
Abstract
α-Synuclein (α-syn) fibrils accumulate in Parkinson’s disease, spreading between cells to template misfolding and drive neurodegeneration. α-Syn fibril entry into healthy neurons is a key step. Here, we comprehensively assessed the membrane proteome for α-syn fibril binding. We identified mGluR4 and NPDC1 as nigral surface proteins binding and internalizing α-syn fibrils. While striatal α-syn fibril injection led to nigral dopamine neuron loss in wild type mice, deletion of either Grm4 or Npdc1 provided protection of dopamine neurons. We observed mGluR4 and Npdc1 to form a complex regulating mGluR4 function. Cultured neurons lacking both Grm4 and Npdc1 fail to bind α-syn fibrils, to accumulate phosphorylated α-syn and to lose synapses. Transheterozygous Grm4, Npdc1 mice showed protection of nigral neurons from α-syn fibrils, demonstrating genetic interaction. For transgenic α-syn A53T mice, double Grm4, Npdc1 heterozygosity increased mouse survival, motor function and spinal motoneuron number. Thus, a cell surface mGluR4–NPDC1 complex participates in α-syn neurodegeneration.
Similar content being viewed by others


Introduction
Parkinson’s disease (PD), Diffuse Lewy Body disease (DLB) and related synucleinopathies are characterized by the accumulation of α-syn amyloid fibrils in Lewy bodies (LB) and Lewy neurites1,2,3. Multiple lines of evidence indicate that misfolded α-syn contributes to degenerative pathophysiology and symptoms4. Specifically, fibrillary α-syn amyloids can serve as templates to induce native α-syn monomers to misfold and propagate aberrant protein conformation5,6,7,8. Importantly, propagation can spread from the intracellular space of one cell to the intracellular space of another cell. Several models for cell spreading of α-syn and aggregates have been proposed, including tunneling nanotubes and bulk micropinocytosis or endocytosis9,10. The most direct and selective model involves high-affinity receptor-mediated uptake of low concentrations of α-syn aggregates from the extracellular space into recipient cells for α-syn templating and neuronal dysfunction. The well-documented ability of focal α-syn fibril injection into the brain to spread misfolding and toxicity along neuronal pathways supports the latter hypothesis5,7,8,11. However, the molecular basis for α-syn amyloid interaction with the neuronal cell surface to trigger aberrant neuronal signaling, internalization, and propagation remains poorly defined.
Previously, we utilized unbiased genome-wide receptor expression cloning to identify cellular prion protein (PrPC) as a high-affinity receptor for misfolded forms of amyloid-ß in Alzheimer’s disease12. A parallel approach limited to 352 cDNAs was applied to identify binding sites for α-syn preformed fibrils (PFF)13, the toxic aggregates prepared from monomeric α-syn protein. This revealed an affinity of α-syn PFF for LAG314, although subsequent studies have indicated that LAG3 is expressed primarily by innate immune cells in the brain and may not be required for α-syn pathology in neurons15. Other potential α-syn fibril receptors have been proposed without unbiased screening. The prion protein itself was tested based on an analogy of misfolded α-syn with misfolded amyloid-ß16, but has yielded conflicting results17,18. The PD risk gene, GPNMB, was postulated to encode an α-syn PFF binding site19, though its primary expression and action may be glial20, similar to LAG3. The microglial Fc-gamma receptor IIB (FcgRIIB) has also been implicated in extracellular α-syn toxicity and the spread of templated misfolding21,22. A recent report ascribed this role to FAM171A223.
Here, we pursued a comprehensive screen for human membrane-associated proteins capable of supporting high-affinity α-syn PFF binding to heterologous cells. A small number of proteins were confirmed as binding sites, and two were prioritized based on neuronal expression in dopamine cells (DA) of the substantia nigra, pars compacta (SNc), namely metabotropic glutamate receptor 4 (mGluR4) and Neuronal Proliferation, Differentiation and Control 1 (NPDC1). We show that these proteins bind α-syn PFF, that PFF binding alters their receptor function, and that they are essential for α-syn-induced neurodegeneration. Unexpectedly, mGluR4 and NPDC1 physically and genetically interact in a complex that has a prominent role in mediating α-syn PFF-triggered neurodegeneration.
Results
Screen for α-syn fibril binding sites identifies nigral membrane proteins mGluR4 and NPDC1
Different membrane proteins have been proposed to act as receptors for α-syn aggregates24 but a comprehensive screen containing the majority of membrane proteins has not been reported. Here, we combined three different cDNA expression libraries encoding more than 80% of all proteins classified by Gene Ontology with the term “plasma membrane” but excluding olfactory receptors (Supplementary Data 1). Expression vectors for a total of 4,401 genes were individually transfected into HEK293T cells. Two days after transfection, binding assays were performed using biotinylated α-syn preformed fibrils (PFF) (Fig. 1a; Supplementary Fig. 1). As a result, we identified 16 novel membrane proteins that bind α-syn PFF (Supplementary Fig. 2). From this list, we deprioritized B3GAT1 and CHST2 because they function primarily as intracellular synthetic enzymes in the secretory pathway. We also deprioritized CX3CL1 because this extracellular ligand is a peripheral membrane protein without a transmembrane segment. The remaining hit genes as well as several literature α-syn PFF binding proteins were assessed for expression in mouse midbrain DA cells using the brain-map.org website. Most of these genes were not expressed in nigral neurons, but Grm4, Npdc1, Prnp and Lrrc4c expression signals were clear (Supplementary Fig. 3)25. A study of human nigral gene expression showed GRM4, NPDC1 and PRNP expression, but not LRRC4C expression26. Since previous studies have disputed a role for PrPC in synucleinopathy, we focused on mGluR4 and NPDC1 here. We validated the expression of Grm4 and Npdc1 by both quantitiative RT-PCR and RNAscope methods in mouse brain (Supplementary Fig. 4). The in situ hybridization signals colocalized in nearly all of tyrosine hydroxylase (TH) positive SNc neurons, with 98% of TH+ cells expressing Grm4 and Npdc1 mRNA. Of note, mGluR4 has also been considered a target for symptomatic treatment of motor symptoms in PD27,28.

a Schematic representation of the screening process. Briefly, HEK293T were plated in 96-well plates. A total of 4,401 cDNAs were used individually to transfect the cells. Two days after transfection, cells were treated with biotinylated α-syn PFF (1000 nM, monomer equivalents) for 2 h at RT and bound α-syn assessed. The micrograph shows that there was no detectable binding to control HEK293T cells. As a result, 16 membrane proteins were confirmed as PFF interactors. b Representative images of HEK293T cells transfected with GRM4 (upper panels) and NPDC1 (lower panels) treated with biotinylated α-syn PFF (1000 nM) for 2 h and stained for α-syn PFF binding (streptavidin, gray) and GRM4 or NPDC1 expression (V5 tag, green). Scale bar, 100 µm. c Graphs show mean ± SEM of the mean intensity of streptavidin (PFF) at different concentrations of α-syn PFF in HEK293T cells transfected with GRM4 or NPDC1. N = 3 independent experiments. d Graphs show mean ± SEM of the mean intensity of streptavidin (PFF) bound to HEK293T cells transfected with GRM4 (upper graph) and NPDC1 (lower graph) after exposure to 3000 nM α-syn monomer or PFF for cell transfected with GRM4 (upper graph) and NPDC1 (lower graph). N = 4 independent experiments.**p = 0.002 for GRM4, ***p = 0.0004 for NPDC1, two tailed t test.
Binding of α-syn PFF to mGluR4 and NPDC1 in HEK293T cells was saturable, with apparent disassociation constants of 33 nM and 80 nM, respectively, expressed as monomer equivalents (Fig. 1b, c). With fibrils sonicated to a length of 50 nm, the concentration of α-syn PFF is at least 100-fold lower than the monomer equivalent, in the picomolar range. Binding was specific for the misfolded α-syn conformation, since cells expressing these proteins did not bind α-syn monomers to an extent greater than background at concentrations up to 3 µM (Fig. 1d). Binding to mGluR4 was specific amongst mGluRs, since mGluR1, 3, 5, 7 and 8, were negative for α-syn PFF interaction (Supplementary Fig. 5). Cells expressing mGluR4 and NPDC1 showed a strong ligand preference for α-syn PFF as compared to Amyloid-ß (Aß) oligomers, while cells expressing most glial α-syn PFF binding proteins also bound Aß (Supplementary Fig. 6). We confirmed that untransfected HEK293T cells do not express endogenous mGluR4 or NPDC1 (Supplementary Fig. 7). Co-immunoprecipitation analysis in HEK293T cells overexpressing mGluR4 and NPDC1 revealed a direct physical interaction of both proteins with α-syn PFF (Supplementary Fig. 8). Thus, a comprehensive unbiased screen of membrane proteins identified 16 previously unrecognized proteins that interact with α-syn PFF. mGluR4 and NPDC1 were selected as candidates for further analysis as potential mediators of α-syn PFF toxicity in nigral dopamine neurons.
α-syn fibrils regulate mGluR4 and NPDC1 cellular functions
Next, we checked whether α-syn PFF binding to mGluR4 and NPDC1 altered physiological receptor function. mGluR4 is a metabotropic glutamate receptor that signals through the inhibition of the cyclic AMP (cAMP) cascade after activation with glutamate ligand29. To analyze the effect of α-syn PFF on mGluR4 function, we performed a calcium assay in HEK293T cells stably expressing mGluR4 and Gαqi5, a chimeric Gq subunit that allows stimulation of phospholipase C by receptors otherwise coupled exclusively to Gi30. Intracellular calcium measurements using L-AP4 as an agonist revealed an increase in calcium response when cells were treated with a low dose of α-syn PFF (62.5 nM monomer equivalent) compared to controls (DPBS and monomeric α-syn) (Fig. 2a). However, when using a higher α-syn PFF dose (1,000 nM monomer equivalent) the outcome was opposite: α-syn PFF blocked calcium signaling. Dose titration indicated that α-syn PFF concentrations above 125 nM blocked subsequent mGluR4 activation by L-AP4 (Fig. 2b). Since endogenous mGluR4 signals primarily via adenylate cycle inhibition, we also examined cAMP level in HEK293T cells expressing mGluR4 without Gαqi5 (Supplementary Fig. 9). L-AP4 attenuated forskolin-stimulated cAMP increases in mGluR4 cells, while preincubation with α-syn PFF (1,200 nM monomer equivalent) prevented the mGluR4-mediated suppression.

a Graph shows the calcium influx as a response to L-AP4 addition (10-5 M) in stable GRM4-Gαqi5 cells treated with DPBS, α-syn monomer or α-syn PFF (62.5 nM and 1000 nM). Mean ± SEM indicated from 9 separate wells. b Graph shows mean ± SEM of the area under the curve in a calcium assay response to different concentrations of L-AP4 in stable GRM4-Gαqi5 cells treated with DPBS, α-syn monomer or PFF at different concentrations. N = 9 wells from 3 separate experiments. c Representative images of HEK293T cells transfected with GRM4 and treated with mGluR4 ligands glutamate, L-AP4 and MSOP at the indicated concentrations before the addition of biotinylated α-syn PFF for 3 h. α-syn PFF bound to the cells are detected with streptavidin. Scale bar, 200 µm. d Graph shows mean ± SEM of the area occupied with PFF in HEK293T treated with biotinylated α-syn PFF for 3 h after the addition of mGluR4 ligands (Glu, L-AP4 and MSOP). One-way ANOVA with Tukey’s post-hoc test. N = 4 independent experiments. ***p < 0.0001 for untreated vs PFF; ***p < 0.0001 for PFF vs PFF Glu; ***p < 0.0001 for PFF vs PFF L-AP4; ***p < 0.0001 for PFF vs PFF MSOP. e Representative images of HEK293T cells transfected or not with NPDC1 and treated with biotinylated α-syn PFF for 3 h. Cells were stained with phalloidin (gray, to detect actin) and V5 (red, to confirm NPDC1 expression). α-syn PFF bound to the cells are detected with streptavidin (green). Scale bar, 10 µm. f Graph shows mean ± SEM of the number of filopodia per µm (top) and filopodia length (bottom) in HEK293T cells treated with biotinylated α-syn PFF for 3 h. One-way ANOVA with Tukey’s post hoc test. N = 3 independent experiments. Filopodia number: ***p = 0.0005 for empty vector untreated vs NPDC1 untreated; **p = 0.0005 for NPDC1 untreated vs empty vector PFF 2000; *p = 0.0423 for NPDC1 vs NPDC1 PFF 1000; *p = 0.0272 for NPDC1 vs NPDC1 2000. Filopodia length: **p = 0.004 for empty vector untreated vs NPDC1 untreated; **p = 0.0047 for NPDC1 untreated vs empty vector PFF 2000; *p = 0.0145 for NPDC1 vs NPDC1 PFF 1000; *p = 0.0267 for NPDC1 vs NPDC1 2000.
We also checked whether the presence of mGluR4 orthosteric agonists (glutamate and L-AP4) or the orthosteric antagonist MSOP affect α-syn PFF binding to the receptor. Binding assays in HEK293T cells overexpressing mGluR4 and treated with the different orthosteric ligands showed a decrease in α-syn PFF binding compared to untreated controls (Fig. 2c, d), indicating antagonism between known mGluR4 ligands and α-syn PFF binding. The reduction in α-syn PFF binding was not due to alterations in the plasma membrane levels of mGluR4 upon ligand addition (Supplementary Fig. 10a, b) nor to changes in total amount of mGluR4 present (Supplementary Fig. 10c, d) even when using high concentrations of drugs (Supplementary Fig. 10e, f). Treatment with the same compounds in NPDC1-overexpressing cells showed no effect in α-syn PFF binding or total NPDC1 expression (Supplementary Fig. 10g–i), consistent with a receptor-specific effect on mGluR4 conformational state.
NPDC1 was originally identified in a neuronal precursor cell line after growth arrest, indicating it may inhibit proliferation31. NPDC1 is a single transmembrane protein where the N-terminal (Nt) part is extracellular and C-terminal (Ct) intracellular32. We deleted two regions of the extracellular domain (Supplementary Fig. 10j) in order to identify the interacting region with α-syn PFF. Both deletions abrogated binding of α-syn PFF (Supplementary Fig. 10k, l) without major changes in NPDC1 expression (Supplementary Fig. 10m).
When performing binding assays in HEK293T cells, we noted that NPDC1-overexpressing cells displayed increased numbers of filopodia, and that these were enriched for filamentous actin detected with rhodamine-phalloidin (Fig. 2e, f). Thus, we assessed whether α-syn PFF binding altered this phenotype induced by NPDC1 expression. Addition of α-syn PFF to NPDC1-overexpressing cells caused a decrease in the number and length of filopodia compared to non-treated cells (Fig. 2e, f). Thus, expression of NPDC1 induces an increase in number and length of filopodia, which is prevented in the presence of α-syn PFF as an NPDC1 ligand.
The binding and signaling assays showed interaction of α-syn PFF with mGluR4 and NPDC1, so we considered whether these cell surface interactions supported endocytosis of α-syn PFF. Cells expressing one of both binding partners were exposed to biotinylated α-syn PFF and incubated at either 4 °C to support binding or 37 °C to support binding and endocytosis (Supplementary Fig. 11a). Subsequently, intact fixed cells were exposed to unlabeled streptavidin to block surface biotin-PFF at 4 °C and then permeabilized and stained with Alexa-555-streptavidin to detect internalized α-syn PFF (Supplementary Fig. 11b). In a temperature-dependent manner, the expression of mGluR4, NPDC1 or both resulted in significant α-syn PFF endocytosis (Supplementary Fig. 11c, d).
While these studies demonstrate α-syn PFF regulation of mGluR4 and NPDC1, activity of the deprioritized candidate LRRC4C was not altered by α-syn PFF. Specifically, binding of a soluble form of the LRRC4C ligand netrin-G1 was not altered by PFF, and the ability of LRRC4C expressed in HEK293T cells to induce presynaptic specializations from co-cultured cortical neurons33 was not altered by the presence of α-syn PFF (L.A.-G. and S.M.S., unpublished observations). Based on these interaction studies, we explored the potential role of mGluR4 and NPDC1 in α-syn-mediated neurodegeneration in mice.
Grm4 is required for nigral dopamine neuron loss induced by α-syn fibril striatal injection
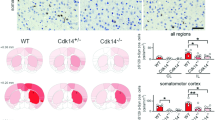
In order to evaluate the role of mGluR4 in α-syn induced dopamine neurodegeneration in vivo, we performed intrastriatal injections of α-syn PFF5 in wt and Grm4−/− mice34 and analyzed histology and behavior 6 months after the injections (Fig. 3a). General mouse health was not altered as body weights were unchanged in both genotypes after α-syn PFF injections (Supplementary Fig. 12a). In the striatum, the injection resulted in local α-syn phosphorylation (p-α-syn) and aggregation in both strains (Fig. 3b, d; Supplementary Fig. 12c). The staining of TH immunoreactive fibers in the striatum was not significantly reduced in the wt mice injected with α-syn PFF as compared to PBS, and was similar in Grm4−/− mice (Fig. 3b, c; Supplementary Fig. 12b).

a Diagram illustrating the experimental design for α-syn PFF injection into wt and Grm4−/− mice striatum. Created in BioRender. Chen, M. (2025) https://BioRender.com/9w6mss6. b Representative images showing TH and p-α-syn co-staining in the caudate-putamen of wt and Grm4−/− mice injected with α-syn PFF. Scale bar, 100 µm. c Graph shows mean ± SEM of TH levels in the striatum of wt and Grm4−/− mice injected with α-syn PFF. N = mouse number is shown above each bar. One-way ANOVA with Tukey’s post hoc test. d Graph shows mean ± SEM of p-α-syn levels in the striatum of wt and Grm4−/− mice injected with α-syn PFF. N = mouse number is shown above each bar. One-way ANOVA with Tukey’s post hoc test. **p = 0.0052 for WT-PBS vs WT-PFF; **p = 0.0098 for Grm4−/− +PBS vs Grm4−/− +PFF. e Representative images of TH staining in the ipsilateral side of the SNc of wt and Grm4−/− mice injected with α-syn PFF. Coronal section with dorsal up and midline to left. Scale bar, 500 µm. f Graph shows mean ± SEM of TH positive neurons in the ipsilateral side of the SNc of wt and Grm4−/− mice injected with α-syn PFF. N = mouse number is shown above each bar. One-way ANOVA with Tukey’s post hoc test. ***p < 0.0001 for WT-PBS vs WT+PFF and WT+PFF vs Grm4−/− +PFF, mean ± SEM. g Graph shows the percentage of wt and Grm4−/− mice injected with α-syn PFF remaining in the inverted grid in the wire screen test. N = mouse number is shown next to the group label. Gehan–Breslow–Wilcoxon test. p = 0.11 for Grm4−/− +PBS vs Grm4−/− +PFF; p = 0.0008 for WT-+PFF vs Grm4−/− +PFF.
Stereological counts of dopaminergic TH+ cells in the SNc revealed a robust decrease in dopaminergic neurons in the ipsilateral site of wt mice injected with α-syn PFF (Fig. 3e, f; Supplementary Fig. 12d). Dopaminergic cell loss was prevented in Grm4−/− injected mice, indicating that mGluR4 is required for cell loss induced by α-syn PFF. Significant neuronal loss was not observed in the contralateral SNc for any cohort (Supplementary Fig. 12d–f).
We also analyzed the effect of mGluR4 knockout on behavioral consequences of α-syn PFF injection. The wire screen test measures motor abilities using an inverted grid. In this test, α-syn PFF injection impaired performance of wt mice, but not of Grm4−/− mice (Fig. 3g), indicating that mGluR4 is required for this α-syn PFF-induced motor impairment. Other tests performed, including vertical pole test, open field, measurement of grip strength and rotarod, did not show a significant effect of α-syn PFF administration on motor behavior in wt or Grm4−/− mice (Supplementary Fig. 12g–m).
To confirm and extend these results we employed a genetic model, the A53T transgenic mouse35, which expresses a A53T missense mutant form of human α-syn under the control of the murine prion promoter. A53T mice present an age-dependent phenotype including progressive motor deficits, intraneuronal inclusion bodies and neuronal loss most prominent in the spinal cord35,36. Thus, we crossbred A53T transgenic mice with Grm4−/− mice in order to obtain double mutants and conducted behavioral tests at 12 months of age (Fig. 4a). A53T mice showed a slight decrease in body weight when compared to wt littermate controls (Supplementary Fig. 12n). Survival analysis of the different genotypes revealed a significant decrease in survival for A53T mice compared to wt littermate controls (Fig. 4b), as reported35,36. Survival of A53T-Grm4 KO was greater than that for A53T mice (Fig. 4b), indicating that mGluR4 participates in the early death of A53T mice.

a Diagram illustrating the experimental design for generating and analyzing A53T-Grm4−/− mice. Created in BioRender. Chen, M. (2025) https://BioRender.com/yecxy34. b Graphs show survival rates for A53T-Grm4−/− mice. N=mouse number is shown next to the group label. Gehan–Breslow–Wilcoxon test. p = 0.0020 for A53T vs A53T-Grm4 + /-; p = 0.011 for A53T vs A53T-Grm4−/−. c Graph shows mean ± SEM of grip strength in A53T-Grm4−/− mice. N = mouse number is shown above each bar. One-way ANOVA with Tukey’s post hoc test. ***p < 0.0001 for WT vs A53T, Grm4−/− vs A53T, and A53T vs A53T-Grm4−/−. d Graph shows mean ± SEM of time to turn in the pole test in A53T-Grm4−/− mice. N = mouse number is shown above each bar. One-way ANOVA with Tukey’s post hoc test. ***p < 0.0001 for Grm4−/− vs A53T; **p = 0.0015 for WT vs A53T. e Graphs show the percentage of A53T-Grm4−/− mice remaining in the inverted grid in the inverted wire screen test. N = mouse number is shown next to the group label. Gehan–Breslow–Wilcoxon test. p = 0.83 for A53T vs A53T-Grm4 + /−; p = 0.061 A53T vs A53T-Grm4−/−.
At 12 months of age, surviving A53T mice showed a reduction in grip strength compared to wt and Grm4−/− mice. The A53T grip strength decrement was alleviated in surviving A53T-Grm4 KO, but not in surviving A53T-Grm4 HET (Fig. 4c). A deficit in motor abilities was also observed in A53T mice when performing the vertical pole test. This A53T motor deficit relative to wt mice was not corrected in A53T-Grm4 HET and A53T-Grm4 KO mice (Fig. 4d). In the wire screen test, the percentage of surviving A53T and A53T-Grm4 HET mice remaining on the grid as a function of time was reduced compared to the other genotypes (Fig. 4e), indicating a motor impairment for these groups. The A53T-Grm4 KO mice remained on the grid for a numerically longer time than A53T mice (p = 0.061). In the open field test, A53T-Grm4 KO mice were slightly faster than the other groups (Supplementary Fig. 12o, p), and no differences were observed among the different genotypes in the rotarod test (Supplementary Fig. 12q). Altogether, these results suggest that mGluR4 deletion reduces a subset of motor abnormalities present on the A53T background as well as protecting nigral DA neurons from α-syn PFF induced degeneration.
Mice lacking Npdc1 protect nigral neurons from α-syn fibril pathology
We used a parallel experimental design to investigate potential Npdc1 effects in α-syn pathology in vivo after α-syn PFF injection. Mice were assessed 6 months after the injections (Fig. 5a). Body weight monitoring showed that wt and Npdc1−/− mice37 remained healthy after α-syn PFF injections (Supplementary Fig. 13a). Similar to the Grm4−/− cohort, α-syn PFF delivery did not decrease striatal TH levels in wt mice injected with α-syn PFF (Fig. 5b, c; Supplementary Fig. 13b). The local striatal increase in α-syn phosphorylation after α-syn PFF injections was similar in wt and Npdc1−/− mice (Fig. 5b, d; Supplementary Fig. 13c).

a Diagram illustrating the experimental design for α-syn PFF injection into wt and Npdc1−/− mice striatum. Created in BioRender. Chen, M. (2025) https://BioRender.com/9w6mss6. b Representative images showing TH and p-α-syn co-staining in the caudate-putamen of wt and Npdc1−/−mice injected with α-syn PFF. Scale bar, 100 µm. c Graph shows mean ± SEM of striatal TH levels in the ipsilateral side of the striatum in wt and Npdc1−/− mice injected with α-syn PFF. N = mouse number is shown above each bar. One-way ANOVA with Tukey’s post hoc test. p = 0.15 for WT + PBS vs WT + PFF. d Graph shows mean ± SEM of striatal p-α-syn levels in the ipsilateral side of the striatum in wt and Npdc1−/− mice injected with α-syn PFF. N = mouse number is shown above each bar. One-way ANOVA with Tukey’s post hoc test. ***p < 0.0001 for WT + PBS vs WT + PFF; *p = 0.020 for Npdc1−/−+PBS vs Npdc1−/−+PPF; p = 0.075 for WT + PFF vs Npdc1−/−+PFF. e Representative images of TH staining in ipsilateral side of the SNc of wt and Npdc1−/− mice injected with α-syn PFF. Coronal section with dorsal up and midline to left. Scale bar, 500 µm. f Graph shows mean ± SEM of TH positive neurons in the ipsilateral side of the SNc of wt and Npdc1−/− mice injected with α-syn PFF. N = mouse number is shown above each bar. One-way ANOVA with Tukey’s post hoc test. ***p < 0.0001 for WT + PBS vs WT + PFF; **p = 0.0014 for WT + PFF vs Npdc1−/−+PPF. g Graph shows the percentage of wt and Npdc1−/− mice injected with α-syn PFF remaining in the inverted grid in the wire screen test. N = mouse number is shown next to the group label. Gehan–Breslow–Wilcoxon test. p = 0.75 for Npdc1−/−+PBS vs Npdc1−/−+PFF; p = 0.054 for WT + PFF vs Npdc1−/−+PFF.
Critically, α-syn PFF injection resulted in dopaminergic neuronal loss from the ipsilateral SNc of these wt mice, but not of Npdc1−/− mice (Fig. 5e, f; Supplementary Fig. 13d). Thus, Npdc1 depletion is protective against the nigral DA cell loss induced by α-syn PFF in vivo. No neuronal loss was observed in the contralateral site for any of the genotypes (Supplementary Fig. 13d–f). In the wire screen test, PFF-injected Npdc1−/− remained on the grid for an intermediate period, not significantly different from PBS-injected Npdc1−/− mice or from PFF-injected wt mice (Fig. 5g). α-syn PFF delivery had no effect in the vertical pole test, open field, measurement of grip strength and rotarod in the control wt mice, or in the Npdc1−/− mice (Supplementary Fig. 13g–k).
Npdc1−/− mice were also crossbred with A53T transgenic in order to obtain double mutants. As for the A53T-Grm4 cohort, behavioral tests were conducted at 12 months of age (Fig. 6a). In this cohort, A53T mice showed a trend to decrease in body weight, not significantly different from wt littermate controls (Supplementary Fig. 13l). Survival analysis of the different genotypes revealed a significant decrease in survival rate of A53T mice compared to control, and this was not improved by Npdc1 deletion (Fig. 6b).

a Diagram illustrating the experimental design for generating and analyzing A53T-Npdc1−/− mice. Created in BioRender. Chen, M. (2025) https://BioRender.com/yecxy34. b Graphs show survival rates for A53T-Npdc1−/− mice. N = mouse number is shown next to the group label. Gehan–Breslow–Wilcoxon test. p = 0.21 for A53T vs A53T-Npdc1 + /-; p = 0.21 for A53T vs A53T-Npdc1−/−. c Graph shows mean ± SEM of grip strength in A53T-Npdc1−/− mice. N = mouse number is shown above each bar. One-way ANOVA with Tukey’s post hoc test. **p = 0.0023 for WT vs A53T; *p = 0.030 for Npdc1−/− vs A53T. d Graph shows mean ± SEM of time to turn in the pole test in A53T-Npdc1−/− mice. N = mouse number is shown above each bar. One-way ANOVA with Tukey’s post hoc test. ***p < 0.0001 for WT vs A53T and Npdc1−/− vs A53T; *p = 0.034 for A53T vs A53T-Npdc1−/−. e Graphs show the percentage of A53T-Npdc1−/− mice remaining in the inverted grid in the wire screen test. N = mouse number is shown next to the group label. Gehan–Breslow–Wilcoxon test. p = 0.75 for A53T vs A53T-Npdc1 + /-; p = 0.054 for A53T vs A53T-Npdc1−/−.
Behaviorally, surviving A53T mice at 12 months of age showed a reduction in grip strength compared to wt and Npdc1−/− mice, and this was not lessened by Npdc1 deletion (Fig. 6c). In contrast, the impairment of A53T mice on the vertical pole test was rescued in A53T-Npdc1 KO mice (Fig. 6d). In the wire screen test, the percentage of A53T and A53T-Npdc1 HET mice remaining on the grid was less than in the rest of the genotypes (Fig. 6e), indicating a motor impairment in these two groups. The percentage of A53T-Npdc1 KO mice was greater than in A53T mice, though the difference was not statistically significant (p = 0.054). No differences were observed among the surviving mice of the different genotypes in the open field or rotarod tests (Supplementary Fig. 13m–o). These data indicate that Npdc1 removal improves a subset of motor deficits present in A53T mice and also rescues nigral DA neurons from α-syn PFF induced degeneration.
mGluR4 and NPDC1 form a complex regulating mGluR4 activation and mediating α-syn fibril action
While the protection of nigral DA neurons from α-syn PFF induced degeneration is robust with either mGluR4 or NPDC1 gene knockout, other phenotypes are only partially protected by single gene deletion. This suggested that the two proteins have partially redundant roles. We also noted that the benefits of Npdc1 deletion on α-syn pathologies in mice are strikingly similar to the improvements seen in Grm4 null mice. This suggested that the two proteins might function cooperatively, as a complex. To explore potential interaction between mGluR4 and NPDC1, we performed co-immunoprecipitation (co-IP) in HEK293T cells overexpressing both receptors. Retention of NPDC1 with V5 beads showed that NPDC1 co-immunoprecipitated with mGluR4, but not mGluR5, another metabotropic glutamate receptor, indicating that the mGluR4–NPDC1 interplay is specific (Fig. 7a, b). Reverse co-IP experiments, retaining mGluR4 with DYDDK beads, confirmed an interaction of Npdc1 with mGluR4, but not mGluR5 (Fig. 7a, b). No available antibody was adequate to assess association of endogenous brain proteins, so we evaluated functional interactions. We performed calcium assays in GRM4-Gαqi5 cells transfected or not with NPDC1. NPDC1 co-expression completely blocked the calcium response of mGluR4 cells to L-AP4 stimulation, indicating that the presence of NPDC1 negatively impacts mGluR4 function (Fig. 7c, Supplementary Fig. 14a, b). To examine whether NPDC1 interacts with mGluR4 function in cis and/or in trans, we performed the same calcium assay but with a mixture of GRM4-Gαqi5 cells with NPDC1-transfected cells. Mixing GRM4-Gαqi5 cells with HEK293T cells transfected with an empty vector did not alter calcium responses (Fig. 7d, Supplementary Fig. 14c, d). However, combining GRM4-Gαqi5 cells with NPDC1-expressing cells produced a significant reduction in calcium response after stimulation at a 1:1 ratio or a complete blockade of the response at a 1:2 ratio (Fig. 7d, Supplementary Fig. 14e, f). Thus, the expression of NPDC1 in adjacent cells is sufficient to block mGluR4 function in trans, although cis interactions may be equally effective.

a Representative immunoblots with anti-V5 and anti-DYDDK of co-IP assays using HEK293T cells expressing GRM4-DYDDK or GRM5-DYDDK, together with V5-tagged NPDC1. b Graphs show mean ± SEM of pulldown by V5 beads (left) and DYDDK beads (right). N = 3 independent experiments. One-way ANOVA with Tukey’s post hoc test. For both V5 and DYDDK pulldown: ***p < 0.0001 for GRM4-DYDDK vs NPDC1-V5 + GRM4-DYDDK and NPDC1-V5 vs GRM4-DYDDK + NPDC1-V5. c Graph shows mean ± SEM of calcium influx curve after L-AP4 stimulation in GRM4-Gαqi5 cells (G4) transfected with an empty vector (Vec, pcDNA) or with NPDC1. N = 3 mean values from 3 independent experiments, each mean value calculated from 3 wells. d (left) Graph shows mean ± SEM of calcium influx curve after L-AP4 stimulation in GRM4-Gαqi5 cells (G4) co-cultured with HEK293T cells transfected with an empty vector (Vec, pcDNA) or with NPDC1 at different ratios. d (right) Graph shows mean ± SEM of calcium response after 100 µM L-AP4 stimulation in GRM4-Gαqi5 cells (G4) co-cultured with HEK293T cells transfected with an empty vector (Vec, pcDNA) or with NPDC1 at different ratios. N = wells numbers from 3 independent experiments shows above the bar. One-way ANOVA with Tukey’s post hoc test. ***p = 0.0009 for G4:Vec, 1:1 vs G4:NPDC1, 1:1; **p = 0.0015 for G4:Vec, 1:2 vs G4:NPDC1, 1:2.
Since mGluR4 and NPDC1 interact physically and functionally, we decided to investigate whether the elimination or reduction of both proteins synergized in neurons with regard to their effect on α-syn PFF binding, the accumulation of aggregated α-syn, and synaptic damage. We crossbred Grm4−/− and Npdc1−/− mice and cultured cortical neurons from wt, double heterozygous (dHET) and double ko (dKO) embryos. Incubation with α-syn PFF for 2 hours at 37 °C or 4 °C revealed a significant decrease in α-syn PFF binding in dHET and dKO compared to wt mice (Fig. 8a, d, Supplementary Fig. 14g, h) without alterations in total neuronal density as measured with MAP2 staining (Supplementary Fig. 14g, i, j). In comparison, single heterozygous Grm4 + /− or Npdc1 + − neurons and homozygous Grm4−/− showed no significant alteration in binding and homozygous Npdc1−/− neurons showed only partial reduction (Supplementary Fig. 15a, 16a). The greater suppression of binding in the double knockout as compared single knockouts supports partially redundant action of these two α-syn PFF binding proteins. More strikingly, the strong reduction of binding to double heterozygous neurons documents a genetic interaction that supports a critical role for the mGluR4 – NPDC1 physical complex in neuronal binding of α-syn PFF.

a Representative images of streptavidin (PFF, green), MAP2 (red), and DAPI (blue) in cultured neurons from WT, dHET and dKO mice after 2 h of exposure with biotinylated α-syn monomer or PFF at 37 °C. Scale bar, 50 µm. b Representative images of p-α-syn (green), MAP2 (red) and DAPI (blue) in cultured neurons from WT, dHET and dKO mice after 7 days of exposure with α-syn monomer or PFF at 37 °C. Scale bar, 50 µm. c Representative images of Synapsin1 (green), Homer1/2 (red), and DAPI (blue) staining in cultured neurons from WT, dHET and dKO mice after 7 days of exposure with α-syn monomer or PFF at 37 °C. Scale bar, 50 µm. d Quantification of streptavidin (PFF) fluorescence intensity within MAP2-positive regions after 2 h of exposure with biotinylated a-syn monomer or PFF at 37 °C. Data represent mean ± SEM from WT(N = 5), dHET(N = 6), and dKO(N = 5) embryos. One-way ANOVA with Tukey’s post hoc test. 150 nM: *p = 0.018 for WT vs dHET, *p = 0.012 for WT vs dKO; 300 nM: ***p < 0.0001 for WT vs dHET and WT vs dKO; 600 nM: ***p < 0.0001 for WT vs dHET and WT vs dKO. e Quantification of p-α-syn fluorescence intensity within MAP2-positive regions after 7 days of exposure with a-syn monomer or PFF. Data represent mean ± SEM from WT(N = 5), dHET(N = 6), and dKO(N = 5) embryos. One-way ANOVA with Tukey’s post hoc test. 150 nM: *p = 0.034 for WT vs dHET, *p = 0.028 for WT vs dKO; 300 nM: ***p < 0.0001 for WT vs dHET and WT vs dKO; 600 nM: ***p < 0.0001 for WT vs dHET and WT vs dKO. f Quantification of Synapsin1 and Homer1/2 double positive dots within MAP2-positive regions after 7 days of exposure with a-syn monomer or PFF. Data represent mean ± SEM from WT(N = 5), dHET(N = 6), and dKO(N = 5) embryos. One-way ANOVA with Tukey’s post hoc test. 300 nM: *p = 0.040 for WT vs dHET; 600 nM: *p = 0.039 for WT vs dHET, **p = 0.0014 for WT vs dKO.
When neurons are exposed to α-syn PFF for 7 days, aggregates of p-α-syn accumulate in these cells. Both dHET and dKO neurons showed a marked reduction in accumulation of p-α-syn (Fig. 8b, e) without changes in neuronal density detected by MAP2 for PFF concentrations up to 600 nM (Supplementary Fig. 14k, l). The single heterozygous Grm4 + /- or Npdc1 + /- neurons exhibited no alteration in p-α-syn accumulation while homozygous Grm4−/− and Npdc1−/− neurons displayed a significant reduction but did not reach the degree of reduction in the double mutants (Supplementary Fig. 15b, 16b). While neuronal cell density measured by MAP2 staining is not altered in this situation, exposure of wt neurons to α-syn PFF for 7 days led to reduced synaptic density as measured by colocalization of puncta stained for synapsin1 and Homer1/2. Neurons of both the dHET and dKO genotype were protected from synapse loss triggered by α-syn PFF (Fig. 8c, f). In contrast, the single heterozygous Grm4 + /- or Npdc1 + /- and homozygous Npdc1−/− neurons showed no rescue of synapse density from α-syn PFF exposure, while homozygous Grm4−/− neurons exhibited partial synapse preservation similar to that for the double mutants (Supplementary Figs. 15c, 16c). In summary, mGluR4 and Npdc1 individually have partially redundant roles in α-syn PFF binding to neurons, as evidenced by comparing dKO with single KO. Furthermore, the physical and genetic interaction of mGluR4 and NPDC1 is required for efficient binding, accumulation of pathological aggregates and neuronal toxicity as shown in the dHET phenotypes despite the absence of a single heterozygous phenotype.
Transheterozygote Grm4, Npdc1 nigral neurons are protected from α-syn fibril striatal injection
Given the genetic interactions of mGluR4 and Npdc1 in culture, we investigated the impact of their combined absence in vivo. Littermate wt, dHET and dKO mice were injected with α-syn PFF in the striatum and analyzed 6 months after injection. No differences in body weight progression were observed among the groups (Supplementary Fig. 17a). In line with the single knockout studies, α-syn PFF generated local accumulation of phosphorylated α-syn in the striatum but no change in striatal TH fibers from wt (Supplementary Fig. 17b–d). The striatal effects of α-syn PFF were similar in all genotypes.
For the wt mice, the striatal α-syn PFF injection generated a pronounced decrease in the number of TH-positive (dopaminergic, DA) cells in the ipsilateral SNc (Fig. 9a, b, Supplementary Fig. 17d). Critically, removal of a single copy (dHET) or two copies (dKO) of both Grm4 and Npdc1 significantly reduced α-syn PFF-induced neurodegeneration in the SNc (Fig. 9a, b, Supplementary Fig. 17e) compared to wt PFF-injected mice. As expected, the decrease of TH-positive nigral cells contralateral to the α-syn PFF injection was partial in wt mice, but there was no cell loss in dHET or dKO mice (Supplementary Fig. 17e–g). Behavioral analysis of this cohort showed no deficit caused by α-syn PFF in the wt group, so any potential benefit of combined mGluR4 and Npdc1 deletion could not be assessed (Supplementary Fig. 17h–k). The protection of nigral DA neurons from α-syn PFF induced neurodegeneration in the transheterozygote state confirms a key role for the interaction of mGluR4 and Npdc1 in synucleinopathy.

a Representative images of TH staining in the ipsilateral side of the SNc of in WT, dHET and dKO mice injected with a-syn PFF. Coronal section with dorsal up and midline to left. Scale bar, 500 µm. b Graph shows mean ± SEM of the number of TH positive neurons in the ipsilateral site of the SNc in WT, dHET and dKO mice injected with a-syn PFF. N=mouse number is shown above each bar. One-way ANOVA with Tukey’s test. ***p < 0.0001 for WT-PBS vs WT-PFF, dHET-PBS vs dHET-PFF, dKO-PBS vs dKO-PFF, and for WT-PFF vs dHET-PPF; **p = 0.0030 for WT-PFF vs dKO-PFF.
Interaction of Grm4 and Npdc1 is required for α-syn A53T transgenic neurodegeneration
To validate these observations in another model, we crossbred Grm4−/− and Npdc1−/− mice with A53T transgenic mice and generated double heterozygous (dHET) and double ko mice (dKO) on the A53T background (A53T-dHET and A53T-dKO, respectively) as well as wt and A53T littermates (Fig. 10a). Body weights were similar among the different genotypes (Supplementary Fig. 18a). The A53T mice displayed reduced survival, and the A53T-dHET mice were protected (Fig. 10b). Behavioral and histological analysis were performed for surviving mice at 12 months of age. The motor deficit of A53T mice was clear when measuring grip strength, the vertical pole test and the wire screen test (Fig. 10c–e). The A53T-dHET and A53T-dKO mice were protected from impairment on each of these motor tasks (Fig. 10c–e). No alterations in A53T mice were detected in the open field and the rotarod test (Supplementary Fig. 18b–d).

a Diagram illustrating the experimental design for generating and analyzing A53T-dHET and A53T-dKO mice. Created in BioRender. Chen, M. (2025) https://BioRender.com/yecxy34. b Graph shows mean ± SEM of the survival rate in A53T-dHET and A53T-dKO mice. N = mouse number is shown next to the group label. Gehan–Breslow–Wilcoxon test. p = 0.0031 for A53T vs A53T-dHET, p = 0.0056 for A53T vs A53T-dKO. c Graph shows mean ± SEM of the forelimbs grip strength test in A53T-dHET and A53T-dKO mice. N = mouse number is shown above each bar. One-way ANOVA with Tukey’s post hoc test. **p = 0.0032 for WT vs A53T; *p = 0.038 for dHET vs A53T; **p = 0.0015 for dKO vs A53T; **p = 0.0094 for A53T vs A53T-dHET; *p = 0.023 for A53T vs A53T-dKO. d Graph shows mean ± SEM of the time to turn in the pole test in A53T-dHET and A53T-dKO mice. N = mouse number is shown above each bar. One-way ANOVA with Tukey’s post hoc test. ***p < 0.0001. e Graph shows the percentage of mice remaining in the inverted grid in the wire screen test. N = mouse number is shown next to the group label. Gehan–Breslow–Wilcoxon test. p < 0.0001 for A53T vs A53T-dHET, p = 0.0039 for A53T vs A53T-dKO. f Representative images of NeuN (red) and DAPI (blue) staining of sections L4-L6 of the spinal cord in A53T-dHET and A53T-dKO mice. Scale bar, 500 µm. g Representative images of ChAT (red), p-α-syn (green) and DAPI (blue) staining in the lumbar anterior horn of the spinal cord in A53T-dHET and A53T-dKO mice. Scale bar, 100 µm. h Graph shows mean ± SEM of the number of large NeuN positive neurons per section in the lumbar anterior horn of the spinal cord in A53T-dHET and A53T-dKO mice. N=mouse number is shown above each bar. One-way ANOVA with Tukey’s post hoc test. **p = 0.0020 for WT vs A53T; *p = 0.028 for A53T vs A53T-dKO. i, Graph shows mean ± SEM of the number of ChAT cells in the lumbar anterior horn of the spinal cord in A53T-dHET and A53T-dKO mice. N=mouse number is shown above each bar. One-way ANOVA with Tukey’s post hoc test. ***p = 0.0006 for WT vs A53T.
After survival and behavioral studies, histological correlates were examined. We focused on the spinal cord and its motoneurons because they are known to be strongly affected in this strain and are likely responsible for the survival and motor function differences. Astrogliosis as detected by anti-GFAP staining was not prominent in A53T mice as compared to wt and was not altered by the dHET or dKO genotype (Supplementary Fig. 18e). In contrast, the A53T mice showed a loss of more than 80% of spinal motoneurons in the ventral horn as compared to wt mice, whether detected as large diameter NeuN positive cells or by staining with choline acetyltransferase (ChAT) (Fig. 10f–i). The transheterozygotes and double knockouts for mGluR4 and Npdc1 prevented neuronal loss in the A53T background (A53T-dHET and A53T-dKO mice), even though phosphorylated α-syn accumulated. These results confirm that partial or complete depletion of mGluR4 and Npdc1 rescues the reduced survival rates, motor impairment and neuronal loss in the A53T mice.
Discussion
We completed a comprehensive expression cloning screen of plasma membrane proteins for high-affinity interactions with α-syn PFF. mGluR4 and NPDC1 were identified as α-syn amyloid binding proteins expressed by nigral dopamine neurons. Pathophysiological relevance of these sites for α-syn amyloid triggered disease is supported by multiple lines of evidence. α-syn PFF titrated the physiological function of both mGluR4 and NPDC1. Cell surface α-syn PFF binding to these receptors supported endocytosis of α-syn aggregates. Null mutations of either mGluR4 and NPDC1 prevented nigral dopamine neuron loss triggered by α-syn PFF injection and reduced behavioral deficits associated with PFF injection or mutant α-syn overexpression. Furthermore, the two α-syn binding proteins associated with one another and interacted genetically to mediate α-syn-triggered pathology in both transgenic and fibril injection models. Thus, the mGluR4 – NPDC1 protein complex is a potential therapeutic intervention target for PD, LBD and related disorders.
The observation that loss of cell surface proteins capable of interacting with α-syn amyloid protects nigral dopamine neurons from neurodegeneration supports the hypothesis that synucleinopathy involves a crucial extracellular phase and is not a purely cell-autonomous intracellular process, even in a transgenic overexpression system. This is consistent with a prion or prion-like mechanism of disease progression. However, neurodegeneration does not correlate directly with the accumulation of p-α-syn aggregates5,7,38. The protection of motoneurons from degeneration triggered by mutant α-syn overexpression and of nigral dopamine neurons from degeneration triggered by striatal PFF injection occurred despite unchanged striatal and spinal cord p-α-syn accumulation. Thus, the presence of mGluR4 and Npdc1 is required for sensitive neurons to detect extracellular aggregates and transduce this information into neuronal loss.
Our studies focused on mGluR4 and NPDC1 because they are expressed by nigral dopamine neurons. Other neuronal binding sites confirmed or identified here include LRRC4C and PRNP. We did not assess their role in neurodegeneration. Both are expressed widely by neurons and may constitute additional neuronal α-syn PFF receptors required for neurodegeneration.
Many of the hits on our screens are expressed primarily or exclusively by microglia and astrocytes. These sites include FCGR1A, FCRL4, SCARF1, LAG3, TRIL, CXCL16, CD72, CD93 and Cdh1. The role of these proteins and glial cells in propagating neuronal inclusions and associated neurodegeneration was not studied here. They may interact with extracellular α-syn amyloids for transfer to neurons, for modification of α-syn protein, for degradation of α-syn, or to stimulate cytokine release. Based on the affinity for Aß oligomers, many are likely to have more general roles in clearing neurodegenerative protein aggregates from the extracellular space. Further study will be required to assess the role of this collection of non-neuronal α-syn PFF binding sites. Given the role of LRRK2 genetic variants in PD risk and the expression of LRRK2 by immune cells39, the interaction of LRRK2 signaling with these receptors will be of great interest.
The mGluR4 protein has previously been implicated in modifying basal ganglia function after experimental alteration of nigral DA function. In rodent and non-human primate, acute administration of mGluR4 agonists led to improved motor function acutely and dose dependently40,41,42,43. This action is distinct from the disease-modifying neuroprotective effect of Grm4 deletion described here. However, it may be related to the ability of α-syn PFF to modulate mGluR4 signaling and the capacity of mGluR4 agonists to displace α-syn PFF from mGluR4. A mGluR4 positive allosteric modulator has been tested in a phase 2 clinical trial examining short-term symptomatic benefit for motor function in PD28. While symptomatic benefit did not reach statistical significance, the compounds were safe in PD subjects. Any potential disease-modifying effect of mGluR4 pharmacology has not yet been examined.
The presence of low concentrations of α-syn PFF positively modulates L-AP4 activation, while higher concentrations are strongly inhibitory. One interpretation of the biphasic action relates to the dimeric nature of mGluR4-PFF binding to one subunit at low concentrations may differ from the effects of binding both subunits at higher concentrations. Another possibility is that low α-syn PFF doses may act as an allosteric positive modulator to enhance mGluR4 function. With saturating α-syn PFF concentrations, receptor desensitization may occur, preventing increased activity after subsequent addition with L-AP4.
An unexpected observation was that the two neuronal α-syn binding proteins mGluR4 and NPDC1 physically associate, and that NPDC1 inhibits mGluR4 agonist action. Moreover, the two binding sites interacted genetically to mediate α-syn PFF effects, such that the transheterozygous state greatly reduced α-syn PFF binding, p-α-syn accumulation and synapse loss in vitro as well as survival, motor deficits and neurodegeneration in vivo. While mGluR4 and NPDC1 can independently bind α-syn PFF to alter signaling and mediate endocytosis, the interaction studies strongly suggest that the complex of these two proteins is most relevant for α-syn PFF neuronal actions (Supplementary Fig. 19). The NPDC1 deletion mapping and the cellular action in trans indicate that extracellular contact between mGluR4 and NPDC1 is responsible for the observations. While glutamate is the endogenous ligand for mGluR4, an extracellular polypeptide partner has precedence. The leucine-rich repeat-containing transmembrane protein, ELFN1, interacts with mGluR4 and other Class III mGluRs in trans with negative allosteric effects44,45.
NPDC1 itself has not been studied in depth previously. In addition to its ability to regulate mGluR4, we observed that NPDC1 overexpressing cells protruded more F-actin-rich filopodia. The short cytoplasmic tail of NPDC1 does not provide a clear hypothesis as to how this cellular effect may occur.
In the A53T model, the deletion of mGluR4 and NPDC1 rescues motoneurons, but phospho-α-syn accumulation is still present in the spinal cord. Because the pathology is driven by a transgene, a fraction of the accumulation is likely independent of extracellular α-syn spreading being cell autonomous. Since motoneuron preservation and mouse survival are strongly dependent on deletion of the mGluR4 and NPDC1 surface α-syn PFF receptors, the data suggest that an extracellular fraction of α-syn aggregates plays a key role in neurodegeneration, even in this transgenic neuronal over-expression model.
Supporting a role for mGluR receptors in PD, GWAS-defined risk was recently shown to depend on variation in GRM746, encoding the mGluR7 protein. While we show here that mGluR7 alone does not support α-syn PFF binding, it is known to form mGluR4/7 heterodimers with altered signaling47. Future studies defining the role of mGluR4 homomers versus heterodimers in synucleinopathy may define a link of mGluR4 to PD genetic risk via mGluR7 modulation. Such heterodimer studies may also have implications for which neurons are susceptible to neurodegeneration based on the relative expression of different subtype combinations.
Of particular interest for the development of disease-modifying treatment for α-syn-driven neurodegeneration, mGluR4 is an intriguing target. Here, deletion alone or in combination with NPDC1 rescued nigral neurons from striatal α-syn PFF and motoneurons from α-syn-A53T overexpression. Clinical studies with mGluR4 positive allosteric modulators in Parkinson’s have some limited symptomatic benefit but any possible disease-modifying action over longer periods was never assessed. Here, we show that orthosteric mGluR4 ligands reduce α-syn PFF binding, but the actions of potent negative and positive allosteric modulators require exploration. It will be of great interest to optimize mGluR4 pharmacologic to slow or reverse the course of disease driven by misfolded α-syn.
Methods
α-syn purification and PFF preparation
Untagged α-syn was purified from recombinant bacteria as previously described48. Full-length human α-syn encoded by the pET21a vector (Addgene, #51486) and full-length mouse α-syn encoded by the pET3a vector (Addgene, #108865) were expressed in BL21(DE3) E. coli (Invitrogen, #C600003). The α-syn monomers were then purified using HiLoad 25/60 Superdex 200 and HR Source Q 20 ml exchange columns (GE Healthcare Life Sciences). Endotoxins were subsequently removed using an endotoxin removal kit (GenScript, #L00338). To generate α-syn PFF (preformed fibrils), the purified α-syn monomers were diluted in phosphate-buffered saline (PBS) to a concentration of 5 µg/µl and agitated for 7 days at 37 °C and 1000 rpm to allow the formation of mature fibrils. The aggregates were collected by centrifugation, resuspended in endotoxin-free PBS and stored in aliquots at −80 ˚C. These aggregates were then sonicated for 1 min and 15 s (1 s on, 1 s off, 30% amplitude) using Sonifier 250 sonicator (Branson) to generate PFF. Sonication was performed immediately before each experiment. PFF generated from human α-syn monomers were used for experiments in cell lines, while PFF from murine monomers were applied to primary neuronal cultures and stereotaxic injections in mice. The concentration of α-syn was measured using a BCA assay (Thermo Fisher Scientific).
For cell cultures, recombinant α-syn monomer was labeled with sulfo-NHS-LC-Biotin (Thermo Scientific, #21335) after purification. Free, unbound biotin was removed from the sample using Amicon Ultra-4 3 kDa tubes (Millipore, #UFC8003). The molar ratio of biotin to α-syn was 2. After biotinylation, α-syn PFF were prepared as mentioned above.
Atomic force microscopy (AFM)
α-syn fibrils and PFF size were analyzed by Atomic Force Microscopy. Briefly, 10 µl of solution were applied to a 10 mm diameter mica discs (Ted Pella, #50) for 5 min, then washed twice with distilled water and let dry before imaging. Imaging was performed using the Dimension FastScan AFM (Bruker) with FASTSCAN-B tips (Bruker) in the tapping mode.
Expression cloning screening process
HEK293T cells (ATCC) were plated onto Poly-D-Lysine(PDL)-coated 96-well plates (Corning, #354461) in DMEM media (GIBCO, #11965-092) supplemented with 10% fetal bovine serum (GIBCO, #16000-044) and Pen Strep (GIBCO, #15140-122). HEK293T cells were individually transfected with 4,401 human cDNAs encoding membrane proteins from three different libraries. A library from Dharmacon contained 875 cDNAs in pLOC-GFP or pLX304-V5 vector (Dharmacon, #SO-2608946G). The second library was from Transomic and contained 365 cDNA expressing for membrane proteins in pLOC vector (Transomic, #TCH3000). We obtained additional plasmids from a 13,579 cDNA collection in pLX304-V5 plasmid (Transomic, #TOH7500). In order to select clones from this library, we cherry-picked a list of cDNAs classified as plasma membrane encoding genes based on Gene Ontology (GO) classification. Selected cDNAs were re-arranged in 96-well plates (NUNC, #VWR 62407-174). The final number of clones resulting from the 13,579 cDNA collection was 3,161 cDNAs.
Briefly, 15 µl of glycerol stocks for each cDNA were grown in 1 ml of LB media with 100 µg/ml carbenicillin in deep 96-well plates (USA Scientific, #1896-2110) for 16-18 h at 37 °C and 230 rpm. DNA extraction was performed using Plasmid Plus 96 Miniprep Kit (Qiagen, #16181).
HEK293T cells were transfected using Lipofectamine 3000 (ThermoFisher, #L3000015). Two days after transfection, biotinylated α-syn PFF (1000 nM) were added to cell media (DMEM with 10% FBS) and incubated for 2 h at RT. Cells were then washed twice with DMEM media and then with PBS 1x (3 times). Cells were fixed in PFA 4% for 12 min and then blocked for 1 h with BSA 2%, 0.1% Triton X-100 in PBS 1x. Protein expression was monitored with GFP signal or with an anti-V5 antibody (Cell Signaling, #13202S), incubated overnight in blocking solution. PFF binding was detected using Alexa-555 streptavidin (ThermoFisher, #S2138), incubated with a goat anti-rabbit secondary antibody to detect V5 (ThermoFisher, #A-11008) for 1 h. Images were acquired using the automated ImageXpress Micro XLS (Molecular Devices) with a 20x air objective. Potential hit genes were identified by qualitative visual inspection, followed by validation with secondary screening. Each of the 16 hits were confirmed in a second round of specific retesting. Hits obtained were further validated by comparing its binding to biotinylated α-syn PFF with biotinylated monomer and responding to increasing concentrations of α-syn PFF.
Synuclein PFF binding to different mGluRs
HEK293T cells were plated onto poly-D-Lysine(PDL)-coated 96-well plates and transfected using Lipofectamine 3000 (ThermoFisher, #L3000015) as described above. The following plasmids were used: rat GRM1 and GRM5 in pcDNA3.1+ (ThermoFisher, #V80020); human GRM3, GRM4 and GRM8 in pLX304 vector (Transomic, #TCH3000, #TOH7500); and human GRM7 with an HA tag was a gift from Xin Lin at Columbia University. Two days after transfection, biotinylated α-syn PFF (1000 nM) were added to cell media (DMEM with 10% FBS) and incubated for 2 h at RT. Cells were then washed, fixed and stained. Protein expression was monitored with anti-V5 antibody (Cell Signaling, #13202S, 1:1000), c-Myc antibody 9B11 (Cell Signaling, #2276, 1:1000) or HA antibody (Cell Signaling, #3724S 1:1000), incubated overnight in blocking solution. PFF binding was detected using Alexa-555 streptavidin (ThermoFisher, #S21381, 1:1000), incubated with a goat anti-rabbit secondary antibody to detect V5 (ThermoFisher, #A-11008, 1:1000) or goat anti-mouse secondary antibody to detect c-Myc (ThermoFisher, #A-11001, 1:1000) for 1 h. Images were acquired using the automated ImageXpress Micro XLS (Molecular Devices) with a 20X air objective.
Aß oligomer preparation
We have described biotinylated Aß oligomer preparation and cellular binding assays12. Lyophilized synthetic biotinylated β-amyloid (1–42) peptide (The ERI Amyloid Laboratory, LLC) was initially dissolved in 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) at a concentration of 10 mg/ml. The solution was then heated at 70 °C for 1 h, divided into 0.5 mg aliquots in microcentrifuge tubes, and left to air-dry overnight at room temperature. The following day, the dried samples were further desiccated using a SpeedVac concentrator for 1 h. To generate oligomers, one of the dried aliquots was resuspended in 40 μl of DMSO, split into two portions, and each was diluted in 1 ml of F12 medium (R&D Systems, M15350) to a final concentration of 55 μM. The mixtures were then incubated at RT overnight to promote oligomer formation. On the subsequent day, samples were centrifuged at 20,817 × g for 15 min using a tabletop centrifuge (Eppendorf 5430) to remove any fibrillar aggregates. The resulting supernatant, assumed to contain 55 μM Aβ monomer equivalent, was subsequently diluted in F12 medium to desired working concentrations. Binding and ligand detection utilized the same method as for biotinylated α-syn PFF.
Quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was extracted from the substantia nigra and cortex using the RNeasy Plus Mini Kit (Qiagen, #74136). RNA concentration was quantified with a NanoDrop 1000 spectrophotometer (Thermo Scientific), and cDNA was synthesized by an equal amount of total RNA (500 ng) via reverse transcription with the iScript Reverse Transcription Supermix (Bio-Rad, 1708840). Quantitative real-time PCR was performed on a CFX96 Real-Time PCR system (Bio-Rad, CA, USA) using the SsoAdvanced Universal SYBR Green Supermix (Bio-Rad, #1725272). Gene expression levels were calculated by the 2^-ΔΔCt method. PCR amplification was conducted for 40 cycles under the following conditions: denaturation at 95 °C for 30 s, annealing at 55 °C for 30 s, and extension at 72 °C for 30 s.
RNAScope
Three-month-old wt mice were euthanized via CO2 inhalation, followed by perfusion with ice-cold DPBS and 4% paraformaldehyde. The brains were quickly dissected and post-fixed for 12 h in 4% paraformaldehyde at 4 °C. Next, the brains were immersed in 30% sucrose for 2 days. For cryosectioning, brains were included in Tissue-Tek O.C.T. compound (Sakura, #4583). 20 µm coronal sections were generated using a Leica CM1900 cryostat and mounted on SuperFrost Plus glass slides (O. Kindle).
RNA was detected using the RNAScope technology (RNAscope Multiplex Fluorescent Reagent Kit v2, BioTechne, #323280) following the manufacturer’s protocol under RNase-free conditions. Briefly, brain cryosections were fixed with 4% PFA for 15 min at 4 °C and dehydrated using an increasing ethanol series (50%, 75%, 100%), for 5 min each. Tissue pretreatment included protease III addition for 30 min at RT. Then, sections were hybridized with the mouse Npdc1-C1 probe (BioTechne, #1123541-C1) and mouse Grm4-C2probe (BioTechne, #480991-C2) for 2 h at 40 °C in a HybEZ II hybridization oven (ACD). Target probe signals were amplified following the manufacturer’s instructions at 40 °C in the HybEZ hybridization oven.
After the RNA detection, tyrosine hydroxylase-positive neurons of the SNc were stained by immunofluorescence. Sections were blocked with 2% BSA (Sigma) with 0.1% Triton X-100 in DPBS for 1 h and then incubated with anti-TH antibody (Novus Bio, #NB300-109, 1:750) overnight. The next day, sections were washed with DPBS and incubated with a goat anti-rabbit secondary antibody to detect TH (ThermoFisher, #A-21244). DAPI ready-to-use solution (BioTechne, included in Kit) was used to counterstain nuclei. Slides were mounted with FluorSave reagent (Millipore, #345789) and imaged in a Fluoview FV10i confocal microscope (Olympus).
NPDC1 deletion mutants
Deletions in human NPDC1 gene in pLX304-V5 plasmid were generated using the Q5 Site-Directed Mutagenesis Kit (NEB, #E0554S) according to the manufacturer’s instructions. The resulting plasmids were transformed into DH5alpha competent cells (Thermofisher Scientific). Integrity of the constructs was verified by sequencing.
In vitro co-immunoprecipitation
HEK293T cells were transfected with GRM4-V5 or NPDC1-V5 using Lipofectamine 3000. Two days after transfection, α-syn monomer or PFF at 1000 nM were added to the cells and incubated for 2 h at 23 °C. Cells were washed twice with media and with PBS 1X and then collected in lysis buffer: 50 mM Tris pH 8.0, 150 mM NaCl, 1% Triton X-100, protease inhibitors cOmplete-mini (Roche, #11836170001). Lysates were centrifuged at 18,000 g for 20 min and the supernatant was collected. Protein quantification was determined using Pierce BCA Protein Assay Kit (ThermoFisher, #23225). Then, 500 µg of protein were incubated with 50 µl of protein G magnetic Dynabeads (ThermoFisher, #10003D) conjugated with 5 µg V5 tag monoclonal antibody (ThermoFisher, #R960-25) or mouse IgG2a Isotype Control (CST, #61656). Incubation was performed for 2 h at 23 °C. Beads were washed 4 times with lysis buffer and eluted with Laemmli buffer at 95 °C for 5 min.
Immunoblot
Protein samples were electrophoresed using precast 4–20% Tris-glycine gels (Bio-Rad, #4561096) and transferred with an iBlot 2 Transfer Device onto nitrocellulose membranes (Invitrogen, #IB23001). The membranes were incubated in blocking buffer (Rockland, #MB-070) for 1 h at room temperature and then incubated overnight at 4 °C in blocking buffer with primary antibody against V5-Tag D3H8Q (CST, 13202). The next day, membranes were washed three times with TBST for 3 min and incubated in secondary antibodies (Li-Cor, IR Dye 680 or 800, 1:10,000) for 1 h at room temperature. After washing three times with TBST for 3 min, proteins were visualized with an Odyssey Infrared imaging system (Li-Cor). The immunoreactive bands were quantified using ImageJ software.
Immunofluorescence of cultures
HEK293T cells and cultured neurons were washed twice with cell media and then 3 times with PBS1x. Cells were fixed in PFA 4% for 12 min and then blocked for 1 h with BSA 2%, 0.1% Triton X-100 in PBS 1x. Cells were then incubated overnight with primary antibodies in blocking buffer at 4 °C: V5 antibody (Cell Signaling, #13202S, 1:1000) or MAP2 (Millipore, # AB5622, 1:1000). The cells were then washed 3 times with DPBS and incubated for 2 h at room temperature in Alexa Fluor 488 donkey anti-rabbit IgG (H + L) (Invitrogen #A21206, 1:1000) in blocking buffer. Biotinylated α-syn PFF binding was detected using streptavidin-555 (ThermoFisher, #S2138, 1:1000). Images were acquired using the automated ImageXpress Micro XLS (Molecular Devices) with a ×20 air objective for HEK293T cells. Stained neurons were imaged using LSM800 confocal microscopy (Zeiss) with ×40 magnification objective lens.
For automated microscopy of primary neurons using ImageXpress® Micro Confocal system, the cells were fixed with 4% paraformaldehyde for 15 min at 4 °C, followed by blocking and permeabilization using a solution of 10% horse serum and 0.1% Triton X-100 for 1 h at room temperature. Subsequently, the cells were incubated overnight at 4 °C in a primary antibody solution consisting of 1% normal horse serum, 0.1% Triton X-100 in DPBS. The following primary antibodies were used: anti-V5 (CST 80076S, 1:500), anti-DYKDDDDK Tag (CST 14793S, 1:500), anti-MAP2 (Abcam AB5392, 1:10000), anti- p-α-syn (Invitrogen PA14686, 1:2000), anti-Synapsin1/2 (Synaptic System 106004, 1:500), and anti-Homer 1b/c (Synaptic System 160111, 1:500). After primary antibody incubation, cells were washed three times with DPBS, then incubated with fluorescent secondary antibodies (Invitrogen Alexa Fluor 1:500) in 1% normal donkey serum and 0.2% Triton X-100 in DPBS for 1–2 h at 23 °C.
For single V5 staining, Alexa Fluor 488 (Invitrogen # A-21202) was used. For V5/Flag co-staining, Alexa Fluor 488 (Invitrogen # A-21202) and Alexa Fluor 647 (Invitrogen # A-31573) were applied. For α-syn-PFF/MAP2 co-staining, Alexa Fluor 555 streptavidin (Invitrogen # S21381) and Alexa Fluor 647 (Invitrogen # A-78952) were used. In the case of p-α-syn /MAP2 co-staining, Alexa Fluor 488 (Invitrogen # A-11008) and Alexa Fluor 647 (Invitrogen # A-78952) were used. For triple staining of Synapsin1/2, Homer1, and MAP2, the secondary antibodies applied were Alexa Fluor 488 (Invitrogen # A-11001), Alexa Fluor 555 (Invitrogen # A-21435), and Alexa Fluor 647 (Invitrogen # A-21450), respectively.
After secondary antibody incubation, nuclei were stained with DAPI (Invitrogen D1306, 1 µg/mL in DPBS) for 15 min at room temperature, followed by three washes with DPBS. Images of HEK293T cells and primary neurons were captured using the ImageXpress® Micro Confocal system (Molecular Devices) with 20X or 40X objective lenses.
For endocytosis assays, HEK293T cells were cultured, transfected and incubated with biotinylated α-syn PFF (1200 nM monomer equivalent) for 2 h at either 4 °C or 37 °C, and then fixed with 4% paraformaldehyde. Intact cells were then incubated with 10 ng/ml streptavidin (ThermoFisher, #434301) in 10% horse serum at 4 °C for 12 h to block cell surface biotin-PFF prior to permeabilization with 0.1% TritonX-100 and detection of internalized biotin-PFF with Alexa-555-streptavidin (Invitrogen #S21381). Imaging and quantitation of fluorescent signal utilized the ImageXpress imaging and quantitation methods for routine binding experiments.
mGluR4 signaling assays
α-syn PFF binding assays in the presence of mGluR4 modulators were conducted in HEK293T cells. Cells were plated onto PDL-coated 96-well plates (Corning, #354461) in DMEM media (GIBCO, #11965-092) supplemented with 10% fetal bovine serum (GIBCO, #16000-044) and Pen Strep (GIBCO, #15140-122). Cells were transfected with GRM4-V5 in a pLX304 plasmid using Lipofectamine 3000 (ThermoFisher, #L3000015). Two days after transfection, media was replaced by DMEM without FBS 30 min before the experiment. Then, mGluR4 modulators were added for 30 min at RT: glutamate (Sigma, #G1626), L-AP4 (Abcam, #ab144481), and MSOP (Tocris, #0103). Cells were then treated with biotinylated α-syn PFF for 2 h at RT. Immunostaining was performed as previously described. Images were acquired using the automated ImageXpress Micro XLS (Molecular Devices) with a 20x air objective
HEK293T cells (passages 5 to 20) were cultured in DMEM GlutaMAX (GIBCO, #10566-016) supplemented with 10% FBS (Seradigm, #1500-500) and 2 mM sodium pyruvate (ThermoFisher Scientific, #11360070) at 37 °C with 5% CO2. mGluR4 Gαqi5 stable HEK293T cells (passages 3 to 20, Multispan, #CG1191) were cultured in DMEM GlutaMAX with 10% dialyzed FBS (ThermoFisher Scientific, #26400044), 2 mM sodium pyruvate, 1 μg/ml puromycin (Invivogen, #ant-pr-5), and 250 μg/ml hygromycin (Invivogen, #ant-hm-5). Cells were transiently transfected with either a synthetic vector encoding pcDNA3.1 or human NPDC1 cDNA in the pLX304 vector using Lipofectamine 3000 (ThermoFisher Scientific, #L3000-008) according to the manufacturer’s protocol.
For experiments involving only mGluR4 Gαqi5 stable HEK293T cells or transfected mGluR4 Gαqi5 stable HEK293T cells, cells were plated onto PDL-coated 96-well Black/Clear Flat Bottom TC-treated Assay Plates (Corning, #354640) at a density of 1 × 105 cells/ml. HEK293T cells were transfected in 6-well plates for 24 h, followed by a media change to DMEM without L-glutamine (HyClone, #SH30285.01) supplemented with 10% dialyzed FBS, and incubated at 37 °C with 5% CO2 for an additional 24 h. The medium was then collected as conditioned medium. Transfected cells were resuspended and mixed with mGluR4 Gαqi5 stable HEK293T cells at a ratio of either 1:1 or 2:1 (maintaining 1 × 104 cells per well for Grm4-HEK293T cells) using DMEM without L-glutamine (10% dialyzed FBS), and plated onto PDL-coated 96-well plates. They were incubated at 37 °C for another 24 h.
For experiments using only mGluR4 Gαqi5 stable HEK293T cells, the medium was changed to DMEM without L-glutamine (10% dialyzed FBS) one day before the FLIPR experiment. Prior to the conditioned medium assay, the medium was replaced with conditioned medium and incubated at 37 °C with 5% CO2 for 2 h. On the day of the FLIPR experiment, FLIPR Calcium 6 dye was diluted with Hanks’ Balanced Salt Solution (HBSS) supplemented with 5 mM Probenecid (Invitrogen, #P36400). Then, 100 µL of the diluted dye was added to each well, and the plate was incubated at 37 °C with 5% CO2 for 2 h. Fluorescence measurements were taken using the FlexStation 2 (Molecular Devices) over 120 s at 2-second intervals. 20 µL of L-AP4 (Hello Bio, HB0370) or DPBS was injected into the wells at 20 s, followed by a similar injection program for Ionomycin (Sigma, #I0634-1MG). The relative fluorescence units (RFU) were calculated by subtracting the basal signal (mean signal from 0 to 20 s) from the raw signal (mean signal from 20 to 180 s).
For mGluR4 cAMP assays in HEK293T cells without chimeric G protein expression, a luminescent reporter was utilized. HEK293T vells were transfected with the pGloSensor™−22F cAMP plasmid (Promega, #E2301) together with pcDNA+pcDNA, NPDC1+pcDNA, GRM4+pcDNA, or NPDC1 + GRM4 in HEK293T cells. Two days after transfection, the culture medium was replaced with DMEM lacking L-glutamine and supplemented with 10% dialyzed FBS. On the following day, the medium was changed to DMEM without L-glutamine containing either α-syn PFF (1200 nM monomer equivalent) or DPBS, and cells were incubated at 37 °C with 5% CO2 for 2 h. The cAMP test was then performed according to the manufacturer’s instructions (GloSensor™ Technology, Promega, #E1291). In brief, cells were pre-equilibrated with GloSensor™ cAMP Reagent for 2 h, after which luminescence was recorded using a VICTOR Nivo Multimode Microplate Reader for 5 min to establish baseline levels. Cells were then treated with L-AP4 (100 µM, Hello Bio, HB0370) or DPBS at 37 °C with 5% CO2 for 5 min, followed by the addition of forskolin (10 µM, MedChem Express, HY15371). Luminescence was immediately measured for 60 min.
Npdc1 modulation of F-Actin
HEK293T cells were plated onto 8-well Lab-Tek chamber slides (Nunc, #154941PK) in DMEM media (GIBCO, #11965-092) supplemented with 10% fetal bovine serum (GIBCO, #16000-044) and Pen Strep (GIBCO, #15140-122). Cells were transfected with NPDC1-V5 in a pLX304 plasmid using Lipofectamine 3000 (ThermoFisher, #L3000015). Two days after transfection, cells were treated with biotinylated α-syn PFF for 3 h at 37 °C. Cells were then washed twice with media and 3 times with PBS 1x. Immunofluorescence was performed as previously indicated. Alexa-555 Phalloidin was used to detect actin filaments (ThermoFisher, #A34055) and α-syn PFF were detected with Alexa-488 streptavidin (ThermoFisher, #S32354) in blocking buffer.
Neuronal culture and PFF exposure
Cortical cells were isolated from the cortices of E16-17 mouse embryos as reported previously49. The embryos were decapitated, and the anterior cortex was dissected out under a stereotactic microscope using BrainBits Hibernate E minus calcium medium, kept on ice. The tissue was then digested with a freshly prepared enzyme solution (Mg/Ca-free HBSS containing 20 U/ml Papain (Worthington LK003178), 3 mM EDTA (AmericanBio), 2 mM CaCl₂ (VWR E506), and 1 mg/ml DNAse (Sigma DN25)) at 37 °C for 30 min. After digestion, the cell suspension was filtered through a 40 µm cell strainer (Corning, #352340), and the cells were counted and diluted to a concentration of 1 × 10⁵ cells/ml. A 100 µl volume of this suspension was plated onto a PDL-coated 96-well Black/Clear Flat Bottom Plate. The plates were incubated at 37 °C with 5% CO₂, and the medium was half-changed every 7 days.
For the binding assay, PFF was added at DIV 17-18 and incubated at 4 °C or 37 °C for 2 h. For the α-syn phosphorylation and synaptic loss assays, PFF were added at DIV 10 and incubated at 37 °C for 7 days.
Mice
All protocols were approved by Yale Institutional Animal Care and Use Committee (IACUC) (2024-07281). Npdc1 knock-out mouse embryos were obtained from EMMA (B6;129×1-Npdc1tm1Ce/Orl, #EM:00211) and rederived at Yale Genome Editing Center. Grm4 knock-out mice with C57BL/6 J background (B6.129-Grm4tm1Hpn/J, #003619) and human α-syn A53T transgenic mice with C57BL/6 J background (B6.Cg-2310039L15RikTg(Prnp-SNCA*A53T)23Mkle/J, #006823) were obtained from the Jackson Laboratory. The mice were grouped and housed 2–5 per cage, adhering to a 12-h light-dark cycle, with constant access to food and water. The room conditions were maintained at a temperature of 21–23 °C and a humidity of 50 ± 20%. Both male and female mice were included in the experiments, with a total of 333 males and 325 females. Sex was not considered as a separate variable because prior studies of the synuclein models have not detected substantial differences.
α-syn PFF stereotaxic injections
Immediately before the intrastriatal injections, fibrils were diluted in sterile PBS and sonicated as indicated above. PBS 1X or recombinant α-syn PFF (5 µg in 2 µl) was stereotactically delivered into the right dorsal striatum using the following coordinates: +0.2 mm Medial-lateral (ML); +2.0 mm antero-posterior (AP) and +2.8 mm dorso-ventral (DV) from bregma. Injections were performed using a 10 µL syringe (Hamilton, #7635-01) and 33-gauge needle, 45° tip (Hamilton, #7803-05) at a rate of 0.4 µl per minute. After delivery, the needle was left in place for 5 min and then withdrawn over 2 min. After surgery, mice received 0.05 mg/kg buprenorphine for 3 days (twice daily, 12 h apart) as analgesic and body weight was monitored once a month.
Animal behavior was performed after 6 months and mice were then euthanized. For histological studies, mice were perfused transcardially with PBS and 4% PFA and brains were removed, followed by overnight fixation in 4% PFA and washed and kept in PBS 1X after.
Immunohistology of brain sections
All histological tests were completed and analyzed by an experimenter blind to genotype and group.
Mice were euthanized via CO2 inhalation, followed by perfusion with ice-cold DPBS and 4% paraformaldehyde in sequence. The brains and spinal cords were quickly dissected and sectioned into 40 μm slices using a Leica WT1000S vibratome. The sections were blocked and permeabilized in 10% normal horse serum with 0.1% Triton X-100 for 1 h at room temperature. Following this, the sections were incubated overnight at 4 °C with primary antibodies diluted in 1% normal horse serum, 0.1% Triton X-100 in DPBS. The primary antibodies used were: anti-TH (EMD Millipore AB152, 1:500), anti-p-α-syn (BioLegend 825701, 1:500), anti-NeuN (EMD Millipore ABN91, 1:500), anti-ChAT (EMD Millipore AB144P, 1:100), and anti-GFAP (DAKO Z0334, 1:500).
After incubation, the sections were washed three times with DPBS and incubated for 1-2 h at room temperature with fluorescent secondary antibodies (Invitrogen Alexa Fluor, 1:500) in 1% normal donkey serum and 0.2% Triton X-100 in DPBS. For TH single staining, Alexa Fluor 488 (Invitrogen # A-11008) was used. For p-α-syn/TH co-staining, Alexa Fluor 488 (Invitrogen # A-11008) and Alexa Fluor 647 (Invitrogen # A-31571) were used. For NeuN/GFAP co-staining, Alexa Fluor 488 (Invitrogen # A-11008) and Alexa Fluor 647 (Invitrogen # A-78952) were employed. For ChAT/GFAP/p-α-syn triple staining, Alexa Fluor 488 (Invitrogen # A-11008), Alexa Fluor 555 (Invitrogen # A-21432), and Alexa Fluor 647 (Invitrogen # A-31571) were used. Following antibody incubation, nuclei were stained with DAPI (Invitrogen D1306, 1 µg/mL in DPBS) for 15 min at room temperature, followed by three DPBS washes. The sections were mounted on glass slides and coverslipped using Vectashield antifade mounting medium (Vector).
Images of TH single staining in nigra sections and NeuN/GFAP co-staining in spinal cord sections were captured using an Olympus VS200 Slide Scanner with a 10X objective lens. Confocal images of p-α-syn/TH co-staining and ChAT/GFAP/p-α-syn triple staining were obtained with a Leica DMi8 confocal microscope using a 40X objective lens.
Image quantification of immunohistochemistry
All images were obtained, processed, and analyzed by a researcher who was unaware of the animal genotypes and group information.
All quantitative image analyses conducted with the Leica DMi8 confocal microscope were processed using ImageJ (National Institutes of Health). For the p-α-Syn/TH co-staining of brain slices, we applied uniform thresholding and binarization across all images, followed by measuring the areas of p-α-Syn and TH. In the ChAT/GFAP/p-α-Syn triple staining of spinal cord slices, we also applied uniform thresholding and binarization, manually counted the ChAT-positive cells, and measured the GFAP intensity in the anterior horn.
Similarly, all quantitative image analyses from the Olympus VS200 Slide Scanner microscope were processed using ImageJ. To evaluate neuronal loss in the substantia nigra stereologically, we selected every sixth serial coronal slice of 40 µm thickness through the entire substantia nigra, applied uniform thresholding and binarization, and counted all TH-positive cells. From this standardized sampling method, we calculated the DA neuron count per SNc. For NeuN staining, we manually counted the larger NeuN-positive cells in the anterior horn.
Quantitative analyses of images acquired using the ImageXpress® Micro Confocal system were processed with MetaXpress software. After uniformly thresholding and binarizing the images, we measured MAP2 intensity and then expanded the MAP2 signal by one pixel using the “Grow Objects” function. The MAP2 signal was set as a mask to measure streptavidin (PFF) intensity on MAP2 for PFF/MAP2 co-staining, and p-α-Syn intensity on MAP2 for p-α-Syn/MAP2 co-staining. Additionally, we quantified the number of “active synapses” on MAP2 by using the “Found Round Objects” tool to detect Synapsin1/2 and Homer1b/c double-positive puncta on MAP2, with diameters ranging from 0.5–2 µm and intensities of 1500 above local background.
Behavioral tests
All behavioral tests were performed and analyzed by experimenter blind to genotype and group.
Pole test
It was performed with a wooden pole of 57 cm in height and 2 cm in width. Animals were acclimatized for two consecutive days prior to the test and each time five trials were given to each animal. During the test, animal was placed near the top of the pole facing upwards. The time taken by each animal to turn downwards was monitored as the pole turn time. Moreover, the time taken by each animal to reach the base of the pole was recorded as the pole climb downtime. The surface of the pole was made rough by covering it with bandage gauze. The maximum time for recording was set as 60 s. If any animal was found to stall for more than 60 s, the test was further performed for that particular animal.
Inverted wire screen test
The inverted wire screen test of motor function was performed by placing the mouse upright on a platform covered with a wire screen and then inverting the platform. The latency of the mouse to fall off of the wire screen was measured, and the better time was taken from two trials. Trials were stopped if a mouse reached a criterion of 120 s.
Open field
Open field test was conducted to look at the locomotor activities of the animals. Locomotor activity was monitored with a camera linked to Noldus system and EthoVisionXT software (Netherlands). The instrument records the overall movement abilities of the animals including total distance moved, velocity, total moving time, resting time, center time, and frequencies of movement and rest. Before the test, mice were placed inside the open field arena for 10 min daily for two consecutive days to train them and record their baseline values. Two days after the training, each mouse was taken from the cage and gently placed in the middle of the open field arena. After releasing the animal, data acquisition was started by the software for the next 5 min and the parameters related to the locomotor activities were collected by the software.
Rotarod
Prior to the rotarod test, mice were placed on the rotarod instrument for 5–10 min daily for consecutive two days to train them. After 2 days of training, mice were placed on the rotating rod, which rotates with a gradually increasing speed of 4–40 rpm. The experiment was ended if the animal slips from the rotating rod to the base of the instrument or just grips the rod to turn in the opposite direction without rotating against the direction of the rotating rod.
Grip strength test
Grip strength was measured using a grip force meter from Columbus Instruments (Columbus, OH, USA). During each assessment, mice were placed on the apparatus and gently pulled parallel to the ground until they could no longer maintain their grip. Each mouse underwent five trials at every time point, with a 10-second rest period between each trial. The highest three scores from the five trials were averaged to calculate the final grip strength score for each mouse.
Statistical analysis
Data are shown as mean ± SEM. One-way ANOVA with Tukey’s correction (for multiple comparisons), Gehan–Breslow–Wilcoxon test, two-way ANOVA with Sidak’s post hoc test, and Kruskal–Wallis test with Dunn’s post hoc test were analyzed using Prism (version 9.0.1) software. Gaussian distributions were assumed based on the previous studies. Separate dots on each figure correspond to N values reflecting different mice. Data are reported as statistically significant for p < 0.05 (*p < 0.05; **p < 0.005; ***p < 0.001).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Source data are provided with this paper.
References
Spillantini, M. G. et al. Alpha-synuclein in Lewy bodies. Nature 388, 839–840 (1997).
Spillantini, M. G., Crowther, R. A., Jakes, R., Hasegawa, M. & Goedert, M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc. Natl. Acad. Sci. USA 95, 6469–6473 (1998).
Dickson, D. W. Neuropathology of Parkinson disease. Parkinsonism Relat. Disord. 46, S30–S33 (2018).
Goedert, M., Jakes, R. & Spillantini, M. G. The Synucleinopathies: twenty Years On. J. Parkinsons Dis. 7, S51–S69 (2017).
Luk, K. C. et al. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338, 949–953 (2012).
Peng, C. et al. Cellular milieu imparts distinct pathological alpha-synuclein strains in alpha-synucleinopathies. Nature 557, 558–563 (2018).
Prusiner, S. B. et al. Evidence for alpha-synuclein prions causing multiple system atrophy in humans with Parkinsonism. Proc. Natl. Acad. Sci. USA 112, E5308–E5317 (2015).
Masuda-Suzukake, M. et al. Prion-like spreading of pathological alpha-synuclein in brain. Brain 136, 1128–1138 (2013).
Neupane, S., De Cecco, E. & Aguzzi, A. The hidden cell-to-cell trail of alpha-synuclein aggregates. J. Mol. Biol. 435, 167930 (2023).
Scheiblich, H. et al. Microglia rescue neurons from aggregate-induced neuronal dysfunction and death through tunneling nanotubes. Neuron 112, 3106–3125 e3108 (2024).
Henderson, M. X. et al. Spread of alpha-synuclein pathology through the brain connectome is modulated by selective vulnerability and predicted by network analysis. Nat. Neurosci. 22, 1248–1257 (2019).
Lauren, J., Gimbel, D. A., Nygaard, H. B., Gilbert, J. W. & Strittmatter, S. M. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 457, 1128–1132 (2009).
Mao, X. et al. Pathological alpha-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 353 (2016).
Gu, H. et al. Lymphocyte activation gene 3 (Lag3) contributes to alpha-synucleinopathy in alpha-synuclein transgenic mice. Front. Cell Neurosci. 15, 656426 (2021).
Emmenegger, M. et al. LAG3 is not expressed in human and murine neurons and does not modulate alpha-synucleinopathies. EMBO Mol. Med. 13, e14745 (2021).
Ferreira, D. G. et al. alpha-synuclein interacts with PrP(C) to induce cognitive impairment through mGluR5 and NMDAR2B. Nat. Neurosci. 20, 1569–1579 (2017).
Thom, T. et al. Cellular prion protein mediates alpha-synuclein uptake, localization, and toxicity in vitro and in vivo. Mov. Disord. 37, 39–51 (2022).
So, R. W. L. et al. alpha-Synuclein strain propagation is independent of cellular prion protein expression in a transgenic synucleinopathy mouse model. PLoS Pathog. 20, e1012517 (2024).
Diaz-Ortiz, M. E. et al. GPNMB confers risk for Parkinson’s disease through interaction with alpha-synuclein. Science 377, eabk0637 (2022).
Brendza, R. et al. Genetic ablation of Gpnmb does not alter synuclein-related pathology. Neurobiol. Dis. 159, 105494 (2021).
Choi, Y. R. et al. FcgammaRIIB mediates the inhibitory effect of aggregated alpha-synuclein on microglial phagocytosis. Neurobiol. Dis. 83, 90–99 (2015).
Choi, Y. R. et al. Prion-like propagation of alpha-synuclein is regulated by the FcgammaRIIB-SHP-1/2 signaling pathway in neurons. Cell Rep. 22, 136–148 (2018).
Wu, K. M. et al. Neuronal FAM171A2 mediates alpha-synuclein fibril uptake and drives Parkinson’s disease. Science 387, 892–900 (2025).
Domingues, R. et al. Extracellular alpha-synuclein: sensors, receptors, and responses. Neurobiol. Dis. 168, 105696 (2022).
Yao, Z. et al. A high-resolution transcriptomic and spatial atlas of cell types in the whole mouse brain. Nature 624, 317–332 (2023).
Griesser, E. et al. Quantitative profiling of the human substantia nigra proteome from laser-capture microdissected FFPE tissue. Mol. Cell Proteom. 19, 839–851 (2020).
Cenci, M. A., Skovgard, K. & Odin, P. Non-dopaminergic approaches to the treatment of motor complications in Parkinson’s disease. Neuropharmacology 210, 109027 (2022).
Rascol, O., Medori, R., Baayen, C., Such, P., Meulien, D. & Group AS. A randomized, double-blind, controlled phase II study of Foliglurax in Parkinson’s disease. Mov. Disord. 37, 1088–1093 (2022).
Conn, P. J. & Pin, J. P. Pharmacology and functions of metabotropic glutamate receptors. Annu. Rev. Pharm. Toxicol. 37, 205–237 (1997).
Conklin, B. R., Farfel, Z., Lustig, K. D., Julius, D. & Bourne, H. R. Substitution of three amino acids switches receptor specificity of Gq alpha to that of Gi alpha. Nature 363, 274–276 (1993).
Galiana, E., Vernier, P., Dupont, E., Evrard, C. & Rouget, P. Identification of a neural-specific cDNA, NPDC-1, able to down-regulate cell proliferation and to suppress transformation. Proc. Natl. Acad. Sci. USA 92, 1560–1564 (1995).
Evrard, C. & Rouget, P. Subcellular localization of neural-specific NPDC-1 protein. J. Neurosci. Res. 79, 747–755 (2005).
Kim, S. et al. NGL family PSD-95-interacting adhesion molecules regulate excitatory synapse formation. Nat. Neurosci. 9, 1294–1301 (2006).
Pekhletski, R. et al. Impaired cerebellar synaptic plasticity and motor performance in mice lacking the mGluR4 subtype of metabotropic glutamate receptor. J. Neurosci. 16, 6364–6373 (1996).
Lee, M. K. et al. Human alpha-synuclein-harboring familial Parkinson’s disease-linked Ala-53 -> Thr mutation causes neurodegenerative disease with alpha-synuclein aggregation in transgenic mice. Proc. Natl. Acad. Sci. USA 99, 8968–8973 (2002).
Martin, L. J. et al. Parkinson’s disease alpha-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J. Neurosci. 26, 41–50 (2006).
Evrard, C., Caron, S. & Rouget, P. Functional analysis of the NPDC-1 gene. Gene 343, 153–163 (2004).
Goedert, M. NEURODEGENERATION. Alzheimer’s and Parkinson’s diseases: the prion concept in relation to assembled Abeta, tau, and alpha-synuclein. Science 349, 1255555 (2015).
Moehle, M. S. et al. LRRK2 inhibition attenuates microglial inflammatory responses. J. Neurosci. 32, 1602–1611 (2012).
Doller, D., Bespalov, A., Miller, R., Pietraszek, M. & Kalinichev, M. A case study of foliglurax, the first clinical mGluR4 PAM for symptomatic treatment of Parkinson’s disease: translational gaps or a failing industry innovation model?. Expert Opin. Investig. Drugs 29, 1323–1338 (2020).
Charvin, D. et al. An mGlu4-positive allosteric modulator alleviates parkinsonism in primates. Mov. Disord. 33, 1619–1631 (2018).
Marino, M. J. et al. Allosteric modulation of group III metabotropic glutamate receptor 4: a potential approach to Parkinson’s disease treatment. Proc. Natl. Acad. Sci. USA 100, 13668–13673 (2003).
Beurrier, C. et al. Electrophysiological and behavioral evidence that modulation of metabotropic glutamate receptor 4 with a new agonist reverses experimental parkinsonism. FASEB J. 23, 3619–3628 (2009).
Tomioka, N. H. et al. Elfn1 recruits presynaptic mGluR7 in trans and its loss results in seizures. Nat. Commun. 5, 4501 (2014).
Dunn, H. A., Patil, D. N., Cao, Y., Orlandi, C. & Martemyanov, K. A. Synaptic adhesion protein ELFN1 is a selective allosteric modulator of group III metabotropic glutamate receptors in trans. Proc. Natl. Acad. Sci. USA 115, 5022–5027 (2018).
Leonard H. L., Global Parkinson’s Genetics P. Novel Parkinson’s disease genetic risk factors within and across european populations. medRxiv, https://doi.org/10.1101/2025.1103.1114.24319455 (2025).
Belkacemi, K., Rondard, P., Pin, J. P. & Prezeau, L. Heterodimers revolutionize the field of metabotropic glutamate receptors. Neuroscience 578, 46–58 (2025).
Volpicelli-Daley, L. A., Luk, K. C. & Lee, V. M. Addition of exogenous alpha-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous alpha-synuclein to Lewy body and Lewy neurite-like aggregates. Nat. Protoc. 9, 2135–2146 (2014).
Hu, F. et al. Sortilin-mediated endocytosis determines levels of the frontotemporal dementia protein, progranulin. Neuron 68, 654–667 (2010).
Acknowledgements
This work was supported by grant 16257 from the Michael J. Fox Foundation to A.P.C., and by grants R01AG034924, R35NS097283, R01AG070926, R01AG066165 and P30AG066508 from the N.I.H. to S.M.S.
Author information
Authors and Affiliations
Contributions
Conceptualization, A.P.C., M.C., and S.M.S.; Methodology, A.P.C., M.C., L.A.G., N.K., E.C.G., and S.M.S.; Investigation, A.P.C., M.C., L.A.G., A.H., S.J.T., and N.K.; Writing—Original Draft, A.P.C., M.C., and S.M.S.; Writing—Review & Editing, all; Funding Acquisition, A.P.C. and S.M.S.; Resources, S.M.S; Supervision, S.M.S.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Perez-Canamas, A., Chen, M., Almandoz-Gil, L. et al. mGluR4–NPDC1 complex mediates α-synuclein fibril-induced neurodegeneration. Nat Commun 17, 994 (2026). https://doi.org/10.1038/s41467-025-67731-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67731-3
