
Abstract
Vitamin D3 is clinically used for the treatment of vitamin D3 deficiency or osteoporosis, partially because of its role in regulating phosphate (Pi) and calcium (Ca2+) homeostasis. The renal sodium-phosphate cotransporter 2a (Npt2a) plays an important role in Pi homeostasis; however, the role of vitamin D3 in hypophosphatemia has never been investigated. We administered vehicle or vitamin D3 to wild-type (WT) mice or hypophosphatemic Npt2a−/− mice. In contrast to WT mice, vitamin D3 treatment increased plasma Pi levels in Npt2a−/− mice, despite similar levels of reduced parathyroid hormone and increased fibroblast growth factor 23. Plasma Ca2+ was increased ~ twofold in both genotypes. Whereas WT mice were able to increase urinary Pi and Ca2+/creatinine ratios, in Npt2a−/− mice, Pi/creatinine was unchanged and Ca2+/creatinine drastically decreased, coinciding with the highest kidney Ca2+ content, highest plasma creatinine, and greatest amount of nephrocalcinosis. In Npt2a−/− mice, vitamin D3 treatment completely diminished Npt2c abundance, so that mice resembled Npt2a/c double knockout mice. Abundance of intestinal Npt2b and claudin-3 (tight junctions protein) were reduced in Npt2a−/− only, the latter might facilitate the increase in plasma Pi in Npt2a−/− mice. Npt2a might function as regulator between renal Ca2+ excretion and reabsorption in response to vitamin D3.
Similar content being viewed by others
Introduction
Active vitamin D3 or 1,25-dihydroxyvitamin D3 (1,25(OH)2D3) is produced via the combined actions of skin, liver and kidneys1. Under normal conditions, it plays an essential role in the regulation of calcium (Ca2+) and phosphate (Pi) homeostasis in the body2. A lack of vitamin D3 can lead to rickets and other potential processes beyond bone health, including immune system dysregulation, development of cancer, or progression of cardiovascular disease3. The actions of vitamin D3 are complex and involve hormones such as parathyroid hormone (PTH) and fibroblast growth factor 23 (FGF23)4,5,6. PTH and FGF23 are both phosphaturic hormones (via action on the renal Na+-Pi transporters, Npt2a and Npt2c) which work collaboratively to maintain Pi homeostasis; however, this process involves a complicated regulatory role of 1,25(OH)2D34,7. 1,25(OH)2D3 has opposing effects on these hormones: it enhances the production of FGF23 in bone, while simultaneously suppressing the synthesis of PTH8,9. In this complex feedback loop, where PTH and FGF23 normally promote renal Pi excretion, 1,25(OH)2D3 may act as a switch between Pi excretion/absorption in order to maintain total body Pi4. The precursor of vitamin D3, previtamin D3, is formed by the skin and subsequently undergoes spontaneous isomerization to vitamin D3, which has a half-life of ~ 26 h10. Any excess vitamin D3 is stored mainly within fat tissue11. In the liver, an initial hydroxylation step takes place which converts vitamin D3 into 25(OH) vitamin D3. After reaching the kidneys via the circulation, 25(OH) vitamin D3 undergoes a second hydroxylation process to form 1,25(OH)2D34,7. PTH is part of a feedback loop that facilitates the production of 1,25(OH)2D3 by inducing the transcription of renal 1α-hydroxylase, the enzyme responsible for this process4. Hormonal signaling of 1,25(OH)2D3 is mediated by activation of the vitamin D receptor (VDR)12 and many of its actions were described by studying VDR knockout (VDR−/−) mice13. When VDR−/− mice mature on a control diet, they develop rickets because of hypophosphatemia, hypocalcemia, elevated plasma 1,25(OH)2D3 and PTH levels14. This was found mainly to be the consequence of impaired intestinal Pi absorption rather than a renal Pi problem. Vice versa, administering 1,25(OH)2D3 to wild-type mice (WT) led to enhanced intestinal Pi absorption15, an effect absent in mice lacking the intestinal Na+-Pi transporter (Npt2b)15 implying a direct effect on transcellular Pi transport.
The importance of Npt2a for renal Pi reabsorption has been demonstrated in Npt2a knockout (Npt2a−/−) mice. These mice are characterized by renal Pi wasting consequently leading to hypophosphatemia, hypoparathyroidism, reduced FGF23 levels and hypercalcemia16,17,18. Of note, effects of Npt2a knockout on plasma 1,25(OH)2D3 have shown to be increased16,18 or unchanged4. This finding might be related to the age when mice are studied because 1,25(OH)2D3 levels decreased over time in Npt2a−/− mice between day 8 to day 3519.
Despite these known factors, the effects of vitamin D3 on Pi and Ca2+ regulation are incompletely understood, especially in the context of renal Npt2a and the involved complex regulatory pathways involving PTH and FGF23. In order to address this question, we treated mice lacking Npt2a (Npt2a−/−) with vitamin D3 and studied the impact on Pi and Ca2+ homeostasis. Our results demonstrate that vitamin D3 plays a distinct role in regulating Pi homeostasis, Ca2+ balance, and suggest the existence of novel regulatory pathways involving functional Npt2a for the regulation of Pi homeostasis.
Results
Lower plasma Pi and a greater increase of plasma Pi in response to vitamin D3 treatment in Npt2a−/− compared to WT mice
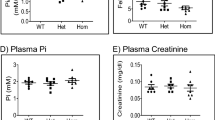
Consistent with previous reports16,20, plasma Pi in Npt2a−/− mice was significantly lower compared to WT mice (Fig. 1a,b). In WT mice, neither vehicle treatment nor vitamin D3 (300,000 IU/kg body weight) treatment affected plasma Pi levels (Fig. 1a). In contrast, plasma Pi significantly increased in Npt2a−/− mice (~ 1.6-fold), whereas vehicle treatment was without effect (Fig. 1b). Under baseline conditions, urinary Pi/creatinine ratios were not significantly different between genotypes (Fig. 1c,d). In WT mice, the urinary Pi/creatinine ratio remained unaltered in response to vehicle treatment but vitamin D3 treatment resulted in a significant increase (~ 1.7-fold) compared to baseline (Fig. 1c). No significant changes were observed in urinary Pi/creatinine ratios in response to vehicle or vitamin D3 treatment in Npt2a−/− mice (Fig. 1d).

Lack of Npt2a unravels a link of vitamin D3 on plasma Pi. Measurements of plasma and urinary Pi and Ca2+ were conducted in WT and Npt2a−/− mice after 4 days of treatment with either a vehicle or vitamin D3 (n = 6–10 per genotype). (a) In WT mice, plasma Pi levels remained unchanged following vitamin D3 treatment. (B) In contrast, lower plasma Pi levels under baseline conditions in Npt2a−/− mice significantly increased in response to vitamin D3 treatment. (c) The urinary Pi/creatinine ratio in WT mice increased significantly in response to vitamin D3 treatment. (d) This ratio in Npt2a−/− mice was unchanged (d). Plasma Ca2+ levels in both WT and Npt2a−/− mice showed a significant increase following vitamin D3 treatment (e & f). In WT mice, the urinary Ca2+ to creatinine ratio significantly increased after vitamin D3 treatment (g). In contrast, this ratio significantly decreased in Npt2a−/− mice (h). Male mice were used in these studies. In addition to single data summary data are shown and are expressed as mean ± SEM and were analyzed by repeated-measures two-way ANOVA followed by Tukey’s multiple comparisons test. *P < 0.05 vs WT same time point, #P < 0.05 vs baseline same genotype, §P < 0.05 vs vehicle same genotype and time point.
Npt2a−/− mice showed significantly higher plasma Ca2+ levels (~ 1.2-fold) compared to WT mice (Fig. 1e,f). Vehicle treatment did not affect plasma Ca2+ levels (Fig. 1e,f) in WT or Npt2a−/− mice. Vitamin D3 treatment significantly increased plasma Ca2+ in WT (~ 2.0-fold) and Npt2a−/− (~ 1.8-fold) mice. Of note, plasma Ca2+ was significantly greater in Npt2a−/− mice compared to WT mice in response to vitamin D3 treatment (Fig. 1e,f). Under baseline conditions, urinary Ca2+/creatinine ratios were significantly greater in Npt2a−/− compared to WT mice (Fig. 1g,h). In WT mice, the urinary Ca2+/creatinine ratio remained unaltered in response to vehicle treatment; however, consistent with increased plasma Ca2+ in response to vitamin D3 treatment, the urinary Ca2+/creatinine ratio was appropriately increased (~ 11-fold). In contrast to WT mice, the urinary Ca2+/creatinine ratio in Npt2a−/− mice was significantly decreased (~ 50%) in response to vitamin D3 treatment; vehicle treatment was without effect (Fig. 1h).
Using a smaller dose of vitamin D3 (3000 IU/kg body weight) showed no significant effects on plasma Pi or urinary Pi/creatinine ratios between genotypes (Supplementary Fig. 1a,b). Whereas plasma Ca2+ and urinary Ca2+/creatinine ratios were not affected by vitamin D3 in WT mice (Supplementary Fig. 1c,d), in Npt2a−/− mice, plasma Ca2+ and urinary Ca2+/creatinine ratios significantly increased (~ 1.05-fold and ~ 2.7-fold, respectively).
Npt2a−/− mice lack PTH responses but FGF23 levels were significantly increased in response to vitamin D3 treatment
Npt2a−/− mice show significantly lower plasma PTH levels under baseline conditions (Fig. 2a,b). In WT mice, plasma PTH showed a small but significant decrease in response to vehicle treatment (Fig. 2a). Vitamin D3 treatment significantly decreased (~ 85%) plasma PTH (Fig. 2a) in WT mice. No significant changes in plasma PTH were observed in response to vehicle or vitamin D3 treatment in Npt2a−/− mice (Fig. 2b). In addition to lower PTH levels in Npt2a−/− mice under baseline conditions, FGF23 levels were also significantly lower (~ 50%) compared to WT mice (Fig. 2c,d). Vehicle treatment did not significantly change FGF23 levels in either genotype (Fig. 2c,d). Vitamin D3 treatment caused a significant increase of FGF23 levels in both genotypes: in WT mice an ~ 80-fold increase was observed, whereas in Npt2a−/− mice a ~ 200-fold increase was observed. The more than double increase of FGF23 in Npt2a−/− compared to WT mice in response to vitamin D3 treatment is the consequence of the significantly lower baseline levels because FGF23 levels were not significantly different in response to vitamin D3 treatment between genotypes.

Vitamin D3 induces divergent responses in plasma PTH and increases FGF23 in both WT or Npt2a−/− mice. Measurements of plasma PTH and FGF23 were performed in WT and Npt2a−/− mice following 4 days of treatment with either vehicle or vitamin D3 (n = 6 per genotype). (a) In WT mice, vitamin D3 treatment led to a decrease in plasma PTH levels. (b) In Npt2a−/− mice, plasma PTH levels were lower and unchanged in response to vitamin D3 treatment. (c & d) FGF23 levels significantly increased in both genotypes in response to vitamin D3 treatment. Male mice were used in these studies. In addition to single data summary data are shown and are expressed as mean ± SEM and were analyzed by repeated-measures two-way ANOVA followed by Tukey’s multiple comparisons test. *P < 0.05 vs WT same time point, #P < 0.05 vs baseline same genotype, §P < 0.05 vs vehicle same genotype and time point.
Vitamin D3 has distinct effects on bone remodeling markers in WT and Npt2a−/− mice
Since vitamin D3 is instrumental for bone remodeling, we determined 2 bone formation markers (osteocalcin and PINP) and 2 bone resorption markers (TRAcP 5b and CTX-1) under baseline conditions and in response to vehicle or vitamin D3 treatment. Under baseline conditions, no significant differences were observed in osteocalcin levels between genotypes, and vehicle and vitamin D3 treatment resulted in a small but significant increase in osteocalcin levels independent of genotype (Fig. 3a,b). Plasma PINP levels were not significantly different between genotypes under baseline conditions, and vehicle treatment did not significantly change PINP levels in either genotype (Fig. 3c,d). Vitamin D3 treatment significantly reduced (~ 60%) PINP levels in WT mice, and a similar reduction (~ 44%) was observed in Npt2a−/− mice. Plasma TRAcP 5b levels were not significantly different between genotypes under baseline conditions, and in both genotypes TRAcP 5b levels slightly but significantly decreased in response to vehicle treatment. Vitamin D3 treatment significantly increased (~ 1.5-fold) TRAcP 5b levels in WT mice but was without significant effect in Npt2a−/− mice (Fig. 3e,f). Plasma CTX-1 levels were not significantly different between genotypes under baseline conditions, and vehicle treatment did not significantly affect CTX-1 levels in either genotype (Fig. 3g,h). Vitamin D3 treatment significantly increased (~ twofold) CTX-1 levels in WT mice and even further increased (~ 3.5-fold) CTX-1 in Npt2a−/− mice. Consequently, CTX-1 levels were significantly higher in Npt2a−/− compared to WT mice after vitamin D3 treatment (Fig. 3g,h).

Npt2a determines the effects of Vitamin D3 on bone remodeling markers. Circulating bone markers, including osteocalcin, PINP, TRAcP 5b, and CTX-1 were measured in WT and Npt2a−/− mice after 4 days of treatment with either a vehicle or vitamin D3 (n = 6 per genotype). (a & b) Both genotypes show a small but significant increase in osteocalcin levels independent of treatment. (c & d) Vitamin D3 decreased plasma PINP independent of genotype. (e & f) In both genotypes, vehicle treatment slightly but significantly decreased plasma TRAcP 5b levels but vitamin D3 only significantly increased TRAcP 5b in WT mice. (g & h) Vitamin D3 treatment increased CTX-1 levels in both genotypes but to a significantly greater extent in Npt2a−/− mice. Male mice were used in these studies. In addition to single data summary data are shown and are expressed as mean ± SEM and were analyzed by repeated-measures two-way ANOVA followed by Tukey’s multiple comparisons test. *P < 0.05 vs WT same time point, #P < 0.05 vs baseline same genotype, §P < 0.05 vs vehicle same genotype and time point.
Acute oral Pi loading results in greater plasma Pi levels in response to vitamin D3 treatment compared to vehicle
In vehicle-treated WT and Npt2a−/− mice, acute oral Pi loading resulted in a significant increase in plasma Pi levels (2.4 ± 0.1 and 3.1 ± 0.1 mmol L−1, respectively). In vitamin D3-treated WT and Npt2a−/− mice, acute oral Pi loading resulted in a significantly greater increase in plasma Pi levels (3.9 ± 0.2 and 4.2 ± 0.3 mmol L−1, respectively) compared to their respective vehicle-treated genotype (Fig. 4).

Similar effects of acute oral Pi loading on plasma Pi levels between genotypes. Plasma Pi levels were measured in WT and Npt2a−/− mice before and one hour after oral Pi loading via gavage (0.5 mol*L−1, 1% of body weight). These measurements were performed following 4 days of treatment with either a vehicle or vitamin D3 (n = 6 per genotype). (a & b) Oral Pi loading significantly increased plasma Pi levels independent of genotype or treatment; however, in vitamin D3-treated mice the increase was significantly greater compared to vehicle-treated mice. Male mice were used in these studies. In addition to single data summary data are shown and are expressed as mean ± SEM and were analyzed by repeated-measures two-way ANOVA followed by Tukey’s multiple comparisons test. *P < 0.05 vs WT same time point, #P < 0.05 vs baseline same genotype, §P < 0.05 vs vehicle same genotype and time point.
Vitamin D3 treatment causes significant Ca2+ accumulation in the kidney of Npt2a−/− mice
Determination of Ca2+ and Pi amounts were conducted on ashed tissue from vehicle and vitamin D3-treated WT and Npt2a−/− mice. Amounts of Pi or Ca2+ in bone were not significantly different between genotypes in response to vehicle or vitamin D3 treatment (Fig. 5A,B). Similarly, kidney Pi levels were not significantly different between genotypes in response to vehicle or vitamin D3 treatment (Fig. 5c). Of note, kidney Ca2+ levels in response to vehicle treatment were significantly greater (~ threefold) in Npt2a−/− compared to WT mice (Fig. 5d). Kidney Ca2+ levels were not significantly different between vehicle- and vitamin D3-treated WT mice. In contrast, vitamin D3 treatment in Npt2a−/− mice resulted in the highest kidney Ca2+ content observed between groups and genotypes and was ~ 3.5-fold greater compared to vitamin D3-treated WT mice.

Vitamin D3 treatment increases kidney Ca2+ levels in Npt2a−/− mice. Measurements of Pi and Ca2+ levels were carried out in bone and kidney tissues of WT and Npt2a−/− mice following 4 days of treatment with either a vehicle or vitamin D3 (n = 6 per genotype). (a & b) There was no difference in the levels of Pi and Ca2+ in bone of either genotype in response to vehicle or vitamin D3 treatment. (c) Kidney Pi levels were not significantly different between genotypes or treatment. (d) The kidney Ca2+ levels were significantly greater in response to vehicle treatment in Npt2a−/− compared to WT mice. Vitamin D3 treatment was not associated with altered kidney Ca2+ levels in WT mice; however, resulted in the highest kidney Ca2+ levels in Npt2a−/− mice. Male mice were used in these studies. In addition to single data summary data are shown and are expressed as mean ± SEM and were analyzed by two-way ANOVA followed by Tukey’s multiple comparisons test. *P < 0.05 vs WT same treatment, §P < 0.05 vs vehicle same genotype.
Vitamin D3 treatment causes signs of impaired renal function associated with severe renal Ca2+-Pi deposits in Npt2a−/− mice
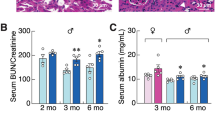
Plasma creatinine shows the highest levels in vitamin D3-treated Npt2a−/− mice, no differences were observed between the other groups (Fig. 6a). Urinary albumin/creatinine ratios also showed significantly increased levels in vitamin D3-treated Npt2a−/− mice compared to vehicle-treated Npt2a−/− mice (Fig. 6b). To visualize Ca2+-Pi deposits, we used von Kossa staining and performed semi-quantitative analysis (Fig. 6c–g). Consistent with plasma creatinine levels, vitamin D3-treated Npt2a−/− mice show the most severe amount (> 50%) of crystal deposits in the tubular lumen. The tubules show evidence of damage with attenuation of the epithelial lining. In contrast to vitamin D3-treated Npt2a−/− mice, the majority of vitamin D3-treated WT mice show only mild (< 10%) crystal deposits similar to vehicle-treated Npt2a−/− mice.

Vitamin D3-treated Npt2a−/− mice show signs of impaired kidney function and greater renal Ca2+-Pi deposits. (a) Plasma creatinine levels were the highest in vitamin D3-treated Npt2a−/− mice. (b) Similarly, urinary albumin/creatinine ratios showed the biggest increase compared to baseline in vitamin D3-treated Npt2a−/− mice. (c) Histological classification of mineral deposits in genotypes with vehicle or vitamin D3 treatment. The majority of vehicle-treated WT mice show no mineral deposits, vehicle-treated Npt2a−/− mice showed mild deposits (< 10%), vitamin D3-treated WT showed greater severity (moderate 10–50%) and only some vitamin D3-treated Npt2a−/− mice showed the greatest number of deposits (> 50%). Representative examples of H&E and von Kossa staining are shown for each condition from mice with no mineral deposits (d), mild deposits (e), moderate deposits (f) and severe deposits (g). Von Kossa staining shows that Ca2+-Pi crystal deposits (black stains) localize within the tubular lumen. The tubules show evidence of damage with attenuation of the epithelial lining. Magnification × 200. Scale bar of 100 µm is shown in each image. Male and female mice were used in these studies. In addition to single data summary data are shown and are expressed as mean ± SEM and were analyzed by two-way ANOVA followed by the two-stage linear step-up procedure of Benjamini, Krieger, and Yekutieli. *P < 0.05 vs WT same treatment, §P < 0.05 vs vehicle same genotype.
Renal mRNA expression
RT-qPCR profiling of genes (all shown in Supplementary Fig. 2) expressed in the kidney confirmed that primers for Slc34a1 (Npt2a) were specific for Npt2a since no mRNA amplification was found in Npt2a−/− mice. Vehicle treatment showed significantly lower CaSR (Ca2+-sensing receptor) and Cldn19 (claudin-19) expression in Npt2a−/− compared with WT mice. In vitamin D3-treated WT mice, significant differences in mRNA expression were found compared to vehicle treatment for Slc34a1, Slc34a3 (Npt2c), Slc8a1 (Na+/Ca2+ exchanger, NCX1), Atp2b4 (ATPase plasma membrane Ca2+ transporting 4), and CaSR. In vitamin D3-treated Npt2a−/− mice, significant differences in mRNA expression were observed compared to vehicle treatment for Slc34a3, Atp2b4, Trpv5 (transient receptor potential cation channel subfamily V member 5), CaSR, Cyp27b1 (25-hydroxyvitamin D-1α-hydroxylase) and Cldn16 (claudin-16). Significant differences between genotypes in response to vitamin D3 treatment were found for CaSR, Cyp27b1, and Cldn16.
Npt2a maintains Npt2c and claudin-3 expression in response to vitamin D3
Confirming the specificity of the Npt2a antibody, no Npt2a band was observed in kidney tissue of Npt2a−/− mice (Fig. 7a). In response to vitamin D3 treatment in WT mice, Npt2a abundance was significantly lower (~ 65%) compared to vehicle-treated mice (Fig. 7a). Consistent with a compensatory response of Npt2c abundance in Npt2a−/− mice, Npt2c abundance was ~ 3.5-fold greater in vehicle-treated Npt2a−/− mice compared to vehicle-treated WT mice (Fig. 7b). In response to vitamin D3 treatment in WT mice, Npt2c abundance was significantly lower (~ 40%) compared to vehicle-treated WT mice. Of note, vitamin D3 treatment completely diminished Npt2c abundance in Npt2a−/− mice. Since vitamin D3 affects intestinal Pi transport, we further analyzed intestinal abundance of Npt2b and claudin-3, the latter being a paracellular tight junction protein involved in Pi transport21. The majority of intestinal Pi uptake in the mouse occurs in the distal small intestine22,23. The abundance of Npt2b in the proximal small intestine was not significantly different between genotypes or treatments (Fig. 8a). No differences were observed in the abundance of Npt2b in the distal small intestine between genotypes in response to vehicle treatment. In WT mice (Fig. 8b), Npt2b abundance was not significantly different in response to vitamin D3 compared to vehicle treatment. In contrast, Npt2b expression in the distal small intestine of Npt2a−/− mice was significantly lower (~ 77%) in response to vitamin D3 compared to vehicle treatment (Fig. 8b).

Npt2c abundance is diminished in Npt2a−/− mice in response to vitamin D3 treatment. Abundance of Npt2a and Npt2c in kidney tissues of WT and Npt2a−/− mice after 4 days of treatment with vehicle or vitamin D3 (n = 4–6 per genotype). (a) In this study we confirmed the specificity of the Npt2a antibody in Npt2a−/− mice, which lack the ~ 75–80 kDa band representing Npt2a. An unspecific band was detected. In WT mice, vitamin D3 treatment showed lower Npt2a expression compared to vehicle treatment. (b) In response to vehicle treatment, Npt2c abundance was significantly greater in Npt2a−/− compared to WT mice. Npt2c abundance was significantly lower in vitamin D3-treated mice; however, the level in Npt2a−/− mice was almost undetectable. Male mice were used in these studies. In addition to single data summary data are shown and are expressed as mean ± SEM and were analyzed by two-way ANOVA followed by Tukey’s multiple comparisons test. *P < 0.05 vs WT same treatment, §P < 0.05 vs vehicle same genotype.

Npt2b abundance is diminished in Npt2a−/− mice in response to vitamin D3 treatment. (a) Abundance of Npt2b in the proximal small intestine was not different between genotypes or treatment. (b) In the distal small intestine, no differences were detected in Npt2b abundance between vehicle-treated genotypes. In WT mice, Npt2b abundance was similar in response to vitamin D3 compared to vehicle treatment. Of note, Npt2b abundance in vitamin D3 -treated Npt2a−/− mice was lower compared to vehicle treatment. (c) Abundance of claudin-3 was somewhat variable in the proximal small intestine and no differences were observed between genotype or treatment. (d) In the distal small intestine, no differences in claudin-3 abundance were detected between vehicle-treated genotypes. In WT mice, claudin-3 abundance was similar in response to vitamin D3 compared to vehicle treatment. Of note, claudin-3 abundance in vitamin D3-treated Npt2a−/− mice was lower compared to vehicle treatment. Male mice were used in these studies. In addition to single data summary data are shown and are expressed as mean ± SEM and were analyzed by two-way ANOVA followed by Tukey’s multiple comparisons test. §P < 0.05 vs vehicle same genotype.
In the proximal small intestine of WT mice, claudin-3 protein abundance was not significantly different between vehicle or treatment groups (Fig. 8c). In the distal small intestine, no significant differences were observed in claudin-3 abundance between genotypes in response to vehicle treatment (Fig. 8d) and in WT mice no significant differences were observed between vehicle and vitamin D3 treatment. In contrast, in Npt2a−/− mice, claudin-3 protein abundance was significantly lower (~ 72%) in response to vitamin D3 compared to vehicle treatment (Fig. 8d).
Discussion
The role of Npt2a in regulating renal Pi transport has been extensively studied. However, there are significant knowledge gaps when it comes to the complex hormonal regulation of this transporter, in particular the role of vitamin D3. To gain further mechanistic insight, we studied Pi and Ca2+ homeostasis when the body is challenged by exogenous administration of a high dose of vitamin D3 in the absence and presence of hyposphosphatemia, the latter caused by lack of Npt2a. Surprisingly, despite lack of Npt2a which should have facilitated renal Pi excretion, these mice show signs of impaired Pi excretion, possibly a results of greater nephrocalcinosis and a reduction of kidney function, in response to vitamin D3 administration (for a summary see Fig. 9).

Summary figure. In vitamin D3-treated Npt2a−/− mice, a circulus vitiosus is observed leading to kidney failure. We hypothesize that vitamin D3 treatment leads to elevated plasma Ca2+ levels (possibly via increased bone resorption as indicated by greater CTX-1 levels and/or increased intestinal Ca2+ absorption) and decreased intestinal Pi absorption (via lower Npt2b and claudin-3 levels). A combination of vitamin D3, elevated plasma Pi, and reduced kidney function causes FGF23 levels to be drastically elevated, subsequently diminishing Npt2c abundance and leading to supersaturation of tubular fluid with Ca2+ and Pi. Formation of Ca2+/Pi crystals lead to renal calcification and reduced kidney function (increase in plasma creatinine and urinary albumin). Consequently, plasma Ca2+ and Pi levels are further increased. Green arrows indicate an increase, red arrows indicate a decrease. The table on the right summarizes the most significant findings observed between vitamin D3 treated mice and vehicle treated mice for both genotypes.
Lack of Npt2a causes hypophosphatemia16,18,20, a finding confirmed in the current study. A similar situation can be induced by administration of a Npt2a inhibitor20,24,25. In WT mice, the administration of vitamin D3 did not affect plasma Pi levels; in contrast, Npt2a−/− mice had a very uniform increase in plasma Pi following administration of vitamin D3. Regarding the former, other studies which administered vitamin D3 at a dose of 400,000 (IU kg−1) to C57Bl/6 mice did not report changes in plasma Pi levels26. It is conceivable that in WT mice, the increase in urinary Pi/creatinine ratio served to stabilize plasma Pi levels. Consistent with this, vitamin D3 treatment resulted in lower Npt2a and Npt2c expression in WT mice.
Under baseline conditions, we did not find a clear Pi wasting phenotype in Npt2a−/− mice. This could be related to a substantial compensatory greater Npt2c expression (seen on the protein but not mRNA level) which might mitigate the Pi wasting phenotype. It is notable that plasma Pi significantly increased in response to vitamin D3 treatment despite the absence of Npt2a. This corroborated with greater urinary albumin/creatinine ratios and greater plasma creatinine levels, possibly implying that reduced kidney function might have contributed to this finding. In addition, Npt2c abundance was also diminished in Npt2a−/− mice under these conditions, both of which should facilitate urinary Pi excretion and prevent a rise in plasma Pi. In terms of renal Npt2 transporter expression in response to vitamin D3, Npt2a−/− mice resemble Npt2a/c double knockout mice. What could explain the lack of increase in urinary Pi/creatinine ratios in response to vitamin D3 in Npt2a−/− mice? One possible explanation could be that urinary Pi excretion has reached a maximum. Consistent with this, we have previously shown in short-term metabolic cage experiments that urinary Pi/creatinine ratios in Npt2a−/− mice were of the same magnitude, and only acute Npt2a inhibition in control mice was able to double urinary Pi/creatinine20, suggesting that in Npt2a−/− mice (chronically), despite the presence of compensatory mechanisms, no further increase in urinary Pi excretion can be achieved.
In terms of Ca2+, our data are consistent with the previously published Npt2a−/− phenotype as well as the role of vitamin D3 in Ca2+ homeostasis16,18. Npt2a−/− mice have higher plasma Ca2+ levels and greater urinary Ca2+/creatinine ratios. Vitamin D3 is a well-known regulator of intestinal Ca2+ absorption27 and knockout of Npt2a is associated with significantly increased intestinal Ca2+ absorption, possibly as a consequence of greater intestinal mRNA expression of epithelial Ca2+ channels (ECaC1 and ECaC2) and the Ca2+ binding protein calbindin-D9k28. Vitamin D3 treatment increased plasma Ca2+ in both genotypes, but to a greater amount in Npt2a−/− mice. Of note, one of the most interesting findings in this study relates to the response of the kidney after administration of vitamin D3. In WT mice, urinary Ca2+/creatinine was appropriately increased possibly as a consequence of significant hypercalcemia. In contrast to WT mice, in Npt2a−/− mice urinary Ca2+/creatinine was reduced in response to vitamin D3 administration, reaching levels seen in WT mice under baseline conditions. Consequently, the reduction of urinary Ca2+/creatinine ratio could have contributed to the greater increase in plasma Ca2+ in Npt2a−/− mice. Despite vitamin D response elements being present in the CaSR gene causing up-regulation of CaSR expression29, our study provides evidence that vitamin D3 treatment can reduce CaSR expression.
Ultrastructural studies in Npt2a−/− mice showed that at early age Ca2+/Pi deposits develop that were purged during later stages of life30,31. Along those lines, our tissue analysis showed that vitamin D3-treated Npt2a−/− mice had the highest kidney Ca2+ content of all studied groups, without significant differences in kidney Pi content. Of note, Npt2a mutations in humans seem fairly common in a large cohort of Ca2+-stone forming pedigrees, but they do not seem to corroborate with clinically significant Pi or Ca2+ handling abnormalities32. Our studies expand this knowledge and show that vitamin D3-treated Npt2a−/− mice show the greatest amount of Ca2+-Pi crystal deposits in the tubule lumen. Of note, vitamin D3-treated WT mice show a similar pattern of Ca2+-Pi crystal deposits compared to vehicle-treated Npt2a−/− mice. Taken together, Npt2a−/− mice have a significant problem in excreting Ca2+ in their urine and, considering also the lack of Npt2c in response to vitamin D3 administration, their kidney Ca2+ content is further consistent with the phenotype of Npt2a/c double knockout mice, which show severe renal calcifications18.
Possibly because of a combination of hypophosphatemia and hypercalcemia in Npt2a−/− mice, PTH and FGF23 levels are significantly lower compared to WT mice16,17,18 which is still present when a high Pi or high Pi/Ca2+ diet is provided17. Under baseline conditions, these findings were confirmed in our study. PTH synthesis and release under these conditions seems to be under a dual control: (1) hypercalcemia inhibits the synthesis and secretion of PTH from the parathyroid gland via activation of the CaSR and (2) active vitamin D3 suppresses the synthesis and release of PTH via activation of the VDR33. Of note, the changes observed cannot explain the paradoxical response of urinary Ca2+/creatinine in Npt2a−/− mice in response to vitamin D3. The situation in vitamin D3-treated Npt2a−/− mice is similar to hereditary hypophosphatemic rickets with hypercalciuria34 a physiology also resembled in Npt2a/c double knockout mice18. One notable difference is the accumulation of Ca2+ in the kidney of vitamin D3-treated Npt2a−/− mice rather than the development of a hypercalciuric response. Along those lines, PTH was already substantially reduced in Npt2a−/− mice under baseline conditions possibly in the face of lower plasma Pi and elevated plasma Ca2+ levels. The decrease of PTH in WT mice in response to vehicle treatment possibly relates to the presence of ethanol which has been shown to decrease PTH levels35,36.
Consistent with previous reports, our study confirms the lower FGF23 levels in Npt2a−/− mice, a possible consequence of lower plasma Pi levels18. Our study did not determine 1,25(OH)2D3 levels but levels were found to be significantly greater in Npt2a−/− compared with WT mice16,18. Our data show that vitamin D3 treatment does not affect Cyp24a1 and Cyp27b1 mRNA expression in WT mice. In chronic kidney disease, expression of Cyp24a1 is increased possibly accounting for decreased 1,25(OH)2D3 due to degradation37. Vehicle treatment in Npt2a−/− mice showed significantly lower expression compared to WT mice, possibly explaining the body’s effort to increase 1,25(OH)2D3 levels. Only in vitamin D3-treated Npt2a−/− mice was Cyp27b1 mRNA expression significantly increased, which is consistent with greater 1,25(OH)2D3 production. Study participants treated with vitamin D3 normally do not show increases in 1,25(OH)2D3 levels38, which is reflected in unchanged Cyp27b1 levels in vitamin D3-treated WT mice compared to vehicle treatment. However, in the absence of Npt2a this can be offset, and our data imply that Cyp27b1 mRNA expression is paradoxically increased.
When VDR are knocked out in chondrocytes of mice, FGF23 expression in osteoblasts and consequently FGF23 plasma levels are significantly reduced, implying that VDR is a prerequisite in this signaling pathway39. Of note, exogenous administration of vitamin D3 is a powerful stimulator of FGF23, leading to ~ 80–200-fold increase; however, the exact signaling pathway(s) causing this increase remain unclear and the relationship between these hormones is complex. Our results dispute the role of PTH being a major determining factor for FGF23 production (which was drastically suppressed in both genotypes), or suggest additional regulatory mechanisms, which has been demonstrated in vivo and in vitro as well as in mice with hyperparathyroidism40,41. Vice versa, our data are consistent with the notion that FGF23 reduces PTH synthesis directly42.
Npt2a−/− mice show a skeletal phenotype characterized by delayed secondary ossifications at 21 days of age which are reversed at 45 days of age and are ultimately overcompensated at > 74 days of age16, these effects are even more exaggerated in Npt2a/c double knockout mice18. Our study used highly sensitive bone remodeling markers as estimators, which, to our knowledge, have never been determined in Npt2a−/− mice. Despite significant differences in Pi and Ca2+ homeostasis between genotypes, our study did not identify changes in any of the bone remodeling markers studied under baseline conditions. This might relate to the fact that Npt2a−/− mice were of adult age when our studies were performed. Osteocalcin is predominantly produced and secreted by osteoblasts during bone formation43. Although low doses of vitamin D3 can stimulate bone turnover, high doses can cause bone resorption44. Despite these results, our study did not provide any differences between treatment or genotype in terms of osteocalcin levels. A possible explanation could be the short-term experimental setup we employed. PINP is considered the most sensitive marker of bone formation45, which has been reported to be under the control of PTH46 and shows an inverse relationship with active vitamin D347. Consistent with this, both genotypes decreased PINP levels after vitamin D3 administration, consistent with a role of reduced bone formation. Of note, this occurred despite a significant decrease of PTH in both genotypes.
TRAcP 5b is an osteoclast-derived marker of bone resorption.48. Our findings show that vitamin D3 treatment increased TRAcP 5b in WT mice. So far, no correlations have been described between FGF23 and TRAcP 5b under normal conditions; however, in patients on evocalcet treatment (CaSR agonist) PTH, FGF23 and TRAcP 5b decreased over the 30 week treatment period49. Our data point toward a role of Npt2a in this process since Npt2a−/− mice lack a response in TRAcP 5b in response to vitamin D3. CTX-1 is a marker for bone remodeling that is released when type 1 collagen is degraded50. The role of vitamin D3 on CTX-1 is ambiguous, with several human studies showing no effect of vitamin D3 supplementation on CTX-1 levels51,52 whereas others show a positive correlation47. This might relate to the pre-existing conditions that were studied, e.g. presence or absence of vitamin D3 deficiency. Of note, and consistent with our study, a study in humans showed a dose-dependent effect of vitamin D3 bolus administration (up to 600,000 IU) on CTX-1 levels 1 day after administration53. This might explain an increase in fracture risk when elderly women are treated annually with a single high dose (500,000 IU) of vitamin D354. In addition, daily doses of 10,000 IU for 3 years also resulted in a significant increase of CTX-1 in healthy adults44. Lack of Npt2a possibly unravels that these mice are more susceptible for disturbed bone remodeling.
The intestine plays a vital role in Pi and Ca2+ absorption in order to regulate homeostasis in the body and vitamin D3 has been implicated in this regulation14,28. Our acute Pi loading experiments confirm these findings: independent of genotype, the intestinal uptake of Pi was significantly greater in vitamin D3-treated mice compared to vehicle-treated mice as evidenced by greater increases in plasma Pi levels in the face of reduced renal Npt2a/c abundance. Our studies on Npt2b abundance also expand the knowledge on spatial regulation, where abundance in response to Npt2b was unaffected in the proximal small intestine, which contrasts with the distal small intestine. Of note, the contribution of transcellular versus paracellular intestinal Pi transport is a highly debated topic15,23, and claudin-3 has been implicated in the paracellular process. Supporting this hypothesis are data from claudin-3 knockout mice, which have enhanced intestinal Pi uptake21. Similar to Npt2b, no regulation of claudin-3 abundance in the proximal small intestine was found in our studies, but in the distal small intestine of Npt2a−/− mice, claudin-3 abundance was significantly reduced, possibly contributing to greater plasma Pi levels in Npt2a−/− mice. In the kidney we find evidence that claudin-16, expressed in the thick ascending limb and distal convoluted tubule, was significantly reduced in vitamin D3-treated Npt2a−/− mice. Claudin-16 inactivating mutations in humans are associated with hypercalciuria and nephrocalcinosis55,56, possibly suggesting that significantly reduced claudin-16 expression in our studies might have contributed to the phenotype of vitamin D3-treated Npt2a−/− mice. Along those lines, claudin-16 interacts with Trpv5 since knockdown of claudin-16 delocalized Trpv5 from the luminal membrane57. In our study, Trpv5 was also significantly reduced in vitamin D3-treated Npt2a−/− mice compared to vehicle-treated Npt2a−/− mice. Our data provide information on the regulation of Atp2b4, which was reduced in response to vitamin D3 in both genotypes; however, knockout of Atp2b4 in mice did not cause a Ca2+ phenotype58.
In summary, our data provide novel insight into the role of vitamin D3 in the regulation of Pi and Ca2+ homeostasis in the context of Npt2a. One limitation of using mice to study Pi homeostasis relates to distinct differences in intestinal and renal Pi handling compared to humans. Despite the vitamin D3 dose used in our studies is supraphysiological, significant differences were observed between genotypes that pinpoint to an important role of Npt2a (and possibly claudin-16) in renal calcification and consequently kidney function decline. It is noteworthy that vitamin D3 treatment in Npt2a−/− mice resulted in a complete loss of Npt2c, and mice in terms of renal Pi transporter expression resembled Npt2a/c double knockout mice. Despite a complete lack of renal Pi transporters, Npt2a−/− mice experience greater plasma Pi levels, possibly a consequence of reduced intestinal claudin-3 abundance. Further, Npt2a−/− mice develop significantly greater plasma Ca2+ levels in response to vitamin D3, possibly a consequence of impaired renal Ca2+ excretion with tissue accumulation of Ca2+, implying that Npt2a can function as a switch between renal Ca2+ excretion and reabsorption. However, the contribution of Npt2c in this process cannot be excluded considering its absence in abundance in response to vitamin D3 treatment in Npt2a−/− mice.
Methods
The animal experiments were conducted in compliance with the NIH Guide for Care and Use of Laboratory Animals, set by the National Institutes of Health (Bethesda, MD), received approval from the Institutional Animal Care and Use Committee (11201R) at the University of South Florida, and are reported in accordance with ARRIVE guidelines. Npt2a−/− mice were obtained from the Jackson Laboratory (strain# 004802, Bar Harbor, ME) and propagated by heterozygote breeding. Mice have been backcrossed to C57BL/6J for 9 generations. Only male WT and Npt2a−/− mice, 3–5 months old, were used for the study. The specific pathogen free mice were group housed and kept in a controlled environment with a 12-h light–dark cycle (light off at 18:00) in isolated ventilated cages. They were provided with free access to standard rodent diet (TD.2018, containing 0.7% Pi and 1% Ca2+, Envigo, Madison, WI) and drinking water. Genotype was determined by PCR amplification of genomic DNA, which was extracted from ear tissue samples. The genotyping was carried out in accordance with protocol # 29530 published on the Jackson Laboratory website.
Vitamin D3 treatment
Wild-type and Npt2a−/− mice were randomized into two treatment groups: one vehicle (5% Ethanol, 5% Cremophor EL, and 90% water) or vitamin D3 (3,000 and 300,000 IU/kg body weight, Alfa Aesar, Haverhill, MA) dissolved in vehicle26. Treatments were administered on 4 consecutive days via subcutaneous injections (2 µL g−1 body weight) by and investigator blinded to genotype and treatment. Blood samples were collected under brief isoflurane anesthesia from the retrobulbar plexus before and after the 4-day treatment period. Spontaneously voided urine was collected at the same time.
Analysis of plasma and urine samples
Clinical chemistry was performed utilizing commercially available assays, adapted for use with small sample volumes20,23. Concentrations of Pi and Ca2+ in both plasma and urine were measured using inorganic phosphorous reagent and calcium arsenazo III reagent respectively, (Pointe Scientific, Canton, MI)59. Urinary creatinine was measured by infinity creatinine liquid stable reagent (Thermo Fisher Scientific, Middletown, VA). Urinary albumin and plasma creatinine were determined as described previously60,61. PTH and intact FGF23 were measured according to the manufacturer instructions (Quidel, San Diego, CA). Markers for bone resorption (tartrate-resistant acid phosphatase isoform 5b [TRAcP 5b, Quidel] and type I collagen cross-linked C-telopeptide [CTX-1, Immunodiagnostic Systems]) and bone formation (procollagen type I N-propeptide [PINP, Immunodiagnostic Systems, Gaithersburg, MD] and osteocalcin [Quidel]) were measured using ELISAs.
Acute hyperphosphatemic model
Four days after the administration of either vehicle or vitamin D3, WT and Npt2a−/− mice were subjected to gavage of 0.5 mol L−1 NaH2PO4, 1% of body weight by an investigator blinded to genotype23,62. Before gavage and 60 min after administration, blood samples were collected under brief isoflurane anesthesia. Plasma Pi was measured as described above.
Determination of Ca2+ and Pi content in the kidney
In another set of WT and Npt2a−/− mice, femurs and kidneys were harvested under terminal isoflurane anesthesia 4 days after the last administration of vehicle or vitamin D3. The collected tissues were dried for 24 h at 50 °C. Following the drying process, the weight of each tissue was determined. Next, the tissues were incinerated at a temperature of 560 °C for 12 h in a muffle furnace (Thermolyne F48015-60, Thermo Fisher Scientific). The ashes from the incineration were dissolved in 0.75 mol L−1 HCl. Concentrations of Ca2+ and Pi in the dissolved samples were determined as described above.
Histological analysis of kidneys
In a separate cohort of mice kidneys were perfused in vivo through the left ventricle with 4% PFA in phosphate buffered saline under isoflurane anesthesia. After kidneys were removed, they were fixed overnight in the same solution and subsequently paraffin embedded and sectioned at 4–6 μm. After deparaffinization and rehydration, sections were stained with hematoxylin and eosin (H&E) and von Kossa (to determine mineral deposits). Sectioning and staining were performed by Reliance Pathology Partners, LLC (Tampa, FL). Quantification of Ca2+-Pi deposits were performed using the following scheme: none, mild (< 10%), moderate (10–50%), or severe (> 50%). The highest score seen in sections was reported for each mouse. All scoring was performed by a pathologist (M.T.) blinded to sample identity.
Isolation of intestinal epithelial cells
Another cohort of mice was randomized to administration of either vehicle or vitamin D3 as described above. Following the 4-day treatment, mice were anesthetized by isoflurane and their kidneys and small intestines removed. Isolation of intestinal epithelial cells (IEC) via Ca2+ chelation was performed as described previously23,63,64. The IEC pellets were prepared for immunoblotting as described below.
Western blotting
Collected IEC and kidneys were homogenized in a buffer composed of 250 mmol L−1 sucrose and 10 mmol L−1 triethanolamine (Sigma-Aldrich, St. Louis, MO) containing Halt protease inhibitor cocktail and Halt phosphatase inhibitor cocktail (both Thermo Fisher Scientific). The homogenate was then subjected to a centrifugation process at 1000×g for 15 min followed by generation of plasma membrane-enriched samples (by centrifugation of the supernatant at 17,000×g) for 30 min. The pellets that emerged from this process were then resuspended and prepared for Western blotting. Protein quantity was determined using a bicinchoninic acid assay (Thermo Fisher Scientific). Samples of equal concentration were made by the addition of Laemmli sample buffer (final concentration of 0.1 mol L−1 SDS and 15 mg L−1 DTT). Samples were heated at 65 0C for 15 min before immunoblotting. The samples were resolved on either NuPAGE 4–12% or 12% Bis–Tris gels in MOPS. Proteins were transferred to polyvinylidene difluoride membranes and immunoblotted with rabbit polyclonal antibodies against Npt2a, Npt2b, Npt2c (each with a dilution of 1:1500, generous gift from M. Levi)23,24,62, rabbit anti claudin-3 (dilution 1:1000, also rabbit-sourced, Thermo Fisher Scientific)23, and mouse anti β-actin (dilution 1:30,000, Sigma-Aldrich). These targets were then detected with secondary antibodies designed for rabbit (IRDye® 800CW donkey anti-rabbit IgG, at a dilution of 1:5000) or mouse (IRDye® 680RD donkey anti-mouse IgG, also at a dilution of 1:5000), using an Odyssey® CLx detection system (LI-COR Biosciences, Lincoln, NE). Quantification of the band intensities was carried out using Image Studio Lite for densitometric analysis (LI-COR Biosciences).
Quantitative polymerase chain reaction from kidney and bone
Total RNA from kidney homogenates was extracted using Tri Reagent (Sigma-Aldrich) using a protocol adapted from the manufacturer’s recommendations. Total RNA was quantified using a Synergy Neo2 plate reader (Agilent, Santa Clara, CA). One thousand ng RNA of kidney sample were used to produce cDNA using a Revert Aid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific). Maxima SYBR Green/ROX qPCR Master Mix (Thermo Fisher Scientific) was used in conjunction with a QuantStudio 6 Pro (Applied Biosystems, Thermo Fisher Scientific) for amplification. Template concentration was 1 ng µl−1 cDNA per 10 µl reaction (performed in triplicate) and used in conjunction with primer pairs specific for Slc8a1, Slc34a1, Slc34a3, Trpv5, Atp2b4, Cyp25a1, Cyp27b1, Cldn2, Cldn14, Cldn16, Cldn19, and CaSR with actin used as a reference gene (all primer sequences are provided in the Supplementary Information). Data analysis used the ΔΔCt method, i.e. cycle thresholds (Ct), were normalized to actin expression, and compared with control.
Statistical analyses
Data are expressed as mean ± S.E.M. Two-way ANOVA or repeated-measures two-way ANOVA followed by Tukey’s multiple comparison tests, or two-way mixed-effects ANOVA followed by the two-stage linear step-up procedure of Benjamini, Krieger, and Yekutieli, as indicated in the figure legends, were used to test for significant differences between genotype and/or treatment. All data were analyzed via GraphPad Prism (Version 10.1, Boston, MA) or SigmaPlot (Version 14, San Jose, CA, USA). Significance was considered at P < 0.05.
Data availability
The data that support the findings of this study are available from the authors upon reasonable request.
References
Dusso, A. S., Brown, A. J. & Slatopolsky, E. Vitamin D. Am. J. Physiol. Renal Physiol. 289, F8–28 (2005). https://doi.org/10.1152/ajprenal.00336.2004
Underland, L., Markowitz, M. & Gensure, R. Calcium and phosphate hormones: Vitamin D, parathyroid hormone, and fibroblast growth factor 23. Pediatr. Rev. 41, 3–11. https://doi.org/10.1542/pir.2018-0065 (2020).
Christakos, S., Dhawan, P., Verstuyf, A., Verlinden, L. & Carmeliet, G. Vitamin D: metabolism, molecular mechanism of action, and pleiotropic effects. Physiol. Rev. 96, 365–408. https://doi.org/10.1152/physrev.00014.2015 (2016).
Bergwitz, C. & Juppner, H. Regulation of phosphate homeostasis by PTH, vitamin D, and FGF23. Annu. Rev. Med. 61, 91–104. https://doi.org/10.1146/annurev.med.051308.111339 (2010).
Thomas, L. et al. Acute adaption to oral or intravenous phosphate requires parathyroid hormone. J. Am. Soc. Nephrol. 28, 903–914. https://doi.org/10.1681/ASN.2016010082 (2017).
Juppner, H. Phosphate and FGF-23. Kidney Int. Suppl. 79, S24-27. https://doi.org/10.1038/ki.2011.27 (2011).
Quarles, L. D. Role of FGF23 in vitamin D and phosphate metabolism: Implications in chronic kidney disease. Exp. Cell Res. 318, 1040–1048. https://doi.org/10.1016/j.yexcr.2012.02.027 (2012).
Demay, M. B., Kiernan, M. S., DeLuca, H. F. & Kronenberg, H. M. Sequences in the human parathyroid hormone gene that bind the 1,25-dihydroxyvitamin D3 receptor and mediate transcriptional repression in response to 1,25-dihydroxyvitamin D3. Proc. Natl. Acad. Sci. USA 89, 8097–8101. https://doi.org/10.1073/pnas.89.17.8097 (1992).
Kolek, O. I. et al. 1alpha,25-Dihydroxyvitamin D3 upregulates FGF23 gene expression in bone: The final link in a renal-gastrointestinal-skeletal axis that controls phosphate transport. Am. J. Physiol. Gastrointest. Liver Physiol. 289, G1036-1042. https://doi.org/10.1152/ajpgi.00243.2005 (2005).
Avioli, L. V., Lee, S. W., McDonald, J. E., Lund, J. & DeLuca, H. F. Metabolism of vitamin D3–3H in human subjects: Distribution in blood, bile, feces, and urine. J. Clin. Invest. 46, 983–992. https://doi.org/10.1172/JCI105605 (1967).
Rosenstreich, S. J., Rich, C. & Volwiler, W. Deposition in and release of vitamin D3 from body fat: Evidence for a storage site in the rat. J. Clin. Invest. 50, 679–687. https://doi.org/10.1172/JCI106538 (1971).
Haussler, M. R. et al. New understanding of the molecular mechanism of receptor-mediated genomic actions of the vitamin D hormone. Bone 17, 33S-38S. https://doi.org/10.1016/8756-3282(95)00205-r (1995).
Song, Y., Kato, S. & Fleet, J. C. Vitamin D receptor (VDR) knockout mice reveal VDR-independent regulation of intestinal calcium absorption and ECaC2 and calbindin D9k mRNA. J. Nutr. 133, 374–380. https://doi.org/10.1093/jn/133.2.374 (2003).
Kido, S., Kaneko, I., Tatsumi, S., Segawa, H. & Miyamoto, K. Vitamin D and type II sodium-dependent phosphate cotransporters. Contrib. Nephrol. 180, 86–97. https://doi.org/10.1159/000346786 (2013).
Hernando, N. et al. 1,25(OH)(2) vitamin D(3) stimulates active phosphate transport but not paracellular phosphate absorption in mouse intestine. J. Physiol. 599, 1131–1150. https://doi.org/10.1113/JP280345 (2021).
Beck, L. et al. Targeted inactivation of Npt2 in mice leads to severe renal phosphate wasting, hypercalciuria, and skeletal abnormalities. Proc. Natl. Acad. Sci. USA 95, 5372–5377. https://doi.org/10.1073/pnas.95.9.5372 (1998).
Li, Y. et al. Response of Npt2a knockout mice to dietary calcium and phosphorus. PLoS One 12, e0176232. https://doi.org/10.1371/journal.pone.0176232 (2017).
Segawa, H. et al. Npt2a and Npt2c in mice play distinct and synergistic roles in inorganic phosphate metabolism and skeletal development. Am. J. Physiol. Renal. Physiol. 297, F671-678. https://doi.org/10.1152/ajprenal.00156.2009 (2009).
Miedlich, S. U., Zhu, E. D., Sabbagh, Y. & Demay, M. B. The receptor-dependent actions of 1,25-dihydroxyvitamin D are required for normal growth plate maturation in NPt2a knockout mice. Endocrinology 151, 4607–4612. https://doi.org/10.1210/en.2010-0354 (2010).
Thomas, L. et al. PF-06869206 is a selective inhibitor of renal P(i) transport: Evidence from in vitro and in vivo studies. Am. J. Physiol. Renal. Physiol. 319, F541–F551. https://doi.org/10.1152/ajprenal.00146.2020 (2020).
Hashimoto, N. et al. Lithocholic acid increases intestinal phosphate and calcium absorption in a vitamin D receptor dependent but transcellular pathway independent manner. Kidney Int. 97, 1164–1180. https://doi.org/10.1016/j.kint.2020.01.032 (2020).
Radanovic, T., Wagner, C. A., Murer, H. & Biber, J. Regulation of intestinal phosphate transport. I. Segmental expression and adaptation to low-P(i) diet of the type IIb Na(+)-P(i) cotransporter in mouse small intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 288, G496-500. https://doi.org/10.1152/ajpgi.00167.2004 (2005).
Xue, J. et al. Enhanced phosphate absorption in intestinal epithelial cell-specific NHE3 knockout mice. Acta Physiol. (Oxf.) 234, e13756. https://doi.org/10.1111/apha.13756 (2022).
Thomas, L. et al. Pharmacological Npt2a inhibition causes phosphaturia and reduces plasma phosphate in mice with normal and reduced kidney function. J. Am. Soc. Nephrol. 30, 2128–2139. https://doi.org/10.1681/ASN.2018121250 (2019).
Clerin, V. et al. Selective pharmacological inhibition of the sodium-dependent phosphate cotransporter NPT2a promotes phosphate excretion. J. Clin. Invest. 130, 6510–6522. https://doi.org/10.1172/JCI135665 (2020).
Luong, T. T. D. et al. Acid sphingomyelinase promotes SGK1-dependent vascular calcification. Clin. Sci. (Lond.) 135, 515–534. https://doi.org/10.1042/CS20201122 (2021).
Christakos, S. Vitamin D: A critical regulator of intestinal physiology. JBMR Plus 5, e10554. https://doi.org/10.1002/jbm4.10554 (2021).
Tenenhouse, H. S. et al. Na/P(i) cotransporter (Npt2) gene disruption increases duodenal calcium absorption and expression of epithelial calcium channels 1 and 2. Pflugers Arch 444, 670–676. https://doi.org/10.1007/s00424-002-0865-2 (2002).
Canaff, L. & Hendy, G. N. Human calcium-sensing receptor gene. Vitamin D response elements in promoters P1 and P2 confer transcriptional responsiveness to 1,25-dihydroxyvitamin D. J. Biol. Chem. 277, 30337–30350. https://doi.org/10.1074/jbc.M201804200 (2002).
Khan, S. R. & Canales, B. K. Ultrastructural investigation of crystal deposits in Npt2a knockout mice: Are they similar to human Randall’s plaques?. J. Urol. 186, 1107–1113. https://doi.org/10.1016/j.juro.2011.04.109 (2011).
Chau, H., El-Maadawy, S., McKee, M. D. & Tenenhouse, H. S. Renal calcification in mice homozygous for the disrupted type IIa Na/Pi cotransporter gene Npt2. J. Bone Miner. Res. 18, 644–657. https://doi.org/10.1359/jbmr.2003.18.4.644 (2003).
Lapointe, J. Y. et al. NPT2a gene variation in calcium nephrolithiasis with renal phosphate leak. Kidney Int. 69, 2261–2267. https://doi.org/10.1038/sj.ki.5000437 (2006).
Ritter, C. S. & Brown, A. J. Direct suppression of Pth gene expression by the vitamin D prohormones doxercalciferol and calcidiol requires the vitamin D receptor. J. Mol. Endocrinol. 46, 63–66. https://doi.org/10.1677/JME-10-0128 (2011).
Bergwitz, C. & Miyamoto, K. I. Hereditary hypophosphatemic rickets with hypercalciuria: Pathophysiology, clinical presentation, diagnosis and therapy. Pflugers Arch 471, 149–163. https://doi.org/10.1007/s00424-018-2184-2 (2019).
Diez, A. et al. Acute effects of ethanol on mineral metabolism and trabecular bone in Sprague-Dawley rats. Calcif. Tissue Int. 61, 168–171. https://doi.org/10.1007/s002239900317 (1997).
Laitinen, K., Tahtela, R. & Valimaki, M. The dose-dependency of alcohol-induced hypoparathyroidism, hypercalciuria, and hypermagnesuria. Bone Miner 19, 75–83. https://doi.org/10.1016/0169-6009(92)90845-5 (1992).
Jones, G., Prosser, D. E. & Kaufmann, M. 25-Hydroxyvitamin D-24-hydroxylase (CYP24A1): Its important role in the degradation of vitamin D. Arch Biochem. Biophys. 523, 9–18. https://doi.org/10.1016/j.abb.2011.11.003 (2012).
Turner, M. E. et al. The metabolism of 1,25(OH)(2)D(3) in clinical and experimental kidney disease. Sci. Rep. 12, 10925. https://doi.org/10.1038/s41598-022-15033-9 (2022).
Masuyama, R. et al. Vitamin D receptor in chondrocytes promotes osteoclastogenesis and regulates FGF23 production in osteoblasts. J. Clin. Invest. 116, 3150–3159. https://doi.org/10.1172/JCI29463 (2006).
Rhee, Y. et al. Parathyroid hormone receptor signaling in osteocytes increases the expression of fibroblast growth factor-23 in vitro and in vivo. Bone 49, 636–643. https://doi.org/10.1016/j.bone.2011.06.025 (2011).
Kawata, T. et al. Parathyroid hormone regulates fibroblast growth factor-23 in a mouse model of primary hyperparathyroidism. J. Am. Soc. Nephrol. 18, 2683–2688. https://doi.org/10.1681/ASN.2006070783 (2007).
Ben-Dov, I. Z. et al. The parathyroid is a target organ for FGF23 in rats. J. Clin. Invest. 117, 4003–4008. https://doi.org/10.1172/JCI32409 (2007).
Manolagas, S. C. Osteocalcin promotes bone mineralization but is not a hormone. PLoS Genet. 16, e1008714. https://doi.org/10.1371/journal.pgen.1008714 (2020).
Burt, L. A. et al. Effect of high-dose vitamin D supplementation on volumetric bone density and bone strength: A randomized clinical trial. JAMA 322, 736–745. https://doi.org/10.1001/jama.2019.11889 (2019).
Gillett, M. J., Vasikaran, S. D. & Inderjeeth, C. A. (2021) The role of PINP in diagnosis and management of metabolic bone disease. Clin. Biochem. Rev. 42, 3–10. https://doi.org/10.33176/AACB-20-0001
Bauer, D. C. et al. Short-term changes in bone turnover markers and bone mineral density response to parathyroid hormone in postmenopausal women with osteoporosis. J. Clin. Endocrinol. Metab. 91, 1370–1375. https://doi.org/10.1210/jc.2005-1712 (2006).
Nair, S. et al. Effect of vitamin D levels on bone remodeling in healthy women. Int. J. Endocrinol. Metab. 18, e100656. https://doi.org/10.5812/ijem.100656 (2020).
Halleen, J. M., Tiitinen, S. L., Ylipahkala, H., Fagerlund, K. M. & Vaananen, H. K. Tartrate-resistant acid phosphatase 5b (TRACP 5b) as a marker of bone resorption. Clin. Lab 52, 499–509 (2006).
Shigematsu, T. et al. Evocalcet with vitamin D receptor activator treatment for secondary hyperparathyroidism. PLoS One 17, e0262829. https://doi.org/10.1371/journal.pone.0262829 (2022).
Naylor, K. & Eastell, R. Bone turnover markers: Use in osteoporosis. Nat. Rev. Rheumatol. 8, 379–389. https://doi.org/10.1038/nrrheum.2012.86 (2012).
Madar, A. A. et al. Effect of vitamin D(3)-supplementation on bone markers (serum P1NP and CTX): A randomized, double blinded, placebo controlled trial among healthy immigrants living in Norway. Bone Rep. 2, 82–88. https://doi.org/10.1016/j.bonr.2015.05.004 (2015).
Jorde, R. et al. Effects of vitamin D supplementation on bone turnover markers and other bone-related substances in subjects with vitamin D deficiency. Bone 124, 7–13. https://doi.org/10.1016/j.bone.2019.04.002 (2019).
Rossini, M. et al. Dose-dependent short-term effects of single high doses of oral vitamin D(3) on bone turnover markers. Calcif. Tissue Int. 91, 365–369. https://doi.org/10.1007/s00223-012-9637-y (2012).
Sanders, K. M. et al. Annual high-dose oral vitamin D and falls and fractures in older women: A randomized controlled trial. JAMA 303, 1815–1822. https://doi.org/10.1001/jama.2010.594 (2010).
Muller, D. et al. A novel claudin 16 mutation associated with childhood hypercalciuria abolishes binding to ZO-1 and results in lysosomal mistargeting. Am. J. Hum. Genet. 73, 1293–1301. https://doi.org/10.1086/380418 (2003).
Weber, S. et al. Novel paracellin-1 mutations in 25 families with familial hypomagnesemia with hypercalciuria and nephrocalcinosis. J. Am. Soc. Nephrol. 12, 1872–1881. https://doi.org/10.1681/ASN.V1291872 (2001).
Hou, J. et al. Phosphorylated claudin-16 interacts with Trpv5 and regulates transcellular calcium transport in the kidney. Proc. Natl. Acad. Sci. USA 116, 19176–19186. https://doi.org/10.1073/pnas.1902042116 (2019).
van Loon, E. P. et al. Calcium extrusion pump PMCA4: A new player in renal calcium handling?. PLoS One 11, e0153483. https://doi.org/10.1371/journal.pone.0153483 (2016).
Thomas, L., Xue, J., Dominguez Rieg, J. A. & Rieg, T. Contribution of NHE3 and dietary phosphate to lithium pharmacokinetics. Eur. J. Pharm. Sci. 128, 1–7. https://doi.org/10.1016/j.ejps.2018.11.008 (2019).
Vallon, V. et al. SGLT2 inhibitor empagliflozin reduces renal growth and albuminuria in proportion to hyperglycemia and prevents glomerular hyperfiltration in diabetic Akita mice. Am. J. Physiol. Renal Physiol. 306, F194-204. https://doi.org/10.1152/ajprenal.00520.2013 (2014).
Vallon, V. et al. A role for the organic anion transporter OAT3 in renal creatinine secretion in mice. Am. J. Physiol. Renal Physiol. 302, F1293-1299. https://doi.org/10.1152/ajprenal.00013.2012 (2012).
Fenton, R. A. et al. Renal phosphate wasting in the absence of adenylyl cyclase 6. J. Am. Soc. Nephrol. 25, 2822–2834. https://doi.org/10.1681/ASN.2013101102 (2014).
Fenton, R. A. et al. Adenylyl cyclase 6 expression is essential for cholera toxin-induced diarrhea. J. Infect. Dis. 220, 1719–1728. https://doi.org/10.1093/infdis/jiz013 (2019).
Xue, J. et al. An inducible intestinal epithelial cell-specific NHE3 knockout mouse model mimicking congenital sodium diarrhea. Clin. Sci. (Lond.) 134, 941–953. https://doi.org/10.1042/CS20200065 (2020).
Funding
This work was supported by the National Institute of Diabetes and Digestive and Kidney Diseases 1R01DK110621 (to T.R.) and VA Merit Review Awards IBX004968A (to T.R.) and IBX004024 (to A.S.). Additional support was provided by a Pilot Project from the USF Microbiomes Institute (to T.R. and J.D.R). The contents do not represent the views of the U.S. Department of Veterans Affairs or the United States Government.
Author information
Authors and Affiliations
Contributions
L.T. and T.R. conceived and designed research; L.T., L.V.D, M.T., S.A.M. and T.R. performed experiments; L.T., L.V.D, M.T. and T.R. analyzed data; L.T., L.V.D, M.T., A.S., J.A.D.R. and T.R. interpreted results of experiments; L.T., M.T. and T.R. prepared figures; L.T. and T.R. drafted manuscript; L.T., L.V.D., M.T., A.S., S.A.M., J.A.D.R., and T.R. edited and revised manuscript; L.T., L.V.D., M.T., A.S., S.A.M., J.A.D.R., and T.R. approved final version of manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Thomas, L., Dissanayake, L.V., Tahmasbi, M. et al. Vitamin D3 suppresses Npt2c abundance and differentially modulates phosphate and calcium homeostasis in Npt2a knockout mice. Sci Rep 14, 16997 (2024). https://doi.org/10.1038/s41598-024-67839-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-67839-4
