular iron content. The results showed that the rate of iron release was significantly reduced in cKO neurons compared to WT (Fig. 5B and Fig. S5A). Additionally, we generated transgenic mice overexpressing Slc22a17 in the brain (Slc22a17 OE/OE mice, OE) by crossing them with the Nestin-Cre mouse line. Using calcein staining, we visualized iron content in organotypic hippocampal slice cultures and found that FAC failed to elevate iron levels in the DG of the OE hippocampus (Fig. 5C), as calcein fluorescence decreases upon Fe³⁺ binding. Immunoblot analysis revealed that overexpression of Slc22a17 resulted in decreased levels of the iron storage protein Ferritin-H (Fig. 5D). These results obtained from both Slc22a17 knockout and overexpressing mice confirm that Slc22a17 plays a role in mediating iron export in the hippocampus.

A Western blot analysis of IREB2, TfR, Slc40a1, Ferritin-H, and Ferritin-L protein levels in WT and cKO hippocampus at P14. Target proteins were normalized to their respective loading controls. B Iron release rate was calculated as the percentage reduction in intracellular iron content measured 2 h after FAC removal in WT and cKO primary neurons (n = 6). C Representative images showing iron contents labeled by the iron probe Calcein in the organotypic brain slice with or without FAC treatment. Scale bar, 50 μm. D Western blotting analysis of Slc22a17 and Ferritin-H protein levels in WT and OE hippocampus at P14. Target proteins were normalized to their respective loading controls. E Phalloidin (green) staining and IF staining of Tuj1 (yellow) and Slc22a17 (red) in primary neuron showing the intracellular location of Slc22a17. Scale bar, 20 μm. F Representative confocal images showing ER-tracker (red) and Slc22a17-GFP (green) co-localizations in HT22 cells. Scale bar, 5 μm. G Residual intracellular iron contents are determined for the indicated periods after iron preload in EV and Slc22a17-OE HEK293 cells (n = 6). H Live-cell imaging showing localization of Slc22a17 (green) to Lcn2 (blue) and recycling endosome marked by Rab11 (red). White arrow indicates the complex shown in the upper enlarged panel, and white arrowhead points to the region shown in the lower enlarged panel. Scale bar, 5 μm. Statistical significance was determined by using Student’s t-test, two-sided. Data are presented as mean ± SEM. Source data are provided as a Source Data file.
Interestingly, we examined the endogenous localization of Slc22a17 in primary neurons, revealing its predominant intracellular distribution (Fig. 5E). This finding contrasts with its previously described role as a cell-surface receptor12 and suggests a potential relationship between the intracellular localization of Slc22a17 and its function in iron efflux. To investigate the subcellular localization of Slc22a17 and its role in iron trafficking, we generated HEK293 and HT22 cell lines overexpressing Slc22a17 tagged with GFP (Fig. S5B). Confocal microscopy confirmed its intracellular localization, and we observed co-localization of recombinant Slc22a17 with an endoplasmic reticulum (ER)-targeting fluorescent probe (Fig. 5F). We then conducted an iron release experiment using Slc22a17-OE cells. Overexpression of Slc22a17 accelerated iron transport out of the cells following 24-h FAC treatment (Fig. 5G), which was consistent with the reduced iron release rate observed in cKO neurons. We also analyzed the expression of two key genes involved in iron transport, Tfrc and Slc40a1, which are up-regulated and down-regulated, respectively, when intracellular iron levels decrease (Fig. S5C). Collectively, these results confirm that a portion of Slc22a17 is localized to the ER, and that overexpression of Slc22a17 facilitates iron export after FAC treatment.
Next, we investigated whether ER-localized Slc22a17 regulates iron export via the recycling endosome pathway, similar to the classical mechanism of Slc22a17-mediated iron trafficking12. To test this hypothesis, we examined our transcriptomic data, which revealed significant changes in the expression of several endosome markers (Fig. S5D). Notably, we observed downregulation of Rab11a, a recycling endosome marker, in cKO neurospheres. We then transfected Slc22a17-OE cells with plasmids encoding mCherry-Rab11 and BFP-Lcn2 fusion proteins, and used long-term live-cell imaging to monitor their behavior. The imaging data showed that BFP-Lcn2 and mCherry-Rab11 colocalized with GFP-Slc22a17 on the ER (Fig. 5H), suggesting that ER-localized Slc22a17 is associated with recycling endosomes.
To further investigate the role of Slc22a17 in cellular iron export, we compared the iron export ability between Slc22a17 and Slc40a1. We performed siRNA-mediated knockdown of SLC22A17 and SLC40A1 in HEK293 cells (Fig. S5E). Cells were exposed to FAC, and extracellular iron levels were measured using a non-heme iron assay at 4, 8, and 24 h after FAC removal. Knockdown of SLC40A1 significantly reduced iron efflux, while SLC22A17 knockdown also impaired iron export, particularly at later time points. Dual knockdown of both genes further blocked iron efflux compared to the siSLC22A17 group, suggesting the cooperative role of FPN in the SLC22A17-mediated iron efflux mechanism (Fig. S5F). siRNAs targeting SLC22A17 or SLC40A1 were co-transfected with a plasmid encoding mCherry into cells, and fluorescence imaging 48 h post-transfection showed that SLC22A17 knockdown led to a moderate decrease in calcein green fluorescence intensity, while SLC40A1 knockdown caused more pronounced fluorescence quenching (Fig. S5G). Additionally, real-time monitoring using calcein green fluorescence revealed that SLC22A17 knockdown accelerated iron accumulation, while SLC40A1 knockdown led to more severe intracellular iron retention (Fig. S5H). Overexpression of Slc22a17 had no significant effect on iron accumulation in the short term, but over a longer period, it showed similar iron export effects as Slc40a1 (Fig. S5I, J). These results indicate that Slc40a1 directly mediates iron export, while Slc22a17 may function upstream, playing a key role in iron transport from the ER to the cytoplasm.
Slc22a17 interacted with p62 to regulate the activity of Nrf2/HO-1 pathway
The deletion of Slc22a17 led to the upregulation of gene sets associated with oxidative stress response and iron homeostasis, as revealed by correlation network analysis (Fig. 6A). Notably, several mRNAs that were significantly up-regulated in cKO neurospheres were encoded by redox-regulated genes. Among them, Hmox1, a gene containing antioxidant response elements (ARE), was identified as a downstream target of the transcription factor Nrf2 (Fig. 6B)42. Further analysis revealed an enrichment of Nrf2-activated genes among the up-regulated genes in cKO neurospheres, providing additional evidence for the activation of the Nrf2 signaling pathway (Fig. 6C). QPCR analysis confirmed a marked reduction in the expression of neurogenesis-related genes, such as Bmp4 and Bdnf (Fig. S6A), and an increase in the expression of oxidative stress response genes, including Hmox1 and Aox1 (Fig. S6B). In addition, ATAC-seq analysis of neurospheres from cKO and WT mice identified 1528 loci with increased chromatin accessibility, the majority of which were localized to promoters, while only seven loci exhibited reduced accessibility (p < 0.05, Fig. S6C–E and Supplementary Data 3). Consistent with our RNA-seq findings, we observed increased chromatin accessibility at the Nfe2l2 and Hmox1 loci in cKO neurospheres (Fig. 6D).

A Pearson correlation network showing genes involved in significantly up- or down-regulated pathways. The blue circles represent the down-regulated pathways (Top left: Cell cycle; Top right: Neuron differentiation; Bottom left: Neural precursor cell proliferation; Bottom right: Nervous system development) and the red represents the up-regulated ones (Top: Response to oxidative stress; Bottom: Iron ion homeostasis). Edges are significant at Pearson’s coefficient > 0.5 and p < 0.05 for a positive correlation. B Volcano plot showing the down-regulated and up-regulated genes measured using RNA-seq analysis. C GSEA of genes targeted and activated by Nrf2 through their promoters are up-regulated upon Slc22a17 deletion. Genes were ranked based on decreasing log2 fold gene expression changes comparing the KO neurosphere to the WT. D Chromatin accessibility at the Nfe2l2 and Hmox1 loci was analyzed by ATAC-seq in WT and cKO neurospheres. E Increased p-Nrf2, Nrf2, HO-1, and p62 levels in the cKO neurosphere. Target proteins were normalized to their respective loading controls. F Increased nuclear Nrf2 and cytosolic HO-1 levels in cKO neurosphere. G Decreased nuclear Nrf2 levels and decreased cytosolic p62 and Ferritin-H levels in Het (Slc22a17 OE/+; Nestin-Cre) and OE (Slc22a17 OE/OE; Nestin-Cre) mice. H Ubiquitination assays showing Nrf2 ubiquitination and degradation in cKO mice. I Confocal microscopy images of NSCs stained for Nrf2 and DAPI. Scale bar, 20 μm. J Co-IP assay with Slc22a17-Flag and p62-HA showing the interaction between Slc22a17 and p62. K Representative live-cell imaging showing localization of Slc22a17-mCherry (red) to p62-GFP (green). A 3D reconstruction of Slc22a17 and p62 droplet is shown in the bottom panels. Scale bar, 2 μm. L Representative confocal images showing localization of p62-GFP (green) to LysoTracker (red) in control and SLC22A17-/- HEK293 cells. Left: Scale bar, 5 μm; Right: Scale bar, 500 nm. Statistical significance was determined by using Student’s t-test, two-sided.
Nrf2 translocates to the nucleus in response to oxidative stress, where it binds to AREs and activates the transcription of target genes, such as Hmox1. This process may facilitate heme degradation and the release of free iron43. We observed increased expression of Nrf2, phosphorylated Nrf2 (p-Nrf2), and HO-1 in cKO neurospheres, accompanied by elevated levels of p62, which binds to Keap1 and promotes Nrf2 nuclear translocation (Fig. 6E). Immunoblot analysis revealed upregulation of nuclear Nrf2 and cytosolic HO-1 protein levels in cKO neurospheres compared to WT controls (Fig. 6F). Next, we examined the expression of proteins involved in the Nrf2/HO-1 pathway in Slc22a17-overexpressing (Slc22a17-OE) cell lines and OE mice. In these models, we observed a significant downregulation of nuclear Nrf2 and cytosolic p62 levels upon Slc22a17 overexpression (Fig. 6G and Fig. S6F). Additionally, we detected reduced Nrf2 ubiquitination in the cKO hippocampus, a finding consistent with enhanced nuclear translocation, as confirmed by confocal microscopy (Fig. 6H, I). This effect resembled the response observed with the known Nrf2 activator, cobalt protoporphyrin (CoPP)44. To further investigate the role of Nrf2 in this context, we infected cKO neurospheres with lentivirus expressing shNrf2 and observed that Nrf2 knockdown significantly reduced the aberrant induction of HO-1 in cKO neurospheres (Fig. S6G).
To identify SLC22A17-interacting proteins, we performed TurboID-based proximity labeling45, followed by quantitative proteomic analysis. This revealed that p62 may serve as a potential interacting partner of SLC22A17 (Fig. S6H and Supplementary Data 4). To validate this interaction, we conducted co-immunoprecipitation (co-IP) analysis. Flag-tagged Slc22a17 efficiently co-immunoprecipitated with p62-HA, confirming their association (Fig. 6J). Confocal imaging further showed that a portion of Slc22a17 colocalized with p62, and this co-localization increased after iron addition, in line with the proteomic results (Fig. 6K and Fig. S6H). Although we observed that p62 was up-regulated in the cKO hippocampus, the mechanism by which Slc22a17 regulates p62 remains unclear. To investigate this further, we expressed GFP-p62 in a CRISPR/Cas9-mediated SLC22A17 knockout cell line (SLC22A17-/-) and labeled lysosomes with LysoTracker Red to track their intracellular localization. Live-cell imaging revealed that, in the control group, p62 predominantly fused with lysosomes, entering the autophagic pathway for degradation. In contrast, p62 showed minimal co-localization with lysosomes in SLC22A17-/- cells, resulting in failure in degradation and accumulation of large p62 droplets (Fig. 6L). p62 droplets are known to act as receptors for autophagy, where autophagosomes form at the droplet surface, mediated by LC3-conjugated membranes46. Using live-cell imaging to explore Slc22a17’s role in autophagy, we observed that Slc22a17 colocalized with both p62 and LC3. Furthermore, Slc22a17 formed small puncta at the surface of p62 droplets, similar to LC3 (Fig. S6I). Additionally, we found that vesicular Slc22a17 colocalized with the late endosome marker Rab7 (Fig. S6J). These findings suggest that Slc22a17 participates in the endocytic-autophagic pathway, facilitating p62 degradation. In the absence of Slc22a17, p62 accumulates, which inhibits Keap1 and activates the Nrf2 signaling pathway. Collectively, these data highlight a functional link between Slc22a17 deficiency and Nrf2/HO-1 pathway activation, which may exacerbate oxidative injury and disrupt neurogenesis.
Reducing iron levels or inhibition of Nrf2/HO-1 signaling rescues learning and memory deficits in Slc22a17 iKO mice
To assess whether iron overload is critical for triggering abnormal neurogenesis and impairing learning and memory, we fed 8-week-old WT and iKO mice diets with varying iron levels. After four weeks, iKO mice on a low-iron diet showed significantly improved performance in MWM tests compared to those on normal- or high-iron diets. Furthermore, Slc22a17 iKO mice treated with membrane-permeable DFO also exhibited recovery in learning and memory responses (Fig. 7A–D). Consistently, the numbers of Ki67⁺, Nestin⁺, NeuroD1⁺, and Dcx⁺ cells in the hippocampus were markedly restored in iKO mice following either a low-iron diet or DFO treatment, indicating a rescue of impaired neurogenesis (Fig. S7A, B). In contrast, WT mice on different iron diets did not show significant performance differences in MWM tests (Fig. S7C–E).

A–D Impaired learning and memory of iKO mice in the MWM test is restored upon DFO and chronic low-iron diet treatment. Latency and distance to the platform at the training trials, time spent in the target quadrant, and the platform crossing times at the probe trials are decreased in iKO mice, and are recovered by DFO and chronic low-iron diet treatment (WT, n = 31; iKO, n = 18, iKO-High, n = 10; iKO-Low, n = 10; iKO-DFO, n = 11). E–H HO-1 inhibitor ZnPP improved learning and memory of Slc22a17 iKO mice. Shown are results of latency to the platform, distance to the platform, time spent in the target quadrant, and crossing times to the platform (WT, n = 34; iKO, n = 18; iKO-ZnPP, n = 10). I–L Nrf2 deletion improved learning and memory of Slc22a17 iKO mice. Shown are results of latency to the platform, distance to the platform, time spent in the target quadrant, and crossing times to the platform (WT, n = 14; iKO, n = 17, Nrf2-/-, n = 9; DKO, n = 7). M Immunostaining for DG with Ki67, Sox2, NeuroD1, Dcx and DAPI in WT, iKO, Nrf2-/- and DKO (iKO; Nrf2-/-) mice. Scale bar, 50 μm. N Quantification of the numbers of Ki67+, Sox2+, NeuroD1+, and Dcx+ cells in WT, iKO, Nrf2-/- and DKO mice (n = 6). Statistical significance in A, B, and E, F, and I, J was determined by using two-way ANOVA and Scheffe’s multiple comparisons test. Statistical significance in C, D, G, H, K, L, and N was determined by using one-way ANOVA and Scheffe’s multiple comparisons test. Data are presented as mean ± SEM. Source data are provided as a Source Data file.
To explore the role of HO-1 in oxidative damage, we treated mice with the competitive HO-1 inhibitor zinc protoporphyrin IX (ZnPP). Chronic ZnPP administration significantly improved learning and memory in iKO mice (Fig. 7E–H). We next examined the regulatory role of Nrf2 in hippocampal structure and functions. To determine whether activation of Nrf2/HO-1 signaling contributed to impaired learning and memory in iKO mice, we bred iKO mice with Nrf2 knockout (Nrf2-/-) mice. The resulting double knockout (Slc22a17 flox/flox, Nestin-CreERT2; Nrf2-/-; DKO) mice showed alleviated learning and memory impairments in MWM tests (Fig. 7I–L). Additionally, the number of Ki67+, Sox2+, NeuroD1+, and Dcx+ cells in the DG was significantly higher in DKO mice compared to iKO mice (Fig. 7M, N). These cumulative findings demonstrate that Slc22a17/Nrf2/HO-1 signaling plays a crucial role in maintaining iron homeostasis, adult hippocampal neurogenesis, and learning and memory. Slc22a17 deficiency promotes nuclear accumulation of Nrf2, thereby enhancing Nrf2-dependent HO-1 expression, which ultimately disrupts neurogenesis in the brain.
Discussion
Our study elucidates the physiological and molecular functions of Slc22a17 in regulating iron metabolism in NSCs, crucial for proper hippocampal neurogenesis. Slc22a17 knockout leads to abnormal iron accumulation, ROS-induced apoptosis in NSCs, and consequently severe neurogenesis impairment and early postnatal death. We identified that Slc22a17 regulates the p62/Nrf2/HO-1 signaling axis through its interaction with p62. Notably, both HO-1 inhibition and Nrf2 knockout significantly rescued neurogenesis in cKO mice, akin to the protective effects observed with DFO treatment. Together, these findings identify Slc22a17 as a key iron sensor in NSCs and reveal that its deletion disrupts neurogenesis through aberrant activation of the Nrf2/HO-1 pathway, thereby uncovering a crucial connection between iron metabolism and neural development.
Among the several cell-type-specific knockout mouse models we generated, only the Nestin-Cre-induced Slc22a17 cKO mice exhibited severe weight loss, early postnatal death, disrupted neurogenesis, and excessive NSC apoptosis, indicating a close association between Slc22a17 and the development of neural stem and progenitor cells. Additionally, the expression pattern of known iron-transporting genes suggests that Slc22a17 is likely the principal regulator of iron homeostasis in hippocampal NSCs. Notably, although CaMKIIα-Cre-driven cKO mice also exhibited spatial learning and memory impairments, they showed no neuronal morphological abnormalities, suggesting distinct functions of Slc22a17 in adult neurogenesis and mature neurons, the latter of which we are currently investigating.
Our findings indicate that the disrupted iron homeostasis resulting from Slc22a17 deletion in NSCs arises from its role in facilitating iron efflux, rather than influx. ER-localized Slc22a17 serves as a critical exit point for iron through the recycling endosome pathway. Subcellular localization analysis reveals that a portion of Slc22a17 is localized to the ER and downstream vesicles. Live-cell imaging demonstrates that Lcn2, which colocalizes with Rab11, is recognized by ER-localized Slc22a17 and subsequently enters the recycling endosome, allowing the transfer of intracellular iron out of the cell. This mechanism, where Slc22a17 localizes to the ER to maintain intracellular iron efflux in NSCs, represents a unique regulatory pathway (Fig. S7I). Meanwhile, our findings indicate that, unlike Slc40a1, which directly mediates iron transmembrane transport, Slc22a17 facilitates iron efflux from the ER to the cytoplasm, playing a crucial upstream role in maintaining iron homeostasis. NSCs have varying iron requirements at different developmental stages. Our results demonstrate that NSCs require iron during embryonic development, but this demand decreases postnatally with the initiation of Slc22a17 expression. Slc22a17 likely helps NSCs precisely regulate intracellular iron levels and ROS, maintaining redox balance and normal cellular function. Redox signaling governs the proliferation and differentiation of NSCs, and excessive ROS can induce NSC death47. The redox state is largely determined by an iron redox couple48. Aberrant intracellular iron accumulation catalyzes ROS generation, leading to oxidative stress and apoptosis49. Although ferroptosis was considered as a potential mechanism, it seems unlikely, as only minimal or no changes were observed in ferroptosis markers such as GPX4. Additionally, we observed that brain-specific deletion of Slc22a17 led to iron dysregulation, which may indirectly enhance hepatic hepcidin expression and cause iron accumulation in peripheral organs. This phenomenon is reminiscent of the iron dysregulation observed in neurodegenerative diseases like Alzheimer’s and Parkinson’s disease, where central iron imbalance may drive peripheral iron accumulation and alter hepcidin expression. For example, a study has reported that participants with a higher amyloid-beta load show significantly elevated serum hepcidin levels50. Taken together, these findings suggest that Slc22a17 may function as a key regulator of the central-peripheral iron axis, responding to both cellular and extracellular iron levels to maintain iron balance and redox homeostasis. Further studies on Slc22a17 in the central nervous system could provide valuable insights into the mechanisms of central-peripheral iron metabolism, which may ultimately aid in the early diagnosis of neurodegenerative diseases.
As the receptor for Lcn2, Slc22a17 knockout mice exhibit distinctive phenotypes and cellular dysfunctions compared to Lcn2-null mice. Previous studies have reported varying impacts of Lcn2 on adult neurogenesis, NSC development, and neuroinflammation under stimuli, acting as an iron chelator51. However, Lcn2-null mice do not show significant weight loss, postnatal mortality, or severe early NSC defects (Fig. S7F–H)35, indicating that Slc22a17 functions independently of Lcn2. This suggests that Slc22a17 is not solely a receptor for Lcn2 but also has unique cellular functions distinct from those of Lcn2. Based on the above findings, we conclude that Slc22a17 functions as the primary iron transporter and sensor in the hippocampus, responding to both cellular and extracellular iron levels to maintain normal physiological iron requirements and redox balance. Slc22a17 likely interacts with Lcn2 to transport iron, depending on its subcellular localization or the iron concentration in the physiological environment.
Nrf2 is a key redox sensor in NSCs52. Most studies investigating the Nrf2/HO-1 pathway have concluded that this pathway primarily serves antioxidant functions53. However, our findings reveal that Nrf2/HO-1 signaling is activated in the NSCs of Slc22a17 cKO mice, serving as a major source of iron-induced ROS. Slc22a17 deletion not only disrupts its interaction with p62, preventing p62 from binding to Keap1 and blocking Keap1’s access to Nrf2, but also impedes iron efflux by inhibiting the release of iron from heme oxidation through the upregulation of HO-1 expression. Slc22a17 deficiency, leading to sustained Nrf2 activation, likely serves as a crucial link between iron overload and p62 accumulation. Additionally, we discovered that Slc22a17 is recruited by p62 and participates in autophagosome formation, a potential mechanism by which Slc22a17 regulates p62 degradation (Fig. S7I). Although Nrf2 activation is generally considered neuroprotective in disease models such as Alzheimer’s disease54,55,56, our results show that its sustained activation in Slc22a17-deficient mice impairs neurogenesis and learning, highlighting a context-dependent role of Nrf2 signaling that may be detrimental under conditions of Slc22a17 deficiency. These findings raise important concerns about therapeutic strategies targeting oxidative stress-related diseases that involve activation of the Nrf2/HO-1 pathway57,58.
In conclusion, our results provide direct evidence that the dysregulation of Slc22a17 leads to molecular, cellular, and behavioral abnormalities, particularly in hippocampal neurogenesis. We show that Slc22a17, a unique iron transporter in the brain, is essential for maintaining hippocampal neurogenesis. Given the pivotal role of iron in the pathogenesis of various neurodegenerative diseases59, this study contributes to a broader understanding of how disruptions in iron homeostasis in the brain can impact neurological health and disorders, including multiple sclerosis and Alzheimer’s disease.
Methods
Animals
All animal experiments were performed in accordance with guidelines of the Institute of Laboratory Animal Research Center of Tsinghua University. All animal procedures used in the study were approved by the Administrative Committee of Experimental Animal Care and Use of Tsinghua University. They licensed by the Science and Technology Commission of Beijing Municipality (SYXK-2014-0024). Mice were housed in a temperature (22 °C RT) and light (12-h light/dark cycle) regulated room in a SPF facility and received food and water ad libitum.
Slc22a17 floxed mice (Slc22a17flox/flox) were purchased from Shanghai Biomodel Organism Science & Technology Development Co., Ltd. Slc22a17flox/flox mice were crossed with the Nestin-Cre (JAX:003771, kindly provided by Bailong Xiao, Tsinghua University, China) line to generate Slc22a17flox/flox; Nestin-Cre (defined as conditional knockout, cKO) mice and Slc22a17flox/flox littermates (defined as Wild-type, WT). Inducible conditional knockout mice were generated by crossing Slc22a17flox/flox mice with the Nestin-CreERT2 (JAX:016261, kindly provided by Qin Shen, Tongji University, China) line carrying a Cre-dependent tdTomato reporter (JAX:007909), followed by tamoxifen administration. Thy1-GFP-M mice (JAX: 007788, kindly provided by Zengqiang Yuan, Beijing Institute of Basic Medical Sciences, China) were crossed with WT or cKO mice to label a variety of neuronal subsets. Slc22a17flox/flox mice were also crossbred with CaMKIIα-Cre (JAX:005359, kindly provided by Yichang Jia, Tsinghua University, China), Emx1-Cre (JAX:005628), VGAT-Cre (JAX:016962), Olig1-Cre (JAX:011105), and Lepr-Cre60 mice to generate neuron or glia-specific knockout mice. To overexpress Slc22a17 in the brain, ROSA26loxP-stop-loxP-Slc22a17-3XFlag mice (Slc22a17OE/OE) were purchased from Cyagen US Inc. and mated with the Nestin-Cre mice to obtain Slc22a17OE/OE; Nestin-Cre mice. The resulting Slc22a17OE/OE (defined as Wild-type, WT) and Slc22a17OE/OE; Nestin-Cre (defined as overexpressed, OE) littermate mice were used for the experiments. Nrf2 knockout (Nrf2-/-) mice (JAX: 017009, kindly provided by Fudi Wang, Zhejiang University, China) were crossbred with cKO mice to generate Slc22a17 and Nrf2 double knockout (defined as double knockout, DKO) mice.
Mice of either sex on C57BL/6 were used in the majority of this study. In experiments involving adult neurogenesis and behavioral assays, we exclusively used male mice. All mice used in behavioral assays adhered to animal welfare guidelines, and data from mice that were injured or died during behavioral assays were removed. The influence of sex was not evaluated in this study. Age is specified in the main text.
Drug administration
Tamoxifen (Sigma-Aldrich, T5648) was prepared in 100 mL corn oil to mixture 10 mg/mL. For adult male mice, Tamoxifen (100 mg/kg) was administered by intraperitoneal injection once a day for five consecutive days.
Mice were intraperitoneally injected with 100 mg/kg deferoxamine (DFO, Sigma-Aldrich) seven days before examination or sacrifice.
Mice were intraperitoneally injected with 10 mg/kg ZnPP (Milipore, 691550) seven days before examination or sacrifice.
Mice were fed with standard rodent laboratory diet (TROPHIC Animal Feed High-Tech Co. Ltd) containing 47ppm iron (standard diet), 12ppm iron (Low-iron-diet), or 912ppm iron (High-iron-diet).
Immunofluorescence staining
Animals were perfused with cold 4% paraformaldehyde (in 0.1 M PBS, pH 7.4) and post-fixed overnight, cryoprotected with 20% and 30% sucrose. For immunofluorescence staining in tissues, cryo-sections (30μm) were permeabilized and blocked in the blocking buffer (5% BSA and 0.4% Triton X-100 in PBS) for 1 h at room temperature (r.t.) and incubated overnight (4 °C) with primary antibody. For staining in cells, cells in confocal dishes were fixed with 4% PFA for 10 min and then permeabilized and blocked in the blocking buffer (5% BSA and 0.2% Triton X-100 in PBS) for 1 h r.t. and incubated overnight (4 °C) with primary antibody.
Antibodies used in the study were: Rabbit anti-Nestin (Abcam, ab221660); Rabbit anti-Neurod1 (Proteintech, 12081); Rabbit anti-Dcx (Abcam, ab207175); Mouse anti-Tuj1 (Biolegend, 801201); Rat anti-BrdU (Abcam, ab6326); Rabbit anti-Ki67 (Abcam, ab15580); Rabbit anti-Pax6 (Abcam, ab195045); Rabbit anti-Sox2 (Abcam, ab92494); Rabbit anti-NeuN (Abcam, ab177487); Rabbit anti-8OHdG (Abnova, MAB6638); Rabbit anti-Cleaved Caspase-3 (Cell Signaling Technology, 9664); Rabbit anti-Slc22a17 (Sigma-Aldrich, SAB3500306); Mouse anti-Nrf2 (Abcam, ab62352); Mouse anti-GFAP (Cell Signaling Technology, 3670S); Mouse anti-Iba1(Abcam, ab283319).
After washing with PBS, sections were incubated with secondary antibody for 1 h r.t.. and counterstained with DAPI for 10 min. Slides were mounted in Fluoromount (SouthernBiotech, 0100-01) and visualized by Leica STELLARIS 8 FALCON confocal microscopy.
For stereological analysis, consecutive brain tissue sections were procured to quantify the number of labeled cells. Quantifications of marker-positive cells were performed in at least three sections containing the Dentate Gyrus (DG) from at least three different animals. The exact value of n is described in the figure legends.
BrdU treatment
For analysis of cell proliferation in the dentate gyrus, mice were injected with BrdU (50 mg/kg body weight, i.p., Sigma-Aldrich) and analyzed 2h later. For analysis of cell differentiation, mice were injected with BrdU and analyzed 7 days later. For BrdU immunostaining, the sections were subjected to DNA denaturation with 1 N HCl for 30 min at 37 °C. The following procedures were immunofluorescence staining.
Golgi-Cox staining
The brains were stored in the dark for 2 days (37 °C) in the Golgi-Cox solution and then sectioned at a thickness of 200 μm in the coronal plane with a vibratome. All the sections were collected and stained with ammonia, Kodak Film Fix, and then dehydrated, cleaned, and mounted using resinous medium.
Fluorescence-activated cell sorting (FACS) of neural progenitor cells
Neural progenitor cells (NPCs) were isolated using fluorescence-activated cell sorting (FACS) based on established protocols described by Liu et al.61. Briefly, NPCs were obtained from mouse brain tissues at multiple developmental stages, including embryonic day 14 (E14), embryonic day 18 (E18), postnatal day 0 (P0), P7, P14, and P21. Tissues were enzymatically dissociated to single-cell suspensions, stained with fluorophore-conjugated antibodies, and sorted for CD24-Thy1-/lo cells using a FACSAria III (BD Biosciences). The following antibodies were used: anti-CD90.2 (Thy-1)-APC (1:400, BD Biosciences, 561641) and anti-CD24-PE (1:400, BD Biosciences, 553262). Dead cells were excluded using Fixable Viability Dye EF506 (Thermo Scientific).
Primary NSCs culture
NSCs were isolated from the hippocampus of P0 WT and Slc22a17 cKO mice (within 24 h after birth) based on previously published methods with some modifications62. Mouse hippocampal tissues were acutely dissected and dissociated in 2 mL of Accutase (GIBCO) for 20 min, and 3 mL of DMEM-F12 (Procell, PM150312) containing 1% B27 (GIBCO), 1% N2 (Invitrogen), 20 ng/mL epidermal growth factor (EGF) (Peprotech, 100-15), 10 ng/mL basic fibroblast growth factor (bFGF) (Peprotech, 100-18B), and penicillin/streptomycin (Beyotime, ST488S) was added into each sample to stop digestion. After filtering through a 70-μm cell strainer (Falcon, 352350) and washing with DMEM-F12 containing penicillin/streptomycin, the single-cell suspension (approximately 5 × 106 per mouse) was collected and cultured with DMEM-F12 containing 1% B27, 1% N2, 20 ng/mL EGF, and 10 ng/mL bFGF in a 5% CO2 incubator at 37 °C. After 14 days of culture, neurospheres were used to both western blot analysis and immunofluorescence. Primary neurospheres were collected after 14 days of culture, dissociated into single cells using Neurocult dissociation kit (StemCell, 05702) and seeded again to obtain secondary NSCs.
Fibronectin-induced neurosphere differentiation assay
24-well plates containing sterile glass coverslips were pre-coated with 0.01% poly-D-ornithine overnight at 4 °C, washed with PBS, and further coated with fibronectin (5 μg/mL, MCE, HY-P3160) for 2 h at 37 °C. Neurospheres (~ 100–200 μm in diameter) were gently dissociated with Accutase at 37 °C for 5–10 min into small clusters and seeded at a density of 1 × 10⁵ cells per well in neurobasal medium (Invitrogen, 21103049) supplemented with B27 (GIBCO), GlutaMax (GIBCO, 35050061). Cultures were maintained for 7–14 days with half-medium changes every 2–3 days, and neuronal differentiation was evaluated by immunostaining for neuronal markers.
Primary neuron culture
Primary mouse hippocampal neurons were acutely dissociated according to the previously described method with some modifications63. Briefly, the hippocampal tissues were dissected from P0 WT and Slc22a17 cKO mice (within 24 h after birth), and dissociated in 0.25% trypsin-EDTA (GIBCO, 1798279) for 15 min to obtain a single-cell suspension. The cell suspension was centrifuged to collect cells after terminating the digestion process. Cells were re-suspended in neurobasal medium (Invitrogen, 21103049) supplemented with B27, GlutaMax (GIBCO, 35050061), and penicillin/streptomycin and cultured on 0.1 mg/mL poly-L-lysine (PLL) (Sigma-Aldrich, P2636) coated plates at 37 °C in a humidified incubator containing 5% CO2. After 14 days of culture, neurons were used for both Western blot analysis and immunofluorescence.
Western blot
Tissues and cells were lysed with RIPA buffer containing Protease Inhibitor for 1 h on ice, then centrifuged at 14,000 × g for 15 min. Collected the supernatant with SDS loading buffer, 100 °C, 10 min. Total protein was loaded into a 10% SDS-PAGE gel and then transferred onto PVDF membranes. Membranes were blocked in 5% nonfat milk in PBST before incubation with primary antibodies overnight at 4 °C. Secondary antibodies used for 1 h r.t.. Antibodies used in the study were: Rabbit anti-Slc22a17 (Abcam, ab124506); Rabbit anti-NeuN (Abcam, ab177487); Rabbit anti-Synapsin I (Abcam, ab64581); Rabbit anti-NMDAR2A (Abcam, ab124913); Rabbit anti-GluR1 (Abcam, ab109450); Rabbit anti-PARP (Cell signaling Technology, 9532); Mouse anti-Cytochrome c (BD Biosciences, 556433); Mouse anti-Caspase-9 (Cell Signaling Technology, 9508); Rabbit anti-Caspase-3 (Cell Signaling Technology, 9662); Rabbit anti-IRP2 (Cell Signaling Technology, 37135); Rabbit anti-Transferrin Receptor (Abcam, ab214039); Rabbit anti-Ferroportin (Thermo Fisher, PA5-22993); Rabbit anti-Ferritin Heavy Chain (Abcam, ab183781); Rabbit anti-Ferritin Light Chain (Abcam, ab109019); Rabbit anti-Nrf2 (phosph S40) (Abcam, ab76026); Mouse anti-Nrf2 (Abcam, ab62352); Rabbit anti-Heme Oxygenase 1 (Abcam, ab68477); Rabbit anti-Keap1 (Cell Signaling Technology, 8047); Rabbit anti-p62 (Proteintech, 18420-1-AP); Rabbit anti-xCT (Cell Signaling Technology, 12691); Rabbit anti-GPX4 (Cell Signaling Technology, 52455); Rabbit anti-NCOA4 (Cell Signaling Technology, 66849); Mouse anti-DMT1 (Abcam, ab123085); Mouse anti-GAPDH (Proteintech, 60004); Mouse anti-Beta Actin (Proteintech, 66009); Mouse anti-Beta Tubulin (Beyotime, AF2835); Rabbit anti-Lamin B1 (Proteintech, 12987-1-AP); Rabbit anti-Histone H3 (Abcam, ab32388). In Western blot quantifications, target proteins were first normalized to their respective loading controls and further normalized by adjusting to the average expression of inner reference proteins across all groups.
Annexin V/PI staining
Neurospheres were collected, washed in cold PBS, and binding buffer. FITC-conjugated Annexin V 5 μL and propidium iodide 50 μg/mL were added to the cells, incubated for 15 min at RT in the dark. Propidium iodide signals were measured using the FACSCaliber2 flow cytometer. Analysis was further performed using the FlowJo 7.6.
Intracellular ROS measurement
Neurospheres were incubated with 10 μM of DCFH-DA dye for 30 min at 37 °C. After washing with DMEM medium, cells were further washed with PBS. The amount of intracellular ROS levels was analyzed on a BD Calibur flow cytometer, and data were analyzed using FlowJo 7.6.
MDA measurement
MDA levels were determined using the thiobarbituric acid method by a commercial kit (Solarbio, BC0025) according to the manufacturer’s instructions.
JC-1 analysis
Mitochondrial membrane potential was measured by the JC-1 fluorescent probe (Beyotime, C2003S). FAC-treated or non-treated cells were analyzed according to the manufacturer’s instructions.
Plasmid construction
GFP-fusion expressed Slc22a17 plasmid (Slc22a17-GFP), mCherry-fusion expressed Slc22a17 plasmid (Slc22a17-mCherry), mCherry-fusion expressed Rab11a plasmid (Rab11a-mCherry), mCherry-fusion expressed Rab7a plasmid (Rab7a-plasmid), BFP-fusion expressed Lcn2 plasmid (Lcn2-BFP), GFP-fusion expressed p62 plasmid (p62-GFP), and BFP-fusion expressed Lc3b plasmid (Lc3b-BFP) were generated by PCR- subcloning. Plasmid vectors were co-transfected into the HEK293 or HT22 cell line using PEI or Lipofectamine 3000 (Invitrogen, L3000001). Live-cell imaging was performed using Zeiss LSM980 Airyscan2 confocal microscopy.
Overexpressing stable cell line
HT22 (Cat# CL-0697, Procell) or HEK293T (CRL-3216, ATCC) cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM, Cytiva) supplemented with 10% FBS at 37 °C with 5% CO2. The gene encoding Slc22a17 or Slc40a1 was cloned into the Pljm1-Puro vector. Cells were co-transfected with Pljm1-Slc22a17 or Pljm1-Slc40a1, psPAX2, and pMD2.G using PEI (Polyscience). Viral supernatants were collected at 72 h post-transfection, filtered through a 0.45 µm filter, and stored at −80 °C. Cells were seeded at 2 × 105 cells/well in 6-well plates and transduced with lentiviral supernatants in the presence of 8 µg/mL polybrene (Yeasen). After 24 h, the medium was replaced with fresh DMEM containing 10% FBS. Selection with 3 µg/mL puromycin (Solarbio) began 24 h post-transduction. Single colonies were isolated and expanded. Total RNA extracted from puromycin-resistant clones was used to quantify overexpression levels of Slc22a17 and Slc40a1 using qPCR.
CRISPR-Cas9-mediated SLC22A17 knockout cell line
CRISPR-Cas9-mediated knockout of SLC22A17 was performed using the pX458 plasmid containing sgRNAs targeting the third and tenth exons of SLC22A17 (SLC22A17-sgRNA1: 5’-ACCTGTCTGCGGGGTAACCC-3’; SLC22A17-sgRNA2: 5’-TAGGATGCTCACAATC CCAC-3’). Cells were co-transfected with the plasmid using PEI. GFP-positive cells were sorted by flow cytometry to isolate single clones 48 h post-transfection. Selected clones were seeded into 96-well plates and cultured. After 3 weeks, cells from selected clones were split: half were expanded into 6-well plates for further culture, and the other half had genomic DNA extracted using the TIANamp Genomic DNA Kit (TIANGEN) for sequencing to confirm SLC22A17 knockout.
Transmission electron microscopy
Tissue processing was carried out as previously described64. Briefly, mice were anesthetized and perfused with 2% PFA/2% glutaraldehyde in PBS. Tissues were dissected and postfixed in 1% OsO4. Ultrathin sections were stained with lead citrate for electron microscopy imaging.
Perl’s staining
DAB-enhanced Perl’s iron staining was performed according to what was described previously65. After deparaffinization, tissue slides were incubated in freshly prepared, acidified 1% potassium ferrocyanide (Sigma-Aldrich) in distilled water (pH ~ 1.0 to 1.5) solution for 40 min r.t.. Afterward, slides were washed three times in distilled water and incubated in methanol containing 0.01 M NaN3 and 0.3% H2O2 for 75 min r.t.. Slides were washed three times in 0.1 M PBS (pH 7.4) and then incubated in PBS containing 0.025% DAB and 0.005% H2O2 for 40 min r.t. in the dark and counterstained with Hematoxylin. As a negative control, one duplicate slide was incubated with non-acidified ferrocyanide solution (pH ~ 8.0), which did not show any staining. Additionally, coverslips with cells permeabilized with Triton X-100 prior to Perl’s staining as a negative control did not show iron staining.
Iron measurement
The brain, liver, spleen, and neurosphere were collected, weighed, and sent for quantitative elemental analysis with inductively coupled plasma-mass spectrometry (ICP-MS) XII by the Department of Physics of Tsinghua University.
Brain non-heme iron was measured as described previously66. Brain tissue samples were digested in NHI acid (10% trichloroacetic acid (Aladdin, T104257) in 3 M HCl) at 65–70 °C for 48 h. Equal volumes of sample or iron standard (500 µg/dl) were incubated with buffer (0.2% thioglycolic acid (Aladdin, T501446) and 0.02% disodium-4,7-diphenyl-1,10-phenanthroline disulfonate (Aladdin, B339500) in 50% saturated NaAc solution) for 10 min r.t.. Absorbance of the samples was read at 535 nm, and unknowns were quantified using a standard curve. Results are reported as micrograms of iron per gram of wet tissue weight.
Intracellular iron was measured using FerroOrange (DOJINDO, F374) and Calcein-AM (Invitrogen, C1430) as described12. Cells were stained with 1 μM FerroOrange or 0.5 mM Calcein-AM in PBS for 10–30 min r.t. and then washed three times with PBS. Fluorescence images were captured using Leica STELLARIS 8 FALCON confocal microscopy and processed using Leica Application Suite X (LAS X).
To dynamically monitor cellular iron uptake, transfected HEK293 cells were seeded into 96-well black-wall clear-bottom plates (Beyotime-FCP965) at a density of 3 × 105 cells/well. After 48 h, cells were incubated with 0.5 μM calcein-AM for 30 min, washed, and immediately subjected to live fluorescence measurement using a Multimode plate reader (PerkinElmer, 494/514 nm). Ferrous chloride (FeCl₂, 100 μM) was added directly to the wells during measurement, and fluorescence intensity was continuously recorded every 10 s for 400 s to assess the kinetics of iron uptake and calcein quenching.
Organotypic slice culture assay
Organotypic cultures were prepared as previously described67. Brains from WT and Slc22a17-OE mice (P14) were dissected and glued (Super Glue Loctite) onto the chuck of an ice-cooled vibratome Leica VT1200S. 300μm coronal sections were cut and collected in chilled 20% DMEM. The coronal slices were cultured in confocal dishes, and FAC (Sigma-Aldrich) was added to a randomly selected group of slices for 2 h. Fluorescence images were acquired using Leica STELLARIS 8 FALCON confocal microscopy.
TurboID-based proximity labeling and mass spectrometry
The pLX304-lgK-V5-TurboID-KDEL plasmid was obtained from Dr. Wei Qin (Tsinghua University). According to the method reported previously68, the TurboID gene was amplified with pLX304-lgK-V5-TurboID-KDEL plasmid as a template. The SLC22A17 gene was amplified with human cDNA as a template. pLX304-lgK-V5-TurboID-KDEL used restriction sites BamHI and AgeI, and the SLC22A17 and TurboID fragments were recombined into the linearized pLX304-lgK-V5-TurboID-KDEL plasmid. The successfully sequenced plasmid was named SLC22A17-TurboID.
The SLC22A17-TurboID plasmid was transfected into HEK293T cells. After 24 h of transfection, cells were treated with 100 μM of DFO for 12 h. Subsequently, cells were treated with DMEM containing 50 μM Biotin for 10 min (Sigma-Aldrich, B4639). After washing with cold PBS (repeated washing twice), the cells were lysed with RIPA to obtain protein samples. Negative controls were included in which either the DFO or the biotin was omitted. Incubate 100 μL streptavidin agarose resin beads (Thermo Scientific, 20353) with 2 mg protein from each sample with rotation overnight at 4 °C. After overnight incubation, the supernatant was discarded, and the beads were washed with 1 mL of RIPA buffer (repeated washing twice). Biotinylated proteins were eluted by boiling each sample in 60 μL of 1× protein loading. Biotinylated proteins were subjected to mass spectrometry identification in the Protein Core Facility of Tsinghua University.
Co-immunoprecipitation
The pLJM1-p62-HA and pCMV-Slc22a17-3xFLAG plasmids were also constructed through homologous recombination. Cell lysates derived from HEK293T cells transiently transfected with plasmids were prepared using a lysis buffer (Beyotime, P0013) and protease inhibitor cocktail (Roche, 04693116001). The anti-Flag magnetic beads (Bimake, B26101) or anti-HA magnetic beads (Bimake, B26201) were incubated with cell lysates with rotation overnight at 4 °C and subsequently washed five times with a washing buffer (TBS buffer, Beyotime, ST661) containing protease Inhibitor cocktail. The immunoprecipitated proteins were eluted with 1× SDS loading buffer.
Nrf2 ubiquitination assay
Tissues were lysed with lysis buffer (TBS buffer containing 0.3% TritonX-100 and protease Inhibitor cocktail) for 1 h on ice, then centrifuged at 14,000 × g for 15 min to collect the supernatant. Protein A/G Immunoprecipitation Magnetic Beads (Bimake, B23201) were incubated with 20 μg/mL of Nrf2 antibodies for 15 min to bind the antibody. Equal amounts of lysates were incubated with Nrf2-beads with rotation overnight at 4 °C and subsequently washed five times with a washing buffer (TBS buffer containing 0.3% TritonX-100 and protease Inhibitor). The immunoprecipitated proteins were eluted with 1× SDS loading buffer, and the ubiquitination levels of Nrf2 were detected by Western Blot.
RNA-seq and data processing
For RNA-seq analysis, total RNA was extracted using the RNeasy Lipid Mini Kit (QIAGEN, 74134). 1 μg RNA sample. RNA quality was determined by Agilent 2100. The RNA samples were sent to Huada (Wuhan) Sequencing Company for RNA sequence establishment. Gene counts were calculated for each transcript and normalized to determine differentially expressed genes using DESeq2 (v1.38.3)69, with significance defined by p < 0.05 and fold change >1.5. GSEA (Gene Set Enrichment Analysis) was performed using the default parameters of the clusterProfiler R package (v4.7.1.2), with significance defined by a false discovery rate (FDR) < 0.25. The list of NRF2-activating genes used in GSEA was obtained from available dataset70. Gene enrichment and interaction network were analyzed using Cytoscape (v3.7.1)71.
ATAC-seq and data processing
ATAC-seq was performed using the previously mentioned protocol72. Briefly, Nuclei isolated from 5 × 104 neural stem cells were used for ATAC-seq. Cells were harvested at 500 × g 4 °C for 5 min, and then cell pellets were re-suspended in 100 μL of lysis buffer (10 mM Tris-HCl, pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.5% NP-40) and then nuclei were centrifuged at 500 × g 4 °C for 15 min. The nuclei pellets were re-suspended in the transposition reaction mix containing Tn5 transposase (Vazyme Biotech, S602). Reactions were incubated for 30 min at 37 °C. The transposed DNA fragments were purified using the MinElute Kit (QIAGEN, 28004,) and the library was amplified using primers with unique barcodes (Vazyme Biotech), which were selected by the AMPureXP beads (Agencourt, 20508200) to establish the library. The library was sequenced on an Illumina HiSeq X Ten sequencer for an average of 20 million unique reads per sample to obtain 150 bp paired-end reads.
ATAC-seq data analysis was performed as described previously72. Reads were trimmed to remove adapter sequences using Trim Galore (v0.6.2) with options ‘-q 25 –paired’, and trimmed reads were aligned to mm10 reference genome using Bowtie2 (v2.3.5.1)73. Alignments were filtered using SAMtools (v1.9)74 to retain only uniquely mapping read pairs and remove duplicates using Sambamba (v0.7.0)75. Peak calling was performed using the MACS2 (v2.1.2)76 callpeak command with parameters ‘-q 0.01’. A matrix of non-nucleosomal Tn5 insertion counts was generated for the common set of peaks using BEDtools (v2.29.2)77. The count matrix was used to identify differentially accessible peaks with DESeq2 (v1.38.3)69. Differential peaks at FDR < 0.05 were retained for analysis. The peaks were annotated to RefSeq (mm10) using ChIPseeker (v1.20.0)78. Mean-normalized Tn5 insertions at differential peaks were computed for pooled replicates in both WT and cKO mice and smoothed over a 100-bp window. Average signal values were plotted using deepTools (v3.3.0)79.
Real-time quantitative PCR (qPCR) analysis
qPCR analyses were performed both on hippocampal tissues and on NSCs. Total RNA extraction using Trizol reagent, reverse transcription, and SYBR green quantitative real-time PCR were performed according to the manufacturer’s instructions using gene-specific primers (listed in Supplementary Data 5).
Behavioral analysis
Morris water maze experiments were performed as previously described with minor modifications64. The animals were released from different positions in the water maze and performed four trials daily for 6 days. On the seventh day, the mice were given a 1 min probe trial in which the platform was removed. Mice were tracked by a video camera, the latency, velocity and distance traveled to the platform were collected and analyzed by software. The platform crossings were recorded. The water temperature was controlled at 22 ± 1°C during the experiments.
Contextual fear conditioning was conducted for 2 days. On the training day, mice were exposed to the conditioning chamber for 7 min. Within the following 3 min, mice were trained with a 1 s, 0.5 mA foot-shocks for three times, separated by a 60 s interval. After the last shock, the mice stayed in the chamber for 1min, then were put back in their cages. Contextual testing was executed 24 h after the training. The mice were placed into the same chamber and stayed there for 7 min with no foot-shock. The percentage of freezing time at each experiment was recorded, and contextual fear memory was measured by the software.
siRNA transfection
HEK293 cells were transfected with siRNAs targeting SLC40A1 and SLC22A17 (designed and purchased from TsingkeBiotechnologyCo., Ltd, Beijing, China) using Lipofectamine 2000 (YEASEN, Cat# 40802ES03) according to the manufacturer’s instructions. To label transfected cells, an mCherry-expressing empty plasmid was co-transfected in the same liposome complex. Transfected cells were incubated for 48 h before further analysis.
Lentivirus construction
Lentivirus was produced as described. HEK293T cells were transfected with the lentiviral packaging plasmid PSPAX2, VSVG, and the target plasmid, using PEI (Polyscience). 48–72 h after transfection, collect viral supernatants and centrifuge at 1000 × g to pellet cellular debris. Add 5XPEG8000 overnight, 2500 × g, 4 °C,30 min to obtain high-titer virus. Pelleted virus was re-suspended in 200 μL phosphate-buffered saline (PBS). Infection of cells by adding the virus to the media supplemented with 8 μg/mL polybrene overnight. Cells were collected after 48 h.
Separation of cytoplasmic and nuclear fraction
Nuclear, cytoplasm, and cell membrane proteins were extracted using the total protein extraction kit for animal cultured cells and tissues (Invent, SC-003).
Statistics and reproducibility
All experiments in this paper were carried out with more than three biological replicates. All experiments were performed under the same conditions, and results were consistently reproducible across independent replicates. Representative data from one of these experiments are shown in the figures. GraphPad Prism 8.0 and ImageJ (v1.53t) were used as statistical analysis software. Comparisons between two groups were assessed using unpaired two-tailed Student’s t-test or one-way ANOVA with Scheffe’s multiple comparisons test, or one-way ANOVA with Tukey’s multiple comparisons test, or two-way ANOVA, or two-way ANOVA with Scheffe’s multiple comparisons test, or Chi-square test with Bonferroni correction, wherever applicable. All data were presented as mean ± SEM. Significance is indicated by an asterisk (*p < 0.05, **p < 0.01, ***p < 0.001).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
RNA Seq and ATAC-seq data generated by this study have been deposited in the Gene Expression Omnibus (GEO), under accession code GSE306965, GSE306966 and GSE306969. Source data are provided with this paper.
References
Gonçalves, J. T., Schafer, S. T. & Gage, F. H. Adult neurogenesis in the hippocampus: From stem cells to behavior. Cell 167, 897–914 (2016).
Zhong, S. et al. Decoding the development of the human hippocampus. Nature 577, 531–536 (2020).
Niklison-Chirou, M. V., Agostini, M., Amelio, I. & Melino, G. Regulation of adult neurogenesis in mammalian brain. Int. J. Mol. Sci. 21, 4869 (2020).
Fuentealba, L. C., Obernier, K. & Alvarez-Buylla, A. Adult neural stem cells bridge their niche. Cell Stem Cell 10, 698–708 (2012).
Wang, Y., Wu, Y., Li, T., Wang, X. & Zhu, C. Iron metabolism and brain development in premature infants. Front. Physiol. 10, 463 (2019).
Ferreira, A. C., Sousa, N., Bessa, J. M., Sousa, J. C. & Marques, F. Metabolism and adult neurogenesis: Towards an understanding of the role of lipocalin-2 and iron-related oxidative stress. Neurosci. Biobehav. Rev. 95, 73–84 (2018).
Stockwell, B. R. et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171, 273–285 (2017).
Wang, B. et al. Ros-induced lipid peroxidation modulates cell death outcome: Mechanisms behind apoptosis, autophagy, and ferroptosis. Arch. Toxicol. 97, 1439–1451 (2023).
Jiang, X., Stockwell, B. R. & Conrad, M. Ferroptosis: mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 22, 266–282 (2021).
Hameed, L. S. et al. Environmental changes in oxygen tension reveal ROS-dependent neurogenesis and regeneration in the adult newt brain. Elife 4, e08422 (2015).
Maraldi, T., Angeloni, C., Prata, C. & Hrelia, S. Nadph oxidases: Redox regulators of stem cell fate and function. Antioxidants 10, 973 (2021).
Devireddy, L. R., Gazin, C., Zhu, X. & Green, M. R. A cell-surface receptor for lipocalin 24p3 selectively mediates apoptosis and iron uptake. Cell 123, 1293–1305 (2005).
Yang, J. et al. An iron delivery pathway mediated by a lipocalin. Mol. Cell 10, 1045–1056 (2002).
Bennett, K. M., Liu, J., Hoelting, C. & Stoll, J. Expression and analysis of two novel rat organic cation transporter homologs, slc22a17 and slc22a23. Mol. Cell. Biochem. 352, 143–154 (2011).
Ling, J. P. et al. Ascot identifies key regulators of neuronal subtype-specific splicing. Nat. Commun. 11, 137 (2020).
Zhang, J. et al. Whole-brain three-dimensional profiling reveals brain region specific axon vulnerability in 5xfad mouse model. Front. Neuroanat. 14, 608177 (2020).
Wei, J., Gao, X., Qin, Y., Liu, T. & Kang, Y. An iron metabolism-related SLC22A17 for the prognostic value of gastric cancer. Onco. Targets Ther. 13, 12763–12775 (2020).
Liu, M. F. et al. Ngal and ngalr are frequently overexpressed in human gliomas and are associated with clinical prognosis. J. Neurooncol. 104, 119–127 (2011).
Chi, Y. et al. Cancer cells deploy lipocalin-2 to collect limiting iron in leptomeningeal metastasis. Science 369, 276–282 (2020).
Chia, W. J., Tan, F. C., Ong, W. Y. & Dawe, G. S. Expression and localisation of brain-type organic cation transporter (boct/24p3r/lcn2r) in the normal rat hippocampus and after kainate-induced excitotoxicity. Neurochem. Int. 87, 43–59 (2015).
Zhang, C. Y. et al. The Nrf2-NLRP3-caspase-1 axis mediates the neuroprotective effects of celastrol in parkinson’s disease. Redox Biol. 47, 102134 (2021).
Bento-Pereira, C. & Dinkova-Kostova, A. T. Activation of transcription factor Nrf2 to counteract mitochondrial dysfunction in parkinson’s disease. Med. Res. Rev. 41, 785–802 (2021).
Baird, L. & Yamamoto, M. The molecular mechanisms regulating the Keap1-Nrf2 pathway. Mol. Cell Biol. 40, e00099–00020 (2020).
Sanchez-Martin, P. et al. Nbr1-mediated p62-liquid droplets enhance the Keap1-Nrf2 system. EMBO Rep. 21, e48902 (2020).
Kim, Y. S., Zhuang, H., Koehler, R. C. & Doré, S. Distinct protective mechanisms of HO-1 and HO-2 against hydroperoxide-induced cytotoxicity. Free Radic. Biol. Med. 38, 85–92 (2005).
Kitagishi, H. et al. Feedback response to selective depletion of endogenous carbon monoxide in the blood. J. Am. Chem. Soc. 138, 5417–5425 (2016).
Schipper, H. M., Song, W., Tavitian, A. & Cressatti, M. The sinister face of heme oxygenase-1 in brain aging and disease. Prog. Neurobiol. 172, 40–70 (2019).
Imai, T., Matsubara, H. & Hara, H. Potential therapeutic effects of Nrf2 activators on intracranial hemorrhage. J. Cereb. Blood Flow. Metab. 41, 1483–1500 (2021).
Rochette, L., Zeller, M., Cottin, Y. & Vergely, C. Redox functions of heme oxygenase-1 and biliverdin reductase in diabetes. Trends Endocrinol. Metab. 29, 74–85 (2018).
Li, L. et al. Overexpression of heme oxygenase 1 impairs cognitive ability and changes the plasticity of the synapse. J. Alzheimers Dis. 47, 595–608 (2015).
Si, Z. & Wang, X. The neuroprotective and neurodegeneration effects of heme oxygenase-1 in alzheimer’s disease. J. Alzheimers Dis. 78, 1259–1272 (2020).
Cioffi, F., Adam, R. H. I., Bansal, R. & Broersen, K. A review of oxidative stress products and related genes in early alzheimer’s disease. J. Alzheimers Dis. 83, 977–1001 (2021).
Wang, Y. et al. Pharmacological modulation of Nrf2/HO-1 signaling pathway as a therapeutic target of parkinson’s disease. Front. Pharmacol. 12, 757161 (2021).
Lawrence, J. M., Schardien, K., Wigdahl, B. & Nonnemacher, M. R. Roles of neuropathology-associated reactive astrocytes: a systematic review. Acta Neuropathol. Commun. 11, 42 (2023).
Ferreira, A. C. et al. Lipocalin-2 regulates adult neurogenesis and contextual discriminative behaviours. Mol. Psychiatry 23, 1031–1039 (2018).
Kim, T. H., Bae, C. H., Yang, S., Park, J. C. & Cho, E. S. Nfic regulates tooth root patterning and growth. Anat. Cell Biol. 48, 188–194 (2015).
Farioli-Vecchioli, S., Ricci, V. & Middei, S. Adult hippocampal neurogenesis in Alzheimer’s disease: an overview of human and animal studies with implications for therapeutic perspectives aimed at memory recovery. Neural Plast. 2022, 9959044 (2022).
Hernandez-Mercado, K. & Zepeda, A. Morris water maze and contextual fear conditioning tasks to evaluate cognitive functions associated with adult hippocampal neurogenesis. Front. Neurosci. 15, 782947 (2021).
Martin, C. et al. Leptin-responsive GABAergic neurons regulate fertility through pathways that result in reduced kisspeptinergic tone. J. Neurosci. 34, 6047–6056 (2014).
Kromrey, M. L. et al. Whole-body r2 * mapping to quantify tissue iron in iron storage organs: Reference values and a genotype. Clin. Radiol. 76, 863 e811–863.e817 (2021).
Hinarejos, I., Machuca-Arellano, C., Sancho, P. & Espinos, C. Mitochondrial dysfunction, oxidative stress and neuroinflammation in neurodegeneration with brain iron accumulation (NBIA). Antioxidants 9, 1020 (2020).
Raghunath, A. et al. Antioxidant response elements: discovery, classes, regulation and potential applications. Redox Biol. 17, 297–314 (2018).
Loboda, A., Damulewicz, M., Pyza, E., Jozkowicz, A. & Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: an evolutionarily conserved mechanism. Cell. Mol. Life Sci. 73, 3221–3247 (2016).
Shan, Y., Lambrecht, R. W., Donohue, S. E. & Bonkovsky, H. L. Role of Bach1 and Nrf2 in up-regulation of the heme oxygenase-1 gene by cobalt protoporphyrin. FASEB J. 20, 2651–2653 (2006).
Qin, W., Cho, K. F., Cavanagh, P. E. & Ting, A. Y. Deciphering molecular interactions by proximity labeling. Nat. Methods 18, 133–143 (2021).
Agudo-Canalejo, J. et al. Wetting regulates autophagy of phase-separated compartments and the cytosol. Nature 591, 142–146 (2021).
Hwang, I., Tang, D. & Paik, J. Oxidative stress sensing and response in neural stem cell fate. Free Radic. Biol. Med. 169, 74–83 (2021).
Gudjoncik, A. et al. Iron, oxidative stress, and redox signaling in the cardiovascular system. Mol. Nutr. Food Res. 58, 1721–1738 (2014).
Behl, T. et al. Mechanistic insights expatiating the redox-active-metal-mediated neuronal degeneration in parkinson’s disease. Int. J. Mol. Sci. 23, 678 (2022).
Chatterjee, P. et al. Serum hepcidin levels in cognitively normal older adults with high neocortical amyloid-beta load. J. Alzheimers Dis. 76, 291–301 (2020).
Lim, D., Jeong, J. H. & Song, J. Lipocalin 2 regulates iron homeostasis, neuroinflammation, and insulin resistance in the brains of patients with dementia: Evidence from the current literature. CNS Neurosci. Ther. 27, 883–894 (2021).
Anandhan, A., Kirwan, K. R., Corenblum, M. J. & Madhavan, L. Enhanced NRf2 expression mitigates the decline in neural stem cell function during aging. Aging Cell 20, e13385 (2021).
Ayer, A., Zarjou, A., Agarwal, A. & Stocker, R. Heme oxygenases in cardiovascular health and disease. Physiol. Rev. 96, 1449–1508 (2016).
Branca, C. et al. Genetic reduction of Nrf2 exacerbates cognitive deficits in a mouse model of alzheimer’s disease. Hum. Mol. Genet. 26, 4823–4835 (2017).
Gergues, M. M., Moiseyenko, A., Saad, S. Z., Kong, A. N. & Wagner, G. C. Nrf2 deletion results in impaired performance in memory tasks and hyperactivity in mature and aged mice. Brain Res. 1701, 103–111 (2018).
Kanninen, K. et al. Intrahippocampal injection of a lentiviral vector expressing Nrf2 improves spatial learning in a mouse model of alzheimer’s disease. Proc. Natl Acad. Sci. USA 106, 16505–16510 (2009).
George, M., Tharakan, M., Culberson, J., Reddy, A. P. & Reddy, P. H. Role of Nrf2 in aging, alzheimer’s and other neurodegenerative diseases. Ageing Res. Rev. 82, 101756 (2022).
Wu, J. et al. The non-canonical effects of heme oxygenase-1, a classical fighter against oxidative stress. Redox Biol. 47, 102170 (2021).
Ward, R., Zucca, F. A., Duyn, J. H., Crichton, R. R. & Zecca, L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 13, 1045–1060 (2014).
Leshan, R. L., Bjornholm, M., Munzberg, H. & Myers, M. G. Jr. Leptin receptor signaling and action in the central nervous system. Obesity 14, 208S–212S (2006).
Liu, D. D. et al. Purification and characterization of human neural stem and progenitor cells. Cell 186, 1179–1194 e1115 (2023).
Chojnacki, A. & Weiss, S. Production of neurons, astrocytes and oligodendrocytes from mammalian CNS stem cells. Nat. Protoc. 3, 935–940 (2008).
Seibenhener, M. L. & Wooten, M. W. Isolation and culture of hippocampal neurons from prenatal mice. J. Vis. Exp. 65, e3634 (2012).
Chen, J. F. et al. Enhancing myelin renewal reverses cognitive dysfunction in a murine model of alzheimer’s disease. Neuron 109, 2292–2307.e2295 (2021).
Zimmer, T. S. et al. Seizure-mediated iron accumulation and dysregulated iron metabolism after status epilepticus and in temporal lobe epilepsy. Acta Neuropathol. 142, 729–759 (2021).
Wang, X. et al. Adipocyte-derived ferroptotic signaling mitigates obesity. Cell Metab. 37, 673–691.e677 (2025).
Fancy, S. P. et al. Axin2 as regulatory and therapeutic target in newborn brain injury and remyelination. Nat. Neurosci. 14, 1009–1016 (2011).
Cho, K. F. et al. Proximity labeling in mammalian cells with turboid and split-turboid. Nat. Protoc. 15, 3971–3999 (2020).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with deseq2. Genome Biol. 15, 550 (2014).
Mitsuishi, Y. et al. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 22, 66–79 (2012).
Shannon, P. et al. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
Hatzi, K. et al. Histone demethylase lsd1 is required for germinal center formation and BCL6-driven lymphomagenesis. Nat. Immunol. 20, 86–96 (2019).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. GigaScience 10, giab008 (2021).
Tarasov, A., Vilella, A. J., Cuppen, E., Nijman, I. J. & Prins, P. Sambamba: Fast processing of NGS alignment formats. Bioinformatics 31, 2032–2034 (2015).
Zhang, Y. et al. Model-based analysis of chip-seq (MACS). Genome Biol. 9, R137 (2008).
Quinlan, A. R. & Hall, I. M. Bedtools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Yu, G., Wang, L. G. & He, Q. Y. Chipseeker: An r/bioconductor package for chip peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383 (2015).
Ramirez, F. et al. Deeptools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–165 (2016).
Acknowledgements
We are very grateful to Dr. Songhai Shi and Dr. Liang Ge at the School of Life Sciences and Dr. Bailong Xiao, Dr. Wei Guo, Ms. Hazel Hua, and Ms. Haneul Gil at the School of Pharmaceutical Sciences at Tsinghua University for their very helpful discussion and for supplying Cre mouse strains. This work is supported by the Ministry of Science and Technology of China National Key R&D Programs (2022YFA0806503 and 2024YFA1306103 to L.C.), National Natural Science Foundation of China (32130048 and 92157301 to L.C.), A private donation from Tsinghua University Education Foundation (202011 to L.C.), A collaborative fund from CytoCraft Biotech Co., Tsinghua-Foshan Innovation Special Fund (20229990133 to L.C.), and Tsinghua Precision Medicine Foundation (2022TS013 to L.C.).
Author information
Authors and Affiliations
Contributions
Conceptualization: Ligong Chen, Mengmeng Ge, Lei Tao, Long Teng. Investigation: Mengmeng Ge, Lei Tao, Long Teng, Yaqi Wang, Chengliang Hu, Jianan Chen, Jingyu Zhao, Lili Cheng, Ligong Chen. Methodology: Lei Tao, Long Teng, Mengmeng Ge, Yaqi Wang, Chengliang Hu, Jianan Chen, Jingyu Zhao, Lili Cheng, Zijuan Zhang, Wei Qin, Yichang Jia, Da Mi, Zengqiang Yuan, Ligong Chen. Visualization: Lei Tao, Mengmeng Ge. Writing, review, and editing: Ligong Chen, Lei Tao, Mengmeng Ge, Da Mi, Zengqiang Yuan. Funding Acquisition: Ligong Chen.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Thomas Bartnikas, Milankumar Prajapati, Phu Tran, and the other anonymous reviewer(s) for their contribution to the peer review of this work. [A peer review file is available].
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tao, L., Teng, L., Ge, M. et al. Slc22a17 governs postnatal neurogenesis by maintaining the iron homeostasis in hippocampus. Nat Commun 16, 11117 (2025). https://doi.org/10.1038/s41467-025-66108-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-66108-w
