
Abstract
A comprehensive understanding of the evolution of the immune landscape in humans across the entire lifespan at single-cell transcriptional and protein levels, during development, maturation and senescence is currently lacking. We recruited a total of 220 healthy volunteers from the Shanghai Pudong Cohort (NCT05206643), spanning 13 age groups from 0 to over 90 years, and profiled their peripheral immune cells through single-cell RNA-sequencing coupled with single T cell and B cell receptor sequencing, high-throughput mass cytometry, bulk RNA-sequencing and flow cytometry validation experiments. We revealed that T cells were the most strongly affected by age and experienced the most intensive rewiring in cell–cell interactions during specific age. Different T cell subsets displayed different aging patterns in both transcriptomes and immune repertoires; examples included GNLY+CD8+ effector memory T cells, which exhibited the highest clonal expansion among all T cell subsets and displayed distinct functional signatures in children and the elderly; and CD8+ MAIT cells, which reached their peaks of relative abundance, clonal diversity and antibacterial capability in adolescents and then gradually tapered off. Interestingly, we identified and experimentally verified a previously unrecognized ‘cytotoxic’ B cell subset that was enriched in children. Finally, an immune age prediction model was developed based on lifecycle-wide single-cell data that can evaluate the immune status of healthy individuals and identify those with disturbed immune functions. Our work provides both valuable insights and resources for further understanding the aging of the immune system across the whole human lifespan.
Similar content being viewed by others


Main
Development and aging run throughout human life, accompanied by gradual maturation and inevitable degeneration of the immune system1,2. After birth, the immune system gradually matures and provides better protection against foreign pathogens3. Conversely, immune aging leads to higher incidences of infection, cardiovascular diseases4 and cancers5. Moreover, it was recently reported that aged immune cells drive the aging of solid organs, such as the heart and liver, which is a probable cause of systemic senescence6. However, it remains unclear when and how the immune system changes during human aging, which typically spans nearly 90 years and encompasses development, maturation and senescence across the entire lifespan.
Human peripheral blood mononuclear cells (PBMCs) are a diverse mixture of highly specialized immune cells that are critical components of the immune system. Previous omics studies of human PBMCs have demonstrated their involvement in immune pathologies (for example, immunosenescence7, autoimmune disorders8 and infectious diseases9) and in diseases of solid organs10,11. Significant age-related changes in PBMC composition and function in healthy individuals have been demonstrated by flow cytometry and bulk sequencing analyses12,13. Recently, single-cell RNA sequencing (scRNA-seq) and single-cell T cell receptor (TCR) and B cell receptor (BCR) sequencing (scTCR/BCR-seq), as well as single-cell mass cytometry (cytometry by time-of-flight, CyTOF) protein profiling have provided opportunities to comprehensively identify previously undefined cell types at single-cell resolution14,15,16. Several scRNA-seq studies on peripheral immune cells at specific ages have yielded valuable findings17,18,19,20,21. Although they provide valuable insights into age-related changes in immune cells at a single-cell resolution, these scRNA-seq studies involved limited age groups and did not cover the full temporal span of human aging. Moreover, none of the scRNA-seq studies provided single-cell protein validation, and human immune-aging datasets across the entire lifespan at single-cell protein levels are currently lacking.
Here, through combined scRNA-seq, scTCR/BCR-seq and CyTOF analyses, we investigated the temporal dynamics of the cell type compositions, transcriptional and protein profiles, cell–cell interactions (CCIs) and immune repertoires of peripheral immune cells during development, maturation and senescence in humans throughout the entire lifespan, using PBMCs from healthy participants aged 0 to ≥90 years, spanning 13 age groups. Using this unique lifecycle-wide dataset, we developed a single-cell immune age (siAge) prediction model to assess an individual’s immune age and describe the immune status in health and disease. Our work provides a rich resource for understanding the evolution of peripheral immune cells during human development, maturation and aging across the whole lifespan. The data are publicly available in a web portal for easy exploration (https://pu-lab.sjtu.edu.cn/shiny/lifespan) and through the preprint version of our data (https://www.biorxiv.org/content/10.1101/2022.07.11.498621v2).
Results
A single-cell atlas of peripheral immune cells across the human lifespan
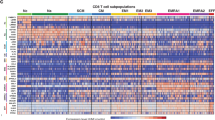
A total of 309 participants from both internal and external cohorts were included in this study. To characterize the dynamics of peripheral immune cell composition and function throughout the human lifespan, 220 healthy volunteers aged 0 years (neonates) to ≥90 years (nonagenarians) were recruited from the Shanghai Pudong Cohort (NCT05206643) (Fig. 1a and Supplementary Table 1). The participants spanned the entire human lifespan and were partitioned into 13 age groups (0-, 1-, 2-, 6-, 12-, 18-, 30-, 40-, 50-, 60-, 70-, 80- and >90-year-olds)22,23,24. We further developed an siAge prediction model based on this lifecycle-wide single-cell dataset and validated the siAge model with an external cohort of 89 additional individuals, including 33 healthy individuals and 56 with a disturbed immune function (Supplementary Table 2). For scRNA-seq analyses, after filtering out low-quality droplets, a total of 538,266 cells were obtained. According to unsupervised clustering and the expression of signature genes for each subset (Supplementary Table 3), 25 distinct PBMC subsets were identified and visualized by uniform manifold approximation and projection (UMAP) (Fig. 1b and Extended Data Fig. 1a; details in the Methods). The representative signature genes of the cell subsets and expression levels along with the number of detected genes and unique molecular identifiers (UMIs) for each subset are shown in heatmaps, feature plots and violin plots (Fig. 1c, Extended Data Fig. 1b–e and Supplementary Table 3).

a, Workflow of the overall experimental design of this study. Panel a created in BioRender. Huang, T. (2024) https://BioRender.com/a58r007. b, UMAP plot illustrating the peripheral immune cell subsets identified within PBMCs from 61 samples. c, Heatmaps showing the expression of signature genes in each cell subset. d, Dot plot showing the age-related variations in proportion of the 25 immune cell subsets across 13 age groups. Kruskal–Wallis one-way analysis was used to identify cell subsets with significant differences across age groups. FDR correction was applied to account for multiple testing and the values are shown above the dot plot. Horizontal dashed line marks the threshold for FDR significance (FDR < 0.05). mDCs, myeloid dendritic cells; pDCs, plasmacytoid dendritic cells; UMAP, uniform manifold approximation and projection; y, years.
Given that cell-type-specific aging analyses largely depend on accurate cell-type annotation, we took multiple approaches to ensure the reliability of our annotation. The cell subset annotation derived from scRNA-seq was first validated at the single-cell protein level using high-throughput CyTOF analyses with a panel of metal isotope-tagged protein markers (Supplementary Table 4) and demonstrated good agreement between the two measures (Extended Data Fig. 2a–f). Additional validations were performed using two other approaches, the automatic annotation tool Azimuth25 (Extended Data Fig. 2g) and gene set enrichment analysis (Extended Data Fig. 2h), with both showing good agreement.
Peripheral immune cell composition dynamics across the lifespan
To obtain a global view of the dynamics of PBMC composition, the proportion of each of the 25 cell subsets in total PBMCs was calculated for each sample from the 13 age groups (Extended Data Fig. 3a). Of the 25 cell subsets, 22 exhibited significant differences in proportions among different age groups (Fig. 1d and Extended Data Fig. 3a) (Kruskal–Wallis test, false discovery rate (FDR) < 0.05). A proportion of certain cell subsets were positively correlated with age, such as CD4_TEM_GNLY cells and CD8_TEM_GNLY cells (Extended Data Fig. 3b) and others were negatively correlated with age, such as CD4_Naive_CCR7 cells, CD8_Naive_LEF1 cells, NK_CD56high cells and B_Naive cells (Extended Data Fig. 3c). Several cell subsets exhibited enrichment in certain age groups. For example, CD14+ monocyte cells were enriched in the umbilical cord blood group, plasma cells were enriched in children (peaking in the 6-year-old group) and CD8_MAIT_SLC4A10 cells were enriched in adolescents (peaking in the 18-year-old group) (Extended Data Fig. 3d). Three cell subsets did not show significant changes in proportion with respect to age (for example, natural killer (NK) proliferating, CD16+ monocytes and plasmacytoid dendritic cells), suggesting that they are relatively stable across the human lifespan (Kruskal–Wallis test, FDR > 0.05). Cell composition dynamics were further analyzed at the single-cell protein level using high-throughput CyTOF and showed good agreement (Extended Data Fig. 3e–g). These results demonstrate differential correlations of different PBMC subsets with age at single-cell resolution.
Peripheral immune cell transcriptional dynamics across the lifespan
To systematically investigate the transcriptome dynamics of PBMC subsets over the entire lifespan, we ranked the 25 PBMC subsets by the number of differentially expressed genes (DEGs) (Fig. 2a). The top ten cell subsets with the highest DEG numbers all belonged to lymphoid lineage cell subsets (that is, T cell, NK cell and B cell types), indicating that these cell subsets were the most strongly affected by age. Among the top ten cell subsets, eight were T cells, and marked differences in the numbers of DEGs were observed among different T cell subsets (Fig. 2a). No myeloid lineage (that is, monocytes, dendritic cells (DCs) and megakaryocytes) had ≥100 DEGs.

a, Bar plot showing the number of DEGs derived from scRNA-seq analyses of all cell subsets. b, Heatmap showing the functional enrichment analyses of DEGs in all cell subsets. P values were determined by hypergeometric test. c, Bar plots showing the frequencies of upregulated and downregulated M-DEGs derived from scRNA-seq analyses observed in all cell subsets. All M-DEGs with a frequency of ≥5 are displayed. d, Line plots showing the expression trajectories of upregulated (left panel) and downregulated (right panel) age-related genes identified by bulk RNA-seq analyses. Data are shown in terms of gene expression z-score. Each gray line represents the expression trajectory of a gene. e, Venn diagrams showing the number of overlapping genes between common M-DEGs identified in scRNA-seq data and age-related genes identified in bulk RNA-seq data. Among the 56 common M-DEG genes defined by scRNA-seq, 13 intersected with the age-related genes defined by bulk RNA-seq. f, Heatmap showing the expression of SELL gene in seven cell subsets by scRNA-seq and CD62L protein (encoded by SELL) in corresponding cell subsets by CyTOF among eight life stages.
To explore the functional dynamics of PBMC subsets across the lifespan, the DEGs in the 25 subsets were subjected to functional enrichment analyses separately. DEGs were enriched in biological processes including pathways related to positive regulation of cell activation (24 cell subsets), lymphocyte-mediated immunity (23 cell subsets), regulation of leukocyte cell–cell adhesion (22 cell subsets) and type II interferon production (22 cell subsets) (Fig. 2b). We then focused on age-related genes that were monotonically correlated with age (monotonical DEGs, M-DEGs). Consistent with results from the DEG analyses, the top ten cell subsets with the highest number of M-DEGs (≥30) were lymphoid lineage cell subsets, supporting that these lymphoid lineage cell subsets are susceptible to aging (Extended Data Fig. 4a,b and Supplementary Table 5). Eight of the top ten were T cell subsets, one was an NK cell subset and one was a B cell subset. The top three cell subsets with the largest number of M-DEGs were T cell subsets: CD4_Naive_CCR7, CD4_TCM_AQP3 and CD8_Naive_LEF1 cells (Extended Data Fig. 4a,b). Functional enrichment analyses showed that the upregulated M-DEGs were enriched in pro-inflammatory biological processes, such as type II interferon production and positive regulation of I-κB kinase/NF-κB signaling (Extended Data Fig. 4c and Supplementary Table 6), whereas the downregulated M-DEGs were enriched in telomere organization and actin filament organization (Extended Data Fig. 4d and Supplementary Table 6).
Next, we ranked the M-DEGs by the number of cell subsets in which they were involved and determined the common M-DEGs shared by multiple cell subsets. M-DEGs shared by greater than or equal to five cell subsets were defined as common M-DEGs. The analyses identified 18 upregulated common M-DEGs and 38 downregulated common M-DEGs (Fig. 2c and Supplementary Table 5). Functional enrichment analyses showed that the upregulated common M-DEGs were enriched in pathways associated with positive regulation of I-κB kinase/NF-κB signaling, positive regulation of cell–cell adhesion and major histocompatibility complex class II protein complex assembly (Extended Data Fig. 4e). Downregulated common M-DEGs were enriched in pathways associated with telomere maintenance, mononuclear cell differentiation and actin filament organization (Extended Data Fig. 4f). In addition to monotonic changes across the life stages, age-related DEGs defined by scRNA-seq exhibited enrichment at specific age ranges. For instance, 144 DEGs from 12 cell types were enriched at the child stage (Extended Data Fig. 4g,i), whereas 90 DEGs from 18 cell types were enriched at the 70–80-year-old (elderly) stage (Extended Data Fig. 4h,j). The scRNA-seq-defined common M-DEGs represent genes that are both widely expressed in different cell subsets and strongly correlated with age. We next sought to validate these scRNA-seq-defined common M-DEGs using traditional bulk RNA-seq in an independent cohort of 34 healthy individuals (0 to ≥90 years old), which provides an average gene expression profile for the entire population of cells26,27. We identified 1,071 age-related genes in PBMCs in the bulk RNA-seq dataset (Fig. 2d, Extended Data Fig. 4k and Supplementary Table 7). Remarkably, among the 56 common M-DEG genes defined by scRNA-seq, 13 intersected with the age-related genes defined by bulk RNA-seq, including four genes upregulated with age and nine downregulated with age (Fig. 2e). This overlap underscores a high concordance between the findings obtained from scRNA-seq, covering different cell subsets, and those from bulk RNA-seq, giving a global view. Of the four upregulated common M-DEGs, S100A6, S100A4 and LGALS1 encode proteins that can promote inflammation and drive tumorigenesis and metastasis28,29,30. Although S100A4 was previously reported to be highly elevated in centenarians21, its potential functional role in aging has not been investigated. Using an in vitro aging model, we found that S100A4 acted as a positive regulator in the aging process (Supplementary Fig. 1 and Supplementary Information Section 1.1). Of the downregulated common M-DEGs, SELL, CCR7 and TMIGD2 encode proteins that maintain immune homeostasis by modulating the balance of immunity and tolerance31,32,33. The age-related common M-DEG SELL (encoding CD62L) was further validated at the single-cell protein level with CyTOF (Fig. 2f). These results suggest that the age-related common M-DEGs shared by different cell subsets may represent potential immune biomarkers across the lifespan.
Immunological processes involve interactions among various immune cell types to coordinate their behavior during an immune response34,35. We thus further investigated the rewiring of CCIs among peripheral immune cells across the lifespan and found that the CCIs displayed different patterns at different stages of the lifespan (Fig. 3a–f). Topological characteristics indicated that immune checkpoints have a more critical role in the age-modulated CCI network than non-immune checkpoints (Fig. 3g–j). The alterations of immune checkpoints across the whole human lifespan mainly occur in T cells and might have essential roles during human development and aging (Fig. 3k,l and Extended Data Fig. 5a,b). Detailed information about CCIs is provided in Supplementary Information Section 1.2.

a, Heatmap showing that CCI probability varied significantly across the lifespan. The probability for each CCI was row-scaled with a z-score. The significantly varied CCIs among eight different human life stages were determined using Kruskal–Wallis one-way analyses. P values were adjusted with FDR, and an adjusted P value (Padj) of <0.0001 was considered statistically significant. b, Bar plot showing that T cells experience the most intensive rewiring in CCIs with age. c–f, Network graphs showing the CCI cluster 1 (c), cluster 2 (d), cluster 3 (e) and cluster 4 (f) from a. The hub cell subset of each cluster is highlighted with a larger node size in c–f. The hub cell subset of each cluster is highlighted with a larger node size. g, Network graph showing the CCI network mediated by differentially expressed ligands and receptors. h, Bar plot showing the top ten biological processes enriched in the differentially expressed ligands and receptors. P values were determined by hypergeometric test. i, Box plot showing the average shortest path length and degree of immune checkpoints and non-immune checkpoints in g. Box plots show median, first (lower hinge) and third (upper hinge) quartiles; whiskers show 1.5× the interquartile range. P values were determined by a two-sided unpaired Wilcoxon rank-sum test. j, Bar plot showing, for each immune checkpoint ligand and receptor, the number of cell subsets in which they were detected as differentially expressed. k, Heatmaps showing the scaled expression of CD27, ICOS and PDCD1 genes across the lifespan in the indicated cell subsets, detected by scRNA-seq. l, Heatmaps showing the scaled expression of CD27, ICOS and PD1 proteins across the lifespan in the indicated cell subsets, detected by CyTOF. UCB, umbilical cord blood.
Clonal expansion of effector T cells across the lifespan
We further explored the clonal expansion in all 11 αβ T cell subsets and their V(D)J gene usages across the entire lifespan by reconstructing TCR sequences from scTCR-seq. We obtained matched TCR and transcriptome information from over 90% of all αβ T cells (Extended Data Fig. 6a). In prior research focused on the adult population, an increased peak in T cell clonal expansion was observed only in the elderly36. However, our lifecycle-wide cohort, with exclusive access to childhood and adolescent data, reveals that high T cell clonal expansions manifest two peaks, occurring during the early life developmental stage (2–12 years old) and the late-life elderly stage (70–90 years old) (Fig. 4a). Further comparison of the V(D)J gene usage profiles among these six age groups showed that 43 genes were differentially used (Kruskal–Wallis test, P < 0.05) (Fig. 4b). No significant differences were observed in the length distribution of the complementarity-determining region-3 (CDR3) amino acid sequences in TCR α-chains and β-chains (Extended Data Fig. 6b). Using hierarchical clustering from the differentially used V(D)J genes, the six age groups were clustered into two distinct groups: the 2–12-year-old group (early life high T cell clonal expansion (high-TCE) group) and the 70–90-year-old group (late-life high-TCE group) (Fig. 4b). The selective usage of V(D)J genes reflects the different features of T cell responses in different age groups and may be associated with human immune development or senescence.

a, Stacked bar graph showing the distribution of T cell clone sizes in the 13 age groups. b, Heatmap showing the row-scaled usage of significantly differentially expressed V-J genes from TCR α-chains and TCR β-chains across six high-TCE groups (2, 6, 12, 70, 80 and 90 years). The six age groups were clustered into two distinct groups: the 2–12-year-old group (early life high-TCE group) and the 70–90-year-old group (late-life high-TCE group). c, Bar plot showing the distribution of T cell clone sizes in each αβ T cell subset. d,e, Box plot showing the clonal expansion levels of total αβ T cell subsets (d) and CD8_TEM_GNLY cells (e) across 13 age groups quantified by the STARTRAC-expa metric. The black line indicates LOESS regression. f, Volcano plot showing the DEGs in high-TCE CD8_TEM_GNLY cells (clone size ≥ 50 cells) in the late-life high-TCE group (70–90 years old) compared with those in the early life high-TCE group (2, 6 and 12 years old). The representative genes are labeled. Dashed lines demarcate two-sided Benjamini–Hochberg-corrected Padj = 0.05 and log2 fold change (FC) = 0.25. g, Bar plots showing the representative enriched biological processes of DEGs upregulated in high-TCE CD8_TEM_GNLY cells in early life (left panel) and in late life (right panel). P values were determined by hypergeometric test. h, Box plots showing the gene module scores for the pathways in high-TCE CD8_TEM_GNLY cells in the early life and late-life high-TCE groups. The two-sided Wilcoxon rank-sum test was used to calculate the P values. i, Rank for TF regulons in high-TCE CD8_TEM_GNLY cells in the early life (left panel) and late-life (right panel) high-TCE groups using the regulon specificity score. In d, e and h, box plots show median, first (lower hinge) and third (upper hinge) quartiles; whiskers show 1.5× the interquartile range. MHC, major histocompatibility complex; TF, transcription factor.
Next, we explored the distribution of high-TCE cells among all 11 αβ T cell subsets. We detected different degrees of clonal expansion in different T cell subsets: high-TCE cells (clone size of ≥50 cells) only appeared in three effector memory T cell subsets (CD8_TEM_GNLY, CD4_TEM_GNLY and CD8_TEM_CMC1 cells) with high STARTRAC expansion scores (Fig. 4c–e and Extended Data Fig. 6c). Among them, CD8_TEM_GNLY cells accounted for the highest proportions of high-TCE cells (Fig. 4c and Extended Data Fig. 6c).
Next, we performed DEG analyses to explore the functional features underlying the clonal expansion of CD8_TEM_GNLY cells in the two high-TCE groups. In the early life high-TCE group, 635 significantly upregulated DEGs were identified, including cytotoxic genes (GZMB and PRF1) and energy metabolism-related genes (COX5B, COX5A, NDUFAB1, NDUFB10, NDUFS6 and NDUFS3) (Fig. 4f). Functional enrichment analyses showed that these DEGs were enriched in the regulation of T cell activation, cell killing, regulation of leukocyte proliferation and oxidative phosphorylation pathways (Fig. 4g). In the late-life high-TCE group, 87 upregulated DEGs were identified, including interferon-related genes (IFITM3 and ISG20) and anti-apoptosis genes (TSC22D3 and IL7R)37 (Fig. 4f). Functional enrichment analyses showed that these DEGs were enriched in the response to virus, response to type II interferon, negative regulation of lymphocyte apoptotic process and response to type II interferon pathways (Fig. 4g). Accordingly, we analyzed the expression of cytotoxicity, oxidative phosphorylation, leukocyte proliferation and type II interferon-related pathways in the clonal expansion of CD8_TEM_GNLY cells in the early life and late-life high-TCE groups. We found that the expressions of cytotoxicity, oxidative phosphorylation and positive regulation of mononuclear cell proliferation were significantly upregulated in early life high-TCE groups, whereas the expression of genes in the response to type II interferon pathway was upregulated in late-life high-TCE groups (Fig. 4h). Thus, the upregulated DEGs in high-TCE CD8_TEM_GNLY cells at the elderly stage were involved in biological processes related to chronic inflammation, which has been linked to aging of solid organs37,38. By contrast, the higher expression of cytotoxic genes in high-TCE CD8_TEM_GNLY cells at the developmental stage indicated that these T cells may possess powerful killing functions against infected and cancerous cells. Furthermore, transcription factor analyses indicated that the transcription factor regulon GATA3 displayed the highest activity in the early life high-TCE group (Fig. 4i), whereas the transcription factor regulon ETS1 displayed the highest activity in the late-life high-TCE group (Fig. 4i). GATA3 regulates T cell development, proliferation and maintenance39,40. ETS1 acts as a regulator of human healthy aging by decreasing ribosomal activity41. Thus, although both groups displayed high clonal expansions of CD8_TEM_GNLY cells, there were distinct functions underlying these expanded T cells between the early life and late-life high-TCE groups.
Different aging patterns in naive CD4+ versus CD8+ T cells
A key characteristic of age-related immune dysfunction is the loss of naive T cells5,42. Previous studies that relied on adult data have concluded that CD8+ cells show a more pronounced decline with age than their CD4+ counterparts36. However, using scRNA-seq-derived cell classification with our full lifespan coverage, we observed that although both naive T cell subsets (CD8_Naive_LEF1 and CD4_Naive_CCR7) exhibited a decreased trend with age (Fig. 5a), their aging trajectories diverge beginning at birth. Specifically, the two naive T cell subsets followed different decreasing trajectories across the human lifespan: naive CD4+ T cells exhibited an abrupt decrease during the early life developmental stage followed by a slower decline after entering adulthood; conversely, naive CD8+ T cells exhibited a gradual decrease through the early life stage followed by a rapid decline in adulthood. The aging patterns of the two naive T cell subsets were further supported by CyTOF data (Fig. 5b).

a,b, Scatter plot illustrating changes in the proportion of CD4+ (CD4_Naive_CCR7; left) and CD8+ naive T cells (CD8_Naive_LEF1; right) in total αβ T cells with aging from scRNA-seq (a) and CyTOF (b). c,d, Heatmap showing the row-scaled expression of M-DEGs in CD4_Naive_CCR7 (c) and CD8_Naive_LEF1 (d) cells across eight life stages (left), and line plots showing the expression trajectories of M-DEGs upregulated with age (right). Expression z-scores are shown. e, Dot plot showing the representative enriched biological processes of M-DEGs upregulated with age in CD4+ and CD8+ naive T cells. f,g, Scatter plots showing TCR repertoire diversities in CD4+ (CD4_Naive_CCR7; left) and CD8+ naive T cells (CD8_Naive_LEF1; right) across eight life stages, as measured by Shannon’s entropy (f) and the inverse Simpson index (g). h–j, Scatter plot illustrating the proportion changes of CD8_MAIT_SLC4A10 cells in total αβ T cells with aging from scRNA-seq (h) and CyTOF (i) analyses and their TCR repertoire diversities as measured by Shannon’s entropy (j) across eight life stages. k, Heatmap showing the expression of all the DEGs (left) and zoom-in on upregulated genes (right) in CD8_MAIT_SLC4A10 cells in adolescents (12–18 years old). l, Bar plots showing the representative enriched biological processes of upregulated (top) and downregulated (bottom) DEGs in CD8_MAIT_SLC4A10 cells in adolescents. m, GSEA enrichment plot for gene set ‘tuberculosis’ in CD8_MAIT_SLC4A10 cells in the adolescent group compared with other groups. n, Schematic diagram showing the design of MAIT cell’s bactericidal capability assay (left) and bar plot showing the bacteria load after MAIT cell co-culture. The P value was determined by two-sided unpaired Wilcoxon rank-sum test (right, n = 9 each). P values in e, l and m were determined by hypergeometric test. P < 0.05 was considered statistically significant in e, l, m and n. The black lines indicate LOESS regression in a, b, f and g. CFU, colony forming unit; GSEA, gene set enrichment analysis.
We next sought to examine the functional dynamics of the two naive T cell subsets and identified 76 upregulated M-DEGs for naive CD4+ T cells and 73 for naive CD8+ T cells. The M-DEGs for naive CD4+ T cells changed rapidly in earlier life and reached a plateau-like state in later life, whereas the M-DEGs for naive CD8+ T cells showed a continuous and straight increase throughout life (Fig. 5c,d). Functional enrichment analyses of the upregulated M-DEGs in naive CD4+ and naive CD8+ T cells revealed several common biological processes shared by the two naive T cell subsets related to pro-inflammatory pathways, including response to type II interferon and regulation of autophagy43 (Fig. 5e).
Next, we examined the dynamic patterns of TCR diversity in naive CD4+ and CD8+ T cells and found that their patterns were also different (Fig. 5f,g and Extended Data Fig. 6d). Again, naive CD4+ T cells exhibited a more abrupt decrease during the early life developmental stage than naive CD8+ cells; conversely, naive CD8+ T cells displayed a more prominent decline in TCR diversity at older ages than naive CD4+ T cells. These results reveal the different aging-associated transcriptional patterns and repertoire dynamics between naive CD4+ and CD8+ T cells.
Expansion of CD8+ MAIT cells during maturation
Mucosal-associated invariant T (MAIT) cells are unique innate-like T lymphocytes that bridge innate and adaptive immunity44. MAIT cells rapidly produce cytokines and cytotoxic effectors through both TCR-dependent and TCR-independent mechanisms45,46 in response to a broad range of bacteria and viruses, such as Mycobacterium tuberculosis47 and hepatitis B virus. We show that CD8_MAIT_SLC4A10 cells exhibit a unimodal pattern of enrichment during adolescence with a peak in the 18-year-old group (Fig. 5h). The results from the CyTOF analyses exhibited a pronounced concordance with those from scRNA-seq (Fig. 5i). Clonal analyses showed that the TCR diversity of CD8_MAIT_SLC4A10 cells also peaked at adolescence (Fig. 5j and Extended Data Fig. 6d,e), implying that CD8_MAIT_SLC4A10 cells at this stage might exhibit a stronger ability to fend off pathogen invasion than those at other life stages.
To characterize the function of CD8_MAIT_SLC4A10 cells during maturation, we compared gene expression profiles of CD8_MAIT_SLC4A10 cells in adolescents with those in other life periods, including children, adults and the elderly. Fig. 5k displays the expression of the upregulated and downregulated DEGs in CD8_MAIT_SLC4A10 cells in adolescents; the expression of the upregulated DEGs reached a peak in adolescents (named ‘upregulated DEGs’; Fig. 5k). The interaction network analyses suggested that 19 out of the 29 upregulated DEGs closely interacted, with KLRB1 identified as a hub gene (Extended Data Fig. 6f). Notably, CyTOF analyses showed that the expression of CD161 protein (encoded by KLRB1) within MAIT cells also reached its peak in the adolescent period (Extended Data Fig. 6g).
Further Gene Ontology enrichment analyses showed that the upregulated DEGs in CD8_MAIT_SLC4A10 cells from individuals during adolescence were enriched in biological processes related to aerobic respiration, lymphocyte proliferation and positive regulation of inflammatory response (Fig. 5l), whereas the downregulated DEGs were related to cell adhesion and the apoptotic pathway (Fig. 5l). We further depicted the functional properties of CD8_MAIT_SLC4A10 cells in the adolescent group by gene set enrichment analysis with all expressed genes. Tuberculosis-related signaling displayed a significant downregulation in CD8_MAIT_SLC4A10 cells at this stage (Fig. 5m). The incidence and prevalence of tuberculosis are the lowest in young people (15–24 years old) according to the World Health Organization 2021 Global Tuberculosis Report (https://www.who.int), in line with our findings of enhanced MAIT cell function during this period. In addition, antimicrobial effector function assays showed that the antimicrobial activities of MAIT cells from young adults (aged 18 years) were significantly higher than those from middle-aged counterparts (aged 50 years) (Fig. 5n). Together, these results indicate that CD8_MAIT_SLC4A10 cells at the maturation stage may have a greater ability to resist pathogen invasion than those in other age groups.
A newly identified cytotoxic B cell subset
Immune cells with cytotoxic functions in the peripheral blood of healthy individuals generally include NK and cytotoxic T cells48. Interestingly, we detected a newly identified and unique B cell subset (B_BCR+GNLY+ cells) that simultaneously expressed B cell markers (such as BCR genes, including immunoglobulin heavy-chain or light-chain coding genes, and JCHAIN) and canonical cytotoxic genes (such as GNLY and NKG7) (Fig. 1b,c and Extended Data Fig. 1b,d). The expression of TCR genes, TRAC and KLRF1 were hardly detected in them, excluding the possibility of these cells being T cells or NK cells (Extended Data Fig. 1b,d). We ruled out the possibility of potential technical artefacts (that is, multiplets) because the number of detected genes and UMIs in B_BCR+GNLY+ cells were lower than those in other subsets (Extended Data Fig. 1e) and a stringent doublet filtration standard was used to exclude potential doublets. B_BCR+GNLY+ cells exhibited a unimodal pattern, with enrichment in children with a peak in the 6-year-old group. These cells were low in neonates and 1-year-old infants and increased in 2–6-year-old children, followed by a gradual decrease at 12 years, reaching a low level in adulthood (Fig. 1d).
To explore the functional differences between B_BCR+GNLY+ cells and the other four B cell subsets, we identified the signature genes of all B cell subsets (Supplementary Table 10). The top pathway enriched by the signature genes of B_BCR+GNLY+ cells was the cytolysis pathway (Fig. 6a). Double-positive cells (J chain and granulysin, IgM and granulysin or IgA and granulysin) were identified in PBMCs from six additional healthy 6-year-olds using immunofluorescence (Fig. 6b). Longitudinal scRNA-seq and scTCR/BCR-seq results showed the expansion of B_BCR+GNLY+ cells from age 1 (0.02%, 0.03% and 0.04% in PBMCs) to age 2 (1.84%, 10.96% and 3.09% in PBMCs) in these children (Fig. 6c). We performed flow cytometry to detect the percentage of double-positive cells (IgM and granulysin) in peripheral blood. The proportions of double-positive cells (IgM and granulysin) were significantly higher in children (4–9 years old; n = 5) than those from adults (24–30 years old; n = 5) or the elderly (64–72 years old; n = 5) (Fig. 6d and Supplementary Fig. 2).

a, Heatmap showing the functional enrichment analyses of signature genes in each B cell subset based on the scRNA-seq analyses. The top ten pathways in each cell subset (ranked by P value) are listed. P values were determined by hypergeometric test. b, Immunofluorescence of B cell markers (IgM, IgA and J chain) and the cytotoxic protein granulysin (GNLY) in PBMCs collected from six healthy 6-year-olds. The experiment was repeated four times with similar results. c, Bar plot showing the percentage of B_BCR+GNLY+ cells in PBMCs from three infants with longitudinal follow-up, examined at the ages of 1 year and 2 years by scRNA-seq analyses. R1, R2 and R3 represent different individuals. d, Flow cytometry analysis showing proportions of IgM+GNLY+ cells in children (4–9 years old), adults (24–30 years old) and the elderly (64–72 years old), without stimulation (control, top left) and day 1 after in vitro stimulation with an expansion mixture consisting of IL-21 (10 ng ml−1), IL-2 (50 IU ml−1), CD40L (50 ng ml−1), anti-BCR (5 mg ml−1) and CpG ODN 2006 (1 mg ml−1) (bottom left) (n = 5 per group). Bar plots showing the percentage of IgM+GNLY+ cells in indicated groups (right panel). e, Flow cytometry analysis showing the proportion of IgM+GNLY+ cells in cultured human B cells on days 1 and 3 after in vitro stimulation with an expansion mixture (left panel) (n = 4 per group). Bar plots showing the percentage of IgM+GNLY+ cells in B cells from each group (right panel). f, Bar plots showing GNLY concentration in culture supernatants of B cells on days 1 and 3 of culture after in vitro stimulation with or without an expansion mixture. The concentrations of GNLY were determined by ELISA (n = 4 per group). In d, e and f, bar plots show the mean ± s.e.m.; P < 0.05 was considered statistically significant. P values in a were determined by hypergeometric test. P values in d were determined by one-way ANOVA followed by the Tukey post hoc test. P values in e and f were determined by two-sided unpaired Wilcoxon rank-sum test. ELISA, enzyme-linked immunosorbent assay.
Cytotoxic T and B cells are induced after in vitro stimulation with expansion mixtures (including cytokines, chemokines, antibodies and adjuvants)49,50,51. Therefore, circulating B cells were isolated from healthy individuals and stimulated with expansion mixtures that activate B cells50. Flow cytometric analyses showed that the proportion of IgM+GNLY+ double-positive B cells was significantly increased in cultured B cells exposed to the expansion mixtures compared with those with vehicle (Fig. 6e). Meanwhile, nanogram levels of granulysin were detected in the culture supernatant of B cells (Fig. 6f). Furthermore, to determine whether the response to stimulation was age-dependent, PBMCs isolated from three different life stages (children, 4–9 years old; adults, 24–30 years old; elderly, 64–72 years old) were subjected to the expansion mixtures. The proportion of IgM+GNLY+ double-positive B cells was significantly higher in children than in the adult and elderly groups (Fig. 6d), in accordance with our scRNA-seq results.
The role of granulysin in B cell-mediated functions has yet to be reported. Therefore, we sought to determine whether B_BCR+GNLY+ cells possess potential cytotoxic activities. Our results indicate that although B_BCR+GNLY+ cells are closest to plasma cells among all the B cell subsets with respect to gene expression profiles, the distribution of immunoglobulin isotypes, BCR clonotypes and transcriptional regulatory mechanisms, this newly identified B cell type has its own distinct characteristics and displays cytotoxic activities. Additional details concerning B_BCR+GNLY+ cells are provided in Supplementary Information Section 1.3 (Extended Data Figs. 8 and 9 and Supplementary Fig. 3).
Single-cell immune age prediction model
The aging immune system contributes to the morbidity and mortality of the elderly5. One significant obstacle in understanding immune system alterations involves the precise quantification of aging20,52. Our unique lifecycle-wide single-cell data provided an opportunity to construct a single-cell immune age clock model (that is, siAge score) to quantify an individual’s immune age and describe their immune status (Fig. 7a). We first built 25 models with data from just one cell subset and another model incorporating all cell subsets. The prediction model integrating all 25 cell subset datasets had the highest predictive score and achieved the best performance (Pearson’s correlation coefficient (PCC) = 0.9940, P < 2.2 × 10−16) among the 26 prediction models (Fig. 7b,c and Supplementary Fig. 4a) and was chosen as the final siAge prediction model.

a, Workflow of the single-cell immune age prediction model construction. b, Dot plot showing the predictive performance of the cell-type-specific immune age prediction models based on our lifecycle-wide single-cell data. The top ten cell types with the highest scores are shown. c, Scatter plot showing the correlation of single-cell immune age (siAge) with chronological age (cAge) in the training set (n = 56 samples). d, Scatter plot showing the correlation of siAge with cAge in the external healthy validation cohort (n = 33 samples). e, Scatter plot showing the correlation of siAge with cAge in the external validation cohort of diseased patients with disturbed immune functions (n = 56 samples). f, Box plot comparing the fit index distribution calculated by the siAge model in the healthy validation cohort with those of the diseased validation cohorts, which include NPC (n = 10 samples), SLE (n = 40 samples) and KD (n = 6 samples) cohorts. Box plots show median, first (lower hinge) and third (upper hinge) quartiles; whiskers show 1.5× the interquartile range. P values were determined by two-sided unpaired Wilcoxon rank-sum test. g, Bar plot showing the top 21 key genes identified by random forest regression. The genes are ranked in descending order of importance with respect to the accuracy of the model. The insert represents a tenfold cross-validation error as a function of the number of input features used to regress against the chronological ages. h, Heatmap showing the row-scaled expression of the top 21 key genes across age in corresponding cell subsets. i, Bar plot showing functional enrichment of the top 21 key genes. P values were determined by hypergeometric test. In c–e, the blue line indicates linear regression; PCC and P values are indicated. The gray shadow covers the 95% confidence interval. In c–f, P < 0.05 was considered statistically significant. KD, Kawasaki diseases; NPC, nasopharyngeal carcinoma; SLE, systemic lupus erythematosus.
To further validate the prediction capability of the siAge prediction model, we measured the prediction model efficacy with scRNA-seq data using several independent validation cohorts (detailed in Methods). In the healthy validation cohort (33 individuals), there was a strong positive correlation between chronological age (cAge) and siAge (PCC = 0.8894, P = 4.557 × 10−12) (Fig. 7d), indicating that the siAge prediction model achieved good performance in healthy individuals. However, in the validation cohort with a compromised immune function (56 PBMC samples), the correlation between cAge and siAge became weaker (PCC = 0.6526, P = 5.008 × 10−8) (Fig. 7e and Supplementary Fig. 4b). The disrupted correlation between the cAge and siAge scores in the diseased indicated the potential ability of our prediction model to present the disrupted immune homeostasis in individuals with immune-related diseases. To better assess the predictive ability of the siAge model, we defined a fit index that was calculated from the ratio between siAge and cAge and used it to evaluate the deviations in siAge and cAge between healthy subjects and patients with a disturbed immune function. Our findings highlight that the siAge prediction model, driven by 21 key genes across T cell subsets, serves as a potential marker to access immune system status and aging-associated changes across the human lifespan (Fig. 7f–i, detailed in Supplementary Information Section 1.4).
Discussion
Our data represent a resource to simultaneously encompass both single-cell transcriptional and protein-level changes of human peripheral immune cells across the entire human lifespan, with the largest age range to date (13 age groups, ranging from age 0 to over 90 years). Using this unique resource, the current study provides important insights into the age-specific alterations in cell-type composition, transcription and protein profiles, CCIs and immune repertoires of PBMCs over the lifespan at a fine temporal and single-cell resolution. Several important observations were made (Extended Data Fig. 10). First, the T cell subsets were the most susceptible to aging among all PBMCs and were characterized by increased pro-inflammatory pathways, reduced telomere organization and markedly altered immune checkpoints. Second, scRNA-seq and immune repertoire sequencing revealed unique age-specific transcriptional and clonal expansion features of T cell subsets: (1) high T cell clonal expansions exhibited peaks at both ends of life: the early life developmental stage (2–12 years old) and the late-life elderly stage (70–90 years old); (2) naive CD4+ and naive CD8+ T cells exhibited distinct aging-associated cell compositions, transcriptional patterns and repertoire dynamics from birth; and (3) CD8_MAIT_SLC4A10 cells exhibited peak abundance and clonal diversity during adolescence, with greater antimicrobial ability than those in other age groups. The findings from scRNA-seq-based and scTCR/BCR-seq-based analyses were further recapitulated by high-throughput CyTOF-based protein profiling. Importantly, we described a newly identified and unique ‘cytotoxic’ B cell subset (B_BCR+GNLY+ cells) that simultaneously expressed BCR genes and cytotoxic genes and was enriched in children. Using this single-cell lifecycle-wide dataset, we developed an immune age prediction model (siAge score) that enabled the identification of a disturbed immune state. Overall, our work provides fine temporal resolution of peripheral immune cell transcriptomes and immune repertoires during development, maturation and senescence in humans across the entire lifespan and will facilitate in-depth exploration of the immune system at each age stage.
The immune system is immature at birth and gradually develops under postnatal environmental stimuli53. Maternally derived antibodies taper off in the infant’s circulation and are almost completely lost by 1 year after birth54,55, whereas self-derived immune mediators—such as T cells from the thymus, and B cells and innate immune cells from the bone marrow—augment over time56, accompanied by external stimulation including routine immunization and environmental microbe and food antigen exposures. Immunoglobulins in infants increase progressively and reach normal adult levels at age 5–9 years57,58. During this critical developmental period, children are more vulnerable to infectious diseases59. We found that certain cell subsets exhibited enrichment in certain age categories during development, including CD14+ monocytes in neonates and plasma cells in children. Importantly, we identified a unique ‘cytotoxic’ B cell type (B_BCR+GNLY+ cells) that expanded significantly in children from 2 to 12 years old, with a peak in 6-year-olds. Given its low proportion in other age groups, it may have been overlooked in previous single-cell studies that did not examine all age categories or perform scBCR-seq analyses. B_BCR+GNLY+ cells are characterized by the simultaneous expression of BCR genes and cytotoxic genes, such as GNLY and NKG7. Traditionally, B cells are thought to be immune cell populations incapable of cytotoxic functions. However, recent studies have reported a ‘cytotoxic’ phenotype of B cell subsets expressing cytotoxic genes in diseased populations51,60,61,62. The enrichment of the B_BCR+GNLY+ cells with cytotoxic signatures before adolescence may represent a potential mechanism for protection against infection before immune maturation. The detailed biological functions of B_BCR+GNLY+ cells require further investigation (detailed in Supplementary Information Section 2.1).
The immune system undergoes dramatic changes during and after adolescence and sexual maturity63,64. Both our scRNA-seq and CyTOF analyses showed that MAIT cells were enriched during adolescence, with a peak in the 18-year-old group, and MAIT cells in adolescents possessed higher TCR diversity and stronger antimicrobial effector function than in other age groups. MAIT cells are a unique T cell type that bridge innate and adaptive immunity44. The involvement of MAIT cells in a large spectrum of pathological conditions makes them attractive therapeutic targets45. Commensal bacteria mediate MAIT cell maturation and recirculation into the thymus65. The dramatic hormonal changes that occur during adolescence lead to alteration of the mucosal membrane microenvironment and variation in commensal bacterial species and numbers66, which may account for the MAIT cell expansion during the period of adolescence seen in our study. However, a systematic understanding of the peripheral circulating MAIT cells in healthy individuals across the life course was lacking at a single-cell resolution. Our findings delineate the potential functional significance of MAIT cells during adolescence by characterizing the transcriptome and immune repertoire of this cell type across the lifespan. The observed transcriptomic and expansion features and antimicrobial activities of MAIT cells during adolescence may represent a key component of immune maturation and competence for protection against infection.
Immune function declines with aging, termed ‘immunosenescence’5. However, different types of immune cells display differential susceptibility to aging. In line with a mouse aging study showing that T cells exhibited a more vulnerable phenotype to senescence than other immune cells6, our results in humans revealed that the functions of T cell subsets were the most susceptible to aging among all PBMCs across the entire lifespan. Previous studies on the adult population (25–88 years old) have concluded that cytotoxic T cells (CD8+), compared to helper T cells (CD4+), show a more pronounced decline with age36. However, with our full lifespan coverage, we found the aging trajectories of CD4+ and CD8+ T cells to diverge beginning at birth, based on both scRNA-seq and CyTOF analyses. During the early life developmental stage, naive CD4+ cells declined more rapidly than naive CD8+ cells, whereas during later life, naive CD8+ cells exhibited more drastic attrition than naive CD4+ cells in terms of frequency and TCR diversity. The collapse of TCR diversity in naive T cells has been considered a strong predictor of poor health in older people67. Our scRNA-seq dataset, covering each age category from birth to old age, revealed, at the single-cell level, the differential dynamics of the alteration of TCR diversity in naive CD4+ T cells and naive CD8+ T cells in both early and late life.
Age-dependent changes in the immune response are the most obvious at the extremes of age, with immune malfunction or dysregulation occurring during the early life development and the late-life elderly stages56. Previous studies that were limited to the adult population have shown elevated T cell clonal expansion only in elderly ages36. However, taking advantage of our lifecycle-wide cohort from birth to old age, we revealed that high T cell clonal expansions actually exhibited two peaks at the extremes of the lifespan, with an early peak in the 2–12-year-old group and a late peak in the 70–90-year-old group. Our results provide insights into the similarities and differences in T cell functions between two age groups, representing immune development and immune aging, respectively (detailed in Supplementary Information Section 2.2). The human lifespan has increased in recent decades, which raises major health issues related to many age-associated pathologies. Using our lifecycle-wide single-cell dataset covering 13 age groups over the extent of the human lifespan, we established a comprehensive cross-age index (siAge score) as a type of machine-learning-based immune aging clock to evaluate an individual’s immune status across different life stages. We demonstrated that the siAge score is highly correlated with the calendar age in healthy individuals but not in those with disturbed immune functions in multiple diseased validation cohorts. Thus, our siAge model holds the potential to effectively quantify a person’s immune status and distinguish a healthy individual from one with an unhealthy immune state. It has been transformed into user-friendly software (https://pu-lab.sjtu.edu.cn/shiny/lifespan), making it easily accessible to end users. Further research is needed to validate the generalization of our findings (detailed in Supplementary Information Section 2.3).
Methods
Participants and ethics
All human samples were obtained in accordance with protocols approved by the Institutional Review Board at Shanghai Jiao Tong University. All donors were enrolled with approval by the Ethics Committee of Ren Ji Hospital (KY2019-136), School of Medicine, Shanghai Jiao Tong University. After providing written informed consent in accordance with the criteria set by the Declaration of Helsinki (2013)68, 220 healthy volunteers aged 0 to ≥90 years were recruited from Shanghai Pudong Cohort (NCT05206643), including 61 healthy participants aged 0 to ≥90 years for scRNA and scTCR/scBCR sequencing; 70 healthy participants aged 0 to ≥90 years for independent CyTOF-based single-cell protein validation experiments; 34 healthy participants aged 0 to ≥90 years for independent bulk RNA-seq validation experiments; and 55 healthy participants aged 2–72 years for validation experiments. The inclusion and exclusion criteria for participants in this study is provided in Supplementary Information Section 3. The participants spanned the entire lifespan, partitioned into 13 age groups (0-, 1-, 2-, 6-, 12-, 18-, 30-, 40-, 50-, 60-, 70-, 80- and over 90-year-olds). To yield more biologically meaningful results, when performing statistical testing to determine the differences among groups, participants were categorized into eight life stages as suggested by developmental biologists22,23,24 as follows: 0 years old, newborn (umbilical cord blood); 1–2 years old, toddler; 6–8 years old, childhood; 12–18 years old, adolescent; 30–40 years old, young adult, 50–60 years old, middle-aged adult; 70–80 years old, elderly; and over 90 years old, elderly-plus.
Single-cell RNA-seq library preparation and sequencing
To produce single-cell gel beads in emulsion, cell suspensions were quantified with 400–600 living cells per µl determined by a Countstar automated cell counter. A Chromium single-cell controller (10× Genomics) was used to prepare cell suspensions as gel beads in emulsion, with a Single Cell 5′ Library and Gel Bead kit (1000006, 10× Genomics) and Chromium Single Cell A Chip kit (120236, 10× Genomics) following the manufacturer’s protocols. Approximately 15,000 cells were added to each channel and the recovered target cells were estimated to be approximately 8,000 cells. scRNA-seq libraries were constructed using the Chromium Single Cell 5′ v.1.1 Reagent (Human T Cell, 1000005; Human B Cell, 1000016; 10×Genomics), and a unique sample index was included in each sequencing library. Finally, sequencing was performed with a sequencing depth of at least 100,000 reads per cell with a paired-end 150 bp reading strategy using the Illumina platform (CapitalBio Technology).
scRNA-seq data processing
Cell Ranger (v.3.0.2) was used to align reads to reference (GRCh38-3.0.0) and generate feature-barcode matrices from the Chromium scRNA-seq output. Filtering steps for high-quality single cells included removal of doublets using Scrublet69 (doublet rate = 0.075, doubletScore < 0.25), cells expressing <400 genes, cells with >2,500 detected genes and cells with a mitochondrial gene count percentage of >10%. Genes appearing in less than three cells were also excluded.
Based on the clean scRNA-seq data after quality control, gene expression profiles were normalized for each cell using the LogNormalize method with the Seurat NormalizeData function. The FindVariableFeatures function was then applied to identify the highly variable genes using the default parameters. Next, we scaled the data using the ScaleData function and performed principal component analysis on the scaled data using the RunPCA function with default parameters. The scVIIntegration algorithm was used for batch correction70. We constructed a shared nearest neighbor graph using the FindNeighbors function and clustered cells using the Louvain algorithm with the FindClusters function based on the scVIIntegration reduction. Finally, the RunUMAP function facilitated the visualization of all cells. A total of 538,266 high-quality cells were derived from 61 samples.
Cell-type annotation and cluster marker identification
The FindAllMarkers function was used to find marker genes for each cluster. Clusters were then classified and annotated based on the marker gene expression of each cell subset. We used a two-round hierarchical approach to cluster and annotate the cells71,72. In brief, the first round of clustering (resolution, 0.8) identified 11 major cell types, including CD4+ T cells, CD8+ T cells, γδ T cells, NK cells, B cells, plasma cells, CD14+ monocytes, CD16+ monocytes, monocyte-derived dendritic cells, plasmacytoid dendritic cells and megakaryocytes. In round two, cells from these major types were subset out and the whole integration and clustering process was applied to them separately. The second round of clustering (resolution, 0.3–1.2) resulted in 25 distinct cell subsets (shown in Fig. 1b and Extended Data Fig. 1a–d). Single cells expressing two sets of well-studied canonical markers of major cell types were labeled as doublets and excluded from subsequent analyses. A full list of canonical and signature genes for each subset is shown in Supplementary Table 3.
CyTOF sample processing and data acquisition
Cryopreserved PBMC samples were rapidly thawed in a 37 °C water bath within 2 min. PBMCs were collected by centrifugation and cell precipitates were resuspended with PBS. A Countstar automated cell counter was used to count the cell numbers, and Trypan Blue was used to verify the viability of the PBMCs. The adequacy of PBMC quantity and quality for subsequent analyses was assessed based on the following criteria: the cell number per <3 × 106 and the viability per sample of >90%.
The total amounts of 41 metal-conjugated antibodies used for CyTOF in this study are listed in Supplementary Table 4. Following sample collection, protein-free PBS was used to wash the samples. Then, PBMCs were stained with cisplatin (Fluidigm) to exclude dead cells. To minimize nonspecific staining, PBMCs were incubated with Fc blocking solution at 4 °C for 30 min. Staining medium containing cell-surface antibodies was then applied to stain the PBMCs in PBS containing 5% goat serum and 30% BSA for 30 min at 4 °C. Then, PBMCs were fixed and permeated overnight using a permeabilization kit (eBioscience). Next, PBMCs were washed with perm buffer (eBioscience) and stained with intracellular antibodies in permeabilization buffer for 30 min at 4 °C. PBMCs were washed twice and resuspended with double distilled water for data acquisition. Before each batch of samples was loaded, the signal strength of each channel was adjusted for the instrument according to the same beads signal (140Ce, 151Eu, 153Eu, 165Ho and 175Lu). The data were acquired using the Helio3 CyTOF Mass Cytometer (Fluidigm).
CyTOF data analyses
Data of each sample were de-barcoded from the raw data using a doublet-filtering scheme with unique mass-tagged barcodes. Each ‘.fcs’ file generated from different batches was normalized using the bead normalization method. We used FlowJo software (v.10.8.1) to exclude debris, dead cells and doublets from the data using step-by-step manual gating, leaving only live single immune cells for subsequent analyses. After strict data cleaning and quality control, a total of 10,887,899 single immune cells were obtained for the subsequent analyses. Clean CyTOF data were processed using the R package cytofWorkflow (v.1.14.0). The X-shift clustering algorithm was used on all cells to partition the cells into distinct phenotypes based on marker expression levels and then each cell cluster was annotated according to its marker expression pattern based on a heatmap of clusters and markers. The dimensional reduction algorithm UMAP was used to visualize the high-dimensional data in two dimensions, including the clustering of the cells and the distribution of the marker genes in different clusters or groups of cells. For both marker expression and cell proportion of the annotated cell populations, their dynamic profiles with age were fitted by generalized LOESS regression. The Spearman correlation coefficient and significance were also calculated from the cor.test function in the stats package. A P value of <0.05 was considered statistically significant.
Bulk RNA-seq library preparation and sequencing
Bulk RNA-seq was performed on PBMCs obtained from an independent cohort of 34 healthy individuals (0 to >90 years old). Total RNA was extracted using Trizol reagent (Invitrogen) following the manufacturer’s procedure. The RNA amount and purity of each sample were quantified using NanoDrop 2000 with OD260/280 = 1.6–1.8. RNA integrity was assessed by Agilent 2100 (Agilent Technologies). All samples had a RIN number of >7.0. RNA-seq libraries were prepared following the manufacturer’s instructions, and sequencing was performed on an Illumina NovaSeq 6000 (CFBiotech) following the vendor’s recommended protocol.
Bulk RNA-seq data analyses
Quality control of the raw reads was performed by Trimmomatic (v.0.39)73 software. Then, the sequencing read quality was verified using FastQC (v.0.11.9) (http://www.bioinformatics.babraham.ac.uk/projects/fastqc, 2010). Raw reads were processed with fastp (v.0.22.0)74. Fastq files were aligned to the human genome (GRCh38) using HISAT2 (v.2.1.0)75. Counts of reads and fragments per kilobase of exon per million (FPKM) were calculated using StringTie (v.2.1.3b)76. Linear regression fitting was used to evaluate the correlation between age and the FPKM of each gene through the lm function. The R package stats was used to calculate Spearman’s correlation coefficient and P value. Age-related genes were further determined using Spearman rho analyses (|ρ| ≥ 0.5, P < 0.01).
TCR and BCR repertoire sequencing and analyses
The scTCR and scBCR repertoire libraries of all samples (10× Genomics) were prepared and profiled with paired-end sequencing (2 × 150 bp) on the Illumina HiSeq2500 platform. The Cell Ranger V(D)J pipeline (10× Genomics, v.3.0.2) was used to mark the TCR and BCR expression and clonal types. The TCR and BCR repertoire sequencing data were filtered with the following criteria: productive is “True”; high_confidence is “True”; umis ≥1; and raw_consensus_id is not “None”. Only T cells with at least one TCR α-chain and one TCR β-chain were used to identify the TCR clonotypes for each T cell. If two or more TCR α-chains or β-chains were assembled in a given T cell, the highest expressed (UMI or reads) TCR α-chain or β-chain was defined as the dominant TCR α-chain or TCR β-chain. A clonotype was defined by a unique TCR α(s)-TCR β(s) pair (CDR3 nucleotide sequences and rearranged VJ genes included) within a T cell. BCR clonotypes were identified in a similar way to TCR clonotypes. In brief, only B cells with at least one productive heavy chain (IGH) and one productive light chain (IGL or IGK) were retained for further analyses. Each unique IGH and IGL/IGK pair within a B cell was defined as a BCR clonotype. If there were two or more IGH and IGL/IGK assembled in a given B cell, the highest expressed IGH and IGL/IGK chain was regarded as the dominant IGH and IGL/IGK chain.
For CDR3 amino acid length analyses, filtered TCR α-chains and β-chains were separately calculated with nchar, as.dat.frame, unlist, table, sum and round functions. For the TCR clonal status analyses, as.dat.frame, unlist and table functions were used to calculate clonotype counts of filtered cells. Cells expressing the same clonotype were counted. The counts were classified into eight classes to identify the cellular clonal expansion status; >100, 51–100, 31–50, 21–30, 11–20, 6–10, 2–5 and a unique clone without expansion.
From barcode mapping, clonal type information from Cell Ranger V(D)J was integrated into the metadata of the Seurat object for further analysis. Shannon’s entropy, inverse Simpson’s index and the Chao1 index were used to estimate the diversity, species abundance and richness of TCR repertories, respectively77.
Shannon’s entropy was measured using the function H of the DescTools (v.0.99.54) package as follows:
Here, the frequency of a given TCR clone in all αβ T cells with TCR identified is represented as p(x). Changes in clone diversity with age were fitted by LOESS regression. The Spearman correlation coefficient and significance were also calculated from the cor.test function in the stats package. P < 0.05 was considered statistically significant.
Inverse Simpson’s index was used for calculating the species abundance of the TCR repertoire36. The calculation of this index was calculated by the repDiversity function of the immunarch (v.1.0.0) package:
where i represents each TCR sequence, N represents the number of TCR sequences for a given sample, pi represents the UMI percentage occupancy for a TCR (UMI count of TCR divided by total UMI counts) and λ represents Simpson’s index.
The Chao1 index of TCR repertoires was calculated by the repDiversity function in the immunarch (v.1.0.0) package, following the formula:
where S represents TCR repertoire richness, that is, the total number of unique TCR clonotypes for a given sample, F1 is the number of TCR clonotypes that counted 1 and F2 is the number of TCR clonotypes that counted 2.
Human B cell isolation and purification
Human PBMCs were separated from whole blood samples using Vacutainer CPT tubes (362761, BD) following the manufacturer’s protocols78. B cells were isolated from PBMCs using the Human Pan B Cell Isolation Kit (130-101-638, Miltenyi Biotec) following the manufacturer’s instructions. The purity of B cells was confirmed by flow cytometry (>95% purity). Human B cells were cultured and expanded in ImmunoCult Human B Cell Expansion Kit (100-0645, STEMCELL Technologies) for further use.
Construction of single-cell immune age prediction model
An age-prediction model for evaluating immune homeostasis at single-cell resolution (siAge) was constructed using DEGs in each cell subset as independent variables. The scRNA-seq dataset, including 13 age groups across the entire lifespan from neonates to nonagenarians, was used as the training set. First, all cell-type-specific DEGs in the training set were used to generate valuable feature variables based on the random forest model through the randomForest (v.4.7-1.1) package79. To examine the contributions of different cell subsets to the performance of age prediction, we trained 26 random-forest-based regression models80,81 to predict immune age, including 25 cell-type-specific prediction models trained using their corresponding 25 cell-type-specific datasets and one prediction model using integrated data from all 25 cell-type-specific training datasets. K-fold cross-validation with cv = 10 was used to improve the generalization ability of gene feature selection through the rfcv function. After sorting the importance of multiple variables with IncMSE, the top 21 key genes (visualized through the pheatmap function) were selected to construct the siAge prediction model using the random forest algorithm through the randomForest R package.
Next, to test the generalization ability of the siAge prediction model, we conducted a comprehensive search in the Gene Expression Omnibus database and the Genome Sequence Archive of the Beijing Institute of Genomics Data Center of data up to 30 May 2022. A combination of the following keywords was used: ‘single-cell’ sequencing’ or ‘scRNA-sequencing’ or ‘single cell’ and ‘peripheral blood mononuclear cells’ or ‘PBMCs’. The inclusion criteria for the published scRNA-seq datasets were as follows: gene expression profiling of human PBMCs by 10× Genomics scRNA-seq, detailed information on the age of participants, fresh PBMC samples and informed consent from authors for raw data. We obtained 77 PBMC samples from published available studies (3,531 published datasets) with scRNA-seq data and detailed information on the age of participants, including 21 PBMC samples from healthy volunteers aged 1.7–58 years (GSE158055 (ref. 71), GSE168732 (ref. 82) and HRA000150 (ref. 83)) and 56 PBMC samples from patients aged 1.6–65 years, including ten PBMC samples from nasopharyngeal carcinoma patients aged 22–65 years (GSE162025 (ref. 84)), six PBMC samples from Kawasaki disease patients aged 1.6–5.4 years (GSE168732 (ref. 82)) and 40 PBMC samples from systemic lupus erythematosus patients aged 10–63 years (GSE135779 (ref. 8)). Detailed information of all datasets used are shown in Supplementary Table 2. A total of 12 healthy participants aged 12–39 years were recruited from the Shanghai Pudong Cohort. Linear regression fitting was used to evaluate the correlation between the cAge and our siAge through the lm function. The PCC85 and P value were calculated through the cor.test function in the stats package. P < 0.05 was considered statistically significant. We defined a fit index to evaluate the deviations between siAge and cAge in healthy individuals and patients with a disturbed immune function. The fit index was calculated following the equation:
Statistical analyses
Statistical analyses were performed using R software (v.4.3.0). The Wilcoxon rank-sum test was used to examine the difference between two groups. The Kruskal–Wallis test was used to determine whether or not there was a statistically significant difference among three or more age groups. The significant differences in cell subset proportions, gene expression and CCIs among all age groups were determined by Kruskal–Wallis tests followed by Dunnett’s post hoc test, and the P values were adjusted with FDR in multiple comparisons. The statistical significance of the differences was determined by two-sided unpaired Wilcoxon rank-sum tests for node degree and average shortest path length of immune checkpoints and non-immune checkpoints, as well as the fit index of siAge of the healthy cohort and the cohort with diseases. The dynamic profiles of variables (that is, cell subset proportions, gene expression, TCR diversity and STARTRAC expansion score) with age were fitted by generalized LOESS regression, and associations between age and these variables were tested using a Spearman’s correlation test. The P value significance thresholds used for each result are described in the corresponding methods and figure legends. For other comparisons, the name and type of the statistical test are stated in the figure legends.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The raw scRNA-sequencing data reported in this study are available from the Genome Sequence Archive for Human under accession number HRA009014. The processed gene expression of scRNA-seq data (Seurat object RDS file), the expression profiles of bulk RNA-seq data and CyTOF single-cell protein data were uploaded into the SYNAPSE database (https://www.synapse.org) under accession numbers syn61609846, syn61853526 and syn61765870, respectively. The machine-learning-based immune age prediction framework has been made publicly available on the online website (https://pu-lab.sjtu.edu.cn/shiny/siage-model). Source data are provided with this paper.
Code availability
The source code of the model is available on GitHub (https://github.com/shanzha9/siAgeModel.git). The codes described in the Methods or other methods used in our study are publicly available upon request to the corresponding author.
References
Mittelbrunn, M. & Kroemer, G. Hallmarks of T cell aging. Nat. Immunol. 22, 687–698 (2021).
Moqri, M. et al. Biomarkers of aging for the identification and evaluation of longevity interventions. Cell 186, 3758–3775 (2023).
Simon, A. K., Hollander, G. A. & McMichael, A. Evolution of the immune system in humans from infancy to old age. Proc. Biol. Sci. 282, 20143085 (2015).
Tong, Z. et al. Single-cell RNA sequencing maps immune cell heterogeneity in mice with allogeneic cardiac transplantation. Cardiovasc. Innov. Appl. 8, 988 (2023).
Nikolich-Zugich, J. The twilight of immunity: emerging concepts in aging of the immune system. Nat. Immunol. 19, 10–19 (2018).
Yousefzadeh, M. J. et al. An aged immune system drives senescence and ageing of solid organs. Nature 594, 100–105 (2021).
Mogilenko, D. A. et al. Comprehensive profiling of an aging immune system reveals clonal GZMK+ CD8+ T cells as conserved hallmark of inflammaging. Immunity 54, 99–115.e12 (2021).
Nehar-Belaid, D. et al. Mapping systemic lupus erythematosus heterogeneity at the single-cell level. Nat. Immunol. 21, 1094–1106 (2020).
Zhu, L. et al. Single-cell sequencing of peripheral mononuclear cells reveals distinct immune response landscapes of COVID-19 and influenza patients. Immunity 53, 685–696.e3 (2020).
Mazzola, A. et al. Using peripheral blood mononuclear cells to determine proteome profiles in human cardiac failure. Eur. J. Heart Fail. 10, 749–757 (2008).
Li, J. et al. PBMC transcriptomics identifies immune-metabolism disorder during the development of HBV-ACLF. Gut 71, 163–175 (2022).
Li, M. et al. Age related human T cell subset evolution and senescence. Immun. Ageing 16, 24 (2019).
Peters, M. J. et al. The transcriptional landscape of age in human peripheral blood. Nat. Commun. 6, 8570 (2015).
Villani, A. C. et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 356, eaah4573 (2017).
Ahmed, R. et al. A public BCR present in a unique dual-receptor-expressing lymphocyte from type 1 diabetes patients encodes a potent T cell autoantigen. Cell 177, 1583–1599.e16 (2019).
Weng, J. et al. Intratumoral PPT1-positive macrophages determine immunosuppressive contexture and immunotherapy response in hepatocellular carcinoma. J. Immunother. Cancer 11, e006655 (2023).
Zhao, Y. et al. Single-cell transcriptomic landscape of nucleated cells in umbilical cord blood. Gigascience 8, giz047 (2019).
Hashimoto, K. et al. Single-cell transcriptomics reveals expansion of cytotoxic CD4 T cells in supercentenarians. Proc. Natl Acad. Sci. USA 116, 24242–24251 (2019).
Luo, O. J. et al. Multidimensional single-cell analysis of human peripheral blood reveals characteristic features of the immune system landscape in aging and frailty. Nat. Aging 2, 348–364 (2022).
Zhu, H. et al. Human PBMC scRNA-seq-based aging clocks reveal ribosome to inflammation balance as a single-cell aging hallmark and super longevity. Sci. Adv. 9, eabq7599 (2023).
Karagiannis, T. T. et al. Multi-modal profiling of peripheral blood cells across the human lifespan reveals distinct immune cell signatures of aging and longevity. EBioMedicine 90, 104514 (2023).
Kivnick, H. Q. & Wells, C. K. Untapped richness in Erik H. Erikson’s rootstock. Gerontologist 54, 40–50 (2014).
Zimmerman, M. & Snow, B. An Introduction to Nutrition (Independent, 2012).
Balasundaram, P. & Avulakunta, I. D. Human Growth and Development (StatPearls Publishing, 2022).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29 (2021).
Patrick, R. et al. Sierra: discovery of differential transcript usage from polyA-captured single-cell RNA-seq data. Genome Biol. 21, 167 (2020).
Khalyfa, A. et al. Transcriptomic changes of murine visceral fat exposed to intermittent hypoxia at single cell resolution. Int. J. Mol. Sci. 22, 261 (2020).
Harada, C. et al. EGFR tyrosine kinase inhibition worsens acute lung injury in mice with repairing airway epithelium. Am. J. Respir. Crit. Care Med. 183, 743–751 (2011).
Oslejsková, L., Grigorian, M., Gay, S., Neidhart, M. & Senolt, L. The metastasis associated protein S100A4: a potential novel link to inflammation and consequent aggressive behaviour of rheumatoid arthritis synovial fibroblasts. Ann. Rheum. Dis. 67, 1499–1504 (2008).
Li, J. M. et al. Upregulation of LGALS1 is associated with oral cancer metastasis. Ther. Adv. Med Oncol. 10, 1758835918794622 (2018).
Janakiram, M., Chinai, J. M., Zhao, A., Sparano, J. A. & Zang, X. HHLA2 and TMIGD2: new immunotherapeutic targets of the B7 and CD28 families. Oncoimmunology 4, e1026534 (2015).
Förster, R., Davalos-Misslitz, A. C. & Rot, A. CCR7 and its ligands: balancing immunity and tolerance. Nat. Rev. Immunol. 8, 362–371 (2008).
Xu, H., Manivannan, A., Crane, I., Dawson, R. & Liversidge, J. Critical but divergent roles for CD62L and CD44 in directing blood monocyte trafficking in vivo during inflammation. Blood 112, 1166–1174 (2008).
Hao, S. et al. Network approaches for dissecting the immune system. iScience 23, 101354 (2020).
Rieckmann, J. C. et al. Social network architecture of human immune cells unveiled by quantitative proteomics. Nat. Immunol. 18, 583–593 (2017).
Sun, X. et al. Longitudinal analysis reveals age-related changes in the T cell receptor repertoire of human T cell subsets. J. Clin. Invest. 132, 158122 (2022).
Sayed, N. et al. An inflammatory aging clock (iAge) based on deep learning tracks multimorbidity, immunosenescence, frailty and cardiovascular aging. Nat. Aging 1, 598–615 (2021).
Ferrucci, L. & Fabbri, E. Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 15, 505–522 (2018).
Wan, Y. Y. GATA3: a master of many trades in immune regulation. Trends Immunol. 35, 233–242 (2014).
Ting, C. N., Olson, M. C., Barton, K. P. & Leiden, J. M. Transcription factor GATA-3 is required for development of the T-cell lineage. Nature 384, 474–478 (1996).
Xiao, F. H. et al. ETS1 acts as a regulator of human healthy aging via decreasing ribosomal activity. Sci. Adv. 8, eabf2017 (2022).
Lu, J. et al. Heterogeneity and transcriptome changes of human CD8+ T cells across nine decades of life. Nat. Commun. 13, 5128 (2022).
Levine, B., Mizushima, N. & Virgin, H. W. Autophagy in immunity and inflammation. Nature 469, 323–335 (2011).
Toubal, A., Nel, I., Lotersztajn, S. & Lehuen, A. Mucosal-associated invariant T cells and disease. Nat. Rev. Immunol. 19, 643–657 (2019).
Provine, N. M. & Klenerman, P. MAIT cells in health and disease. Annu. Rev. Immunol. 38, 203–228 (2020).
Legoux, F., Salou, M. & Lantz, O. MAIT cell development and functions: the microbial connection. Immunity 53, 710–723 (2020).
Sakai, S. et al. MAIT cell-directed therapy of Mycobacterium tuberculosis infection. Mucosal Immunol. 14, 199–208 (2021).
Taylor, M. K. & Cohen, J. J. Cell-mediated cytotoxicity. Curr. Opin. Immunol. 4, 338–343 (1992).
Coffman, R. L., Sher, A. & Seder, R. A. Vaccine adjuvants: putting innate immunity to work. Immunity 33, 492–503 (2010).
Chesneau, M. et al. Efficient expansion of human granzyme B-expressing B cells with potent regulatory properties. J. Immunol. 205, 2391–2401 (2020).
Hagn, M. et al. CD5+ B cells from individuals with systemic lupus erythematosus express granzyme B. Eur. J. Immunol. 40, 2060–2069 (2010).
Huang, H. et al. Decoding aging clocks: new insights from metabolomics. Cell Metabol. 37, 34–58 (2025).
Brodin, P. & Davis, M. M. Human immune system variation. Nat. Rev. Immunol. 17, 21–29 (2017).
Saso, A. & Kampmann, B. Vaccine responses in newborns. Semin. Immunopathol. 39, 627–642 (2017).
Kollmann, T. R., Kampmann, B., Mazmanian, S. K., Marchant, A. & Levy, O. Protecting the newborn and young infant from infectious diseases: lessons from immune ontogeny. Immunity 46, 350–363 (2017).
Goronzy, J. J. et al. in Clinical Immunology 5th edn (eds Rich, R. R. et al.) 535–543 (Elsevier, 2019).
Blanco, E. et al. Age-associated distribution of normal B-cell and plasma cell subsets in peripheral blood. J. Allergy Clin. Immunol. 141, 2208–2219.e16 (2018).
Liu, Z. et al. A reference interval for serum IgG subclasses in Chinese children. PLoS ONE 13, e0192923 (2018).
Hill, D. L. et al. Immune system development varies according to age, location, and anemia in African children. Sci. Transl. Med. 12, eaaw9522 (2020).
Boldrini, V. O. et al. Cytotoxic B cells in relapsing-remitting multiple sclerosis patients. Front. Immunol. 13, 750660 (2022).
Arabpour, M., Rasolmali, R., Talei, A., Mehdipour, F. & Ghaderi, A. Granzyme B production by activated B cells derived from breast cancer-draining lymph nodes. Mol. Immunol. 114, 172–178 (2019).
Xu, W. et al. Human plasma cells express granzyme B. Eur. J. Immunol. 44, 275–284 (2014).
Vallejo, A. N. Immunological hurdles of ageing: indispensable research of the human model. Ageing Res. Rev. 10, 315–318 (2011).
Brenhouse, H. C. & Schwarz, J. M. Immunoadolescence: neuroimmune development and adolescent behavior. Neurosci. Biobehav Rev. 70, 288–299 (2016).
Legoux, F. et al. Microbial metabolites control the thymic development of mucosal-associated invariant T cells. Science 366, 494–499 (2019).
Kundu, P., Blacher, E., Elinav, E. & Pettersson, S. Our gut microbiome: the evolving inner self. Cell 171, 1481–1493 (2017).
Qi, Q. et al. Diversity and clonal selection in the human T-cell repertoire. Proc. Natl Acad. Sci. USA 111, 13139–13144 (2014).
World Medical Association. World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA 310, 2191–2194 (2013).
Wolock, S. L., Lopez, R. & Klein, A. M. Scrublet: computational identification of cell doublets in single-cell transcriptomic data. Cell Syst. 8, 281–291.e9 (2019).
Gayoso, A. et al. A Python library for probabilistic analysis of single-cell omics data. Nat. Biotechnol. 40, 163–166 (2022).
Ren, X. et al. COVID-19 immune features revealed by a large-scale single-cell transcriptome atlas. Cell 184, 1895–1913.e19 (2021).
Tong, R. et al. Characterizing the cellular and molecular variabilities of peripheral immune cells in healthy recipients of BBIBP-CorV inactivated SARS-CoV-2 vaccine by single-cell RNA sequencing. Emerg. Microbes Infect. 12, e2187245 (2023).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Chiffelle, J. et al. T-cell repertoire analysis and metrics of diversity and clonality. Curr. Opin. Biotechnol. 65, 284–295 (2020).
Corkum, C. P. et al. Immune cell subsets and their gene expression profiles from human PBMC isolated by vacutainer cell preparation tube (CPT) and standard density gradient. BMC Immunol. 16, 48 (2015).
Breiman, L. Random forests. Mach. Learn. 45, 5–32 (2001).
Charbonneau, M. R. et al. A microbial perspective of human developmental biology. Nature 535, 48–55 (2016).
Davie, K. et al. A single-cell transcriptome atlas of the aging Drosophila brain. Cell 174, 982–998.e20 (2018).
Wang, Z. et al. Single-cell RNA sequencing of peripheral blood mononuclear cells from acute Kawasaki disease patients. Nat. Commun. 12, 5444 (2021).
Zhang, J. Y. et al. Single-cell landscape of immunological responses in patients with COVID-19. Nat. Immunol. 21, 1107–1118 (2020).
Liu, Y. et al. Tumour heterogeneity and intercellular networks of nasopharyngeal carcinoma at single cell resolution. Nat. Commun. 12, 741 (2021).
Lehallier, B. et al. Undulating changes in human plasma proteome profiles across the lifespan. Nat. Med. 25, 1843–1850 (2019).
Acknowledgements
We thank the volunteers from the Shanghai Pudong Cohort for their contribution to the study. This work was supported by grants from the Noncommunicable Chronic Diseases—National Science and Technology Major Project (2023ZD0503204 to J.P.), Innovative Research Group Project of China’s National Natural Science Foundation (82421001 to J.P.), the National Natural Science Foundation of China (82230014 to J.P., 81930007 to J.P., 823B2005 to Y.C. and 32100426 to R.L.); National Science Fund for Distinguished Young Scholars (81625002 to J.P.); Shanghai Municipality Science and Technology Commission (22JC1402100 to J.P., 22DZ2292400 to J.P. and 21YF1445000 to R.L.); Shanghai Municipal Health Commission (2022JC013, 2023ZZ02021, 2022ZZ01008 and GWVI-11.1-26 to J.P.); Shanghai Municipal Education Commission (SHSMU-ZDCX20210700 to J.P.); and the Postdoctoral Fellowship Program of the China Postdoctoral Science Foundation (2023TQ0219, GZB20230434 and 2024M752015 to T.C.). We thank Z. Zhang, L.-S. Yeap, C. Huang and L. Guan Ng for their helpful discussions and Z. Yu, B. Kang and L. Zheng for their statistical and technical assistance.
Author information
Authors and Affiliations
Contributions
J.P. conceived the study. Y.W., R.T. and Z.L. recruited volunteers. Y.W., R.T., Z.L. and M.S. collected samples from donors. R.T., T.C. and M.S. completed the experiments. R.L., Y.W., R.T., L.L. and L. Wei performed the data analyses. R.T., R.L. and Y.W. drafted the initial version of the manuscript. Y.W., Y.Z., C.Z. and Y.C. proofed the manuscript. J.P., B.S., L. Wang, F.W., W.L., B.L., L.C., J.L., A.F.C., K.Q. and H.C. provided guidance and constructive comments for the final version. All the authors read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Immunology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editor: S. Houston, in collaboration with the Nature Immunology team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Basic characteristics of the integrated dataset and signature genes of 25 peripheral immune cell subsets covering 13 age groups.
a. UMAP plots showing the 25 peripheral immune cell subsets covering 13 age groups across the entire lifespan. Cell subsets are indicated in different colors. b. UMAP plots showing the distribution of expression of selected signature genes for 25 cell subsets. c. UMAP plots showing the distribution of expression of selected signature genes for 12 T cell subsets. d. Violin plots showing the distribution of expression of selected signature genes for 25 cell subsets. e. Violin plots showing the distribution of UMI counts (left panel) and gene counts (right panel) per cell type. B_BCR+GNLY+ cells are circled with dashed circle in (a) and (b).
Extended Data Fig. 2 Validation of cell annotation derived from scRNA-seq data by CyTOF, Azimuth, and GSEA.
a. UMAP of integrated CyTOF data of peripheral immune cell subsets identified within the PBMCs. b. Heatmap showing the relative mean cluster gene and protein expressions in major immune cell types from scRNA-seq (left panel) and CyTOF (right panel) analyses, respectively. c. Heatmap showing the correlation between scRNA-seq-derived major cell types and CyTOF-derived major cell types, computed from expression of scRNA-seq variable genes and corresponding CyTOF protein markers. d. UMAP of T cell subsets identified in the integrated CyTOF data. e. Heatmap showing the relative mean cluster gene and protein expressions in T cell subsets from scRNA-seq (left panel) and CyTOF (right panel) analyses, respectively. f. Heatmap showing the correlation between the scRNA-seq-derived T cell subsets and the CyTOF-derived T cell subsets, computed from expression of scRNA-seq variable genes and corresponding CyTOF protein markers. g. Heatmap showing strong consistency of our cell annotations based on scRNA-seq and cell annotations predicted by the automated annotation tool, Azimuth. h. GSEA plots showing immunological signature gene sets enriched by the marker genes of each T cell subset, illustrating that our categorization of T cells based on scRNA-seq aligned with established findings.
Extended Data Fig. 3 Cell composition dynamics of 25 peripheral immune cell subsets across 13 age groups.
a. Stacked bar graph showing the average proportion of each cell subset within total PBMCs based on scRNA-seq analyses across 13 age groups. b–d. Scatter plots showing the correlation between age and the proportion of each cell subset based on the scRNA-seq analyses in Fig. 1d. (b) Cell subsets significantly positively correlated with age, (c) cell subsets significantly negatively correlated with age, (d) cell subsets enriched in certain age groups. e–g. Scatter plots showing the correlation between age and the proportion of each cell subset based on the CyTOF analyses. (e) Cell subsets significantly positively correlated with age, (f) cell subsets significantly negatively correlated with age, (g) cell subsets enriched in certain age groups. In box plots b–g, median (center line), first and third quartiles (box), and extra 1.5× interguartile ranges (whiskers) are shown. In b, c, e, and f, Spearman correlation coefficient ρ and P values are indicated. A P value < 0.05 was considered statistically significant.
Extended Data Fig. 4 Functional enrichment analyses of M-DEGs in peripheral immune cell subsets.
a. Bar plot showing the numbers of total M-DEGs defined by scRNA-seq in each cell subset. b. Bar plot showing the numbers of upregulated (left panel) and downregulated (right panel) M-DEGs in each cell subset. c. Heatmap showing the functional enrichment analyses of upregulated M-DEGs in different peripheral immune cell subsets. d. Heatmap showing the functional enrichment analyses of downregulated M-DEGs in different peripheral immune cell subsets. e. Functional enrichment network of the common M-DEGs in Fig. 2c. f. Functional enrichment network of the common M-DEGs in Fig. 2d. g, h. The DEGs were classified into distinct clusters by Mfuzz based on the temporal trajectories of their expression. The DEG clusters whose expression peaked in children (144 genes, g) and the elderly (90 genes, h) are shown. i. Heatmap showing the functional enrichment analyses of the genes whose expression peaked in children in (g). j. Heatmap showing the functional enrichment analyses of the genes whose expression peaked in the elderly in (h). k. The positive correlation of LGALS1 expression with age and the negative correlation of CCR7 and TMIGD2 expression with age via bulk RNA-seq analyses (n = 34 samples). The blue line indicates linear regression. The gray shadow covers the 95% confidence interval. Spearman correlation coefficient ρ and P values are indicated. A P value < 0.05 was considered statistically significant. P values were determined by hypergeometric test in c–f,i and j. DEG, differentially-expressed genes; M-DEGs, monotonically differentially expressed genes.
Extended Data Fig. 5 Expression dynamics of differentially expressed immune checkpoints within immune cell subsets among the different life stages.
a. Heatmaps showing the row-scaled expression of immune checkpoint ligands in the indicated cell subsets across the eight life stages. b. Heatmaps showing the row-scaled expression of immune checkpoint receptors in the indicated cell subsets across the eight life stages.
Extended Data Fig. 6 Characteristics of T cell subsets across the entire lifespan.
a. Bar plot showing the percentage of TCR detection in each αβ T cell subset and in total αβ T cells. b. Bar plot showing the frequency of CDR3 amino acid length from TCR α-chains (top panel) and TCR β-chains (bottom panel) across eight life stages. Error bar shows the mean ± SEM. c. Box plots showing the clonal expansion levels of each αβ T cell subset quantified by STARTRAC-expa metric in αβ T cell subsets across 13 age groups. d. Box plots showing TCR repertoire diversity in CD4+ naive T cells (CD4_Naive_CCR7; left panel), CD8+ naive T cells (CD8_Naive_LEF1; middle panel) and MAIT cells (CD8_MAIT_SLC4A10; right panel) across eight life stages. The diversity of TCR repertoires was measured by Chao1 method. e. Box plots showing TCR repertoire diversity in CD8_MAIT_SLC4A10 cells across eight life stages. The diversity of TCR repertoires was measured by Inverse Simpson Index. f. Representative gene interaction network of upregulated genes in (Fig. 5k) using the STRING database (https://string-db.org/). Edge thickness represents the strength of data support from the STRING database. g. Box plots showing normalized protein expression of CD161 (encoded by KLRB1) in CD8_MAIT cells in children (1–6 years old), adolescents (12–18 years old), adults (30–60 years old), and the elderly (70–90 years old) based on the CyTOF analyses. In box plots c–e and g, median (center line), first and third quartiles (box), and extra 1.5× interquartile ranges (whiskers) are shown, and the black line indicates LOESS regression. CDR3, complementarity-determining region-3.
Extended Data Fig. 7 Functional difference between B_BCR+GNLY+ cells and other peripheral immune cells with cytotoxic functions.
a. Upset plots showing the number of shared upregulated DEGs in B_BCR+GNLY+ cells compared with that in seven cytotoxic T cell subsets including CD4_TEM_GNLY, CD8_TCM_HAVCR2, CD8_TEM_ZNF683, CD8_TEM_CMC1, CD8_TEM_GNLY, CD8_MAIT_SLC4A10, and γδ T cells. b. Upset plots showing the number of shared upregulated DEGs in B_BC +GNLY+ cells compared with that in three cytotoxic NK cell subsets including NK_CD56low, NK_CD56high, and NK_Proliferating cells. c. Dot plots showing the representative enriched biological processes of upregulated DEGs in B_BCR+GNLY+ cells compared with those in seven cytotoxic T cell subsets. d. Dot plots showing the representative enriched biological processes of upregulated DEGs in B_BCR+ GNLY+ cells compared with those in three cytotoxic NK cell subsets. e. Box plots showing the expression score of cytotoxicity in B_BCR+GNLY+ cells and seven cytotoxic T cell and three cytotoxic NK cell subsets. Box plots show median (center line), first and third quartiles (box), and extra 1.5× interquartile ranges (whiskers) and the black line indicates LOESS regression. The dashed line indicates the median score of cytotoxicity in B_BCR+GNLY+ cells. P values were determined by the two-sided unpaired Wilcoxon rank-sum test. A P value < 0.05 was considered statistically significant. In c and d, P values were determined by hypergeometric test.
Extended Data Fig. 8 Features of BCR across the human lifespan.
a. Bar plot showing the percentage of BCR detection in all peripheral immune cell subsets. b. UMAP plots showing selected shared BCR clones among different B cell subsets. c. Box plot showing the clonal expansion levels of total B cells (top left) and five B cell subsets across eight age stages quantified by STARTRAC-expa metric. d. Box plots showing the CDR3 amino acid length from BCR heavy chains (left panel) and BCR light chains (right panel) across eight life stages. After controlling for donor, Kruskal-Wallis test reveals no significant differences in CDR3 amino acid length across eight life stages (P > 0.05). In box plots c and d, median (center line), first and third quartiles (box), and extra 1.5× interquartile ranges (whiskers) are shown, and the black line indicates LOESS regression.
Extended Data Fig. 9 Transcriptional regulatory characteristics of cell differentiation in the B cell subsets.
a. Heatmap showing the area under the curve values of TF regulons in each B cell subset. Numbers between brackets represent the regulon counts for respective TFs. The value for each TF regulon is a row-scaled Z-score. b. Rank for TF regulons in each B cell subset by the regulon specificity score. c. Heatmap showing the row-scaled expression of the top 10 TFs in each B cell subset shown in (b). TF, transcription factor.
Extended Data Fig. 10 A graphic summary of the novel insights from this study.
We developed a comprehensive single-cell atlas of human peripheral immune cells across the lifespan from newborns to the elderly. Single-cell multi-omics analyses were performed to capture the diversity and dynamic trajectories of peripheral immune cells, revealing key age-specific features: a newly identified cytotoxic B-cell population enriched in children, expansion of mucosal-associated invariant T (MAIT) cells during adolescence, distinct aging patterns in naive CD4+ and CD8+ T cells, and clonal expansions of CD8+ T cells in both young and elderly individuals. Additionally, we constructed an immune age prediction model, which estimates immune age and identifies potential immune dysfunction. Created in BioRender. Huang, T. (2024) https://BioRender.com/a58r007.
Supplementary information
Supplementary Information
Supplementary Results, Discussions, Methods, Figs. 1–4, Table list and References.
Supplementary Table 1
Characteristics of healthy participants across the entire human lifespan.
Supplementary Table 2
Characteristics of the healthy and diseased validation cohorts for siAge prediction model.
Supplementary Table 3
Marker genes of 25 distinct PBMC subsets defined by scRNA-seq.
Supplementary Table 4
Antibody panels used for high-throughput CyTOF analyses in this study.
Supplementary Table 5
Monotonical differentially expressed genes associated with age in 25 cell subsets.
Supplementary Table 6
Functional enrichment analyses of monotonical differentially expressed genes associated with age.
Supplementary Table 7
Age-related genes in bulk RNA-seq data.
Supplementary Table 8
Differentially expressed ligands and receptors across the entire lifespan.
Supplementary Table 9
Dynamics of differentially expressed immune checkpoint expression in 25 cell subsets across the entire lifespan.
Supplementary Table 10
Signature genes of each B cell subset.
Supplementary Table 11
The information on PCR primers.
Supplementary Data 1
Statistical source data for Supplementary Fig. 1a–f.
Supplementary Data 2
Statistical source data for Supplementary Fig. 3b–g,i.
Supplementary Data 3
Statistical source data for Supplementary Fig. 4a–d.
Source data
Source Data Fig. 1
Statistical source data for Fig. 1c,d.
Source Data Fig. 2
Statistical source data for Fig. 2a–f.
Source Data Fig. 3
Statistical source data for Fig. 3a–k.
Source Data Fig. 4
Statistical source data for Fig. 4a–i.
Source Data Fig. 5
Statistical source data for Fig. 5a–l,n.
Source Data Fig. 6
Statistical source data for Figs. 6a,c–f.
Source Data Fig. 7
Statistical source data for Fig. 7b–i.
Source Data Extended Data Fig. 1
Statistical source data for Extended Data Fig. 1e.
Source Data Extended Data Fig. 2
Statistical source data for Extended Data Fig. 2b,c,e–g.
Source Data Extended Data Fig. 3
Statistical source data for Extended Data Fig. 3a–g.
Source Data Extended Data Fig. 4
Statistical source data for Extended Data Fig. 4a–d,h–k.
Source Data Extended Data Fig. 5
Statistical source data for Extended Data Fig. 5.
Source Data Extended Data Fig. 6
Statistical source data for Extended Data Fig. 6a,b,d–e,g.
Source Data Extended Data Fig. 7
Statistical source data for Extended Data Fig. 7a–e.
Source Data Extended Data Fig. 8
Statistical source data for Extended Data Fig. 8a,c,d.
Source Data Extended Data Fig. 9
Statistical source data for Extended Data Fig. 9a–c.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, Y., Li, R., Tong, R. et al. Integrating single-cell RNA and T cell/B cell receptor sequencing with mass cytometry reveals dynamic trajectories of human peripheral immune cells from birth to old age. Nat Immunol 26, 308–322 (2025). https://doi.org/10.1038/s41590-024-02059-6
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41590-024-02059-6
This article is cited by
-
Liquid biopsies in precision oncology for older adults with cancer
npj Precision Oncology (2026)
-
iPSCs derived sEVs ameliorate NK cell senescence by targeting CISH-STAT3
Stem Cell Research & Therapy (2025)
-
Developing a novel aging assessment model to uncover heterogeneity in organ aging and screening of aging-related drugs
Genome Medicine (2025)
-
Interaction and biological pathway analysis of proteomic products in patients with premature coronary artery disease
Clinical Proteomics (2025)
-
Immunosenescence in aging and neurodegenerative diseases: evidence, key hallmarks, and therapeutic implications
Translational Neurodegeneration (2025)
