
Abstract
Alzheimer’s disease (AD) is the most common neurodegenerative disease characterized by memory loss. Our recent study has demonstrated that thioredoxin-1 (Trx-1) could protect neurons via repressing NLRP1‑mediated neuronal pyroptosis in AD models. However, whether Trx-1 could inhibit NLRP3 activation is largely unknown. Here, we found that AAV-mediated Trx-1 overexpression significantly inhibited NLRP3-mediated pyroptosis in the hippocampus of APP/PS1 mice. A mouse hippocampal neuron HT22 cell line overexpressing Trx-1 was successfully obtained through lentivirus transfection. Further study showed that Trx-1 overexpression protected HT22 cells against the neurocytotoxicity of Aβ25−35. Consistently with the results of in vivo experiments, overexpression of Trx-1 remarkedly inhibited the activation of NLRP3. In the contrary, knockdown of Trx-1 by siRNA transfection further aggravated the activation of NLRP3. Mechanistically, Trx-1 overexpression significantly inhibited the increase of thioredoxin-interacting protein (TXNIP) in in vivo and in vitro and weakened the interaction between TXNIP and NLRP3. Taken together, Trx-1 inhibits NLRP3-mediated pyroptosis by regulating TXNIP expression and its interaction with NLRP3 in AD models.
Similar content being viewed by others


Introduction
Alzheimer’s disease (AD) is the commonest type of neurodegenerative disorders and is the leading causation of dementia1. “The World Alzheimer’s Disease Report” indicates that more than 46.8 million people worldwide are affected by AD2. According to “The China Alzheimer Report 2022”, AD is listed as the fifth leading cause of death for patients over age 65 in China3. The rising number of AD patients is causing significant social and economic pressure. Prevenient investigations have clarified that amyloid β (Aβ) is closely related with the progress of AD4. Early studies have shown that the neurotoxicity of Aβ is mediated by the reactive oxygen species (ROS) family5. Under natural conditions, the production and clearance of ROS is highly controlled and keep balance. The accumulation of ROS can occur through the overproduction of ROS or the insufficient elimination of ROS.
Thioredoxin 1 (Trx-1), a small molecule protein with multiple functions, is primarily localized in the cytoplasm and is imported into the nucleus in some cases6. The active center of Trx-1 contains two cysteine residues (-Cys-X-X-Cys-), which renders it redox regulating activity7. Generally, Trx-1 acts critical roles in many kinds of biological processes, including cellular proliferation, redox regulation, inhibition of apoptosis, gene expression and DNA synthesis8. Importantly, Trx-1 plays neuroprotection in neurodegenerative diseases, such as Parkinson disease and AD9. One of prevenient investigations from our lab has demonstrated that Trx-1 defended dopaminergic neurons from MPP+/MPTP-elicited endoplasmic reticulum stress via restraining the activation of inositol-requiring enzyme 1α (IRE 1α)10. Our recent study further clarified that Trx-1 inhibited the activation of IRE 1α by upregulating Hsp90/p-Cdc37 in PD models11. Our study also revealed that Trx-1 decreased the levels of α-synuclein through promoting autophagy-lysosome pathway in MPTP models12. What’s more, the actions of Trx-1 in AD are also defined13. The protein levels of Trx-1 were markedly decreased in the brains of AD model animals and patients14,15, which leads to ROS accumulation followed by neuronal loss and ultimately neurodegeneration. Our previous study has demonstrated that Trx-1 promotes mitochondrial biogenesis through upregulating the AMPK/SIRT1/PPARGC1A signaling in the brain of FAD4T APP/PS1 mice16. In the past few years, pyroptosis was considered as the key player in the neuronal loss in AD17. We reported for the first time the inhibition of Trx-1 on NLR family pyrin domain-containing 1 (NLRP1)-mediated pyroptosis in PC12 cells incubated with Aβ25−3518. Our recent study has demonstrated that Trx-1 could repress NLRP1‑mediated neuronal pyroptosis in AD models19. However, the inhibition of Trx-1 on NLRP3, another pyroptosis mediator, and the accurate mechanism in AD need to be further elucidated.
In the present study, it’s clarified that overexpression of Trx-1 remarkedly inhibited the activation of NLRP3 through inhibiting the increase of thioredoxin-interacting protein (TXNIP) in in vivo and in vitro AD models and weakening the interaction between TXNIP and NLRP3.
Materials and methods
Materials
DH5α competent cell (CB101-02) was purchased from TIANGEN. DNA polymerase (P505-D1) was obtained from Vazyme. Plasmid DNA purification (740412) was obtained from MACHEREY-NAGEL. Gel DNA purification (GK2041) was obtained from Generay. ReverTra Ace® qPCR RT Kit (FSQ-301) and SYBRGreen real-time RCP Master Mix (QPK-201) were purchased from TOYOBO. Primary caspase-3 antibody (#9661) was obtained from CST (Danvers, USA). Primary GSDMD antibody (sc-393581) was purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). Primary Trx-1 antibody (14999-1-AP) and NLRP3 primary antibody (19771-1-AP) were obtained from Proteintech (Wuhan, China). Primary TXNIP antibody (JM60-35) was purchased from Hua’an Biotecchnology Co., Ltd (Hangzhou, China). Primary IL-1β antibody (AF5103) were purchased from Affinity Biosciences Co., Ltd (Changzhou, China). Primary Aβ (TA500973S), caspase-1 (TA383869) and β-actin antibody (TA811000) were purchased from Origene (Rockville, USA).
Animals and stereotaxical injection of adeno-associated virus
The animals used in this study was from our previous study19. Briefly, male 2-month-old FAD4T APP/PS1 mice and wild type (WT) littermates were purchased from GemPharmtech Co., Ltd. (Nanjing, Jiangsu, China) and kept in plastic cages with free access to water and food and on a 12-hour dark-light cycle. Stereotaxical Injection of adeno-associated virus (AAV) in hippocampus was the same as our previous study19. All experimental protocols were approved by the Experimental Animal Ethics Committee of Jiaxing University Medical College (JUMC2021-144). All methods are reported in accordance with ARRIVE guidelines (https://arriveguidelines.org) for the reporting of animal experiments.
Morris water maze test
Morris Water Maze (MWM) tests were employed to evaluate the learning and memory of mice when they were 5-months old. The mice were subjected to a training test for 5 days (Day 1–5) to seek the hidden platform. On Day 6, the mice were subjected to a probe test to obtain the escape latency (time for getting on the platform). After removing the platform, record the times for crossing platform in 2 min.
Immunohistochemistry staining
After MWM tests, the mouse brains were isolated rapidly, immersed in 4% paraformaldehyde for 24 h, and then dehydrated in sucrose solution (25%) until the tissues sank to the bottom. Brain tissues were cut coronally into Sect. (8 μm) using a Leica frozen microtome after embedded in OCT. The sections were incubated with 0.25% Triton X-100 for 10 min at room temperature, blocked in 5% BSA for 1 h at 37℃, and then incubated with primary antibody (1:200) overnight at 4℃. After incubation with secondary antibody (1:1000) conjugated with biotin at 37℃ for 1 h, the sections were blocked with DAB. Finally, the staining images was obtained by using a Digital Slice Scan System (HS6, SOPTOP, Ningbo, China).
Western blot (WB)
Protein lysates were prepared using the RIPA lysate (CWBIO, Taizhou, China) complemented with 1% phosphatase inhibitor and 1% protease inhibitor. The concentration of protein samples was measured using BCA method (CWBIO, Taizhou, China). Equal protein content was split by SDS-PAGE, and then transformed onto a PVDF membrane. The membrane was labeled with the related primary antibody (1: 1000) overnight at 4 °C after being immersed in sealed liquid. Then the membrane was labeled with HRP-conjugated anti-rabbit IgG (1: 10000) (CWBIO, Taizhou, China). The protein bands were analyzed with ImageJ.
Cell culture and construction of HT22 overexpressing Trx-1
Rat pheochromocytoma PC12 cells were obtained from National Collection of Authenticated Cell Cultures (Shanghai, China) and were cultured in RPMI 1640 medium (Invitrogen, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS, 10091-148, Gibco) and 1% penicillin/streptomycin solution. Human neuroblastoma SH-SY5Y cells were obtained from Wuhan Pricella Biotechnology Co., Ltd. (China) and were cultured in MEM/F12 supplemented with 15% FBS and 1% penicillin/streptomycin solution. Mouse hippocampal HT22 neurons were obtained from Shanghai Fuheng Biotechnology Co., Ltd. (China) and were cultured in DMEM medium complemented with 10% FBS and 1% penicillin/streptomycin solution. All cells were cultured in a 37℃ humid incubator containing 5% CO2.
Construction of HT22 overexpressing Trx-1
The human Trx-1 mRNA was chemically synthesized according to the transcript (NM_003329.4). RT-PCR was performed to amplify cDNA for human Trx-1 with the forward and reverse primers and the PCR product was inserted into the lentiviral vector (pHBLV-CMV-MCS-3xFlag-EF1-ZsGreen-T2A-PuroR). The lentivirus was transfected into HT22 cells for 48 h and then the cells were harvested. Subsequently, the transfected cells were screened with puromycin (2.5 µg/ml) to establish HT22 cell line stably overexpressing Trx-1.
qRT-PCR
qRT-PCR was used to detect the levels of Trx-1 mRNA. Trizol reagent (15596018, Invitrogen Life Technologies) was employed to extract the total RNA of control HT22 cells and HT22 cells overexpressing Trx-1. A NanoDrop 2000 spectrophotometer (Thermo Forma, USA) was used to measure the purity of RNA and agarose gel electrophoresis was employed to determine the quantity of RNA. The RNA was reversely transcribed into cDNA by using ReverTra Ace® qPCR RT Kit (FSQ-301, TOYOBO). qRT-PCR was performed by using SYBRGreen real-time RCP Master Mix (QPK-201, TOYOBO). The cycle threshold (∆∆CT) method was used for quantification. GAPDH was employed as an internal control. The sequence of h-TXN primer is: Forward, 5’-GCCTTTCTTTCATTCCCTC-3’; Reverse, 5’-TTCACCCACCTTTTGTCC-3’. The sequence of mGAPDH primer is: Forward, 5’-TTCCTACCCCCAATGTGTCC-3’; Reverse, 5’-GGTCCTCAGTGTAGCCCAAG-3’. PCR reactions were performed using the primers in Table 1 with the cDNA as the template, and the PCR cycling conditions are shown in Table 1.
Preparation of Aβ25−35
Sterile ultrapure water was used to dissolve Aβ25−35 (Chinapeptides, Shanghai, China) at a concentration of 2 mM and the Aβ25−35 solution was then incubated for 7 days in a 37 °C water-bath to produce oligomers.
Detection of cellular apoptosis
Annexin V-mCherry Apoptosis Detection Kit (C1070M, Beyotime, Shanghai, China) was employed to detect cellular apoptosis. Control HT22 cells and HT22 cells overexpressing human Trx-1 were added into a 6-well plate (2 × 105/well) overnight. The cells were incubated with Aβ25−35 (40 µM) for 48 h and then incubated with Annexin V-mCherry in a dark box for 20 min at room temperature. The red color indicated the apoptotic cells, which was captured with a fluorescence microscope.
Cell viability
HT22 cells and HT22 cells with overexpressing human Trx-1 were added in a 96-well plate (2 × 104 cells/well). The cells were exposed to Aβ25−35 (40 µM) for 48 h and then incubated with MTT (Solarbio, Beijing, China) for 4 hours in a 37 °C cell incubator. The precipitation was then dissolved in DMSO. The absorbance value at 570 nm of each well was determined by using a microplate reader. The cell viability was presented as (absorbance value in experimental group/mean absorbance value in control group) × 100%.
Release of lactate dehydrogenase
Lactate dehydrogenase (LDH) Cytotoxicity Assay Kit (C0017, Beyotime, Shanghai, China) was employed to detect the release of LDH. Control HT22 cells and HT22 cells overexpressing human Trx-1 were added into a 96-well plate (2 × 104/well) overnight. The cells were incubated with Aβ25−35 (40 µM) for 48 h and then the supernatant was incubated with LDH detection working solution in a dark box for 30 min at room temperature. The absorbance value at 490 nm of each well was determined by using a microplate reader. The LDH content was presented as (absorbance value in experimental group/mean absorbance value in control group) × 100%.
Immunofluorescence staining
HT22 cells and HT22 cells with overexpressing human Trx-1 were added on a cover glass put in a 24-well plate (5 × 104 cells/well) overnight. The cells were incubated with Aβ25−35 (40 µM) for 48 h. The cells were fixed with 4% PFA for 30 min and then incubated with Triton X-100 (0.25%) at room temperature for 10 min. After blocking in goat serum at room temperature for 30 min, the cells were immersed within the unconjugated antibody targeting NLRP3 (1:50 in PBS) or TXNIP (1:50 in PBS) and then were deposited in a humid container to incubate overnight at 4°C. The secondary antibody (AlexaFluor 647-conjugated goat anti-mouse antibody, ab150075, Abcam, Cambridge, England, 1:100 in PBS) was added on each cover glass. The slides were placed in a humid container again to incubate at room temperature for 30 min. The immunofluorescence was observed using a Laser Scanning Confocal Microscope (FV3000, Olympus, Tokyo, Japan).
Transfection of SiRNA
Mouse Trx-1 siRNA was chemically synthesized by GenePharma (Shanghai, China). The sequences of negative control were: sense, 5’-UUCUCCGAACGUGUCACGUTT-3’; anti-sense, 5’-ACGUGACACGUUCGGAGAATT-3’. The sequences of Trx-1 siRNA were: sense, 5′-GAUCGAGAGCAAGGAAGCUTT-3’; anti-sense, 5′-AGCUUCCUUGCUCUCGAUCTT-3′. Trx-1 siRNA or negative control (50 nM) was transfected into HT22 neurons by using siRNA Transfection Mate (G04002, GenePharma, Shanghai, China). HT22 cells were administrated with Aβ25−35 (40 µM) for 48 h after siRNA transfection for 24 h.
Co-immunoprecipitation
An immunoprecipitation kit with protein A + G magnetic beads (P2179, Beyotime Biotechnology, Shanghai, China) was employed to detect the protein interaction. HT22 cells and HT22 cells with overexpressing human Trx-1 were added in a 6-well plate (2 × 105 cells/well) overnight. The cells were incubated with Aβ25−35 (40 µM) for 48 h. Cells were harvested in co-immunoprecipitation buffer (50 mM Tris·HCl pH7.4, 0.25% sodium deoxycholate,1% NP-40, 1 mM Na3VO4, 150 mM NaCl, 1 mM EDTA,1 mM NaF, 1 mM PMSF plus protease inhibitor cocktail). Primary Trx-1 or TXNIP antibody were incubated with the lysates at 4℃ overnight on a slightly shaking shaker. Protein A + G magnetic beads were added into the lysates to capture the protein and antibody complex. Finally, the immunoprecipitants were washed for 5 times with cold PBS buffer, collected and subjected to WB analysis. Mouse IgG was used as a negative control. Equal volume lysate was loaded as input samples.
Statistical analysis
The detection in each experiment was duplicated for more than 3 times with independent samples. All data were analyzed with GraphPad Prism 9.5.0 statistical software and the values were presented as mean ± SD. For the comparison of the, Student’s t-test and one-way ANOVA was respectively used to compare the values from two groups and multiple groups. P value no more than 0.05 was considered as significant differences.
Results
Trx-1 expression was decreased in AD models
The expression of Trx-1 was detected in AD cellular and animal models. Aβ25−35 (40 µM) was used to induced AD cellular models in various cell lines (HT22, SH-SY5Y and PC12 cells). As shown in Fig. 1A-C, the expression of Trx-1 was significantly decreased in Aβ25−35-treated HT22, SH-SY5Y or PC12 cells. The APP/PS1 mice were used as AD animal model. The expression of Trx-1 was also reduced significantly in the hippocampus of APP/PS1 mice compared to that of WT mice (Fig. 1D). These data suggest that Trx-1 expression was decreased in AD models.

Trx-1 expression was decreased in AD models. (A) Trx-1 expression in HT22 cells treated with Aβ25−35 (40 µM) for 48 h. (B) Trx-1 expression in SH-SY5Y cells treated with Aβ25−35 (40 µM) for 48 h. (C) Trx-1 expression in PC12 cells treated with Aβ25−35 (40 µM) for 48 h. (D) The expression of Trx-1 in the hippocampus of APP/PS1 mice. Statistical analysis was performed by t-test and the statistical results were represented as means ± SD, *P < 0.05.
AAV-mediated Trx-1 overexpression decreased the Aβ deposition and improved the learning and memory of APP/PS1 mice
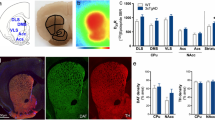
In our recent study, AAV-mediated Trx-1 overexpression in the hippocampus decreased the Aβ deposition and improved the learning and memory of APP/PS1 mice19. The schedule of animal experiment was showed in Fig. 2A. AAV-Vector or AAV-Trx-1 were injected into the hippocampus of mice and fully expressed after 4 weeks (Fig. 2B). Result from Western blot analysis demonstrated overexpression of Trx-1 in the hippocampus (Fig. 2C). The effect of Trx-1 overexpression on the deposition of Aβ and the learning and memory of APP/PS1 mice were further determined. As shown in Fig. 2D-F, immunohistochemistry analysis revealed a significant increase of Aβ deposition and Western Blots analysis showed a marked increase of Aβ expression in the hippocampus of APP/PS1 mice compared to that of WT mice; Both changes were reversed by overexpression of Trx-1. MWM test recorded the increased escape latency and decreased platform-crossing times of APP/PS1 mice compared to that of WT mice, which also was significantly reversed by Trx-1 overexpression (Fig. 2G-J). These data suggest that local Trx-1 overexpression in the hippocampus mitigate effectively the pathological features and cognitive function of AD mice.

Trx-1 overexpression decreased the Aβ deposition and improved the learning and memory of APP/PS1 mice. (A) The schedule of animal experiments. (B) AAV injection into the hippocampus of mice and fluorescence observation of frozen sections after 4 weeks. (C) Western blot analysis of Trx-1 overexpression after AAV injection for 4 weeks. (D) Immunohistochemical staining of Aβ deposition in the brains. scale bar = 200 μm in up row, scale bar = 50 μm in down row. (E) APP and Aβ expression in the hippocampus of mice (n = 5 in WT + AAV-Trx-1 and WT + AAV-Vector groups, n = 4 in APP/PS1 + AAV-Vector and APP/PS1 + AAV-Trx-1 groups). (F) Quantitative analysis of APP and Aβ in the hippocampus (n = 5 in WT + AAV-Trx-1 and WT + AAV-Vector groups, n = 4 in APP/PS1 + AAV-Vector and APP/PS1 + AAV-Trx-1 groups). (G) The trajectories of mice to get on the target platform in MWM test (n = 6 in WT + AAV-Trx-1 and WT + AAV-Vector groups, n = 5 in APP/PS1 + AAV-Vector and APP/PS1 + AAV-Trx-1 groups). (H) Statistical analysis of the escape latency of mice to get on the target platform (n = 6 in WT + AAV-Trx-1 and WT + AAV-Vector groups, n = 5 in APP/PS1 + AAV-Vector and APP/PS1 + AAV-Trx-1 groups). (I) The trajectories of mice to cross the target platform area in MWM test (n = 6 in WT + AAV-Trx-1 and WT + AAV-Vector groups, n = 5 in APP/PS1 + AAV-Vector and APP/PS1 + AAV-Trx-1 groups). (J) Statistical analysis of the platform-crossing times of mice in MWM test (n = 6 in WT + AAV-Trx-1 and WT + AAV-Vector groups, n = 5 in APP/PS1 + AAV-Vector and APP/PS1 + AAV-Trx-1 groups). Statistical analysis was performed by one or two-way ANOVA with Tukey’s multiple comparisons test and the statistical results were represented as means ± SD, *P < 0.05, **P < 0.01, ***P < 0.001.
Trx-1 overexpression inhibited the activation of NLRP3 in the hippocampus of APP/PS1 mice
We further investigated the effect of Trx-1 on NLRP3 using this batch of tissue samples. The WB analysis showed that the protein levels of NLRP3, were remarkedly upregulated in the hippocampus of APP/PS1 mice, which was significantly reversed by Trx-1 overexpression (Fig. 3A and B). Furthermore, the increase of caspase-1 and IL-1β in the hippocampus of APP/PS1 mice was also inhibited by Trx-1 overexpression (Fig. 3A, C and D). These data suggest that Trx-1 overexpression inhibits the NLRP3-mediated pyroptosis in AD.

Trx-1 overexpression inhibited the activation of NLRP3 in the hippocampus of APP/PS1 mice. (A) The WB detection of NLRP3, caspase-1 and IL-1β in the hippocampus of APP/PS1 mice. (B) Gray scale analysis of NLRP3. (C) Gray scale analysis of caspase-1. (D) Gray scale analysis of IL-1β. Statistics were calculated using one-way ANOVA with Tukey’s multiple comparisons test and are presented as mean ± SD from three independent experiments (n = 5 in WT + AAV-Trx-1 and WT + AAV-Vector groups, n = 4 in APP/PS1 + AAV-Vector and APP/PS1 + AAV-Trx-1 groups). **P < 0.01, ***P < 0.001.
Construction of HT22 cell line stably overexpressing human Trx-1
In order to investigate the mechanism of Trx-1 inhibiting NLRP3 in vitro, HT22 cells were infected with LV-Trx-1 (Fig. 4A) for 24 h to obtain a stable cell line. Afterward, puromycin screening was performed to obtain HT22 cells stably overexpressing human Trx-1. As shown in Fig. 4B, the green fluorescence showed a good status of LV-Trx-1 expression in both groups. RNA agarose gel electrophoresis showed that the three rRNA bands were 5 S, 18 S and 28 S (Fig. 4C). The band brightness of 5 S was the weakest and 28 S was the brightest, indicating a good RNA integrity. qRT-PCR results demonstrated that the content of Trx-1 mRNA in Trx-1-overexpressed HT22 cells is 144,930 folds of control HT22 cells (Fig. 4D). The protein levels of human Trx-1 in LV-Trx-1 HT22 cells were significantly increased compared with control HT22 cells (Fig. 4E and F). In conclusion, we have successfully constructed the hippocampal HT22 cell line overexpressing human Trx-1.

A stable cell line overexpressing human Trx-1 was successfully constructed. (A) The structure diagram of LV-Trx-1. (B) HT22 cells were transfected with LV-Vector and LV-Trx-1 and then screened with puromycin (2 µg/ml). HT22 cells stably overexpressing human Trx-1 were obtained. (C) RNA agarose gel electrophoresis for RNA extraction. (D) The mRNA expression of human Trx-1 was measured by qRT-PCR. (E) The protein expression levels of human Trx-1 and mouse Trx-1 were assessed by WB. (F) Gray scale analysis of human Trx-1 and mouse Trx-1 expression in HT22 cells overexpressing Trx-1 or treated with lentivirus alone; The quantitative analysis was based on three independent experiments. Statistics were calculated using one-way ANOVA with Tukey’s multiple comparisons test and are presented as mean ± SD from three independent experiments. ***P < 0.001.
Trx-1 overexpression in HT22 cells resisted the neurotoxicity of Aβ25−35
Control HT22 cells and Trx-1-overexpressing HT22 cells were stimulated with Aβ25−35 (40 µM) for 48 h. In bright fields, Aβ25−35 treatment prominently changed the morphology of HT22 cells, and this change was restored by stable overexpression of Trx-1 (Fig. 5A). The MTT analysis was used to further evaluate the cytoprotective effect of Trx-1 against Aβ25−35 neurotoxicity. The viability of HT22 cells was markedly decreased by Aβ25−35 treatment, which was reversed by Trx-1 overexpression (Fig. 5B). In addition, the increase in the extracellular LDH content caused by Aβ25−35 was also inhibited by Trx-1 overexpression in HT22 cells (Fig. 5C).

Trx-1 overexpression in HT22 cells resisted the neurotoxicity of Aβ25−35. (A) Cell morphology of HT22 cells overexpressing human Trx-1 treated for 48 h with Aβ25−35 (40 µM). (B) Cell viability was measured by MTT analysis. Trx-1 overexpression reversed the viability of HT22 cells. (C) The increase in the extracellular LDH content was detected with LDH Cytotoxicity Assay Kit. Trx-1 overexpression inhibited the release of LDH. (D) Overexpression of Trx-1 inhibited Aβ25−35-induced apoptosis in HT22 cells (red fluorescence). Annexin V-mCherry can be coupled to phosphatidylserine on the outer membrane of apoptotic cells, and mCherry labeled with red fluorescent protein can indicate apoptotic cells. (E) Statistical analysis of Aβ25−35-induced apoptosis in HT22 cells. Red fluorescence intensity was quantified by using ImageJ. (F) Overexpression of Trx-1 inhibited the Aβ25−35-induced caspase-3 activation in HT22 cells. (G) Gray scale analysis of caspase-3 expression in HT22 cells; The quantitative analysis was based on three independent experiments. Statistics were calculated using one-way ANOVA with Tukey’s multiple comparisons test and are presented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001.
The effect of Trx-1 against Aβ25−35-induced apoptosis was further examined. A Annexin V-mCherry apoptosis detection kit was employed to analyze cellular apoptosis. Annexin V can specifically bind the phosphoserine on the outer membrane surface of apoptotic cells. The fluorescence intensity of Annexinv-conjugated mCherry reflects the extent of apoptosis. Treatment with Aβ25−35 dramatically augmented the apoptosis of HT22 cells, while overexpression of Trx-1 suppressed apoptosis induced by Aβ25−35 (Fig. 5D and E). Furthermore, it is showed that caspase-3 activation was prominently incremental in Aβ25−35 treated HT22 cells, and this effect was remarkably suppressed by overexpressing Trx-1 (Fig. 5F and G).
Trx-1 inhibited the activation of NLRP3 by Aβ25−35 in HT22 cells
Whether Trx-1 overexpression could inhibit NLRP3-mediated pyroptosis in HT22 model of AD was detected by WB analysis and immunofluorescence staining. The results showed that the expression of NLRP3 was prominently increased after Aβ25−35 incubation, which was reversed in Trx-1 overexpressing HT22 cells (Fig. 6A, B and F). Similarly, Trx-1 overexpression reversed the elevation of caspase-1, GSDMD and IL-1β expression in HT22 cells treated with Aβ25−35 (Fig. 6A, C-E). In the contrary, knockdown of Trx-1 expression in HT22 cells by siRNA transfection further aggravated the activation of NLRP3 (Fig. 6G and H). Human recombinant Trx-1 (rhTrx-1) treatment also suppressed the increase of NLRP3 (Fig. 6I). These data suggest that Trx-1 overexpression diminished the NLRP3-mediated pyroptosis in vitro.

Trx-1 inhibited the activation of NLRP3 by Aβ25−35 in HT22 cells. (A) The WB detection of NLRP3, caspase-1, GSDMD and IL-1β in WT HT22 and Trx-1 overexpressing HT22 cells treated by Aβ25−35 (40 µM). (B) Gray scale analysis of NLRP3. (C) Gray scale analysis of caspase-1. (D) Gray scale analysis of GSDMD. (E) Gray scale analysis of IL-1β. (F) Immunofluorescence staining of NLRP3 in WT HT22 and Trx-1 overexpressing HT22 cells treated by Aβ25−35 (40 µM). (G) Transfection with Trx-1 siRNA for 24 h downregulated the protein levels of Trx-1 in HT22 cells. (H) Trx-1 knockdown in HT22 cells by siRNA transfection further aggravated the activation of NLRP3. (I) The increase of NLRP3 by Aβ25−35 was abolished in SH-SY5Y cells treated with rhTrx-1 (50 µg/ml). Statistics were calculated using one-way ANOVA with Tukey’s multiple comparisons test and are presented as mean ± SD from three independent experiments. The quantitative analysis was based on three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
Trx-1 overexpression inhibited the increase of TXNIP in vivo and in vitro
TXNIP plays a critical role in the activation of the NLRP3 inflammasome9,17, thus the expression of TXNIP in in vivo and in vitro AD models was detected. The WB analysis and immunofluorescence staining results showed that the expression of TXNIP was prominently increased in the hippocampus of APP/PS1 mice and in the HT22 cells administrated with Aβ25−35, which were significantly downregulated by Trx-1 overexpression (Fig. 7A-C). In order to detect the roles of TXNIP upregulation in AD pathology, the specific inhibitor of TXNIP, SRI37330, was employed. As shown in Fig. 7D, SRI37330 significantly reversed the decreased viability of HT22 cells by Aβ25−35 treatment, suggesting that TXNIP contributes to the loss of AD neurons.

Trx-1 overexpression inhibited the increase of TXNIP in vivo and in vitro. (A) The WB analysis and the gray scale analysis of TXNIP in the hippocampus of APP/PS1 mice (n = 5 in WT + AAV-Trx-1 and WT + AAV-Vector groups, n = 4 in APP/PS1 + AAV-Vector and APP/PS1 + AAV-Trx-1 groups). (B) The WB detection and gray scale analysis of TXNIP in WT HT22 and Trx-1 overexpressing HT22 cells treated by Aβ25−35 (40 µM). The quantitative analysis was based on three independent experiments. (C) Immunofluorescence staining of TXNIP in WT HT22 and Trx-1 overexpressing HT22 cells treated by Aβ25−35 (40 µM). (D) MTT analysis of cell viability of HT22 cells treated by Aβ25−35 (40 µM) with or without pretreatment with TXNIP inhibitor SRI37330 (5 µM); Statistics were calculated using one-way ANOVA with Tukey’s multiple comparisons test and are presented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001.
Trx-1 overexpression weakened the interaction between NLRP3 and TXNIP
Considering that Trx-1 overexpression decreased the expression of NLRP3 and TXNIP in in vivo and in vitro AD models, we further detected the interaction between NLRP3 and TXNIP in HT22 cells treat with Aβ25−35 by employing co-IP. When Trx-1 antibody was selected to capture the protein-protein complex, the endogenous interaction between Trx-1 and TXNIP was decreased in Aβ25−35-treated HT22 cells, which is reversed by Trx-1 overexpression (Fig. 8A). Notably, although the interaction between NLRP3 and TXNIP was also decreased in Aβ25−35-treated HT22 cells, the interplay between them displayed no significant change in Trx-1 overexpressing HT22 cells (Fig. 8A). When TXNIP antibody was applied to perform co-IP assay, the interaction between NLRP3 and TXNIP was increased and the interaction between Trx-1 and TXNIP was decreased in Aβ25−35-treated HT22 cells; while Trx-1 overexpression reversed the interaction between NLRP3 and TXNIP (Fig. 8B).

Trx-1 overexpression weakened the interaction between NLRP3 and TXNIP. WT HT22 and Trx-1 overexpressing HT22 cells treated by Aβ25−35 (40 µM). (A) Primary Trx-1 antibody was employed to perform the co-IP analysis. (B) Primary TXNIP antibody was employed to perform the co-IP analysis. IgG was used as a negative control.
Discussion
In this study, we have demonstrated that overexpression of Trx-1 blocked the pyroptosis in in vivo and in vitro models of AD via inhibiting TXNIP/NLRP3 pathway. Trx-1 inhibits NLRP3-mediated pyroptosis by regulating TXNIP expression and its interaction with NLRP3 in AD models. Simultaneously, we provide the possibility to in vitro study the neuroprotective mechanisms of Trx-1 in cellular model of AD by constructing the HT22 cell line stably overexpressing Trx-1.
AD is the most frequent cause of dementia and exhibits progressive changes in cognition and behavior20,21. Despite extensive research, there is no effective cure for AD due to its complex pathogenesis. Clinicians commonly use cholinesterase inhibitors and N-methyl-D-aspartate receptor antagonists to treat AD, such as Rivastigmine22 and Memantine23, but these drugs can only alleviate the symptoms of AD and are accompanied by large side effects. The newly launched Lecanemab is only a humanized monoclonal antibody for the Treatment of early AD patients24. Therefore, it is imperative to develop new and effective prevention and control strategies25,26,27. The Aβ deposition and the tau nerve fiber tangle are two major pathophysiological features of AD28. Aβ in AD leads to the abnormal phosphorylation of Tau by inducing the activation of p38 mitogen-activated protein kinase in cells. The latter causes the aggregation of paired helical filaments within neurons with neurofibrillary tangles, destabilizing microtubules, and causing subsequent loss of neuronal function25,26. Oxidative stress is an important component of the AD procession and is closely related to Aβ pathology. The oxidative damage of neurons in AD patients highlights the potential therapeutic role of antioxidants in inhibiting AD progression29,30.
Trx-1 has multiple biological functions, including anti-oxidation and anti-apoptosis31. Trx-1 plays an important neuroprotective role in AD and is a potential therapeutic target9. Our previous study has showed that Trx-1 inhibited Aβ-induced oxidative stress and promoted the mitochondrial biogenesis through activating the AMPK/Sirt1/PGC1α Pathway in AD models16. Trx-1 decreased the levels of p-Tau in the hippocampus of streptozotocin-induced AD mice32. As a reductase, Trx-1 plays important roles in inhibition of oxidative aggregation of proteins33. Another study from our group has demonstrated that AAV-mediated overexpression of human Trx-1 in the hippocampus of FAD4T APP/PS1 mice showed a neuroprotective role16. In the past few years, pyroptosis was considered as the key player in the neuronal loss in AD and targeting NLRP1 and NLRP3 inflammasome- mediated pyroptosis is beneficial for the development of novel therapies for AD17. And our latest study has reported that Trx-1 overexpression protected the hippocampal neurons in APP/PS1 mice through inhibiting NLRP1-mediated pyroptosis19. In this study, it was further elucidated that Trx-1 overexpression significantly attenuated the NLRP3-mediated pyroptosis in the hippocampus of APP/PS1 mice (Fig. 2). Our results are consistent with the previous study, in which the authors found that echinacoside (a phenylethanoid glycoside) treatment upregulated the expression of Trx-1 and downregulated the expression of NLRP3, as well as improved the cognitive impairment of APP/PS1 Mice34.
In order to in vitro study the inhibitory mechanism of Trx-1 on NLRP3 in AD, a hippocampal cell line overexpressing Trx-1 should be constructed. HT22 is a widely used mouse hippocampal neuronal cell line that has been extensively studied in AD35,36. In this study, LV-Trx-1 was designed (Fig. 4A) and transfected into HT22 cells (Fig. 4B). After puromycin screening, qPCR and WB were used to verify that LV-Trx-1 transfection of HT22 cells could achieve stable intracellular overexpression of Trx-1 mRNA and protein (Fig. 4C-F). The cytoprotection of LV-mediated Trx-1 overexpression against Aβ was estimated by using MTT analysis. After Aβ25−35 treatment, the viability of HT22 cells was significantly decreased and cell morphology was also changed. These changes were significantly restored by Trx-1 overexpression (Fig. 5A and B). LDH release by Aβ25−35 treatment was also inhibited in Trx-1 overexpressing HT22 cells (Fig. 5C). Apoptosis plays a crucial role in cell survival, cell homeostasis, and maintenance of optimal functional status37. Neurodegenerative manifestations can occur when they are abnormal and apoptosis may be involved in AD-related neural cell death38. Annexin V-mCherry staining showed that the quantity of apoptotic cells was markedly incremental after Aβ25−35 incubation, which was dramatically suppressed by overexpression of Trx-1 (Fig. 5D and E). What’s more, Aβ25−35 incubation prominently enhanced the activation of caspase-3, which was restored by Trx-1 overexpression (Fig. 5F and G), which are consistent with the results obtained in Aβ25−35-treated PC12 cells overexpressing Trx-116. These data suggest that Trx-1 overexpression play neuroprotective roles in AD neurons. Compared with differential PC12 cell line overexpressing Trx-1 used in our previous study19, the HT22 neuronal cell line overexpressing Trx-1 is a better tool to in vitro research the neuroprotective role of Trx-1 in AD model since it is a hippocampal neuronal cell line and can more accurately reflect the response of hippocampal neurons in AD. This newly established cell line will provide us opportunity to continue the exploration of the exact neuroprotective mechanisms of Trx-1 in AD.
It was further verified that Trx-1 overexpression inhibited NLRP3 activation in Aβ25−35-treated HT22 cells. As shown in Fig. 4A, B and F, Aβ25−35 treatment significantly increased the protein levels of NLRP3, which was reversed by Trx-1 overexpression. Besides that, Trx-1 inhibited the increase of downstream molecules of NLRP3, such as caspase-1, GSDMD and IL-1β (Fig. 6). These data suggest that Overexpression of Trx-1 can also exert inhibitory effects on NLRP3 activation in in vitro model of AD. Mechanically, TXNIP, an endogenous activator, can interact with NLRP3 and activate the NLRP3 inflammasome, mediate proinflammatory responses and lead to pyroptosis17. Therefore, the in vivo and in vitro expression of TXNIP was firstly examined by using WB and immunofluorescence staining. The TXNIP expression was significantly elevated in both the hippocampus of APP/PS1 mice and Aβ25−35-treated HT22 cells, which was inhibited by AAV- or LV-mediated Trx-1 overexpression (Fig. 7). The interaction between NLRP3 and TXNIP was further studied by employing co-IP. The results showed that Trx-1 overexpression weakened the interaction between TXNIP and NLRP3 in AD (Fig. 8). TXNIP acts as a natural inhibitor of Trx-1 in the cells39; Thus, Trx-1 overexpression can naturally enhance the interaction with TXNIP. Simultaneously, the expression of TXNIP was attenuated in Trx-1 overexpressing AD cells. These two effects synergistically reduced the activation of NLRP3 by TXNIP. Regarding the inhibitory effects of Trx-1 on NLRP3 activation by regulating TXNIP, only several natural products were reported in recent years34,40,41,42. Our findings more intuitively and specifically demonstrate the inhibitory effect of Trx-1 on NLRP3, as Trx-1 is expressed endogenously in the present study rather than induced exogenously.
Taken together, our findings demonstrate that Trx-1 inhibits the activation of NLRP3 through downregulating the levels of TXNIP and decreasing its interaction with NLRP3 and plays neuroprotective roles in AD. Considering the important neuroprotection of Trx-1, upregulating the expression of Trx-1 may be a potential therapeutic strategy for AD. Fortunately, several compounds, such as resveratrol, salidroside, estrogen and Dl-3-n-butylphthalide, have been reported to be Trx-1 inducers and have presented potential against AD13. Obviously, pharmacological activation of Trx-1 may be a feasible alternative to gene therapy for PD treatment.
Data availability
Data will be made available at the corresponding author (Xiansi Zeng) on reasonable request.
References
Crous-Bou, M., Minguillon, C., Gramunt, N. & Molinuevo, J. L. Alzheimer’s disease prevention: from risk factors to early intervention. Alzheimers Res. Ther. 9, 71. https://doi.org/10.1186/s13195-017-0297-z (2017).
Mantzavinos, V. & Alexiou, A. Biomarkers for Alzheimer’s disease diagnosis. Curr. Alzheimer Res. 14, 1149–1154. https://doi.org/10.2174/1567205014666170203125942 (2017).
Ren, R. et al. The China alzheimer report 2022. Gen. Psychiatr. 35, e100751. https://doi.org/10.1136/gpsych-2022-100751 (2022).
Knopman, D. S. et al. Alzheimer disease. Nat. Rev. Dis. Primers. 7, 33. https://doi.org/10.1038/s41572-021-00269-y (2021).
Benzi, G. & Moretti, A. Are reactive oxygen species involved in Alzheimer’s disease? Neurobiol. Aging. 16, 661–674. https://doi.org/10.1016/0197-4580(95)00066-n (1995).
Holmgren, A. Thioredoxin. Annu. Rev. Biochem. 54, 237–271. https://doi.org/10.1146/annurev.bi.54.070185.001321 (1985).
Zeng, X. S., Geng, W. S., Chen, L. & Jia, J. J. Thioredoxin as a therapeutic target in cerebral ischemia. Curr. Pharm. Des. 24, 2986–2992. https://doi.org/10.2174/1381612824666180820143853 (2018).
Jia, J. J., Geng, W. S., Wang, Z. Q., Chen, L. & Zeng, X. S. The role of thioredoxin system in cancer: strategy for cancer therapy. Cancer Chemother. Pharmacol. 84, 453–470. https://doi.org/10.1007/s00280-019-03869-4 (2019).
Jia, J. et al. Advances in the functions of thioredoxin system in central nervous system diseases. Antioxid. Redox Signal. 38, 425–441. https://doi.org/10.1089/ars.2022.0079 (2023).
Zeng, X. S., Jia, J. J., Kwon, Y., Wang, S. D. & Bai, J. The role of thioredoxin-1 in suppression of Endoplasmic reticulum stress in Parkinson disease. Free Radic Biol. Med. 67, 10–18. https://doi.org/10.1016/j.freeradbiomed.2013.10.013 (2014).
Zeng, X. et al. Thioredoxin-1 inhibits the activation of IRE1 by targeting Hsp90/p-Cdc37 chaperone complex in Parkinson disease. Ageing Res. Rev. 90, 102000. https://doi.org/10.1016/j.arr.2023.102000 (2023).
Gu, R. et al. Thioredoxin-1 decreases alpha-synuclein induced by MPTP through promoting autophagy-lysosome pathway. Cell. Death Discov. 10, 93. https://doi.org/10.1038/s41420-024-01848-0 (2024).
Jia, J., Zeng, X., Xu, G. & Wang, Z. The potential roles of redox enzymes in Alzheimer’s disease: focus on thioredoxin. ASN Neuro. 13, 1759091421994351. https://doi.org/10.1177/1759091421994351 (2021).
Akterin, S. et al. Involvement of glutaredoxin-1 and thioredoxin-1 in beta-amyloid toxicity and Alzheimer’s disease. Cell. Death Differ. 13, 1454–1465. https://doi.org/10.1038/sj.cdd.4401818 (2006).
Di Domenico, F. et al. Protein levels of heat shock proteins 27, 32, 60, 70, 90 and thioredoxin-1 in amnestic mild cognitive impairment: an investigation on the role of cellular stress response in the progression of alzheimer disease. Brain Res. 1333, 72–81. https://doi.org/10.1016/j.brainres.2010.03.085 (2010).
Jia, J. et al. Thioredoxin-1 promotes mitochondrial biogenesis through regulating AMPK/Sirt1/PGC1alpha pathway in Alzheimer’s disease. ASN Neuro. 15, 17590914231159226. https://doi.org/10.1177/17590914231159226 (2023).
Hu, B. et al. NLRP3/1-mediated pyroptosis: beneficial clues for the development of novel therapies for Alzheimer’s disease. Neural Regen Res. 19, 2400–2410. https://doi.org/10.4103/1673-5374.391311 (2024).
Jia, J., Zhang, X., Xu, G., Zeng, X. & Li, L. Thioredoxin-1 inhibits amyloid-beta25–35-induced activation of NLRP1/caspase-1/GSDMD pyroptotic pathway in PC12 cells. Mol. Biol. Rep. 49, 3445–3452. https://doi.org/10.1007/s11033-022-07177-8 (2022).
Jia, J., Liu, H., Sun, L., Xu, Y. & Zeng, X. Thioredoxin-1 protects neurons through inhibiting NLRP1-Mediated neuronal pyroptosis in models of Alzheimer’s disease. Mol. Neurobiol. 61, 9723–9734. https://doi.org/10.1007/s12035-024-04341-y (2024).
Grundke-Iqbal, I. et al. Abnormal phosphorylation of the microtubule-associated protein Tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. U S A. 83, 4913–4917. https://doi.org/10.1073/pnas.83.13.4913 (1986).
Hanseeuw, B. J. et al. Association of amyloid and Tau with cognition in preclinical alzheimer disease: A longitudinal study. JAMA Neurol. 76, 915–924. https://doi.org/10.1001/jamaneurol.2019.1424 (2019).
Khoury, R., Rajamanickam, J. & Grossberg, G. T. An update on the safety of current therapies for Alzheimer’s disease: focus on Rivastigmine. Ther. Adv. Drug Saf. 9, 171–178. https://doi.org/10.1177/2042098617750555 (2018).
Folch, J. et al. Memantine for the treatment of dementia: A review on its current and future applications. J. Alzheimers Dis. 62, 1223–1240. https://doi.org/10.3233/JAD-170672 (2018).
Boxer, A. L. & Sperling, R. Accelerating Alzheimer’s therapeutic development: the past and future of clinical trials. Cell 186, 4757–4772. https://doi.org/10.1016/j.cell.2023.09.023 (2023).
Feng, L., Li, J. & Zhang, R. Current research status of blood biomarkers in Alzheimer’s disease: diagnosis and prognosis. Ageing Res. Rev. 72, 101492. https://doi.org/10.1016/j.arr.2021.101492 (2021).
Gupta, G. L. & Samant, N. P. Current druggable targets for therapeutic control of Alzheimer’s disease. Contemp. Clin. Trials. 109, 106549. https://doi.org/10.1016/j.cct.2021.106549 (2021).
Breijyeh, Z. & Karaman, R. Comprehensive review on Alzheimer’s disease: causes and treatment. Molecules 25, 5789. https://doi.org/10.3390/molecules25245789 (2020).
Weller, J. & Budson, A. Current understanding of Alzheimer’s disease diagnosis and treatment. F1000Res 7 (F1000 Faculty Rev-1161). https://doi.org/10.12688/f1000research.14506.1 (2018).
Tarozzi, A. Oxidative stress in neurodegenerative diseases: from preclinical studies to clinical applications. J. Clin. Med. 9, 1223. https://doi.org/10.3390/jcm9041223 (2020).
Valko, M., Morris, H. & Cronin, M. T. Metals, toxicity and oxidative stress. Curr. Med. Chem. 12, 1161–1208. https://doi.org/10.2174/0929867053764635 (2005).
Haendeler, J. Thioredoxin-1 and posttranslational modifications. Antioxid. Redox Signal. 8, 1723–1728. https://doi.org/10.1089/ars.2006.8.1723 (2006).
Guo, Y. et al. Thioredoxin-1 is a target to attenuate Alzheimer-Like pathology in diabetic encephalopathy by alleviating Endoplasmic reticulum stress and oxidative stress. Front. Physiol. 12, 651105. https://doi.org/10.3389/fphys.2021.651105 (2021).
Awan, M. U. N. et al. The functions of thioredoxin 1 in neurodegeneration. Antioxid. Redox Signal. 36, 1023–1036. https://doi.org/10.1089/ars.2021.0186 (2022).
Qiu, H. & Liu, X. Echinacoside improves cognitive impairment by inhibiting Abeta deposition through the PI3K/AKT/Nrf2/PPARgamma signaling pathways in APP/PS1 mice. Mol. Neurobiol. 59, 4987–4999. https://doi.org/10.1007/s12035-022-02885-5 (2022).
Caldwell, J. D., Shapiro, R. A., Jirikowski, G. F. & Suleman, F. Internalization of sex hormone-binding Globulin into neurons and brain cells in vitro and in vivo. Neuroendocrinology 86, 84–93. https://doi.org/10.1159/000107072 (2007).
Murphy, T. H., Miyamoto, M., Sastre, A., Schnaar, R. L. & Coyle, J. T. Glutamate toxicity in a neuronal cell line involves Inhibition of cystine transport leading to oxidative stress. Neuron 2, 1547–1558. https://doi.org/10.1016/0896-6273(89)90043-3 (1989).
Sharma, V. K., Singh, T. G., Singh, S., Garg, N. & Dhiman, S. Apoptotic pathways and Alzheimer’s disease: probing therapeutic potential. Neurochem Res. 46, 3103–3122. https://doi.org/10.1007/s11064-021-03418-7 (2021).
Behl, C. Apoptosis and Alzheimer’s disease. J. Neural Transm (Vienna). 107, 1325–1344. https://doi.org/10.1007/s007020070021 (2000).
Billiet, L. et al. Thioredoxin-1 and its natural inhibitor, vitamin D3 up-regulated protein 1, are differentially regulated by PPARalpha in human macrophages. J. Mol. Biol. 384, 564–576. https://doi.org/10.1016/j.jmb.2008.09.061 (2008).
Xu, Z. et al. Sinensetin, a polymethoxyflavone from citrus fruits, ameliorates LPS-induced acute lung injury by suppressing Txnip/NLRP3/Caspase-1/GSDMD signaling-mediated inflammatory responses and pyroptosis. Food Funct. 15, 7592–7604. https://doi.org/10.1039/d4fo01704h (2024).
Lv, H. et al. Enhanced Keap1-Nrf2/Trx-1 axis by Daphnetin protects against oxidative stress-driven hepatotoxicity via inhibiting ASK1/JNK and Txnip/NLRP3 inflammasome activation. Phytomedicine 71, 153241. https://doi.org/10.1016/j.phymed.2020.153241 (2020).
Wang, C. Y. et al. Dl-3-n-Butylphthalide inhibits NLRP3 inflammasome and mitigates Alzheimer’s-Like pathology via Nrf2-TXNIP-TrX Axis. Antioxid. Redox Signal. 30, 1411–1431. https://doi.org/10.1089/ars.2017.7440 (2019).
Funding
This study was supported by the National Natural Science Foundation of China (82401428), Zhejiang Provincial Natural Science Foundation of China (Grant Nos.: LQ22H090003 and LTGY23C090001), the Sci-Tech Planning Project of Jiaxing (Nos.: 2021AY30001, 2022AY30020), and Jiaxing University Student Research and Training Program (8517241007).
Author information
Authors and Affiliations
Contributions
Conceptualization: Xiansi Zeng and Hongjun Liu. Experimental operation: Jinjing Jia, Zixuan Sheng, Yuqian Zhang, Lunxi Guo, Zhuo’er Chen and Dongsheng Zhu. Data analysis: Xiansi Zeng. Writing-original draft: Jinjing Jia, Zixuan Sheng and Yuqian Zhang. Funding acquisition: Jinjing Jia and Xiansi Zeng. Writing-review and editing: Xiansi Zeng and Hongjun Liu. All authors have approved the submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
The experiments were performed in accordance with guidelines for the use of Experimental Animal Ethics Committee of Jiaxing University Medical College (JUMC2021-144).
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jia, J., Sheng, Z., Zhang, Y. et al. Thioredoxin-1 inhibits NLRP3-mediated pyroptosis by regulating TXNIP in models of Alzheimer’s disease. Sci Rep 15, 16551 (2025). https://doi.org/10.1038/s41598-025-01636-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-01636-5
