
Abstract
Human prenatal skin is populated by innate immune cells, including macrophages, but whether they act solely in immunity or have additional functions in morphogenesis is unclear. Here we assembled a comprehensive multi-omics reference atlas of prenatal human skin (7–17 post-conception weeks), combining single-cell and spatial transcriptomics data, to characterize the microanatomical tissue niches of the skin. This atlas revealed that crosstalk between non-immune and immune cells underpins the formation of hair follicles, is implicated in scarless wound healing and is crucial for skin angiogenesis. We systematically compared a hair-bearing skin organoid (SkO) model derived from human embryonic stem cells and induced pluripotent stem cells to prenatal and adult skin1. The SkO model closely recapitulated in vivo skin epidermal and dermal cell types during hair follicle development and expression of genes implicated in the pathogenesis of genetic hair and skin disorders. However, the SkO model lacked immune cells and had markedly reduced endothelial cell heterogeneity and quantity. Our in vivo prenatal skin cell atlas indicated that macrophages and macrophage-derived growth factors have a role in driving endothelial development. Indeed, vascular network remodelling was enhanced following transfer of autologous macrophages derived from induced pluripotent stem cells into SkO cultures. Innate immune cells are therefore key players in skin morphogenesis beyond their conventional role in immunity, a function they achieve through crosstalk with non-immune cells.
Similar content being viewed by others

Main
Human skin organogenesis begins after gastrulation from two primary germ layers. The epidermis, the most superficial layer of the skin, melanocytes and neural cells arise from the ectoderm. The dermis, which is separated from the epidermis by the basement membrane and contains endothelial and mural cells, derives from the mesoderm (apart from facial and cranial skin, where it arises from ectoderm-derived neural crest cells)2. The skin appendages, which include hair follicles (HFs) and sebaceous glands, form in a cephalocaudal direction3. Prenatal HFs start forming between 11 and 14 post-conception weeks (PCW)3, initiated by interactions between epidermal placodes (focal sites of epidermal layer thickening) and dermal condensates (aggregates of dermal fibroblasts), whereas sebaceous glands develop from around 16 PCW3. There is, however, a paucity of information about the precise cellular composition of human prenatal skin over these developmental periods and whether cells interact in functional microanatomical niches that support skin morphogenesis.
Prenatal skin interfaces with the amniotic fluid in a sterile environment4. However, immune cells such as macrophages seed the skin as early as 6 PCW and express a range of pro-inflammatory genes, although genes relating to antigen presentation (for example, major histocompatibility complex class II (MHCII)) are only upregulated after 11 PCW5. Decoupling of the expression of pro-inflammatory genes from MHCII genes before 11 PCW5 suggests that antigen presentation may not be a key function of human macrophages during early gestation. Together with evidence of their role in tissue homeostasis6,7 and healing in mouse models8,9, this raises the question of whether macrophages contribute to human early skin morphogenesis.
Our current study provides a comprehensive multi-omics cell atlas of 7–17 PCW human prenatal skin. We profiled human prenatal skin using single-cell RNA sequencing (scRNA-seq), spatial transcriptomics and multiplex RNA in situ hybridization to decode the dynamic cellular and molecular changes across gestation that regulate skin and HF morphogenesis. We leveraged adult healthy skin and HF datasets10,11 to compare and assess developmental-specific features that contribute to scarless skin healing and cues that guide de novo HF formation. We used a hair-bearing SkO model1 to validate the role of macrophages in prenatal skin vascular network formation.
Single-cell atlas of human prenatal skin
To characterize the role of distinct lineages and cell states during human prenatal skin development, we obtained single-cell suspensions of skin from 7 to 17 PCW, spanning the first and second trimesters (Fig. 1a). We performed fluorescence-activated cell sorting (FACS) to isolate live, single immune (CD45+) and non-immune (CD45–) populations and to enhance keratinocyte and endothelial cell capture before scRNA-seq profiling (Extended Data Fig. 1a and Supplementary Tables 1 and 2). Single-cell αβ T cell receptor (TCR) sequencing data were generated to accurately resolve T cell subsets. Spatial validation was carried out using multiplex RNA in situ hybridization (RNAscope), newly generated spatial transcriptomics (Visium) data from embryonic facial and abdominal skin, and published Visium data from embryonic limb from which only skin areas were analysed12 (Fig. 1a and Supplementary Table 2). In addition, we integrated new and published single-cell datasets of adult skin10 and of a hair-bearing SkO model derived from human embryonic stem (ES) cells and induced pluripotent stem (iPS) cells1 for comparative analysis (Fig. 1a and Supplementary Table 2). We also compared in vivo prenatal and organoid HFs with scRNA-seq data of adult HFs11. Our data can be interactively explored through our WebAtlas-based13 portal (https://developmental.cellatlas.io/fetal-skin). The analysis software for this study is archived at Zenodo (https://doi.org/10.5281/zenodo.8164271)14.

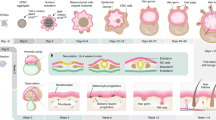
a, Experimental overview demonstrating the generation of scRNA-seq data from dissociated prenatal skin cells (n = 18, 7–17 PCW). Datasets of adult HF11, adult healthy skin10 and hair-bearing SkO1 were integrated for comparison. Spatial experiments were carried out using RNAscope, immunofluorescence and Visium analyses. Findings of the study were functionally validated using a SkO co-culture. b, Uniform manifold approximation and projection (UMAP) visualization of the prenatal skin dataset with broad annotation of cell states, as denoted by colour and number in the legend. c, Milo beeswarm plot showing the differential abundance of neighbourhoods in prenatal skin across gestation time, annotated by broad cell labels. Red and blue neighbourhoods are significantly enriched in earlier or later gestation, respectively. Colour intensity denotes degree of significance. d, Dotplot showing spatial microenvironments (MEs). Cell type to microenvironment coefficients are normalized by cell type sums, and cell type to microenvironment assignment is shown by colour. ME5, which shows co-locating macrophages and endothelial cells, is highlighted (grey). e, UMAP visualizations of the integrated prenatal skin, adult skin10 and SkO1 datasets, coloured by broad cell lineages. ASDC, Axl+Siglec6+ dendritic cells; DC, dendritic cells; HSC, haematopoietic stem cells; LC, Langerhans cells; LE, lymphatic endothelium; LTi, lymphoid tissue inducer cell; MEMP, megakaryocyte-erythroid-mast cell progenitor; NK cell, natural killer cells; pDC, plasmacytoid dendritic cells. The images in a were created using BioRender (https://biorender.com).
Our prenatal skin scRNA-seq dataset comprised 534,581 cells, of which 433,961 cells passed quality control (Extended Data Fig. 1b). Broad cell labels (epidermis, dermal stroma, immune and endothelium) and fine-grained annotations of cell states were assigned on the basis of differentially expressed genes (DEGs) (Fig. 1b, Extended Data Fig. 1c and Supplementary Table 3). Differential abundance analysis testing revealed how different cell populations varied across gestation. Among ectoderm-derived cells, neural cells and the periderm, which constitutes the first skin permeability barrier2, were enriched in early gestation, whereas suprabasal epidermal and HF cells were mainly observed in later gestation (Fig. 1c, Extended Data Fig. 1d and Supplementary Table 4). Mesoderm-derived cells, including skin fibroblasts and endothelial cells, and immune cells were present throughout gestation (Fig. 1c, Extended Data Fig. 1d and Supplementary Table 4). Innate immune cells, such as macrophages and innate lymphoid cells (ILCs), were present from early gestation, whereas B cells and T cells emerged later, accompanying thymus, bone marrow and spleen formation from around 10 PCW (Fig. 1c, Extended Data Fig. 1d and Supplementary Table 4). Some subsets of macrophages, ILCs and fibroblasts exhibited distinct gene expression profiles between early and late gestation, which suggested that functional maturation or dual waves of production occur during development (Fig. 1c and Extended Data Fig. 1d).
To locate cells identified from scRNA-seq data in situ, we performed Cell2location15 analysis of spatial transcriptomics data of facial and abdominal skin (10 PCW) and embryonic lower limb skin (6–8 PCW)12 (Supplementary Table 2). We assessed cell type co-location using non-negative matrix factorization (NMF) to computationally predict microenvironments in conjunction with correlation analyses. Co-location was indicated by a high proportion of two or more cell types sharing a microenvironment (Fig. 1d) and/or by a positive correlation coefficient between cell pairs (Extended Data Fig. 1e,f). NMF can predict significant cellular co-locations that are not readily evident in conventional histopathology analyses5,15. Our analysis predicted distinct microenvironments in prenatal skin comprising epidermal, dermal, vascular and neural cells, each including specific types of immune cells (Fig. 1d and Extended Data Fig. 1e,f). Macrophages co-located with endothelial and neural cells in ‘early and late neurovascular microenvironments’ (ME1 and ME5, respectively), whereas pre-dermal condensate (pre-Dc) cells co-located with dendritic and lymphoid cells based on correlation analyses (Fig. 1d and Extended Data Fig. 1e,f). These observations indicated that immune cells may occupy defined microanatomical niches where they have non-immune functions during early gestation.
We next integrated and compared human prenatal and adult skin data10 with the SkO model1. The aim was to determine the extent to which the SkO model recapitulates human skin differentiation at a molecular level and its potential utility to functionally assess the role of immune cells in skin morphogenesis (Fig. 1e and Extended Data Fig. 2a,b). Broadly, cell states were conserved among SkO, prenatal and adult skin, but SkO cell states matched prenatal skin more closely than adult skin across culture duration (Extended Data Fig. 2c,d and Supplementary Table 5). However, the tempo of differentiation varied across the distinct skin cell lineages. Even after 19 weeks of culture, fibroblasts, mural and Schwann cells had a low probability of correspondence to adult skin cell states (Extended Data Fig. 2d and Supplementary Table 5). By contrast, accelerated differentiation was observed in keratinocytes and melanocytes, with alignment to adult cell states seen as early as 4 weeks of SkO culture (Extended Data Fig. 2d and Supplementary Table 5). Notably, the SkO model recapitulated the different components of prenatal skin HF, interfollicular epidermis, neural cells and dermal fibroblasts, but immune cells were not represented and endothelial cells were markedly reduced.
Epidermal placode and matrix formation
The precise mechanisms of de novo HF formation in human embryonic development are largely inferred from mouse studies16. Human studies have primarily focused on morphological descriptions during development17 or cycling HFs in adult skin18. Our single-cell dataset captured the onset of HF formation, which enabled direct comparison between prenatal developing HFs and adult cycling HFs.
Prenatal skin up to 8 PCW consisted of a layer of epidermal cells overlying the dermal stroma, with the periderm seen sloughing from 11 PCW (Fig. 2a). At 14–15 PCW, budding of basal cells (hair placode and germ cells) and elongation of HFs (hair pegs) were observed (Fig. 2a). At 17 PCW, hair pegs were evident beneath a stratified epidermal layer (Fig. 2a).

a, Representative images of prenatal skin and HF morphogenesis (stained with haematoxylin and eosin). Scale bars, 200 µm. b, Average proportions of prenatal skin epidermal cell states. c, Large-area (top) and magnified peri-follicular (bottom left) and inter-follicular (bottom right) RNAscope images of prenatal skin (15 PCW) demonstrating the ORS (SLC26A7), matrix (SHH) and the Dp (NDP) with Treg cells (FOXP3) around HFs. Scale bars, 100 µm (top) or 20 µm (bottom). d,e, Inferred pseudotime trajectory of prenatal skin and SkO epidermal cells (d) and fibroblasts (e). UMAP overlaid with partition-based graph abstraction (PAGA) (default threshold), coloured by cell state, showing connectivities (dashed) and transitions (arrows). f, Spatial distribution of WNT2+ fibroblasts and HOXC5+ early fibroblasts. Predicted cell abundances shown as the sum of two-colour gradients per spot (left; scale bars, 100 µm) or averaged across all spots located at the same distance from the tissue border (right; mean (line) ± 2 s.e.m. (shaded area)). Dotted line indicates the tissue border. g, CellPhoneDB-predicted mesenchymal–epithelial interactions (early, immature basal; late, DPYSL2+ basal, POSTN+ basal, placode, matrix, ORS, CL, IRS and cuticle/cortex). The top ten interactions per prenatal skin cell pair are shown (top), and the same interactions are plotted for SkO (bottom). Colour scale represents mean expression values of ligand–receptor pairs. CellPhoneDB-computed significance used empirical shuffling and were adjusted for false discovery rate (FDR). h, RNAscope images showing ACKR3 and CXCL12 expression in early epithelium (SERPINB7+) and pre-Dc cells (PDGFD+) (top) and co-expression at the dermo–epidermal junction (arrows, bottom right). Scale bars, 20 µm. i, Alignment of SkO and prenatal skin pseudotime trajectories considering 15 equispaced pseudotime points. Left, heatmap shows the number of matching TFs (colour scale) for each pair of organoid–prenatal pseudotime points and average alignment path (white line) across all TFs along pseudotime (the diagonal path represents the best-matched pseudotime point–pairs, and vertical and horizontal paths indicate mismatches). Right, average alignment mapping visualized against cell type composition per pseudotime point. For details on statistics and reproducibility, see Methods.
Consistent with our histological observations, we identified HF cells from 14 PCW in our scRNA-seq data, which comprised placode, matrix (SHH+), outer root sheath (ORS) (SLC26A7+), companion layer (CL), inner root sheath (IRS) and cuticle and cortex cells (cuticle/cortex; part of the inner layers of the HF) (Fig. 2a–c, Extended Data Fig. 3a,b and Supplementary Table 6). In addition, we observed immature and mature interfollicular epidermal (IFE) cells. Immature IFE cells, including periderm, immature basal and immature suprabasal cells, were present from 7 PCW and decreased after 11 PCW, during the transition from embryonic to fetal skin (Fig. 2b). Mature basal (DPYSL2+) and suprabasal IFE cells increased after 11 PCW, whereas POSTN+ basal cells were present throughout gestation (Fig. 2b and Extended Data Fig. 3b). Sebaceous and apocrine gland cells, which mature after 16 PCW19, were not captured at these stages. Accordingly, sebocyte progenitors are present from day 133 of SkO differentiation1. Within the dermal compartment, we observed HF-specialized fibroblasts, the dermal condensate (Dc) and dermal papilla (Dp), from 12 PCW (Extended Data Fig. 3c).
We evaluated hair matrix cells, which arise from the epidermal placode, a prenatal-specific cell state absent in established adult HFs20 (Extended Data Fig. 3d,e). Compared with adult HFs, prenatal skin matrix cells had increased expression of genes involved in chemotaxis, such as CXCL14, a chemokine previously reported to recruit regulatory T (Treg) cells21, and in control of autoimmunity (CD24)22. This result highlighted the potential role of Treg cell accumulation and immune protection in the early stages of matrix differentiation (Extended Data Fig. 3f). Treg cells are known to localize around the HF in late second trimester (around 21 PCW) and in postnatal skin23,24. RNAscope (FOXP3+) and immunofluorescence staining (FOXP3+) showed that Treg cells were primarily located within and around HFs compared to interfollicular skin as early as 15 PCW (Fig. 2c and Extended Data Fig. 3g,h).
Inferred trajectory and pseudotime analysis of epidermal cells in the integrated prenatal skin and SkO data predicted the differentiation of POSTN+ basal cells into two paths: ORS/CL trajectory, comprising DPYSL2+ basal cells, ORS and CL; and IRS trajectory, involving placode, matrix, cuticle/cortex and IRS (Fig. 2d, Extended Data Fig. 4a and Supplementary Table 7). Along the ORS/CL trajectory, we identified new genes upregulated by DPYSL2+ basal cells, such as AGR2, and previously reported genes related to ORS differentiation (BARX2 and SOX9)25,26 (Extended Data Fig. 4b,c and Supplementary Table 7). AGR2 was downregulated along the IRS trajectory, whereas known matrix markers such as SHH and WNT10B25,26 were upregulated (Extended Data Fig. 4b,c and Supplementary Table 7). Loss of AGR2, which functions in the assembly of cysteine-rich receptors enriched in HFs, promotes cell migration27. Our findings suggest that increased cellular migration in POSTN+ basal cells may be involved in placode specification and dermal invagination.
HF mesenchymal differentiation
We delineated the dermal cell types involved in crosstalk with epidermal cells during HF development and captured the precursors of the human Dc (Extended Data Figs. 2b and 3c). In mice, transitional PDGFRA+FOXD1+SOX2low fibroblasts termed pre-Dc cells aggregate to form the Dc (FOXD1+SOX2+), which abuts the epithelial hair placode26,28. Using orthologous marker genes, we annotated pre-Dc cells and the Dc in human prenatal skin28,29. Following HF invagination, the Dc becomes encapsulated at its base as the Dp (NDP+, SOX2+)26,28 (Fig. 2c and Extended Data Fig. 3h).
To infer the origin of pre-Dc cells and the Dc and Dp, we performed trajectory and pseudotime analysis of integrated prenatal skin and SkO fibroblast clusters (Fig. 2e, Extended Data Fig. 4d and Supplementary Table 7). We excluded FRZB+ fibroblasts, which were primarily observed in one sample from the earliest gestation stage (7 PCW) (Extended Data Fig. 3c). Although rare in prenatal skin, FRZB-expressing fibroblasts were present in several other developing organs (Extended Data Fig. 4e). Inferred trajectory analysis predicted that HOXC5+ early fibroblasts (located in the upper dermis (Fig. 2f) and abundant before 11 PCW (Extended Data Fig. 3c)) differentiated along two paths: the first (hair fibroblast trajectory) formed hair-specialized fibroblasts (pre-Dc cells, the Dc and Dp) and the second (dermal fibroblast trajectory) formed WNT2+ fibroblasts and PEAR1+ fibroblasts (abundant after 11 PCW) (Fig. 2e, Extended Data Figs. 3c and 4d and Supplementary Table 7). Along the hair fibroblast pseudotime, genes involved in regulation of cell adhesion (ADAMST1), cell–cell contacts (CLDN11) and directed migration (CXCL12) were upregulated as pre-Dc cells migrated towards the epidermis, which indicated a process of collective migration30,31,32 (Extended Data Fig. 4f and Supplementary Table 7). Genes implicated in collagen fibril formation and cell adhesion (COL6A3, MFAP4 and PTK7) were expressed as the pre-Dc cells aggregated into the Dc (Extended Data Fig. 4f and Supplementary Table 7). Formation of the Dp was characterized by genes (RSPO3 and WNT5A) (Extended Data Fig. 4f and Supplementary Table 7) that coordinate the differentiation of adjacent hair matrix cells33,34.
We explored the mesenchymal–epithelial interactions that instruct early HF formation. Receptor–ligand analysis predicted interactions between CXCL12 expressed by pre-Dc cells (Extended Data Fig. 4g,h) with ACKR3 on epidermal basal cells (Fig. 2g and Supplementary Table 8). RNAscope analysis confirmed that these two genes co-located (Fig. 2h). This result suggests that CXCL12 probably interacts with ACKR3 to mediate the migration of pre-Dc cells31,35. Notably, lymphoid tissue inducer and ILC3 cells were also predicted to co-locate and interact with pre-Dc cells through ligand–receptor signals implicated in the regulation of cellular adhesion and migration (CXCL12–CXCR4 and CXCL12–DPP4)35,36 (Extended Data Figs. 1f and 5a and Supplementary Table 8), which suggested that innate immune cells may support pre-Dc cell migration during early HF development. Additional experiments are required to functionally validate these interactions in prenatal hair formation.
The Dc, for which formation is accompanied by invagination of the placode, expressed FAM3C and EFNB1, which were, respectively, predicted to interact with LAMP1 or CXADR and EPHB6 on the placode, and have been reported to promote cell migration and invasion37,38 (Fig. 2g, Extended Data Fig. 5b and Supplementary Table 8). Finally, RSPO3 from the Dp was predicted to interact with LGR4 and LGR6 (Fig. 2g, Extended Data Fig. 5b and Supplementary Table 8) in overlying matrix cells to contribute to the proliferation of HF epithelial cells33. Notably, the highlighted interactions were conserved between the mesenchymal and epithelial cells of the SkO model for corresponding stages during HF formation (Fig. 2g, Extended Data Fig. 5c and Supplementary Table 8). These results provide orthogonal validation of our findings and reinforce the utility of the SkO as an accurate model of prenatal skin development.
We further evaluated the differentiation trajectory alignment between prenatal skin and the SkO model using the Genes2Genes analysis framework39 to compare the expression of transcription factors (TFs) along the hair fibroblast trajectory. Overall, we observed a high number of matching TFs across pseudotime, which indicated that there are similar activated gene regulatory programs between prenatal skin and SkO during HF fibroblast differentiation (Fig. 2i and Supplementary Table 9). TFs that were mismatched across pseudotime or drove misalignment in early and late pseudotime (for example, HOXA7 and BARX1) were largely attributable to the different origins of dermal cells between prenatal skin (trunk and limb) and SkO (neural crest differentiation)40 (Extended Data Fig. 5d,e and Supplementary Table 9).
We also assessed the expression profiles of genes previously reported in mouse HF formation. Similar signalling pathways were upregulated, including WNT and EDA for hair placode specification, bone morphogenetic protein (BMP) and noggin to inhibit hair formation in IFE cells, and PDGFA and TGFβ signalling for HF down growth26 (Extended Data Fig. 5f). Additionally, similar to fibroblast differentiation in mouse skin, pre-Dcs, the Dc and Dp and dermal fibroblasts in human prenatal skin also originated from a common fibroblast progenitor (HOXC5+ early fibroblast) (Fig. 2e and Extended Data Fig. 4d). However, cross-species data integration of human and mouse HF41 showed that human pre-Dc cells and the Dc aligned not only to their counterparts in mouse skin but also to fibroblasts in earlier stages of differentiation (Extended Data Fig. 5g–i and Supplementary Tables 10 and 11). This result suggests that for corresponding cell types, HF fibroblasts are in a more differentiated state in mouse compared to human prenatal skin. Additionally, dermal fibroblast differentiation into histologically defined subsets (papillary and reticular) has been reported to occur early in mice (about embryonic day 12.5)42. Our human prenatal skin fibroblasts did not significantly express papillary fibroblast markers (for example, COL13A1)43 (Extended Data Fig. 5j), which suggested that the distinction between papillary and reticular fibroblasts emerges after 17 PCW. These distinctions between human and mouse skin may be attributed to organismal differences in gestation lengths and tempo of differentiation. Cellular differentiation occurs at a quicker pace in mice44, whereas the longer gestation period in humans permits more advanced maturation to take place in utero.
Genetic hair and skin disorders
Having mapped the differentiation of prenatal skin HFs, we leveraged this information to assess the extent to which genetic hair diseases have their roots in utero. Genes harbouring mutations known to cause reduced hair growth (hypotrichosis) or abnormally shaped hair (for example, pili torti) (Supplementary Table 12) were expressed along the ORS/CL trajectory, IRS trajectory and hair fibroblast trajectory pseudotimes (Extended Data Figs. 4f and 6a,b) and in prenatal HF cell states (Extended Data Fig. 6c). This finding suggested that these disorders result from dysfunctional HF development.
Genes causing epidermolysis bullosa (EB), an inherited blistering skin disorder characterized by skin fragility secondary to structural defects in the epidermis and adjacent dermoepidermal junction45, were highly expressed in prenatal epidermal cells and at the dermoepidermal junction (Extended Data Fig. 6d,e). Gene therapy studies for dystrophic EB have identified fibroblasts expressing COL7A1 as a promising therapeutic strategy46. We observed COL7A1 expression across several fibroblast subsets in prenatal skin and SkOs (Extended Data Fig. 6d), which lends support to the gene therapy approaches. The expression of genes implicated in congenital ichthyoses, a group of disorders resulting from abnormal epidermal differentiation47, were primarily confined to keratinocytes (Extended Data Fig. 6f).
Notably, we observed similar gene expression patterns across prenatal skin and SkO for the above described genetic hair and skin disorders (Extended Data Fig. 6c,d,f), which supported the value of the SkO as a model to study congenital diseases. Although we found that expression of genes implicated in these disorders are confined to structural cells, disease manifestations are often associated with immune infiltration, which implicates skin–immune crosstalk during pathogenesis48,49.
Scarless healing and potential macrophage contribution
Prenatal human skin is able to heal without scarring but loses this capacity after 24 PCW50. Scars result from aggregation of collagen produced by dermal fibroblasts and failure of the overlying epidermis to completely regenerate51. To identify the cellular and molecular mechanisms that may endow early prenatal skin with scarless healing properties, we investigated the temporal changes in composition and transcriptional profile of the dermal fibroblast subsets (Extended Data Figs. 3c and 7a). We first compared prenatal skin dermal fibroblasts with healthy adult skin fibroblasts10. All adult fibroblast subsets expressed high levels of inflammatory cytokines and receptors (for example, IL6 and IL1RA) and genes involved in antigen presentation (for example, HLA-A), innate immune and inflammatory responses (for example, CD55 and PTGES) and cellular senescence (CDKN1A) (Fig. 3a and Supplementary Tables 13–15). By contrast, prenatal skin fibroblasts had upregulated genes involved in immune suppression (CD200), regulation of inflammation (for example, RAMP2) and tissue regeneration (MDK) (Fig. 3a and Supplementary Tables 13–15).

a, Dot plot showing variance-scaled, mean expression (dot colour) and per cent of expressing cells (dot size) of DEGs by prenatal (anti-inflammatory and immune suppression) and adult skin fibroblasts (pro-inflammatory and immune activation)10. b, Matrix plot showing variance-scaled, mean expression (colour) of Milo-generated DEGs (grouped by function) by gestational age (grouped PCW) in WNT2+ fibroblasts. Labels in bold indicate genes selectively referenced in text. c, Bar plot showing cell type co-location, indicated by positive Pearson correlation coefficients calculated between per-spot normalized cell type abundances, for selected cell type pairs (macrophages and WNT2+ fibroblasts). Pearson correlation coefficients were calculated across all skin-covered spots of Visium samples; each sample is shown by an individual bar. d, Spatial distribution of LYVE1+ macrophages and WNT2+ fibroblasts (top two rows) and of TML macrophages and WNT2+ fibroblasts (bottom two rows) (representative 8 PCW samples). Predicted cell abundances shown as the sum of two-colour gradients per spot (left; scale bars, 100 µm) or averaged across all spots located at the same distance from tissue border (right; mean (line) ± 2 s.e.m. (shaded area)). Dotted line indicates tissue border. e, Immunofluorescence (representative 10 PCW prenatal skin cryosections) showing LYVE1+ macrophage (CD45+LYVE1+) co-locating with fibroblasts (VIM+). Scale bars, 50 µm. f, Dot plot showing variance-scaled, mean expression (dot colour) and per cent of expressing cells (dot size) of genes (grouped by function) upregulated by TML macrophages in prenatal and adult skin macrophages. Labels in bold indicate genes selectively referenced in text. g, Bar plot showing cell type co-location, indicated by positive Pearson correlation coefficients calculated between per-spot normalized cell type abundances, for selected cell type pairs (TML macrophage and neural cells). Pearson correlation coefficients were calculated across all skin spots of Visium samples; each sample is shown by an individual bar. For details on statistics and reproducibility, see Methods.
The adult fibroblast gene expression profile was increased in WNT2+ and PEAR1+ prenatal fibroblasts, which were abundant in later gestation (Fig. 3a and Extended Data Figs. 3c and 7b). Genes associated with a pro-inflammatory fibroblast phenotype (APOE, IGFBP7 and ITM2A)43,52 were also upregulated during the transition from HOXC5+ fibroblasts into PEAR1+ fibroblasts (Extended Data Fig. 7c). In addition to transcriptomics differences between fibroblast subsets enriched in early versus late gestation, we observed differences within the WNT2+ fibroblast population across gestation time (Extended Data Fig. 1d). Late gestation WNT2+ fibroblasts had upregulated genes related to extracellular matrix and collagen deposition (for example, COL1A1), whereas early WNT2+ fibroblasts had DEGs involved in cellular growth and differentiation (for example, SFRP1) (Fig. 3b, Extended Data Fig. 7d and Supplementary Tables 16–18). Notably, WNT2+ and PEAR1+ prenatal fibroblasts expressed several genes involved in cellular senescence (CDKN1A), cytokine pathways (for example, IL1R1) and collagen deposition (for example, POSTN) (Fig. 3a,b), which are highly expressed in pathogenic fibroblasts of fibrotic skin disorders53. These results provide further support for our finding of progressive acquisition of scar-promoting genes in later gestation, consistent with the clinical observation of scarring in third trimester skin54.
The role of macrophages in promoting wound healing has been described in postnatal mouse skin and in adult human skin55. In prenatal skin, macrophage subsets (Extended Data Fig. 7e,f) were predicted to co-locate with fibroblasts, neural cells and vascular cells in distinct tissue microenvironments in early gestation (Fig. 1d). Specifically, LYVE1+ macrophages co-located with WNT2+ fibroblasts (Fig. 3c–e) and were predicted to interact through platelet-derived growth factors (PDGFs) and corresponding receptors (PDGFRα and PDGFRβ) expressed on fibroblasts (Extended Data Fig. 7g and Supplementary Table 8). Interactions between macrophages and fibroblasts maintain tissue homeostasis in diverse organs such as spleen, peritoneum and heart56. Our identification of additional growth factor interactions (IGF1–IGF1R and GRN–EGFR) (Extended Data Fig. 7g and Supplementary Table 8) suggests that LYVE1+ macrophages play a part in the maintenance of prenatal skin dermal fibroblasts.
We recently identified yolk-sac derived TREM2+ macrophages that share an expression profile (P2RY12, CX3CR1 and OLFML3) with microglia-like macrophages from other developing organs, such as the brain, prenatal skin and gonads57,58 (Extended Data Fig. 7e,f). Prenatal skin TREM2+ microglia-like (TML) macrophages were highly correlated with embryonic brain microglia57 (Extended Data Fig. 8a,b) and co-expressed immunomodulatory genes, including immune-inhibitory receptors (for example, CX3CR1) and regulators of IL-6 production (for example, SYT11)59 (Fig. 3f, Extended Data Fig. 8c and Supplementary Tables 19–23). Downregulation of inflammation and IL-6 confers anti-fibrogenic properties in mouse skin transplants and in fetal wounds60,61. TML macrophages were predicted to co-locate with WNT2+ fibroblasts in early prenatal skin (6–8 PCW) (Fig. 3c,d) and WNT2+ fibroblasts had downregulated IL6 expression compared with adult fibroblasts (Fig. 3a). This led us to infer a potential contribution of macrophages in scarless healing in prenatal skin. Additionally, GAS6, expressed by TML macrophages and LYVE1+ macrophages, was predicted to interact with AXL receptors on WNT2+ fibroblasts (Extended Data Fig. 7g and Supplementary Table 8), and these interactions can induce immunosuppression and tissue repair62,63.
We further compared prenatal skin fibroblasts and macrophages to their counterparts in reindeer skin from antlers, which heal without scarring, and in back skin, which scars64. Early-gestation human skin fibroblasts had a higher probability of correspondence to pro-regenerative reindeer fibroblasts, whereas in later gestation, the probability of matching to pro-fibrotic fibroblasts was higher (Extended Data Fig. 8d and Supplementary Table 19). Accordingly, several pro-regenerative genes (for example, CRABP1 and MDK) were downregulated in late gestation prenatal skin (Extended Data Fig. 8e and Supplementary Table 20). Notably, prenatal skin macrophages resembled ‘early macrophages’ that are enriched in reindeer antler skin but not macrophages in back skin (Extended Data Fig. 8f and Supplementary Table 21). Using a scratch assay of SkO-derived fibroblasts cultured with or without iPS cell-derived macrophages, we demonstrated that scratch wound width closure was improved when fibroblasts were co-cultured with macrophages over 72 h (Extended Data Fig. 8g).
Collectively, our findings suggest that prenatal skin fibroblasts in early gestation downregulate genes involved in extracellular matrix formation, collagen deposition and inflammation, which may favour tissue regeneration over scarring. Based on our data and previous studies, we also posit a potential role for early skin macrophages in conferring the distinct property of scarless healing in early prenatal skin. However, further studies are required to fully elucidate the interactions between macrophages and fibroblasts in human prenatal skin and to conclusively establish their role in scarless healing.
Macrophages in cutaneous neural differentiation
TML macrophages were also predicted to co-locate with Schwann cells in prenatal skin (‘early neurovascular microenvironment’, ME1) (Figs. 1d and 3g) and expressed genes related to cell migration and neural development (Fig. 3f, Extended Data Fig. 8c and Supplementary Tables 22 and 23), which mirrored the functions of brain microglia and peripheral nerve-associated macrophages in mouse skin65. TML macrophages were predicted to interact with Schwann cells, contributing to synapse formation and axon guidance (VEGFA–NRP1, VEGFA–NRP2, SEMA3C–NRP2 and SEMA3E–PLXND1)66 (Extended Data Fig. 8h and Supplementary Table 8). These findings suggest that prenatal skin macrophages may support the establishment of the skin peripheral nervous system during early gestation, as previously reported in mouse skin65.
Macrophages support prenatal skin angiogenesis
Macrophages have been implicated in angiogenesis during prenatal organ development and in the postnatal setting such as cancer-related angiogenesis67,68. Furthermore, macrophages expressing pro-angiogenic genes have been observed in diverse tissues during human development69. Visium deconvolution analysis predicted co-location of prenatal skin macrophages with endothelial cells (‘early and late neurovascular microenvironments’, ME1 and ME5) (Fig. 1d and Extended Data Fig. 1e). Gene ontology analysis showed that the four macrophage subsets (LYVE1+, MHCII+, TML and iron-recycling) expressed gene programs that drive angiogenesis (Supplementary Tables 23–26). Gene module expression profiles suggested that sprouting angiogenesis (growth of new vessels) was promoted by LYVE1+ and TML macrophages, blood vessel morphogenesis by LYVE1+ macrophages and endothelial cell chemotaxis by iron-recycling macrophages (Extended Data Fig. 8i and Supplementary Table 27). Consistent with this finding, multiplex RNAscope and immunofluorescence staining showed LYVE1+ and TML macrophages in close proximity to endothelial cells (Fig. 4a and Supplementary Video 1). Predicted ligand–receptor interactions were consistent with reciprocal communication between macrophages and endothelial cells to support angiogenesis, chemotaxis and cell migration (for example, CXCL8–ACKR1 and CCL8–ACKR1)10,70 (Extended Data Fig. 9a and Supplementary Table 28).

a, Close proximity of endothelium with macrophages shown in prenatal skin. Left, RNAscope images of cryosections with endothelium (CDH5), TML macrophage (P2RY12) and macrophages (CD68). Scale bars, 100 µm. Centre, immunofluorescence images of cryosections with LYVE1+ macrophages (LYVE1+CD45+) and endothelial cells (CD31+). Scale bars, 10 µm. Right, three-dimensional rendering of co-localized areas (magenta) of endothelial cells (CD31+) and LYVE1+ macrophages (LYVE1+) from whole-mount immunostaining. Scale bars, 80 µm (top) or 5 µm (bottom). b, UMAP visualization of endothelial cell states in prenatal skin and SkO. c, Average proportions of prenatal skin endothelial cell states (bar colours) across gestation. d, Inferred pseudotime trajectory of prenatal skin endothelial cell states differentiating along the arteriolar trajectory and the venular trajectory: UMAP overlaid with PAGA (default threshold), coloured by cell state showing connectivities (dashed) and transitions (arrows). e, Schematic showing differences between prenatal skin and SkOs in pro-angiogenic and anti-angiogenic factors and corresponding receptors. f, Representative whole-mount immunofluorescence images of SkO without (top) and with (bottom) macrophage co-culture at day 47 showing macrophages (CD45), endothelium (CD31) and DAPI nuclei stain (blue). Scale bars, 100 µm. g, Quantification of endothelial cell coverage in SkOs (day 47) cultured with (n = 5, magenta) and without macrophages (n = 5, grey) from 2 batches of differentiation of SkOs and macrophages. Data are mean ± s.d. and statistics generated from unpaired t-test. h, z projection immunofluorescence of cryosections of day 47 SkO co-cultured with macrophages (day 35 of co-culture) demonstrating close interactions of macrophages (CD45, in magenta) and endothelium (CD31, in green). Scale bar, 50 µm. ECs, endothelial cells; LE, lymphatic endothelium. For details on statistics and reproducibility, see Methods. The images in e were created using BioRender (https://biorender.com).
Our data suggested that macrophages contribute to prenatal skin angiogenesis. Consistent with this hypothesis, we observed fewer and less heterogeneous endothelial cells in the immunodeficient SkOs compared to prenatal skin, despite the formation of well-developed HFs, epidermis and neural cells (Fig. 4b,c, Extended Data Fig. 9b and Supplementary Table 29). Inferred trajectory analysis showed that early endothelial cells in prenatal skin differentiated into either an arteriolar pathway (capillaries, capillary arterioles and arterioles) or venular pathway (postcapillary venules and venules), with expression of characteristic genes (for example, GJA5 for arteriolar and PLVAP for venular) (Fig. 4d and Extended Data Fig. 10a–c). Unlike SkO capillary arteriole cells, prenatal skin capillary arteriole cells could further differentiate into arterioles (Fig. 4d and Extended Data Fig. 10a). Additional comparison with a human ES cell-derived and iPS cell-derived blood vessel organoid71, which also lacked immune cells, further demonstrated the limited vasculature differentiation of this mesoderm-geared blood vessel organoid model (Extended Data Fig. 10d). This result confirms that immune cells are required to fully recapitulate in vivo endothelial cell development.
We next investigated additional mechanisms for failed expansion and differentiation of SkO endothelial cells. Expression of genes and gene modules related to blood flow and hypoxia were lower in SkO than in prenatal skin (Extended Data Fig. 10e,f and Supplementary Table 29). However, sprouting angiogenesis potential, assessed by scoring the ‘tip’ cell state, was increased in both SkO capillary arteriole cells and prenatal skin arteriole, capillary arteriole and capillary cells (Extended Data Fig. 10g,h). This suggests that despite strong expression of the sprouting angiogenesis gene signature, SkO capillary arteriole cells are unable to guide stalk cells for new blood vessel formation.
Anti-angiogenic genes (for example, WNT5A) and corresponding receptors were highly expressed in SkO, whereas pro-angiogenic genes (for example, CXCL8) were upregulated in prenatal skin and primarily expressed by macrophages (Extended Data Fig. 11a and Supplementary Tables 30–34). Although expression of vascular endothelial growth factors (VEGF), VEGFA and VEGFB were increased in SkO cells, their receptors (KDR and FLT1) on SkO capillary arterioles were downregulated compared to prenatal skin (Extended Data Fig. 11b,c). These receptors are known downstream targets of GATA2, which has a key role in angiogenesis during development and regulates VEGF-induced endothelial cell migration and sprouting in vitro72. Regulon analysis showed that GATA2 and related regulons (for example, NFATC1) were downregulated in SkO capillary arterioles (Extended Data Fig. 11d,e). Several target genes of GATA2 and NFATC1 (for example, VWF), which were expressed across the venular trajectory pseudotime and are involved in endothelial cell differentiation73, were downregulated in the SkO capillary arterioles compared to prenatal skin (Extended Data Fig. 11c,f). An orthogonal approach (NicheNet74) identified macrophage-expressed VEGFA as one of the top upstream ligands that regulate differences in GATA2 expression between prenatal skin and SkO endothelial cells (Extended Data Fig. 11g and Supplementary Tables 35–37). These findings suggest that high VEGF production in the SkO cannot compensate for missing macrophage-related factors that drive GATA2 activity and downstream VEGF receptor expression (Fig. 4e).
We next introduced autologous iPS cell-derived macrophages (Extended Data Fig. 11h) in the early stages of SkO differentiation and assessed the endothelial network on day 35 of co-culture. Macrophages co-localized with blood vessels even after 5 weeks of culture (Fig. 4f–h). A more elaborate and organized vascular network was seen in SkOs co-cultured with macrophages compared with control SkOs without macrophages (Fig. 4f,g). Control SkOs displayed a mesh-like aggregation of endothelial cells, quantified as a higher density of endothelial cell coverage of SkO volume, which was absent in SkOs co-cultured with macrophages (Fig. 4f,g). This disorganized vascular mesh may have prevented the isolation of endothelial cells for scRNA-seq analysis (Fig. 4b). We also observed a visibly more refined network and a trend towards reduced endothelial density in a two-dimensional angiogenesis assay of iPS cell-derived endothelial cells cultured with and without macrophages over 72 h (Extended Data Fig. 11i). Collectively, our findings demonstrate that interactions between macrophages and endothelial cells are required to support angiogenesis through blood vessel remodelling.
Discussion
In this study, we characterized the dynamic composition of human prenatal skin during the early stages of de novo HF formation and highlighted the crucial skin immune and non-immune crosstalk that contributes to skin morphogenesis, results that are in line with emerging evidence in animal and human studies69,75. Our atlas indicated that macrophages contribute to scarless skin repair, fibroblast homeostasis and neurovascular development. This is in part contributed to by yolk-sac derived TML macrophages, which suggests that these cells have broader functions outside the central nervous system in early gestation. The presence of TML macrophages has previously been identified in several prenatal organs5,58,69.
Successful co-culture with immune cells has been demonstrated in some organoid systems76,77. We identified a crucial role of macrophages in vascular network remodelling after adding macrophages to hair-bearing iPS cell-derived SkOs. Not only is this important for our understanding of the diverse cellular interactions that mediate morphogenesis but it also has practical implications for in vitro models, which commonly fail to vascularize78. Our study provides further insights into human HF formation and the origin of the CL, which seems to develop along the same trajectory as the ORS. These findings are consistent with recent results from mouse studies26,79, which showed that CL development occurs before hair matrix formation79 and there is greater transcriptional similarity of CL cells to ORS cells25. Although we note similarities between human and mouse in the signalling pathways co-ordinating HF formation, our study reveals key cross-species distinctions in the differentiation tempo of HF mesenchymal cells. Future studies are required to fully delineate the features that distinguish human skin development.
A combination of fibroblast and macrophage-associated molecular features potentially contribute to the ability of prenatal skin to heal without scarring, including the presence of fibroblast progenitors, a downregulated immune milieu and reduced collagen expression. However, we found progressive ‘ageing’ and acquisition of the adult ‘pro-inflammatory’ phenotype as early as 9 PCW, which could be targeted in fibroblasts to guide postnatal scarless healing. Future studies that align human fibroblast subsets across the lifespan are required to investigate the dynamics of scarless healing and the roles of mechanical forces, microbiota and environmental exposure on fibroblast functions.
Our prenatal human skin atlas represents a valuable resource to explore genes that cause congenital hair and skin disorders and is freely accessible from our web portal (https://developmental.cellatlas.io/fetal-skin). We found that implicated genes are indeed expressed during prenatal skin development and HF differentiation, thereby supporting an in utero origin for these disorders. Our systematic prenatal skin–SkO comparison provides a blueprint to guide more faithful in vitro SkO generation, which can facilitate future studies of interactions with the microbiota, the pathogenesis of congenital skin disorders, and hair and skin engineering for therapeutic applications, including hair regeneration and skin transplant.
Methods
Tissue acquisition and processing
Human developmental tissue samples used for this study were obtained from the MRC– Wellcome Trust-funded Human Developmental Biology Resource (http://www.hdbr.org) with approval from the Newcastle and North Tyneside NHS Health Authority Joint Ethics Committee (08/H0906/21+5) and East of England–Cambridge Central Research Ethics Committee (NHS REC 96/085). Prenatal skin for immunofluorescence imaging of proteins causing congenital skin disorders were obtained with approval from the Guy’s and St Thomas’ Hospital Trust Ethics Committee. For samples used for 3D rendering in the Koehler Laboratory, fetal tissue specimens were obtained from the Birth Defects Research Laboratory at the University of Washington (UW) with approval from the UW institutional review board Committee, and the study was performed in accordance with ethical and legal guidelines of the Boston Children’s Hospital institutional review board. All samples were collected following either elective termination of pregnancy or miscarriages, with informed written consent, following all relevant rules and regulations.
Tissues were processed into single-cell suspensions immediately after receipt for single-cell transcriptomic profiling. Tissue was first transferred to a sterile 10 mm2 tissue culture dish and cut in <1 mm3 segments using a scalpel. It was then digested with type IV collagenase (final concentration of 1.6 mg ml–1; Worthington) in RPMI (Sigma-Aldrich) supplemented with 10% heat-inactivated FBS (Gibco) at 37 °C for 30 min with intermittent agitation. Digested tissue was then passed through a 100 μm cell strainer. For 2 samples (F220, F221), 500 µl of 0.25% trypsin (Sigma-Aldrich) was further added to any remaining unfiltered tissue and incubated at room temperature for 5 min. Cells were collected by centrifugation (500g for 5 min at 4 °C). Cells were treated with 1× RBC lysis buffer (eBioscience) for 5 min at room temperature and washed once with flow buffer (PBS containing 5% (v/v) FBS and 2 mM EDTA) before cell counting and antibody staining. Single-cell suspensions were generated from skin of 18 donors with ages spanning from 7 PCW to 17 PCW.
scRNA-seq experiment
Dissociated cells were stained with anti-CD45 antibody (1:20, PE, clone HI30, BD Biosciences (samples F220 and F221) or 1:33, BUV395, clone HI30, BD Biosciences (other sorted samples)) on ice in the dark for 30 min, except for one sample (F217) for which no cell sorting was performed. To improve capture of less abundant cell populations from the CD45– fraction, such as keratinocytes and endothelial cells, additional staining was carried out to separate them from the abundant CD34+ stromal cells in a subset of samples. For samples F220 and F221, anti-CD34 (1:25, APC/Cy7, clone 581, BioLegend) antibodies was used; for samples F69 and F71, additional staining included anti-CD34 (1:25, APC/Cy7, clone: 581, BioLegend) and anti-CD14 antibodies (1:33, PE-CF594, clone MφP9, BD Biosciences). Immediately before sorting, cells were passed through a 35 µm filter (Falcon), and DAPI (Sigma-Aldrich) was added at a final concentration of 3 μM. Sorting by flow cytometry was performed with a BD FACSAria Fusion flow cytometer. The CD45+ fraction was sorted from the DAPI–CD45+ gate, and the CD45– fraction was sorted from the DAPI–CD45– gate. CD45 gating was contiguous so that no live cells were lost in sorting. Live CD45+ and CD45– cells were sorted into separate chilled FACS tubes coated with FBS. For samples F220 and F221, CD34+ and CD34– fractions were sorted from CD45–CD34+ and CD45–CD34– gates, respectively. For samples F69 and F71, in addition to the live CD45+ and CD45– cells, we isolated all cells from the CD45– fraction that were not within the CD34+CD14– gate and collected them into a separate chilled FACS tubes coated with FBS (Extended Data Fig. 1a).
FACS sorted cell suspensions were counted and loaded onto a 10x Genomics Chromium Controller to achieve a maximum yield of 10,000 cells per reaction. Either Chromium single-cell 3′ reagent kits (v.2) or Chromium single-cell V(D)J kits from 10x Genomics were used. Cells were loaded onto each channel of the Chromium chip following the manufacturer’s instructions before droplet encapsulation on the Chromium Controller. Gene expression and TCR libraries were generated according to the manufacturer’s instructions. The gene expression libraries were sequenced to achieve a minimum target depth of 20,000 reads per cell and the TCR libraries were sequenced to achieve a minimum target depth of 5,000 reads per cell using Illumina sequencing instruments.
Statistics and reproducibility
Images of haematoxylin and eosin-stained skin sections (Fig. 2a) were taken from 13 independent samples from the following gestational ages: 6 PCW (n = 1 and 3 sections), 8 PCW (n = 3), 11 PCW (n = 2), 14 PCW (n = 1 and 2 sections), 15 PCW (n = 4) and 17 PCW (n = 2).
Image analysis of multiplex RNAscope and immunofluorescence staining was performed on independent biological and/or technical replicates for each experiment: n = 5 biological replicates for RNAscope slides with FOXP3, SHH, SLC26A7 and NDP probes (Fig. 2c); n = 1 biological replicate with 4 technical replicates for RNAscope slides with ACKR3, CXCL12, PDGFD and SERPINB7 probes (Fig. 2h); n = 3 biological replicates with 2 technical replicates for immunofluorescence slides with anti-FOXP3, anti-SOX2 and anti-KRT14 (Extended Data Fig. 3h); n = 1 biological replicate with n = 2 technical replicates for immunofluorescence slides with anti-LYVE1, anti-CD45 and anti-VIM (Fig. 3e); n = 3 biological replicates for RNAscope slides with CDH5, CD68, P2RY12 and ELAVL3 probes (Fig. 4a); n = 1 biological replicate with 4 technical replicates for immunofluorescence slides with anti-CD45, anti-LYVE1 and anti-CD31 (Fig. 4a); n = 15 biological replicates with a minimum of n = 2 technical replicates for prenatal skin immunofluorescence slides with each of the following antibodies: anti-KRT1, anti-KRT14, anti-plectin, anti-BP180, anti-laminin-332 and anti-type VII collagen (Extended Data Fig. 6e); n = 3 biological replicates for prenatal skin whole-mount immunofluorescence with anti-CD31 and anti-LYVE1 (Fig. 4a); whole-mount immunostaining of SkO co-culture with and without macrophages was performed on n = 5 SkOs without macrophages and n = 5 SkOs with macrophages (Fig. 4f); immunostaining of cryosections of SkOs co-cultured with macrophages was performed on n = 2 SkOs (Fig. 4h).
Visium spatial transcriptomic data were generated from n = 4 biological samples from 3 different sites, with 2 or 3 technical replicates each (Figs. 2f and 3d).
Scratch wound assays were performed on SkO-derived fibroblasts: n = 3 and each experiment included technical replicates in 3–6 wells (Extended Data Fig. 8g). Data represented as the mean ± s.d. and statistics generated with two-way analysis of variance (ANOVA) with Tukey’s multiple comparisons test. Endothelial cells and macrophages were generated across two independent differentiation batches. Endothelial cell and macrophage co-culture for angiogenesis assays were performed on n = 6 wells (Extended Data Fig. 11i). Data represented as the mean ± s.d. and statistics generated with an unpaired t-test.
Human iPS and ES cell line information
The iPS cell line Kolf2.1S was obtained from the HipSci Initiative under a material transfer agreement. This line was not independently authenticated. Details about the generation and characterization of the line at the time of derivation is available from the HipSci website (https://www.hipsci.org/#/lines/HPSI0114i-kolf_2).
The WTC-mEGFP-DSP-cl65 iPS cell line and the WA25 ES cell lines were obtained under a material transfer agreement with the Coriell or WiCell Institute. These lines were determined to have a normal karyotype before SkO differentiation.
All cell lines tested negative for mycoplasma before experiments.
Scratch wound assay of fibroblasts in co-culture with macrophages
Fibroblasts were isolated from Kolf2.1S-derived SkOs (n = 10) at day 76. In brief, SkOs were washed with dPBS then incubated with dispase and a ROCK inhibitor for 40 min at 37 °C. The epidermis and dermis layers of the SkO were separated using forceps, and the dermis was transferred to collagenase for 40 min at 37 °C. Collagenase was neutralized with fibroblast medium, and the single-cell suspension was filtered through a 40 µm cell strainer. After centrifugation at 180g for 3 min, the fibroblasts were resuspended and seeded in fibroblast medium then cultivated as primary fibroblasts. Macrophages were differentiated from Kolf2.1S iPS cells as previously described80.
For the scratch assay, fibroblasts and macrophages were seeded in 48-well plates at 5:1 ratio then incubated for 24 h at 37 °C. The next day, the scratch was generated using a p1000 tip down the centre of each well. The assay was imaged using Incucyte S3, whole well module and analysed using the ImageJ Wound_healing_size_tool_updated macro81. Two-way ANOVA was carried out to assess statistics in GraphPad. Scratch assays performed were n = 3 independent experiments with 3–6 replicates per experiment.
Visium spatial data generation
Prenatal facial (n = 1, replicate = 2) and abdominal skin (n = 1, replicate = 2) samples from a single donor at 10 PCW were embedded in optimal cutting temperature (OCT) medium and flash-frozen in isopentane cooled with dry ice. Cryosections (10 µm) from the OCT blocks were cut onto 10x Genomics Visium slides. Sections were stained with haematoxylin and eosin and imaged at ×20 magnification on a Hamamatsu Nanozoomer. These sections were then processed according to the 10x Genomics Visium protocol, using a permeabilization time of 12 min found through a previous tissue optimization step. Dual-indexed libraries were prepared as per the manufacturer’s protocol, pooled at 2.8 nM and sequenced in 8 samples per Illumina Novaseq S4 flow cell with the following run parameters: read 1: 28 cycles; i7 index: 10 cycles; i5 index: 10 cycles; read 2: 90 cycles.
Endothelial cell and SkO co-culture with macrophages
Endothelial cell culture co-culture and image acquisition
Endothelial cells were derived from Kolf2.1S iPS cells cultured on Matrigel-coated plates in mTeSR1 medium with ROCK inhibitor at 4.5 × 104 cells per cm2. iPS cells were differentiated through lateral mesoderm into CD144+ endothelial cells as previously described82. Macrophages and SkOs were also derived from Kolf2.1S iPS cells according to previously published methods1,80. The angiogenesis assay was carried out by culturing iPS cell-derived endothelial cells and macrophages separately or in co-culture in 15-well 3D chambered µ-slide (ibidi, 81506). This was done using a three-layered sandwich method, whereby layer one was 10 µl Matrigel (Corning, 354230), layer two was supplemented StemPro medium (Gibco, 10639011) + 10% Matrigel with and without the endothelial cells and layer three was supplemented StemPro medium with and without macrophages. The endothelial cells were left to settle for 4 h at 37 °C before addition of macrophages. The assay was imaged 2 h after initial culture and then every 24 h for 3 days using an EVOS 7000 microscope, and images were analysed using Fiji distribution of the ImageJ software (v.2.14.0)83. Before co-culture, iPS cell-derived macrophages were phenotyped using flow cytometry (Extended Data Fig. 11h). Macrophages were collected using TrypLE (Gibco) at 37 °C, 5% CO2 for 5 min, and cells were collected by centrifugation (300g for 6 min). Cells were washed once with cell staining buffer (BioLegend) before cell counting and antibody staining. Nonspecific bindings were blocked using Human TruStain FcX (Fc receptor blocking solution, BioLegend) for 10 min on ice and then stained using a Fixable Blue Dead Cell Stain kit for 10 min on ice (1:500 in PBS, ThermoFisher). Cells were washed twice with cell staining buffer. Single-staining was performed on cells with anti-CD206 antibody (1:200, PE, clone 19.2, ThermoFisher), anti-CD16 antibody (1:50, PE-Cyanine7, clone eBioCB16, ThermoFisher), anti-CD14 antibody (1:100, PerCP-Cyanine5.5, clone 61D3, ThermoFisher), anti-CD1c antibody (1:25, Pacific Blue, clone L161, BioLegend), anti-CD45 (1:300, BV480, clone HI30, BD Biosciences) and anti-human Lineage Cocktail (1:100, CD3, CD19, CD20, CD56, clones UCTH1, HIB19, 2H7, 5.1H11, BioLegend) on ice in the dark for 30 min. Before acquiring on the analyzer, cells were washed once in cell strained buffer and passed through a 35 µm filter (Falcon). Acquisition by flow cytometry was performed using a Cytek Aurora. Live single CD16+, CD14+, CD206+, CD45+, CD1c– and Lin– cells were analysed using FlowJo (v.10.9.0).
SkO co-culture and image acquisition
The co-culture was performed by adding the macrophages to the SkOs on day 12 of culture, with a 1:5 ratio. SkOs were transferred to a low attachment 96-well plate (Nunclon Sphera, Life Technologies) in SkO maturation medium1 containing 20% Matrigel (Corning). Macrophages were added to the SkOs and the co-culture was centrifuged at 100g for 6 min 1 acc, 0 dec. On day 3 of co-culture, the cells were transferred to a low-attachment 24-well plate, and Matrigel was diluted with fresh SkO maturation medium. On day 35 of co-culture (day 47 of SkO differentiation), the SkOs were fixed in a 2 ml tube with 4% paraformaldehyde (PFA) overnight for whole-mount serial staining (2 batches of differentiation, SkOs with macrophages n = 5, SkOs without macrophages n = 5). The co-culture was then permeabilized in blocking buffer (0.3% (v/v) Triton X-100, 1% (v/v) normal goat serum based on the antibodies and 1% BSA (v/v) dissolved in 1× PBS) for 8 h at room temperature on a shaker. Cells were then incubated overnight at 4 °C on a shaker (65 r.p.m.) with the first primary antibody, anti-CD45 (1:100, clone YAML501.4, ThermoFisher) for macrophages, for 48 h. The morning after, cells were washed and then incubated with the first secondary antibody overnight (goat anti-rat IgG, Alexa Fluor Plus 647, ThermoFisher). The morning after, SkOs were washed and incubated with the second primary antibody, anti-CD31 (1:100, clone JC70A, Dako) for endothelial cells for 48 h. The SkOs were then washed and incubated with the second secondary antibody (goat anti-mouse IgG1, Alexa Fluor 568, ThermoFisher) and DAPI overnight on a shaker. Cells were washed and placed in 50% glycerol for 30 min on a shaker at room temperature. Cells were then transferred to 70% glycerol overnight on a shaker at room temperature. The following morning, the co-culture was mounted and imaged using a custom 4-camera spinning disk confocal microscope. The microscope consists of an OpenFrame microscope frame connected to a CrestOptics X-Light V3 spinning disk confocal module that has four Teledyne Photometrics Kinetix cameras mounted to it. It was assembled by Cairn Research UK. All of the organoids were imaged in tiled stacks 800 µm deep using an Olympus ×10, 0.3 NA air objective with 5 µm z steps. The tiles were then stitched using Bigstitcher84 to produce the final image. As the sample holder was transparent on both sides, each organoid was imaged twice, once from each direction.
Image analysis of endothelial cells and SkOs co-cultured with macrophages
Image analysis of endothelial cell culture
To quantify the area covered by endothelial cells in the 2D angiogenesis assay with and without macrophages, phase-contrast images of the wells at 24, 48 and 72 h of culture were analysed using the Fiji distribution of the ImageJ software (v.2.14.0)83. The endothelial area was estimated by measuring the area of the wells covered by all cells (that is, endothelial cells alone or endothelial cells with macrophages). To obtain the endothelial density (in per cent) the area covered by cells was measured in pixels after segmentation using intensity thresholding and normalized to the total imaged area (constant, 2,115,570 pixels).
Image analysis of SkO culture
To quantify the endothelial area covering organoids with and without macrophages, maximum intensity z projections of confocal stacks of CD31+ staining were analysed using the Fiji distribution of the ImageJ software (v.2.14.0)83. The CD31+ endothelial area was measured in µm2 after segmentation using intensity thresholding and normalized to the organoid area (in µm2) to obtain the endothelial density (in per cent). Each dot represents the endothelial density of one organoid. The analysed stacks contained either 161 or 201 slices, each measuring 1 µm in the z dimension and up to 1,415 µm by 1,415 µm in the x and y dimensions. Their maximum intensity z projections covered a total area ranging from 1.86 to 14.74 million of µm2 per organoid.
Whole-mount immunostaining of human prenatal skin sample
For the prenatal tissue specimens, a single PBS rinse was followed by fixation in a freshly prepared 4% PFA solution in 1× PBS at room temperature for half an hour on a shaker. After three PBS washes, specimens were placed in a cold 12.5% SHIELD epoxy solution (LifeCanvas Technologies, SH-Ex) within SHIELD buffer (LifeCanvas Technologies, SH-BS) and gently shaken for 2 days at 4 °C. Next, specimens were moved to a SHIELD-ON warming solution (LifeCanvas Technologies, SH-ON) for 2 h at 37 °C on a gentle shaker. Following extensive washing in fresh 1× PBS for 8 h (with hourly refreshment) and a 24-h delipidation step at 55 °C in SHIELD Delipidation buffer (LifeCanvas Technologies, DB), specimens were rinsed in room temperature PBST (PBS with 0.1% Triton X-100 and 0.02% sodium azide) for a day. Anti-CD31 (1:100, clone C31.3, Novus Biologicals) and anti-LYVE1 (1:50, polyclonal, Novus Biologicals) primary antibodies were then applied overnight in a 0.1% PBST buffer on a room temperature shaker. Following three 0.1% PBST washes over 3 h, the specimens were incubated for 4 h at room temperature on a shaker with the following secondary antibodies: goat anti-mouse IgG1, Alexa Fluor 488 (ThermoFisher) and goat anti-Rabbit IgG, Alexa Fluor 647 (ThermoFisher). This was then followed by another trio of 0.1% PBST washes. Before imaging, the specimens were conditioned in a 1:1 solution of Easy-Index Matching solution (LifeCanvas Technologies, EI-Z1001) and 1× PBS for 4 h at 37 °C, which was subsequently replaced with a 100% immersion medium for a minimum of 6 h at 37 °C. Imaging was performed using a Nikon A1R HD25 confocal microscope system.
3D rendering
3D volume rendering and segmentation was created using Imaris 10 software at the Boston Children’s Hospital Cellular Imaging Core. For Supplementary Video 1, CD31+ vasculature and LYVE1+ macrophages were processed using the Imaris ‘Surfaces’ module. Co-localization of the CD31 and LYVE1 channels were processed using the ‘Coloc’ feature, generating a separate channel for overlapping signals. The parameters used in the Coloc feature depended on signal overlap and close contact of the CD31 and LYVE1 channels, leading to larger areas labelled as co-localized surfaces than the actual contact points of vessels and macrophages. Classification was based on estimated size and machine learning training. The following build parameters were used for CD31+ endothelial cells: area above 11.0 µm2; for LYVE1+ macrophages: ‘number of voxels Img=1’ above 953; for co-localized surfaces: area above 1,004 µm2; filter type ‘overlapped volume ratio to surfaces surfaces=LYVE1’ threshold=0.00976.
Immunofluorescence of prenatal skin and SkO cryosections
Cryosections (10 µm) were obtained from prenatal skin samples or iPS cell-derived SkOs frozen in OCT (Tissue-Tek OCT). The acquired slides were stored at −80 °C until use. On the day of the experiment, the slides were thawed and dried at room temperature, then fixed for 10 min in 4% PFA solution in 1× PBS (Alfa Aesar, J61899). Slides were washed with 1× PBS (Gibco, 10010-015) and incubated for 1 h at room temperature with 120 µl per slide of blocking solution (3% goat serum prepared in 1× PBS containing 0.1% Triton X-100 (Millipore, 648466)). A volume of 120 µl per slide of primary antibodies was then applied overnight at 4 °C in the blocking solution (a list of antibodies is supplied in Supplementary Table 38). The following day, the slides were washed 3 times with 1× PBS and then incubated for 1–2 h at room temperature with 120 µl per slide of secondary antibodies prepared in blocking solution (Supplementary Table 38). Slides were washed three times with 1× PBS and incubated with 1 µg ml–1 of DAPI solution prepared in 1× PBS. Following a final wash with 1× PBS, slides were coverslipped with ProLong Gold Antifade mountant (ThermoFisher, P36930). Slides were dried overnight in the dark at room temperature and imaged using a Leica SP8 Confocal microscope.
Multiplex RNAscope staining and image analysis
Prenatal skin tissue (8, 10 and 15 PCW) was frozen in OCT compound (Tissue-Tek OCT). 4-plex smFISH was performed using a RNAscope Multiplex Fluorescent Detection kit v.2 (ACDBio, 323100) or a RNAscope LS Multiplex Fluorescent Reagent kit v.2 assay and a RNAscope LS 4-Plex Ancillary Kit for LS Multiplex Fluorescent (Advanced Cell Diagnostics (ACD), bio-techne) according to the manufacturer’s instructions. The standard pretreatment for fresh frozen sections of 10–20 μm and permeabilization with Protease IV for 30 min at room temperature were performed.
Human probes against FOXP3, SHH, SLC26A7, NDP, CDH5, CD68, P2RY12, ACKR3, CXCL12, PDGFD and SERPINB7 transcripts were used (all from ACDBio catalogue probes). Opal dyes (Akoya Biosciences) were used at a dilution of 1:1,000 for the fluorophore step to develop each channel: Opal 520 Reagent Pack (FP1487001KT), Opal 570 Reagent Pack (FP1488001KT) and Opal 650 Reagent Pack (FP1496001KT) and Atto-425. Finally, the slides were counterstained with DAPI and coverslipped for imaging with ProLong Gold Antifade mountant (ThermoFisher, P36930).
4-plex RNAscope slides with FOXP3, SHH, SLC26A7, NDP, CDH5, CD68 and P2RY12 probes were imaged on a Perkin Elmer Opera Phenix Plus High-Content Screening System using a ×40 (NA 1.1, 0.149 μm per pixel) water-immersion objective with a 2 µm z step. The following channels were used: DAPI (excitation (ex.) 375 nm, emission (em.) 435–480 nm); Atto 425 (ex. 425 nm, em. 463–501 nm); Opal 520 (ex. 488 nm, em. 500–550 nm); Opal 570 (ex. 561 nm, em. 570-630 nm); and Opal 650 (ex. 640 nm, em. 650–760 nm). Confocal image stacks were stitched as 2D maximum intensity projections using proprietary Acapella scripts provided by Perkin Elmer and visualized using OMERO Plus (Glencoe Software).
4-plex RNAscope slides with ACKR3, CXCL12, PDGFD and SERPINB7 probes were imaged on the same custom spinning disk confocal microscope used for 3D imaging of the organoids. The objective used was a ×40 Nikon CFI Plan Apochromat Lambda D (NA 0.95). Imaging was performed with a 1.5 µm z step and stitched with the Bigstitcher Fiji plugin to generate a final z-projected image from individual tiles for analysis.
Quantification of FOXP3 coverage was carried out using QuPath image analysis software (v.0.5.1)85. Two-pixel classifiers were trained: one to segment the tissue from the image background and the other to segment out the FOXP3 spots against the background. All of the HF regions were manually segmented out of the whole skin section image. A new segmentation mask was automatically generated from the difference between the whole skin tissue mask and the HF masks. FOXP3 coverage was then calculated separately for the HF regions and the skin tissue by calculating the percentage of the masks that were taken up by segmented FOXP3 spots.
scRNA-seq data analysis
Alignment, quality control, clustering and annotation of prenatal skin dataset
The gene expression data were mapped using CellRanger (v.2.1.1 and v.2.0.2) to an Ensembl 84-based GRCh38 reference (10x Genomics–distributed v.1.2.0). The Python package emptydrops (v.0.0.5) was used to detect cells in each sample. Potential doublets were flagged using Scrublet (v.0.2.1)86 as previously described87. Low-quality cells were filtered out first by using a median + (X × MAD) score (where MAD is the median absolute deviation) of the median score for the mitochondrial UMI fraction (5 × MAD), maximum number of UMIs (8 × MAD), followed by strict cut-off values (minimum number of genes = 200, maximum number of UMIs = 50,000, maximum mitochondrial UMI fraction = 0.20). Possible maternal contamination (total of 118 cells) was identified using the souporcell pipeline (v.2.4.0)88 as previously described5,58. In brief, samples were pooled on a per-donor basis and processed with souporcell. The common GRCh38 variants file (SNPs with ≥2% frequency from 1k genomes) from souporcell authors was used. The pipeline was run twice, with genotype clusters set to 1 and 2 to obtain models for no maternal contamination and possible maternal contamination. The better model was identified using Bayesian information criterion (BIC), calculated using the formula BIC = kn log(m) − 2l, where k is the number of genotype clusters set for each souporcell run, n denotes the number of loci used for genotype deconvolution, m is the cell count for a given donor, and l is the log likelihood obtained after running the pipeline with each k. The cells with the minor genotype were identified as possible maternal contaminants where identified. Data pre-processing was performed using scanpy (v.1.4.3)89. After pooling data from all samples, genes detected in fewer than three cells were removed, and data were normalized to 1 × 104 UMI per cell and log1p transformed.
Highly variable genes were selected on the basis of normalized dispersion (scanpy.pp.highly_variable_genes with flavor = “seurat”, min_mean = 0.0125, max_mean = 3, min_dispersion = 0.5). Dimensionality reduction was done using principal component analysis and the first 50 principal components were used to compute the nearest-neighbour graph (scanpy.pp.neighbors with n_neighbors = 15). scVI module within scvi-tools (v.0.19.0) was used to correct for donor and 10x kit version batch effects (HVG = 15 000, dropout_rate = 0.2, n_layer = 2)90. Leiden algorithm was used to cluster cells based on the corrected graph with a relatively low resolution (scanpy.tl.leiden with resolution = 0.3) into coarse clusters that were manually annotated into broad lineages using known marker genes.
For each broad lineage, the data were re-processed starting from highly variable gene selection to better reveal finer heterogeneity. At this level, we used Harmony (v.0.0.5)91 and scVI from scvi-tools (v.0.19.0) in parallel for batch correction (again treating each donor as a separate batch) for every broad lineage and observed highly consistent embedding and clustering (data provided on the portal). Leiden clusters at the highest resolution were manually annotated using marker genes identified through the literature search, and their expression of distinctive DEGs specific to each cluster, such as WNT2 expression in WNT2+ fibroblasts. The full list of DEGs for each cluster is provided in Supplementary Table 3. DEGs were calculated using the sctk (Single Cell analysis Tool kit) package (https://github.com/Teichlab/sctk), where filtering is carried out followed by a two-sided Wilcoxon rank-sum test using pass-filter genes only in a one-versus-all fashion. The sctk package also carries out comparisons between the group of interest (one with highest expression) and the next group (second highly expressed), where the maximum proportion of cells expressing the gene in question in the second most highly expressed group was 0.2. For epidermal annotations, we created a combined embedding of prenatal skin and SkO data1, integrated using the Harmony pipeline, as well as integration with adult HF to check annotations, as described below. Harmony-corrected principal components were used to compute the batch-corrected nearest neighbourhood graph, and the Leiden algorithm was used to cluster the integrated data. The sctk package was then used to derive DEGs for each Leiden cluster. Annotation was carried out on the clusters based on marker genes and refined annotations in the SkO data1.
Clusters of doublets were manually flagged and removed, taking into account markers genes and previously calculated scrublet scores. To have a final global visualization of the atlas, a doublet-free UMAP was generated (Fig. 1b).
Processing, clustering and annotation of SkO dataset
Organoid data were pre-processed, filtered, clustered and annotated separately before integration with prenatal skin. In brief, cells filtered by CellRanger (CellRanger 2.1.0 with GRCh38-1.2.0 and CellRanger 3.0.2 with GRCh38-3.0.0) from SkO samples (2 strains, each with 4 time points) were pooled and quality control thresholds for UMI counts, gene counts, percentage of mitochondrial genes and top 50 highly expressed genes were established by fitting Gaussian mixture models to the distribution of each metric respectively. The following thresholds were used: minimum number of genes = 450, maximum number of genes = 5,731, minimum number of UMIs = 1,063, maximum number of UMIs = 25,559, maximum mitochondrial UMI fraction = 0.133, minimum cumulative percentage of counts for 50 most expressed genes in a cell = 23.7%, maximum cumulative percentage of counts for 50 most expressed genes in a cell = 56.6%. Highly variable gene selection, dimensionality reduction and KNN graph construction were done using the same method and parameters as prenatal skin. BBKNN (v.1.3.390)92 was used for batch-correction treating combinations of strains and 10x kit versions as batches. Broad lineages were annotated based on known markers. Each broad lineage was then re-processed in the same way as prenatal skin to annotate cell types at higher resolution.
Integration of prenatal skin and SkO datasets
Prenatal skin cells and organoid cells were integrated using Harmony (v.0.0.5)91, treating datasets as batches (prenatal skin or organoid) and within dataset batches as covariates (donor for prenatal skin, strain for SkO, and 10x kit version for both datasets). Leiden clusters were annotated using known markers.
Comparison of prenatal skin, adult skin and SkO datasets: distance-based analysis
Prenatal skin, adult skin and SkO cells were integrated using Harmony (v.0.0.5)91, treating datasets as batches and within-dataset batches as covariates (donor for prenatal and adult skin and strain for organoid, 10x kit version for all datasets). The principal component vectors of the downsampled Harmony-integrated object were then used to transform the gene expression matrix (NumPy (v.1.23.4) function ‘linalg.lstsq’, rcond = ‘warn’) of all cells in the non-downsampled pooled data and project for UMAP visualization (Fig. 1e and Extended Data Fig. 2a). The median transformed gene expression was used to compute the Euclidean distance between prenatal skin, adult skin and SkO for each broad cell cluster, using ‘spatial.distance_matrix’ function in SciPy (v.1.9.3), which was then plotted as a heatmap (Extended Data Fig. 2c).
Time-encoded cell state predictions: prenatal skin, adult skin and SkO datasets
The median probability of class correspondence between gene expression matrices in single-cell datasets was carried out using a logistic regression (LR) framework as previously described93, based on a similar workflow to CellTypist tool94. Annotated raw scRNA-seq datasets (prenatal skin, adult skin and SkO) were first concatenated, normalized and log-transformed. Linear variational autoencoder (VAE) latent representations were computed using the LDVAE module within scvi-tools (hidden layers = 256, dropout-rate = 0.2, reconstruction-loss = negative binomial) with dataset and chemistry information taken as technical covariates. ElasticNet LR models were built using the linear_model.LogisticRegression module in the sklearn package (v.0.22). The models were trained on SCVI batch-corrected low-dimensional LDVAE representation of the training data (prenatal and adult skin) using time-encoded labels (age_cell category). Regularization parameters (L1-ratio and alpha) were tuned using the GridSearchCV function in sklearn (v.1.1.3). The test grid was designed with five l1_ratio intervals (0.05, 0.2, 0.4, 0.6 and 0.8), five alpha (inverse of regularization strength) intervals (0.05, 0.2, 0.4, 0.6 and 0.8) at five train–test splits and three repeats for cross-validation. The unweighted mean over the weighted mean squared errors (MSEs) of each test fold (the cross-validated MSE) was used to determine the optimal model. The resultant model was used to predict the probability of correspondence between trained time-encoded labels and pre-annotated time_encoded clusters (week of culture_cell category) in the target dataset (SkO). The median probability of training label assignments per predesignated class overall (all cell groups) and for individual broad cell categories were computed (Supplementary Table 5). For visualization, the adult skin dataset was randomly downsampled to 10% (overall or by cell lineage) and resultant LR probabilistic relationship between labels of the training and target datasets were plotted as heatmaps (Extended Data Fig. 2d).
Differential abundance analysis
Differences in cell abundance associated with gestational age were tested using Milo (v.1.0.0)95, correcting for CD45+ and CD45– FACS isolation strategies. We first re-embedded cells into a batch-corrected latent space with a dimension of 30 using scVI model as implemented in scvi-tools considering donor and chemistry as batches. The model was trained using the 15,000 most highly variable genes that were selected based on dispersion (min_mean = 0.001, max_mean = 10) as previously described58. Where FACS correction was applied, we calculated a FACS isolation correction factor for each sample s sorted with gate i as (fs = log(piS/Si)) where pi is the true proportion of cells from gate i and S represents the total number of cells from both gates58. A KNN graph of cells was constructed based on distances in the latent space and cells assigned to neighbourhoods using the milopy.core.make_nhoods function (prop = 0.1). The number of cells of each cell type was then counted in each neighbourhood (milopy.core.count_nhoods). Labels were assigned to each neighbourhood based on majority voting (milopy.utils.annotate_nhoods) of cell labels by frequency (>50%). To test for differential abundance across gestational age, prenatal skin data were split into 4 age bins (7–8 PCW, 9–10 PCW, 11–13 PCW and 15–17 PCW), and cell counts were modelled using a negative binomial generalized model, with Benjamini–Hochberg weighted correction as previously described5,58, to test the effects of age (age bins) together with cell sorting correction (milopy.core.DA_nhoods). Significantly differentially abundant neighbourhoods were detected using (SpatialFDR < 0.1, log(fold change) < 0) for early enriched neighbourhoods and (SpatialFDR < 0.1, log(fold change) > 0) for late neighborhoods (Supplementary Table 4).
Cell state predictions: adult HFs, embryonic macrophages, blood vessel organoid, reindeer skin
To compare prenatal skin cells with external datasets (adult HF, embryonic macrophages, blood vessel organoid, reindeer skin)11,57,64,71, the datasets were downsampled to have roughly balanced cell counts per annotated cell type before integration with Harmony (v.0.0.5)91, treating datasets as batches and within dataset batches as covariates (donor for prenatal skin, site for embryonic macrophages, group (cell line: day of culture) for blood vessel organoid, chemistry for reindeer skin).
Comparison of cell type correspondence between datasets and probability prediction was carried out using a LR framework similar to the CellTypist package94. A model was built using the implementation of the linear_model.LogisticRegression module from sklearn package (v.1.1.3) (parameters: penalty: L2, solver: saga, regularization strength C = 0.1) and trained on the gene expression matrix of the training dataset using all genes that passed quality control. The resulting model was used to predict the classes in the target dataset. The correspondence between predicted and original classes in the target dataset was computed as the Jaccard index and median probability predictions and visualized as heatmaps. For comparison of the prenatal skin macrophages with embryonic macrophages, the embryonic macrophage dataset was used as training data and prenatal skin macrophages as query; for comparison of the blood vessel organoid with prenatal skin, the prenatal skin dataset (downsampled to maximum of 500 cells per cell type) was used as training data and the blood vessel organoid data as query; for comparison of HF data, merged prenatal and organoid data were used as training data and adult dataset as query; for comparison of the prenatal skin fibroblasts and macrophages with reindeer skin fibroblasts and macrophages, the reindeer skin data subsets were used as training data and prenatal skin data subsets as query.
Cross-species comparison: prenatal skin and mouse skin datasets
The median probability of class correspondence between human and mouse skin single cell datasets was carried out using a LR framework as previously described93, based on a similar workflow to the CellTypist tool94. Annotated raw scRNA-seq datasets (human prenatal skin and mouse embryonic skin41) were first concatenated, normalized, log-transformed and subsetted to retain the top 5,000 highly variable genes (batch_key=dataset) by dispersion. VAE latent representations were computed using scvi-tools (max epochs = 400, batch size = 512) with species, dataset and chemistry information taken as categorical covariates. ElasticNet LR models were built using the linear_model.LogisticRegression module in the sklearn package (v.0.22). The models were trained on SCVI batch-corrected low-dimensional VAE representation of the training data (prenatal skin) for broad cell groupings and refined cell annotations. The resultant models were used to predict the probability of correspondence between trained prenatal skin labels and pre-annotated clusters (broad cell groupings and refined annotations) in the target mouse skin data. The median probability of training label assignments was computed (Supplementary Tables 10 and 11). For visualization, resultant LR probabilistic relationship between labels of the training and target datasets were plotted as heatmaps.
FRZB comparison across developing organs
To compare gene expression of FRZB in fibroblasts across developing organs, a scRNA-seq stromal dataset from multiple developing organs5 (available from the Human Developmental Cell Atlas (https://developmental.cellatlas.io/fetal-immune)) was used, which also includes our prenatal skin scRNA-seq data. The data were normalized to 1 × 104 counts per cell (scanpy.pp.normalize_total), log1p transformed (scanpy.pp.log1p) and subsetted to fibroblast cell types only to plot expression of FRZB by organ across gestation time.
Trajectory analysis
The CellRank package96 (v.1.5.2) was used to define cell transition matrices, lineage drivers and rank fate probabilities of terminal state transitions across annotated lineages in a combined embedding of prenatal skin and SkO for keratinocytes and fibroblasts and in the prenatal skin for endothelial cells. Using pp.moments (n_pcs=10, n_neighbours=30) from the scVelo package (v.0.3.0), first order kinetics matrices were imputed. Using the palantir kernel and the velocity kernel in CellRank96, a mixed probability transition matrix was computed with the palantir kernel weighing 70% and the velocity kernel 30%. Schur matrix Eigen decomposition (n_components=25, method=‘brandts’) identified macrostates, terminal stages and initial stages. Lineage drivers were then computed for each state using compute_lineage_drivers from CellRank and pseudotime and latent time computed in scVelo (Supplementary Table 7).
In vivo–in vitro trajectory alignment analysis
We used Dynamic Programming-based alignment to evaluate agreement between the single-cell trajectories of prenatal skin and SkO fibroblasts, which describe the in vivo and in vitro differentiation lineages from HOXC5+ early fibroblasts to the Dp. Genes2Genes (G2G)39 is a Bayesian Information-theoretic Dynamic Programming framework that consistently captures matches and mismatches between two trajectories at both the gene level and the cell level. G2G outputs an optimal trajectory alignment that describes a nonlinear mapping of in vivo and in vitro pseudotime points in sequential order. This is based on the cost of matching or mismatching the gene expression distributions of each pair of organoid–reference time points, computed as a statistic of entropy difference between the two hypotheses under the minimum message length97 criterion. This statistic is a Shannon information distance, calculated in the unit of information, nits. Given any gene set, G2G runs Dynamic Programming alignment for each gene, outputting a five-state alignment string over matches (one-to-one/one-to-many/many-to-one) and mismatches (insertions and deletions–gaps) between the in vivo and in vitro pseudotime points in sequential order, which is analogous to a DNA/protein alignment output. It then computes an alignment similarity measure (that is, the percentage of matches across the alignment string) for each gene (Supplementary Table 9). These alignment strings enabled us to identify different clusters of genes with different alignment patterns. G2G also generates an aggregated cell-level alignment across all gene-level alignments, resulting in an overall alignment similarity measure as well. This aggregated alignment reflects whether the time points are matched or mismatched across genes on average (that is, if there is a higher proportion of gene-level alignments at which a specific time point pair between the two trajectories is matched, then the average alignment includes them as a match).
Using G2G, we examined the in vivo reference versus in vitro query alignment in terms of 1,369 human TFs98. These TFs were taken after filtering zero expressed genes and genes expressed in fewer than ten cells. Given the reference and organoid log1p normalized gene expression matrices of cells and their pseudotime estimates computed using CellRank96, G2G generated fully descriptive TF-level alignments, as well as an aggregated cell-level alignment across those TF-level alignments. Before alignment, the reference and organoid trajectories were discretized over the pseudotime axis in equal length intervals. Note that the number of discrete pseudotime points was determined as 15 based on the optimal binning structures predicted over their pseudotime estimates distributions using the OptBinning99 python package. Also note that at each alignment, these are the discrete time points that are getting matched or mismatched. For each discrete time point of a TF trajectory in a single dataset, G2G estimates its expression distribution as a Gaussian, with a weighted mean and weighted variance computed using all the cells (that is, a Gaussian-kernel-weighted interpolation approach for each cell’s contribution towards this estimation is based on their distance to the particular time point). Next, after interpolating both reference and organoid trajectories using the 15 discrete time points, the Dynamic Programming alignment was run for each TF, and the TF clusters of different alignment patterns (that is, early mismatches, mid mismatches, late mismatches, and complete mismatches) were identified using the G2G function that runs agglomerative hierarchical clustering over the TF-level alignment outputs.
Cell–cell interaction analysis
CellPhoneDB (v.3.0.0) package100 was used to infer cell–cell interactions within the prenatal skin scRNA-seq dataset overall and in early/late gestation and within the SkO scRNA-seq dataset overall. In the overall analysis, we randomly subsampled each cell type into no more than 200 cells 3 separate times, using all cells if a population contained fewer than 200 cells and excluding populations with fewer than 10 cells. The subsampled dataset were analysed separately using the permutation-based method to establish significance (P value cut-off = 0.05). For the analysis by early/late gestation, the prenatal skin scRNA-seq dataset was first split into early (≤11 PCW) and late (≥12 PCW) gestation datasets, which were then subsampled (no more than 200 cells per cell type) and individually analysed (P value cut-off = 0.05). A summary output file was created for each analysis run, compiling the interactions for each cell pair (P < 0.05) and adjusting P values for multiple testing (FDR set at 0.05) (Supplementary Tables 8 and 28). Circos plots (Circlize package (v.0.4.15)101) were used for downstream visualizations of selected significant (adjusted P value < 0.05) interactions between co-locating cell types for interactions found in all subsampled analyses.
To explore inferred interactions between macrophage subsets and endothelial cells (Extended Data Fig. 9a), we aggregated the interactions predicted for each macrophage subset and the different subtypes of endothelial cells (early endothelial cells, arterioles, capillary arterioles, capillaries, postcapillary venules and venules) by averaging the means and using the minimum of the adjusted P values as previously described5. A curated list of aggregated interactions were plotted for visualization using ggplot2 (v.3.3.6). A similar approach was adopted for assessing interactions between HF dermal and epidermal cells in prenatal skin: for each subset of hair follicle dermal cells, the interactions with early epithelial cells (≤11 PCW; immature basal) or late epithelial cells (≥12 PCW; DPYSL2+ basal, POSTN+ basal, placode, matrix, ORS, CL, IRS, cuticle/cortex) were aggregated, and the top 10 interactions per cell pair visualized using a heatmap (Fig. 2g). The same analysis was performed to obtain the top 10 interactions in SkO HFs (Extended Data Fig. 5c), defining early/late to match corresponding cell states as in prenatal skin. The top 10 interactions identified in prenatal skin HFs were also plotted within the SkO data to highlight similarities and differences between the two (Fig. 2g).
Comparison with adult fibroblasts
Integrated scRNA-seq data from prenatal skin and adult healthy skin (with original annotations)10 was subsetted to the cell group of interest (fibroblasts). DEGs between the adult and prenatal skin fibroblasts were derived using the Wilcoxon rank-sum test implementation in scanpy and adjusted for multiple testing using the Benjamini–Hochberg method (scanpy.tl.rank_genes_groups, method = “wilcoxon”, corr_method = “benjamini-hochberg”). Gene set enrichment analysis was performed using the top 1,000 genes in each group ranked by scores (Supplementary Table 13) and the implementation of the Enrichr workflow102 in the Python package GSEApy (https://gseapy.readthedocs.io/; v.0.10.7) with Gene Ontology Biological Process (2021) as the query database (Supplementary Tables 14, 15). A selected list of genes was plotted to highlight differences between prenatal and adult skin fibroblasts.
Gene set enrichment analysis
To determine the significantly overexpressed genes for gene set enrichment analysis, we first identified the DEGs between cell types for each cell group of interest (myeloid cells) using the Wilcoxon rank-sum test implementation in Scanpy (scanpy.tl.rank_genes_groups, method = “wilcoxon”). Genes with differential expression log(fold change) > 1.5 and adjusted P value < 0.01 were considered as significantly overexpressed (Supplementary Table 22). Gene set enrichment analysis was performed using the implementation of the Enrichr workflow102 in the Python package GSEApy (https://gseapy.readthedocs.io/; v.0.10.7) with Gene Ontology Biological Process (2023) and Molecular Signatures Database (MSigDB) Hallmark (2020) as query databases (Supplementary Tables 23–26).
For comparison between early and late cell states, for cell types of interest (WNT2+ fibroblast), we first identified the index cells belonging to early neighbourhoods (SpatialFDR < 0.1, log(fold change) < 0) and late neighbourhoods (SpatialFDR < 0.1, log(fold change) > 0) based on Milo95 differential abundance testing as described above (Supplementary Table 4). DEGs between early and late cell states were computed using the Wilcoxon rank-sum test implementation in scanpy (scanpy.tl.rank_genes_groups, method = “wilcoxon”). Genes with differential expression log(fold change) > 1 and adjusted P value < 0.01 were considered as significantly overexpressed (Supplementary Table 16) for gene set enrichment analysis using GSEApy (https://gseapy.readthedocs.io/; v.0.10.7), with Gene Ontology Biological Process (2023) as query the database (Supplementary Tables 17 and 18).
Gene module scoring
Gene module scoring was performed using the sc.tl.score_genes function in scanpy. For angiogenesis gene modules, pre-defined gene sets from the Gene Ontology Biological Process Database (2021) in Enrichr libraries103 were used (downloaded from Enrichr (https://maayanlab.cloud/Enrichr/#libraries)). For endothelial cell modules, gene sets defining tip, stalk, arteriole, venule and lymphatic, capillary (Extended Data Fig. 10g,h) were derived from published literature73,104,105, and for the hypoxia score, a hallmark hypoxia gene list was used. The list of genes for each gene module is provided in Supplementary Tables 27 and 29. The score for each module is the average expression of the gene set provided subtracted with the average expression of a reference set of genes. The reference set comprised 100 genes (ctrl_size=100), which were randomly sampled from all genes in the dataset (default gene_pool) with 25 expression level bins (n_bins=25) used for sampling. For angiogenesis modules, the mean module scores were computed for each cell type of interest (for example, LYVE1+ macrophage) and z score normalized for visualization.
Gene regulatory network analysis
The PySCENIC package106 (v.0.11.2) and pipeline were used to identify TFs and their target genes in the combined prenatal skin and SkO scRNA-seq datasets. The ranking database (hg38 refseq-r80 500bp_up_and_100bp_down_tss.mc9nr.feather), motif annotation database (motifs-v9-nr.hgnc-m0.001-o0.0.tbl) were downloaded from the Aert’s laboratory GitHub page. The tool was run 10 times, with a dataset comprising at most 1,000 cells per cell type × tissue pair (where tissue is prenatal skin or SkO). For each run, an adjacency matrix of TFs and their targets was generated and pruned using the Aert’s group suggested parameters. Only regulons present in at least 6 out of 10 runs were used in the analysis. PySCENIC was used to calculate the regulon specificity score for each cell type × tissue pair using the aucell function. An average was computed over the multiple runs. These average scores were used to compare regulon activity between prenatal skin and SkO. A gene interaction network was first built by querying the STRING database with GATA2 target genes, then pruned to only keep genes reported as associated with GATA2. The list was further truncated to 12 genes, by keeping genes that met the following criteria: (1) TFs in the five most active regulons detected in fetal skin; and/or (2) organoid capillary arterioles; and/or (3) associated with pseudotime (that is, in trajectories); and/or (4) VEGF receptors; and/or (5) in the selected gene ontology terms chosen for their role in angiogenesis, extracellular matrix organization, or cell migration, communication, proliferation, or death (GO:0045765, GO:0001568, GO:0030334, GO:0010646, GO:0001936, GO:0045446, GO:0002040, GO:0030155, GO:0010941 and GO:0030198).
Comparison of pro-angiogenic and anti-angiogenic genes between prenatal skin and SkO
The prenatal skin and SkO datasets were integrated using Harmony (v.0.0.5)91 as described above. Differential expression analysis was performed between prenatal skin and SkO cells (all cell types) using the standard scanpy workflow (scanpy.tl.rank_genes_groups, method = “wilcoxon”). Identified DEGs were filtered to only retain those coding for secreted proteins107 (Supplementary Table 39). Gene set enrichment analysis was performed on downregulated and upregulated genes separately, using the implementation of the Enrichr workflow102 in the Python package GSEApy (https://gseapy.readthedocs.io/) with Gene Ontology Biological Process (2021) as the query database. Significant gene ontology terms (adjusted P value < 0.05) (Supplementary Tables 33 and 34) were filtered based on their relevance to vasculature. Only DEGs involved in pathways thereby selected were chosen and their role in prenatal skin angiogenesis checked in the literature.
NicheNet analysis
We used NicheNet74 (v.1.1.1) to infer ligand–target gene links by combining scRNA-seq data (prenatal skin and SkO) of interacting cells (sender and receiver cells) with existing knowledge on signalling and gene regulatory networks. An open-source R implementation including integrated data sources used in the analysis are available at GitHub (https://github.com/saeyslab/nichenetr). NicheNet’s ligand–activity analysis first assesses and ranks ligands in the sender cell type (macrophage subsets), which best predict observed changes in expression of target genes of interest in the receiver cell types (endothelial cells) compared with background genes. Potential ligands were defined as all ligands in the NicheNet model that were expressed in at least 10% of cells in each macrophage (sender) cluster and had at least one specific receptor expressed in at least 10% of endothelial (receiver) cells. Target genes of interest were identified as DEGs between conditions (prenatal skin versus SkO) in receiver cells using FindMarkers function in NicheNet (adjusted P value ≤ 0.05 and average log2(fold change) > 0.25, expressed in at least 10% of endothelial cells). Background genes were all genes in the NicheNet model that were expressed in at least 10% of receiver cells.
Ligands were prioritized based on ligand activity scores, calculated as the Pearson correlation coefficient between a ligand’s target predictions and the observed target gene expression (Supplementary Table 35). The top 20 ligands were used to predict active target genes (top 200 overall) and construct the active ligand–target links (Supplementary Table 36). Receptors of the top-ranked ligands were identified from the NicheNet model, which filters for only bona fide ligand–receptor interactions documented in the literature and publicly available databases (Supplementary Table 37). To infer signalling paths, we defined our ligand (VEGFA, in red) and target genes (GATA2, in blue) of interest. NicheNet identifies which TFs best regulate the target genes and are most closely downstream of the ligand based on weights of the edges in its integrated ligand signalling and gene regulatory networks. The shortest paths between these TFs and the defined ligand are selected and genes along these paths are considered as relevant signalling mediators (in grey). Signalling mediators are prioritized by cross-checking the interactions between the defined ligand, target genes and identified TFs and signalling mediators across the different integrated data sources in NicheNet.
Spatial data analysis
Spatial transcriptomics data were mapped using Space Ranger (v.2.0.1) using GRCh38-1.2.0 reference. In parallel, we manually selected skin-overlapping spots in embryonic limb data12, comprising samples from the following ages: 6 PCW (n = 2, replicate = 2 each) and 8 PCW (n = 1, replicate = 3). To map cell types identified by scRNA-seq in the profiled spatial transcriptomics slides, we used the Cell2location (v.0.1) method15. First, we trained a negative binomial regression model to estimate reference transcriptomic profiles for all the cell types profiled with scRNA-seq in the organ (n = 15 samples). We excluded very lowly expressed genes using the filtering strategy recommended by the authors of Cell2location (cell_count_cutoff=5, cell_percentage_cutoff2=0.03, nonz_mean_cutoff=1.12). Cell types for which fewer than 20 cells were identified in samples ≤10 PCW were excluded from the reference. Individual 10x samples were considered as a batch, donor and chemistry type information was included as categorical covariate. Training was performed for 250 epochs and reached convergence according to manual inspection. Next, we estimated the abundance of cell types in the spatial transcriptomics slides using reference transcriptomic profiles of different cell types. All slides were analysed jointly. The following Cell2location hyperparameters were used: (1) expected cell abundance (N_cells_per_location) = 30; (2) regularization strength of detection efficiency effect (detection_alpha) = 20. The training was stopped after 50,000 iterations. All other parameters were used at default settings. Cell2location estimates the posterior distribution of cell abundance of every cell type in every spot. Posterior distribution was summarized as 5% quantile, representing the value of cell abundance that the model has high confidence in, and therefore incorporating the uncertainty in the estimate into values reported in the paper and used for downstream co-location analysis.
To identify microenvironments of co-locating cell types, we used NMF. We first normalized the matrix of estimated cell type abundances by dividing it by per-spot total abundances. Resulting matrix Xn of dimensions n × c, where n is the total number of spots in the Visium slides and c is the number of cell types in the reference was decomposed as Xn = WZ, where W is a n × d matrix of latent factor values for each spot and Z is a d × c matrix representing the fraction of abundance of each cell type attributed to each latent factor. Here latent factors correspond to tissue microenvironments defined by a set of co-localized cell types. We use the NMF package for R108, setting the number of factors d = 10 and using the default algorithm109. NMF coefficients were normalized by a per-factor maximum. We ran NMF 100 times and constructed the coincidence matrix. Then we selected the best run based on the lower mean silhouette score calculated on the coincidence matrix. If more than one run had the minimal mean silhouette, we selected one with smaller deviance (as reported by NMF function).
For cell type abundance correlation analysis, we used a per-spot normalized Xn matrix. Pearson correlation coefficient was calculated for each pair of cell types (all possible pairs computed) and each sample. For visualization of correlation analysis, selected cell pairs were plotted, guided by NMF results and which cell groups or categories formed microenvironments. For example, macrophages formed microenvironments with endothelial cells (ME1 and ME5), with neural cells (ME1 and ME5) and fibroblasts (ME1, ME4 and ME5) in Fig. 1d (Supplementary Table 40).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The datasets generated and/or analysed during the current study are available in the following repositories. Prenatal scRNA-seq skin data are available from ArrayExpress under accession numbers E-MTAB-11343, E-MTAB-7407 and E-MTAB-13071; accompanying prenatal skin TCR sequencing data are available under accession E-MTAB-13065. SkO scRNA-seq data are available from the Gene Expression Omnibus (GEO) under accession numbers GSE147206, GSE188936 and GSE231607. Visium limb data are available under E-MTAB-10367. Visium facial and abdominal data are deposited in ArrayExpress under E-MTAB-13024. Embryonic macrophage scRNA-seq data are available from GEO under accession numbers GSE13345 and GSE137010. All of the blood vessel organoid scRNA-seq data analysed as part of this study are included in a pre-print article71. Adult healthy skin scRNA-seq data are available from ArrayExpress under E-MTAB-8142. Adult HF scRNA-seq data are accessible from GEO under GSE129611. Mouse and reindeer skin scRNA-seq data are available from E-MTAB-11920 and GSE168748, respectively. Processed data can be accessed on our web portal (https://developmental.cellatlas.io/fetal-skin). Resource databases used in this study include GRCh38 reference, Ensembl Biomart, Gene Ontology Biological Process 2021 and 2023, MSigDB Hallmark 2020, ranking (hg38 refseq-r80 500bp_up_and_100bp_down_tss.mc9nr.feather) and motif annotation databases (motifs-v9-nr.hgnc-m0.001-o0.0.tbl) for PySCENIC analysis and STRING (v12.0). Source data are provided with this paper.
Code availability
Single-cell and spatial data were processed and analysed using publicly available software packages. Python, R code and notebooks for reproducing these analyses are publicly available at Zenodo (https://doi.org/10.5281/zenodo.8164271)14.
References
Lee, J. et al. Hair-bearing human skin generated entirely from pluripotent stem cells. Nature 582, 399–404 (2020).
Sadler, T. W. & Langman, J. Langman’s Medical Embryology (Lippincott Raven, 2000).
Muller, M., Jasmin, J. R., Monteil, R. A. & Loubiere, R. Embryology of the hair follicle. Early Hum. Dev. 26, 159–166 (1991).
Kennedy, K. M. et al. Questioning the fetal microbiome illustrates pitfalls of low-biomass microbial studies. Nature 613, 639–649 (2023).
Suo, C. et al. Mapping the developing human immune system across organs. Science 376, eabo0510 (2022).
Li, D. et al. VCAM-1 macrophages guide the homing of HSPCs to a vascular niche. Nature 564, 119–124 (2018).
Yanez, D. A., Lacher, R. K., Vidyarthi, A. & Colegio, O. R. The role of macrophages in skin homeostasis. Pflugers Arch. 469, 455–463 (2017).
Botting, R. A. & Haniffa, M. The developing immune network in human prenatal skin. Immunology 160, 149–156 (2020).
Xu, Y. et al. Single-cell transcriptome analysis reveals the dynamics of human immune cells during early fetal skin development. Cell Rep. 36, 109524 (2021).
Reynolds, G. et al. Developmental cell programs are co-opted in inflammatory skin disease. Science 371, eaba6500 (2021).
Takahashi, R. et al. Defining transcriptional signatures of human hair follicle cell states. J. Invest. Dermatol. 140, 764–773.e4 (2020).
Zhang, B. et al. A human embryonic limb cell atlas resolved in space and time. Nature https://doi.org/10.1038/s41586-023-06806-x (2023).
Li, T. et al. WebAtlas pipeline for integrated single cell and spatial transcriptomic data. Preprint at bioRxiv https://doi.org/10.1101/2023.05.19.541329 (2023).
Horsfall, D. et al. haniffalab/FCA_fetal_skin: 0.1.2. Zenodo https://doi.org/10.5281/zenodo.8164271 (2023).
Kleshchevnikov, V. et al. Cell2location maps fine-grained cell types in spatial transcriptomics. Nat. Biotechnol. 40, 661–671 (2022).
de Groot, S. C., Ulrich, M. M. W., Gho, C. G. & Huisman, M. A. Back to the future: from appendage development toward future human hair follicle neogenesis. Front. Cell Dev. Biol. 9, 661787 (2021).
Robins, E. J. & Breathnach, A. S. Fine structure of bulbar end of human foetal hair follicle at stage of differentiation of inner root sheath. J. Anat. 107, 131–146 (1970).
Randall, V. A. & Botchkareva, N. V. in Cosmetics Applications of Laser & Light-Based Systems (ed. Ahluwalia, G. S.) 3–35 (Elsevier, 2009).
Shamloul, G. & Khachemoune, A. An updated review of the sebaceous gland and its role in health and diseases part 1: embryology, evolution, structure, and function of sebaceous glands. Dermatol. Ther. 34, e14695 (2021).
Grubbs, H., Nassereddin, A. & Morrison, M. Embryology, Hair. StatPearls [Internet] https://www.ncbi.nlm.nih.gov/books/NBK534794/ (2022).
Lee, H.-T. et al. A crucial role of CXCL14 for promoting regulatory T cells activation in stroke. Theranostics 7, 855–875 (2017).
Zheng, C., Yin, S., Yang, Y., Yu, Y. & Xie, X. CD24 aggravates acute liver injury in autoimmune hepatitis by promoting IFN-γ production by CD4 T cells. Cell. Mol. Immunol. 15, 260–271 (2018).
Ali, N. et al. Regulatory T cells in skin facilitate epithelial stem cell differentiation. Cell 169, 1119–1129.e11 (2017).
Dhariwala, M. O. et al. Developing human skin contains lymphocytes demonstrating a memory signature. Cell Rep. Med. 1, 100132 (2020).
Joost, S. et al. The molecular anatomy of mouse skin during hair growth and rest. Cell Stem Cell 26, 441–457.e7 (2020).
Saxena, N., Mok, K.-W. & Rendl, M. An updated classification of hair follicle morphogenesis. Exp. Dermatol. 28, 332–344 (2019).
Martisova, A. et al. Identification of AGR2 gene-specific expression patterns associated with epithelial–mesenchymal transition. Int. J. Mol. Sci. 23, 10845 (2022).
Mok, K.-W. et al. Dermal condensate niche fate specification occurs prior to formation and is placode progenitor dependent. Dev. Cell 48, 32–48.e5 (2019).
Abbasi, S. et al. Distinct regulatory programs control the latent regenerative potential of dermal fibroblasts during wound healing. Cell Stem Cell 28, 581–583 (2021).
Li, C.-F. et al. Snail-induced claudin-11 prompts collective migration for tumour progression. Nat. Cell Biol. 21, 251–262 (2019).
Dillenburg-Pilla, P. et al. SDF-1/CXCL12 induces directional cell migration and spontaneous metastasis via a CXCR4/Gαi/mTORC1 axis. FASEB J. 29, 1056–1068 (2015).
Biggs, L. C. et al. Hair follicle dermal condensation forms via Fgf20 primed cell cycle exit, cell motility, and aggregation. eLife 7, e36468 (2018).
Hagner, A. et al. Transcriptional profiling of the adult hair follicle mesenchyme reveals R-spondin as a novel regulator of dermal progenitor function. iScience 23, 101019 (2020).
Hu, B. et al. Control of hair follicle cell fate by underlying mesenchyme through a CSL–Wnt5a–FoxN1 regulatory axis. Genes Dev. 24, 1519–1532 (2010).
Belmadani, A., Jung, H., Ren, D. & Miller, R. J. The chemokine SDF-1/CXCL12 regulates the migration of melanocyte progenitors in mouse hair follicles. Differentiation 77, 395–411 (2009).
Ghersi, G. et al. The protease complex consisting of dipeptidyl peptidase IV and seprase plays a role in the migration and invasion of human endothelial cells in collagenous matrices. Cancer Res. 66, 4652–4661 (2006).
Yin, S., Chen, F., Ye, P. & Yang, G. Overexpression of FAM3C protein as a novel biomarker for epithelial–mesenchymal transition and poor outcome in gastric cancer. Int. J. Clin. Exp. Pathol. 11, 4247–4256 (2018).
Nunan, R. et al. Ephrin-Bs drive junctional downregulation and actin stress fiber disassembly to enable wound re-epithelialization. Cell Rep. 13, 1380–1395 (2015).
Sumanaweera, D. et al. Gene-level alignment of single cell trajectories informs the progression of in vitro T cell differentiation. Preprint at bioRxiv https://doi.org/10.1101/2023.03.08.531713 (2023).
Quinonez, S. C. & Innis, J. W. Human HOX gene disorders. Mol. Genet. Metab. 111, 4–15 (2014).
Jacob, T. et al. Molecular and spatial landmarks of early mouse skin development. Dev. Cell 58, 2140–2162.e5 (2023).
Driskell, R. R. et al. Distinct fibroblast lineages determine dermal architecture in skin development and repair. Nature 504, 277–281 (2013).
Solé-Boldo, L. et al. Single-cell transcriptomes of the human skin reveal age-related loss of fibroblast priming. Commun. Biol. 3, 188 (2020).
Halley, A. C. The tempo of mammalian embryogenesis: variation in the pace of brain and body development. Brain Behav. Evol. 97, 96–107 (2022).
Has, C. et al. Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br. J. Dermatol. 183, 614–627 (2020).
Goto, M. et al. Fibroblasts show more potential as target cells than keratinocytes in COL7A1 gene therapy of dystrophic epidermolysis bullosa. J. Invest. Dermatol. 126, 766–772 (2006).
Vahlquist, A., Gånemo, A. & Virtanen, M. Congenital ichthyosis: an overview of current and emerging therapies. Acta Derm. Venereol. 88, 4–14 (2008).
Huitema, L., Phillips, T., Alexeev, V. & Igoucheva, O. Immunological mechanisms underlying progression of chronic wounds in recessive dystrophic epidermolysis bullosa. Exp. Dermatol. 30, 1724–1733 (2021).
Akiyama, M. Understanding immune profiles in ichthyosis may lead to novel therapeutic targets. J. Allergy Clin. Immunol. 149, 1210–1212 (2022).
Larson, B. J., Longaker, M. T. & Lorenz, H. P. Scarless fetal wound healing: a basic science review. Plast. Reconstr. Surg. 126, 1172–1180 (2010).
Darby, I. A. & Desmoulière, A. in Textbook on Scar Management: State of the Art Management and Emerging Technologies (eds Téot, L. et al.) (Springer, 2020).
Ascensión, A. M., Fuertes-Álvarez, S., Ibañez-Solé, O., Izeta, A. & Araúzo-Bravo, M. J. Human dermal fibroblast subpopulations are conserved across single-cell RNA sequencing studies. J. Invest. Dermatol. 141, 1735–1744.e35 (2021).
Gur, C. et al. LGR5 expressing skin fibroblasts define a major cellular hub perturbed in scleroderma. Cell 185, 1373–1388.e20 (2022).
Longaker, M. T. & Adzick, N. S. The biology of fetal wound healing: a review. Plast. Reconstr. Surg. 87, 788–798 (1991).
Wynn, T. A. & Vannella, K. M. Macrophages in tissue repair, regeneration, and fibrosis. Immunity 44, 450–462 (2016).
Buechler, M. B., Fu, W. & Turley, S. J. Fibroblast–macrophage reciprocal interactions in health, fibrosis, and cancer. Immunity 54, 903–915 (2021).
Bian, Z. et al. Deciphering human macrophage development at single-cell resolution. Nature 582, 571–576 (2020).
Goh, I. et al. Yolk sac cell atlas reveals multiorgan functions during human early development. Science 381, eadd7564 (2023).
Du, C. et al. Synaptotagmin-11 inhibits cytokine secretion and phagocytosis in microglia. Glia 65, 1656–1667 (2017).
Henn, D. et al. Xenogeneic skin transplantation promotes angiogenesis and tissue regeneration through activated Trem2 macrophages. Sci. Adv. 7, eabi4528 (2021).
Liechty, K. W., Adzick, N. S. & Crombleholme, T. M. Diminished interleukin 6 (IL-6) production during scarless human fetal wound repair. Cytokine 12, 671–676 (2000).
Alivernini, S. et al. Distinct synovial tissue macrophage subsets regulate inflammation and remission in rheumatoid arthritis. Nat. Med. 26, 1295–1306 (2020).
Ortmayr, G. et al. Immunological aspects of AXL/GAS-6 in the context of human liver regeneration. Hepatol. Commun. 6, 576–592 (2022).
Sinha, S. et al. Fibroblast inflammatory priming determines regenerative versus fibrotic skin repair in reindeer. Cell 185, 4717–4736.e25 (2022).
Kolter, J., Kierdorf, K. & Henneke, P. Origin and differentiation of nerve-associated macrophages. J. Immunol. 204, 271–279 (2020).
Cariboni, A. et al. Neuropilins and their ligands are important in the migration of gonadotropin-releasing hormone neurons. J. Neurosci. 27, 2387–2395 (2007).
Gu, X., Li, S.-Y. & DeFalco, T. Immune and vascular contributions to organogenesis of the testis and ovary. FEBS J. 289, 2386–2408 (2022).
Fantin, A. et al. Tissue macrophages act as cellular chaperones for vascular anastomosis downstream of VEGF-mediated endothelial tip cell induction. Blood 116, 829–840 (2010).
Wang, Z. et al. An immune cell atlas reveals the dynamics of human macrophage specification during prenatal development. Cell 186, 4454–4471.e19 (2023).
Shen, H., Schuster, R., Stringer, K. F., Waltz, S. E. & Lentsch, A. B. The Duffy antigen/receptor for chemokines (DARC) regulates prostate tumor growth. FASEB J. 20, 59–64 (2006).
Nikolova, M. T. et al. Fate and state transitions during human blood vessel organoid development. Preprint at bioRxiv https://doi.org/10.1101/2022.03.23.485329 (2022).
Coma, S. et al. GATA2 and Lmo2 control angiogenesis and lymphangiogenesis via direct transcriptional regulation of neuropilin-2. Angiogenesis 16, 939–952 (2013).
Li, Q. et al. Single-cell transcriptome profiling reveals vascular endothelial cell heterogeneity in human skin. Theranostics 11, 6461–6476 (2021).
Browaeys, R., Saelens, W. & Saeys, Y. NicheNet: modeling intercellular communication by linking ligands to target genes. Nat. Methods 17, 159–162 (2020).
Mass, E., Nimmerjahn, F., Kierdorf, K. & Schlitzer, A. Tissue-specific macrophages: how they develop and choreograph tissue biology. Nat. Rev. Immunol. https://doi.org/10.1038/s41577-023-00848-y (2023).
Zhang, W. et al. Microglia-containing human brain organoids for the study of brain development and pathology. Mol. Psychiatry 28, 96–107 (2023).
Huang, M. et al. Self-assembled human skin equivalents model macrophage activation of cutaneous fibrogenesis in systemic sclerosis. Arthritis Rheumatol. 74, 1245–1256 (2022).
Strobel, H. A., Moss, S. M. & Hoying, J. B. Vascularized tissue organoids. Bioengineering https://doi.org/10.3390/bioengineering10020124 (2023).
Mesler, A. L., Veniaminova, N. A., Lull, M. V. & Wong, S. Y. Hair follicle terminal differentiation is orchestrated by distinct early and late matrix progenitors. Cell Rep. 19, 809–821 (2017).
Alsinet, C. et al. Robust temporal map of human in vitro myelopoiesis using single-cell genomics. Nat. Commun. 13, 2885 (2022).
Suarez-Arnedo, A. et al. An image J plugin for the high throughput image analysis of in vitro scratch wound healing assays. PLoS ONE 15, e0232565 (2020).
Patsch, C. et al. Generation of vascular endothelial and smooth muscle cells from human pluripotent stem cells. Nat. Cell Biol. 17, 994–1003 (2015).
Rueden, C. T. et al. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinformatics 18, 529 (2017).
Hörl, D. et al. BigStitcher: reconstructing high-resolution image datasets of cleared and expanded samples. Nat. Methods 16, 870–874 (2019).
Bankhead, P. et al. QuPath: open source software for digital pathology image analysis. Sci. Rep. 7, 16878 (2017).
Wolock, S. L., Lopez, R. & Klein, A. M. Scrublet: computational identification of cell doublets in single-cell transcriptomic data. Cell Syst. 8, 281–291.e9 (2019).
Pijuan-Sala, B. et al. A single-cell molecular map of mouse gastrulation and early organogenesis. Nature 566, 490–495 (2019).
Heaton, H. et al. Souporcell: robust clustering of single-cell RNA-seq data by genotype without reference genotypes. Nat. Methods 17, 615–620 (2020).
Wolf, F. A., Angerer, P. & Theis, F. J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018).
Lopez, R., Regier, J., Cole, M. B., Jordan, M. I. & Yosef, N. Deep generative modeling for single-cell transcriptomics. Nat. Methods 15, 1053–1058 (2018).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019).
Polański, K. et al. BBKNN: fast batch alignment of single cell transcriptomes. Bioinformatics 36, 964–965 (2020).
Jardine, L. et al. Blood and immune development in human fetal bone marrow and Down syndrome. Nature 598, 327–331 (2021).
Domínguez Conde, C. et al. Cross-tissue immune cell analysis reveals tissue-specific features in humans. Science 376, eabl5197 (2022).
Dann, E., Henderson, N. C., Teichmann, S. A., Morgan, M. D. & Marioni, J. C. Differential abundance testing on single-cell data using k-nearest neighbor graphs. Nat. Biotechnol. 40, 245–253 (2022).
Lange, M. et al. CellRank for directed single-cell fate mapping. Nat. Methods 19, 159–170 (2022).
Wallace, C. S. Statistical and Inductive Inference by Minimum Message Length (Springer Science & Business Media, 2005).
Lambert, S. A. et al. The human transcription factors. Cell 172, 650–665 (2018).
Navas-Palencia, G. Optimal binning: mathematical programming formulation. Preprint at https://doi.org/10.48550/arXiv.2001.08025 (2020).
Efremova, M., Vento-Tormo, M., Teichmann, S. A. & Vento-Tormo, R. CellPhoneDB: inferring cell–cell communication from combined expression of multi-subunit ligand–receptor complexes. Nat. Protoc. 15, 1484–1506 (2020).
Gu, Z., Gu, L., Eils, R., Schlesner, M. & Brors, B. circlize implements and enhances circular visualization in R. Bioinformatics 30, 2811–2812 (2014).
Chen, E. Y. et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128 (2013).
Gene-set Library (Enrichr); https://maayanlab.cloud/Enrichr/#libraries (accessed 16 April 2022).
Schupp, J. C. et al. Integrated single-cell atlas of endothelial cells of the human lung. Circulation 144, 286–302 (2021).
He, Y. et al. Novel blood vascular endothelial subtype-specific markers in human skin unearthed by single-cell transcriptomic profiling. Cells 11, 1111 (2022).
Van de Sande, B. et al. A scalable SCENIC workflow for single-cell gene regulatory network analysis. Nat. Protoc. 15, 2247–2276 (2020).
protein_class:Predicted secreted proteins (The Human Protein Atlas); https://www.proteinatlas.org/search/protein_class%3APredicted+secreted+proteins (accessed 5 October 2022).
Gaujoux, R. & Seoighe, C. A flexible R package for nonnegative matrix factorization. BMC Bioinformatics 11, 367 (2010).
Brunet, J.-P., Tamayo, P., Golub, T. R. & Mesirov, J. P. Metagenes and molecular pattern discovery using matrix factorization. Proc. Natl Acad. Sci. USA 101, 4164–4169 (2004).
Acknowledgements
We thank A. Maartens for scientific writing support; K. A. Aldinger, D. Doherty, I. G. Phelps, J. C. Dempsey, K. J. Lee and L. A. Cort for assistance in procuring fetal samples; Y. Liu and H. Gao of the Indiana University School of Medicine Center for Computational Biology and Bioinformatics for assistance with SkO single-cell data acquisition; staff at the Newcastle University Flow cytometry core facility; staff at Wellcome Sanger SGP for help with skin sample processing and tissue staining, imaging and imaging analyses; staff at the Wellcome Sanger Institute core facility for sequencing and data alignment; and staff at the Newcastle University NovoPath for support with tissue staining and imaging. We acknowledge funding from the Wellcome Human Developmental Biology Initiative (WT215116/Z/18/Z) and CIFAR MacMillan Multiscale Human programme. M.H. is funded by Wellcome (WT107931/Z/15/Z), The Lister Institute for Preventive Medicine and the NIHR-Newcastle Biomedical Research Centre. S.A.T. is funded by Wellcome (WT206194) and the ERC Consolidator Grant DEFINE. K.R.K. is funded by the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIH; R01AR083139 and R01AR075018). L.J. is funded by a Newcastle Health Innovation Partners Lectureship. N.H.G. is funded by an MRC Clinical Research Training Fellowship (MR/W015625/1). B.O. is funded by a Wellcome 4Ward North Clinical Training Fellowship. Some of this research was supported by the NIHR Cambridge Biomedical Research Centre (NIHR203312). F.P. is funded by Wellcome (226579/Z/22/Z). The views expressed are those of the authors and not necessarily those of the NIHR or the Department of Health and Social Care and Wellcome (203151/Z/16/Z). Fetal tissue specimens used in the Koehler Laboratory at Boston Children’s Hospital were obtained from the University of Washington Birth Defects Research Laboratory, which is supported by NIH award number 5R24HD000836 from the Eunice Kennedy Shriver National Institute of Child Health and Human Development to I.A.G. This publication is part of the Human Cell Atlas (http://www.humancellatlas.org/publications/[humancellatlas.org]).
Author information
Authors and Affiliations
Contributions
M.H., S.A.T. and K.R.K. conceived and supervised the research. M.H., S.A.T. and K.R.K. acquired funding. For prenatal skin scRNA-seq data, skin sample processing was done by N.H.G., R. A. Botting, E.S., M.-U.M., K.S., J.E. and S.Li.; flow cytometry was done by R. A. Botting and E.S., supported by A.F.; library preparation was done by E.S., M.-U.M., K.S., J.E. and V.R. For prenatal skin spatial data, skin sample processing was done by F.T. and B.R., supported by K.R., N.-J.C. and V.R. Data alignment was done by P. Mazin. Computational analyses were performed by N.H.G., E.W., B.O., N.H., D.S., I.G., L.S., W.M.T., P. Moghimi, D.-M.P., S.B., V.S. and P. Mazin, with support from K.A., A.R. and J.-E.P. Tissue staining, imaging and imaging analyses were performed by C.A., A.R.F., R. A. Botting, F.T., A.P.L., J.K., L.V., C.G., K.Y.G., S.A.S., K.A.K., T.L., B.R., C.L.T., K.R., S.P., I.M., J.F., E.P., A.G., J.M., C.B.H., J.A.M., F.P., J.L. and L.G. SkO experiments were carried out by C.A., A.R.F., F.T., A.P.L., J.K., D.A., K.Y.G., S.A.S., C.D., J.L., C.B.H. and L.G. Flow cytometry experiments of iPS cell-derived macrophages were done by E.W. and C.A. Scratch assays were undertaken by A.R.F. and D.A. Angiogenesis assays were undertaken by C.A., A.R.F. and D.A. Provision of additional resources such as tissue samples and external datasets were from A.P.L., J.K., K.Y.G., S.A.S., C.D., K.A., R. A. Barker, I.A.G., J.-E.P., M.T.N., P.H., J.E.G.L., J.A.M., B.T., M.K., J.L. and K.R.K. Web portal development was undertaken by T.L., D.B.L., J.M., D.H. and O.B., with disease information assembled by B.O. and E.A.O. Writing of the original draft was by N.H.G., E.W., B.O., C.A., A.R.F., N.H., F.T., D.S., B.R., S.B., P. Mazin, L.G. and M.H. Writing, review and editing was done by N.H.G., E.W., B.O., D.S., A.P.L., J.K., E.S., C.G., S.A.S., L.J., G.R., E.P., A.D., N.R., E.A.O., M.K., F.P., J.L., L.G., K.R.K., S.A.T. and M.H. Visualization was done by N.H.G., E.W., B.O., C.A., A.P.L., J.K., L.V., K.Y.G., S.A.S., M.-U.M., K.A.K., T.L., D.B.L., K.S., A.G., R.V.-T., J.M., D.H., J.A.M., O.B., F.P., L.G., K.R.K. and M.H. C.A. and A.R.F. contributed equally to this work as second co-authors.
Corresponding authors
Ethics declarations
Competing interests
J.L. and K.R.K., with the Indiana University Research and Technology Corporation, have a patent relating to the methodology and composition of SkOs (PCT/US2016/058174). K.R.K. is a consultant for StemCell Technologies. All other authors declare no competing interests.
Peer review
Peer review information
Nature thanks Elvira Mass, Michael Rendl and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Temporal and spatial composition of human prenatal skin.
(a) Prenatal skin cells isolation by fluorescence-activated cell sorting into CD45+ and CD45− fractions (n = 18); from the CD45− fraction we further isolated all cells that were not within the CD34+/CD34+CD14− gate (n = 4) to enrich for endothelial cells and keratinocytes. Representative data from n = 1 is shown as mean percentage +/− SD values. (b) Quality control plots showing frequency distribution of UMI counts (log1p-transformed) and percent of UMI counts in mitochondrial genes per sample fraction. (c) Dot plot showing variance-scaled, mean expression (dot colour) and percent of expressing cells (dot size) of defining genes for cell states corresponding to Fig. 1b. (d) Milo beeswarm plot showing differential abundance of neighbourhoods in prenatal skin across gestation time, annotated by refined cell labels. Red/blue neighbourhoods are significantly enriched in earlier/later gestation respectively. Colour intensity denotes degree of significance. (e) Bar plot showing cell type co-location, indicated by positive Pearson correlation coefficients calculated between per-spot normalised cell type abundances, for selected cell type pairs (macrophage and endothelial cells). Pearson correlation coefficients were calculated across all skin-covered spots of Visium samples; each sample is shown by an individual bar. (f) Bar plot showing cell type co-location, indicated by positive Pearson correlation coefficients calculated between per-spot normalised cell type abundances, for selected cell type pairs (pre-dermal condensate and immune cells: DC1, DC2, LTi and ILC3). Pearson correlation coefficients were calculated across all skin-covered spots of Visium samples; each sample is shown by an individual bar. ASDC, Axl+Siglec6+ dendritic cells; CD45en, CD45 negative fraction enriched for keratinocyte/endothelial cells, CD45N, CD45 negative; CD45NCD34N, CD45 negative-CD34 negative; CD45NCD34P, CD45 negative-CD34 positive; CD45P, CD45 positive; DC, dendritic cells; HSC, hematopoietic stem cells; LC, Langerhans cells; LTi, lymphoid tissue inducer cells; MEMP, megakaryocyte-erythroid-mast cell progenitor; pDC, plasmacytoid dendritic cells.
Extended Data Fig. 2 Comparison of the skin organoid with prenatal and adult skin.
(a) UMAP visualisation of the integrated prenatal skin, adult skin10 and SkO1 scRNA-seq datasets, coloured by broad cell types for each dataset. (b) UMAP visualisations of integrated data from prenatal skin and SkO, coloured by epidermal (left) and dermal (right) cell types. (c) Heatmap showing conserved cell states (measured by distance in principal component space) between prenatal skin, adult skin10 and SkO1 for broad cell categories. (d) Heatmap showing prediction probabilities (overall and per broad cell category) for a logistic regression model trained on time-encoded prenatal skin and adult skin data (y-axis)10 and projected onto time-encoded SkO data1 (x-axis). Colour scale indicates median prediction probabilities. DC, dendritic cells.
Extended Data Fig. 3 Prenatal skin epidermal cell composition and comparison with adult hair follicles.
(a) Schematic of stages of HF formation. (b) Dot plot showing variance-scaled, mean expression (dot colour) and percent of expressing cells (dot size) of DEGs between gestational stage groups (grouped PCW) (right) and expression of the same genes by different epithelial cell states in prenatal skin (left). (c) Bar plot showing the average proportions of stromal cell states across gestational age in prenatal skin. Bar colours represent cell states. (d) Heatmap showing the correspondence (measured by Jaccard index) between prenatal skin/SkO (y-axis) and adult (x-axis) epidermal and HF cell states from a logistic regression model trained on adult HF data11, projected onto integrated prenatal skin/SkO data. (e) UMAPs showing clustered cell states in integrated data from adult HFs11 and prenatal/SkO, coloured by prenatal skin/SkO cell types (left) and adult cell types (right). (f) Volcano plot showing differentially expressed genes between prenatal matrix cells and adult matrix cells using Wilcoxon rank-sum, two-sided, Benjamini-Hochberg adjusted11. (g) Percentage of FOXP3 coverage in HF regions and non-HF regions across five prenatal skin samples. Data are mean ± SD and statistics (p = 0.0131) generated with an unpaired t-test. (h) Immunostained human prenatal skin at 15PCW for Tregs with FOXP3 (red; red arrows), epithelial keratinocytes with Keratin 14 (yellow) and dermal papilla with SOX2 (cyan; cyan arrows). Scale bar: 100 µm. Tregs, Regulatory T cells. For details on statistics and reproducibility, see Methods. The images in a were created using BioRender (https://biorender.com).
Extended Data Fig. 4 Differentiation of prenatal hair follicle epithelial cells.
(a) Inferred pseudotime trajectory of prenatal skin and SkO epidermal cell states, differentiating along the ‘ORS/CL’ and ‘IRS’ trajectories, coloured by gestational age (PCW) for prenatal skin (middle) and days of culture for SkO (bottom). UMAP overlaid with cell directionality (arrows) as inferred over the cell-to-cell transition probability matrix from CellRank and coloured by pseudotime (top). (b) Inferred pseudotime trajectory of prenatal skin epidermal cell states differentiating along the ‘ORS/CL’ and ‘IRS’ trajectories, coloured by gene expression (log-transformed). (c) Dot plot showing variance-scaled, mean expression (dot colour) and percent of expressing cells (dot size) in prenatal skin of genes expressed along the ‘ORS/CL’ and ‘IRS’ trajectories. (d) Inferred pseudotime trajectory of prenatal skin and SkO fibroblasts differentiating along the ‘hair’ and ‘dermal’ trajectories, coloured by gestational age (PCW) for prenatal skin (middle) and days of culture for SkO (bottom). UMAP overlaid with cell directionality (arrows) as inferred over the cell-to-cell transition probability matrix from CellRank and coloured by pseudotime (top). (e) Dot plot showing variance-scaled, mean expression (dot colour) and percent of expressing cells (dot size) of FRZB gene in fibroblasts from developing organs. Gestational ages during which individual organs are present are highlighted. (f) Heatmap showing differentially expressed genes across pseudotime along the ‘hair fibroblast trajectory’. Gene associated with genetic hair disorders is highlighted in grey. (g) Violin plot showing expression of CXCL12 in hair mesenchymal cells (violin width proportional to counts). (h) Violin plot showing expression of CXCL12 in hair mesenchymal cells by gestation (PCW) (violin width proportional to counts).
Extended Data Fig. 5 Differentiation of the prenatal hair follicle mesenchyme.
(a) Circos plot showing selected significant (adjusted p-value<0.05, significance calculated in CellphoneDB using empirical shuffling and FDR-adjusted) predicted interactions between pre-dermal condensate and ILC3 and LTi cells in prenatal skin. Arrows represent directionality of interactions (ligand to receptor); connection width is proportional to the CellphoneDB mean value for each ligand-receptor pair. (b) Schematic representation of mesenchymal-epithelial signalling and cellular processes during hair formation. (c) Heatmap showing significant (adjusted p-value <0.05, significance calculated in CellphoneDB using empirical shuffling and FDR-adjusted) predicted interactions between hair mesenchymal cells and epithelial cells (early: Immature basal; late: DPYSL2+ basal, POSTN+ basal, placode, matrix, ORS, CL, IRS, Cuticle/cortex) in SkO. Top 10 interactions per cell pair are shown. Colour scale represents the mean expression values of each ligand-receptor pair in corresponding cell pairs. (d) Heatmap on the left shows interesting trends of distributional distances in the expression of selected differentially expressed TFs across pseudotime between prenatal skin (reference) and SkO. The distributional distance is a Shannon information measure of dissimilarity (unit: nits), and the heatmap visualises these distances across time for each TF after log-transformation and smoothening using a Gaussian kernel (σ = 2) for highlighting their trends. Heatmaps in the middle and right show the interpolated and z-normalised mean expression of those selected TFs across pseudotime in prenatal skin and SkO respectively. (e) Gene expression plots for representative genes in prenatal skin (green) and SkO (blue) across pseudotime. Left column: the interpolated log1p transformed expression (y-axis) against pseudotime (x-axis). The lines represent mean expression trends; the faded data points are 50 random samples from the estimated expression distribution at each time point. Right two columns: actual log1p transformed expression (y-axis) against pseudotime (x-axis) where each point represents a cell. (f) Dot plot showing variance-scaled, mean expression (dot colour) and percent of expressing cells (dot size) of known genes involved in hair formation26. (g) UMAP co-embedding of human prenatal (left) and mouse embryonic (E12.5, E13.5 and E14.5) (right) skin coloured by broad cell cluster annotations. (h) Heatmap showing prediction probabilities from a logistic regression model trained on human prenatal skin (x-axis), projected onto mouse embryonic skin (y-axis) for broad cell groupings. Colour scale indicates median prediction probabilities. (i) Heatmap showing prediction probabilities from a logistic model trained on human prenatal skin (x-axis), projected onto mouse embryonic skin (y-axis) for refined cell clusters (only fibroblast sub-populations shown from all cell types). (j) Dot plot showing variance-scaled, mean expression (dot colour) and percent of expressing cells (dot size) of fibroblast marker genes. DC, dendritic cells; Fib, fibroblast; LTi, lymphoid tissue inducer cells; Vessel BECs, Vessel blood endothelial cells; Vessel LECs, Vessel lymphatic endothelial cells. The images in b were created using BioRender (https://biorender.com).
Extended Data Fig. 6 Genetic hair and skin disorders.
(a) Heat map showing differentially expressed genes across pseudotime along the ‘Inner root sheath trajectory’. Genes associated with genetic hair disorders are highlighted in grey. (b) Heat map showing differentially expressed genes across pseudotime along the ‘Outer root sheath/ Companion layer trajectory’. Genes associated with genetic hair disorders are highlighted in grey. (c) Dot plot showing variance-scaled, mean expression (dot colour) and percent of expressing cells (dot size) of genes implicated in genetic hair diseases in prenatal skin and SkO1. (d) Dot plot showing variance-scaled mean expression (dot colour) and percent of expressing cells (dot size) of genes causing Epidermolysis Bullosa in prenatal skin and SkO1. (e) Indirect immunofluorescence of 15-17 PCW prenatal skin with antibodies against keratin 14, plectin, BP180 (type XVII collagen), laminin-332, type VII collagen, keratin 1. Scale bars = 25 µm. (f) Dot plot showing variance-scaled, mean expression (dot colour) and percent of expressing cells (dot size) of genes causing congenital ichthyoses in prenatal skin and SkO1. ARCI, Autosomal Recessive Congenital Ichthyosis; EKV, Erythrokeratodermia Variabilis. For details on statistics and reproducibility, see Methods.
Extended Data Fig. 7 The role of early dermal fibroblasts in prenatal skin.
(a) UMAP visualisation showing stromal cells found in prenatal skin, coloured by cell state (left) and by gestational age (PCW) (right). (b) Dot plot showing variance-scaled, mean expression (dot colour) and percent of expressing cells (dot size) of ‘pro-inflammatory and immune activation’ genes (as shown in Fig. 3a) in prenatal skin fibroblasts by grouped gestational age (PCW). (c) Heat map showing differentially expressed genes across pseudotime along the ‘Dermal fibroblast trajectory’. (d) Gene set enrichment analysis results for differentially expressed genes (wilcoxon, two-sided, Benjamini-Hochberg adjusted) in Milo-defined early- and late-specific neighbourhoods of WNT2+ fibroblasts. Each plot shows the top 10 enriched gene sets (using Gene Ontology Biological Process 2023). The x-axis shows the negative log10 of the adjusted p-value (Fisher’s exact test, Benjamini-Hochberg correction for multiple testing); dot size is proportional to the number of genes associated with the gene set and colour represents the combined Enrichr score calculated within GSEApy. (e) UMAP visualisation of the myeloid cells in prenatal skin data, coloured by cell state. (f) Dot plot showing variance-scaled, mean expression (dot colour) and percent of expressing cells (dot size) of marker genes5 used to annotate macrophage subsets in prenatal skin. (g) Circos plot visualisation of representative significant (adjusted p-value <0.05, significance calculated in CellphoneDB using empirical shuffling and FDR-adjusted) predicted interactions between macrophages (LYVE1+ and TML macrophage) and co-localising WNT2+ fibroblasts in prenatal skin. Arrows represent directionality of interactions (ligand to receptor); connection width is proportional to the CellphoneDB mean value for each ligand-receptor pair. ASDC, Axl+Siglec6+ dendritic cells; DC, dendritic cells; LC, Langerhans cells.
Extended Data Fig. 8 The role of macrophages in prenatal skin neurovascular development.
(a) UMAP showing clustered cell states in integrated data from embryonic immune cells57 and prenatal skin myeloid cell subset. (b) Heatmap showing the correspondence (measured by Jaccard index) between embryonic immune cells (x-axis) and prenatal skin (y-axis) myeloid cell states for a logistic regression model trained on embryonic data57 and projected onto prenatal skin myeloid cell subset. TML macrophage had the highest proportion prediction to Mac4 (embryonic brain microglia). (c) Gene set enrichment analysis results of over-expressed genes (wilcoxon, two-sided, Benjamini-Hochberg adjusted) in macrophage subsets (TML, Iron-recycling, LYVE1+ and MHCII+, macrophages). Each plot shows the top 10 enriched gene sets (using Gene Ontology Biological Process 2023 (left) and MSigDB Hallmark 2020 (right) databases). The x-axis shows the negative log10 of the adjusted p-value (Fisher’s exact test, Benjamini-Hochberg correction for multiple testing); dot size is proportional to the number of genes associated with the gene set and colour represents the combined Enrichr score calculated within GSEApy. (d) Heatmap showing prediction probabilities from a logistic regression model trained on classes of reindeer fibroblasts (pro-regenerative, pro-inflammatory and mixed populations) (x-axis), projected onto prenatal skin fibroblasts grouped by age (y-axis). Colour scale indicates median prediction probabilities. (e) Dot plot showing the variance-scaled, mean expression (dot colour) and percent of expressing cells (dot size) of selected pro-regenerative and pro-fibrotic genes in prenatal skin fibroblasts by gestational age (grouped PCW). (f) Heatmap showing prediction probabilities from a logistic regression model trained on reindeer macrophage clusters (x-axis), projected onto prenatal skin macrophage subsets (y-axis). Colour scale indicates median prediction probabilities. (g) Percentage scratch width closure (y-axis) quantified over time (x-axis) for fibroblasts cultured with macrophages (green) or in isolation (black) in n = 3 independent experiments. Data represented as percentage values ±SD. Statistics were generated with two-way ANOVA with Tukey’s multiple comparisons test (p-values shown at 6hrs: 0.0480, 12hrs: 0.0035, 18hrs: 0.0001, 24hrs: 0.0042, 66hrs: 0.0118, 72hrs: <0.0001). (h) Circos plot visualisation of selected significant (adjusted p-value <0.05, significance calculated in CellphoneDB using empirical shuffling and FDR-adjusted) predicted interactions between TML macrophages and co-localising neural cells in prenatal skin. Arrows represent directionality of interactions (ligand to receptor); connections are coloured by sender cell type with width proportional to the CellphoneDB mean value for each ligand-receptor pair. (i) Heatmap of normalised (z-score) mean expression of angiogenesis gene modules in prenatal skin macrophages. CD7hiP: CD7high progenitors; CD7loP: CD7low progenitors; ErP, erythroid progenitors; GMP, granulocyte-monocyte progenitors; HSPC, haematopoietic stem and progenitor cells; LC, Langerhans cells; Mac1-4, macrophages 1-4; MkP, megakaryocte progenitors; YSMP, yolk-sac derived myeloid-biased progenitors. For details on statistics and reproducibility, see Methods.
Extended Data Fig. 9 Endothelial cell heterogeneity and interactions with macrophages.
(a) Dot plot visualisation of selected significant (adjusted p-value<0.05, significance calculated in CellphoneDB using empirical shuffling and FDR-adjusted) CellphoneDB-predicted interactions between macrophage subsets and co-localising vascular endothelial cells in prenatal skin, grouped by function. Right: Ligand (first gene in each gene pair) is expressed by macrophages; Left: Ligand (first gene in each gene pair) is expressed by endothelial cells. Dot colour represents the mean expression values of each ligand-receptor pair for the corresponding cell pairs, dot size represents -log10(adjusted p-value). (b) Violin plots of gene module scores in prenatal skin and SkO endothelial cells. Scores were derived from marker genes for the different endothelial cell groups. LE, lymphatic endothelium.
Extended Data Fig. 10 Factors driving angiogenesis and endothelial cell differentiation.
(a) Inferred pseudotime trajectory of prenatal skin endothelial cell states coloured by gestational age (PCW). UMAP overlaid with cell directionality (arrows) as inferred over the cell-to-cell transition probability matrix from CellRank (left) and coloured by pseudotime (right). (b) Heat map showing differentially expressed genes across pseudotime along the ‘arteriolar’ differentiation trajectory. (c) Heat map showing differentially expressed genes across pseudotime along the ‘venular’ differentiation trajectory. (d) Heatmap showing the correspondence (measured by Jaccard index) between prenatal skin (x-axis) and blood vessel organoid cell states71 (y-axis) for a logistic regression model trained on prenatal skin data. The top 10 predicted prenatal cell states were retained for visualisation. (e) Dot plot showing variance-scaled, mean expression (dot colour) and percent of expressing cells (dot size) of blood flow-related genes in prenatal skin and SkO capillary arteriole cells. (f) Heatmap of normalised (z-score) mean expression of hypoxia gene module in prenatal skin and corresponding cell categories in SkO. (g) Violin plots of ‘Tip’ and ‘Stalk’ cell module scores in prenatal skin and SkO endothelial cells. (h) UMAP visualisation of the ‘Tip’ cell module score in prenatal skin (arterioles, capillaries, capillary arterioles, PROX1hi LE, early endothelial cells, LYVE1hi LE, postcapillary venules, venules) and SkO endothelial cells (capillary arterioles). LE, lymphatic endothelium; MEMP, megakaryocyte-erythroid-mast cell progenitor.
Extended Data Fig. 11 Macrophages support prenatal skin and skin organoid angiogenesis.
(a) Dot plot showing variance-scaled, mean expression (dot colour) and percent of expressing cells (dot size) of pro- and anti-angiogenic factors and of corresponding receptors in prenatal skin and SkO endothelial cells. Genes encoding the main pro-angiogenic factors secreted by macrophages in prenatal skin are highlighted. (b) Dot plot showing variance-scaled, mean expression (dot colour) and percent of expressing cells (dot size) of vascular endothelial growth factors in prenatal skin and SkOs. (c) Dot plot showing variance-scaled, mean expression (dot colour) and percent of expressing cells (dot size) of genes (vascular endothelial growth factor receptors and endothelial differentiation) in prenatal skin and SkO capillary arteriole cells. (d) Comparison of regulon activity between prenatal skin (x-axis) and SkO (y-axis) capillary arterioles. (e) Gene regulation network for regulons with high specificity score in prenatal skin and/or SkO capillary arterioles. Arrows indicate the direction of regulation from transcription factor to target gene. Edges show the proportion of genes shared by two regulons (colour for proportion in the larger regulon and thickness for proportion in the smaller regulon). (f) Gene network for four regulons with high specificity score in prenatal skin and/or SkO capillary arterioles (GATA2, GATA1, NFATC1, SOX7), and selected GATA2 target genes. The proportion of red in the ring around nodes indicates the proportion of gene ontology terms associated with angiogenesis in the gene set enrichment analysis performed with genes in the network. (g) Tree diagram showing network of interactions (NicheNet) linking the ligand VEGFA (red) to GATA2 as target gene (blue) through identified signalling mediators and transcriptional regulators (grey). Edges representing signalling interactions are coloured red and gene regulatory interactions in blue; edge thickness is proportional to the weight of the represented interaction. (h) Gating strategy used on iPSC-derived macrophages before co-culture (n = 3 batches) to isolate single live cells, analyse expression of macrophage markers (CD45, CD14, CD16, CD206) and exclude dendritic cells (CD1c). (i) Representative images of angiogenesis assay of endothelial cells without (top) and with macrophages (bottom), red arrows indicate disorganised network. Quantification of endothelial density in 2D cultures of iPSC-derived endothelial cells without (n = 6, grey) and with macrophages (n = 6; magenta) at 24, 48 and 72 h of culture from 2 independent differentiation batches. Data are mean ± SD and statistics generated with an unpaired t-test. For details on statistics and reproducibility, see Methods
Supplementary information
Supplementary Information (download DOCX )
This file contains Supplementary Discussion and Supplementary References
Supplementary Tables (download ZIP )
Supplementary Tables 1–40.
Supplementary Video 1 (download MP4 )
3D view of co-localization of endothelial cells and macrophages with putative contact points. 3D rendering of co-localized areas (magenta) of endothelial cells (CD31+, green) and LYVE1+ macrophages (LYVE1+, yellow) from whole-mount immunostaining of a 15 PCW prenatal skin sample. DAPI nuclear stain is in blue. Scale bar, 100 µm.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Gopee, N.H., Winheim, E., Olabi, B. et al. A prenatal skin atlas reveals immune regulation of human skin morphogenesis. Nature 635, 679–689 (2024). https://doi.org/10.1038/s41586-024-08002-x
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41586-024-08002-x
This article is cited by
-
Generation and characterization of vestibular inner ear organoids from human pluripotent stem cells
Nature Protocols (2026)
-
Spatial architecture of development and disease
Nature Reviews Genetics (2026)
-
Piezoelectric-immunomodulatory electrospun membrane for enhanced repair of refractory wounds
Journal of Nanobiotechnology (2025)
-
Causes and therapeutic limitations of clinical alopecia and the advent of human pluripotent stem cell follicular transplantation
Stem Cell Research & Therapy (2025)
-
The application of organoids in investigating immune evasion in the microenvironment of gastric cancer and screening novel drug candidates
Molecular Cancer (2025)
