
Abstract
Adverse outcomes including myocardial infarction (MI) and stroke render coronary artery disease (CAD) a leading cause of death worldwide. DNA methylation markers may alert such adversity ahead of the events. We profiled DNA methylation of blood leukocytes in 933 Chinese CAD patients with up-to-13-year follow-up from three centers, identifying 70 differentially methylated sites (DMPs) associated with future death. These DMPs correlated with inflammation markers, left ventricular functions and high-density lipoprotein cholesterol, and impacted gene expression in immune response and cellular scenesence. Notably, cg25563198 and cg25114611 were discovered to regulate FKBP5, whose upregulation persisted during MI and stroke. Fkbp5 knockout in male mice partially rescued MI by reducing infarct size and improving heart function, confirming its critical function. Finally, our prognostic model of 10 methylation sites and 5 clinical features outperformed clinical models. Our study highlights the value of DNA methylation in predicting prognosis in CAD and provides tools for clinical translation.
Similar content being viewed by others

Introduction
Coronary artery disease (CAD) is life-threatening and represents a universal leading cause of death. Studies of the last century suggested a 15-year survival rate of 48–70%1,2. Despite the remarkable amelioration in the recent 30 years in managing its clinical risk factors and the secondary and tertiary preventions, CAD is associated with 17.8 million annual deaths worldwide3. Beyond mortality, other major adverse cardiovascular events (MACE) include myocardial infarction (MI), stroke, and revascularization, casting a heavy burden to the healthcare system. Identifying patients with greater risk of poor prognosis enables closer medical supervision and therefore opportunities for better clinical outcomes. Numerous genetics-based research reported novel targets and tools for predicting adverse outcomes in CAD patients. Indeed, CAD has an estimated heritability of 38–66% for incidence4 and 38–57% for mortality5. However, towards which direction it progresses is multifactorial determined by the combined effects of genetic and environmental factors, therefore we reason that considering multiple layers of information, such as genetics and epigenetics, will better identify patients susceptible to poor prognostic outcomes.
DNA methylation on CpG (cytosine-phosphate-guanine) dinucleotides reflects both genetic regulation and environmental influence, enabling exploration of their integrated effects on diseases6. Epigenome-wide association studies (EWAS) suggested DNA methylation as a feasible biomarker for CAD. Two recent large-scale EWAS surveyed multiple cohorts of various ancestries and collectively reported 85 DNA methylation sites in blood leukocytes to be associated with incident CAD or MI7,8. Comprehensive studies also report association between DNA methylation and the risk factors of CAD including aging9, smoking10, blood lipids11, inflammation12, hypertension13, and diabetes mellitus (DM)14. Furthermore, initial EWAS studies identified strong signals that predicted all-cause death of cardiovascular diseases15,16, albeit its biological mechanisms remained to be explored. As such, DNA methylation indicates not only the risk of CAD incidence but also its progression.
Here, we profiled DNA methylation of blood leukocytes in 933 Chinese CAD patients at baseline and interrogated its association with future death, in up to 13 years of follow-up. We aimed to discover differentially methylated CpG positions (DMPs) which serve as biomarkers for predicting the CAD prognosis. Furthermore, we inferred molecular mechanisms that drive CAD progression. Our results suggest that DNA methylation of blood leukocytes are robust biomarkers and provide rich insights into the prognosis of CAD.
Results
Baseline characteristics
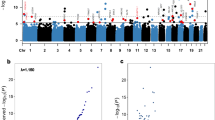
We adopted a two-stage multicenter design for studying DNA methylation related to CAD prognosis (Fig. 1a). Initially, over 5000 CAD patients were enrolled in a medical center in China and followed for medical outcomes, from which 405 patients were selected by the nested case-control study design to form a discovery set for this study. In a follow-up period of up to 13 years, 217 deaths were recorded. For the validation set, we enrolled 528 CAD patients from three medical centers in China. In about three years after enrollment, 25 deaths were recorded. In both study sets, patients in the death group were older and had a lower rate of aspirin use, and higher blood levels of aspartate aminotransferase (AST) and creatinine (Table 1).

a Study design. Patients in the discovery and the validation sets were enrolled from one and three medical centers in China, respectively. The sample size for discovery and validation cohorts were 405 and 528, respectively. Baseline characteristics were collected during enrollment. DNA methylation of peripheral blood leukocytes was measured by Illumina MethylationEPIC 850 K BeadChip. Differential methylation sites associated with death were identified, prognostic risk models were built, and lastly, biological mechanisms were inferred. This graph was created in BioRender. Pan, C. (2025) https://BioRender.com/mao524f. In the boxplot of panels, hinges indicate the 25th, 50th, and 75th percentiles, whiskers indicate 1.5× interquartile ranges. b EWAS of death performed in the discovery set. Red line and blue line mark the P-value thresholds, with the former from Bonferroni correction, and the latter from false discovery rate correction. CAD coronary artery disease, PLR platelet-lymphocyte ratio, FIB fibrinogen, SII systemic immune-inflammation index, HDLC high-density lipoprotein cholesterol, LVEF left ventricular ejection fraction, LVMI left ventricular mass index, DMP differential methylation probe, TF transcriptional factor, IL interleukin.
Differentially methylated CpGs associated with CAD prognosis
We profiled DNA methylation of blood leukocytes collected at enrollment (baseline) via the Illumina Infinium MethylationEPIC 850 K BeadChip. After stringent quality control, 733,737 high-quality CpG probes in the discovery set and 738,366 probes in the validation set were obtained (Supplementary Fig. 1). Subsequently, through COX survival model-based EWAS, using sex, age, smoking status, percutaneous coronary intervention, heart failure, hypertension, arrhythmia, hyperlipidemia, type 2 diabetes and medications as covariates, a total of 333 DMPs were initially identified in the discovery set to be associated with death (PFDR < 0.05) (Fig. 1b and Supplementary Fig. 2A). Most of these were hypermethylation (Supplementary Fig. 2B). Notably, 54% of them (180/333) have been recorded in the EWAS Catalog17 and EWAS Atlas18 to be associated with a variety of traits and disorders (Supplementary Table 1), including Crohn’s disease and inflammatory bowel disease (38 DMPs), smoking (36 DMPs), drinking (18 DMPs), aging (14 DMPs), weight or body mass index14,19 (8 DMPs), death risk20 (5 DMPs), and C-Reactive Protein (2 DMPs)12. In the validation set, 70 of the DMPs were replicated with consistent directions of effect (P < 0.05, Table 2). Noteworthily, 2 DMPs (cg25114611 and cg25563198) mapped to FKBP5 (FK506 binding protein 5) were significantly associated with the death risk of CAD20, among which cg25114611 was also reported to be associated with acute MI21.
Pathways and mediating phenotypes inferred by DMPs
Half of the 70 DMPs reside in gene regulatory elements (Supplementary Fig. 3). Overlap with histone modification chromatin immunoprecipitation (ChIP) peaks and the 15 chromatin states in Roadmap22 revealed strong enrichment of enhancers specific to blood monocytes, adipocytes, myoepithelial cells, fibroblasts, left ventricle, and right atrium (P < 0.05, Fig. 2a, b), most of which are characteristic of the heart and cell types known to play critical roles in CAD. Given that both DNA methylation and RNA transcription in blood were distinct from solid organs23, our results suggest that DNA methylation in leukocytes carried pathophysiological features.

a Enrichment of tissue and cell types by referencing to the histone modification peaks, H3K36me3 (a mark for near transcription termination site) and H3K4me1 (a mark for active enhancers). b Enrichment of tissue and cell types by enhancers and transcription start sites, referencing to the 15 chromatin states in RoadMap Epigenomics. Enh: Enhancer, TxWk: Weak transcription. a, b Statistical enrichment analysis was performed using a binomial test against an array-specific background. c Enrichment of Gene Ontology terms among the prognosis genes. The P-value was computed from the Fisher exact test. d Association of the DMPs with inflammation indices, lipids, and heart functions. Scaled methylation beta values were presented. Significant correlations (P < 0.05) were marked by *. Linear regression was used to identify the relationship between DMPs and clinical phenotypes with age, sex, smoking, and percutaneous coronary intervention adjusted. WBC whole plasma cell count, LMR: lymphocyte-monocyte ratio, NLR neutrophil-lymphocyte ratio, PLR platelet-lymphocyte ratio, FIB fibrinogen, SII systemic immune-inflammation index, LDLC low-density lipoprotein cholesterol, HDLC high-density lipoprotein cholesterol, CHOL total cholesterol, TRIG triglycerides, LVEF left ventricular ejection fraction, LVMI left ventricular mass index. Source data were provided as a Source Data file.
By annotating the 70 DMPs to the nearest genes, we uncovered 69 prognosis genes. These genes were enriched for phosphorylation in signal transduction, stress response, apoptosis, and inflammatory response (Fig. 2c). Separately, we associated the DMPs to nearby enhancers (<=2500 bp)24 and their target genes via a chromatin interaction model, Association by Contact (ABC)25. As such, 53 of the 70 DMPs (75.7%) were connected to 468 genes (Supplementary Fig. 4A), which confirmed a strong enrichment in inflammatory response and senescence (Supplementary Fig. 4B). Notably, the two DMPs of FKBP5, cg25563198 and cg25114611, were mapped to a super-enhancer reported active in CAD relevant tissues, such as blood, lymphoid, adipose tissue, heart ventricle, and aorta26. By ABC model, this super-enhancer connects with >50 genes, including FKBP5, which are strongly enriched for senescence (Supplementary Fig. 4C). Furthermore, through enhancer-cell type specificity mapping, we inferred that the function of the DMPs could be most strongly enriched in monocytes and dendritic cells (Supplementary Fig. 5).
We further verified the association of the DMPs with inflammation and lipids using clinical measurements (Fig. 2d). A strong connection to inflammation markers was observed, particularly those involving platelets, i.e., systemic immune-inflammation index (SII), fibrinogen (FIB), and platelet-lymphocyte ratio (PLR). SII measures the proportion of platelets and neutrophils among the lymphocytes27, fibrinogen is an index for chronic low-grade inflammation28, and PLR serves as a marker for acute inflammation and prothrombotic status29. Furthermore, we observed that a significant proportion of the DMPs (19 out of 70 DMPs, 27%) displayed associations with high-density lipoprotein cholesterol (HDLC) levels in both discovery and validation sets (P < 0.05, Fig. 2d), but not with low-density lipoprotein cholesterol (LDLC), total cholesterol (TC), or triglycerides (TG). The association directions of these 19 DMPs are consistently opposite to HDLC and death (Supplementary Fig. 6), i.e., they are all positively associated with death risk and negatively associated with HDLC levels, in line with the protective role of HDLC (a proxy for “good cholesterols”) in cardiovascular outcomes. Finally, associations between the DMPs and left ventricle (LV) function were found, including its ejection fraction (LVEF) and mass index (LVMI).
Contribution of genetic regulation on DNA methylation
Methylation can be regulated genetically by methylation quantitative trait loci (meQTL), thus providing a tool for investigating how genetics influences CAD prognosis. We queried the DMPs against a meQTL dataset derived from 3523 East Asians30. Notably, a large proportion of the DMPs (53/70, 76%) were paired with over 7000 meQTLs from East Asians (Supplementary Fig. 7A, B). Indeed, compared with all CpGs on the array, the DMPs were enriched for both cis (n = 42, P < 0.0001) and trans meQTLs (n = 31, P < 0.0001, all identified trans-meQTLs resided on chromosomes distinct from their target CpGs), although most associations were weak (Supplementary Fig. 7C, D). Strikingly, cg16500036, closest to AUTS2, which encodes for Activator of Transcription and Developmental Regulator, was associated with more than 1000 cis-meQTLs. The two DMPs for FKBP5, cg25563198 and cg25114611, had the largest number of trans-meQTLs (Supplementary Fig. 7E). These results suggest that the important prognosis genes were regulated genetically.
Contribution of DNA methylation on gene expression
To validate the impact of DMPs on gene expression, we conducted expression quantitative trait methylation (eQTM) analysis. 54.4% DMPs (37/68) correlated with expression levels of their proximal genes (Pearson correlation, P < 0.05, Supplementary Table 2), with a majority (25/37) displaying an inverse correlation between the methylation levels and gene expression levels. The strongest correlations were observed for cg19526450 with ARHGAP26 transcript (cor = −0.49, P = 4.81E-25) and cg26869211 with USP32 transcript (cor = −0.41, P = 2.50E-17).
DNA methylation levels of the two DMPs for FKBP5, cg25114611 and cg25563198, were inversely correlated with the expression level of FKBP5 (Fig. 3a, b). From published transcriptome31,32, we confirmed that the expression of FKBP5 was significantly elevated in the peripheral blood of MI patients (Fig. 3c) and stroke patients (Fig. 3d). Strikingly, FKBP5 was upregulated in nearly all cell types of the heart tissue with MI progression, as demonstrated in the single-cell transcriptome of myogenic (nonischemic zone), ischemic (MI lesion zone), and fibrotic (advanced MI tissue zone) regions of the heart33 (Fig. 3e).

a The correlation between DNA methylation level of cg25114611 and FKBP5 gene expression in whole blood (n = 391). Light green shading represents the 95% confidence intervals. b The correlation between DNA methylation level of cg25563198 and FKBP5 gene expression in whole blood (n = 391). Light green shading represents the 95% confidence intervals. a, b P-values were calculated using a two-sided Pearson’s correlation coefficient assays. c Differential expression analysis of FKBP5 gene between normal and myocardial infarction participants in peripheral blood (ncontrol = 10, nMI = 7). In the boxplot of panels, hinges indicate the 25th, 50th, and 75th percentiles, whiskers indicate 1.5× interquartile ranges. d Differential expression analysis of FKBP5 gene between control and ischemic stroke participants in peripheral blood (ncontrol = 24, nStroke = 39). In the boxplot of panels, hinges indicate the 25th, 50th, and 75th percentiles, whiskers indicate 1.5× interquartile ranges. c, d P-values were calculated using a two-sided Wilcoxon test. e Gene expression of FKBP5 in various cell types as the disease progression of myocardial infarction in heart tissue. MI myocardial infarction. Source data were provided as a Source Data file.
Downregulation of FKBP5 improves left ventricular functions and reduces inflammation responses
Our findings collectively suggest that FKBP5 plays a critical role in CAD prognosis. To further investigate its role in MI, a severe outcome of CAD, we generated the knockout (KO) mice of Fkbp5−/− and subjected them to either sham surgery (KO-sham) or myocardial ischemia/reperfusion (KO-MI/R) injury surgery (Supplementary Fig. 8 and Supplementary Table 3). First, we verified that the Fkbp5 expression level was significantly upregulated in wild-type (WT) mice with MI/R surgery compared with WT-sham mice using RT-qPCR (Fig. 4a). Reducing the Fkbp5 expression did not seem to cause difference in heart functions in the sham surgery groups, as no difference was observed by echocardiography between the WT-sham and the KO-sham mice (Fig. 4b–d). Next, we verified that the MI/R surgery successfully induced MI, as LVEF and LV fractional shortening (LVFS) were significantly reduced in the WT-MI/R compared with the WT-sham mice (Fig. 4b–d). Furthermore, cross-comparison revealed that knocking out of Fkbp5 exerted a protective effect against MI, as LVEF and LVFS were significantly increased in the KO-MI/R mice compared with the WT-MI/R (Fig. 4b–d). Additionally, Evans Blue/TTC staining showed that the infarct size in KO-MI/R was significantly reduced compared with WT-MI/R mice (Fig. 4e–g). The immune marker PLR was significantly lower in MI/R mice with Fkbp5−/− compared with that in WT-MI/R mice (Fig. 4h), consistent with our previous finding that in patients with lower level of inflammation, hypermethylation of FKBP5 were found, which was indicative of lower expression of FKBP5 (Fig. 2d). Our results confirmed an essential role of FKBP5 in MI.

a Relative Fkbp5 mRNA expression in heart tissue of wild-type and Fkbp5−/− mice (n = 3 for each group, N = 3 experiments, *P = 0.018, **P = 0.007). b Representative echocardiogram images of wild-type and Fkbp5−/− mice with sham or myocardial ischemia-reperfusion operation. c Left ventricular ejection fraction in wild-type and Fkbp5−/− mice with sham or myocardial ischemia-reperfusion operation (nWT-sham = 7, nKO-sham = 7, nWT-MI/R = 10, nKO-MI/R = 8, ****P < 0.0001). d Left ventricular fractional shortening in wild-type and Fkbp5−/− mice with sham or myocardial ischemia-reperfusion operation (nWT-sham = 7, nKO-sham = 7, nWT-MI/R = 10, nKO-MI/R = 8, ***P = 0.0002, **P = 0.008). e Representative images of heart sections by TTC/Evans Blue staining depicting the infarcted area. f, g Cardiac injury was evaluated by the ratio of infarct size (INF) to area at risk (AAR) (nWT-MI/R = 4, nKO-MI/R = 4, *P = 0.029). h Platelet-to-lymphocyte ratio of wild-type and Fkbp5−/− mice with sham or myocardial ischemia-reperfusion operation (nWT-sham = 6, nKO-sham = 6, nWT-MI/R = 6, nKO-MI/R = 5, ***P = 0.0003, *P = 0.039). WT wild type, KO knockout, LVEF left ventricular ejection fraction, LVFS left ventricular fractional shortening, AAR: area at risk, INF infarct size, LV left ventricular, PLR platelet-lymphocyte ratio. Data are presented by mean ± SD for each group. Statistical significance was performed by two-way ANOVA with Šídák’s test for multiple comparisons. ns no significance, *P < 0.05, **P < 0.01, ***P < 0.005, ****P < 0.0001. Source data were provided as a Source Data file.
Taken together, we propose that hypomethylation, such as those on cg25114611 and cg25563198, leads to higher expression of FKBP5, which plays critical roles in driving the poor prognosis of CAD. Downregulating Fkbp5 was shown in mouse models to improve the LV dysfunction caused by myocadiac injury, suggesting the potential of FKBP5 in treating and improving CAD outcomes.
Prognostic models for death in CAD
Based on the 15 DMPs that showed epigenome-wide Bonferroni significance in the discovery set, we selected ten DMPs with the biggest effects, as measured by the variance importance value (VIMP) in a random survival forest algorithm based on bootstrap resampling of 1000 repetitions, to construct prognosis models of death in CAD (Supplementary Fig. 9). Subsequently, using COX regression algorithm and 1000 times cross-validation, our model based on the 10 DMPs (the CG model) achieved an area under the curve (AUC) of 0.71 (Fig. 5a). When combining two common risk factors of cardiovascular diseases, i.e., age and sex, the updated model achieved an AUC of 0.81. We also built prognostic models based on the mediating phenotypes. Although not all the clinical features were equally powerful in predicting the adverse outcomes (Supplementary Fig. 10A–C), we found that the Ensemble model combining the 10 DMPs, sex, age, fibrinogen, HDLC, and LVEF achieved an AUC of 0.83 (Fig. 5a), which was a significant increase compared with the clinical model that include only sex, age, fibrinogen, HDLC, and LVEF (ΔAUC = 0.034, P = 3.95E-07). Notably, the Ensemble model could also identify CAD patients who survived severe adverse events such as coronary revascularization, stroke, and myocardial infarction (Fig. 5b), suggesting it indeed captured the essential signals of prognosis. When applying our prognosis model to the independent validation set, a good stratification of the patients by the presence or absence of future death event was observed (Figs. 5c and 4d), albeit there was a drop of 10% in sensitivity and 4% in specificity in this independent validation (P < 3 × 10−4, Table 3).

a ROC curves of the prognostic models of death as constructed based on the discovery set. CG refers to the selected 10 DMPs for model construction. The Ensemble model is composed of 10 DMPs + sex + age + FIB + HDLC + LVEF, and the clinical model is composed of sex + age + FIB + HDLC + LVEF. b Prediction scores when applying the prognostic models of death to the discovery set, with patients categorized by different clinical outcomes (nno adverse outcome = 160, nRevascularization = 34, nStroke = 27, nMI = 26, nDeath = 158). c Kaplan-Meier curves when applying the Ensemble model to the validation set. Patients having all the modeling features were considered. d Prediction scores in the validation set, with patients categorized by clinical outcomes (nSurvival = 503, nDeath = 25). b, d In the boxplot of panels, hinges indicate the 25th, 50th, and 75th percentiles, whiskers indicate 1.5× interquartile ranges. The two-sided P-values were calculated by Kruskal-Wallis test. FIB fibrinogen, HDLC high-density lipoprotein cholesterol, LVEF left ventricular ejection fraction.
Age is a known strong risk factor for CVD. Observing the chronological age predicted closely to the CG models, we explored the performance of several DNA methylation clocks34,35,36,37 for predicting death in CAD patients. DNA methylation clocks have been shown to better represent one’s aging status. Indeed, most clock models achieved better prediction than the chronological age models for death (Supplementary Fig. 10D) and performed equally well or even better than the CG prognostic models. As these clocks comprised dozens to hundreds of CpG sites, one to two orders of magnitude more than the maximal 10 CpG sites in our models, our CG prognostic models are more succinct and specific.
Discussion
In this study, we analyzed about 733,000 DNA methylation sites in 933 Chinese CAD patients for their association with death. Our prognostic model based on 10 DMPs and numerous clinical features reached AUC of 0.83, could be validated in an independent set of patients, and performed superior to the models solely built on clinical features.
A strong association with platelets-involved inflammation, the functions of the left ventricle, and HDLC was observed. As platelets and cholesterol were essential components of thrombosis, our results suggest that early thrombo-inflammation and heart contraction malfunction mediated the adverse outcomes in CAD. Notably, HDL participates in reverse cholesterol transport, interacts with platelets, and exerts an antithrombotic function by suppressing the coagulation cascade and stimulating clot fibrinolysis38. The observations that (1) DMPs were associated strongly with HDLC but not other lipids and (2) HDLC displayed better predictability in adverse outcomes than other lipids suggest that the ability to remove cholesterol, rather than its accumulation, was more relevant to CAD adverse outcomes. A recent study discovered that LDLC, compared to the inflammation index C-reactive protein, was less effective in predicting future cardiovascular events and death39. Our study suggests that HDLC, not LDLC, may be a more relevant predictor. Therefore, our methylation study may inspire new research for clinical translation.
A total of 70 DMPs were replicated in both patient sets to be associated with future death in CAD. Given that many of these DMPs have been reported in European ancestry-centric studies to be associated with autoimmune and cardiovascular risk-related traits, as recorded in the EWAS Catalog and EWAS Atlas, our DMPs are functionally relevant and possess generalizability across populations. We note that 76% of these DMPs were located on or near enhancers (<= 2500 bp), indicating a tight connection to gene expression, particularly those genes in premature senescence and inflammation. We observed cell type specificity per the regulatory elements that the DMPs overlapped with, particularly enhancers, and discovered the DMPs were prone to occur in regions characteristic of heart traits. Interestingly, the characteristic premature senescence and inflammation signals were robust in monocytes and dendric cells, warranting further studies. Furthermore, we observed a significant genetic regulation of the DMPs, with a striking proportion (76%) of the DMPs mapped to known meQTLs. Important prognosis genes such as FKBP5 and AUTS2 own the largest number of meQTLs for their DMPs. The fact that 10 s to 1000 s of meQTLs regulating one single DMP, each with a weak strength, indicated that the trickling of little genetic signals had mounted to significant epigenetic outcomes, which resembled polygenic models in complex traits40.
Among the 69 prognosis genes we identified, most displayed subtle expression changes at the occurrence of adverse outcomes, such as ischemic stroke and myocardial infraction. This can be attributed to the nature of this study that the biomarkers were discovered at the baseline and mainly served as early alarms. They represent proceeding events months to years before the occurrence of the adverse outcomes. That said, however, several prognosis genes such as FKBP5 and AUTS2 displayed drastic expression changes during the adverse events and occurred repetitively as the most significant findings along various analyses.
FKBP5 is an immunophilin protein that binds to immunosuppressive drugs. In our study, FKBP5 had numerous DMPs associated with inflammation markers and heart functions. One of them is cg25114611, which has been reported in acute MI21, death risk20, inflammatory bowel disease41, Crohn’s disease42, maternal BMI43, and diabetes mellitus44. FKBP5 expression was reported to be significantly altered in dilated cardiomyopathy after heart transplantation and suggested as a prognostic marker45. In our analysis of MI and ischemic stroke, FKBP5 appeared as a most highly regulated gene. Its elevation in the MI lesion site was most drastic in both innate immune lymphoid cells and adipocytes of the epicardial fat of the left ventricle. These results align with the recent finding that DNA demethylation led to increased expression of FKBP5, which in turn promoted NF-κB signaling in immune cells, resulting in a proinflammatory response and increased cardiovascular risk46. For AUTS2, its genetic variation was reported in blood pressure47, body mass index47, type 2 diabetes48, and mild heart defects49. This ample evidence strongly supports the roles that these prognosis genes play in CAD progression.
Importantly, our study contributes to the growing evidence that DNA methylation sites not only illuminate mechanistic changes in disease pathology but also serve as predictive biomarkers for clinical outcomes. For example, Zhang et al. identified a panel of DNA methylation markers in peripheral blood that robustly predicted all-cause mortality in large prospective cohorts, with some CpGs located in genes linked to inflammation and cardiometabolic regulation16. Similarly, Chybowska et al. constructed EpiScores based on 45 proteins to predict cardiovascular risks independently of traditional clinical factors50. While prior studies have explored the predictive value of DNA methylation markers for the onset of cardiovascular diseases, i.e. focusing on primary prevention, our study specially addresses treatment outcomes. We present streamlined models comprising just only 10 DMPs that effectively forecast prognostic risks in CAD, targeting secondary prevention. Compared to models that incorporate hundreds of methylation sites, our concise model enhances feasibility for clinical translation.
There are several limitations in our study. First, most adverse events occurred to our CAD patients were within the first 5 years of ascertainment, therefore our study captured signals for short-term to intermediate timespan. Given a longer interrogation time, the DMPs predicting longer-term adversity will be better uncovered. Second, methylation profiling often comes with strong batch effects, therefore, our two sets of patients cannot be simply combined for a larger EWAS test, thus limiting the discovery power. Under this constriction, we adopted a “discover and replicate” strategy to ensure that our reported DMPs were replicated. Third, the relatively small number of death cases in the validation set may limit the statistical power of our study. Nevertheless, the consistent effect sizes observed between the discovery and validation sets strengthen our confidence in the robustness of our findings. Future research of larger prospective cohorts is warranted for confirm these findings. Fourth, although 850 K EPIC array could assess CpG methylation genome-wide, many CpG sites were not probed and therefore leaves a large room for future discovery of prognostic markers. Finally, our murine model employed a full Fkbp5 knockout, which offers clear mechanistic insight but represents a more extreme perturbation than the modest epigenetic downregulation observed in human cohorts. Still, the protective phenotype in knockout mice provides converging evidence that downregulation of FKBP5 is mechanistically linked to ameliorated myocardial injury.
To conclude, our study displays the value of leveraging DNA methylation of peripheral blood in predicting future adverse events in CAD patients. Further studies are warranted to investigate the roles of the methylation sites, genes, pathways, and mediating phenotypes implicated in our study for a mechanistic understanding of the CAD adverse outcomes.
Methods
Study design
This study was approved by the Medical Research Ethics Committee of Guangdong Provincial People’s Hospital, Xiangya Hospital, Central South University and The First Affiliated Hospital of Sun Yat-Sen University (approval number: GDREC2017071H, 201701012, and 2017024) and complied with the Declaration of Helsinki. All patients provided written informed consents.
We recruited over 5000 CAD patients from Guangdong Provincial People’s Hospital between January 2010 to December 2017, and by a nested case-control study design, selected 405 patients therein to form the discovery set. For the validation set, 528 patients were recruited from 2017 to 2018 from three medical centers in two areas of China, namely Guangdong Provincial People’s Hospital, First Affiliated Hospital of Sun Yat-sen University, and Xiangya Hospital of Central South University51. All participants were identified by either a history of coronary artery bypass graft operation or a new diagnosis by coronary angiography to have ≥50% obstruction in minimally one main coronary artery, as assessed by the luminal diameter. The inclusion criteria were: (1) aged over 30 years old, (2) no history of renal transplantation or dialysis, (3) no cirrhosis, (4) not pregnant nor breastfeeding, (5) no malignancy, (6) no history of haemodialysis; (7) no history of thyroid problems, not using antithyroid drugs nor thyroid hormone medication in the past week, and (8) completed the follow-up surveys.
Sample and information collection at baseline
Participants were admitted to the hospitals, and after overnight fasting, blood samples were drawn at 7AM in the morning. Clinical laboratory tests were performed, and detailed clinical surveys, including medical history, family history, smoking status, and medication intake, were collected.
Counts of platelets and circulating leucocytes, including white blood cell (WBC), monocytes, lymphocytes, and neutrophils, were estimated using an automated blood cell counter (LH780; Beckman Coulter, Brea, CA, USA). Plasma fibrinogen levels were measured using a clot-based turbidimetric detection system. Lymphocyte-monocyte ratio (LMR) was calculated as the ratio of lymphocyte count (*109 /L) to monocyte count (*109 /L). Neutrophil-lymphocyte (NLR) ratio was calculated as the ratio of neutrophil count (*109 /L) to lymphocyte count (*109 /L). Platelet-lymphocyte ratio was calculated as the ratio of platelet count (*109 /L) to lymphocyte count (*109 /L). System immune-inflammation index was calculated using SII = platelet count × neutrophil count/lymphocyte count. Echocardiography was used to determine the function and structure of the left ventricle at the time of the baseline. Left ventricular ejection fraction was evaluated by two cardiologists using the modified Simposon’s rule with the Philips iE33 color Doppler ultrasound diagnostic system. Left ventricular mass index was calculated as left ventricular mass (g) / body surface area (m²).
All patients were followed up by telephone every six months by medical staff for inquiring the occurrences of all-cause death or MACE, with the latter defined as nonfatal myocardial infarction, coronary revascularization, stroke, and death.
DNA extraction from blood leukocytes
Whole blood was collected in EDTA-K2 anticoagulant tubes and immediately separated into plasma and hemocyte by centrifuging at 1000 g for 10 min at 4 °C. Genomic DNA was extracted from hemocyte and transferred to cryopreservation tubes, which were stored at −80 °C for subsequent experiments.
Genome-wide DNA methylation profiling and data preprocessing
DNA quality was assessed by ultraviolet spectrophotometer (Thermo Scientific, NanoDrop 2000). Briefly, about 500 ng of DNA was treated with sodium bisulfite for converting unmethylated nucleotide C to U, using the EZ DNA Methylation Kit (Zymo Research). After the conversion, methylation levels of more than 850,000 CpG sites were quantified using the Illumina Infinium MethylationEPIC BeadChip, which was run on an Illumina iScan Systems according to the manufacturer’s standard protocol. DNA methylation profiling was serviced by Genenergy Inc. The experimental operator was blind to the group information and randomly assigned the samples to different chips and plates.
Raw signal intensities of DNA methylation were stored in.idat files and imported to the R software using the “ChAMP” package52. Analysis was performed separately for the discovery set and the validation set. Methylation level of each probe, i.e., beta value, was defined as Meth/(Meth + Unmeth + 100), where Meth was the signal intensity of the CpG site in methylated form and Unmeth was that in unmethylated form. Beta values ranged from 0 to 1, with a larger value indicating a higher level of methylation. Probes were excluded if meeting one of the following criteria: (1) detection P-value >= 0.01, (2) bead count <3 in at least 5% of samples, (3) DNA methylation occurring to non-CpG dinucleotides, (4) aligning to multiple locations53, (5) located on chromosome X or Y. In total, 733,638 probes in the discovery set and 738,366 probes in the validation set were retained.
The qualified probes were normalized with the BMIQ method54 to correct for signal bias caused by type-I and type-II probes on the array. Next, we used the method in Houseman et al.55 to estimate relative proportions of blood cells, including CD8 lymphocytes, CD4 lymphocytes, natural killer cells, B cells, monocytes, and granulocytes. We also leveraged 224 positive control probes to evaluate the impact of technical confounders, which were generally referred to as batch effects, on the DNA methylation values. Briefly, we computed the principal components (PCs) of these positive control probes and assessed the association between the first 20 PCs and several technical parameters, including the indices for bisulfite conversion batch, plates, sample wells, and chip. Methylation residuals were then obtained via linear regression, with independent variables set as the beta value of each probe and dependent variables set as age, sex, smoking status, estimated white-blood-cell proportions, and the top 10 PCs of the positive control probes.
Epigenome-wide association analysis
Cox regression-based survival analysis was employed to explore the association between each methylation residual and the trait, i.e., all-cause death. We performed such EWAS for the discovery set and the validation set, respectively. In each EWAS, we adjusted for age, sex, smoking status, percutaneous coronary intervention, arrhythmia, heart failure, hypertension, hyperlipidemia, and diabetes mellitus, and medication intake including β-receptor blocker, angiotensin converting enzyme inhibitors, calcium channel blocker, proton pump inhibitor, clopidogrel, statin, and aspirin. A strict epigenome-wide significance threshold by Bonferroni correction was set as P < 6.83 × 10−8 and a moderate threshold by Benjamin & Hochberg correction was set as PFDR < 0.05. The differentially methylated site was considered validated when the association showed a consistent direction of effect in both sets and obtained a PFDR < 0.05 in the discovery set and P < 0.05 in the validation set.
Construction of prognostic models for death
Prognosis models were constructed based on the discovery set and tested in the validation set. In the discovery set, DMPs passing the Bonferroni-corrected epigenome-wide significance threshold were pruned by a random survival forest approach56 (feature pruning), and those retained were fit by the multivariate Cox regression to derive the final model (weight tuning). For ‘feature pruning’, variable importance value (VIMP) was calculated using the out of bag data based on permutation with bootstrap resampling by 1000 repetitions. The top 10 DMPs with the largest VIMP, which denoted the contribution of each input feature to the model, were retained. For ‘weight tuning’, the retained DMPs were fit by multivariate Cox regression in the R package “survival”. To derive robust AUC (the area under the receiver operating characteristic curve) values, we adopted a process of 80:20 data split and 1000 times cross-validation. The final model was obtained by combining all patients in the discovery set. For constructing models with both DMPs and clinical features, we repeated the same process described above by inputting both the DMPs and the selected clinical features to the Cox regression. The enhancements in model performance and discrimination were assessed by comparing the AUC value and conducting a likelihood ratio test.
The models were evaluated in the validation set by means of Kaplan-Meier curves and the difference of prediction risk scores between death and survival groups. Wilcoxon test was used to assess whether the prediction scores between the two groups of patients, i.e., with and without death events, were significantly different. Sensitivity and specificity of the models were computed using the ConfusionMatrix function from the R package “caret”.
Prognosis models of six inflammatory indices, four blood lipids, two left ventricular indices, and four DNA methylation clocks for predicting death in CAD were constructed by Cox regression using the R package “survival”. AUC values were computed.
Characterizing genomic features of DMPs
Genomic locations of DMPs were annotated by Annovar57. Overlap with regulatory elements was assessed against the ENCODE Encyclopedia version 5 (ENCODE5) cCRE catalog58, including insulators, promoters, distal enhancers, and proximal enhancers. Enrichment against tissue- and cell type-specific regulatory elements was performed based on histone modification chromatin immunoprecipitation peaks (ChIP) (H3K4me1, H3K4me3, H3K27me3, H3K36me3, H3K9me3, and H3K27ac marks) and regions of 15 chromatin states across 299 cell types and tissues from Roadmap Epigenomics22,59 using eFORGE v2.0 (https://eforge.altiusinstitute.org/).
Target gene predictions
Target genes impacted by the DMPs were predicted by two methods. For one, the annotation file provided by Illumina was queried, which assigned each CpG site to its nearest gene. For the other, the activity-by-contact (ABC) model developed by Nasser et al.25 was referenced, which identified active enhancers in a particular cell type and predicted their target genes based on chromatin states and their three-dimensional contacts. To identify the ABC enhancers that overlap with DMPs, we adopted the locus annotation approach by Zhang K et al.24 by looking up the ±2500 bp surrounding regions of the DMPs and overlapping them with the ABC enhancers of 131 human cell types. We adopted the original ABC score thresholds, i.e., ≥0.015 for distal element-gene connections and ≥0.1 for proximal promoter-gene connections, to define DMP – enhancer – target gene connections.
Differential gene expression analysis in myocardial infarction and stroke
Single-nucleus RNA sequencing data from 19 patients with acute MI as cases and four non-transplanted heart donors as controls were obtained from Kuppe et al.33 These included a total of 191,795 nuclei from 31 tissue samples, including ten major cardiac cell types. We performed differential gene expression analysis between the MI patients and controls, as well as among three tissue zones, namely myogenic, ischemic, and fibrotic zones. We also assessed differences between groups by cell types. Wilcoxon tests implemented in the FindMarkers function of the R package “Seurat” were used. Genes passing the Bonferroni-corrected P-value of 0.05 were considered differentially expressed. Bulk RNA sequencing of peripheral blood from patients of MI31 and patients of ischemic stroke32 were obtained. Differential gene expression analysis was performed between patients and controls using the R package “limma”.
meQTLs for the DMPs
Relationship between CpG sites and methylation quantitative trait loci in the East Asian population60 was queried to obtain overlap with the DMPs. cis-meQTL was defined as within 1 Mbp of the methylation site. trans-meQTL was defined as >5 Mbp away from the methylation site or located on a different chromosome than the methylation site. meQTL enrichment in the DMPs versus in all probes tested by the EPIC array was compared using Wilcoxon test in the R software.
Expression quantitative trait methylation analysis
The relationships between 70 prognosis-associated DMPs and the expression of their nearby or targeted gene were evaluated to determine whether these DMPs were expression quantitative trait methylation. The DNA methylation level of the 70 DMPs were available from the whole blood of 391 participants of the Progression of Early Subclinical Atherosclerosis (PESA) study (GSE220622), and the mRNA expression levels of their nearby or targeted genes were extracted from the same 391 participants of PESA study (GSE221615). Genes not detected in the PESA study were extracted from another transcriptome dataset with 340 samples from the Grady Trauma Project (GSE58137), and the DNA methylation data of the corresponding samples were obtained from GSE72680. cg21223135 was excluded because it failed quality control in the DNA methylation analysis, and cg06872019 was also excluded because its target gene LINC01066 was not detected in both of the transcriptome datasets. Pearson correlation test was used to evaluate the relationship between the levels of DNA methylation and gene expression. A two-sided P < 0.05 was considered statistically significant.
Animal knockouts
All experiments were randomized, and all animal experiments complied with the guidelines from directive 2010/63/EU of the European Parliament and were approved by the Animal Care and Use Committee of Guangdong Provincial People’s Hospital (number KY2023-1002-01). All experimental mice were maintained in a standard laboratory environment, with 70% relative humidity, 22 °C, and a 12:12- hour light–dark cycle. Both strains of Fkbp5 knockout (KO) and wild-type (WT) control mice were male, with a C57BL/6 J background, and were purchased from Cyagen Biological Co., LD (Suzhou, China). Briefly, the KO mice were generated using a conventional CRISPR/Cas9-mediated deletion of exon3 in the Fkbp5 gene. Homozygous KO mice (Fkbp5–/–) were born from a heterozygous (Fkbp5+/–) intercross and used for phenotypic analyses in parallel with WT littermates as a control group.
Myocardial ischemia-reperfusion model
A total of 32 male mice aged 8-12 weeks were included in this study and randomly assigned to myocardial ischemia-reperfusion (MI/R) group or sham sham-operated group. Throughout the entire procedure, mice were placed on a thermostatically controlled heating pad maintained at 37 °C. Anesthesia was induced using 1% sodium pentobarbital, and the mice were subsequently intubated and ventilated using a MiniVent rodent ventilator (Harvard Apparatus) throughout the surgical procedure. Ventilation parameters were set at a tidal volume of 250 μL and a respiratory rate of 150 breaths per minute, in accordance with standard murine physiology under anesthesia. In the MI/R group, a longitudinal incision approximately 1–1.5 cm in length was made along the 3rd-4th intercostal space on the left side of the sternum. Subsequently, the left anterior descending (LAD) coronary artery was ligated with 7-0 silk suture. Ischemia was confirmed by observing blanching and hypokinesia of the anterior wall of left ventricle, along with ST-segment elevation on electrocardiogram. After 30 minutes of the sustained ligation, the coronary artery was released for reperfusion by removing the filament for 24 h, which resulted in a rapid restoration of pink color to the affected myocardium. During these post-surgical steps, no medication was given. Mice in the sham group underwent the same surgical steps without ligating the LAD coronary artery. The surgical success rate was 100%, with no perioperative mortality observed in either the KO or WT control groups.
Echocardiography evaluation
For echocardiography, using 1.0–1.5% isoflurane (Sigma-Aldrich, St Louis, USA) to anesthetize all experimental mice. Echocardiographic evaluation was performed using a Visual Sonics Vevo 2100 (VisualSonics, Toronto, Canada) with a linear probe working at a frequency of 40 MHz. After anesthesia, the hair on the anterior chest was trimmed, and the mice were placed on a heating pad to maintain body temperature at 37 °C. Two-dimensional targeted M-mode trajectories were recorded from the parasternal short-axis view at the level of the mid-papillary muscle and the parasternal long-axis view at the level just below the papillary muscle. Heart rate and the left ventricular parameters including left ventricular end-systolic volume (LVESV), left ventricular end-diastolic volume (LVEDV), left ventricular internal dimension at end-diastole (LVIDd), and left ventricular internal dimension at systole (LVIDs) were analyzed based on M-mode recordings. The data are presented as the average of measurements of three consecutive beats. LVEF was calculated as LVEF = (LVEDV-LVESV)/LVEDV×100%61 and LV fractional shortening (FS) was calculated as FS = (LVIDd-LVIDs)/LVIDd×100%61, respectively.
Evans Blue/TTC staining
Evans blue-triphenyltetrazolium chloride (TTC) double-staining was performed to assess myocardial infarct size in four mice randomly selected from the KO-MI/R and WT-MI/R groups, respectively. At 24 h after reperfusion injury, mice were re-anesthetized and the LAD was re-tightened from the original ligation to occlude the artery. A small incision was then made in the right atrial appendage and injected with 2% Evans Blue dye (Solarbio, Beijing, China) via the left ventricular apex. After adequate dye perfusion, hearts were excised, rinsed in cold PBS to remove excess dye, and embedded in a cardiac slicing mold. Hearts were snap-frozen in the mold at −20 °C for 20 min, then sectioned transversely into 5–6 slices of equal thickness. Following this, the slices were incubated in 2% TTC (Solarbio, Beijing, China) for 15-20 min at 37 °C. The heart slices were then transferred to 4% paraformaldehyde (Servicebio, Wuhan, China) for 4 h prior to imaging. A stereo-microscope (ECLIPSE, Nikon, Japan) was used to observe staining and take images of the stained slices. The percentage of infarct area was calculated using Image Pro-Plus 6.0 software. Blue staining represents non-ischemic and normally perfused myocardium, red staining represents viable myocardium within ischemic areas, and the white area represents infarcted myocardium within high-risk areas (INF). The area at risk (AAR) was calculated as the sum of red and white regions, and the infarct size was expressed as the percentage of white area relative to the AAR (INF/AAR).
RT-PCR quantification of Fkbp5 mRNA
Total RNA was extracted from cardiac tissue with TRIzol Reagent (Life Technologies, USA) according to the manufacturer’s instructions. Gene-specific primers were used to amplify Fkbp5 (5_-GGTTTTGGAGAAGCCGGGAA-3_ and 5_-CAGCCTGCGTCAGCTTTTCT-3_) and GAPDH (5_-GGCCTTCCGTGTTCC-3_ and 5_-CGCCTGCTTCACCACCTTC-3_). The extracted RNA was reverse-transcribed into cDNA with the PrimeScript™ RT Master Mix (TaKara), and RT-qPCR was carried out using the SYBR Premix Ex Taq™ II (TaKara), with GAPDH as the internal control. All the RT-qPCR analyses were performed on an Applied Biosystems StepOnePlus Real-Time PCR System, according to the manufacturer’s protocol. The experiment was repeated three times as independent experiments.
Statistical tests
Baseline demographic and clinical characteristics were presented as mean ± standard deviation for continuous variables and counts (%) for categorical variables. Cox regression-based survival analysis was employed for assessing the association between the features and outcomes. Linear regression was used to explore the relationships between DMPs and six inflammatory markers, four blood lipids and two left ventricular indices. Enrichment analysis of biological pathways was carried out by R package “enrichR” and terms with a P-value smaller than 0.05 was considered as significant. Unless stated, P-values derived from multiple tests were corrected by methods of FDR or Bonferroni correction. Wilcoxon test was used to assess if the difference of continuous variables between two groups were statistically significance. For counts, chi-square tests were used. Animal experimental data were presented as mean ± standard error (SE) and were performed statistical analysis with Graph Pad Prism 9 (San Diego, CA). Student’s t-test was used to evaluate the statistical difference between two groups, and two-way ANOVA with Šídák’s test was used to multiple comparisons as appropriate.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data supporting the findings of this study are available within the article, its Supplementary Information files, and from the corresponding author upon reasonable request. Publicly available datasets used in this study include: Gene regulatory elements from the ENCODE5 catalog (https://www.encodeproject.org/). Enhancer–gene predictions by ABC models from the Engreitz Lab (https://www.engreitzlab.org/abc/). CpG sites associated with diseases and traits from the EWAS Catalog (http://www.ewascatalog.org/) and EWAS Atlas (https://ngdc.cncb.ac.cn/ewas/atlas). meQTL summary statistics from Pan-mQTL (https://www.biosino.org/panmqtl/home). Expression quantitative trait methylation data from the GEO database (GSE220622, GSE221615, GSE58137, and GSE72680). Single-nuclei RNA sequencing data of myocardial infarction patients from the Zenodo data archive (https://zenodo.org/record/6578047). Blood-derived bulk RNA sequencing data from the GEO database (GSE61144 and GSE16561). DNA methylation age estimations (GrimAge, Hannum, Horvath, and PhenoAge clocks) calculated using the DNA Methylation Age Calculator (https://dnamage.genetics.ucla.edu/home). Due to restrictions related to patient informed consent and institutional policies of the three participating medical centers, the Illumina EPIC DNA methylation data generated in this study are subject to controlled access. These data are available from the corresponding author upon reasonable request (contact: gdph_zhongsl@gd.gov.cn and zhongsl@hotmail.com). Requests will be reviewed and responded to within approximately two weeks. Data will be shared for non-commercial academic research purposes only, under a data use agreement that ensures compliance with patient confidentiality and institutional regulations. Source data are provided with this paper.
Code availability
Most analysis were carried out by R. Code used to infer confounders for DNA methylation data, EWAS analysis, and construct prognostic model with 5-folds 1000 times cross-validation is available at (https://github.com/qinmin23/DNA-methylation-of-prognosis-of-CAD). Image Pro-Plus 6.0 software was used to calculated the percentage of infarct area.
References
Cole, J. H., Miller, J. I. 3rd, Sperling, L. S. & Weintraub, W. S. Long-term follow-up of coronary artery disease presenting in young adults. J. Am. Coll. Cardiol. 41, 521–528 (2003).
Proudfit, W. J., Bruschke, A. V., MacMillan, J. P., Williams, G. W. & Sones, F. M. Jr Fifteen year survival study of patients with obstructive coronary artery disease. Circulation 68, 986–997 (1983).
Ananth, C. V. et al. Epidemiologic trends and risk factors associated with the decline in mortality from coronary heart disease in the United States, 1990-2019. Am. Heart J. 263, 46–55 (2023).
Drobni, Z. D. et al. Heritability of Coronary Artery Disease: Insights From a Classical Twin Study. Circ. Cardiovasc Imaging 15, e013348 (2022).
Zdravkovic, S. et al. Heritability of death from coronary heart disease: a 36-year follow-up of 20 966 Swedish twins. J. Intern Med 252, 247–254 (2002).
Smith, Z. D., Hetzel, S. & Meissner, A. DNA methylation in mammalian development and disease. Nat. Rev. Genet. 26, 7–30 (2025).
Agha, G. et al. Blood Leukocyte DNA Methylation Predicts Risk of Future Myocardial Infarction and Coronary Heart Disease. Circulation 140, 645–657 (2019).
Navas-Acien, A. et al. Blood DNA Methylation and Incident Coronary Heart Disease: Evidence From the Strong Heart Study. JAMA Cardiol. 6, 1237–1246 (2021).
Zhang, W., Song, M., Qu, J. & Liu, G. H. Epigenetic Modifications in Cardiovascular Aging and Diseases. Circ. Res 123, 773–786 (2018).
Joehanes, R. et al. Epigenetic Signatures of Cigarette Smoking. Circ. Cardiovasc Genet. 9, 436–447 (2016).
Dekkers, K. F. et al. Blood lipids influence DNA methylation in circulating cells. Genome Biol. 17, 138 (2016).
Ligthart, S. et al. DNA methylation signatures of chronic low-grade inflammation are associated with complex diseases. Genome Biol. 17, 255 (2016).
Richard, M. A. et al. DNA Methylation Analysis Identifies Loci for Blood Pressure Regulation. Am. J. Hum. Genet. 101, 888–902 (2017).
Wahl, S. et al. Epigenome-wide association study of body mass index, and the adverse outcomes of adiposity. Nature 541, 81–86 (2017).
Abdulrahim, J. W. et al. Epigenome-Wide Association Study for All-Cause Mortality in a Cardiovascular Cohort Identifies Differential Methylation in Castor Zinc Finger 1 (CASZ1). J. Am. Heart Assoc. 8, e013228 (2019).
Zhang, Y. et al. DNA methylation signatures in peripheral blood strongly predict all-cause mortality. Nat. Commun. 8, 14617 (2017).
Battram, T. et al. The EWAS Catalog: a database of epigenome-wide association studies. Wellcome Open Res 7, 41 (2022).
Li, M. et al. EWAS Atlas: a curated knowledgebase of epigenome-wide association studies. Nucleic Acids Res 47, D983–D988 (2019).
Demerath, E. W. et al. Epigenome-wide association study (EWAS) of BMI, BMI change and waist circumference in African American adults identifies multiple replicated loci. Hum. Mol. Genet. 24, 4464–4479 (2015).
Colicino, E. et al. Blood DNA methylation sites predict death risk in a longitudinal study of 12,300 individuals. Aging (Albany NY) 12, 14092–14124 (2020).
Fernandez-Sanles, A. et al. DNA methylation biomarkers of myocardial infarction and cardiovascular disease. Clin. Epigenetics 13, 86 (2021).
Roadmap Epigenomics, C. et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015).
Oliva, M. et al. DNA methylation QTL mapping across diverse human tissues provides molecular links between genetic variation and complex traits. Nat. Genet. 55, 112–122 (2023).
Boix, C. A., James, B. T., Park, Y. P., Meuleman, W. & Kellis, M. Regulatory genomic circuitry of human disease loci by integrative epigenomics. Nature 590, 300–307 (2021).
Nasser, J. et al. Genome-wide enhancer maps link risk variants to disease genes. Nature 593, 238–243 (2021).
Wang, Y. et al. SEdb 2.0: a comprehensive super-enhancer database of human and mouse. Nucleic Acids Res 51, D280–D290 (2023).
Yang, Y. L. et al. Systemic immune-inflammation index (SII) predicted clinical outcome in patients with coronary artery disease. Eur. J. Clin. Invest 50, e13230 (2020).
Hahn, J. et al. DNA methylation analysis is used to identify novel genetic loci associated with circulating fibrinogen levels in blood. J. Thromb. Haemost. 21, 1135–1147 (2023).
Balta, S. & Ozturk, C. The platelet-lymphocyte ratio: A simple, inexpensive and rapid prognostic marker for cardiovascular events. Platelets 26, 680–681 (2015).
Peng, Q. et al. Analysis of blood methylation quantitative trait loci in East Asians reveals ancestry-specific impacts on complex traits. Nat. Genet. 56, 846–860 (2024).
Park, H. J. et al. Assessment and diagnostic relevance of novel serum biomarkers for early decision of ST-elevation myocardial infarction. Oncotarget 6, 12970–12983 (2015).
Barr, T. L. et al. Genomic biomarkers and cellular pathways of ischemic stroke by RNA gene expression profiling. Neurology 75, 1009–1014 (2010).
Kuppe, C. et al. Spatial multi-omic map of human myocardial infarction. Nature 608, 766–777 (2022).
Lu, A. T. et al. DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging (Albany NY) 11, 303–327 (2019).
Hannum, G. et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 49, 359–367 (2013).
Levine, M. E. et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging (Albany NY) 10, 573–591 (2018).
Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 14, R115 (2013).
van der Stoep, M., Korporaal, S. J. & Van Eck, M. High-density lipoprotein as a modulator of platelet and coagulation responses. Cardiovasc Res. 103, 362–371 (2014).
Ridker, P. M. et al. Inflammation and cholesterol as predictors of cardiovascular events among patients receiving statin therapy: a collaborative analysis of three randomised trials. Lancet 401, 1293–1301 (2023).
Shi, H., Kichaev, G. & Pasaniuc, B. Contrasting the Genetic Architecture of 30 Complex Traits from Summary Association Data. Am. J. Hum. Genet. 99, 139–153 (2016).
Ventham, N. T. et al. Integrative epigenome-wide analysis demonstrates that DNA methylation may mediate genetic risk in inflammatory bowel disease. Nat. Commun. 7, 13507 (2016).
Somineni, H. K. et al. Blood-Derived DNA Methylation Signatures of Crohn’s Disease and Severity of Intestinal Inflammation. Gastroenterology 156, 2254–2265 e2253 (2019).
Sharp, G. C. et al. Maternal BMI at the start of pregnancy and offspring epigenome-wide DNA methylation: findings from the pregnancy and childhood epigenetics (PACE) consortium. Hum. Mol. Genet. 26, 4067–4085 (2017).
Antoun, E. et al. Maternal dysglycaemia, changes in the infant’s epigenome modified with a diet and physical activity intervention in pregnancy: Secondary analysis of a randomised control trial. PLoS Med 17, e1003229 (2020).
Wei, Y., Cao, H., Peng, Y. Y. & Zhang, B. Alterated gene expression in dilated cardiomyopathy after left ventricular assist device support by bioinformatics analysis. Front Cardiovasc Med. 10, 1013057 (2023).
Zannas, A. S. et al. Epigenetic upregulation of FKBP5 by aging and stress contributes to NF-kappaB-driven inflammation and cardiovascular risk. Proc. Natl. Acad. Sci. USA 116, 11370–11379 (2019).
Sakaue, S. et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat. Genet 53, 1415–1424 (2021).
Spracklen, C. N. et al. Identification of type 2 diabetes loci in 433,540 East Asian individuals. Nature 582, 240–245 (2020).
Beunders, G. et al. A detailed clinical analysis of 13 patients with AUTS2 syndrome further delineates the phenotypic spectrum and underscores the behavioural phenotype. J. Med Genet. 53, 523–532 (2016).
Chybowska, A. D. et al. Epigenetic Contributions to Clinical Risk Prediction of Cardiovascular Disease. Circ. Genom. Precis Med. 17, e004265 (2024).
Wang, Z. et al. Genome-wide association study of metabolites in patients with coronary artery disease identified novel metabolite quantitative trait loci. Clin. Transl. Med 11, e290 (2021).
Tian, Y. et al. ChAMP: updated methylation analysis pipeline for Illumina BeadChips. Bioinformatics 33, 3982–3984 (2017).
Nordlund, J. et al. Genome-wide signatures of differential DNA methylation in pediatric acute lymphoblastic leukemia. Genome Biol. 14, r105 (2013).
Teschendorff, A. E. et al. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics 29, 189–196 (2013).
Houseman, E. A. et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinforma. 13, 86 (2012).
Ishwaran, H., Kogalur, U. B., Blackstone, E. H. & Lauer, M. S. Random survival forests. Ann. Appl. Stat. 2, 841–860 (2008).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38, e164 (2010).
Consortium, E. P. et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583, 699–710 (2020).
Bernstein, B. E. et al. The NIH Roadmap Epigenomics Mapping Consortium. Nat. Biotechnol. 28, 1045–1048 (2010).
Qianqian Peng, X. L., Jing, H., Li, W., Gao, X., Luo, Q. <Comprehensive_mechanistic_characterization_of_mQTL.pdf>. Nat. Genet. (2023).
Teichholz, L. E., Kreulen, T., Herman, M. V. & Gorlin, R. Problems in echocardiographic volume determinations: echocardiographic-angiographic correlations in the presence of absence of asynergy. Am. J. Cardiol. 37, 7–11 (1976).
Acknowledgements
This study was supported by the National Natural Science Foundation of China (grant numbers 82274016, 81872934, 82574497 and 82274016 to S.Z., grant numbers32270626 to C.P., grant number 82504917 to MQ, grant number 82304623 to X.T., grant number 82204525 to M.Y., grant number 82404766 to Q.Z.), the Key-Area Research and Development Program of Guangdong Province, China (grant number 2019B020229003 to S.Z.), Greater Bay Area Research Institute of Precision Medicine (Guangzhou) Research Grants (grant numbers I0005 and R2001 to C.P.), National key research and development program (grant number 2017YFC0909301 to S.Z.), Leading Talent of Guangdong Special Support Program (grant number 0720240120 to S.Z.), the Science and Technology Program of Guangzhou (grant number 2023B03J1251 to S.Z.) and the natural science foundation of the provincial education department of Anhui (grant number 2023AH051994 to M.Y.).
We thank the CAD patients for their willingness to consent and participate in this scientific study. We thank medical staffs of the Guangdong Provincial People’s Hospital, particularly the nurses, for their diligent and careful follow-up of the patients. We thank Dr. Sijia Wang for providing early access to the methylation QTL datasets for our research. We also thank members of the Guangdong Provincial Key Laboratory of Coronary Heart Disease Prevention and members of the Laboratory of Intelligent Computing in Biomedicine in the Greater Bay Area Institute of Precision Medicine (Guangzhou) for insightful discussions and suggestions.
Author information
Authors and Affiliations
Contributions
S.Z. and C.P. designed the study. S.Z. supervised and coordinated the overall study and C.P. supervised the data analysis. S.Z., X.F., B.Z., X.C., M.Y. and C.L. assembled the study cohort. M.Q., X.T., Q.Z., X.W., X.C., M.Y. and C.L. consented the patients and supervised the patient follow-up. M.Q., Q.W., X.T. and H.L. collected samples and prepared them for DNA methylation array. X.T. and M.Q. designed and performed the experiment of Fkbp5 knockout mice. M.Q. and C.P. performed bioinformatic and statistical analyses and generated the figures and tables. C.P. and M.Q. drafted the manuscript. All authors contributed to the result interpretation and discussions. C.P., M.Q., and S.Z. critically reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Anthony Zannas, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Qin, M., Tian, X., Wu, Q. et al. DNA methylation predicts adverse outcomes of coronary artery disease. Nat Commun 16, 11396 (2025). https://doi.org/10.1038/s41467-025-66204-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-66204-x
