
Abstract
Nucleophosmin (NPM1), a nucleolar protein frequently mutated in hematopoietic malignancies, is overexpressed in several solid tumors with poorly understood functional roles. Here, we demonstrate that Npm1 is upregulated after APC loss in WNT-responsive tissues and supports WNT-driven intestinal and liver tumorigenesis. Mechanistically, NPM1 loss induces ribosome pausing and accumulation at the 5’-end of coding sequences, triggering a protein synthesis stress response and p53 activation, which mediate this antitumorigenic effect. Collectively, our data identify NPM1 as a critical WNT effector that sustains WNT-driven hyperproliferation and tumorigenesis by attenuating the integrated stress response and p53 activation. Notably, NPM1 expression correlates with elevated WNT signaling and proliferation in human colorectal cancer (CRC), while CRCs harboring NPM1 deletions exhibit preferential TP53 inactivation, underscoring the clinical relevance of our findings. Being dispensable for adult epithelial homeostasis, NPM1 represents a promising therapeutic target in p53-proficient WNT-driven tumors, including treatment-refractory KRAS-mutant CRC, and hepatic cancers.
Similar content being viewed by others


Main
The response to specific drivers of proliferation and oncogenic transformation varies greatly among tissues1. This tissue-specific ‘permissiveness’ is exemplified by selective mutations in pathway components driving cancers of distinct origins. For instance, the WNT pathway is crucial for tumor initiation in several tissues. However, mutations in CTNNB1 are prevalent in hepatocellular carcinomas (HCCs), but less frequent in colorectal cancer (CRC)2. Conversely, inactivating mutations in the tumor suppressor adenomatous polyposis coli (APC) are rare in HCC, but among the most frequent in CRC, occurring in ~80% of cases. These mutations result in ligand-independent WNT-pathway activation and tumor initiation2,3. APC-deficient intestinal cells rely on mechanistic target of rapamycin complex 1 (mTORC1) to promote translation elongation and hyperproliferation, which can be effectively suppressed by rapamycin4. However, acquisition of additional mutations, such as oncogenic KRAS, found in ~40% of patients with CRC, renders rapamycin ineffective5,6. This highlights the need for additional druggable target identification to tackle WNT-driven disease.
Nucleophosmin (NPM1) is a multifunctional nucleolar protein essential for embryonic development7,8. It has established roles in cell proliferation, survival, genomic integrity, ribosome biogenesis and posttranscriptional ribosomal RNA modifications among others7,8,9,10,11. It also functions as a molecular chaperone, preventing protein aggregation within the confined nuclear space and during stress12,13. NPM1 is the most frequently mutated gene in acute myeloid leukemia (AML)14. These mutations typically produce a truncated, cytoplasmic form of NPM1 that facilitates increased expression of genes driving leukemogenesis15,16,17. In solid tumors, NPM1 is rarely mutated but it is frequently overexpressed in multiple cancers, including CRC, hepatic and lung18,19,20,21,22. Despite extensive research in hematological malignancies, any roles of NPM1 in solid tumors, its impact on adult tissue homeostasis and the potential for repurposing emerging AML-targeted NPM1 therapies to benefit more patients remain unclear.
Pharmacological inhibition of WNT signaling in tumor cells dependent on WNT ligands for growth (for example, with RNF43 loss-of-function mutations) induces MYC-dependent downregulation of NPM1 and other ribosome biogenesis and rRNA processing factors, identifying NPM1 as a key effector in the WNT–MYC axis23. NPM1 has also been identified as both a MYC target gene and a regulator of MYC activity24,25,26, indicating a potential role in tumorigenesis through MYC.
Furthermore, NPM1 has been shown to regulate tumor suppressor pathways. Under stress conditions, NPM1 may activate p53 by directly binding to p53 or its ubiquitin ligase MDM2, thereby stabilizing p53 expression27,28. Conversely, Npm1 deletion can also result in increased p53 levels and p53-dependent growth arrest and apoptosis7,8. While part of these effects may be indirect, they highlight a context-dependent NPM1-p53 relationship.
In this study, we identify Npm1 as a key gene upregulated in organs permissive to WNT-driven oncogenic growth and demonstrate its relevance to human disease. Using genetically engineered mouse models, we show that while NPM1 is dispensable for epithelial tissue homeostasis, it is essential for WNT-driven transformation by attenuating a protein synthesis stress response and posttranscriptional p53 activation. Given that NPM1 is already a therapeutic target in hematopoietic cancers, our findings suggest that targeting NPM1 could also benefit patients with WNT-driven solid tumors, such as KRAS-mutant CRC and hepatic cancers, which remain challenging to treat.
Results
Npm1 is upregulated in WNT-permissive tissues
Genetic alterations in the WNT signaling pathway are common in gastrointestinal tract tumors (‘WNT-permissive tissues’), while less frequent in other tissues2,29. We sought to identify genes that are commonly upregulated across multiple WNT-permissive tissues after global hyperactivation of WNT. This was achieved by acutely deleting Apc (ApcloxP/loxP) within multiple adult mouse tissues using a tamoxifen-inducible Cre recombinase driven by the ubiquitously expressing Rosa26 promoter (R26-CreERT2), followed by transcriptomic analysis (Fig. 1a). The small intestine (SI) was the only tissue exhibiting a pronounced phenotype, displaying crypt expansion (Extended Data Fig. 1a). Nuclear β-catenin in ApcloxP/loxP animals confirmed successful WNT activation in tissues (Extended Data Fig. 1b). The SI, esophagus, colon and stomach had the strongest activation of a WNT transcriptional program, confirming their WNT-permissive status, while the bladder, kidneys, lungs and spleen had comparatively minimal activation, reflecting their nonpermissiveness (Fig. 1b). We identified only 107 genes upregulated in at least four of the WNT-permissive tissues (Fig. 1c). These included known WNT mediators and negative feedback regulators of WNT, for example, RNF43, ZNRF3, AXIN2, TCF7 and NOTUM, confirming successful activation of the WNT transcriptional program in WNT-permissive tissues (Fig. 1c). We also confirmed enrichment in MYC transcriptional signatures, which is consistent with the roles of MYC downstream of APC (Extended Data Fig. 1c).

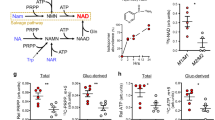
a, Schematic representation of the experimental strategy used to identify targets commonly upregulated in tissues with WNT signaling activation after Apc deletion. Cre recombinase was expressed from the Rosa26 locus (R26-CreERT2); ApcloxP/loxP alleles underwent Cre-mediated recombination upon tamoxifen administration. Multiple tissues were collected four days after induction and subjected to RNA-seq. b, Heatmap displaying GSEA in WNT-permissive and nonpermissive tissues for the indicated Reactome and published WNT activation signatures in the ApcloxP/loxP tissues compared to WT (see Methods for more details on the signatures used). Enrichment level is based on each group average. c, Venn diagram depicting the number of unique and commonly upregulated genes with log2(fold change) > 1 among WNT-permissive tissues (top), and list of upregulated genes in at least four of the permissive tissues (bottom). Known common regulators of WNT signaling are highlighted in the green boxes. d, Npm1 relative expression in WNT-permissive and nonpermissive tissues, highlighted in green and blue, respectively (for esophagus, mid-colon, stomach corpus, liver, bladder, kidney, lung and spleen; Apc+/+ n = 4; ApcloxP/loxP n = 5; for proximal SI n = 3 per group; for stomach antrum n = 4 per group). Statistical significance was assessed using multiple two-sided t-tests, with P values adjusted using the Holm–Šidák method. Adjusted P values are shown. e, TCGA cancer types sorted according to median NPM1 expression in tumor and median NPM1 expression in adjacent normal. n denotes the number of samples. The P values shown above the boxes assessing the tumor-normal difference were obtained using a two-sided t-test. f, Scatter plot showing the correlation between the mean tumor-normal difference of the GSVA score of the signature (WNT signature a) compared to the mean tumor-normal difference of NPM1 expression across different tumor types in the TCGA dataset. g, Scatter plot showing the correlation between NPM1 expression and the GSVA score of the signature (WNT signature a) among COAD/READ tumors (n = 375) in the TCGA dataset. f,g, Data were statistically assessed using a two-sided Pearson correlation. The correlation coefficient (r) is displayed to indicate the degree of association. h, Representative examples of low-intensity and high-intensity NPM1 immunohistochemistry (IHC) staining from a human tissue microarray (TMA) of patients with CRC. i, Plot of CRC tumors ranked according to high or low NPM1 protein levels in the CRC TMA and their respective percentage in Ki-67+ cells. Each point corresponds to an individual patient with CRC; the mean ± s.e.m. is shown (n = 279 patients with low and n = 193 patients with high NPM1 expression within tumors). Data were statistically assessed using an unpaired two-tailed Mann–Whitney U-test. j, NPM1 expression in the GSE39582 patient cohort of fresh-frozen CRC microarray data (tumor: n = 566; normal: n = 19). Data were statistically assessed using a two-sided Wilcoxon rank-sum test. k, Scatter plot showing the correlation between NPM1 expression and the proliferation index in the GSE39582 CRC patient cohort. Data were statistically assessed using a two-sided Pearson correlation. A linear regression line is shown with the gray bands representing the 95% confidence intervals; the correlation coefficient (r) is displayed to indicate the degree of association. l, NPM1 expression across PDS in the GSE39582 CRC cohort (PDS1 n = 186; PDS2 n = 140; PDS3 n = 122). Data were statistically assessed using a two-sided Wilcoxon rank-sum test. In all box plots, the boxes extend from the 25th to 75th percentiles, the whiskers extend to the minimum and maximum values, the line in every box is plotted at the median and outliers as dots outside whiskers. Statistically significant P values are shown in red. a, Illustrations created in BioRender.com (https://BioRender.com/s46am60 and https://BioRender.com/8kuycvt). h, Scale bar, 100 μm.
Given its association with MYC and altered expression in tumors, NPM1 was a target of interest (Fig. 1c). Npm1 was strongly upregulated in WNT-permissive tissues, except the liver, where the APC-MYC module is uncoupled (see below) (Fig. 1d). Npm1 was also moderately increased in the bladder and kidney, both of which were enriched in translation-related signatures, a process where NPM1 and MYC have important roles (Fig. 1d and Extended Data Fig. 1c).
NPM1 is deregulated in CRC and correlates with WNT/MYC signaling
To assess if this link between NPM1 and WNT/MYC signaling was detectable in human cancer, we first examined human tumors from The Cancer Genome Atlas (TCGA). Indeed, NPM1 is significantly overexpressed in many human cancers compared to adjacent normal tissues, but the strongest increase is within colon adenocarcinoma (COAD) and rectal adenocarcinoma (READ) (Fig. 1e). Gene set enrichment analysis (GSEA) of WNT signatures revealed a significant correlation between WNT activation and NPM1 expression across tumor types, and within COAD/READ tumors, the cancer subtype exhibiting the strongest WNT activation (Fig. 1f,g and Extended Data Fig. 1d,e). NPM1 expression was also highly correlated with MYC, supporting its status as a MYC target gene (Extended Data Fig. 1f).
We next compared proliferation across a cohort of patients with CRC and found significantly higher Ki-67 positivity in samples with high NPM1 expression (Fig. 1h,i). Microarray data from an independent CRC patient cohort confirmed significantly higher NPM1 expression in tumors compared to adjacent normal tissue and a significant correlation with proliferative index (Fig. 1j,k). Consistent with this, NPM1 expression was significantly higher in the stem-cell-rich, highly proliferative, pathway-derived subtype 1 (PDS1) of the PDS classification30 (Fig. 1l). Taken together, these data support a role for NPM1 in WNT-driven disease with human relevance. Therefore, we next investigated its functions during homeostasis and WNT-driven tumorigenesis.
NPM1 is critical for WNT-driven hyperproliferation, not homeostasis
As Npm1 deletion is embryonically lethal, we investigated whether its loss is tolerated in adult mice. To this end, we generated mice with conditional Npm1 alleles (Npm1loxP/loxP) (Extended Data Fig. 1g) and crossed them with R26-CreERT2 mice. Long-term NPM1 loss across multiple tissues caused no signs of ill health or gross tissue abnormalities (Extended Data Fig. 2a,b). Consistent with what has been described previously, the only anomaly detected was differences in erythropoiesis, with Npm1loxP/loxP animals having fewer and larger circulating red blood cells, and more immature reticulocytes, compared to wild-type (WT)7,31,32 (Extended Data Fig. 2c–n). This confirms NPM1’s roles in hematopoietic differentiation but, importantly, demonstrates that its loss does not cause immediate tissue toxicity. Moreover, proliferative organs like the intestine and skin retained Npm1 deletion without gross phenotypes, suggesting that although associated with cancer cell proliferation, NPM1 is dispensable for normal homeostatic proliferation, implying probable tolerance for drug targeting.
To investigate whether Npm1 upregulation after APC loss has functional significance, we crossed Npm1loxP/loxP with R26-CreERT2ApcloxP/loxP mice to achieve concurrent deletion of both genes in vivo. Apc deletion induced robust proliferation in WNT-permissive tissues (SI, stomach and liver), while proliferation remained unchanged in WNT-nonpermissive tissues (lungs, kidneys and pancreas) (Extended Data Fig. 3a–i). Notably, concurrent Npm1 deletion significantly attenuated this response, highlighting NPM1 as a key mediator of WNT-driven hyperproliferation.
NPM1 is dispensable for murine intestinal epithelium homeostasis
Given this functional association of NPM1 downstream of APC loss and its marked deregulation in CRC, we investigated its role further using murine CRC models. We generated villin-CreERT2Npm1loxP/loxP mice and confirmed efficient, tissue-specific Npm1 deletion within the intestinal epithelium (Extended Data Fig. 4a). Long-term NPM1 loss was tolerated without disrupting tissue architecture, intestinal stem or differentiated cell populations (for example, goblet and Paneth cells) or proliferation (Extended Data Fig. 4b,c). Despite being a nucleolar factor with roles in ribosome biogenesis33,34,35, NPM1 depletion did not alter 47S pre-rRNA production (Extended Data Fig. 4d,e). Consistent with this, global protein synthesis remained unaffected in ex vivo-induced Npm1loxP/loxP intestinal organoids, in line with previous findings9 (Extended Data Fig. 4f,g). Nucleolar number was also unchanged after Npm1 deletion, although fibrillarin staining revealed a minor variation in nucleolar circularity (Extended Data Fig. 4h–q).
NPM1 loss limits proliferation and extends survival in WNT-high CRC
We next investigated whether Npm1 upregulation depends specifically on high WNT signaling after APC loss, rather than other oncogenic mutations unrelated to the WNT pathway. Notably, Npm1 overexpression was observed exclusively in models with Apc deletion but not in those harboring other CRC-relevant mutations such as Braf, Kras or Trp53 (Fig. 2a and Extended Data Fig. 5a–c). Myc co-deletion prevented Npm1 overexpression, suggesting MYC-dependent regulation (Fig. 2a). NPM1 depletion did not abrogate activation of the BRAF-associated fetal-like transcriptional program36 (Extended Data Fig. 5d,e), further supporting its association with high WNT. Consistent with this, APC mutant human CRCs exhibit enhanced WNT signaling and significantly higher NPM1 expression compared to APC WT counterparts (Extended Data Fig. 5f–h).

a, Npm1 relative expression in SI and colon tissues from WT mice and a range of genetically engineered murine models of CRC (GSE309379; n = 5: WT (SI and colon), VilCreERT2BrafV600E/+ (colon), VilCreERT2BrafV600E/+Trp53loxP/loxPALK5loxP/loxPR26LSL-N1icd/+ (SI); n = 4: VilCreERT2 (SI and colon), VilCreERT2BrafV600E/+Trp53loxP/loxPALK5loxP/loxP (SI), VilCreERT2ApcloxP/loxPKRasG12D/+ (colon), VilCreERT2ApcloxP/loxPKRasG12D/+Trp53loxP/loxP (SI), VilCreERT2ApcloxP/loxPTrp53loxP/loxP (SI), AhCreERT2ApcloxP/loxPMycloxP/loxP (SI); n = 3: VilCreERT2KRasG12D/+Trp53loxP/loxP (SI), VilCreERT2ApcloxP/loxPKRasG12D/+Trp53loxP/loxP (colon); n = 7: VilCreERT2BrafV600E/+Trp53loxP/loxP (SI and colon), VilCreERT2KRasG12D/+Trp53loxP/loxP R26LSL-N1icd/+ (SI); n = 10: VilCreERT2ApcloxP/loxP (SI); n = 12: VilCreERT2ApcloxP/loxP (colon); n = 8: VilCreERT2ApcloxP/loxPKRasG12D/+ (SI); n = 6: AhCreERT2ApcloxP/loxP (SI)). Data were statistically assessed by one-way analysis of variance (ANOVA) followed by Dunnett’s multiple comparisons test. b, Representative micrographs from ApcloxP/loxPNpm1+/+ (n = 5) and ApcloxP/loxPNpm1loxP/loxP (n = 5) SI sections stained with hematoxylin and eosin (H&E) and anti-bromodeoxyuridine (BrdU) from mice collected 4 days after induction. The red bars indicate the expanded crypt depth after APC loss. c, Quantification of BrdU+ cells in SI half-crypts of animals from the groups shown in a (n = 5 per group). Data were statistically assessed using an unpaired, two-tailed Mann–Whitney U-test. d, Survival curves of ApcloxP/+ (n = 14) and ApcloxP/+Npm1loxP/loxP (n = 16) mice sampled at the clinical endpoint. Median survival in days is indicated in parentheses. Censored mice are denoted as tick marks at the indicated times after induction. The P value was obtained using a log-rank (Mantel–Cox) test. e, Tumor numbers from ApcloxP/+ (n = 8) and ApcloxP/+Npm1loxP/loxP (n = 9) mice sampled at the clinical endpoint. Data were statistically assessed using an unpaired, two-tailed t-test. f, Representative staining for NPM1 on SI tissue sections from ApcloxP/+Npm1loxP/loxP animals at the clinical endpoint (n = 5). g, Quantification of the percentage of tumors being positive, negative or mosaic for NPM1 expression in each ApcloxP/+Npm1loxP/loxP animal at the clinical endpoint (n = 5). h, Survival curves of Lgr5-CreERT2ApcloxP/loxP (n = 13) and Lgr5-CreERT2ApcloxP/loxPNpm1loxP/loxP (n = 11) mice sampled at the clinical endpoint. Median survival in days is indicated in parentheses. Censored mice are denoted as tick marks at the indicated times after induction. The P value was obtained using a log-rank (Mantel–Cox) test. i, Tumor numbers from Lgr5-CreERT2ApcloxP/loxP (n = 11) and Lgr5-CreERT2ApcloxP/loxPNpm1loxP/loxP (n = 9) mice sampled at the clinical endpoint. Data were statistically assessed using an unpaired, two-tailed Mann–Whitney U-test. j, Representative staining for NPM1 on SI tissue sections from Lgr5-CreERT2ApcloxP/loxP (n = 3) and Lgr5-CreERT2ApcloxP/loxPNpm1loxP/loxP (n = 7) animals at the clinical endpoint. k, Quantification of the percentage of tumors being positive, negative or mosaic for NPM1 expression in each Lgr5-CreERT2ApcloxP/loxPNpm1loxP/loxP animal at the clinical endpoint (n = 7). l, Representative micrographs from ApcloxP/loxPKrasG12D/+Npm1+/+ (n = 4) and ApcloxP/loxPKrasG12D/+Npm1loxP/loxP (n = 4) SI sections stained with H&E and anti-BrdU from mice collected 3 days after tamoxifen induction. The red bars indicate the expanded crypt depth after APC loss and KRASG12D activation. m, Quantification of BrdU+ cells in the SI of animals from the groups shown in l (n = 4 per group). Data were statistically assessed using an unpaired, two-tailed t-test. n, Survival curves of ApcloxP/+KrasG12D/+ (n = 17) and ApcloxP/+KrasG12D/+Npm1loxP/loxP (n = 20) mice sampled at the clinical endpoint. Median survival in days is indicated in parentheses. The P value was obtained using a log-rank (Mantel–Cox) test. o, Tumor numbers from ApcloxP/+KrasG12D/+ (n = 17) and ApcloxP/+KrasG12D/+Npm1loxP/loxP (n = 20) mice sampled at the clinical endpoint. Data were statistically assessed using an unpaired, two-tailed Mann–Whitney U-test. p, Representative staining for NPM1 on colon tissue sections from ApcloxP/+KrasG12D/+ and ApcloxP/+KrasG12D/+Npm1loxP/loxP animals at the clinical endpoint. q, Quantification of the percentage of tumors being positive, negative or mosaic for NPM1 expression in each ApcloxP/+KrasG12D/+Npm1loxP/loxP animal at the clinical endpoint (n = 9) compared to that of ApcloxP/+Npm1loxP/loxP animals (n = 5) presented in Fig. 2g. Data were statistically assessed using a two-way ANOVA with Šidák’s correction for multiple comparisons. The bar charts present data as the mean ± s.e.m.; the boxes in the box plots extend from the 25th to 75th percentile, the whiskers extend to the minimum and maximum values, and the line in every box is plotted at the median. Statistically significant P values are shown in red. b,l, Scale bar, 50 μm. f, Scale bar, 1 mm. j, Scale bar, 300 μm. p, Scale bar, 500 μm.
Apc deletion in villin-CreERT2-expressing cells triggered intestinal hyperproliferation, which was markedly suppressed by concurrent Npm1 deletion (Fig. 2b,c). Interestingly, the Olfm4+ stem cell population remained unaffected (Extended Data Fig. 5i,j). Stochastic loss of the second Apc allele in villin-CreERT2ApcloxP/+ mice promotes tumor formation. Npm1 deletion significantly extended survival, although most tumors escaped recombination and retained NPM1 expression (Fig. 2d–g and Extended Data Fig. 5k). Notably, the surrounding normal epithelium maintained Npm1 deletion, suggesting a tumor-specific requirement for NPM1 during initiation. Given that the stem cell population appeared unaffected, we investigated whether tumorigenesis driven by Apc deletion in Lgr5-expressing cells (Lgr5-CreERT2ApcloxP/loxP) was suppressed by NPM1 loss. Remarkably, Npm1 deletion suppressed this rapid adenoma model, extending survival threefold, while tumors again retained NPM1 expression (Fig. 2h–k and Extended Data Fig. 5l).
Additionally, NPM1 loss significantly reduced hyperproliferation without affecting stem cells after oncogenic Kras activation (KrasG12D/+) in villin-CreERT2ApcloxP/loxP mice (Fig. 2l,m and Extended Data Fig. 5m,n). KRAS mutations, found in ~40% of cases with CRC, often follow APC loss, confer therapy resistance and accelerate disease progression6. NPM1 loss also significantly extended survival in villin-CreERT2ApcloxP/+KrasG12D/+ mice (Fig. 2n). Unlike with APC loss alone, oncogenic Kras enabled the establishment of NPM1− tumors, although their number was significantly reduced (Fig. 2o–q and Extended Data Fig. 5o). Taken together, our data suggest that NPM1 is required for WNT-driven intestinal cell proliferation and tumor initiation; this dependency persists after oncogenic KRAS activation, which drives resistance to epidermal growth factor receptor and mTOR inhibition.
p53 mediates the antitumorigenic effects of Npm1 deletion
Given the marked phenotypic suppression after Apc deletion, we examined transcriptional changes to uncover how NPM1 loss mediates this effect. Surprisingly, RNA sequencing (RNA-seq) revealed minimal transcriptional differences (Extended Data Fig. 6a–c). Previous studies linked Npm1 embryonic knockout (KO) to increased p53 and p21 levels7,8, prompting us to investigate a potential p53 response in NPM1-depleted intestines. Both p53 and p21 were upregulated in intestinal crypts, irrespective of oncogenic KRASG12D activation or APC loss (Fig. 3a–i). Increased p53 levels were also evident in the crypts and tumors of ApcloxP/+KrasG12D/+ mice collected at the clinical endpoint (Fig. 3j,k and Extended Data Fig. 6d). Consistent with our transcriptomic data, the mRNA levels of Trp53, cyclin-dependent kinase inhibitor 1a (Cdkn1a, encoding p21) and several p53 target genes (for example, Bax, BCL2 binding component 3 (Bbc3), phorbol-12-myristate-13-acetate-induced protein 1 (Pmaip1)) remained unchanged (Extended Data Fig. 6e,f), suggesting posttranscriptional regulation of p53 and p21 after NPM1 loss.

a, Representative p53 (yellow) staining from SI tissue sections of Npm1+/+ and Npm1loxP/loxP (n = 4 per group) animals collected 4 days after induction. Nuclei (blue) are visualized with 4′,6-diamidino-2-phenylindole (DAPI). b, Quantification of p53+ cells in the crypts from the groups in a. Data were statistically assessed using an unpaired, two-tailed Mann–Whitney U-test. c, Representative p21 staining from SI tissue sections of Npm1+/+ (n = 4) and Npm1loxP/loxP (n = 3) animals collected 120 days after induction (top row) and after additional activation of KRASG12D (KrasG12D/+ n = 3; KrasG12D/+Npm1loxP/loxP n = 3) collected 30 days after induction (bottom row). d,e, Quantification of p21+ cells in SI half-crypts of Npm1+/+ and Npm1loxP/loxP animals without (d) and with (e) additional KRASG12D activation from the groups presented in c. Data were statistically assessed using an unpaired, two-tailed t-test. f, Representative staining for NPM1 and p21 on SI tissue sections from ApcloxP/loxP and ApcloxP/loxPNpm1loxP/loxP animals collected 4 days after induction (n = 6 per group). The red dotted line indicates the outer edges of the intestinal crypts. g, Quantification of p21+ cells in the SI half-crypts of animals from the groups in f. Data were statistically assessed using an unpaired, two-tailed t-test. h, Representative staining for p21 (top) and p53 (yellow, bottom) on SI tissue sections from ApcloxP/loxPKrasG12D/+ and ApcloxP/loxPKrasG12D/+Npm1loxP/loxP animals collected 3 days after induction. The red dotted line indicates the outer edges of the intestinal crypts. Nuclei (blue) were visualized with DAPI. i, Quantification of p21+ cells in SI half-crypts and p53+ cells in the crypts of animals from the groups in h (ApcloxP/loxPKrasG12D/+ (n = 3 for p21, n = 4 for p53); ApcloxP/loxPKrasG12D/+Npm1loxP/loxP (n = 4)). Data were statistically assessed using an unpaired, two-tailed t-test. j,k, Quantification of p53+ cells in SI crypts (j) and within tumors (k) of ApcloxP/+KrasG12D/+ and ApcloxP/+KrasG12D/+Npm1loxP/loxP animals at the clinical endpoint. Data were statistically assessed using an unpaired, two-tailed t-test. Related to Extended Data Fig. 6d (n = 4 per group). l, Representative images of SI tissue sections stained with H&E, anti-BrdU and anti-NPM1 from ApcloxP/loxPTrp53loxP/loxP (n = 4) and ApcloxP/loxPTrp53loxP/loxPNpm1loxP/loxP (n = 5) animals sampled 4 days after induction. The red bars indicate crypt depth. m, Quantification of BrdU+ cells in the SI half-crypts of animals from the groups shown in l and statistically compared to that of animals without Trp53 deletion presented in Fig. 2c. Data were statistically assessed using a one-way ANOVA followed by Tukey’s multiple comparisons test. n, Survival curves of ApcloxP/+ (n = 14) and ApcloxP/+Npm1loxP/loxP (n = 16) mice presented as dotted lines (also shown in Fig. 2d), compared to that of ApcloxP/+Trp53loxP/loxP (n = 14) and ApcloxP/+Trp53loxP/loxPNpm1loxP/loxP (n = 15) mice sampled at the clinical endpoint. Median survival in days is indicated in parentheses. P values were obtained using a log-rank (Mantel–Cox) test. o, Tumor number of ApcloxP/+ (n = 8) and ApcloxP/+Npm1loxP/loxP (n = 9) (also shown in Fig. 2e), plotted with that of ApcloxP/+Trp53loxP/loxP (n = 8) and ApcloxP/+Trp53loxP/loxPNpm1loxP/loxP (n = 14) mice sampled at the clinical endpoint. Data were statistically assessed using a one-way ANOVA followed by Tukey’s multiple comparisons test. p, Representative staining for NPM1 on SI tissue sections from ApcloxP/+Npm1loxP/loxP (n = 5) (also shown in Fig. 2f) and ApcloxP/+Trp53loxP/loxPNpm1loxP/loxP (n = 5) mice at the clinical endpoint. q, Quantification of the percentage of tumors being positive, negative or mosaic for NPM1 expression in ApcloxP/+Trp53loxP/loxPNpm1loxP/loxP mice at the clinical endpoint (n = 5) compared to that of ApcloxP/+Npm1loxP/loxP animals (n = 5) presented in Fig. 2g. Data were statistically assessed using a two-way ANOVA followed by Šidák’s multiple comparisons test. r, NPM1 expression in WT and mutant (Mut) TP53 COAD/READ tumors in the TCGA dataset (n = 375). Data were statistically assessed using a two-sided t-test. s,t, Table (s) and graphical representation (t) of COAD/READ tumors in the TCGA dataset (n = 375) with shallow NPM1 deletions and TP53 mutations. Data were analyzed using a two-sided Fisher’s exact test. All bar charts present data as the mean ± s.e.m.; the boxes in the box plots extend from the 25th to 75th percentile, the whiskers extend to the minimum and maximum values, the line in every box is plotted at the median and outliers as dots outside the whiskers. Statistically significant P values are shown in red. a, Scale bar, 20 μm. c,l, Scale bar, 50 μm. f, Scale bar, 100 μm. h, Scale bar, 100 μm (top), 20 μm (bottom). p, Scale bar, 1 mm.
Concurrent conditional deletion of Trp53 (Trp53loxP/loxP) alongside Npm1 and Apc restored proliferation in the ApcloxP/loxPTrp53loxP/loxPNpm1loxP/loxP intestinal epithelium, confirming a functional relationship between p53 and NPM1 (Fig. 3l,m). Furthermore, p53 loss negated the survival benefit of NPM1 depletion in Apc-driven CRC models, while most tumors retained NPM1 loss (Fig. 3n–q and Extended Data Fig. 6g–l). To corroborate the role of the p53 pathway, we deleted Cdkn1a in the Lgr5-CreERT2ApcloxP/loxP tumor model. Like p53 loss, co-deletion of Cdkn1a and Npm1 significantly reduced survival compared to p21-proficient Npm1loxP/loxP mice, and enabled the establishment of significantly more NPM1− tumors (Extended Data Fig. 7a–e).
To probe a CRC model without the Apc mutation, we investigated NPM1 loss in an aggressive model driven by oncogenic Kras, active NOTCH signaling and p53 loss37. Npm1 deletion had no impact (Extended Data Fig. 7f,g). In a less aggressive model driven by KRAS and p53 loss, Npm1 deletion slightly but significantly accelerated tumorigenesis (Extended Data Fig. 7h,i). These data confirm NPM1 dependency in WNT-driven models and the requirement for p53.
Given this relationship with p53, we analyzed human tumors and found that TP53 WT samples from the TCGA COAD/READ dataset exhibited significantly higher NPM1 expression than the TP53-mutated samples (Fig. 3r). Among the n = 375 COAD/READ tumors, 76 harbored shallow NPM1 deletions (none harbored NPM1 deep deletions). Of these, 69 (~91%) also carried TP53 mutations, which is significantly higher than the 177 of 299 TP53-mutated tumors without NPM1 deletions (Fig. 3s,t). This suggests preferential TP53 inactivation in CRCs with NPM1 deletions, mirroring murine data where p53 loss is essential for establishing Npm1-null tumors. Together, our results demonstrate that functional p53 signaling is essential for mediating the tumor-suppressive effects of NPM1 loss. However, it is important to note that the modest p53 induction after Npm1 deletion in the WT intestine did not perturb normal homeostatic proliferation. This suggests that NPM1 loss triggers a specific stress after APC loss, rather than a broad effect on proliferation per se.
NPM1 depletion induces ribosome pauses during translation
Given the minimal transcriptional changes after NPM1 loss despite the increase in p53/p21 protein levels, we investigated potential posttranscriptional regulation by performing ribosome profiling in intestinal epithelia from ApcloxP/loxPKrasG12D/+ mice. Library quality control confirmed good ribosome protected fragment (RPF) quantification, with high enrichment in protein-coding sequences; principal component analysis showed separation between sample groups (Extended Data Fig. 8a–g). NPM1 loss induced significant changes in RPFs, but, strikingly, had minimal impact on total RNA levels, which is consistent with the bulk RNA-seq data (Fig. 4a and Extended Data Fig. 8h–k). To distinguish transcriptional from translational changes, we compared differentially expressed transcripts at the total RNA and RPF levels. We identified 239 transcripts with decreased and 43 transcripts with increased RPF abundance, without corresponding changes in total cytoplasmic RNA (Fig. 4a). Interestingly, only few transcripts changed at both levels (eight decreased, three increased) with none changing exclusively at the mRNA level.

Data comparing ApcloxP/loxPKrasG12D/+Npm1loxP/loxP and ApcloxP/loxPKrasG12D/+ intestinal epithelial cells, sampled 3 days after induction (n = 4 per group) a, Translation efficiency scatter with differentially expressed transcripts at the total cytoplasmic RNA level (x axis) compared to changes at the RPF level (y axis). Transcripts are color-coded depending on their change being significant at the total and RPF level (both up and both down), or exclusively at the RPF level (RPF up and RPF down) (Padj < 0.1). No transcript was detected to be changed exclusively at the total cytoplasmic RNA level. b, GSEA for Hallmark gene sets on transcripts ranked according to changes at the RPF level. c, GSEA for Hallmark gene sets on genes ranked according to changes at the proteome level. d, Enrichment profiles from the GSEA for the Hallmark sets E2F targets, G2M checkpoint and MYC targets V1. Left, Enrichment profiles of RPFs. Right, Enrichment profiles of proteomics data. Enrichment scores were calculated using the weighted Kolmogorov–Smirnov test implemented in the fGSEA package. Significance and normalization were assessed using empirical permutation testing; P values were adjusted using the Benjamini–Hochberg method. Normalized enrichment scores (NES) and Padj values are shown in the plots. e, Quantification of genes shared by the leading edges of both RPFs and proteomics (within the same pathway) are displayed as violins (Npm1+/+ (WT) condition in gray, Npm1loxP/loxP (KO) condition in red). Each plot represents scaled abundances of RPF-normalized reads and protein intensities; the number of genes in the shared leading edge is displayed above each panel. The box plots within the violins extend from the 25th to 75th percentile, with the lines plotted at the median. The whiskers extend to the minimum and maximum values, no further than 1.5 times the interquartile range from the hinges. Outliers beyond the end of the whiskers are not plotted. f, Ribosome pause site distribution across transcript CDS, with induced sites in red, resolved sites in blue and maintained sites in gray. g, Ribosome pause site distribution across genes belonging to the leading edges of the pathways indicated. For each pathway, the pause distribution is shown for those genes belonging to the leading edge of both RPFs and proteins, or to the leading edge exclusively for proteins (only protein) or RPFs (only RPFs). The number of genes in each of these categories is indicated above the plots. Color coding of the pause sites as in f. Note that the number of genes in the shared category is different than in e as we are displaying only transcripts for which ribosomes pause sites were confidently identified. h, Metagene plot of all transcripts detected in the Riboseq experiment with more than 50 average reads aligned to their CDS. The plot represents the variation in ribosome occupancy across the 5′ untranslated region (UTR) (first panel), their CDS (second panel) or their 3′ UTR (third panel). The gray shaded area represents the standard deviation of the delta ribosome occupancy across the four biological replicates assayed per group.
To identify pathways significantly affected by NPM1 loss, we performed GSEA based on RPF-level changes. Among the top positively enriched pathways (Fig. 4b), we confirmed DNA repair activation by increased γH2AX positivity in NPM1-depleted cells and tumors of ApcloxP/+KrasG12D/+ mice, without a corresponding increase in apoptosis, as assessed by cleaved-PARP levels. Interestingly, NPM1 expression was significantly correlated with DNA repair and replication stress signatures in human CRCs (Extended Data Fig. 8l–u). However, the positive enrichment of pathways such as E2F targets, G2M checkpoint and MYC targets (v1) was in stark contrast to the reduced proliferation phenotype observed upon NPM1 loss (Fig. 4b).
To complement ribosome profiling, we quantified protein abundance in the same samples using mass spectrometry (MS). Notably, while E2F targets, G2M checkpoint and MYC targets were positively enriched in RPFs, they were significantly negatively enriched at the protein level (Fig. 4b,c). Comparing the normalized abundance of RPFs and proteins for the genes belonging to the leading edge subsets of these pathways further confirmed this opposite enrichment (Fig. 4d,e). To interrogate this discrepancy, we exploited the positional resolution of RPF data. Across the transcriptome, NPM1 loss caused a striking induction of ribosome pausing in the first half of coding sequences (CDS) (Fig. 4f). Interestingly, genes participating in the leading edge subset of the RPF enrichment exhibited more pause inductions, while genes participating in the leading edge of the proteomics were devoid of them (Fig. 4g). Further positional analyses across all transcripts revealed a pronounced shift in ribosome occupancy after Npm1 deletion, characterized by increased footprint density at the 5′-end and reduced at the 3’-end of the CDS (Fig. 4h). Notably, Trp53 and Cdkn1a transcripts lacked this 5′-end occupancy increase, while NPM1 loss promoted resolution of ribosome pausing sites on Trp53, suggesting more efficient transcript translation that may contribute to the increased p53 protein levels (Extended Data Fig. 8v–x).
NPM1 depletion triggers the integrated stress response
The widespread ribosome pauses induced upon Npm1 deletion prompted us to investigate a potential response to protein synthesis stress. We examined key translation pathway components in the tumors and crypts of ApcloxP/+KrasG12D/+ mice, which develop NPM1-deficient tumors. Phosphorylation of the translation activators eukaryotic initiation factor 4E-binding protein 1 (p-4E-BP1) and eukaryotic initiation factor 4E (p-eIF4E), and elongation inhibitory phosphorylation of eukaryotic elongation factor 2 (p-eEF2), were upregulated after NPM1 loss, while ApcloxP/loxPKrasG12D/+ NPM1-deficient organoids exhibited reduced protein synthesis (Fig. 5a–d and Extended Data Fig. 9a–e). These contradictory signals suggest disrupted translation regulation, potentially inducing proteostatic stress. Therefore, we examined phosphorylation of the integrated stress response (ISR) marker eukaryotic initiation factor-2α (p-eIF2α), an indicator of deregulated proteostasis. p-eIF2α levels were elevated in the tumors and intestinal crypts of Npm1-depleted ApcloxP/+KrasG12D/+ mice, along with upregulation of the endoplasmic reticulum (ER) stress response pathway in the proteomics data (Fig. 5e–h). Only a modest upregulation was evident in the homeostatic Npm1loxP/loxP gut epithelium (Extended Data Fig. 9f–h). Ex vivo analysis of ApcloxP/loxP and ApcloxP/loxPKrasG12D/+ intestinal cells and organoids revealed comparable p-eIF2α levels between genotypes and even a slight decrease in downstream ISR targets like activating transcription factor 4 in NPM1-depleted cells, while p53/p21 upregulation persisted (Extended Data Fig. 9i–l). ER stress induced by tunicamycin in ApcloxP/loxPKrasG12D/+ organoids showed that NPM1-depleted cells could activate the ‘canonical’ stress response, upregulating p-eIF2α, activating transcription factor 4, C/EBP homologous protein and growth arrest and DNA damage-inducible protein 34; however, p53/p21 upregulation was lost under these conditions. These data suggest that NPM1 loss causes dysregulated translation and global protein synthesis stress in response to hyperproliferative signaling, particularly after APC loss and KRASG12D activation. However, this stress may be distinct from the ‘canonical’ ISR.

a, Representative staining for p-4E-BP1 (Thr37/46) on SI tissue sections from ApcloxP/+KrasG12D/+ (n = 6) and ApcloxP/+KrasG12D/+Npm1loxP/loxP (n = 5) animals collected at the endpoint. The red dotted lines indicate intestinal adenomas. b, HALO H-score quantification normalized to the average control value of p-4E-BP1 expression in tumors (left) and crypts (right) of the groups shown in a. c, Representative staining for p-eEF2 (Thr56) on SI tissue sections from ApcloxP/+KrasG12D/+ and ApcloxP/+KrasG12D/+Npm1loxP/loxP animals collected at the endpoint (n = 6 per group). The red dotted lines indicate intestinal adenomas. d, HALO H-score quantification normalized to the average control value of p-eEF2 expression in tumors (left) and crypts (right) of the groups shown in c. e, Representative staining for p-eIF2α (Ser51) on SI tissue sections from ApcloxP/+KrasG12D/+ (n = 4) and ApcloxP/+KrasG12D/+Npm1loxP/loxP (n = 5) animals collected at the endpoint. The red dotted lines indicate intestinal adenomas. f, HALO H-score quantification of p-eIF2α expression in the tumors (left) and crypts (right) of the groups shown in e. g, HALO quantification of p-eIF2α stain intensity in the tumors (left) and crypts (right) of the groups shown in e. h, Enrichment profile for the pathway response to ER stress (from the Gene Ontology Biological Process subset) from the proteomics data. The NES and Padj value were calculated as in Fig. 4d and are shown in the plot. b,d,f, All data were statistically assessed using an unpaired, two-tailed t-test. g, Statistical significance was assessed using multiple two-sided t-tests, with P values adjusted using the Holm-Šidák method. Padj values are shown. The boxes in box plots extend from the 25th to 75th percentile, the whiskers extend to the minimum and maximum values, and the line in every box is plotted at the median. Statistically significant P values are shown in red. a,c, Scale bar, 300 μm. e, Scale bar, 200 μm.
Inhibition of the ISR restores proliferation after NPM1 loss
To investigate which ISR step is critical for the phenotypes after NPM1 loss, we treated mice with the small-molecule inhibitor of the integrated stress response (ISRIB), which reverses the effects downstream of p-eIF2α38. ISRIB rescued proliferation in the ApcloxP/loxPNpm1loxP/loxP intestines, with proliferating cells expanding further into villi compared to vehicle-treated controls (Fig. 6a,b). Similarly, ISRIB restored proliferation in ApcloxP/loxPKrasG12D/+Npm1loxP/loxP intestines to levels matching Npm1-proficient controls (Fig. 6c,d). Interestingly, ISRIB treatment did not restore protein synthesis in NPM1-depleted ApcloxP/loxPKrasG12D/+ organoids, suggesting that reduced proliferation after NPM1 loss is more specific to NPM1’s roles than a general protein synthesis defect (Extended Data Fig. 9m,n).

a, Representative BrdU staining on SI tissue sections from ApcloxP/loxPNpm1loxP/loxP animals treated with vehicle or ISRIB (n = 4 per group) and collected 4 days after induction. b, Quantification of BrdU+ cells in SI half-crypts of animals from the groups shown in a, separated by a dashed line from untreated ApcloxP/loxPNpm1+/+ and ApcloxP/loxPNpm1loxP/loxP (n = 5 per group) animals used for comparison and also shown in Fig. 2c. Data were statistically assessed using a one-way ANOVA followed by Tukey’s multiple comparisons test. c, Representative BrdU staining on SI tissue sections from ApcloxP/loxPKrasG12D/+Npm1+/+ (n = 4) and ApcloxP/loxPKrasG12D/+Npm1loxP/loxP (n = 3) animals treated with ISRIB and collected 3 days after induction. d, Quantification of BrdU+ cells in SI half-crypts of animals from the groups shown in c, separated by a dashed line from untreated ApcloxP/loxPKrasG12D/+Npm1+/+ and ApcloxP/loxPKrasG12D/+Npm1loxP/loxP (n = 4 per group) animals used for comparison and also presented in Fig. 2m. Data were statistically assessed using a one-way ANOVA followed by Tukey’s multiple comparisons test. e, Representative BrdU (top) and p-eIF2α (Ser51) (bottom) staining on SI tissue sections from ApcloxP/loxPNpm1loxP/loxP animals treated with vehicle or PERK inhibitor (PERKi) (n = 4 per group) and collected 4 days after induction. f, Quantification of BrdU+ cells in the SI half-crypts of animals from the groups shown in e, separated by a dashed line from untreated ApcloxP/loxPNpm1+/+ (n = 5) and ApcloxP/loxPNpm1loxP/loxP (n = 5) animals used for comparison and also presented in Fig. 2c. Data were statistically assessed using a one-way ANOVA followed by Tukey’s multiple comparisons test. g, Representative BrdU staining on SI tissue sections from ApcloxP/loxPKrasG12D/+Npm1+/+ (n = 4) and ApcloxP/loxPKrasG12D/+Npm1loxP/loxP (n = 3) animals treated with PERKi and collected 3 days after induction. h, Quantification of BrdU+ cells in the SI half-crypts of animals from the groups shown in g, separated by a dashed line from untreated ApcloxP/loxPKrasG12D/+Npm1+/+ (n = 4) and ApcloxP/loxP KrasG12D/+Npm1loxP/loxP (n = 4) animals used for comparison and also presented in Fig. 2m. Data were statistically assessed using a one-way ANOVA followed by Tukey’s multiple comparisons test. i, Representative BrdU staining on SI tissue sections from untreated ApcloxP/+KrasG12D/+ (n = 5) and ApcloxP/+KrasG12D/+Npm1loxP/loxP (n = 6) animals, or ISRIB-treated ApcloxP/+KrasG12D/+ (n = 4) and ApcloxP/+KrasG12D/+Npm1loxP/loxP (n = 6) animals. ISRIB treatment lasted for 72 h before sampling and started once animals were showing cancer symptoms. The red dotted lines indicate intestinal adenomas. j, Quantification of BrdU+ cells in tumors (left) and crypts (right) from the groups shown in i. Data were statistically assessed using a one-way ANOVA followed by Tukey’s multiple comparisons test (left) and an unpaired, two-tailed t-test (right). k, Representative p-eIF2α (Ser51) staining on SI tissue sections from ISRIB-treated ApcloxP/+KrasG12D/+ (n = 4) and ApcloxP/+KrasG12D/+Npm1loxP/loxP (n = 7) animals. ISRIB treatment lasted for 72 h before sampling and started once animals were showing cancer symptoms. The red dotted lines indicate intestinal adenomas. l, Quantification of p-eIF2α H-score in tumors (left) and crypts (right) from the groups shown in k. Data were statistically assessed using an unpaired, two-tailed t-test. m, Representative NPM1 (left), p21 (middle) and p53 (right) staining on SI tissue sections from ApcloxP/loxPNpm1loxP/loxP animals treated with vehicle or ISRIB (n = 4 per group) and collected 4 days after induction. The red dotted line indicates the outer edges of the intestinal crypts. n, Quantification of p21+ cells in the SI half-crypts of animals from the groups shown in m, separated by a dashed line from untreated ApcloxP/loxPNpm1+/+ (n = 6) and ApcloxP/loxPNpm1loxP/loxP (n = 6) animals used for comparison and also presented in Fig. 3g. Data were statistically assessed using a one-way ANOVA followed by Tukey’s multiple comparisons test. o, Quantification of p53+ cells in the SI half-crypts of animals from the groups shown in m. Data were statistically assessed using an unpaired, two-tailed t-test. All bar charts present data as the mean ± s.e.m. The boxes in the box plots extend from the 25th to 75th percentile, the whiskers extend to the minimum and maximum values, and the line in every box is plotted at the median. Statistically significant P values are shown in red. a,e, Scale bar, 50 μm. c,g,i,k,m, Scale bar, 100 μm.
To confirm the role of p-eIF2α downstream of NPM1 loss, we inhibited the eIF2α protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK) using the small-molecule inhibitor GSK2606414 (ref. 39). This inhibited eIF2α phosphorylation and phenocopied the ISRIB effects, restoring proliferation of NPM1-depleted intestinal cells in both ApcloxP/loxP and ApcloxP/loxPKrasG12D/+ models (Fig. 6e–h). Given the known pancreatic toxicity of GSK2606414 (ref. 40), and lack of long-term ISRIB tolerance41, we used the licensed antidepressant trazodone hydrochloride for long-term studies. While not a specific p-eIF2α inhibitor, trazodone mimics ISRIB in counteracting p-eIF2α-mediated effects and is well tolerated in vivo42. Like ISRIB and PERK inhibition, trazodone hydrochloride treatment restored short-term proliferation in ApcloxP/loxPNpm1loxP/loxP intestinal cells and significantly reduced the survival benefit from Npm1 deletion in the ApcloxP/+KrasG12D/+ CRC model (Extended Data Fig. 9o–r). GSK2606414 exhibits off-target effects and related inhibitors can induce eIF2α kinases43,44. To address these limitations, we used ISRIB in subsequent experiments as a more specific tool to probe the ISR.
Given that ISR and p-eIF2α may have pro-tumorigenic effects in tumors45,46, to complement our findings on targeting ISR during the early proliferative burst after Apc deletion, we treated mice with ISRIB when harboring established tumors. ISRIB supressed proliferation in NPM1-proficient tumors, but significantly increased proliferative capacity in NPM1-deficient tumors, suggesting that both NPM1 and the ISR contribute toward tumor maintenance (Fig. 6i,j). Proliferation within intestinal crypts and p-eIF2α levels in crypts and tumors were comparable in ApcloxP/+KrasG12D/+ and ApcloxP/+KrasG12D/+Npm1loxP/loxP mice after ISRIB treatment (Fig. 6j–l).
Lastly, to examine whether ISRIB modulates the induction of p53 and p21, which are crucial mediators of tumor suppression after NPM1 loss, we quantified their expression in ISRIB-treated mice (Fig. 6m–o). Notably, ISRIB treatment prevented an increase in p21/p53 levels within NPM1-depleted intestinal crypts. Together, these data demonstrate a functional role for protein synthesis stress and the ISR in reducing WNT-mediated hyperproliferation upon NPM1 depletion, through posttranscriptional p53 activation.
Npm1 deletion improves survival in WNT-driven HCC
HCC is the second most commonly WNT-mutated cancer, with ~44% of cases exhibiting high WNT signaling primarily because of β-catenin-activating mutations47. In the liver, WNT-pathway mutations drive hepatocyte differentiation altering hepatic zonation48, rather than promoting tumorigenesis49. Furthermore, unlike CRC and other cancers, the WNT–MYC module is uncoupled in the liver and Myc deletion does not rescue the acute effects of APC loss48,50. Therefore, efficient tumorigenesis in mice requires ectopic MYC expression to synergize with β-catenin to drive HCC; MYC amplification is common in β-catenin-mutated human HCC49,51.
Npm1 expression was marginally changed after APC loss or β-catenin activation (Ctnnb1+/(Dex3)) in the liver, but significantly enhanced after conditional MYC transgene overexpression (R26lsl-MYC/lsl-MYC) (Fig. 1d and Extended Data Fig. 10a). Nevertheless, Npm1 deletion suppressed short-term hepatocyte hyperproliferation after Apc loss (Extended Data Fig. 3d), confirming a WNT effector role in WNT-permissive tissues. Similarly, NPM1 is overexpressed in human HCC (Fig. 1e), with higher expression in MYC-amplified cases; it correlates with WNT activation and is associated with significantly worse prognosis (Extended Data Fig. 10b–h).
Npm1 deletion in adult liver did not disrupt homeostasis. Liver tissue exhibited normal histology, zonation, hepatocyte proliferation and function after long-term NPM1 loss (Extended Data Fig. 10i–l). To model WNT-driven HCC, we generated mice with an activating β-catenin mutation (Ctnnb1+/(Dex3)) combined with a MYC transgene (R26lsl-MYC/lsl-MYC) to induce hepatocyte proliferation and HCC formation. Induction with high AAV8.TBG.Cre viral titers triggered recombination across hepatocytes, leading to increased proliferation and hepatomegaly, both of which were suppressed after concurrent NPM1 depletion (Fig. 7a–d). NPM1 loss modestly suppressed WNT-driven hepatocyte differentiation, indicated by reduced glutamine synthetase staining, while reduced proliferation after Apc deletion was further confirmed (Extended Data Fig. 10m–q). In an oncogenic BRAF-driven model, proliferation remained unchanged by NPM1 depletion 4 days after induction, and modestly reduced by 8 days (with p21 levels increased), confirming NPM1 as a key effector of elevated WNT signaling (Extended Data Fig. 10r–v).

a, Representative staining for BrdU, NPM1 and p21 on liver tissue sections of Ctnnb1+/(Dex3)R26lsl-MYC/lsl-MYC animals with or without Npm1loxP/loxP collected at the indicated time points (four days, n = 3 per group, or ten days, n = 5 per group after high-dose AAV8.TBG.Cre viral induction). b, Quantification of BrdU+ cells in ten ×10 fields of view (FOVs) per biological replicate of Ctnnb1+/(Dex3)R26lsl-MYC/lsl-MYC animals with or without Npm1loxP/loxP sampled 4 days after induction (n = 3 per group). Data were statistically assessed using an unpaired, two-tailed t-test. c, Quantification of BrdU+ cells in ten ×10 FOVs per biological replicate, d, Percentage of liver-to-body weight ratios. e, HALO quantification of the percentage of p21+ hepatocytes from Ctnnb1+/(Dex3)R26lsl−MYC/lsl-MYC animals with or without Npm1loxP/loxP collected 10 days after induction (n = 5 per group except in c where n = 4 for the Npm1loxP/loxP group). f, Significantly positively enriched Reactome pathways in RNA-sequenced Ctnnb1+/(Dex3)R26lsl−MYC/lsl−MYCNpm1loxP/loxP mice compared to controls, sampled 10 days after induction (n = 4 per group). The fraction of regulated genes within each pathway is indicated by the gene ratio and the gene number (according to circle size); the circle color indicates the significance of enrichment. Overrepresentation analysis was conducted using a hypergeometric model implemented in the ReactomePA package, with significance assessed using a one-sided Fisher’s exact test. g,h, Survival curves of male (g) and female (h) Ctnnb1+/(Dex3)R26lsl-MYC/lsl-MYC animals induced with low-dose AAV8.TBG.Cre and sampled at the clinical endpoint. Median survival in days is indicated in parentheses. Censored mice are denoted as tick marks at the indicated times after induction. P values were obtained using a log-rank (Mantel–Cox) test (n = 13 Npm1+/+ and 15 Npm1loxP/loxP in g, and n = 15 per group in h). i, Percentage of Ctnnb1+/(Dex3)R26lsl-MYC/lsl-MYC (n = 11) and Ctnnb1+/(Dex3)R26lsl-MYC/lsl-MYCNpm1loxP/loxP (n = 18) animals with lung metastases at the endpoint. Data were compared using a two-sided Fisher’s exact test. j,k, Percentage of liver-to-body weight ratios at the endpoint for the male and female animals shown in g,h (n = 12 Npm1+/+ and 14 Npm1loxP/loxP in j, and n = 14 per group in k). l,m, Tumor number from male and female animals presented in g,h at the endpoint (n = 13 Npm1+/+ and 14 Npm1loxP/loxP in l, and n = 14 per group in m). n, Representative NPM1 staining on liver tissue sections of animals at the endpoint (n = 4 per group). The red dotted lines indicate individual tumors. o, Quantification of percentage of tumors negative for NPM1 expression in Npm1loxP/loxP animals at the endpoint (n = 4). p, Representative BrdU staining on liver tissue sections of Ctnnb1+/(Dex3)R26lsl-MYC/lsl-MYC animals with or without Npm1loxP/loxP treated with ISRIB and collected 4 days after induction (n = 3 per group). q, Quantification of BrdU+ cells in ten ×10 FOVs from liver sections of animals from the groups shown in p, separated by a dashed line from untreated animals of the same genotypes (n = 3 per group), used for comparison and also presented in b. b–e,j–k,m, Data were statistically assessed using an unpaired, two-tailed t-test. l, Data were statistically assessed using an unpaired, two-tailed Mann–Whitney U-test. q, Data were statistically assessed using a one-way ANOVA followed by Tukey’s multiple comparisons test. b–e, The bar charts present data as the mean ± s.e.m.; the boxes in the box plots extend from the 25th to 75th percentile, the whiskers extend to the minimum and maximum values, and the line in every box is plotted at the median. Statistically significant P values are shown in red. a,p, Scale bar, 100 μm. n, Scale bar, 500 μm.
NPM1 depletion induced p21 expression in the liver irrespective of β-catenin/MYC or oncogenic BRAF activation (Fig. 7a,e and Extended Data Fig. 10t,v). In the β-catenin/MYC model, pathways related to p53 regulation, DNA repair and the cell cycle were among the most enriched after NPM1 loss (Fig. 7f and Extended Data Fig. 10w), indicating a conserved response to NPM1 depletion in both the gut and liver. Consistent with this, TP53 is more frequently inactivated in HCCs with NPM1 deletions compared to those with intact NPM1, recapitulating our observations in human CRC (Extended Data Fig. 10x,y).
Low AAV8.TBG.Cre viral titers induce recombination in few hepatocytes, permitting clonal outgrowth and tumor formation. NPM1 loss significantly extended survival in mice collected at the endpoint, with comparable liver mass and reduced lung metastases (Fig. 7g–k). Npm1loxP/loxP mice also developed fewer tumors, with only ~25% of them being NPM1− (Fig. 7l–o). Finally, to assess whether ISR activation directly mediates reduced proliferation after NPM1 loss in the liver, we treated β-catenin/MYC mice with ISRIB (Fig. 7p). Consistent with the gut findings, ISRIB restored hepatocyte hyperproliferation in Npm1loxP/loxP animals (Fig. 7p,q), suggesting that ISR activation underlies the proliferative defect induced by NPM1 loss in both tissues.
To conclude, we describe an overarching mechanism whereby NPM1 expression is upregulated upon oncogenic insult and is essential to support WNT-driven hyperproliferation and tumor initiation. Mechanistically, this relies on attenuation of both the p53 pathway and the ISR, which become activated upon NPM1 depletion. Importantly, this holds true for multiple WNT-permissive tissues with increased mutational load, solidifying NPM1 as a promising therapeutic target.
Discussion
MYC, a central oncogenic driver1 and WNT mediator, is challenging to target directly. In this study, we identified its target gene Npm1 as a therapeutic candidate in WNT-permissive tissues. While essential during development, NPM1 proved dispensable in adult tissues but critical for WNT-driven hyperproliferation and tumorigenesis. Notably, NPM1 depletion suppressed Kras-mutant WNT-driven CRC and restricted WNT-driven liver cancer, both high-mortality diseases with limited treatment options4,6,52.
NPM1 loss disrupted translation in WNT-activated cells by inducing ribosome pausing on mRNA CDS. While the mechanism remains unclear, NPM1 association with active ribosomes53,54 suggests it may directly modulate translation. This could be crucial during hyperproliferative signaling, where increased translational demand risks proteotoxic stress and growth arrest. Our data highlight NPM1 as a potential key regulator of translational homeostasis under these conditions.
NPM1 also binds small nucleolar RNAs and mediates site-specific 2-O′-methylation of rRNAs via fibrillarin, reducing translation of select mRNAs9. Fibrillarin was also upregulated within WNT-permissive tissues (Fig. 1c), suggesting a potential link between dysregulated rRNA modifications and oncogenic signaling. p53 represses fibrillarin expression, preventing rRNA methylation changes that enhance oncogene translation (for example, MYC) by affecting ribosome fidelity55. Conceivably, NPM1 depletion may activate p53 to maintain translation control, thus abrogating transformation.
ISR inhibition restored proliferation and suppressed the p53 response after NPM1 loss in WNT-activated tissues. However, this may reflect a noncanonical ISR, as typical targets downstream of p-eIF2α were not upregulated and ISRIB treatment failed to restore protein synthesis in NPM1-depleted organoids. Recent studies highlight the plasticity of ISR activation and downstream effects56. This may extend to NPM1-related ISR, where Trp53 notably escapes ribosome pausing after NPM1 loss. Trp53 deletion independently rescued NPM1 loss phenotypes, implicating ISR activation causally. The PERK-regulated p53 isoform (p47), identified under ER stress, supports this functional connection57. After NPM1 loss, p53 activation is subtle, avoiding full p53 transcriptional program activation, permitting homeostatic proliferation but restraining the hyperproliferation required for transformation. p53 stabilization upon NPM1 loss has been observed previously, and concomitant loss of both proteins accelerates leukemogenesis in mice7,8,32. Our findings propose a link between p53 and NPM1, mediated by proteostatic stress from increased protein synthesis. Of note, p53-deficient, KRAS-mutant non-small-cell lung cancer cells were sensitive to NPM1 depletion, indicating that this interplay may be context-dependent21.
NPM1-mutated leukemia is generally associated with favorable patient outcomes and improved treatment response58,59. Disrupting NPM1 oligomerization sensitizes cells to DNA-damaging agents60. NPM1 is involved in the DNA damage response22, with our data demonstrating increased γH2AX after NPM1 loss. We found that NPM1 depletion particularly affects transformed cells, suggesting that aside from suppressing hyperproliferation, targeting NPM1 could also enhance susceptibility to additional stress, which could be exploited therapeutically in combination therapies.
Lastly, biosynthetic capacity via increased activity of RNA polymerase I in tumor cells that are enriched in nucleolar, protein folding and ribosome biogenesis pathways, is critical for sustained growth in CRC61. Standard therapies already target RNA synthesis and nucleolar function62,63,64,65. The enrichment of core nucleolar factors, NPM1 and fibrillarin, in WNT-permissive tissues, suggests an increased reliance on protein synthesis in WNT-driven CRC. Co-targeting translation control components can sensitize KRAS-mutant CRC to standard treatment6. As deregulated translation and protein synthesis emerge as a therapeutic strategy, NPM1 stands out as a target, particularly affecting transformed cells that are highly dependent on these processes for rapid growth. Our data revealed that NPM1 couples major oncogenic and tumor suppressor pathways via translational control, offering a potential avenue toward targeting difficult-to-treat cancer drivers and tumor heterogeneity.
Methods
All animal experiments were performed in accordance with UK Home Office regulations (project licenses 70/8646 and PP3907577), with the approval, and under the oversight, of the animal welfare and ethical review board of the University of Glasgow. All experiments performed adhered to Institutional guidelines in full. Key experimental procedures are outlined below; details of additional methods are available in the Supplementary Note.
In vivo studies
Mice were maintained in conventional cages in a specific pathogen-free facility of constant temperature between 19 °C and 23 °C, and 55 ± 10% humidity, under a 12 h light–12 h dark cycle, and ad libitum access to food and water. Genotyping was performed on ear punch biopsies by TransnetYX. All experiments were conducted on mice that had been bred on a C57BL/6 background for at least three generations (n ≥ 3) for the homeostasis experiments, and at least four generations (n ≥ 4) for the CRC models. No formal randomization was performed and researchers were not blinded to the experimental groups.
For the intestinal experiments, male and female mice were induced between 8 and 15 weeks of age or once they had reached 20 g of body weight. Recombination under the villin promoter (referred to as Villin-CreERT2) was induced via intraperitoneal injection of 2 mg tamoxifen (cat. no. T5648, Sigma-Aldrich) dissolved in corn oil (cat. no. C8267, Sigma-Aldrich)66. Mice for the short-term experiments received two doses of tamoxifen on consecutive days and were sampled at 4 days after induction, with the exception of mice bearing two Apc floxed alleles67 and oncogenic KrasG12D (ref. 68) activation (ApcloxP/loxPKrasG12D/+), which were induced with a single tamoxifen injection and sampled after 3 days. The tumorigenesis experiments were induced by a single 2-mg tamoxifen intraperitoneal injection and samples were collected either at predetermined time points or when they reached the clinical endpoint as determined by hunching, paling or weight loss in the case of tumor-bearing mice. Clinical endpoints were not exceeded at any point during this study. Recombination under the Lgr5 promoter (Lgr5-CreERT2) was induced with a 3-mg tamoxifen intraperitoneal injection69; samples were collected once they had reached the clinical endpoint as defined above. The Trp53 floxed70, Cdkn1a KO71, intracellular expression of the NOTCH1 transgene (Rosa26N1icd)72, BrafV600E (ref. 73) and BrafLSL-V637E (ref. 74) alleles have been described previously.
Whole-body Npm1 depletion in adult tissues was achieved using CreERT2 expression induced from the Rosa26 locus (Rosa26CreERT2)75, and an initial 3-mg tamoxifen followed by three doses of 2-mg tamoxifen intraperitoneal injections on consecutive days. For whole-body deletion of floxed Apc alleles76 a single 3-mg intraperitoneal injection was used. For hepatocyte-specific genetic recombination of the Npm1loxP/loxP alleles, male and female mice between 2 and 4 months of age were induced with adeno-associated virus (AAV) expressing Cre under the control of the thyroxine binding globulin (TBG) promoter (AAV8.TBG.Cre). AAV.TBG.PI.Cre.rBG was a gift from J. M. Wilson (viral prep 107787-AAV8, Addgene; http://n2t.net/addgene:107787; research resource identifier: Addgene_107787). Viral delivery was conducted via intravenous tail vein injections of 2 × 1011 genome copies (GCs) per mouse. To drive liver proliferation and tumorigenesis, β-catenin was activated via conditional excision of exon 3 (Ctnnb1+/(Dex3))77, in combination with human MYC transgene expression from the Rosa26 locus (R26lsl-MYC/lsl-MYC)78. Short-term experiments in the liver tumor models were performed only in male mice because of large cell proliferation variability between the sexes. Samples were collected at specified time points, usually at 4 or 10 days after induction with 2 × 1011 AAV8.TBG.Cre GC per mouse. For the long-term experiments assessing tumor formation, mice of both sexes were induced with a lower dose of 6.4 × 108 GC per mouse, monitored for tumor growth; samples were collected at the clinical endpoint as determined by abdominal swelling, loss of body conditioning or weight. In all tumor model cohorts, mice that had to be euthanized for reasons other than tumor burden (for example, epidermal wounds) or significantly surpassed median survival were censored.
Cell proliferation was assessed using BrdU (cat. no. RPN201, Amersham Biosciences) incorporation, which was administered via intraperitoneal injection (250 μl) 2 h before sampling. Complete blood count analysis was carried out on blood samples collected from animals under terminal anesthesia, cervically dislocated, by cardiac puncture and in tubes containing EDTA anticoagulant (Sarstedt) using the ProCyte Dx (IPU v.00-33-Build51) hematology analyzer (IDEXX). Blood plasma biochemistry was performed on blood samples collected in lithium-heparin-coated tubes (Sarstedt) and after plasma separation using centrifugation at 2,350g for 10 min at room temperature stored at −80 °C.
Npm1 floxed allele generation
A conditional allele of the Npm1 gene (Ensembl ID: ENSMUSG00000057113 in genome assembly GRCm39; Ensembl Release 110) was generated using gene targeting in mouse embryonic stem cells (mESCs). This modified allele introduces loxP sites flanking exons 2–6 (ENSMUSE00000473268, ENSMUSE00000472357, ENSMUSE00000463763, ENSMUSE00000134295 and ENSMUSE00000251772) of the mouse Npm1 gene (Ensembl Transcript ID: Npm1-201; ENSMUST00000075641.10) (see the schematic depiction in Extended Data Fig. 1g).
To generate this allele, a targeting vector for the Npm1 locus (PG00256_Z_5_E12) was imported from EUMMCR79. The imported construct was linearized and used to transfect 129/P2-derived HM1 mESCs80 by electroporation. After transfection, mESCs were selected under G418 (250 μg ml−1) and resistant colonies were picked and screened for the correct targeting of the Npm1 gene using long-range PCR (Expand Long Template PCR System, Roche). Screening was carried out at both the 5′ and 3′ ends of the targeting vector, from within the neomycin selection cassette to endogenous Npm1 sequences outside the homology arms. Oligonucleotide sequences used to screen cells, and to confirm the presence of the isolated 3′ loxP site using PCR, are provided in Supplementary Table 1.
After identification of correctly targeted clones for the Npm1 allele, mouse lines were derived by injection of targeted mESCs cells into C57BL/6J blastocysts according to standard protocols. After breeding of chimeras, germline offspring were identified according to coat color; the presence of the modified allele was confirmed with the 3′ loxP primers described above. Mice were subsequently crossed with a C57Bl/6J mouse strain expressing FLPe (Tg(ACTFLPe)9205Dym) to delete the selectable marker by recombination at the FRT sites81. Deletion of the selectable marker was confirmed using PCR across the remaining FRT site, using the oligonucleotides provided in Supplementary Table 1. After successful validation of the mouse strain carrying the Npm1 target allele, genotyping was subsequently carried out by the commercial genotyping service provider (TransnetYX).
Drug treatments
ISRIB (cat. no. SML0843, Sigma-Aldrich) was prepared as a 1 mg ml−1 stock solution in dimethylsulfoxide and subsequently prepared as a 1:20 dilution in a vehicle consisting of 50% PEG 400 and 45% 0.9% saline solution. A 0.25 mg kg−1 dose was administered in vivo once daily via intraperitoneal injection. Trazodone hydrochloride (cat. no. T6154, Sigma-Aldrich) was dosed once daily via intraperitoneal injection at 40 mg kg−1, dissolved in 0.9% saline solution. Mice were dosed with PERKi (GSK2606414, cat. no. S7307, Selleck Chemicals) twice daily via oral gavage at 50 mg kg−1, which was dissolved in 0.5% hydroxypropyl methylcellulose/0.1% Tween-80 vehicle.
TMA
NPM1 protein expression was assessed in a retrospectively collected cohort of patients with stage I–IV CRC (n = 787). The cohort consisted of patients with CRC undergoing surgical resection with curative intent within Greater Glasgow and Clyde National Health Service between 1997 and 2013. Tissue was previously constructed into TMA consisting of 0.6-mm cores in triplicate for each patient to account for tumor heterogeneity. Patients were excluded from the study due to mortality within 30 days of surgery or administration of neoadjuvant therapy. Data are deposited with the Glasgow Safehaven (no. GSH21ON009) and ethical approval was in place for the study (no. MREC/01/0/3).
Immunohistochemical staining was performed as described in the Supplementary Note. Staining was assessed in tumor cells semi-quantitively using HALO (v.3.6.4134) (Indica Labs) via weighted histoscore. Scores were validated through correlation analysis with manual scores by two observers (G.K., K.P.) for 10% of cases. Averaged scores were input into a clinical database in SPSS (v.28, IBM Corporation). The optimal cutoff point for high-expression and low-expression groups was determined using the Survminer package in RStudio (v.2023.12.1, Posit) based on cancer-specific survival.
Human tumor data and analyses
For the TCGA pan-cancer analysis in Fig. 1e,f and Extended Data Fig. 1d,f, the COAD/READ analysis in Figs. 1g and 3r–t, Extended Data Figs. 1e and 5f–h, and the LIHC analysis in Extended Data Fig. 10b–g,x,y, the harmonized TCGA Pan-Cancer Atlas data was downloaded from UCSC Xena82. For the GSEA, the gsva function from the Python package GSEApy83 was used, which reimplemented the gene set variation analysis84. Specifically, when comparing between tumors and adjacent normal samples (Fig. 1e,f and Extended Data Fig. 1d), gsva was run on the combined expression matrix containing both tumor and adjacent normal samples for each tumor type separately. For Fig. 1g, and Extended Data Figs. 1e, 5f–h and 10d–g, gsva was run on the expression matrix containing only tumor samples from the corresponding tumor type to focus on the variation within tumor samples alone. For stratifying tumors based on APC mutation status, we defined damaging mutations as nonsense mutations or frameshift indels. For copy number status, we used the absolute copy number values calculated by GISTIC2 (ref. 85) and interpreted them as follows: −2 (deep deletion); −1 (shallow deletion); 0 (diploid); 1 (low-level gain); 2 (high-level amplification). Of note, NPM1 is never deep-deleted in COAD/READ or LIHC. Survival analysis of patients with HCC with high NPM1 mRNA expression presented in Extended Data Fig. 10h was performed using Kaplan–Meier Plotter86.
The microarray dataset (GSE39582)87 from patients with colon cancer consisting of n = 566 primary tumors and n = 19 adjacent normal tissue samples was downloaded from the Gene Expression Omnibus (GEO), followed by probe-to-gene collapse using maxMean method WGCNA88 (v.1.70-3) R package, which takes the probe with the highest average score across the samples as gene expression. Tumor samples were called for PDS classification with the PDSclassifier30 (v.1.0.0) R package. To measure the proliferative index, the ProliferativeIndex89 (v.1.0.1) R package was used. The gene signature DNA repair (Hallmark) was accessed using the msigdbr90 (v.7.4.1) R package to obtain the single-sample GSEA score using the GSVA84 (v.1.42.0) R package. The replication stress score was generated as stated previously30. For visualizations, a list of R packages was used, which consists of ggplot2 (v.3.5.1), ggpubr (v.0.6.0) and ggbeeswarm (v.0.7.2) within R (v.4.3.3).
RNA-seq and pathway enrichment analyses
RNA quality was tested using an Agilent 220 TapeStation with RNA screentape and only samples with an RNA integrity number > 6 were used for the downstream analysis. Libraries for cluster generation and RNA-seq were prepared according to a previously described method91, using a TruSeq RNA Sample Prep Kit v2 (Illumina), then run on an Illumina NextSeq system using the 75 cycles High Output Kit (2 × 36 cycles, paired-end reads, single index). Raw sequence quality was assessed using FastQC (v.0.11.8), then sequences were trimmed to remove adapter sequences and low-quality base calls, defined as those with a Phred score of less than 20, using Trim Galore (v.0.6.4). Trimmed sequences were aligned to the mouse genome build GRCm38.98 using HISAT2 (v.2.1.0); raw counts per gene were determined using FeatureCounts (v.1.6.4). Differential expression analysis was performed using the R package DESeq2 (v.1.22.2), using a negative binomial generalized linear model, with significance assessed using a Wald test and Benjamini–Hochberg multiple testing correction. Reactome pathway enrichment was performed using enrichPathway function from the R package ReactomePA (v.1.36.0). For GSEA in the Rosa26CreERT2Apc+/+ versus Rosa26CreERT2ApcloxP/loxP, the following Reactome or Hallmark pathways are shown: Hallmark Myc V1, Hallmark Myc V2, 5991099_Translation, 5991458_Translation_initiation_complex_formation, 5991097_Cap-dependent_Translation_Initiation, 5991098_Eukaryotic_Translation_ Initiation, 5991746_WNT_mediated_activation_of_DVL, 5991743_TCF_dependent_ signaling_in_response_to_WNT and 5991561_Signaling_by_Wnt. The additional WNT signatures are as Watanabe Wnt92, Michel a Wnt93, Van der Flier a Wnt and Van der Flier b Wnt94, and Sansom Wnt95. The last two were also referred to throughout the manuscript as ‘WNT signature a’ and ‘WNT signature b’, respectively.
Ribosome profiling (Riboseq)
Pellets for Riboseq from intestinal epithelial extracts were prepared as described in the Supplementary Note, were placed at −20 °C for 30 min, then on ice for 5 min before being lysed in 550 μl of ice-cold lysis buffer (15 mM Tris-Cl, pH 7.5, 150 mM NaCl, 15 mM MgCl2, 100 μg ml−1 cycloheximide, 1% Triton X-100, 0.05% Tween-20, 2% n-dodecyl-β-D-maltopyranoside detergent, 0.5 mM dithiothreitol, 1× cOmplete mini (cat. no. 04693124001, Roche)) and 1,000 U ml−1 Ribolock (cat. no. EO0382, Thermo Fisher Scientific). Lysis was achieved by resuspension and subsequent mechanical stress by shear force, passing the lysate 5× through a 21-G needle, while keeping the tube on ice as much as possible. Lysates were then cleared using centrifugation (16,000g, 5 min, 4 °C). An aliquot of 25 µl undigested cleared lysates was supplemented with 1 ml TRIzol (cat. no. 15596026, Invitrogen) for total cytoplasmic RNA extraction. TRIzol extraction was performed according to the manufacturer’s instruction.
Then, 450 μl of cleared lysate were transferred to a fresh tube and digested with 5 μl RNase I (cat. no. AM2295, Ambion) at 22 °C for 15 min with gentle agitation (600 r.p.m.). Digestion was stopped with 10 μl of SUPERase In (cat. no. AM2696, Invitrogen). Digested lysates were loaded onto a 10–50% sucrose gradient and ultracentrifuged for 2 h at 256,800g at 4 °C in a SW 40 Ti rotor (Beckman Coulter). Spun samples were fractionated on a Triax Gradientmaster and fractions corresponding to monosomes were extracted in acid-phenol:chloroform, pH 4.5 (with IAA, 125:24:1, Invitrogen), washed twice with chloroform and precipitated with 2 µl glycogen, 1/10th volume of 3 M NaOAc (pH 5.2) and an equal volume of isopropanol. RNA was size-selected on a 15% Urea-TBE gel. rRNA depletion was performed with a custom set of complementary biotinylated DNA oligonucleotides (Supplementary Table 1), subsequently captured with Dynabeads MyOne Streptavidin C1 beads (cat. no. 65001, Invitrogen). After treatment with T4 Polynucleotide Kinase (cat. no. M0201L, New England Biolabs) and final purification with acid-phenol:chloroform and isopropanol precipitation as above. RPF libraries were prepared using the NEXTflex Small RNA Kit v3 (PerkinElmer) according to the manufacturer’s instructions, using all purified material and 13 PCR cycles in the amplification step. Alternative step F and step H2 were used. Total cytoplasmic RNAs, extracted from undigested lysate, were depleted of rRNA with RiboCop V2 (Lexogen); then libraries were prepared using the CORALL kit V1 (Lexogen), all according to the manufacturer’s instructions; 900 μg of total cytoplasmic RNA were used as input for the RiboCop. Thirteen PCR cycles were used in the library amplification stage.
RPF and total libraries were quantified using DNA High Sensitivity Qubit assays (Invitrogen) and size-checked using D1000 High-sensitivity ScreenTape (Agilent Technologies). After equimolar pooling, libraries were single-end sequenced on a NextSeq 500 High Output 75 cycle kit (Illumina).
Riboseq analysis
Riboseq analysis was performed according to the publicly available pipeline at the Bushell’s lab GitHub page (https://github.com/Bushell-lab/Ribo-seq). The custom scripts listed below are available on the GitHub page. All analyses relative to transcript isoforms and their characteristics were performed using the Gencode vM27 dataset for reference. Briefly, both total cytoplasmic RNA reads and RPF reads had adapter sequences removed with cutadapt and unique molecular identifiers were appended to FASTA headers using UMItools96. Then, total RNA reads were aligned to protein-coding transcripts using Bowtie 2 (ref. 97) and subsequently deduplicated, again using UMItools. Isoform quantification for each gene was performed using rsem-calculate-expression97; then, the most abundant transcript (MAT) per gene was extracted with a custom R script. A FASTA file for the MATs was then generated with a custom Python script. RPF reads were aligned to rRNA, tRNA and mitochondrial sequences to assess contamination of input RNA in the library preparation procedure using BBMap (sourceforge.net/projects/bbmap/). The remaining reads were aligned to the MAT FASTA with BBMap and then deduplicated using UMItools. Reads of length 27–38 were extracted and quality-checked using custom Python and R scripts. Finally reads for the protein CDS were extracted for each read length identified as correctly representing RPFs on the basis of quality control (Extended Data Fig. 8).
Differential expression analysis was performed with DESeq2 (ref. 98). During differential expression analysis, batch correction was performed, where the batch corresponded to the day in which the epithelial extractions were processed (from lysis to acid-phenol:chloroform extraction). Any transcript with average reads across all samples lower than ten was excluded from differential expression analysis and downstream processing. Of the 20,622 transcripts in the MAT table, 10,861 were retained for cytoplasmic RNA and 11,279 were retained for RPFs (Supplementary Tables 2–5). Merging the data tables to evaluate changes at RPF and cytoplasmic RNA concomitantly retained 10,261 transcripts. Given that we had previously selected a unique transcript per gene, during the MAT analysis, 10,261 unique genes are represented each by one transcript. Functional analyses were performed with the fGSEA package99. Pause analysis was performed as described previously100. For each condition, a pause site was defined as an RPF peak whose height was ten times greater than the average RPF peak height (excluding zeros) on the same mRNA. The change in peak height between conditions was calculated as the difference between RPFs normalized to mRNA abundance. Pause sites were then classified as ‘resolved’ or ‘induced’ if the decrease or increase, respectively, in their peak height was ten times greater than the average change across the same mRNA. Pauses that were neither induced nor resolved were classified as ‘sustained’. For subsequent analyses, pauses occurring at the translation start site were excluded.
Proteomics
Pellets from epithelial extracts (Supplementary Note) from the same animals used for Riboseq were lysed in 500 μl of 4% SDS, 100 mM Tris-HCl, pH 7.5, prepared in mass spectrmetry-grade water. Samples were boiled for 60 s at 95 °C, then passed through 23-G needles 7× times to fully lyse cells. Samples were then sonicated for five cycles 10 s on/10 s off with an immersion probe at 50% (FB 50, Thermo Fisher Scientific), then centrifuged at 16,000g for 10 min at room temperature. The supernatant was collected in a fresh tube and protein concentration quantified with a bicinchoninic acid assay (Pierce BCA Protein Assay Kit, Thermo Fisher Scientific).
Then, 10 μg of proteins per sample were reduced with 10 mM dithiothreitol and subsequently alkylated in the dark with 55 mM iodoacetamide at room temperature. Alkylated proteins were precipitated by adding four volumes of acetone at −20 °C overnight. Washed pellets were reconstituted in 50 µl of 200 mM HEPES and digested first with endoproteinase Lys-C (1:33 ratio enzyme:lysate) for 1 h, followed by trypsin, overnight (1:33 ratio enzyme:lysate). The digested peptides from each experiment were differentially labeled using the TMTpro 16plex reagent (Thermo Fisher Scientific). The reaction was carried out at room temperature for 2 h. Fully labeled samples were mixed in equal amounts and desalted using a 50-mg Sep Pak C18 reverse-phase solid-phase extraction cartridges (Waters). TMT-labeled peptides were fractionated using high-pH reverse-phase chromatography on a C18 column (150 × 2.1 mm i.d.; Kinetex EVO (5 μm, 100 Å)) on a high-performance liquid chromatography system (LC 1260 Infinity II, Agilent Technologies). A two-step gradient was applied, from 1–28% B in 42 min, then from 28–46% B in 13 min to obtain a total of 21 fractions for MS analysis.
Peptides resulting from all samples were separated using nanoscale C18 reverse-phase liquid chromatography using an EASY-nLC II 1200 (Thermo Fisher Scientific) coupled to an Orbitrap Q Exactive HF (Thermo Fisher Scientific). Elution was carried out at a flow rate of 300 nl min−1 using a binary gradient with buffer A (2% acetonitrile) and B (80% acetonitrile), both containing 0.1% formic acid. Samples were loaded with 6 µl of buffer A into a 50-cm fused silica emitter (New Objective) packed in-house with ReproSil-Pur C18-AQ, 1.9 μm resin (Dr. Maisch). For both systems, the packed emitter was kept at 50 °C using a column oven (Sonation) integrated into the nanoelectrospray ion source (Thermo Fisher Scientific); the Xcalibur software (v.4.1.31.9, Thermo Fisher Scientific) was used for data acquisition. Peptides were eluted using different gradients optimized for three sets of fractions: 1–7; 8–15; and 16–21 (ref. 101). Each fraction was acquired for a duration of 190 min. A full scan over mass range of 375–1400 m/z was acquired at 60,000 resolution at 200 m/z, with a target value of 3 × 106 ions for a maximum injection time of 20 ms. Higher energy collisional dissociation fragmentation was performed on the 20 most intense ions selected within an isolation window of 0.8 m/z. Peptide fragments were analyzed in the Orbitrap at a 45,000 resolution.
Proteomics data analysis
The MS raw data were processed with the MaxQuant software102 (v.1.6.14.0) and searched with Andromeda search engine103 querying SwissProt104 Mus musculus (63,668 entries). First and main searches were performed with precursor mass tolerances of 20 p.p.m. and 4.5 p.p.m., respectively, and MS/MS tolerance of 20 p.p.m. The minimum peptide length was set to six amino acids; specificity for trypsin cleavage was required, allowing up to two missed cleavage sites. The peptide, protein and site false discovery rate was set to 1%. Modification by iodoacetamide on cysteine residues (carbamidomethylation) were specified as fixed, whereas methionine oxidation and N-terminal acetylation modifications were specified as variable. For the proteome analysis, MaxQuant was set to quantify on ‘Reporter ion MS2’; TMT16plex was chosen as the isobaric label. Interference between TMT channels was corrected by MaxQuant using the correction factors provided by the manufacturer. The ‘Filter by PIF’ option was activated and a ‘reporter ion tolerance’ of 0.003 Da was used.
The proteinGroups.txt file from the MaxQuant output was used for protein quantitation analysis using the Perseus software105 (v.1.6.15.0). The ‘reverse’, ‘potential contaminants’ and ‘only identified by site’ protein, as specified in MaxQuant, were removed, as well as protein groups identified with no unique peptides. Only proteins robustly quantified in all replicates in at least one group were allowed in the list of quantified proteins. Significantly different proteins were selected using a permutation-based Student’s t-test with the false discovery rate set at 5%.
Functional analyses were performed with the fgsea package as for Riboseq, preranking genes by test difference (Supplementary Tables 2–6). If a protein group contained more than one protein, all available gene symbols were kept tied in the ranking.
Statistics and reproducibility
Statistical analyses were performed with Prism (v.7.0.4, GraphPad Software). To ensure appropriate statistical comparison test selection, data were initially assessed to determine whether they followed a Gaussian distribution. Sample sizes of eight or more were assessed using the D’Agostino-Pearson omnibus K2 test and those less than eight with the Shapiro–Wilk test. Statistical significance of the parametric data was assessed using a two-tailed t-test, while nonparametric data were compared using a two-tailed Mann–Whitney U-test. Comparisons involving more than two groups were performed using an ANOVA, followed by appropriate post hoc tests as indicated. Survival data were statistically compared using the log-rank (Mantel–Cox) test. The null hypothesis was rejected and statistical significance was assumed for P < 0.05. Data are presented as the mean ± s.e.m. unless otherwise indicated. Sample sizes, specific P values and tests performed for each experiment are indicated in the corresponding figures or figure legends. From the TMA cohort, patients were excluded from the study because of mortality within 30 days of surgery or administration of neoadjuvant therapy. As part of qaulity control in the Riboseq analysis, any transcript with average reads across all samples lower than ten was excluded from any downstream analysis. No other data were excluded from the analyses. The experiments were not randomized. The investigators were not blinded to allocation during the experiments and outcome assessment. No formal statistical methods were used to predetermine sample sizes as part of this study. All in vivo cohort sizes were determined based on power analyses in studies previously carried out in similar models, respecting the limited use of animals in line with the 3R system: replacement, reduction, refinement.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The RNA-seq data generated in this study have been deposited at the GEO under accession numbers GSE230110, GSE309379 and GSE250047. The ribosome profiling data have been deposited at GEO under the accession number GSE249958. The proteomics data have been deposited at the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD062969. The murine liver Npm1 expression data presented in Extended Data Fig. 10a were derived from RNA-seq data available at the GEO under accession number GSE230137. Human cancer analyses were conducted using data that are in whole or part based on data generated by the TCGA Research Network (www.cancer.gov/tcga), as well as the publicly available colon cancer microarray dataset GSE39582 (ref. 87). Source data are provided with this paper.
Code availability
The code used in this project for the Ribo-seq analysis and integration with the proteomics data is available on the project-specific Zenodo repository page (https://doi.org/10.5281/zenodo.17187088)106. The code is based on the Bushell lab pipeline available on the lab’s GitHub page (https://github.com/Bushell-lab/Ribo-seq).
References
Sack, L. M. et al. Profound tissue specificity in proliferation control underlies cancer drivers and aneuploidy patterns. Cell 173, 499–514 (2018).
Groenewald, W., Lund, A. H. & Gay, D. M. The role of WNT pathway mutations in cancer development and an overview of therapeutic options. Cells 12, 990 (2023).
Muzny, D. M. et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature 487, 330–337 (2012).
Faller, W. J. et al. mTORC1-mediated translational elongation limits intestinal tumour initiation and growth. Nature 517, 497–500 (2015).
Di Nicolantonio, F. et al. Deregulation of the PI3K and KRAS signaling pathways in human cancer cells determines their response to everolimus. J. Clin. Invest. 120, 2858–2866 (2010).
Knight, J. R. P. et al. MNK inhibition sensitizes KRAS-mutant colorectal cancer to mTORC1 inhibition by reducing eIF4E phosphorylation and c-MYC expression. Cancer Discov. 11, 1228–1247 (2021).
Grisendi, S. et al. Role of nucleophosmin in embryonic development and tumorigenesis. Nature 437, 147–153 (2005).
Colombo, E. et al. Nucleophosmin is required for DNA integrity and p19(Arf) protein stability. Mol. Cell. Biol. 25, 8874–8886 (2005).
Nachmani, D. et al. Germline NPM1 mutations lead to altered rRNA 2′-O-methylation and cause dyskeratosis congenita. Nat. Genet. 51, 1518–1529 (2019).
Ugolini, I. et al. Chromatin localization of nucleophosmin organizes ribosome biogenesis. Mol. Cell 82, 4443–4457 (2022).
Murano, K., Okuwaki, M., Hisaoka, M. & Nagata, K. Transcription regulation of the rRNA gene by a multifunctional nucleolar protein, B23/nucleophosmin, through its histone chaperone activity. Mol. Cell. Biol. 28, 3114–3126 (2008).
Saluri, M. et al. A “grappling hook” interaction connects self-assembly and chaperone activity of nucleophosmin 1. PNAS Nexus 2, pgac303 (2023).
Szebeni, A. & Olson, M. O. Nucleolar protein B23 has molecular chaperone activities. Protein Sci. 8, 905–912 (1999).
Ranieri, R. et al. Current status and future perspectives in targeted therapy of NPM1-mutated AML. Leukemia 36, 2351–2367 (2022).
Brunetti, L. et al. Mutant NPM1 maintains the leukemic state through HOX expression. Cancer Cell 34, 499–512 (2018).
Wang, X. Q. D. et al. Mutant NPM1 hijacks transcriptional hubs to maintain pathogenic gene programs in acute myeloid leukemia. Cancer Discov. 13, 724–745 (2023).
Uckelmann, H. J. et al. Mutant NPM1 directly regulates oncogenic transcription in acute myeloid leukemia. Cancer Discov. 13, 746–765 (2023).
Grisendi, S., Mecucci, C., Falini, B. & Pandolfi, P. P. Nucleophosmin and cancer. Nat. Rev. Cancer 6, 493–505 (2006).
Wong, J. C. T. et al. Nucleophosmin 1, upregulated in adenomas and cancers of the colon, inhibits p53-mediated cellular senescence. Int. J. Cancer 133, 1567–1577 (2013).
Yun, J.-P. et al. Increased expression of nucleophosmin/B23 in hepatocellular carcinoma and correlation with clinicopathological parameters. Br. J. Cancer 96, 477–484 (2007).
Li, F. et al. Epigenetic CRISPR screens identify Npm1 as a therapeutic vulnerability in non-small cell lung cancer. Cancer Res. 80, 3556–3567 (2020).
Sekhar, K. R. & Freeman, M. L. Nucleophosmin plays a role in repairing DNA damage and is a target for cancer treatment. Cancer Res. 83, 1573–1580 (2023).
Madan, B. et al. Temporal dynamics of Wnt-dependent transcriptome reveal an oncogenic Wnt/MYC/ribosome axis. J. Clin. Invest. 128, 5620–5633 (2018).
Boon, K. et al. N-myc enhances the expression of a large set of genes functioning in ribosome biogenesis and protein synthesis. EMBO J. 20, 1383–1393 (2001).
Li, Z. L., Boone, D. & Hann, S. R. Nucleophosmin interacts directly with c-Myc and controls c-Myc-induced hyperproliferation and transformation. Proc. Natl Acad. Sci. USA 105, 18794–18799 (2008).
Zeller, K. I. et al. Characterization of nucleophosmin (B23) as a Myc target by scanning chromatin immunoprecipitation. J. Biol. Chem. 276, 48285–48291 (2001).
Colombo, E., Marine, J.-C., Danovi, D., Falini, B. & Pelicci, P. G. Nucleophosmin regulates the stability and transcriptional activity of p53. Nat. Cell Biol. 4, 529–533 (2002).
Kurki, S. et al. Nucleolar protein NPM interacts with HDM2 and protects tumor suppressor protein p53 from HDM2-mediated degradation. Cancer Cell 5, 465–475 (2004).
Jackstadt, R., Hodder, M. C. & Sansom, O. J. WNT and beta-catenin in cancer: genes and therapy. Annu. Rev. Cancer Biol. 4, 177–196 (2020).
Malla, S. B. et al. Author correction: Pathway level subtyping identifies a slow-cycling biological phenotype associated with poor clinical outcomes in colorectal cancer. Nat. Genet. 56, 1321 (2024).
Vassiliou, G. S. et al. Mutant nucleophosmin and cooperating pathways drive leukemia initiation and progression in mice. Nat. Genet. 43, 470–475 (2011).
Morganti, C. et al. NPM1 ablation induces HSC aging and inflammation to develop myelodysplastic syndrome exacerbated by p53 loss. EMBO Rep. 23, e54262 (2022).
Maggi, L. B. Jr et al. Nucleophosmin serves as a rate-limiting nuclear export chaperone for the mammalian ribosome. Mol. Cell. Biol. 28, 7050–7065 (2008).
Mitrea, D. M. et al. Self-interaction of NPM1 modulates multiple mechanisms of liquid–liquid phase separation. Nat. Commun. 9, 842 (2018).
Okuwaki, M., Saito, S., Hirawake-Mogi, H. & Nagata, K. The interaction between nucleophosmin/NPM1 and the large ribosomal subunit precursors contribute to maintaining the nucleolar structure. Biochim. Biophys. Acta Mol. Cell Res. 1868, 118879 (2021).
Leach, J. D. G. et al. Oncogenic BRAF, unrestrained by TGFβ-receptor signalling, drives right-sided colonic tumorigenesis. Nat. Commun. 12, 3464 (2021).
Jackstadt, R. et al. Epithelial NOTCH signaling rewires the tumor microenvironment of colorectal cancer to drive poor-prognosis subtypes and metastasis. Cancer Cell 36, 319–336 (2019).
Sidrauski, C., McGeachy, A. M., Ingolia, N. T. & Walter, P. The small molecule ISRIB reverses the effects of elF2α phosphorylation on translation and stress granule assembly. eLife 4, e05033 (2015).
Axten, J. M. et al. Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1H-indol-5-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). J. Med. Chem. 55, 7193–7207 (2012).
Moreno, J. A. et al. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci. Transl. Med. 5, 206ra138 (2013).
Halliday, M. et al. Partial restoration of protein synthesis rates by the small molecule ISRIB prevents neurodegeneration without pancreatic toxicity. Cell Death Dis. 6, e1672 (2015).
Halliday, M. et al. Repurposed drugs targeting eIF2α-P-mediated translational repression prevent neurodegeneration in mice. Brain 140, 1768–1783 (2017).
Szaruga, M. et al. Activation of the integrated stress response by inhibitors of its kinases. Nat. Commun. 14, 5535 (2023).
Mahameed, M. et al. The unfolded protein response modulators GSK2606414 and KIRA6 are potent KIT inhibitors. Cell Death Dis. 10, 300 (2019).
Ghaddar, N. et al. The integrated stress response is tumorigenic and constitutes a therapeutic liability in KRAS-driven lung cancer. Nat. Commun. 12, 4651 (2021).
Smith, R. C. L. et al. Translation initiation in cancer at a glance. J. Cell Sci. 134, jcs248476 (2021).
Ally, A. et al. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell 169, 1327–1341 (2017).
Burke, Z. D. et al. Liver zonation occurs through a β-catenin-dependent, c-Myc-independent mechanism. Gastroenterology 136, 2316–2324 (2009).
Bisso, A. et al. Cooperation between MYC and beta-catenin in liver tumorigenesis requires Yap/Taz. Hepatology 72, 1430–1443 (2020).
Reed, K. R. et al. B-catenin deficiency, but not Myc deletion, suppresses the immediate phenotypes of APC loss in the liver. Proc. Natl Acad. Sci. USA 105, 18919–18923 (2008).
Cairo, S. et al. Hepatic stem-like phenotype and interplay of Wnt/β-catenin and Myc signaling in aggressive childhood liver cancer. Cancer Cell 14, 471–484 (2008).
Llovet, J. M. et al. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 7, 6 (2021).
Bartsch, D. et al. mRNA translational specialization by RBPMS presets the competence for cardiac commitment in hESCs. Sci. Adv. 9, eade1792 (2023).
Simsek, D. et al. The mammalian ribo-interactome reveals ribosome functional diversity and heterogeneity. Cell 169, 1051–1065 (2017).
Marcel, V. et al. p53 acts as a safeguard of translational control by regulating fibrillarin and rRNA methylation in cancer. Cancer Cell 24, 318–330 (2013).
Chen, C.-W. et al. Plasticity of the mammalian integrated stress response. Nature 641, 1319–1328 (2025).
Fusée, L. et al. The p53 endoplasmic reticulum stress-response pathway evolved in humans but not in mice via PERK-regulated p53 mRNA structures. Cell Death Differ. 30, 1072–1081 (2023).
Papaemmanuil, E. et al. Genomic classification and prognosis in acute myeloid leukemia. N. Engl. J. Med. 374, 2209–2221 (2016).
Becker, H. et al. Favorable prognostic impact of NPM1 mutations in older patients with cytogenetically normal de novo acute myeloid leukemia and associated gene- and microRNA-expression signatures: a Cancer and Leukemia Group B study. J. Clin. Oncol. 28, 596–604 (2010).
Jian, Y. et al. RNA aptamers interfering with nucleophosmin oligomerization induce apoptosis of cancer cells. Oncogene 28, 4201–4211 (2009).
Morral, C. et al. Zonation of ribosomal DNA transcription defines a stem cell hierarchy in colorectal cancer. Cell Stem Cell 26, 845–861 (2020).
Chen, J. K. et al. An RNA damage response network mediates the lethality of 5-FU in colorectal cancer. Cell Rep. Med. 5, 101778 (2024).
Schmidt, H. B. et al. Oxaliplatin disrupts nucleolar function through biophysical disintegration. Cell Rep. 41, 111629 (2022).
Simms, N. & Knight, J.R.P. RNA damage: the forgotten target of clinical compounds. Front. RNA Res. 1 https://doi.org/10.3389/frnar.2023.1248236 (2023).
Sutton, E. C. & DeRose, V. J. Early nucleolar responses differentiate mechanisms of cell death induced by oxaliplatin and cisplatin. J. Biol. Chem. 296, 100633 (2021).
El Marjou, F. et al. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis 39, 186–193 (2004).
Shibata, H. et al. Rapid colorectal adenoma formation initiated by conditional targeting of the APC gene. Science 278, 120–123 (1997).
Jackson, E. L. et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 15, 3243–3248 (2001).
Barker, N. et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 449, 1003–1007 (2007).
Marino, S., Vooijs, M., van Der Gulden, H., Jonkers, J. & Berns, A. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev. 14, 994–1004 (2000).
Deng, C. X., Zhang, P. M., Harper, J. W., Elledge, S. J. & Leder, P. Mice Lacking P21C/P1/Waf1 undergo normal development, but are defective in G1 checkpoint control. Cell 82, 675–684 (1995).
Murtaugh, L. C., Stanger, B. Z., Kwan, K. M. & Melton, D. A. Notch signaling controls multiple steps of pancreatic differentiation. Proc. Natl Acad. Sci. USA 100, 14920–14925 (2003).
Mercer, K. et al. Expression of endogenous oncogenic V600EB-raf induces proliferation and developmental defects in mice and transformation of primary fibroblasts. Cancer Res. 65, 11493–11500 (2005).
Rad, R. et al. A genetic progression model of BrafV600E-induced intestinal tumorigenesis reveals targets for therapeutic intervention. Cancer Cell 24, 15–29 (2013).
Hameyer, D. et al. Toxicity of ligand-dependent Cre recombinases and generation of a conditional Cre deleter mouse allowing mosaic recombination in peripheral tissues. Physiol. Genomics 31, 32–41 (2007).
Colnot, S. et al. Liver-targeted disruption of Apc in mice activates β-catenin signaling and leads to hepatocellular carcinomas. Proc. Natl Acad. Sci. USA 101, 17216–17221 (2004).
Harada, N. et al. Intestinal polyposis in mice with a dominant stable mutation of the β-catenin gene. EMBO J. 18, 5931–5942 (1999).
Kruspig, B. et al. The ERBB network facilitates KRAS-driven lung tumorigenesis. Sci. Transl. Med. 10, eaao2565 (2018).
Skarnes, W. C. et al. A conditional knockout resource for the genome-wide study of mouse gene function. Nature 474, 337–342 (2011).
Magin, T. M., Mcwhir, J. & Melton, D. W. A new mouse embryonic stem-cell line with good germ line contribution and gene targeting frequency. Nucleic Acids Res. 20, 3795–3796 (1992).
Rodríguez, C. I. et al. High-efficiency deleter mice show that FLPe is an alternative to Cre-loxP. Nat. Genet. 25, 139–140 (2000).
Goldman, M. J. et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 38, 675–678 (2020).
Fang, Z., Liu, X. & Peltz, G. GSEApy: a comprehensive package for performing gene set enrichment analysis in Python. Bioinformatics 39, btac757 (2023).
Hanzelmann, S., Castelo, R. & Guinney, J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 14, 7 (2013).
Mermel, C. H. et al. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 12, R41 (2011).
Menyhárt, O., Nagy, Á. & Győrffy, B. Determining consistent prognostic biomarkers of overall survival and vascular invasion in hepatocellular carcinoma. R. Soc. Open Sci. 5, 181006 (2018).
Marisa, L. et al. Gene expression classification of colon cancer into molecular subtypes: characterization, validation, and prognostic value. PLoS Med. 10, e1001453 (2013).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559 (2008).
Ramaker, R. C. et al. RNA sequencing-based cell proliferation analysis across 19 cancers identifies a subset of proliferation-informative cancers with a common survival signature. Oncotarget 8, 38668–38681 (2017).
Liberzon, A. et al. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425 (2015).
Fisher, S. et al. A scalable, fully automated process for construction of sequence-ready human exome targeted capture libraries. Genome Biol. 12, R1 (2011).
Watanabe, K. et al. Integrative ChIP-seq/microarray analysis identifies a CTNNB1 target signature enriched in intestinal stem cells and colon cancer. PLoS ONE 9, e92317 (2014).
Michels, B. E. et al. Human colon organoids reveal distinct physiologic and oncogenic Wnt responses. J. Exp. Med. 216, 704–720 (2019).
Van Der Flier, L. G. et al. The intestinal Wnt/TCF signature. Gastroenterology 132, 628–632 (2007).
Sansom, O. J. et al. Loss of Apc in vivo immediately perturbs Wnt signaling, differentiation, and migration. Genes Dev. 18, 1385–1390 (2004).
Smith, T., Heger, A. & Sudbery, I. UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res. 27, 491–499 (2017).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics12, 323 (2011).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Korotkevich, G. et al. Fast gene set enrichment analysis. Preprint at bioRxiv https://doi.org/10.1101/060012 (2021).
Gillen, S. L. et al. Differential regulation of mRNA fate by the human Ccr4-Not complex is driven by coding sequence composition and mRNA localization. Genome Biol. 22, 284 (2021).
Cao, X. F. et al. The mammalian cytosolic thioredoxin reductase pathway acts via a membrane protein to reduce ER-localised proteins. J. Cell Sci. 133, jcs241976 (2020).
Cox, J. & Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 (2008).
Cox, J. et al. Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 (2011).
Apweiler, R. et al. The Universal Protein Resource (UniProt) in 2010. Nucleic Acids Res. 38, D142–D148 (2010).
Tyanova, S. et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740 (2016).
Giacomelli, C. Nucleophosmin role in colorectal cancer. Zenodo https://doi.org/10.5281/zenodo.17187088 (2025).
Acknowledgements
We thank the Core Research Services at the Cancer Research UK (CRUK) Scotland Institute, particularly the Biological Services Unit, Histology and Molecular Technologies services for technical support. We also thank C. Winchester for critical assessment of the manuscript. This work was supported by: CRUK core funding to the CRUK Scotland Institute (no. A31287); CRUK core funding to the CRUK Glasgow Centre (no. A25142); CRUK core funding to the CRUK Scotland Centre (no. CTRQQR-2021\100006); CRUK core funding to the lab of C.M. (no. A29801); CRUK core funding to the lab of M.B. (no. A29252); and CRUK core funding to the lab of O.J.S. (nos. A21139 and DRCQQR-May21\100002). The J.L.Q. lab is funded by the Mazumdar Shaw Chair; CSO fellowships TCS/22/02 and CSO EPD/22/13 (K.P.); the SPECIFICANCER Cancer Grand Challenge funded by CRUK and The Mark Foundation for Cancer Research (no. A29055) (O.J.S., P.J.P.); and the International Accelerator Award, ACRCelerate, jointly funded by CRUK (nos. A26825 and A28223), FC AECC (no. GEACC18004TAB) and AIRC (no. 22795) (A.D.C., P.D.D., O.J.S.).
Author information
Authors and Affiliations
Contributions
G.K. conceptualized the study, carried out the investigation, devised the methodology, wrote the original manuscript draft, and reviewed and edited the manuscript under the supervision of O.J.S. C.G. reviewed and edited the manuscript, and carried out the investigation (analyzed the ribosome profiling and proteomics data) with support from J.M., who performed the ribosome profiling. P.H. and J.A.W. carried out the investigation (performed the pause analysis), and reviewed and edited the manuscript, under the supervision of M.B. A.R. performed the whole-body Apc deletion experiments, and reviewed and edited the manuscript. N.V. performed the 35S-methionine labeling experiments and supported the vivo and in vitro investigation along with C.A., P.P.C. and R.A.R. under the supervision of O.J.S. H.J. and S.B.M. analyzed the publicly available human cancer data, under the supervision of P.J.P. and P.D.D., respectively. N.N. and R.L.B. analyzed the IF-IHC experiments, with support from L.O.-J., under the supervision of J.L.Q. K.G. carried out the RNA-seq analysis. K.P. and J.E. sourced and curated the TMA, which was under the governance of the NHS Greater Glasgow and Clyde Biorepository. K.P. assisted with its analysis. H.H. analyzed publicly available human liver cancer data under the supervision of C.M. D.S. and S.B. generated the Npm1 floxed mouse line. S.L. and S.Z. acquired the MS data. C.N., V.M. and the CRUK Scotland Institute histology services performed the histology processing and tissue staining experiments. J.R.P.K. and A.D.C. supported the investigation, and reviewed and edited the manuscript. M.B. reviewed and edited the manuscript, and acquired the funding. O.J.S. conceptualized the study, reviewed and edited the manuscript, and acquired the funding.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Genetics thanks Charles Mullighan, Ivan Topisirovic, and Louis Vermeulen for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 NPM1 expression correlates with WNT and MYC activation in cancer patients.
a, b, Representative micrographs of sections of the indicated tissues stained with H&E (a) and anti-BrdU (b) derived from R26-CreER-T2Apc+/+ and R26-CreER-T2Apcfl/fl (n = 3 per group) mice harvested four days post-induction. Red bars indicate the expanded crypt depth in the small intestine (SI) following APC loss. Scale bars, 100 μm in (a) and 50 μm in (b). c, Heat map displaying gene set enrichment analysis in WNT permissive and non-permissive tissues for the indicated HALLMARK MYC and Reactome translation signatures in the Apcfl/fl tissues compared to wild-type (see methods for more details on the signatures used). Enrichment level is based on each group average. d, Scatterplot showing the correlation between mean tumor-normal difference of GSVA score of the signature (WNT signature b) compared to mean tumor-normal difference of NPM1 expression across different tumor types in TCGA dataset. e, Scatterplot showing the correlation between NPM1 expression and the GSVA score of the signature (WNT signature b) among NPM1 copy neutral COAD/READ tumours (n = 255) in the TCGA dataset. f, Scatterplot showing the correlation between mean tumor-normal difference of MYC compared to mean tumor-normal difference of NPM1 expression across different tumor types in TCGA dataset. In (d-f) data were statistically assessed by two-sided Pearson correlation. Correlation coefficient (r) is displayed to indicate the degree of association. Statistically significant p-values are shown in red. g, Schematic of the floxing strategy used to generate a conditional Npm1 knockout allele in mice. See methods for more information.
Extended Data Fig. 2 Whole-body Npm1 deletion is tolerated in adult mouse tissues.
a, Representative micrographs of sections of the indicated tissues stained with H&E and anti-NPM1 derived from animals harvested 60 days post-induction of Npm1 deletion by CRE expression from the Rosa26 locus (R26-CreER-T2) (n = 5 per group). Scale bars, 100 μm for all apart from the brain and spleen H&E sections, which are 400 μm. b, c, Liver and spleen to body weight ratios from the groups presented in (a). d-n, Complete red blood cell (RBC) count (d), distribution width (RDW) of RBCs (e), RBC mean corpuscular volume (MCV) (f), RBC mean corpuscular haemoglobin (MCH) (g), haemoglobin (HGB) levels (h), haematocrit (HCT) value (i), reticulocyte count (j), and immature reticulocytes fraction (IRF) (k), with the reticulocyte maturity indices also shown inside the box (l-n) (LFR: low fluorescence ratio – least immature reticulocytes; MFR: medium fluorescence ratio; HFR: high fluorescence ratio – most immature reticulocytes), in circulating blood from the groups shown in (a) (n = 5 per group). Data, mean ± SEM. All data statistically assessed by unpaired two-tailed t tests. Statistically significant p-values are shown in red.
Extended Data Fig. 3 NPM1 is required for WNT-driven proliferation in WNT-permissive tissues.
a, Representative micrographs of indicated tissues from R26-CreER-T2 (WT, n = 3), R26-CreER-T2Apcfl/fl (n = 5) and R26-CreER-T2Apcfl/flNpm1fl/fl mice collected four days post-induction and stained for BrdU. Red bars indicate the expanded crypt depth following APC loss in the small intestine (SI). Scale bars, 100 μm. b-i, Quantification of BrdU+ cells for the tissues and sample sizes shown in (a). Boxes in boxplots extend from the 25th to 75th percentiles, whiskers extend to the minimum and maximum values, and the line in every box is plotted at the median. Data statistically assessed by one-way ANOVA followed by Tukey’s multiple comparisons test. Statistically significant p-values are shown in red.
Extended Data Fig. 4 NPM1 is dispensable for adult intestinal tissue homeostasis.
a, Representative images of NPM1-stained (SI) (left panels) and colon (right panels) sections from Npm1+/+ (n = 4) and Npm1fl/fl (n = 5) animals collected four days post-induction. Scale bars, 50 μm. b, Representative staining with H&E, AB/PAS, anti-lysozyme, anti-BrdU and in situ hybridisation (ISH) for Olfm4 of SI and colon of Npm1+/+ (n = 5) and Npm1fl/fl (n = 5) animals collected 120 days post-induction. Scale bars, 100 μm. c, Quantification of BrdU+ cells in the SI of Npm1+/+ (n = 5) and Npm1fl/fl (n = 4) animals collected four days post-induction. Data, mean ± SEM, statistically assessed by unpaired two-tailed t test. d, e, Representative micrographs of 47S rRNA ISH in sections from intestinal tissue of Npm1+/+ and Npm1fl/fl mice (n = 4 per group) sampled four days post-induction with HALO quantification in (e). Scale bar, 50 μm. Data, mean ± SEM, statistically assessed by unpaired two-tailed Mann Whitney test. f, Representative images of Npm1fl/fl SI organoids induced with ethanol (EtOH) or 4-OHT in culture, taken six days post-induction (n = 3 independent lines derived from individual mice). Scale bar, 100 μm. g, Protein synthesis rate quantification by 35S-methionine incorporation in the organoid lines presented in (f) (n = 3 per group). Data, mean ± SEM, statistically assessed by unpaired two-tailed t test. h-q, Representative fibrillarin (yellow) staining from SI tissue sections of Npm1+/+ and Npm1fl/fl (n = 4 per group) animals collected four days post-induction, and Apcfl/flKrasG12D/+ (n = 4) and Apcfl/flKrasG12D/+Npm1fl/fl (n = 3) animals collected three days post-induction. Nuclei (blue) visualised with DAPI. Scale bars, 20 μm. Quantification of average number of nucleoli per nucleus based on fibrillarin stain, mean fibrillarin area, mean fibrillarin intensity and mean nucleolar circularity based on fibrillarin stain is shown. Data in (i-l) and (n-q) statistically assessed by unpaired two-tailed t tests. Boxes in boxplots extend from the 25th to 75th percentiles, whiskers extend to the minimum and maximum values, and the line in every box is plotted at the median. Statistically significant p-values are shown in red.
Extended Data Fig. 5 NPM1 is upregulated and necessary for WNT-driven tumourigenesis.
a, Representative micrographs of NPM1-stained normal intestinal tissue sections and tumours from Apcfl/+ (n = 3) (high WNT-driven tumours) and BrafV600E/+Alk5fl/fl (n = 6) (low WNT-driven tumours) mice harvested at clinical endpoint. b, c, NPM1 staining HALO H-score quantification of the groups shown in (a), normalised to the mean normal epithelium value. d, e, Anxa1 ISH in proximal colon tissue sections from Villin-CreER-T2 (n = 3) sampled 10 days, Villin-CreER-T2BrafLSL-V637E/+(n = 4) and Villin-CreER-T2BrafLSL-V637E/+Npm1fl/fl (n = 4) sampled 7 days post-induction, and HALO quantification shown in (e). Data statistically assessed by one-way ANOVA followed by Tukey’s multiple comparisons test. f, NPM1 expression in wild-type (WT) and mutant (Mut) APC COAD/READ tumours in the TCGA dataset (n = 375). g, h, GSVA score of WNT signatures a and b in WT and Mut APC TCGA COAD/READ tumours. i, Olfm4 ISH and HALO rendering on intestinal tissue sections from Apcfl/fl mice with or without Npm1fl/fl and HALO quantification in (j) (n = 4 per group). k, Tumor number distribution within equidistant proximal to distal SI and colon sections from Apcfl/+ (n = 8) and Apcfl/+Npm1fl/fl (n = 9) mice sampled at clinical endpoint. Data statistically assessed by two-way ANOVA followed by Sidak’s multiple comparisons test. l, Tumor number distribution within equidistant proximal to distal SI and colon sections from Lgr5-CreER-T2Apcfl/fl (n = 11) and Lgr5-CreER-T2Apcfl/flNpm1fl/fl (n = 9) mice sampled at clinical endpoint. Data statistically assessed by two-way ANOVA followed by Sidak’s multiple comparisons test. m, Olfm4 ISH and HALO rendering on intestinal tissue sections from Apcfl/flKrasG12D/+ mice with or without Npm1fl/fl and HALO quantification in (n) (n = 4 per group). o, Tumor number distribution within equidistant proximal to distal SI and colon sections from Apcfl/+KrasG12D/+ (n = 17) and Apcfl/+KrasG12D/+Npm1fl/fl (n = 20) mice sampled at clinical endpoint. Data statistically assessed by two-way ANOVA followed by Sidak’s multiple comparisons test. Bar chart data, mean ± SEM. Boxes in boxplots extend from the 25th to 75th percentiles, whiskers extend to the minimum and maximum values, and the line in every box is plotted at the median. All data in bar charts and boxplots statistically assessed by unpaired two-tailed t tests. Statistically significant p-values are shown in red. Scale bars, 100 μm.
Extended Data Fig. 6 Post-transcriptional activation of p53 mediates the survival benefit of NPM1 loss in WNT-driven CRC models.
a-c, Volcano plots of SI total RNA differential expression comparing Apcfl/flNpm1fl/fl with Apcfl/fl (a) and Apcfl/flKrasG12D/+Npm1fl/fl with Apcfl/flKrasG12D/+ (b) (n = 4/group). Significantly deregulated transcripts (adj p < 0.05, log2FC < -1 or >1) shown in red. DESeq2 with two-sided Wald test and Benjamini–Hochberg correction used. Npm1 and Npm1 pseudogenes highlighted. c, Number of deregulated transcripts. d, p53 staining in Apcfl/+KrasG12D/+ and Apcfl/+KrasG12D/+Npm1fl/fl (n = 4/group) SI crypts and tumours. Arrows indicate p53+ cells. Scale bar, 50 μm. Quantification in Fig. 3j,k. e, f, qPCR analysis of p53 target gene expression in Apcfl/fl (e) and Apcfl/flKrasG12D/+ (f) mice ±Npm1fl/fl (n = 4/group; Pmaip1 in Apcfl/fl n = 3). Multiple two-sided t-tests with Holm–Sidak correction. g, Tumor distribution within equidistant proximal to distal SI and colon sections from Apcfl/+ (n = 8), Apcfl/+Npm1fl/fl (n = 9) (also shown in Extended Data Fig. 5k), Apcfl/+Trp53fl/fl (n = 8) and Apcfl/+Trp53fl/flNpm1fl/fl (n = 14) endpoint mice. Two-way ANOVA with Sidak’s test. Colour combinations denote groups compared. Black p-values indicate non-significance. h, Survival curves of Lgr5-CreER-T2Apcfl/fl (n = 13), Lgr5-CreER-T2Apcfl/flNpm1fl/fl (n = 11) (also shown in Fig. 2h and Extended Data Fig. 7a), Lgr5-CreER-T2Apcfl/flTrp53fl/fl (n = 14) and Lgr5-CreER-T2Apcfl/flTrp53fl/flNpm1fl/fl (n = 15) endpoint mice. Median survival (days) indicated in brackets. P-values obtained by Log-rank (Mantel-Cox) tests. Censored mice denoted as tick marks. i, Endpoint tumor numbers from Lgr5-CreER-T2Apcfl/fl (n = 11), Lgr5-CreER-T2Apcfl/flNpm1fl/fl (n = 9) (also shown in Fig. 2i and the Npm1fl/fl cohort in Extended Data Fig. 7b), Lgr5-CreER-T2Apcfl/flTrp53fl/fl (n = 8) and Lgr5-CreER-T2Apcfl/flTrp53fl/flNpm1fl/fl (n = 15) mice. One-way ANOVA with Tukey’s test. j, Tumor distribution within equidistant proximal to distal SI and colon sections from groups in (i). Two-way ANOVA with Sidak’s test. Colour combinations denote groups compared. Black p-values indicate non-significance. k, NPM1-stained SI from Lgr5-CreER-T2Apcfl/flNpm1fl/fl (n = 7) (also shown in Extended Data Fig. 7d) and Lgr5-CreER-T2Apcfl/flTrp53fl/flNpm1fl/fl (n = 8) endpoint animals. Scale bar, 1 mm. l, Quantification of NPM1 + /−/mosaic tumours in endpoint Lgr5-CreER-T2Apcfl/flTrp53fl/flNpm1fl/fl (n = 8) compared to Lgr5-CreER-T2Apcfl/flNpm1fl/fl (n = 7) (also presented in Fig. 2k. and Extended Data Fig. 7e) animals. Two-way ANOVA with Sidak’s test. Data mean ± SEM (bar charts); boxplots boxes extend from 25th-75th percentiles, whiskers from minimum to maximum values, and the line denotes the median. Significant p-values in red.
Extended Data Fig. 7 Cdkn1a deletion abrogates the survival benefit of NPM1 loss in WNT-driven CRC models.
a, Survival curves of Lgr5-CreER-T2Apcfl/fl (n = 15), Lgr5-CreER-T2Apcfl/flNpm1fl/fl (n = 11, also shown in Fig. 2h and Extended Data Fig. 6h), Lgr5-CreER-T2Apcfl/flCdkn1a−/− (n = 13) and Lgr5-CreER-T2Apcfl/flCdkn1a−/−Npm1fl/fl (n = 18) mice. Median survival (days) indicated in brackets. b, Tumor numbers from Lgr5-CreER-T2Apcfl/fl (n = 15), Lgr5-CreER-T2Apcfl/flNpm1fl/fl (n = 9, also shown in Fig. 2i and Extended Data Fig. 6i), Lgr5-CreER-T2Apcfl/flCdkn1a−/− (n = 7) and Lgr5-CreER-T2Apcfl/flCdkn1a−/−Npm1fl/fl (n = 7) mice at clinical endpoint. Data, mean ± SEM. Data statistically assessed by one-way ANOVA followed by Tukey’s multiple comparisons test. c, Tumor distribution within equidistant proximal to distal SI and colon sections from the groups shown in (b). Data statistically assessed by two-way ANOVA followed by Sidak’s multiple comparisons test. Colour combinations denote groups compared. P-values in black indicate no statistical significance between groups. d, Representative staining for NPM1 on SI tissue sections from Lgr5-CreER-T2Apcfl/flNpm1fl/fl (n = 7 – also shown in Fig. 2j and Extended Data Fig. 6k) and Lgr5-CreER-T2Apcfl/flCdkn1a−/−Npm1fl/fl (n = 7) animals at clinical endpoint. Scale bars, 100 μm (top panels) and 1 mm (bottom panels). e, Quantification of percentage of tumours being positive, negative or mosaic for NPM1 expression in Lgr5-CreER-T2Apcfl/flCdkn1a−/−Npm1fl/fl animals at clinical endpoint (n = 7) compared to that of Lgr5-CreER-T2Apcfl/flNpm1fl/fl (n = 7) (also shown in Fig. 2k and Extended Data Fig. 6l). Data statistically assessed by two-way ANOVA followed by Sidak’s multiple comparisons test. Boxes in boxplots extend from the 25th to 75th percentiles, whiskers extend to the minimum and maximum values, and the line in every box is plotted at the median. f, Survival curves of villin-CreER-T2KrasG12D/+Trp53fl/flRosa26N1icd/+ and villin-CreER-T2KrasG12D/+Trp53fl/flRosa26N1icd/+Npm1fl/fl (n = 16 per group). g, tumor number distribution within equidistant proximal to distal SI and colon sections from cohorts shown in (f) (Npm1+/+, n = 12; Npm1fl/fl, n = 15). h, Survival curves of villin-CreER-T2KrasG12D/+Trp53fl/fl (n = 15) and villin-CreER-T2KrasG12D/+Trp53fl/flNpm1fl/fl (n = 16). i, tumor number distribution within equidistant proximal to distal SI and colon sections from cohorts shown in (h) (n = 13 in both groups). P-values in all survival data obtained by Log-rank (Mantel-Cox) tests. Censored mice in survival curves denoted as tick marks at indicated times post-induction. Statistically significant p-values are shown in red.
Extended Data Fig. 8 NPM1 loss induces translational changes in Apc-deficient, Kras-mutant CRC model.
a,b, Protein-coding aligned (pc) unique reads for ribosome-protected fragments (RPFs) (a) and total cytoplasmic RNA (b) per replicate (n = 4/group; WT=Apcfl/flKrasG12D/+, KO=Apcfl/flKrasG12D/+Npm1fl/fl SI). All RNA reads used for downstream analyses. c-e, RPFs quality check: c, RPF read length distribution/replicate (pc unique reads/nucleotide length). d, Read coverage across transcript regions/read length: coding sequences (CDS-yellow), 5’-untranslated region (5’UTR-teal), 3’UTR (blue). >75% of reads align to CDSs. e, Read frame analysis/length. Lengths 27-28 lacked frame preference and were discarded, 29-31 had predominantly f0 and extracted with -12 offset, and 32-37 had f2 preference and extracted with -13 offset to correct codon position. Shading indicates lengths extracted for analysis. f, g, RPFs and total cytoplasmic RNA principal component analysis. Samples coloured by genotype and shaped by replicate. h, i, Correlation of rlog-normalised reads for total RNA, and RPFs across replicates. j, k, Volcano plots showing differentially expressed transcripts in total RNA (j) and RPFs (k) upon NPM1 loss. Significant transcripts indicated in red (adj. p-value < 0.1). Red/grey arrows and table indicate transcripts increased/decreased by NPM1 loss at the RNA level. l, m, Scatterplots showing correlations between NPM1 expression and replication stress or DNA repair status in human CRC tumours (n = 566, GSE39582 cohort). Two-sided Pearson correlation. Regression lines with 95% confidence intervals shaded and correlation coefficients (r) displayed. n, γH2AX staining on SI tissue from Npm1+/+ and Npm1fl/fl mice sampled four days post-induction, and Apcfl/+KrasG12D/+ and Apcfl/+KrasG12D/+Npm1fl/fl endpoint mice (n = 5/group). o-q, γH2AX staining quantification in crypts and tumours of groups in (n). Unpaired two-tailed t test (o) and two-tailed Mann Whitney tests (p,q). r-u, Cleaved PARP staining and quantification as in (n-q). Unpaired two-tailed t tests. v, Metagene plots of Trp53 and Cdkn1a ribosome occupancy across 5’UTR, CDS and 3’UTR. Shaded area indicates standard deviation across replicates (n = 4/group). w, Pause site plot for Trp53 with resolved sites marked by arrow. x, Boxplots of p53 levels across total RNA, RPFs and proteomics. Boxplots boxes extend from 25th-75th percentiles, whiskers from minimum to maximum values, and the line denotes the median. Significant p-values in red. Scale bars, 100 μm.
Extended Data Fig. 9 NPM1 loss affects the translation pathway.
a, b, p-eIF4e(Ser209) staining in SI from Apcfl/+KrasG12D/+ and Apcfl/+KrasG12D/+Npm1fl/fl endpoint mice (n = 5/group), and quantification in crypts/tumours. Unpaired two-tailed t tests. c, d, p-eIF4e(Ser209) staining and quantification (crypts) in SI from Npm1+/+ and Npm1fl/fl mice four days post-induction (n = 5/group). Unpaired two-tailed t test. e, 35S-methionine incorporation to assess protein synthesis in Apcfl/flKrasG12D/+ (n = 3) and Apcfl/flKrasG12D/+Npm1fl/fl (n = 4) organoids. Data, mean ± SEM; unpaired two-tailed t test. f-h, p-eIF2α(Ser51) staining in SI from Npm1+/+ and Npm1fl/fl mice four days post-induction (n = 4/group), and H-score/stain intensity quantification. Two-tailed Mann Whitney test (g); multiple two-sided t-tests with Holm–Sidak correction (h). i, j, Western blot for translation pathway components in epithelial extracts from Apcfl/fl, Apcfl/flNpm1fl/fl, Apcfl/flKrasG12D/+ and Apcfl/flKrasG12D/+Npm1fl/fl mice (n = 4/group), and quantification (j). Data, mean ± SEM. Unpaired two-tailed t tests. k, l, Western blot in Apcfl/flKrasG12D/+ and Apcfl/flKrasG12D/+Npm1fl/fl untreated or DMSO/tunicamycin-treated organoids (n = 3/group), and quantification (l). Data, mean ± SEM. Unpaired two-tailed t tests. m, n, Western blot for puromycin incorporation in Apcfl/flKrasG12D/+ and Apcfl/flKrasG12D/+Npm1fl/fl ± DMSO/ISRIB-treated organoids (n = 3/group), and quantification of boxed areas (n). Cyclohexamide used as negative control. Data, mean ± SEM. One-way ANOVA with Tukey’s test. o, BrdU staining in SI from Apcfl/flNpm1fl/fl mice ±vehicle/trazodone treatment (n = 4/group). p, BrdU+ cell quantification in SI half-crypts from groups in (o), compared to untreated Apcfl/flNpm1+/+ (n = 5) and Apcfl/flNpm1fl/fl (n = 5) mice (also presented in Fig. 2c). Data, mean ± SEM. One-way ANOVA with Tukey’s test. q, Survival curves of untreated Apcfl/+KrasG12D/+ (n = 17) and Apcfl/+KrasG12D/+Npm1fl/fl (n = 20) (also shown in Fig. 2n), and trazodone-treated mice (Apcfl/+KrasG12D/+ n = 7, Apcfl/+KrasG12D/+Npm1fl/fl n = 10). Median survival (days) indicated in brackets. Log-rank (Mantel-Cox) tests. Censored mice denoted as tick marks. r, tumor distribution within equidistant proximal to distal SI and colon sections from untreated Apcfl/+KrasG12D/+ (n = 17) and Apcfl/+KrasG12D/+Npm1fl/fl (n = 20) (also shown in Extended Data Fig. 5o) and trazodone-treated Apcfl/+KrasG12D/+ (n = 5) and Apcfl/+KrasG12D/+Npm1fl/fl (n = 8) endpoint mice. Two-way ANOVA with Sidak’s test. Colour combinations denote groups compared. P-values in black indicate no significance. Other significant p-values in red. Boxplots boxes extend from 25th-75th percentiles, whiskers from minimum to maximum values, and the line denotes the median. Scale bars (a,c,f), 100 μm; (o), 50 μm.
Extended Data Fig. 10 NPM1 is overexpressed in HCC and required for WNT-driven hepatocyte proliferation.
a, Npm1 expression in murine livers of indicated genotypes and timepoints normalised to wild-type (GEO data: GSE230137) (wild-type(d10), R26lsl-MYC/lsl-MYC(d4), Ctnnb1+/(Dex3)(d10), Ctnnb1+/(Dex3)R26lsl-MYC/lsl-MYC(d10), n = 3; Ctnnb1+/(Dex3)(d4), Apcfl/fl(d10), R26lsl-MYC/lsl-MYC(d10), n = 4; Ctnnb1+/(Dex3)R26lsl-MYC/lsl-MYC(d4), Apcfl/flR26lsl-MYC/lsl-MYC(d10), n = 5). One-way ANOVA with Tukey’s test. b, c, NPM1 expression in TCGA-LIHC tumours stratified by MYC amplification or CTNNB1 mutation status. d, e, WNT signatures GSVA scores in wild-type and mutant CTTNB1 TCGA-LIHC tumours. f, g, NPM1 expression and WNT signatures GSVA scores correlation scatterplots in TCGA-LIHC tumours. Two-sided Pearson correlation. h, HCC patient survival with high/low NPM1 (n = 249 low, n = 115 high). Log-rank test; tick marks: censored subjects. i, H&E, NPM1, glutamine synthetase (GS) and BrdU staining in Npm1+/+ (n = 3) and Npm1fl/fl (n = 5) livers 120 days post high dose AAV8.TBG.Cre. Scale bars, 50 μm. j-l, Liver-to-body weight ratios, BrdU+ cells (9-13 10X fields of view (FOV)) and blood alanine transaminase (ALT) levels from groups in (i). m-o, GS staining, GS+ cells and H-score quantification in Ctnnb1+/(Dex3)R26lsl-MYC/lsl-MYC±Npm1fl/fl livers ten days post high dose AAV8.TBG.Cre (n = 5/group). Scale bars, 1 mm. p, q, BrdU staining and quantification (six 10X FOV) in Apcfl/fl and Apcfl/flNpm1fl/fl livers ten days post high dose AAV8.TBG.Cre (n = 3/group). Scale bars, 100 μm. r, s, BrdU and NPM1 staining, and BrdU+ cell quantification (ten 10X FOV) in BrafLSL-V637E/+ (n = 5) and BrafLSL-V637E/+Npm1fl/fl (n = 4) livers four days post high dose AAV8.TBG.Cre. Scale bars, 100 μm. t, BrdU, NPM1 and p21 staining in BrafLSL-V637E/+ and BrafLSL-V637E/+Npm1fl/fl livers (n = 4/group) eight days post high dose AAV8.TBG.Cre. Scale bars, 100 μm. u, BrdU+ (ten 10X FOV) and v, p21+ hepatocytes from groups in (t). Unpaired two-tailed Mann Whitney test. w, RNAseq comparing Ctnnb1+/(Dex3)R26lsl-MYC/lsl-MYC±Npm1fl/fl livers ten days post-induction (n = 4/group). Two-sided Wald test, Benjamini–Hochberg correction. Significant (adj p.value < 0.05, log2FC < -1 or >1) transcripts in red. x, y, Shallow NPM1 deletion frequency and/or TP53 mutations in TCGA-LIHC (n = 351). Two-sided Fisher’s exact test. Data (bar charts), mean ± SEM. Boxplots boxes extend from 25th-75th percentiles, whiskers from minimum to maximum values; the line denotes the median. Statistics in (b-e), (j-l), (n-o), (q), (s), (u): unpaired two-tailed t tests. Significant p-values in red.
Supplementary information
Supplementary Information (download PDF )
Supplementary Note.
Supplementary Tables 1–6 (download XLSX )
Oligonucleotide sequences used in the study and lists of peptides and transcripts for proteomics and Riboseq data analysis.
Source data
Source Data for Figures and Extended Data Figures (download XLSX )
Statistical source data for Figs. 1, 2, 3, 5, 6 and 7, and Extended Data Fig. 1–10.
Source Data Extended Data Fig. 9 (download PDF )
Unprocessed immunoblots.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kanellos, G., Giacomelli, C., Raven, A. et al. Nucleophosmin supports WNT-driven hyperproliferation and tumor initiation. Nat Genet 58, 100–115 (2026). https://doi.org/10.1038/s41588-025-02408-7
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41588-025-02408-7
