
Abstract
Dual-objective 4Pi single-molecule localization microscopy (4Pi-SMLM) offers isotropic nanoscale resolution; however, its broader adoption is limited by instrumental complexity and stringent alignment requirements. Here we introduce mirror-enhanced 4Pi-SMLM (me4Pi-SMLM), a single-objective configuration that uses mirror-based retroreflection of the illumination beam to generate phase-tunable interference fringes. This design improves the axial resolution of astigmatism-based methods by approximately fivefold, delivering performance comparable to conventional 4Pi-SMLM while greatly reducing system complexity and maintenance. me4Pi-SMLM achieves near-isotropic localization precision of 2–3 nm in biological samples, enabling clear and unambiguous visualization of diverse ultrastructural features. Furthermore, it achieves sub-15 nm isotropic resolution in brain slices and facilitates high-fidelity two-colour imaging, nanoscale whole-cell reconstruction and live-cell imaging. me4Pi-SMLM can be seamlessly integrated into existing 3D-SMLM systems, enhancing performance with minimal cost and effort.
Similar content being viewed by others


Main
The considerable improvement in resolution of fluorescence microscopy has transformed biology by enabling the visualization of subcellular ultrastructure and detailed analysis of native protein complexes1,2. Among these advances, SMLM has gained widespread adoption owing to its exceptional spatial resolution and single-molecule sensitivity3. However, conventional three-dimensional SMLM (3D-SMLM) is limited by anisotropic resolution, with axial resolution typically two to three times poorer than lateral resolution4,5,6. Interferometric approaches, such as 4Pi-SMLM, address this limitation by enhancing axial localization precision approximately fivefold through dual-objective coherent detection, achieving isotropic 3D resolution of 10–15 nm for multicolour imaging throughout whole cells7,8,9,10,11. Despite these achievements, the broader application of 4Pi-SMLM remains constrained by its complex instrumentation, stringent alignment requirements and high cost. Although ROSE-Z simplifies this architecture by using excitation interference with a single objective12, it still relies on two-beam interference between extended optical paths, which is inherently susceptible to phase instability arising from thermal fluctuations, mechanical vibrations and drift. These persistent challenges highlight the need for a more accessible, robust and user-friendly interferometric solution for routine biological imaging.
Mirror-based interference in excitation13 or emission14 offers a practical alternative to dual-objective architectures for improving axial resolution. This strategy has recently been integrated with various imaging modalities, such as stimulated emission depletion microscopy15, structured illumination microscopy16 and nanoparticle tracking17, to further enhance axial performance. Inspired by these advances, we developed me4Pi-SMLM, a simplified single-objective configuration that matches the performance of ROSE-Z and rivals dual-objective 4Pi-SMLM, while greatly reducing complexity, maintenance and cost. We demonstrate that me4Pi-SMLM achieves near-isotropic 2–3 nm localization precision, clearly resolving fine ultrastructural features such as the hollow structure of bundled microtubules, distinct subunits within nuclear pore complexes and the sheet-like morphology of the endoplasmic reticulum (ER). The method further enables simultaneous two-colour imaging and whole-cell reconstructions across diverse samples, as well as 3D live-cell imaging and single-molecule tracking at high spatiotemporal resolution. Moreover, it achieves sub-15 nm isotropic resolution in brain tissue slices. Collectively, these capabilities underscore the potential of me4Pi-SMLM for elucidating cellular architecture and dynamics at the molecular scale.
Results
Implementation and characterization of me4Pi-SMLM
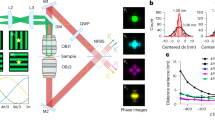
The basic principle of me4Pi-SMLM is illustrated in Fig. 1a, with the optomechanical design detailed in Extended Data Figs. 1 and 2 and Supplementary Video 1. In contrast to conventional 4Pi-SMLM, which relies on a complex dual-objective configuration, our design uses a single objective by eliminating the upper beam path. This architecture circumvents the need for expensive objective pairing and intricate dual-objective alignment. Instead, a mirror positioned above the sample retroreflects the illumination beam to directly generate interference fringes. For precise axial localization, a piezoelectric actuator rapidly translates the mirror within milliseconds, shifting the interference patterns with sub-nm precision to sequentially acquire three phase-shifted images with 2π/3 increments (φ1, φ2 and φ3) (Extended Data Fig. 3). This self-referenced interference renders the system highly resistant to mechanical vibrations and obviates the need for a dedicated, real-time focus-lock module10,18,19. To resolve periodic ambiguity, slight astigmatism is introduced by a cylindrical lens, enabling coarse axial localization and reliable phase unwrapping (Fig. 1b and Extended Data Fig. 4). Collectively, this architecture streamlines mechanical design, optoelectronic synchronization, optical alignment and image processing, greatly simplifying overall operation. The result is an accessible, cost-effective platform that retains the high-resolution advantages of 4Pi-SMLM while removing associated technical barriers.

a, Simplified schematic of me4Pi-SMLM. The laser is focused onto the back focal plane of the objective to illuminate the sample. After passing through the objective and the sample, the laser beam is reflected by a protected silver mirror, generating an axial standing-wave interference pattern. A piezoelectric actuator rapidly translates the mirror within milliseconds, shifting the interference pattern with sub-nm precision. Fluorescence signals are collected by the objective and imaged onto an sCMOS camera. L, lens; M, mirror; DM, dichroic mirror; OBJ, objective; TL, tube lens; CL, cylindrical lens; CAM, camera. b, Top, simulated PSFs at various axial positions and under illumination patterns with different phases; bottom, intensities of three sub-images at different axial locations. c, Distributions of the relative 3D positions of two 40 nm fluorescent beads, reconstructed with me4Pi-SMLM and 3D-SMLM. The 3D-SMLM result was obtained by performing astigmatic Gaussian fitting on the summed phase images. The two beads yielded photon counts of 5,527 ± 358 and 5,091 ± 246 (mean ± s.d.), respectively. d, Histograms of the centred distance between two 40 nm fluorescent beads in c in each direction, reconstructed with me4Pi-SMLM. For comparison, the result in the axial direction reconstructed with 3D-SMLM is also shown.
We implemented the me4Pi-SMLM localization pipeline, building on established algorithms for interferometric SMLM (Extended Data Fig. 5)7,9,12. Unlike previous methods that require multiple camera regions9,12 or several cameras7, our approach sequentially acquires three phase-shifted images on a single camera region, thereby eliminating the need for channel registration. Single molecules were detected in summed images, and astigmatism-based 2D Gaussian fitting20 was applied to estimate lateral positions (x, y) and the point spread function (PSF) widths (σx, σy). The PSF widths were used to determine coarse axial positions based on a calibration curve generated from fluorescent beads on coverslips4 (Extended Data Fig. 4a,b). Subsequently, sub-image intensities were analysed to extract phase and modulation depth along the z direction12 (Supplementary Note 1). Finally, a ridge-finding algorithm was used to unwrap the phases and retrieve precise z positions9 (Supplementary Fig. 1 and Supplementary Note 1). For me4Pi-SMLM, the precise z position was used, whereas for 3D-SMLM, the coarse z position was used.
We characterized me4Pi-SMLM by imaging 40 nm fluorescent beads. Calibration showed an interference period of 210 nm for 552 nm excitation (Extended Data Fig. 4c). By scanning the piezoelectric actuator, we determined the voltage corresponding to a 2π/3 shift in the interference fringes (Supplementary Fig. 2). The modulation depth was estimated to be approximately 0.9 (Supplementary Fig. 3). Under these conditions, me4Pi-SMLM achieved an approximately fivefold improvement in axial localization precision compared to conventional 3D-SMLM, reaching 2–3 nm localization precision in all dimensions with a photon budget of ~5,300 (Fig. 1c,d).
me4Pi-SMLM resolved subcellular architecture
We then evaluated the performance of me4Pi-SMLM on biological samples. Given that me4Pi-SMLM requires three sequential phase images for precise axial localization, we used DNA point accumulation for imaging in nanoscale topography (DNA-PAINT) to mitigate intensity fluctuations caused by fluorophore photoblinking for optimal performance21,22,23. To accommodate the relatively long on-time of DNA-PAINT, imaging was conducted in slow imaging mode, with a cycle time of 45 ms (Extended Data Fig. 3a). Additionally, nanobodies were used instead of conventional antibody pairs when available to reduce label size24,25. As a benchmark, microtubules in COS-7 cells were labelled by overexpression of ALFA-tagged Ensconsin, immunolabelled with NbALFA-conjugated DNA docking strands and imaged with DNA-PAINT (Fig. 2a). The enhanced axial resolution of me4Pi-SMLM resolved the ring structure of individual and bundled microtubules more effectively than 3D-SMLM (Fig. 2b). Localization analysis of molecules emitting ten or more frames showed a 2–3 nm localization precision for me4Pi-SMLM in cells with a photon budget of ~5,500 (Fig. 2c,d). Additionally, the 3D localization precision determined by the distance between adjacent-frame localizations (DAFL)26 was estimated to be 2.6 nm (Supplementary Fig. 4a), while the Fourier ring correlation (FRC)27 resolution was determined to be better than 10 nm in all dimensions (Supplementary Fig. 4b). Quantitative analysis using the rolling FRC method28 confirmed uniform resolution across the field of view (Extended Data Fig. 6). Moreover, we verified that me4Pi-SMLM was fully compatible with conventional immunolabelling for imaging endogenous proteins (Supplementary Fig. 5).

a, me4Pi-SMLM image of microtubules in a fixed COS-7 cell. b, 200 nm-thick cross-sections at locations denoted by numbers in a, reconstructed with me4Pi-SMLM and 3D-SMLM. c, Distributions of 3D localization results from molecules emitting ten frames or more. The photon count for these molecules is 5,550 ± 1,332 (mean ± s.d). d, Histograms of the distributions in c, fitted with Gaussian functions, and the standard deviations are reported. e, me4Pi-SMLM image of Nup96 in a fixed U-2 OS cell. f, Zoomed-in image of the blue boxed region in e and x–z view at the yellow region, reconstructed with me4Pi-SMLM and 3D-SMLM. g, 3D particle averaging results of 500 NPCs from the nucleus shown in e. The lower-right shows the 3D visualization of the averaged NPC after cluster analysis. h, me4Pi-SMLM image of ER in a fixed COS-7 cell. i, A 50 nm-thick x–y slice of the yellow boxed region in h, and a 100 nm-thick x–z cross-section along the dashed line, reconstructed with me4Pi-SMLM and 3D-SMLM. j, Zoomed-in image of the blue boxed region in h, and 100 nm-thick x–z cross-sections along the dashed line denoted by numbers, reconstructed with me4Pi-SMLM and 3D-SMLM. Colour bar denotes depth. Representative images are shown from two to five independent experiments.
Next, we imaged the nuclear pore complex (NPC) in fixed U-2 OS cells. Nup96, an NPC protein exhibiting eightfold symmetry, forms two rings: a nucleoplasmic ring and a cytoplasmic ring29,30. We performed DNA-PAINT imaging on a homozygous knock-in U-2 OS cell line23 expressing endogenously ALFA-tagged Nup96 (Fig. 2e and Supplementary Video 2). me4Pi-SMLM resolved both the nucleoplasmic ring and cytoplasmic ring with greater clarity than 3D-SMLM (Fig. 2f). Through particle averaging of 500 NPCs with advanced algorithms31,32, me4Pi-SMLM visualized nearly all 32 copies of Nup96 (Fig. 2g), with quality comparable to conventional 4Pi-SMLM31 and Ångström-precision MINSTED33. In the structural average, me4Pi-SMLM resolved two adjacent Nup96 proteins, separated by 10 nm laterally and 3 nm axially34, demonstrating a 3D resolution better than 10 nm (Supplementary Fig. 6).
Additionally, we imaged the ER in fixed COS-7 cells. me4Pi-SMLM revealed the ER as a connected network of hollow tubes (60–100 nm in diameter) and sheet-like structures (Fig. 2h). Both x–y and x–z cross-sections displayed detailed 3D membrane contours that are challenging to resolve with conventional super-resolution techniques18,35 (Fig. 2i). Notably, the high axial resolution of me4Pi-SMLM enabled clear visualization of the sheet-like structures with thickness of 30–50 nm, which are thinner than ER tubules and consistent with values obtained by 4Pi-SMLM35 (Fig. 2j and Supplementary Video 3). These observations, together with previous live-cell super-resolution imaging results18, support the existence of peripheral ER sheets, whose functions remain to be elucidated.
Simultaneous two-colour me4Pi-SMLM imaging
Although the high resolution of me4Pi-SMLM is instrumental for resolving subcellular architecture, multicolour imaging is essential for interrogating the spatial organization and interactions within organelles and protein complexes. To enable this capability, we integrated a previously established salvaged fluorescence method11 with me4Pi-SMLM for simultaneous two-colour imaging. We identified Alexa Fluor 568 (AF568) and ATTO Rho11 as spectrally compatible dyes for pairing with Cy3B in DNA-PAINT under 552 nm excitation (Extended Data Fig. 7). Microtubules and ER were co-labelled in COS-7 cells by stable ALFA-Ensconsin expression and transient mEmerald-Sec61β overexpression, followed by immunolabelling with anti-mEmerald and anti-ALFA nanobodies, respectively (Fig. 3a,b). me4Pi-SMLM achieved robust spectral separation with minimal crosstalk, resolving the hollow centres of both ER tubules and microtubules laterally and axially (Fig. 3c,d). For further validation, we labelled the ER outer membrane (Sec61β) and inner lumen (KDEL) using the corresponding nanobodies (Fig. 3e and Supplementary Video 4). The high resolution effectively distinguished these closely spaced compartments and resolved distinct ER substructures (Fig. 3f–i).

a, Two-colour me4Pi-SMLM image of ER and microtubules in a fixed COS-7 cell. The lower-left corner shows the depth colour-coded image, while the upper-right corner shows the merged image of the two labels. b, Zoomed-in image of the yellow boxed region in a. c, A 10 nm-thick x–y slice of the blue boxed region in a. d, A 500 nm-thick y–z cross-section at the dashed region and a 100 nm-thick x–z cross-section along the dashed line in b. e, Merged two-colour me4Pi-SMLM image of ER membrane and lumen in a fixed COS-7 cell. f, Zoomed-in image of the yellow boxed region denoted in e. The lower-left and upper-right corners show the KDEL labelling and Sec61β labelling, respectively. g. A 50 nm-thick x–y slice of the yellow boxed region denoted in e. h, The 200 nm-thick x–z cross-sections at locations denoted by the numbers in g. i, Intensity profile along the white line in g. Colour bar denotes depth. a.u., arbitrary units. Representative images are shown from two to three independent experiments.
me4Pi-SMLM imaging in whole cells
me4Pi-SMLM overcomes axial ambiguity by introducing astigmatism through a cylindrical lens, enabling high-resolution imaging within thick specimens. Given that the objective used in our system has a depth of field of ~1.2 μm, imaging volumes exceeding this thickness require axial sample scanning. To ensure sufficient overlap between each optical section for subsequent stitching, we typically translated the sample axially in 500 nm steps. To illustrate its whole-cell imaging capability, we visualized synaptonemal complexes in mouse spermatocyte nuclei. These essential supramolecular structures mediate chromosome synapsis, recombination and faithful segregation during meiosis. Following immunolabelling of the scaffold protein SYCP3, me4Pi-SMLM resolved fully synapsed chromosomes throughout the nucleus, clearly revealing the characteristic double-helical architecture of the synaptonemal complex substructures irrespective of their spatial orientation or depth (Fig. 4a–e and Supplementary Video 5). Additionally, we imaged the mitochondria outer membrane in HeLa cells to a depth of 4.5 μm, reconstructing the volume from seven optical sections to resolve membrane contours and complex interconnections with high clarity and no detectable ghost artefacts (Fig. 4f,g and Supplementary Video 6). Quantitative analysis confirmed that me4Pi-SMLM maintained a high DAFL localization precision of 3–5 nm and an FRC resolution of 10–15 nm in all dimensions throughout the entire imaging volume (Extended Data Fig. 8).

a, 3D rendering of computationally isolated synaptonemal complexes in a fixed mouse spermatocyte imaged with me4Pi-SMLM. b, y–z cross-sections at the yellow dashed region in a. c, y–z view of a. d, Zoomed-in images of 19 synaptonemal complexes, computationally isolated from the entire mouse spermatocyte nuclei. e, Representative 3D views of two synaptonemal complexes shown in d. f, 3D whole-cell imaging of mitochondria in HeLa cells imaged with me4Pi-SMLM. g, x–y projection of f and the cross-sections at locations denoted by the numbers. The colour bar denotes depth. All cross-section slices are 100 nm thick. Representative images are shown from two independent experiments.
me4Pi-SMLM imaging at higher acquisition speeds
The primary limitation of me4Pi-SMLM is the requirement for molecules to remain fluorescent across three consecutive phase images, necessitating relatively long on-times. Although compatible with DNA-PAINT, this constraint is suboptimal for techniques relying on intrinsic fluorophore photophysics, such as (d)STORM36,37. To address this limitation, we implemented a fast imaging mode using reprogrammed synchronization (Extended Data Fig. 3b). We validated this capability by imaging microtubules in fixed COS-7 cells with fluorogenic DNA-PAINT probes, which exhibit faster binding kinetics than conventional variants. With a cycle time of 9 ms, fivefold faster than the slow imaging mode, we obtained an FRC resolution of ~8 nm and resolved the hollow centres of microtubules with image quality comparable to that of the slow mode (Extended Data Fig. 9).
This increased acquisition speed renders our approach suitable for dSTORM applications. As a demonstration, we imaged immunolabelled mitochondria in fixed COS-7 cells using antibodies conjugated to Alexa Fluor 647 (AF647) with a 15 ms cycle time (Extended Data Fig. 10). The resulting images resolved the membrane contours and interconnections of mitochondria with high fidelity. By aligning me4Pi-SMLM with conventional SMLM frame rates (typically 10–20 ms), this fast imaging mode ensures compatibility with dSTORM and a broad range of DNA-PAINT variants, including speed-optimized22, fluorogenic21, self-quenching38 and TDI39, thereby greatly expanding its versatility for biological studies.
me4Pi-SMLM imaging in living cells
Live-cell imaging using conventional 4Pi-SMLM approaches has remained undemonstrated to date, owing to several technical challenges. The inherent system complexity demands stringent optical alignment, exacerbates mechanical and thermal drift, diminishes fluorescence collection efficiency and precludes standard sample mounts, such as dishes or multi-well chambers. By contrast, me4Pi-SMLM features reduced optical complexity, improved stability, enhanced photon efficiency and compatibility with confocal dishes, thus holding potential for live-cell imaging applications.
As a proof of concept, we generated a U-2 OS cell line stably expressing Sec61β-Emerald-HaloTag and labelled it with a live-cell-compatible dye, PA-JF549-HaloTag40. We imaged live cells in confocal dishes with me4Pi-SMLM at a cycle time of 9 ms, capturing ER dynamics over 10 min (Fig. 5a). At a temporal resolution of 10 s, comparable to prior 3D-SMLM studies41, the reconstructions revealed the dynamic tubule network of ER at the cell periphery (Fig. 5b). Representative snapshots depicted ER tubule growth (Fig. 5c), as well as the fusion and disappearance of two multi-junctions (Fig. 5d). ER tubule diameters were measured to be 60–70 nm (Fig. 5e,f), consistent with prior 2D-SMLM reports41. The FRC resolutions were determined to be 40–60 nm in each dimension (Fig. 5g).

a, Super-resolution image of the ER in a live U-2 OS cell recorded over 10 min. Colour bar denotes time. b, 3D super-resolution reconstruction of the ER from images acquired over 10 s. Colour bar denotes depth. c,d, Time-lapse images of ER dynamics from the boxed regions denoted in b. e,f, Lateral and axial intensity profiles along the magenta line denoted in b were fitted with Gaussian functions to estimate the full width at half maximum (FWHM) of the ER tubule. g, FRC analysis of the image shown in b. h, Single-molecule image of the ER in a live U-2 OS cell. Green circles indicate the positions of single molecules. i, Cumulative image of single-molecule trajectories over 30 s. Tracks comprising fewer than ten frames are not displayed. Colour bar denotes time. j, Representative 3D single-molecule trajectories showing different molecular mobility. k, Distribution of localization precision in each dimension. l, Histogram of track duration. m, Histogram of total displacement. n, Histogram of mean speed. o, Histogram of confinement ratio. Representative images are shown from four to five independent experiments.
Recently, 4Pi-SIM demonstrated isotropic 3D resolution of 100 nm in live cells at a volumetric rate of ~1 Hz18. By comparison, the localization-based nature of me4Pi-SMLM limits its temporal resolution for capturing rapid organelle dynamics. To leverage its high spatial precision, we instead applied me4Pi-SMLM to 3D single-molecule tracking. By using the same cells described above, we combined a reduced concentration of PA-JF549-HaloTag with pulsed activation to maintain an optimal molecular density for tracking (Fig. 5h and Supplementary Video 7). Cumulative trajectories over 30 s effectively reconstructed the ER tubular network (Fig. 5i), while individual 3D tracks revealed heterogeneous molecular mobility (Fig. 5j). Notably, we achieved a localization precision of ~6–7 nm in each direction (Fig. 5k), enabling quantitative analysis of diverse molecular behaviours at the nanometre scale (Fig. 5l–o). In particular, the confinement ratio peaked at 0.2, consistent with Sec61β molecules confined within ER membranes (Fig. 5o). With its sub-10 nm spatial precision and sub-10 ms temporal resolution, me4Pi-SMLM promises to be a powerful approach for investigating molecular dynamics at inter-organelle contact sites.
me4Pi-SMLM imaging in brain slices
Thus far in this study, super-resolution imaging has been confined to cells cultured on coverslips, an experimentally convenient but highly artificial environment that does not recapitulate the complex architecture of native tissues. Extending such imaging to tissue samples is particularly challenging for conventional 4Pi-SMLM, which relies on coherent interference of photons emitted by single molecules and collected through two opposing objectives. In practice, refractive-index inhomogeneities, scattering and depth-dependent aberrations in tissue disrupt the phase relationships required for interference, degrading localization performance and limiting imaging depth. By contrast, me4Pi-SMLM uses a detection configuration equivalent to that of a single-objective 3D-SMLM microscope, making it inherently more compatible with tissue imaging. Motivated by this advantage, we evaluated me4Pi-SMLM for high-resolution imaging in tissue slices.
First, we imaged mitochondria in 30 µm-thick mouse brain slices at multiple depths (Fig. 6a and Supplementary Fig. 7). Across the tissue volume, we observed distinct morphologies of mitochondria, including granule-like, rod-like and elongated forms varying at different regions and depths (Fig. 6b–e). Notably, these mitochondrial architectures differed markedly from the extended tubular networks typically observed in cultured cells (Fig. 4f). Quantitative analysis confirmed that we obtained DAFL 3D precision ~6 nm (Fig. 6f) and FRC resolution ~ 12 nm, both maintained to an imaging depth of ~ 20 µm (Fig. 6g).

a, me4Pi-SMLM image of mitochondria in a 30 μm-thick mouse brain slice at a depth of ~20 μm. b,c, 3D rendering of the boxed regions denoted in a. d,e, 3D rendering of the boxed regions from tissue imaging at depths of 0 μm and 10 μm, shown in Supplementary Fig. 7. Cross-sections at the dashed lines denoted by numbers in b, c, d and e. f, DAFL 3D precision of the mitochondria image at each depth. g, FRC resolution (averaged from xy, xz and yz) of the mitochondria image at each depth. h, me4Pi-SMLM image of ChR2 in a 30 μm-thick mouse brain slice at a depth of ~20 μm (Supplementary Video 8). i, 3D rendering of the boxed regions denoted in h. j,k, 3D rendering of the boxed regions from tissue imaging at depths of 0 μm and 10 μm, shown in Supplementary Fig. 8. l, Cross-sections at the dashed line denoted by numbers in i, j and k. m, DAFL 3D precision of the ChR2 image at each depth. n, FRC resolution (averaged from xy, xz and yz) of the ChR2 image at each depth. Colour bar denotes depth. Representative images are shown from two to three independent experiments.
Next, we examined the nanoscale organization of channelrhodopsin 2 (ChR2), a light-gated ion channel essential for optogenetic modulation. Imaging 30 μm brain slices revealed discrete, membrane-associated ChR2 clusters distributed along axons (Fig. 6h, Supplementary Fig. 8 and Supplementary Video 8). These assemblies appeared as hollow tubular features and blob-like structures throughout the axonal membrane (Fig. 6i–l). With ~ 6 nm DAFL 3D precision (Fig. 6m) and sub-15 nm FRC resolution, we reconstructed the 3D molecular organization of ChR2 (Fig. 6n). This capacity to resolve molecular-level details within tissue slices bridges the gap between ultrastructure and physiological function, establishing me4Pi-SMLM as a transformative tool for advanced neuroscience research.
Discussion
We have developed me4Pi-SMLM, a simple solution for 3D-SMLM that achieves sub-10 nm resolution in three dimensions while accounting for label size in biological samples (Supplementary Table 1). me4Pi-SMLM delivers performance on par with ROSE-Z and is comparable to conventional 4Pi-SMLM, but with greatly reduced hardware complexity and alignment effort. Notably, almost any existing 3D-SMLM setup can be upgraded to me4Pi-SMLM by simply adding a mirror and an affordable piezoelectric actuator. This stands in sharp contrast to conventional 4Pi-SMLM, which demands extensive expertise and substantial financial investment19, restricting its use to only a few specialized laboratories worldwide.
In fast imaging mode, the acquisition speed of me4Pi-SMLM is ultimately constrained by the camera readout and the piezo actuator’s step-and-settle time (~1 ms in this study). Using a faster piezo actuator combined with a resonant mirror12 or electro-optic modulator42 to rapidly project phase images onto separate camera regions would enable even shorter exposure times and broaden compatibility with a wider range of biological applications. Moreover, deep learning based approaches43 have enabled dense single-molecule localization, offering the potential to improve the temporal resolution of me4Pi-SMLM in live-cell imaging.
In this study, tissue imaging was limited to a depth of ~20 µm as a proof of concept. Past this depth, sample-induced optical aberrations dramatically degrade image quality, compromising reconstruction fidelity, particularly along the axial dimension. However, recent advances in 3D-SMLM have demonstrated effective correction of such aberrations using sophisticated algorithms and adaptive optics44,45. Integrating these approaches into me4Pi-SMLM could extend imaging depth to approximately 50 µm. Furthermore, complementary techniques such as tissue clearing and expansion microscopy46,47 could be used to minimize optical aberrations; in such cases, background fluorescence would probably become the dominant factor constraining imaging depth.
Historically, SMLM was developed using widefield illumination3, whereas MINFLUX pioneered the use of patterned illumination to enhance localization precision48,49. Although MINFLUX-like techniques are inherently limited in throughput owing to their reliance on single doughnut-shaped illumination, parallelized strategies using striped patterns have subsequently been used to improve lateral50,51,52 or axial resolution12,42, albeit with reduced resolution gains. In this study, me4Pi-SMLM uses axial illumination interference exclusively to achieve superior axial localization but can be seamlessly integrated with lateral illumination interference for enhanced resolution in all dimensions. This strategy offers two key advantages: molecular localization in all dimensions is determined directly by molecular photon emissions relative to the illumination patterns, rather than by PSF patterns, making it inherently resistant to optical aberrations; and given that localization is defined by the illumination wavelength, chromatic aberrations are eliminated in multicolour imaging. Combined with advanced labelling strategies that minimize probe size and improve efficiency, me4Pi-SMLM enables multiplexed, molecular-scale imaging across a diverse range of biological contexts.
Methods
Microscope setup
The complete optomechanical configuration is detailed in Extended Data Figs. 1 and 2 and Supplementary Video 1. The microscope was constructed on an inverted frame (ECLIPSE Ti2-E, Nikon). Four excitation lasers, 405 nm (OBIS LX 100 mW, Coherent), 488 nm (OBIS LS 150 mW, Coherent), 552 nm (2RU-VFL-P-2000-552, MPB Communications) and 642 nm (2RU-VFL-P-2000-642, MPB Communications), were combined using dichroic mirrors (DM1, LM01-613-25; DM2, LM01-503-25; DM3, LM01-427-25; all from Semrock) and modulated by an acousto-optical tunable filter (AOTF) (AOTFnC-400.650-TN, AA Opto-Electronic). The combined beam was coupled into a single-mode fibre (P1-488PM-FC-2, Thorlabs) by a coupling lens (PAF2P-A10A, Thorlabs). The fibre output was collimated by lens L1 (f = 50 mm; AC254-050-A, Thorlabs), expanded by lenses L2 (f = 100 mm; AC254-100-A, Thorlabs) and L3 (f = 250 mm; ACT508-250-A, Thorlabs) and focused onto the back focal plane of the objective by lens L4 (f = 400 mm; ACT508-400-A, Thorlabs) for sample illumination. An oil-immersion objective (UPLAPO100XOHR, ×100/1.5 NA, Olympus) was used for imaging structures near the coverslip, whereas a silicone oil-immersion objective (UPLSAPO100XS, ×100/1.35 NA, Olympus) was used for whole-cell and tissue imaging. Sample positioning was controlled by an automated stage (PZ-2000FT, Applied Scientific Instrumentation), and precise axial positioning of the objective was driven by a PIFOC piezo scanner (P-721.12Q, Physik Instrumente). After traversing the objective and sample, the excitation beam was retroreflected back by a protected silver mirror, generating the axial standing-wave interference pattern at the sample plane.
Fluorescence detection
Emitted fluorescence was separated from the excitation light using a dichroic mirror (DM4: ZT543rdc, Chroma) and filtered with a longpass emission filter (F1: ET560lp, Chroma). For single-colour imaging, signals exited through the right port of the microscope, passed through a cylindrical lens (CL1: f = 1000 mm; LJ1516RM-A, Thorlabs) to induce slight astigmatism for coarse axial localization and were focused onto an sCMOS camera (CAM1: ORCA-Fusion; C14440-20UP, Hamamatsu) with an additional emission filter (F2: ET560lp, Chroma (for cell imaging); ET605/70 m, Chroma (for tissue imaging)) placed in front of the camera. For two-colour imaging, signals exited through the left port, passed through a cylindrical lens (CL2: f = 1,000 mm; LJ1516RM-A, Thorlabs) and were relayed by a lens pair (L5 and L6/L7: f = 200 mm; 49364, Edmund). A dichroic mirror (DM5: ZT561rdc, Chroma) spectrally separated the light: conventional fluorescence passed through a bandpass filter (F3: ET605/70 m, Chroma), while salvaged fluorescence passed through a separate filter (F4: FF01-572/28-25, Semrock). A right-angle prism mirror (RAP120-RA-A, LBTEK) projected both channels onto distinct regions of a second sCMOS camera (CAM2: ORCA-Fusion; C14440-20UP, Hamamatsu). For dSTORM imaging of AF647, an alternative filter set was used: DM4 (ZT405/488/561/640rpcv2, Chroma), F1 (ZET405/488/561/640mv2, Chroma) and F2 (ET700/75m, Chroma).
Axial interference illumination module
The module design is illustrated in Extended Data Fig. 2 and Supplementary Video 1. To ensure a stable axial standing-wave interference pattern, the module was mechanically isolated from the microscope frame using a custom mount secured directly to the optical platform, positioned above the sample stage for optimal alignment. A 10 mm diameter protected silver mirror (M11; 34-386, Edmund) was attached to a piezoelectric actuator (S23.Z10K, CoreMorrow) for precise axial positioning. This assembly was mounted on a kinematic mirror mount (MT-AM1, LBTEK) to allow for coarse tip and tilt adjustments. A piezo linear stage (N-565.260, Physik Instrumente) provided coarse axial positioning and sufficient clearance for sample replacement.
Data acquisition
Two synchronization control modes were implemented. Slow imaging mode (Extended Data Fig. 3a) was driven by an NI-DAQ card (USB-6363, BNC, National Instruments) and the LabVIEW 2022 DAQ module. A periodic voltage waveform consisting of three 15 ms phases was generated by an analogue output port to control the piezoelectric actuator. Simultaneously, the sCMOS camera operated in external edge trigger mode, initiated by the rising edge of a digital output signal. To provide sufficient mechanical settling time for the mirror, the digital output signal was delayed by 3 ms relative to the analogue output signal. The camera exposure time was set to 10 ms, and the AOTF was maintained in continuous transmission mode throughout image acquisition. For fast imaging mode (Extended Data Fig. 3b), the sCMOS camera operated in internal trigger mode. A field-programmable gate array (PXIe-7867, National Instruments) monitored the camera readout signal and transmitted synchronized control signals to the AOTF to dictate the effective exposure time, as well as to the piezoelectric actuator to drive cyclic motion to predefined axial positions.
System calibration
To generate the calibration file for astigmatic localization, a sparse sample of 40 nm fluorescent beads (F8793, 580/605 nm, Invitrogen) was axially scanned over a range of 1 µm using the piezo stage at three distinct phases to acquire a 3D image stack. PSF widths were calculated to estimate the coarse z positions, revealing a linear relationship with the actual axial displacement. To calibrate the interference fringe period, the intensities of each phase were measured, and the positional and phase data were then fitted to determine the axial period of the interference fringes (Extended Data Fig. 4c). To determine the voltage of the piezoelectric actuator corresponding a 2π/3 shift in the interference fringe, a sparse sample of 40 nm beads was imaged while the sample remained stationary. The piezoelectric actuator was actuated using a 4–6 V analogue output signal. Frame-by-frame intensity measurements were fitted to a sine function to determine the oscillation period (2π), and the voltage corresponding to a 2π/3 shift was identified (Supplementary Fig. 2).
Reconstruction
The algorithm workflow is illustrated in Extended Data Fig. 5. We adopted previously developed pipelines9,12 with minor modifications. Sub-images (Z1, Z2, Z3) corresponding to the three phase-shifted illumination patterns were summed. Two-dimensional Gaussian fitting was used to estimate the lateral (x–y) positions and PSF widths (σx, σy) for each molecule. For 3D-SMLM, σx and σy were used to calculate coarse axial positions. For me4Pi-SMLM, the intensities of the sub-images were analysed to determine the phase and modulation depth along the z direction (Supplementary Note 1), and a ridge-finding algorithm was applied to unwrap the phases and determine precise z positions9 (Supplementary Fig. 1 and Supplementary Note 1). Colour assignment followed established protocols11. All images and videos were rendered using Vutara SRX software (Bruker). For live-cell imaging, data processing followed the same pipeline. For image reconstruction, localizations acquired within a defined time window (10 s in this study) were combined to generate a super-resolved image. For single-molecule tracking, 2D detection, localization and tracking were performed using TrackMate53. Subsequently, the axial position of each molecule was estimated using the aforementioned method and integrated with the 2D data to generate 3D trajectories. Trajectories shorter than ten frames were excluded from subsequent mobility analysis.
Drift correction
Given that me4Pi-SMLM uses interference illumination to achieve precise axial localization, both positional and phase drift can affect the final localization accuracy. To correct for phase drift, we adopted a previously developed cross-correlation approach54. In brief, localized molecules were divided into temporal segments, typically corresponding to a time window of 1,000–3,000 frames. A series of 2D histogram images representing phase and normalized metric of the molecules was generated and Gaussian-blurred (Supplementary Note 1). Cross-correlation was then used to estimate phase drift between images, which was subsequently corrected by spline interpolation. This approach substantially reduces phase drift, thereby enabling robust phase unwrapping and the reliable conversion of phase data into axial coordinates (Supplementary Fig. 1). For 3D sample drift correction, the AIM (adaptive intersection maximization)55 and DME (drift at minimum entropy)56 algorithms were applied sequentially, during which any residual phase drift was also addressed.
Buffers
The following buffers were used for DNA-PAINT or dSTORM imaging:
-
Blocking buffer A: PBS (1× PBS; Gibco, 10010023), 3% BSA (Jackson ImmunoResearch, 001-000-162), 0.2% Triton-X100 (Sigma-Aldrich, T8787);
-
Blocking buffer B: PBS, 3% BSA, 0.25% Triton-X100;
-
Blocking buffer C: PBS, 3% BSA, 1% Triton-X100;
-
Dilution buffer A: PBS, 1% BSA, 0.2% Triton-X100;
-
Dilution buffer B: PBS, 1% BSA, 1% Triton-X100;
-
Wash buffer: PBS, 0.1% Triton-X100;
-
Buffer C: PBS, 1 mM EDTA (Invitrogen, 91222915), 500 mM NaCl (Sigma-Aldrich, S5886-5KG), 0.02% Tween-20 (Sigma-Aldrich, P1379-500ML), pH 7.4;
-
Buffer D: 5 mM TCEP (Sigma-Aldrich, C4706-2G) + PBS + 1 mM EDTA, pH 6.8;
-
Trolox (100×) (Sigma-Aldrich, 238813-1 G): 100 mg Trolox, 430 µl 100% methanol (Sigma-Aldrich, 322415-100 ML), 480 µl 1 M NaOH (Sigma-Aldrich, S5881-500G) in 3.2 ml water;
-
Buffer E: PBS, 500 mM NaCl, 20 mM Na2SO3 (Sigma-Aldrich, 71988-250 G), 1 mM Trolox, pH 7.4;
-
Storage buffer (50% glycerol (Sigma-Aldrich, V900122), PBS);
-
Cytoskeletal buffer (CB buffer: 10 mM MES (Sigma-Aldrich, M3671-50G), 150 mM NaCl, 5 mM MgCl2 (Sigma-Aldrich, M2393-500G), 5 mM EGTA (Sigma-Aldrich, 03777-10 g), 5 mM glucose (Sigma-Aldrich, G8270-1kg), pH 6.1);
-
Base buffer (44% glycerol, 50 mM Tris pH 8.0 (Thermo Fisher Scientific, 15568025), 10 mM NaCl, 10% glucose).
DNA-PAINT docking and imager sequences
HPLC-purified azide-labelled, dye-labelled and quencher-labelled oligonucleotides were obtained from Ningbo Karebay Biochem and GENEray Biotechnology. Three docking strands (L5R2, S4 and LS4) and their corresponding imager strands were used in this study. Specifically, L5R2 was paired with LR2; S4 with P10, P33 and P81; and LS4 with LP10 and LP71. To minimize non-specific binding, the ‘L-Oligo’ sequences (L5R2, LS4, LR2, LP10 and LP71) were synthesized using left-handed DNA57. S4 and LS4 were used as orthogonal pairs for two-colour imaging. The nucleotide sequences for all docking and imager strands used in this study are detailed below:
Oligonucleotide sequence (5′–3′)
Docking strands
L5R2: azide_ACCACCACCACCACCACCA
S4: CCTTCAACATTTCTTCTAC_azide
LS4: CCTTCAACATTTCTTCTAC_azide
Imager strands
LR2: TGGTGGT_Cy3B
P10: Cy3B_AGAAGTAATGTGGAA_BHQ2
P33: ATTO643_AGAAGTAATGTGGAA_BBQ650
P81: ATTO Rho11_AGAAGTAATGTGGAA_BHQ2
LP10: Cy3B_AGAAGTAATGTGGAA_BHQ2
LP71: AF568_AGAAGTAATGTGGAA_BHQ2
Conjugation of secondary antibodies with docking strands
Docking strands were conjugated to goat anti-mouse (Jackson ImmunoResearch, 115-005-146, lot no. 171417) and donkey anti-rabbit (Jackson ImmunoResearch, 711-005-152, lot no. 159871) secondary antibodies as previously described21. In brief, 250 µg of antibody was prepared using 100 kDa MWCO Amicon Ultra centrifugal filters (Sigma-Aldrich, UFC5100) and reacted with a 20-fold molar excess of DBCO-sulfo-NHS ester crosslinker (Sigma-Aldrich, 762040) overnight in the dark at 4 °C to label surface-exposed lysine residues. Unreacted crosslinker was subsequently removed using a ZEBA spin desalting column (7 kDa MWCO, Thermo Fisher Scientific, 89882), followed by three washes with PBS using the 100 kDa MWCO centrifugal filters. DBCO-functionalized antibodies were recovered by filter inversion and centrifugation. Next, copper-free click chemistry was used to attach the azide-modified DNA oligonucleotides (S4 and LS4). A 20-fold molar excess of the respective azide-DNA was added to the DBCO-conjugated antibodies, gently mixed and incubated in the dark at room temperature (20−25 °C) for 1 h. The resulting DNA-antibody conjugates (goat anti-mouse-S4 and donkey anti-rabbit-LS4) were purified from free DNA through five PBS washes using 100 kDa MWCO centrifugal filters. The final recovered conjugates were adjusted to a concentration of 20 µM in storage buffer and stored at −20 °C.
Nanobody–DNA conjugation
Nanobodies engineered with ectopic amino-terminal and carboxy-terminal cysteines targeting ALFA (clone 1G5, N1502, lot no. 15220402), GFP (clone 1H1, N0302, lot no. 022307) and RFP (clone 2B12, N0402, lot no. 012309) were ordered from NanoTag Biotechnologies and conjugated to azide-modified DNA following established protocols22,58. In brief, nanobodies were diluted in buffer D for 30 min in the dark at 4 °C. After removing TCEP with buffer exchange into PBS using 10 kDa Amicon filters (Sigma-Aldrich, UFC5010; 10 kDa MWCO), A 20-fold molar excess of DBCO-PEG₄-maleimide (Sigma-Aldrich, 760676) was added. Following a 4 h incubation in the dark at 4 °C on a shaker, unreacted crosslinker was removed, and a fivefold molar excess of azide-modified DNA was added for overnight click conjugation at 4 °C. The resulting conjugates (NbALFA-L5R2, NbALFA-LS4, NbALFA-S4, NbGFP-S4 and NbRFP-LS4) were purified from free DNA and unreacted nanobodies using size-exclusion (Superdex 75 10/300 GL GE Healthcare) and anion-exchange (Resource Q 1 ml column Cytiva) chromatography on an ÄKTA Pure system and then stored at −20 °C in storage buffer.
Plasmid construction
The following base plasmids were used: mEmerald-Sec61β, mEmerald-Ensconsin and EGFP-OMP25 (ref. 18). For microtubule labelling, oxStayGold-ALFA-Ensconsin was constructed by replacing the mEmerald sequence in mEmerald-Ensconsin with oxStayGold from pcDNA3/er-(n2)oxStayGold(c4) (Addgene, plasmid no. 185822) and inserting an ALFA tag at the N terminus of Ensconsin. The resulting fragment was cloned into the pSin vector using SpeI and BamHI digestion. For ER or mitochondria labelling, ALFA tags were inserted at the N termini of Sec61β and OMP25 within their respective base plasmids to generate mEmerald-ALFA-Sec61β and EGFP-ALFA-OMP25. Both constructs were subsequently subcloned into the pSin vector for stable cell line generation.
Cell culture and transfection
COS-7 (CRL-1651), HeLa (CCL-2), HEK293T (CRL-3216) and U-2 OS (HTB-96) cell lines were purchased from the American Type Culture Collection. COS-7, HEK293T and HeLa cells were cultured in DMEM (Gibco, C11965500CP) and U-2 OS cells in McCoy’s 5A medium (Gibco, 16600-082). All media were supplemented with 10% FBS (Sigma-Aldrich, F8318) and 1% penicillin–streptomycin (Gibco, 15140122). Before cell seeding, coverslips (Marienfeld, 0117650) and confocal dishes (MatTek, P35G-1.5-14-C) were cleaned by immersion in 1 M KOH (Sigma-Aldrich, 484016) and sonicated for 15 min, subjected to three 10 min sonication cycles in ddH2O, sterilized with 100% ethanol (J&K Scientific, 258449) for 1 min and coated with poly-L-lysine (Sigma-Aldrich, P4707). Approximately 1 × 105 cells were seeded per coverslip for transient transfections with Lipofectamine 3000 (Thermo Fisher Scientific, L3000015) according to the manufacturer’s instructions.
Stable cell line generation
Lentiviruses were produced by co-transfecting HEK293T cells with packaging plasmids psPAX2 (Addgene, plasmid no. 12260) and pMD2.G (Addgene, plasmid no. 12259) along with the appropriate transfer plasmid (pSin-oxStayGold-ALFA-Ensconsin, pSin-mEmerald-ALFA-Sec61β or pSin-EGFP-ALFA-OMP25) using Lipofectamine 3000. Transfection medium was replaced with fresh medium after 12–24 h. At 48–72 h post transfection, viral supernatants were collected, clarified by centrifugation (600g, 5 min), mixed at a 1:4 ratio with a virus concentration reagent (Biodragon, BF06205) and incubated overnight at 4 °C. Viruses were pelleted by centrifugation and resuspended in fresh medium. Target COS-7 and HeLa cells were transduced with the concentrated virus supplemented with 8 µg ml−1 polybrene (Beyotime, C0351). After 4–6 h at 37 °C, the medium was replaced. Following a 24 h recovery, stably transduced cells were selected using 1 µg ml−1 puromycin (Beyotime, ST551) for 3–5 days and expanded for imaging.
Microtubule labelling
COS-7 cells (transfected with oxStayGold-ALFA-Ensconsin for nanobody labelling) were cultured on coverslips. Cells were pre-fixed with pre-warmed 0.3% glutaraldehyde (Electron Microscopy Sciences, 16020) and 0.25% Triton X-100 in cytoskeletal buffer for 2 min, followed by fixation with pre-warmed 2% glutaraldehyde in CB buffer for 10 min. After fixation, cells were quenched with 0.1 M NH4Cl (Sigma-Aldrich, A9434) in PBS for 5 min. After rinsing three times with PBS, cells were blocked and permeabilized with blocking buffer A for 1 h at room temperature. For nanobody labelling, cells were labelled with anti-ALFA nanobodies with docking strands L5R2 or LS4 (NbALFA-L5R2 or NbALFA-LS4) in dilution buffer A at 4 °C overnight. Cells were washed three times with wash buffer for 5 min each, followed by two washes with PBS for 10 min each. Before adding the imager solution (LR2 in buffer C and LP10 in buffer E), samples were washed with buffer C for 5 min. For antibody labelling, cells were incubated with mouse anti-TUBA4A primary antibody (Sigma-Aldrich, clone B-5-1-2, T6074, lot no.0000419295; 1:500) in dilution buffer A, washed three times with wash buffer and subsequently incubated with goat anti-mouse-S4 secondary antibody in dilution buffer A at room temperature for 1 h. After washing, samples were washed with buffer E for 5 min and introduced into the imager solution (P33 in buffer E).
NPC labelling
The U-2 OS NUP96-ALFA-mEGFP CRISPR knock-in cell line was generated in a previous study23. Cells were cultured on coverslips for 48 h, fixed with pre-warmed 2.4% paraformaldehyde (PFA; Electron Microscopy Sciences, 15710) in PBS for 30 min, quenched with 0.1 M NH4Cl in PBS for 5 min and washed four times with PBS for 30 s, 60 s and 2 × 5 min. Permeabilization and blocking were performed simultaneously in blocking buffer B for 2 h at room temperature. For labelling, home-made NbALFA-L5R2 probes (50 nM) in dilution buffer A were added to the samples at 4 °C overnight. After washing, samples were prepared for imaging by adding imager strand LR2 in buffer C.
ER labelling
COS-7 cells stably expressing mEmerald-ALFA-Sec16β were cultured on poly-L-lysine-coated coverslips for 24 h. Cells were fixed with 3% PFA and 0.1% glutaraldehyde in PBS for 15 min at room temperature, quenched with freshly prepared 0.1 M NH4Cl for 5 min. Cells were washed three times with PBS for 5 min each. After incubating with blocking buffer A for 1 h at room temperature, cells were incubated with NbALFA-L5R2 in dilution buffer A at 4 °C overnight. Cells were then washed sequentially with wash buffer (three 5 min washes) and PBS (two 10 min washes), rinsed in buffer C (5 min) and mounted in imager solution (strand LR2 in buffer C).
Two-colour sample labelling
For dual microtubule and ER labelling, COS-7 cells stably expressing oxStayGold-ALFA-Ensconsin were transfected with mEmerald-Sec61β and cultured for 24 h. Samples were processed similarly to ER labelling, except cells were incubated with NbALFA-LS4 and NbGFP-S4 probes in dilution buffer A at 4 °C overnight. After washing, cells were washed with buffer E for 5 min and introduced into the imager solution (LP10 and P81 in buffer E). For dual ER membrane and lumen labelling, COS-7 cells stably expressing mEmerald-ALFA-Sec61β were transfected with mCherry-KDEL11 and cultured for 24 h. Following standard fixation and blocking, cells were incubated with NbALFA-S4 and NbRFP-LS4 probes in dilution buffer A overnight at 4 °C. After washing, cells were washed with buffer E for 5 min and introduced into the imager solution (P10 and LP71 in buffer E).
Mitochondrion labelling
HeLa cells stably expressing EGFP-ALFA-OMP25 and COS-7 cells were processed similarly to samples for ER labelling. For DNA-PAINT, HeLa cells were incubated with NbALFA-LS4 in dilution buffer A overnight at 4 °C. After washing, samples were washed with buffer E for 5 min and introduced into the imager solution (imager strand LP10 in buffer E). For dSTORM, COS-7 cells were incubated with rabbit anti-TOMM20 primary antibody (HUABIO, clone ST04-72, ET1609-25, lot no. H641562031; 1:500) in dilution buffer A, washed three times and incubated with goat anti-rabbit-AF647 secondary antibody (Thermo Fisher Scientific, A21245, lot no. 2390714; 1:1,000) in dilution buffer A at room temperature for 1 h. After washing, samples were imaged in freshly prepared dSTORM buffer containing 1 kU ml−1 catalase (Sigma-Aldrich, C40-100MG), 0.135 kU ml−1 glucose oxidase (Sigma-Aldrich, G2133-50KU) and 35 mM MEA (Sigma-Aldrich, M6500) in base buffer.
Live-cell labelling
U-2 OS cells stably expressing Sec61β-Emerald-HaloTag (a gift from D. Yang, Westlake University) were seeded in confocal dishes. Cells were incubated with 500 µl of PA-JF549-HaloTag ligand (a gift from L. Lavis, Janelia Research Campus) in phenol-red-free DMEM (Gibco, 21063-029) supplemented with 10% FBS for 1 h at 37 °C with 5% CO2. The ligand concentration was 100 nM for live-cell imaging and 25 nM for single-molecule tracking. After three washes with PBS and two washes with live-cell imaging medium (FluoroBrite DMEM; Gibco, A1896701), cells were incubated in the same medium for 1 h at 37 °C to remove unreacted dye. Imaging was performed at room temperature in fresh imaging medium.
Mice
Male C57BL/6J mice (JAX, 000664) were bred in the laboratory or purchased from the animal source centre at Westlake University for synaptonemal complex samples. The Rosa26-GFP-OMP25 knock-in line was generated by inserting a GFP-OMP25 cassette into the Rosa26locus (Shanghai Model Organisms Center). The ChAT-Cre-ChR2-YFP line was generated by crossing ChAT-IRES-Cre (JAX, 006410) and RCL-ChR2(H134R)/EYFP (JAX, 024109). All strains were maintained on a C57BL/6J × ICR background. All experimental procedures were approved by the Institutional Animal Care and Use Committee at Westlake University. Mice were housed in a specific-pathogen-free facility (22–26 °C, 40–70% humidity, 12 h light–dark cycle) with ad libitum access to food and water.
Synaptonemal complex samples
Spermatocyte preparation was adapted from a previous study18. Seminiferous tubules from 3-week-old male C57BL/6J mice were incubated in buffer I (137 mM NaCl, 2.7 mM KCl, 8 mM Na2HPO4, 2 mM KH2PO4; Sigma-Aldrich) for 10 min at room temperature. Released spermatocytes were mixed 1:1 with buffer II (100 mM sucrose, Sigma-Aldrich), passed through a 70 µm strainer and spread onto coverslips presoaked in buffer III (1% PFA, 0.15% Triton X-100, pH 9.2). After drying for 4 h in a humidified chamber and washing, samples were treated with Image-iT (Invitrogen, I36933) for 30 min. Samples were washed three times, blocked with 3% BSA in PBS for 30 min at room temperature and incubated overnight at 4 °C with mouse anti-SCP-3 (Santa Cruz Biotechnology, clone D-1, sc-74569, lot no. J2221; 1:1,000 in 1% BSA/PBS). Following three washes, samples were incubated overnight at 4 °C with goat anti-mouse-S4 (1:500). After a final three washes, samples were mounted in buffer E containing imager strand P10.
Transcardial perfusion and tissue labelling
Solutions were freshly prepared daily. Adult Rosa26-GFP-OMP25 and ChAT-Cre-ChR2-YFP mice were anaesthetized (intraperitoneal 1% sodium pentobarbital, 200 μl) and transcardially perfused at 10 ml min−1 with 50 ml PBS followed by 50 ml 4% PFA in PBS. Brains were excised, post-fixed in 4% PFA at 4 °C for 8–12 h with agitation and rinsed three times for 20 min in cold PBS. Vibratome sections (30 μm; Leica VT1200 S), coronal for Rosa26-GFP-OMP25 mice and sagittal for ChAT-Cre-ChR2-YFP mice, were stored at 4 °C in PBS with 0.05% ProClin 300 (BIOSS, D10200). Before labelling, sections were washed twice in PBS (10 min each), quenched with 100 mM glycine in PBS, washed twice more in PBS and permeabilized or blocked in blocking buffer C for 2 h at room temperature. Sections from Rosa26-GFP-OMP25 mice were incubated overnight at 4 °C with NbGFP-LS4 in dilution buffer B. After three washes in wash buffer and two in PBS (10 min each), sections were washed in buffer E (10 min) and mounted in imager solution (LP10 in buffer E). Sections from ChAT-Cre-ChR2-YFP mice were incubated overnight at 4 °C with rabbit anti-GFP (Thermo Fisher Scientific, A11122, lot 2901498,1:500) in dilution buffer B, washed three times in wash buffer and incubated overnight at 4 °C with donkey anti-rabbit-LS4 (1:500). After identical washing steps, samples were mounted in LP10/buffer E for imaging.
Sample mounting
For fixed-cell imaging, samples were rinsed with fresh imaging solution, placed in a custom holder with 100 μl of imaging solution and covered with a clean coverslip. Excess solution was removed, and the assembly was sealed with two-component silicone glue (Twinsil, Picodent), curing for 10–20 min before imaging. For live-cell imaging, the solution in the confocal dishes was replaced with 300 μl of live-cell imaging medium before covering with a coverslip and sealing in the identical manner.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
An example dataset is available on figshare at https://doi.org/10.6084/m9.figshare.31265875 (ref. 59). Owing to their extensive size (>50 TB), the full image datasets cannot be hosted online but are available from the corresponding authors upon request.
Code availability
The me4Pi-SMLM reconstruction code is available at https://github.com/zhanglab-srm/me4Pi (ref. 60).
References
Liu, S., Hoess, P. & Ries, J. Super-resolution microscopy for structural cell biology. Annu. Rev. Biophys. 51, 301–326 (2022).
Sigal, Y. M., Zhou, R. & Zhuang, X. Visualizing and discovering cellular structures with super-resolution microscopy. Science 361, 880–887 (2018).
Lelek, M. et al. Single-molecule localization microscopy. Nat. Rev. Methods Primers 1, 39 (2021).
Huang, B., Wang, W., Bates, M. & Zhuang, X. Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science 319, 810–813 (2008).
Juette, M. F. et al. Three-dimensional sub-100 nm resolution fluorescence microscopy of thick samples. Nat. Methods 5, 527–529 (2008).
Pavani, S. R. et al. Three-dimensional, single-molecule fluorescence imaging beyond the diffraction limit by using a double-helix point spread function. Proc. Natl Acad. Sci. USA 106, 2995–2999 (2009).
Shtengel, G. et al. Interferometric fluorescent super-resolution microscopy resolves 3D cellular ultrastructure. Proc. Natl Acad. Sci. USA 106, 3125–3130 (2009).
Aquino, D. et al. Two-color nanoscopy of three-dimensional volumes by 4Pi detection of stochastically switched fluorophores. Nat. Methods 8, 353–359 (2011).
Huang, F. et al. Ultra-high resolution 3D imaging of whole cells. Cell 166, 1028–1040 (2016).
Bates, M. et al. Optimal precision and accuracy in 4Pi-STORM using dynamic spline PSF models. Nat. Methods 19, 603–612 (2022).
Zhang, Y. et al. Nanoscale subcellular architecture revealed by multicolor three-dimensional salvaged fluorescence imaging. Nat. Methods 17, 225–231 (2020).
Gu, L. et al. Molecular-scale axial localization by repetitive optical selective exposure. Nat. Methods 18, 369–373 (2021).
Bailey, B., Farkas, D. L., Taylor, D. L. & Lanni, F. Enhancement of axial resolution in fluorescence microscopy by standing-wave excitation. Nature 366, 44–48 (1993).
Schnitzbauer, J., McGorty, R. & Huang, B. 4Pi fluorescence detection and 3D particle localization with a single objective. Opt. Express 21, 19701–19708 (2013).
Yang, X. et al. Mirror-enhanced super-resolution microscopy. Light Sci. Appl. 5, e16134 (2016).
Li, X. et al. Three-dimensional structured illumination microscopy with enhanced axial resolution. Nat. Biotechnol. 41, 1307–1319 (2023).
Liu, Y. et al. Axial localization and tracking of self-interference nanoparticles by lateral point spread functions. Nat. Commun. 12, 2019 (2021).
Ouyang, Z. et al. Elucidating subcellular architecture and dynamics at isotropic 100-nm resolution with 4Pi-SIM. Nat. Methods 22, 335–347 (2025).
Wang, J. et al. Implementation of a 4Pi-SMS super-resolution microscope. Nat. Protoc. 16, 677–727 (2021).
Smith, C. S., Joseph, N., Rieger, B. & Lidke, K. A. Fast, single-molecule localization that achieves theoretically minimum uncertainty. Nat. Methods 7, 373–375 (2010).
Chung, K. K. H. et al. Fluorogenic DNA-PAINT for faster, low-background super-resolution imaging. Nat. Methods 19, 554–559 (2022).
Strauss, S. & Jungmann, R. Up to 100-fold speed-up and multiplexing in optimized DNA-PAINT. Nat. Methods 17, 789–791 (2020).
Wang, Q. et al. 4Pi-SIMFLUX: 4Pi single-molecule localization microscopy with structured illumination. Nat. Methods 23, 175–182 (2026).
Gotzke, H. et al. The ALFA-tag is a highly versatile tool for nanobody-based bioscience applications. Nat. Commun. 10, 4403 (2019).
Ries, J., Kaplan, C., Platonova, E., Eghlidi, H. & Ewers, H. A simple, versatile method for GFP-based super-resolution microscopy via nanobodies. Nat. Methods 9, 582–584 (2012).
Dai, M., Jungmann, R. & Yin, P. Optical imaging of individual biomolecules in densely packed clusters. Nat. Nanotechnol. 11, 798–807 (2016).
Nieuwenhuizen, R. P. et al. Measuring image resolution in optical nanoscopy. Nat. Methods 10, 557–562 (2013).
Zhao, W. et al. Quantitatively mapping local quality of super-resolution microscopy by rolling Fourier ring correlation. Light Sci. Appl. 12, 298 (2023).
Schuller, A. P. et al. The cellular environment shapes the nuclear pore complex architecture. Nature 598, 667–671 (2021).
Thevathasan, J. V. et al. Nuclear pores as versatile reference standards for quantitative superresolution microscopy. Nat. Methods 16, 1045–1053 (2019).
Heydarian, H. et al. 3D particle averaging and detection of macromolecular symmetry in localization microscopy. Nat. Commun. 12, 2847 (2021).
Wang, W. et al. Particle fusion of super-resolution data reveals the unit structure of Nup96 in Nuclear Pore Complex. Sci. Rep. 13, 13327 (2023).
Weber, M. et al. MINSTED nanoscopy enters the Ångström localization range. Nat. Biotechnol. 41, 569–576 (2023).
Reinhardt, S. C. M. et al. Angstrom-resolution fluorescence microscopy. Nature 617, 711–716 (2023).
Schroeder, L. K. et al. Dynamic nanoscale morphology of the ER surveyed by STED microscopy. J. Cell Biol. 218, 83–96 (2019).
Rust, M. J., Bates, M. & Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 3, 793–795 (2006).
Heilemann, M. et al. Subdiffraction-resolution fluorescence imaging with conventional fluorescent probes. Angew. Chem. Int. Ed. Engl. 47, 6172–6176 (2008).
Kessler, L. F. et al. Self-quenched fluorophore-DNA labels for super-resolution fluorescence microscopy. J. Phys. Chem. B 128, 6751–6759 (2024).
Ghosh, A. et al. Decoding the molecular interplay of CD20 and therapeutic antibodies with fast volumetric nanoscopy. Science 387, eadq4510 (2025).
Grimm, J. B. et al. Bright photoactivatable fluorophores for single-molecule imaging. Nat. Methods 13, 985–988 (2016).
Takakura, H. et al. Long time-lapse nanoscopy with spontaneously blinking membrane probes. Nat. Biotechnol. 35, 773–780 (2017).
Jouchet, P. et al. Nanometric axial localization of single fluorescent molecules with modulated excitation. Nat. Photonics 15, 297–304 (2021).
Speiser, A. et al. Deep learning enables fast and dense single-molecule localization with high accuracy. Nat. Methods 18, 1082–1090 (2021).
Zhang, P. et al. Deep learning-driven adaptive optics for single-molecule localization microscopy. Nat. Methods 20, 1748–1758 (2023).
Xu, F. et al. Three-dimensional nanoscopy of whole cells and tissues with in situ point spread function retrieval. Nat. Methods 17, 531–540 (2020).
Shi, X. et al. Label-retention expansion microscopy. J. Cell Biol. 220, e202105067 (2021).
Zwettler, F. U. et al. Molecular resolution imaging by post-labeling expansion single-molecule localization microscopy (Ex-SMLM). Nat. Commun. 11, 3388 (2020).
Balzarotti, F. et al. Nanometer resolution imaging and tracking of fluorescent molecules with minimal photon fluxes. Science 355, 606–612 (2017).
Scheiderer, L., Marin, Z. & Ries, J. MINFLUX achieves molecular resolution with minimal photons. Nat. Photonics 19, 238–247 (2025).
Cnossen, J. et al. Localization microscopy at doubled precision with patterned illumination. Nat. Methods 17, 59–63 (2020).
Reymond, L. et al. SIMPLE: structured illumination based point localization estimator with enhanced precision. Opt. Express 27, 24578–24590 (2019).
Gu, L. et al. Molecular resolution imaging by repetitive optical selective exposure. Nat. Methods 16, 1114–1118 (2019).
Ershov, D. et al. TrackMate 7: integrating state-of-the-art segmentation algorithms into tracking pipelines. Nat. Methods 19, 829–832 (2022).
Liu, S. & Huang, F. Enhanced 4Pi single-molecule localization microscopy with coherent pupil based localization. Commun. Biol. 3, 220 (2020).
Ma, H., Chen, M., Nguyen, P. & Liu, Y. Toward drift-free high-throughput nanoscopy through adaptive intersection maximization. Sci. Adv. 10, eadm7765 (2024).
Cnossen, J., Cui, T. J., Joo, C. & Smith, C. Drift correction in localization microscopy using entropy minimization. Opt. Express 29, 27961–27974 (2021).
Geertsema, H. J. et al. Left-handed DNA-PAINT for improved super-resolution imaging in the nucleus. Nat. Biotechnol. 39, 551–554 (2021).
Sograte-Idrissi, S. et al. Nanobody detection of standard fluorescent proteins enables multi-target DNA-PAINT with high resolution and minimal displacement errors. Cells 8, 48 (2019).
Yu, Z., Zheng, B. & Zhang, Y. Example dataset for “Mirror-enhanced 4Pi-SMLM with one objective enables isotropic nanoscale imaging”. figshare https://doi.org/10.6084/m9.figshare.31265875 (2026).
Yu, Z., Zheng, B. & Zhang, Y. Mirror-enhanced 4Pi-SMLM with one objective enables isotropic nanoscale imaging. Source Code. GitHub https://github.com/zhanglab-srm/me4Pi (2026).
Acknowledgements
We thank D. Yang at Westlake University for sharing the U-2 OS cell line expressing Sec61β-Emerald-HaloTag and L. Lavis at Janelia Research Campus for sharing PA-JF549-HaloTag. Y. Zhang acknowledges support from the National Science Foundation of China (32150015, 32471516) and the National Key Research and Development Program of China (2022YFC3400600). C.L., L.H. and X. Wu acknowledge support from the National Science Foundation of China (32371010, 32370885, 32471303). X.Y. acknowledges support from the ‘Pioneer’ and ‘Leading Goose’ R&D Program of Zhejiang (2024SSYS0033). This work was also supported by the State Key Laboratory of Gene Expression, the Westlake Laboratory of Life Sciences and Biomedicine, the Westlake Education Foundation and the Research Center for Industries of the Future at Westlake University.
Author information
Authors and Affiliations
Contributions
Y. Zhang conceived the idea and supervised the project. Z.Y. designed and built the optical system and performed the imaging. B.Z. developed and optimized DNA-PAINT imaging. Z.Y. and Q.D. generated the stable COS-7 cell line expressing Ensconsin. Z.Y. generated the CRISPR-edited and all other stable cell lines. X. Wu, B.Z., Y. Zhan and Q.D. optimized and prepared biological samples. L.C., X.Y. and B.Z. prepared the synaptonemal complex samples. Y.C., Z.W., B.Z., W.Z., J.T., T.X., L.H. and C.L. prepared mouse tissue samples. Y. Zhang, X. Wang and S.L. implemented the software and performed data analysis. Y. Zhang, Z.Y. and B.Z. wrote the manuscript with input from all authors.
Corresponding author
Ethics declarations
Competing interests
Y. Zhang, Z.Y. and B.Z. have filed a patent application on me4Pi-SMLM described in this work. Y. Zhang is co-inventor of a US patent (US11209367B2) related to the salvaged fluorescence method used in this work. The other authors declare no competing interests.
Peer review
Peer review information
Nature Biotechnology thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Optical layout of me4Pi-SMLM.
See the Methods section for a detailed description. Laser 1: 405 nm (OBIS LX 100 mW, Coherent); Laser 2: 488 nm (OBIS LS 150 mW, Coherent); Laser 3: 552 nm (2RU-VFL-P-2000-552, MPB Communications); Laser 4: 642 nm (2RU-VFL-P-2000-642, MPB Communications). AOTF: acousto-optic tunable filter; SMF: single-mode fiber; DM1–DM5: dichroic mirrors; F1–F4: emission filters; L1–L7: lenses; M1–M14: mirrors; CL1 and CL2: cylindrical lenses; CAM1 and CAM2: sCMOS cameras.
Extended Data Fig. 2 SolidWorks design of me4Pi-SMLM.
a–c, Top (a), rear (b), and front (c) view of the me4Pi-SMLM SolidWorks model. d, Mechanical design of the axial interference illumination module.
Extended Data Fig. 3 Synchronization diagram of me4Pi-SMLM.
a, Slow imaging mode. The sCMOS camera operated in external trigger mode. The position of the mirror and piezoelectric actuator was controlled using three voltages, each applied for 15 ms. The sCMOS camera trigger signal was delayed by 3 ms relative to the piezoelectric actuator signal, and the camera exposure time was set to 10 ms. AOTF remained open throughout the imaging. One imaging cycle is 45 ms. b, Fast imaging mode. The sCMOS camera operated in internal trigger mode, with the total exposure time, Tall, set manually. Upon initiating sensor readout, the camera generated a Vsync signal with an adjustable pulse width corresponding to Toff. An FPGA monitored the Vsync signal in real-time and transmitted an inverted signal to the AOTF to modulate the laser. In this configuration, the effective exposure time was defined as Ton = Tall - Toff. Simultaneously, the FPGA sent a control signal to the piezoelectric actuator, triggering cyclic movement to specific positions upon the rising edge of the Vsync signal. Although the example diagram illustrates a cycle time of 9 ms, this parameter could be adjusted according to the sample type and field of view. The first frame of the raw data was discarded prior to analysis.
Extended Data Fig. 4 Axial localization calibration.
a, PSF width of a 40 nm bead as a function of axial position. b, Measured z-position versus the piezo z-position. c, Axial intensity oscillations of the three interference phases, showing a period of ~210 nm.
Extended Data Fig. 5 Flowchart for me4Pi-SMLM reconstruction.
Refer to the Methods section for a detailed description. Three sequential sub-images were acquired, corresponding to phase-shifted illumination patterns. 2D astigmatic Gaussian fitting was used to determine lateral positions (x, y) and PSF widths, which were then used to estimate coarse axial positions. Intensities across the three sub-images were analyzed to extract interferometric phase information. After phase-drift correction, a ridge-finding algorithm was used for axial phase unwrapping to convert phase data into precise axial positions. A 3D drift-correction algorithm compensated for both sample drift and residual system drift. Molecules that did not exhibit modulation across all three frames were excluded based on modulation depth (Step 1), followed by further exclusion based on photon count and localization precision (Step 2). Final reconstructions used the precise z positions for me4Pi-SMLM, whereas coarse z positions were used for the 3D-SMLM comparison.
Extended Data Fig. 6 Quantitative resolution analysis of microtubules using rFRC.
a, rFRC map of the microtubule data shown in Fig. 2a, showing the lateral resolution. b, Lateral localization precision (CRLBxy) map. c, Modulation depth map. d, Axial localization precision (CRLBz) map. e, Slice-by-slice x–z rFRC map. Scale bars: 5 μm (a-d), 2.5 μm (e).
Extended Data Fig. 7 Transmission profiles for conventional and salvaged channels.
a, Transmission spectra of the dichroic beamsplitter and emission filters used for one-color imaging, together with the emission spectrum of Cy3B. The red shaded region indicates the effective detection band for Cy3B, given by the combined transmission of the dichroic and emission filters. b, Transmission spectra of the dichroic beamsplitters and emission filters used for two-color imaging. Red and green shaded regions indicate the effective detection bands of the conventional and salvaged fluorescence paths, respectively. c-d, Emission spectra of Cy3B and AF568 (c), or Cy3B and ATTO Rho11 (d), overlaid with the effective detection bands of the conventional and salvaged fluorescence paths.
Extended Data Fig. 8 Resolution analysis for me4Pi-SMLM in whole-cell mitochondrial imaging.
a–c, Lateral localization precision (a), axial localization precision (b) and modulation depth (c) for the mitochondria image shown in Fig. 4f. d, DAFL 3D precision for molecules from each optical section. e, FRC resolution for each optical section. f, 3D localization distributions for molecules emitting ≥10 frames. The photon count for these molecules is 6,422 ± 2,362 (mean ± s.d.). g, Histograms of the distributions in f were fit with Gaussian functions, with fitted standard deviations reported.
Extended Data Fig. 9 me4Pi-SMLM imaging of microtubules with fast imaging mode.
a, me4Pi-SMLM image of microtubules in a fixed COS-7 cell recorded with a cycle time of 9 ms. Color bar denotes depth. b, Cross-sections at locations denoted by the numbers in a, reconstructed with me4Pi-SMLM and 3D-SMLM. c, DAFL localization precision in 3D for the data in a. d, FRC analysis in each dimension of the data in a. Scale bars: 2 μm (a), 50 nm (b). Representative images are shown from six independent experiments.
Extended Data Fig. 10 dSTORM imaging of mitochondria with me4Pi-SMLM.
a, me4Pi-SMLM image of mitochondria in a fixed COS-7 cell immunolabeled with AF647-conjugated antibodies. b, A 100-nm-thick x–y slice of the boxed region in a. c,d, 100-nm-thick x–z and y–z cross-sections along the dashed lines denoted in a. Color bar denotes depth. Scale bars: 2 μm (a), 1 μm (b-d). Representative images are shown from five independent experiments.
Supplementary information
Supplementary Information (download PDF )
Supplementary Figs. 1–8, Supplementary Table 1 and Supplementary Note 1.
Supplementary Video 1 (download MP4 )
The video shows the SolidWorks model of the me4Pi-SMLM microscope (see also Extended Data Fig. 2). A detailed description of the optical components can be found in the Methods.
Supplementary Video 2 (download MP4 )
The video shows the 3D rendering of the immunolabeled NPC in a fixed U-2 OS cell (see also Fig. 2e–g). A zoomed-in sub-region compares the axial cross-sections of me4Pi-SMLM and 3D-SMLM.
Supplementary Video 3 (download MP4 )
The video shows the 3D rendering of the immunolabeled ER in a fixed COS-7 cell (see also Fig. 2h–j). A zoomed-in sub-region compares the axial cross-sections of me4Pi-SMLM and 3D-SMLM.
Supplementary Video 4 (download MP4 )
Video rendering of the data shown in Fig. 3e–i. Two-color images of ER membrane (mEmerald-ALFA-Sec61β labeled with NbALFA; green) and ER lumen (mCherry-KDEL labeled with NbRFP; magenta).
Supplementary Video 5 (download MP4 )
The video shows the 3D rendering of computationally isolated synaptonemal complexes (SYCP3) in a fixed mouse spermatocyte (see also Fig. 4a–e).
Supplementary Video 6 (download MP4 )
The video shows the 3D rendering of the immunolabeled mitochondrial outer membrane in a fixed HeLa cell (see also Fig. 4f,g).
Supplementary Video 7 (download MP4 )
The video shows single-molecule tracking of ER (Sec61β-Emerald-HaloTag) labeled with PA-JF549-HaloTag in a live U-2 OS cell (see also Fig. 5h).
Supplementary Video 8 (download MP4 )
The video shows the 3D rendering of ChR2 in 30-μm-thick mouse brain slices (see also Fig. 6h–l).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yu, Z., Zheng, B., Zhan, Y. et al. Mirror-enhanced 4Pi-SMLM with one objective enables isotropic nanoscale imaging. Nat Biotechnol (2026). https://doi.org/10.1038/s41587-026-03083-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41587-026-03083-7
