
Abstract
Impaired neuroplasticity is a one of the key pathological mechanism of depression. Sirtuin 1 plays a crucial role in neuroplasticity; however, its precise mechanisms remain unclear. This study examined whether sirtuin 1 regulates dendritic outgrowth and spine formation via mTORC1 signaling in rat primary cortical cells under dexamethasone-induced neurotoxic conditions. Cortical cells were treated with SRT2104 (0.1, 1, and 10 µM), a selective sirtuin 1 activator, in the presence of dexamethasone (500 µM). Protein levels of sirtuin 1, mTORC1 signaling components, and synaptic markers (PSD-95 and GluA1) were analyzed by Western blotting, while dendritic outgrowth and spine density were assessed via immunofluorescence. SRT2104 significantly increased sirtuin 1 expression and ERK1/2 (a downstream target of sirtuin 1) phosphorylation. SRT2104 led to a substantial augmentation in the phosphorylation levels of mTORC1, as well as 4E-BP1 and p70S6K, which are downstream targets of mTORC1. Furthermore, SRT2104 led to an increase in dendritic outgrowth and spine density. Conversely, sirtuin 1 knockdown by siRNA transfection markedly reduced ERK1/2 and mTORC1 phosphorylation, as well as dendritic complexity and spine formation. These results suggest that sirtuin 1 promotes neuroplasticity by activating mTORC1 signaling, providing potential therapeutic implications for depression treatment.
Similar content being viewed by others


Introduction
Depression is a prevalent mental disorder that is pervasive in contemporary society. The condition is typified by symptoms including a depressed mood, a lack of motivation and interest, a sense of hopelessness, and suicidal thoughts. The prescription of antidepressants has been a continuous phenomenon since the 1950 s, with the majority of antidepressants medications functioning by increasing neurotransmitters such as serotonin and norepinephrine. Antidepressants have been proven to be effective, but these drugs have some limitations. Fifty to 60% of patients do not respond adequately to their first antidepressant treatment, and it takes weeks to months for antidepressants to show therapeutic effects1. This underscores the pressing need for the development of novel antidepressant agents that can exhibit a swift and potent response. Despite extensive research, the precise mechanisms underlying the pathophysiology of depression remain elusive. However, mounting evidence points to impaired neuroplasticity as a key contributor to the pathophysiology2,3,4.
Sirtuin 1 is a nicotinamide adenine dinucleotide (NAD+)-dependent histone deacetylase that plays a role in various metabolic and pathophysiological processes, including stress resistance, cell differentiation, apoptosis, autophagy, inflammation, and circadian rhythms5. In the brain, sirtuin 1 has been shown to play a role in learning, memory, and neuroplasticity6,7. Furthermore, sirtuin 1 has been shown to possess neuroprotective properties, which are effective against ischemic damage to the hippocampus8 and neurodegenerative diseases such as Alzheimer’s and Huntington’s disease9,10.
Recent studies have indicated the significance of sirtuin 1 in the aetiology of depression. In a whole-genome association analysis of 11,670 depressed patients, the sirtuin 1 gene has been identified at a genome-wide significant locus that contributes to the risk of depression11. Polymorphisms in the sirtuin 1 gene have been linked to depression and suicidal behavior12. Furthermore, a decline in the expression of sirtuin 1 mRNA has been observed in the peripheral blood of depressed patients13. In animal studies, stress reduced the activity of sirtuin 1 in the hippocampus, and genetic knockout of sirtuin 1 resulted in depression-like behavior14. Furthermore, sirtuin 1 activation has been demonstrated to prevent depression-like behavior and neuronal atrophy through the activation of its downstream regulator, extracellular signal-regulated protein kinase 1/2 (ERK1/2), suggesting that increased sirtuin 1 activity enhances neuroplasticity14. However, other studies have yielded conflicting results. For instance, brain-specific sirtuin 1 knockout mice exhibited reduced anxiety and increased resilience to stress, while mice with sirtuin 1 overexpression in the brain demonstrated heightened anxiety and vulnerability to stress15. Consequently, further research is necessary to elucidate the mechanisms through which sirtuin 1 impacts neuroplasticity.
In a clinical study, ketamine administration was found to improve depressive symptoms in patients with treatment-resistant depression, with antidepressant effects that were observed to last for a minimum of one week16,17. Li et al. reported that ketamine increases the expression of synaptic proteins and synaptogenesis through activation of the mechanistic target of rapamycin complex 1 (mTORC1) signaling in the prefrontal cortex (PFC) of mice, resulting in antidepressant effects18. The mechanism of action of mTORC1 involves the phosphorylation of eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1) and p70 ribosomal protein S6 kinase (p70S6K), which are downstream proteins of mTORC1 and regulate synaptic protein synthesis2,19. This signaling cascade has been shown to increase the expression of synaptic proteins, including postsynaptic density-95 (PSD-95) and the AMPA receptor GluA1 subunit, which have been demonstrated to enhance neuroplasticity2,19.
SRT2104 is a selective small-molecule activator of sirtuin 1 that binds to an allosteric N-terminal domain of the enzyme, thereby enhancing its deacetylase activity20. Preclinical studies have demonstrated that SRT2104 confers anti-inflammatory and neuroprotective effects in models of neurodegenerative and metabolic disorders, and clinical trials report that it is well tolerated in humans20. Notably, in rodent models of chronic stress-induced depression, SRT2104 treatment produced antidepressant-like effects, preventing the development of depression-related phenotypes14 while attenuating neuroinflammation and shifting microglial activation toward an anti-inflammatory (M2) phenotype21. These findings underscore the utility of SRT2104 as a pharmacological tool for investigating sirtuin 1-mediated mechanisms underlying neuroplasticity and mood disorders.
We hypothesized that sirtuin 1 may affect neuroplasticity through the activation of the mTORC1 signaling pathway. To order to ascertain this, we investigated whether SRT2104, a selective sirtuin 1 activator, affects neuroplasticity mediated by mTORC1 signaling in primary cortical cells under dexamethasone conditions. We also investigated whether the knockdown of sirtuin 1 in neurons would leads to changes in mTORC1 activity, dendritic outgrowth, and spine formation.
Results
Effects of SRT2104 on Sirtuin 1 signaling in primary cortical cells treated with dexamethasone
To establish a concentration range of SRT2104 that was not cytotoxic, cell viability was assessed using MTT assay prior to further analysis. SRT2104 was treated at various concentrations (0.1–50 µM) with or without dexamethasone (500 µM). No significant changes were observed in cortical cells treated with 0.1–10 µM of SRT2104, whereas treatment with 50 µM reduced cell viability by 64% (P < 0.001) in normal conditions and 48% (P < 0.001) in dexamethasone conditions (F[9, 70] = 45.900, P < 0.001, Fig. 1). Furthermore, treatment with 100 µM SRT2104 resulted in an even greater reduction in viability, with a 65% decrease under normal conditions and a 60% decrease under dexamethasone conditions (both P < 0.001, Supplementary information Fig. S1). Thus, 50 µM of SRT2104 was excluded from subsequent experiment.

Effects of SRT2104 on cell viability in primary cortical cells. Cortical cells were treated with SRT2104 (0.1, 1, 10, and 50 µM) or DMSO (non-drug treated control, final concentration 1%) for 4 days in the presence or absence of dexamethasone (500 µM). An MTT assay was performed (n = 8). The values represent the mean ± SEM expressed as a percentage of the control cells (DMSO-treated, non-dexamethasone-treated cells). ***P < 0.001 vs. control cells; †††P < 0.001 vs. DMSO-treated, dexamethasone-treated cells.
The effect of 0.1–10 µM of SRT2104 on sirtuin 1 expression was examined under dexamethasone conditions. One way-ANOVA revealed a significant effect on sirtuin 1 level (F[4, 35] = 4.320, P = 0.006, Fig. 2A). Sirtuin 1 expression was reduced by dexamethasone treatment (52% of control, P = 0.006), and this reduction was restored by SRT2104 with a significant increase at 1 µM (94% of control, P = 0.022). Based on these results, 0.1 and 1 µM of SRT2104 was used in the following experiment. Next, we examined the activity of ERK1/2, a downstream regulator of sirtuin 1. One way-ANOVA showed a significant effect on the phosphorylation level of ERK1/2 (F[3, 28] = 9.290, P < 0.001, Fig. 2B). Dexamethasone treatment reduced the level of phosphorylated ERK1/2 (63% of control, P < 0.001) and this reduction was blocked by treatment with 0.1 and 1 µM of SRT2104 (84% of control, P = 0.028 for 0.1 µM SRT2104; 88% of control, P = 0.007 for 1 µM SRT2104).

Effects of SRT2104 on the levels of sirtuin 1 expression and ERK1/2 phosphorylation in dexamethasone-treated cells. Cortical cells were treated with SRT2104 (0.1, 1, and 10 µM) or DMSO (non-drug treated control, final concentration 1%) for 4 days in the presence of dexamethasone (500 µM). The levels of sirtuin 1 (A) and phospho-Thr202/Tyr204-ERK1/2 (B) were determined by Western blotting (n = 8). Full-length uncropped gels for all Western blots are available in the Supplementary Information. The values represent the mean ± SEM expressed as a percentage of the control cells (DMSO-treated, non-dexamethasone-treated cells). ***P < 0.001 vs. control cells; †P < 0.05 vs. DMSO-treated, dexamethasone-treated cells.
Effects of SRT2104 on mTORC1 signaling in primary cortical cells treated with dexamethasone
To investigate the effects of SRT2104 on mTORC1 signaling under dexamethasone conditions, the phosphorylation levels of mTORC1, 4E-BP1, and p70S6K, as well as the expression levels of the synaptic protein PSD-95 and GluA1, were assessed by Western blotting.
One-way ANOVA revealed significant differences in the phosphorylated levels of mTORC1 (F[3, 28] = 4.520, P = 0.010, Fig. 3A), 4E-BP1 (F[3, 20] = 5.050, P = 0.009, Fig. 3B), and p70S6K (F[3, 28] = 10.100, P < 0.001, Fig. 3C). Dexamethasone resulted in significant decreases in the phosphorylated levels of mTORC1 (P = 0.040), 4E-BP1 (P = 0.016), and p70S6K (P < 0.001). These effects were significantly prevented by SRT2104 1 µM (119% of control, P = 0.006 for phosphor-Ser2448-mTORC1; 98% of control, P = 0.031 for phosphor-Thr37/46–4E-BP1; 99% of control, P = 0.013 for phosphor-Thr389-p70S6K).

Effects of SRT2104 on the levels of mTORC1, 4E-BP1 and p70S6K phosphorylation in dexamethasone-treated cells. Cortical cells were treated with SRT2104 (0.1 and 1 µM) or DMSO for 4 days in the presence of dexamethasone. The levels of phospho-Ser2448-mTORC1 (A), phospho-Thr37/46–4E-BP1 (B) and phospho-Thr389-p70S6K (C) were determined by Western blotting (n = 6–8). Full-length uncropped gels for all Western blots are available in the Supplementary Information. The values represent the mean ± SEM expressed as a percentage of control cells (DMSO-treated, non-dexamethasone-treated cells). *P < 0.05 or ***P < 0.001 vs. control cells; †P < 0.05, ††P < 0.01 or †††P < 0.001 vs. DMSO-treated, dexamethasone-treated cells.
Figure 4 shows effects of SRT2104 (0.1 and 1 µM) on the expression of PSD-95 (F[3, 20] = 8.920, P < 0.001, Fig. 4A) and GluA1 (F[3, 28] = 9.250, P < 0.001, Fig. 4B). The expression levels of PSD-95 and GluA1 were significantly reduced by dexamethasone (65% of control, P = 0.005 for PSD-95; 70% of control, P = 0.005 for GluA1), and these reductions were significantly reversed by SRT2104 1 µM (96% of control, P = 0.014 for PSD-95; 105% of control, P = 0.001 for GluA1).

Effects of SRT2104 on the levels of PSD-95 and GluA1 expression in dexamethasone-treated cells. Cortical cells were treated with SRT2104 (0.1 and 1 µM) or DMSO for 4 days in the presence of dexamethasone. The levels of PSD-95 (A) and GluA1 (B) were determined by Western blotting (n = 6). Full-length uncropped gels for all Western blots are available in the Supplementary Information. The values represent the mean ± SEM expressed as a percentage of control cells (DMSO-treated, non-dexamethasone-treated cells). **P < 0.01 vs. control cells; †P < 0.05 or ††P < 0.01 vs. DMSO-treated, dexamethasone-treated cells.
Effects of SRT2104 on dendritic outgrowth and spine density in primary cortical cells treated with dexamethasone
To investigate whether SRT2104 increases neuroplasticity, SRT2104 (0.1 and 1 µM) was added to cortical cells treated with dexamethasone and cultured for 5 days. Dendritic outgrowth and spine density were measured using antibodies against the dendritic marker MAP-2 and the spine marker phalloidin, respectively.
One-way ANOVA revealed significant effects on all measured morphological parameters: neurite length (F[3, 2396] = 26.600, P < 0.001; Fig. 5A-a), number of neurites (F[3, 2396] = 21.700, P < 0.001; Fig. 5A-b), branch points (F[3, 2396] = 9.360, P < 0.001; Fig. 5A-c), and soma area (F[3, 2396] = 46.700, P = 0.002; Fig. 5A-d). Dexamethasone exposure significantly reduced neurite length (169.44 μm vs. 142.85 μm), neurite number (4.27 vs. 3.49), branch points (3.31 vs. 2.56), and soma area (1254 μm² vs. 837 μm²) compared to control cells (all P < 0.001). Treatment with SRT2104 attenuated these deficits in a concentration-dependent manner. At 0.1 µM, partial recovery was observed across all parameters, while 1 µM SRT2104 fully restored neurite length, neurite number, complexity, and soma size to levels not significantly different from controls. Compared to the dexamethasone alone, 1 µM SRT2104 significantly increased neurite length (142.85 μm vs. 170.88 μm), neurite number (3.5 vs. 4.2), branch points (2.6 vs. 3.2), and soma area (837 μm² vs. 1290 μm²; all P < 0.001), indicating effective reversal of glucocorticoid-induced neuronal atrophy.

Effects of SRT2104 on dendritic outgrowth and spine density in dexamethasone-treated cells. Cortical cells were treated with SRT2104 (0.1 and 1 µM) or DMSO for 5 days in the presence of dexamethasone. In total, 600 cells from each group were analyzed using anti-MAP2 to assess neurite length (A-a), neurite number (A-b), branch points (A-c), and soma area (A-d). In total, 35 dendritic segments from each group were analyzed using phalloidin dye to assess spine formation (B). All data are expressed as the mean ± SEM. ***P < 0.001 vs. control cells (DMSO-treated, non-dexamethasone-treated cells); †††P < 0.001 vs. DMSO-treated, dexamethasone-treated cells.
Changes similar to those observed in dendritic outgrowth were observed for spine density (F[3,136] = 35.000, P < 0.001, Fig. 5B). Spine formation was significantly reduced in dexamethasone-treated cells in comparison with control cells (4.45 vs. 1.81, P < 0.001). Furthermore, SRT2104 significantly increased spine density in dexamethasone-treated cells (3.51 for 1 µM SRT2104, P < 0.001).
Effect of Sirtuin 1 knockdown on mTORC1 signaling, dendritic outgrowth and spine density
To explore the role of sirtuin 1 in the regulation of neuroplasticity mediated by mTORC1 signaling, cortical cells were induced to express sirtuin 1 knockdown. Sirtuin 1 expression was reduced by 42% in sirtuin 1 siRNA-transfected cells (t = 11.900, P < 0.001, Fig. 6A). Sirtuin 1 knockdown significantly decreased the phosphorylation levels of ERK1/2 (t(10) = 5.980, P < 0.001, Fig. 6B) and mTORC1 (t(10) = 7.440, P < 0.001, Fig. 6C) by 17% and 29%, respectively.

Effects of sirtuin 1 knockdown on the levels of ERK1/2 and mTORC1 phosphorylation in primary cortical cells. Cortical cells were transfected with fluorescein-labeled control or sirtuin 1 siRNA (100 nM) for 24 h. The levels of sirtuin 1 (A), phospho-Thr202/Tyr204-ERK1/2 (B) and phospho-Ser2448-mTORC1 (C) were determined by Western blotting (n = 6). Full-length uncropped gels for all Western blots are available in the Supplementary Information. The values represent the mean ± SEM expressed as a percentage of control cells (control siRNA-transfected cells). ***P < 0.001 vs. control cells.
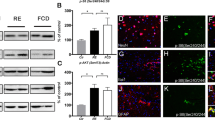
Figure 7A illustrates the morphological consequences of sirtuin 1 knockdown in primary neurons. Compared to control cells, sirtuin 1 siRNA significantly reduced total neurite length (231.97 μm vs. 188.15 μm; t(1198) = 11.900, P < 0.001; Fig. 7A-a), the number of primary neurites per cell (5.77 vs. 5.40; t(1198) = 2.590, P = 0.010; Fig. 7A-b), and dendritic branch points (8.50 vs. 6.94; t(1198) = 6.340, P < 0.001; Fig. 7A-c). Soma area was also significantly reduced following sirtuin 1 knockdown (1606 μm² vs. 1406 μm²; t(1198) = 2.860, P = 0.004; Fig. 7A-d). Moreover, dendritic spine density was markedly decreased in sirtuin 1 siRNA-transfected neurons relative to controls (3.39 vs. 2.31; t(88) = 3.190, P = 0.002; Fig. 7B), indicating that sirtuin 1 plays a critical role in maintaining synaptic architecture as well as overall neuronal complexity.

Effects of sirtuin 1 knockdown on dendritic outgrowth and spine density in primary cortical cells. Cortical cells were transfected with fluorescein-labeled control or sirtuin 1 siRNA (green) for 24 h. In total, 600 cells from each group were analyzed using anti-MAP2 (red) to assess neurite length (A-a), neurite number (A-b), branch points (A-c), and soma area (A-d). In total, 45 dendritic segments from each group were analyzed using phalloidin dye (red) for spine formation (B). All data are expressed as the mean ± SEM. All data are expressed as the mean ± SEM. *P < 0.05, **P < 0.01 or ***P < 0.001 vs. control cells (control siRNA-dtransfected cells).
Discussion
In this study, the activation of sirtuin 1 in primary cortical cells was found to enhance impaired neuroplasticity induced by dexamethasone. Specifically, this action of sirtuin 1 was associated with the activation of ERK1/2-mTORC1 signaling. Treatment with SRT2104, a selective activator of sirtuin 1, resulted in the activation of ERK1/2-mTORC1 signaling and increased dendritic outgrowth and spine density. On the other hand, reduction of endogenous sirtuin 1 led to decreased levels of ERK1/2 and mTORC1 phosphorylation as well as impaired dendritic outgrowth and spine density. These findings suggest that sirtuin 1/mTORC1 signaling in neurons is involved in the regulation of neuroplasticity, which may contribute to mechanisms of depression and antidepressant action.
Sirtuin 1 is distributed at high levels in the hippocampus and cortex22. The present study was conducted in cortical cells. Preclinical study has shown that the inhibition of sirtuin 1 via viral-mediated gene transfer in the hippocampus results in depression-like behavior in behavioral tests such as the social interaction test, forced swimming test, and novelty-suppressed feeding test, whereas activation of sirtuin 1 ameliorates these behaviors induced by chronic mild stress, suggesting that sirtuin 1 shows antidepressant-like effects14. Furthermore, activation of sirtuin 1 in the medial prefrontal cortex or hippocampus of mice by microinjection of SRT2104 reversed anhedonia and behavioral despair induced by chronic stress23,24. Conversely, genetic activation of sirtuin 1 in the nucleus accumbens led to anxiety- and depression-like behaviors25. The differential effects of sirtuin 1 activity may be dependent on specific brain regions.
Resveratrol, a nonspecific sirtuin activator, has been reported to ameliorate depression-like behaviors induced by lipopolysaccharide by increasing hippocampal neurogenesis9. These effects were associated with increased ERK1/2 activity and BDNF levels, which are implicated in mechanisms of antidepressant action24. Abe-Higuchi et al. reported that treatment with resveratrol promotes neurite outgrowth in Neuro2a cells grown in low serum conditions, and this effect is blocked by transfection with siRNAs against sirtuin 1 to knockdown the expression of sirtuin 114. Furthermore, activation of hippocampal sirtuin 1 by treatment with SRT2104 enhanced total dendritic length and dendritic spine along with an increase in ERK1/2 phosphorylation under chronic stress, which is consistent with finding of this study. The activation and inhibition of ERK1/2 via gene transfer in the hippocampus led to antidepressant- and depression-like behaviors, respectively14,26. The aforementioned studies underscore the role of ERK1/2 as a potential downstream target of sirtuin 1 on neuroplasticity. It is well established that ERK1/2 is associated with neuroplasticity27,28. Consequently, the present study hypothesized that sirtuin 1 may positively regulate mTORC1 signaling via downstream ERK1/2 to enhance neuroplasticity. In this study, activation of sirtuin 1 by SRT2104 increased phosphorylation of mTORC1 and downstream proteins 4E-BP1 and p70S6K under dexamethasone conditions, resulting in increased expression of synaptic proteins PSD-95 and GluA1. Moreover, sirtuin 1 knockdown using siRNA reduced ERK1/2 and mTORC1 phosphorylation, along with decreases in dendritic outgrowth and spine density. Consistently with this hypothesis, initial study has shown that sirtuin 1 overexpression activates ERK1/2-p70S6K signaling to antagonize cellular senescence in human diploid fibroblasts29. These findings also support a mechanistic pathway whereby sirtuin 1 activation enhances neuroplasticity via ERK1/2-dependent activation of mTORC1. It is well established that ERK1/2 promotes mTORC1 signaling by phosphorylating and inactivating TSC2, a negative regulator of mTORC1 activity30,31. In our previous study, mTORC1 activation induced by antidepressant treatment was completely abolished by co-administration of the ERK1/2 inhibitor PD98059, confirming that ERK1/2 signaling is required for mTORC1 phosphorylation in a comparable neuropharmacological context32. These results strengthen the proposed model of sirtuin 1-ERK1/2-mTORC1 signaling as a molecular mechanism underlying neuroplasticity in stress-induced neuronal impairment.
On the other hand, sirtuin 1 can regulate AMPA or PI3K pathways, potentially modulating mTORC1 indirectly. Sirtuin 1 has been shown to activate the energy sensor AMPK via deacetylation of the upstream kinase LKB133, and as a consequence mTORC1 signaling is inhibited under low-energy conditions by activated AMPK34. Sirtuin 1 can also modulate insulin/IGF signaling; sirtuin 1-dependent deacetylation of insulin receptor substrate-2 (IRS2) has been reported to increase its activity and thereby enhance PI3K/Akt signaling35. This interplay is further complicated by a feedback mechanism in which mTORC1-activated S6K1 phosphorylates IRS1, leading to attenuation of upstream PI3K/Akt signaling36. Although we did not directly investigate AMPK activation or conduct PI3K/Akt inhibition experiments such as LY294002 treatment in this study, these mechanisms are recognized as important contexts for understanding the crosstalk between sirtuin 1 and mTORC1 and should be thoroughly explored in future study.
Conversely, sirtuin 1 has been identified as a negative regulator of mTORC1 signaling via TSC1/2 complex in mouse embryonic fibroblast and HeLa cells37. Furthermore, in neuronal cultures isolated from human sirtuin 1-overexpressing transgenic mice, sirtuin 1 overexpression enhanced dendritic outgrowth and cell survival via negative regulation of mTORC1 signaling38. The potential explanations for these discrepancies may be attributable to the use of rodent models of varying strains and genetic background, in conjunction with the diverse experimental methods employed to assess sirtuin 1 activity. Further studies with antidepressants and mTORC1 inhibitors are needed to provide stronger evidence for a positive relationship between sirtuin 1 and mTORC1.
The PI3K/Akt pathway has been identified as a key regulator of cell survival and protein synthesis, often acting in parallel with mTORC1 signaling39. Previous studies have shown that inhibition of PI3K/Akt, ERK1/2, or mTORC1 each leads to a reduction in the phosphorylation of S6K and 4E-BP1, indicating that these pathways converge on shared downstream targets32. Given these interactions, it is possible that PI3K/Akt activity could compensate for mTORC1 inhibition under certain conditions. Although this study did not assess PI3K/Akt signaling, the presence of such compensatory mechanisms should be considered when interpreting the effects of SRT2104 on neuroplasticity. Future research should investigate the interplay between sirtuin 1, PI3K/Akt, and mTORC1 to further clarify their roles in synaptic adaptation.
In addition to modulating mTORC1 signaling, sirtuin 1 may exert neuroprotective effects through anti-inflammatory mechanisms. This NAD+-dependent deacetylase has been shown to deacetylate the NF-κB/RelA p65 subunit, thereby suppressing the transcription of pro-inflammatory cytokines40,41. Consistent with this, the selective sirtuin 1 activator SRT2104 significantly reduced levels of interleukin-6 (IL-6) and IL-1β while elevating the anti-inflammatory cytokine IL-10 in previous study24. Although inflammation-related endpoints were not measured in the present study, such anti-inflammatory actions may have contributed to the observed enhancement of neuroplasticity. Future studies should explore this pathway in greater detail to elucidate the role of sirtuin 1-mediated anti-inflammatory effects in neuroplasticity.
In this study, primary cortical cells were treated with dexamethasone, a synthetic glucocorticoid, to mimic hypothalamus-pituitary-adrenal (HPA) axis dysregulation characterized by glucocorticoid hypersecretion. The HPA axis is known to be activated by a feedback loop in response to stress, as follows: the hypothalamus secretes corticotropin-releasing hormone (CRH), which stimulates the pituitary gland to release adrenocorticotrophic hormone (ACTH), and ACTH in turn stimulates glucocorticoid release secreted from adrenal gland42. Depression has been associated with over-activation of HPA axis, including glucocorticoid hypersecretion due to disruption of the negative feedback loop43.
Antidepressants such as serotonin reuptake inhibitors have been demonstrated to inhibit the activation of the HPA axis44. Changes in neuroplasticity are mediated by high glucocorticoid levels45. In our previous studies, we demonstrated that a 500 µM dexamethasone concentration reduced neuroplasticity via inhibition of mTORC1 signaling in primary hippocampal cultures46,47. In present study, dexamethasone conditions resulted in a reduction in sirtuin 1 expression, which was restored by treatment with SRT2104. These results were consistent with a study showing that decreased expression and activity of sirtuin 1 in the hippocampus of chronically stressed mice was prevented by chronic imipramine treatment14.
Sirtuin 1 has been demonstrated to play a significant role in learning, memory, and cognitive function. Cognitive dysfunction is a known feature of depression. Overexpression of sirtuin 1 in the hippocampal neurons has been shown to result in an increase in dendritic arborization, suggesting that sirtuin 1 may play an important role in interconnected neurons and signal transduction in the neuronal network48. Sirtuin 1 levels were found to be decreased in aged neurons, a finding that may contribute to memory impairments49. Overexpression of sirtuin 1 in animal model of Alzheimer’s disease prevented memory impairment and enhanced cognition50,51. Similarly, overexpression of sirtuin 1 in a rat hippocampus attenuated propofol-induced cognitive dysfunction52. Particularly, resveratrol has been shown to prevent cognitive decline induced by chronic stress, with this effect being associated with increased CREB/BDNF expression through sirtuin 1 signaling53. Conversely, sirtuin 1 knockout mice exhibited impaired cognitive function, including reduced short-term memory, long-term memory, and spatial learning and exhibited reduced dendritic outgrowth and spine density in the hippocampal CA1 neurons6. Similar to these findings, the present study demonstrated that sirtuin 1 siRNA knockdown via RNA interference in primary cortical cells resulted in a reduction in dendritic outgrowth and spine density. In this regard, we highlight a potential link between sirtuin 1 and mTORC1. Further in vivo studies are required to fully elucidate the role of sirtuin 1 downstream signaling in cognitive function.
BDNF and CREB play critical roles in neuroplasticity by regulating synaptic function and neuronal survival54. Sirtuin 1 and ERK1/2 activation have also been linked to increased BDNF and CREB activity53. Previous study has shown that srituin 1 activation enhances BDNF expression, potentially via the deacetylation of transcriptional regulators such as PGC-1α and FOXO3a5. Additionally, mTORC1 has been implicated in promoting CREB phosphorylation, which in turn facilitates BDNF synthesis and synaptic plasticity55. Given that both pathways are key modulators of neuroplasticity, it is possible that SRT2104-induced sirtuin 1 activation influenced these targets. However, BDNF mRNA/protein levels and CREB phosphorylation were not directly assessed in this study, representing a limitation. Future studies should investigate these molecular changes to further elucidate the role of sirtuin 1 in neuroplasticity.
This study has several limitations. Although we primarily focused on mTORC1 signaling, mTORC2, through its core component Rictor, also plays a key role in neuroplasticity by regulating actin cytoskeleton remodeling and phosphorylating Akt at Ser47356,57. Previous studies have indicated that sirtuin 1 may influence mTORC2 indirectly through its interaction with mTORC1-linked pathways58but this was not addressed in the present work. Furthermore, functional synaptic analyses such as PSD-95/synaptophysin immunofluorescence, CDC42 activity assays, and calcium imaging were not conducted. To better understand the mechanisms by which sirtuin 1 influences synaptic remodeling, future studies should investigate the involvement of mTORC2 and incorporate direct assessments of synaptic function.
Our findings demonstrate that SRT2104 exhibits a hormetic dose-response, wherein low concentrations confer beneficial effects whereas higher concentrations are cytotoxic59,60. Specifically, exposure to 100 µM SRT2104 resulted in over 50% loss of cell viability (Supplementary information Fig. S1), consistent with previous reports that high concentrations of SRT2104 (50–100 µM) markedly reduce cell viability while sub-10 µM doses can enhance cell survival or proliferation60. Mechanistically, excessive sirtuin 1 activation has been reported to paradoxically increase reactive oxygen species (ROS) production and impair mitochondrial function, leading to oxidative damage61. In addition, sirtuin 1 interacts with mitochondrial sirtuins such as sirtuin 3 and sirtuin 6, which are essential for regulating mitochondrial homeostasis and oxidative stress responses62. Off-target effects involving mTORC2 may also play a role, as mTORC2 regulates cytoskeletal integrity and cellular survival via Akt Ser473 phosphorylation58. Moreover, apoptotic pathways may contribute to the observed cytotoxicity, as high-dose sirtuin 1 activation has been linked to increased caspase-3/7 activity and alterations in the Bcl-2/Bax ratio63. Collectively, these results underscore the importance of dose optimization and highlight the need for further studies to elucidate the mechanistic basis of high-dose effects of SRT2104 and to define its safe therapeutic window.
In conclusion, this study describes one of the molecular mechanisms that affect impaired neuroplasticity, which is known to be a potential mechanism underlying depression. The activation of sirtuin 1 has been shown to enhance neuroplasticity through the upregulation of mTORC1 signaling, thereby increasing dendritic outgrowth and spine density in dexamethasone-induced neuronal cells. These results provide meaningful evidence linking sirtuin 1 activation, mTORC1 signaling, and neuroplasticity. These findings also underscore a potential interplay between sirtuin 1 and mTORC1 pathways in regulating neuroplasticity. Further studies are needed to explore additional mechanisms that may contribute to sirtuin 1-mediated neuroplasticity, such as sirtuin 1-neuroinflammation crosstalk, compensatory pathways (e.g., PI3K/Akt), and functional synaptic adaptation. Such investigation will help to define the therapeutic potential of sirtuin 1 activation.
Materials and methods
Primary cortical culture
All procedures were conducted in accordance with the guidelines for the Institutional Animal Care and Use Committee (IACUC), Inje University, Republic of Korea, and followed the ARRIVE guidelines64. The animal experiments were approved by the IACUC of the College of Medicine Inje University (approval no. 2023-001). Primary cortical cells were cultured using a method similar to that developed by Kaech and Banker65. Pregnant Sprague-Dawley rats (Hanna Biotech, Pyeongtaek, Korea) were euthanized using carbon dioxide (CO2) gas. The brains were harvested from rat fetuses (embryonic day 19), and the meninges were removed. The cerebral cortex was then dissected and reacted in the Neurobasal™ (NB) culture medium (Gibco BRL, Waltham, MA, USA) with 0.03% Trypsin-EDTA (0.05%, phenol red, Gibco BRL) for 20 min at room temperature. The cells were suspended in NB culture medium containing the following components: 10% B27™ supplement (B27, serum free, Gibco BRL), 1% fetal bovine serum (FBS, Gibco BRL), 1% horse serum (HS, Gibco BRL), 0.25% L-glutamine (Gibco BRL), and 1% Penicillin-Streptomycin (Pen/Strep, Gibco BRL) and subsequently seeded onto culture dishes coated with 100 ng/ml of Poly-D-lysine (PDL, Sigma-Aldrich, St. Louis, MO, USA). For cell viability analyses, cells were seeded at a density of 1 × 104 cells per 96-well dish. For Western blotting analyses, cells were seeded at 2 × 105 cells per 6-well dish. For immunofluorescence, cells were seeded on 18 × 18 mm coverslips in 12-well dishes at a density of 2 × 104 (dendritic outgrowth) and 5 × 103 cells (spine density). Cells were then cultured at 37 °C and 5% CO2 for 10 days.
Drug treatment
SRT2104 (10 mM; Selleckchem, Houston, TX, USA) and dexamethasone (100 mM; Sigma-Aldrich) were dissolved in dimethyl sulfoxide (DMSO) and subsequently diluted to concentrations ranging from 0.1 to 50 µM for SRT2104 and from 500 µM for dexamethasone, using a final concentration of DMSO of less than 1%. Following a 10-day incubation period, the cells were exposed to SRT2104 (0.1, 1, and 10 µM) in the presence of dexamethasone (500 µM) for a further 4 days (for cell viability and Western blotting analyses) and 5 days (for immunofluorescence). The culture medium and these drugs were changed every 2 days. The choice of 500 µM dexamethasone was informed by prior studies demonstrating 75–80% cell viability at this concentration47. The concentrations of SRT2104 used in this study were determined on the basis of observations indicating that concentrations of 0.1, 1, and 10 µM exhibited no evidence of toxicity (see Fig. 1). However, concentrations of 50 µM and above were found to result in a reduction in cell viability. Furthermore, SRT2104 at concentrations ranging from 0.1 to 10 µM was observed to enhance Sirtuin 1 expression, while lower concentrations (less than 0.1 µM) exhibited no discernible effect on Sirtuin 1 expression levels (data not shown).
Cell viability
To determine the cytotoxic effect of SRT2104, cell viability was analyzed using 3-[4,5-dimethylthiazol-2-yl]−2,5-diphenyltetrazolium bromide (MTT; Amresco, Solon, OH, USA). MTT of 0.5 mg/ml was added and reacted for 4 h in a 37 °C. Following this, the MTT was removed, and DMSO (GenDEPOT, Katy, TX, USA) was added to dissolve the formazan produced by the reaction. The dissolved solution was transferred to 96-well dishes, and the absorbances were measured at a wavelength of 570 nm using a microplate reader (SpectraMax M2e; Molecular Devices, Cridersville, OH, USA).
Knockdown of Sirtuin 1 using small interference RNA (siRNA)
Sirtuin 1 knockdown using siRNA was performed as follow. Chemically synthesized double-stranded AccTarget™ fluorescein-labeled siRNAs were purchased from Bioneer (Daejeon, Korea). The catalog numbers of sirtuin 1 specific and control (non-silencing RNA) siRNAs were 93759-3 (sense: 5’-GACCGUUUAAUGACUGGAU-3’; antisense: 5’-AUCCA GUCAUUAAACGGUC-3’) and SN-1022 (sense: 5’-CCUACGCCACCAAUUUCGU-3’; antisense: 5’-ACGAAAUUGGUGGCGUAGG-3’), respectively. Primary cortical cells that had reached 60–80% confluence in a 12-well dish were subjected to transfection with each siRNA at a concentration of 100 nM using Lipofectamine™ RNAiMAX and Opti-MEM (Gibco BRL) according to the manufacturer’s protocol (Invitrogen, Carlsbad, CA, USA). The culture medium was changed after 6 h. Western blotting and immunofluorescence were performed 2 days after siRNA transfection.
Western blotting analyses
Cortical cells were lysed in RIPA cell lysis buffer (GenDEPOT) with protease inhibitor cocktail solution (GenDEPOT) and phosphatase inhibitor cocktail solution (GenDEPOT). Protein concentration was then measured using the Bradford protein assay plus kit (GenDEPOT). The total protein lysates of 20 µg were then subjected to electrophoresis using 8% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The polyvinylidene difluoride membranes (Millipore, Billerica, MA, USA) were then used to transfer the protein samples. The membranes were then blocked in 5% skim milk powder blocking solution [0. 15% Tween-20, triple-buffered saline (TBS-T, GenDEPOT)] for 1 h. Immunoblotting was performed with following primary antibodies: anti-sirtuin1(07–131) (Millipore, USA), anti-phospho-Thr202/Tyr204-ERK1/2 (#9101), anti-total ERK1/2 (#4695), anti-phospho-Ser2448-mTORC1 (#2971), anti-total-mTORC1 (#2972), anti-phospho-Thr37/46−4E-BP1 (#2855), anti-total 4E-BP1 (#9452), anti-phospho-Thr389-p70S6K (#9205), anti-total-p70S6K (#9202), anti-PSD-95 (#3450), and anti-GluA1 (#13185) (Cell signaling, Danvers, MA, USA) [1: 1,000], anti-α-tubulin (T9026, Sigma) [1:2,000] in TBS-T at 4 °C overnight. The membranes were washed in TBS-T for 10 min. These were reacted for 1 h in TBST-T containing horseradish peroxidase-conjugated secondary antibodies: mouse-anti-rabbit IgG (1:2,000, Santa Cruz, Dallas, TX, USA, sc-2357) and anti-mouse IgG (1:10,000, Sigma-Aldrich, A4416) at room temperature. Protein bands were visualized on an Amersham™ Imager 600 (AI600) (GE Healthcare, Chicago, IL, USA) using Clarity Western enhanced chemiluminescence solution (GenDEPOT) for 1 min and quantified using Multi-Gauge Software (Fuji Photo Film Co, Ltd., Tokyo, Japan).
Immunofluorescence
The cells were fixed in 4% paraformaldehyde (Biosesang, Seongnam, Korea) for 20 min at room temperature and then permeabilized in phosphate-buffered saline (PBS, GenDEPOT) containing 0.1% triton X-100 for 3 min. The cells were blocked using PBS containing 4% bovine serum albumin for 2 h. For dendritic outgrowth, the cells were treated with anti-microtubule-associated protein-2 (MAP-2) antibody (1:1,000, MAB341, Millipore) for 2 h as primary antibody. Alexa Fluor™ 568 goat anti-mouse IgG (1: 1,000, A-11031, Invitrogen) was used as a secondary antibody, and this was reacted for 1 h. For nuclear staining, Hoechst 33,258 (1:10,000, H21491, Invitrogen) was reacted for 5 min. To immunostain the spine, cells were treated with anti-Alexa Fluor™ 488 Phalloidin (1:40, A12379, Invitrogen) or anti-Alexa Fluor™ 594 Phalloidin (1:40, A12381, Invitrogen) for 4 h. Stained cells were mounted with mounting solution (Biфmeda, Electron Microscopy Sciences, Foster City, CA, USA). Dendritic outgrowth was observed and imaged using a fluorescence in situ hybridization (FISH) microscope (BX51, Olympus, Tokyo, Japan). Spine formation was observed by means of a confocal laser microscope (A1+, Nikon, Tokyo, Japan). The images were obtained with a resolution of 2048 × 2048 pixels for dendritic outgrowth and 1024 × 1024 pixels for spine density.
Immunostaining analyses for dendritic outgrowth and spine density
To analyze the total dendritic length, five fields were randomly selected from each group. The total number of neurons in these fields was counted. Neurite outgrowth was quantified using the automated image-analysis software MetaMorph® (Molecular Devices, USA)47,66. The following morphological parameters were measured for each neuron: total neurite length (µm), number of primary processes, number of branches, and soma area (µm²). Total neurite length was defined as the cumulative length of all neurites extending from the soma. The number of processes referred to the primary neurites emerging directly from the soma, while the number of branches indicated the total number of secondary and higher-order branches. The soma area represented the two-dimensional area of the neuronal cell body. This measure was then averaged across each group. At least 200 cells were analyzed in 5 fields by a researcher blinded to the groups. To analyze spine formation, we differentiated between spines and filopodia based on shape and length. Spines were defined as being less than 3 μm long and round or mushroom-shaped. Filopodia were defined as being 3–10 μm long and narrow in shape. Thus, less than 3 μm were counted47. Ten neurons were randomly selected from each group. Two 50 μm dendritic segments per neuron were analyzed by a researcher blinded to the groups. To calculate the average spine density of a 10 μm dendrite segment, the number of spines in 50 μm dendritic segment was divided by 5.
Statistical analysis
The data from all experiments were recorded from at least three independent cultures and expressed as mean ± standard error of the mean (SEM). Statistical analyses were performed using GraphPad Prism software (version 10.0.2; GraphPad software, Inc., San Diego, CA, USA). One-way ANOVAs was used and significant effects were evaluated with Tukey’s post hoc tests. The unpaired t-test was used to compare the two groups. A value of P < 0.05 was considered to be statistically significant.
Data availability
Data is available on request from the corresponding authors.
References
Artigas, F. Limitations to enhancing the speed of onset of antidepressants-are rapid action antidepressants possible? Hum. Psychopharmacol. 16, 29–36. https://doi.org/10.1002/hup (2001).
Duman, R. S. & Aghajanian, G. K. Synaptic dysfunction in depression: potential therapeutic targets. Science 338, 68–72. https://doi.org/10.1126/science.1222939 (2012).
Malykhin, N. V. & Coupland, N. J. Hippocampal neuroplasticity in major depressive disorder. Neuroscience 309, 200–213. http://doi.org/ (2015).
McEwen, B. S., Eiland, L., Hunter, R. G. & Miller, M. M. Stress and anxiety: structural plasticity and epigenetic regulation as a consequence of stress. Neuropharmacology 62, 3–12. https://doi.org/10.1016/j.neuropharm.2011.07.014 (2012).
Michan, S. & Sinclair, D. Sirtuins in mammals: insights into their biological function. Biochem. J. 404, 1–13. https://doi.org/10.1042/BJ20070140 (2007).
Gao, J. et al. A novel pathway regulates memory and plasticity via SIRT1 and miR-134. Nature 466, 1105–1109. https://doi.org/10.1038/nature09271 (2010).
Michán, S. et al. SIRT1 is essential for normal cognitive function and synaptic plasticity. J. Neurosci. 30, 9695–9707. https://doi.org/10.1523/JNEUROSCI.0027-10.2010 (2010).
Raval, A. P. et al. Resveratrol and ischemic preconditioning in the brain. Curr. Med. Chem. 15, 1545–1551. https://doi.org/10.2174/092986708784638861 (2008).
Pallàs, M. et al. Modulation of SIRT1 expression in different neurodegenerative models and human pathologies. Neuroscience 154, 1388–1397. https://doi.org/10.1016/j.neuroscience (2008). 2008.04.065.
Qin, W. et al. Neuronal SIRT1 activation as a novel mechanism underlying the prevention of alzheimer disease amyloid neuropathology by calorie restriction. J. Biol. Chem. 281, 21745–21754. https://doi.org/10.1074/jbc.M60290920 (2006).
CONVERGE consortium. Sparse whole-genome sequencing identifies two loci for major depressive disorder. Nature 523, 588–591. https://doi.org/10.1038/nature14659 (2015).
Nivoli, A. et al. Association between Sirtuin 1 gene rs10997870 polymorphism and suicide behaviors in bipolar disorder. Neuropsychobiology 74, 1–7. https://doi.org/10.1159/000446921 (2016).
Luo, X. J. & Zhang, C. Down-Regulation of SIRT1 gene expression in major depressive disorder. Am. J. Psychiatry. 173, 1046. https://doi.org/10.1176/appi.ajp.2016.16040394 (2016).
Abe-Higuchi, N. et al. Hippocampal Sirtuin 1 signaling mediates Depression-like behavior. Biol. Psychiatry. 80, 815–826. https://doi.org/10.1016/j.biopsych.2016.01.009 (2016).
Libert, S. et al. SIRT1 activates MAO-A in the brain to mediate anxiety and exploratory drive. Cell 147, 1459–1472. https://doi.org/10.1016/j.cell.2011.10.054 (2011).
Zarate, C. A. Jr. et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch. Gen. Psychiatry. 63, 856–864. https://doi.org/10.1001/archpsyc.63.8.856 (2006).
Coyle, C. M. & Laws, K. R. The use of ketamine as an antidepressant: a systematic review and meta-analysis. Hum. Psychopharmacol. 30, 152–163. https://doi.org/10.1002/hup.2475 (2015).
Li, N. et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 329, 959–964. https://doi.org/10.1126/science.1190287 (2010).
Duman, R. S. & Voleti, B. Signaling pathways underlying the pathophysiology and treatment of depression: novel mechanisms for rapid-acting agents. Trends Neurosci. 35, 47–56. https://doi.org/10.1016/j.tins.2011.11.004 (2012).
Schultz, M. B., Rinaldi, C., Lu, Y., Amorim, J. A. & Sinclair, D. A. Molecular and cellular characterization of SIRT1 allosteric activators. Methods Mol. Biol. 1983, 133–149. https://doi.org/10.1007/978-1-4939-9434-2_8 (2019).
Chang, N. et al. Emerging roles of SIRT1 activator, SRT2104, in disease treatment. Sci. Rep. 14, 5521. https://doi.org/10.1038/s41598-024-55923-8 (2024).
Zakhary, S. M. et al. Distribution analysis of deacetylase SIRT1 in rodent and human nervous systems. Anat. Rec (Hoboken). 293, 1024–1032. https://doi.org/10.1002/ar.21116 (2010).
Lei, Y. et al. SIRT1 in forebrain excitatory neurons produces sexually dimorphic effects on depression-related behaviors and modulates neuronal excitability and synaptic transmission in the medial prefrontal cortex. Mol. Psychiatry. 25, 1094–1111. https://doi.org/10.1038/s41380-019-0352-1 (2020).
Duan, C. M. et al. SRT2104 attenuates chronic unpredictable mild stress-induced depressive-like behaviors and imbalance between microglial M1 and M2 phenotypes in the mice. Behav. Brain Res. 378, 112296. https://doi.org/10.1016/j.bbr.2019.112296 (2020).
Kim, H. D. et al. SIRT1 mediates Depression-Like behaviors in the nucleus accumbens. J. Neurosci. 36, 8441–8452. https://doi.org/10.1523/JNEUROSCI.0212-16.2016 (2016).
Ferland, C. L. et al. Sirtuin activity in dentate gyrus contributes to chronic stress-induced behavior and extracellular signal-regulated protein kinases 1 and 2 cascade changes in the hippocampus. Biol. Psychiatry. 74, 927–935. https://doi.org/10.1016/j.biopsych.2013.07 (2013).
Alonso, M. et al. Mitochondrial extracellular signal-regulated kinases 1/2 (ERK1/2) are modulated during brain development. J. Neurochem. 89, 248–256. http://doi.org/10.1111/j. 1471-4159.2004.02323.x (2004).
Impey, S., Obrietan, K. & Storm, D. R. Making new connections: role of ERK/MAP kinase signaling in neuronal plasticity. Neuron 23 (00), 11–14. https://doi.org/10.1016/s0896-6273 (1999).
Huang, J. et al. SIRT1 overexpression antagonizes cellular senescence with activated ERK/S6k1 signaling in human diploid fibroblasts. PLoS One. 3, e1710. https://doi.org/10.1371/journal.pone.0001710 (2008).
Carrière, A. et al. Oncogenic MAPK signaling stimulates mTORC1 activity by promoting RSK-mediated raptor phosphorylation. Curr. Biol. 18, 1269–1277. http://doi.org/10.1016/j. cub.2008.07.078 (2008).
Dunkerly-Eyring, B. L. et al. Single Serine on TSC2 exerts biased control over mTORC1 activation mediated by ERK1/2 but not Akt. Life Sci. Alliance. 5, e202101169. https://doi.org/10.26508/lsa.202101169 (2022).
Park, S. W. et al. Differential effects of antidepressant drugs on mTOR signalling in rat hippocampal neurons. Int. J. Neuropsychopharmacol. 17, 1831–1846. https://doi.org/10.1017/S1461145714000534 (2014).
Ruderman, N. B. et al. AMPK and SIRT1: a long-standing partnership? Am. J. Physiol. Endocrino L Metab. 298, E751–E760. https://doi.org/10.1152/ajpendo.00745.2009 (2010).
Bond, P. Regulation of mTORC1 by growth factors, energy status, amino acids and mechanical stimuli at a glance. J. Int. Soc. Sports Nutr. 13, 8. https://doi.org/10.1186/s12970-016-0118-y (2016).
Pillai, V. B., Sundaresan, N. R. & Gupta, M. P. Regulation of Akt signaling by sirtuins: its implication in cardiac hypertrophy and aging. Circ. Res. 114, 368–378. https://doi.org/10.1161/CIRCRESAHA.113.300536 (2014).
Melemedjian, O. K. et al. mTORC1 inhibition induces pain via IRS-1-dependent feedback activation of ERK. Pain 154, 1080–1091. (2013). https://doi.org/10.1016/j.pain.2013.03.021
Ghosh, H. S., McBurney, M. & Robbins, P. D. SIRT1 negatively regulates the mammalian target of Rapamycin. PLoS One. 5, e9199. https://doi.org/10.1371/journal.pone.0009199 (2010).
Guo, W. et al. Sirt1 overexpression in neurons promotes neurite outgrowth and cell survival through Inhibition of the mTOR signaling. J. Neurosci. Res. 89, 1723–1736. https://doi.org/10.1002/jnr.22725 (2011).
Manning, B. D., Toker, A. A. K. T. P. K. B. & Signaling Navigating the network. Cell 169, 381–405. https://doi.org/10.1016/j.cell.2017.04.001 (2017).
Yeung, F. et al. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 23, 2369–2380. https://doi.org/10.1038/sj.emboj.7600244 (2004).
Yang, H. et al. SIRT1 activators suppress inflammatory responses through promotion of p65 deacetylation and Inhibition of NF-κB activity. PLoS One. 7, e46364. https://doi.org/10.1371/journal.pone.0046364 (2012).
Joseph, J. J. & Golden, S. H. Cortisol dysregulation: the bidirectional link between stress, depression, and type 2 diabetes mellitus. Ann. N Y Acad. Sci. 1391, 20–34. https://doi.org/10.1111/nyas.13217 (2017).
Troubat, R. et al. Neuroinflammation and depression: A review. Eur. J. Neurosci. 53, 151–171. https://doi.org/10.1111/ejn.14720 (2021).
Paslakis, G., Heuser, I., Schweiger, U. & Deuschle, M. A single DEX/CRH test in male drug-free depressed patients is associated with the clinical response to treatment with Fluoxetine. J. Psychiatr Res. 44, 1154–1157. https://doi.org/10.1016/j.jpsychires.2010.04 (2010).
Hill, A. R. & Spencer-Segal, J. L. Glucocorticoids and the brain after critical illness. Endocrinology 162, bqaa242. https://doi.org/10.1210/endocr/bqaa242 (2021).
Seo, M. K. et al. AMPA receptor-mTORC1 signaling activation is required for neuroplastic effects of LY341495 in rat hippocampal neurons. Sci. Rep. 10, 993. https://doi.org/10.1038/s41598-020-58017-3 (2020).
Park, S. W. et al. Liraglutide activates mTORC1 signaling and AMPA receptors in rat hippocampal neurons under toxic conditions. Front. Neurosci. 12, 756. https://doi.org/10.3389/fnins.2018.00756 (2018).
Codocedo, J. F., Allard, C., Godoy, J. A., Varela-Nallar, L. & Inestrosa, N. C. SIRT1 regulates dendritic development in hippocampal neurons. PLoS One. 7, e47073. https://doi.org/10.1371/journal.pone.0047073 (2012).
Quintas, A., de Solís, A. J., Díez-Guerra, F. J., Carrascosa, J. M. & Bogónez, E. Age-associated decrease of SIRT1 expression in rat hippocampus: prevention by late onset caloric restriction. Exp. Gerontol. 47, 198–201. https://doi.org/10.1016/j.exger.2011.11.010 (2012).
Corpas, R. et al. SIRT1 overexpression in mouse Hippocampus induces cognitive enhancement through proteostatic and neurotrophic mechanisms. Mol. Neurobiol. 54, 5604–5619. https://doi.org/10.1007/s12035-016-0087-9 (2017).
Wang, R., Zhang, Y., Li, J. & Zhang, C. Resveratrol ameliorates Spatial learning memory impairment induced by Aβ1–42 in rats. Neuroscience 344, 39–47. http://doi.org/10.1016/j. neuroscience.2016.08.051 (2017).
Ma, L. H. et al. Hippocampal SIRT1-Mediated synaptic plasticity and glutamatergic neuronal excitability are involved in prolonged cognitive dysfunction of neonatal rats exposed to Propofol. Mol. Neurobiol. 59, 1938–1953. https://doi.org/10.1007/s12035-021-02684-4 (2022).
Shen, J., Xu, L., Qu, C., Sun, H. & Zhang, J. Resveratrol prevents cognitive deficits induced by chronic unpredictable mild stress: Sirt1/miR-134 signalling pathway regulates CREB/BDNF expression in hippocampus in vivo and in vitro. Behav. Brain Res. 349, 1–7. https://doi.org/10.1016/j.bbr.2018.04.050 (2018).
Park, H. & Poo, M. M. Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci. 214, 7–23. https://doi.org/10.1038/nrn3379 (2013).
Moya-Alvarado, G. et al. BDNF/TrkB signaling endosomes in axons coordinate CREB/ mTOR activation and protein synthesis in the cell body to induce dendritic growth in cortical neurons. Elife 12, e77455. https://doi.org/10.7554/eLife.77455 (2023).
Sarbassov, D. D. et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 14, 1296–1302. https://doi.org/10.1016/j.cub.2004.06.054 (2004).
Huang, W. et al. mTORC2 controls actin polymerization required for consolidation of long-term memory. Nat. Neurosci. 16, 441–448. https://doi.org/10.1038/nn.3351 (2013).
Cohen, H. Y. et al. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science 305, 390–392. https://doi.org/10.1126/science.1099196 (2004).
Sapuleni, J., Szymanska, M. & Meidan, R. Diverse actions of sirtuin-1 on ovulatory genes and cell death pathways in human granulosa cells. Reprod. Biol. Endocrinol. 20, 104. https://doi.org/10.1186/s12958-022-00970-x (2022).
Miyaji, N. et al. Inhibition of knee osteoarthritis progression in mice by administering SRT2014, an activator of silent information regulator 2 ortholog 1. Cartilage 13, 1356S–1366S. https://doi.org/10.1177/1947603519900795 (2021).
Kozako, T. et al. SRT1720 induces SIRT1-independent cell death in adult T-cell leukemia/ lymphoma. FEBS J. 289, 3477–3488. https://doi.org/10.1111/febs.16353 (2022).
Kitada, M., Ogura, Y., Monno, I. & Koya, D. Sirtuins and type 2 diabetes: role in inflammation, oxidative stress, and mitochondrial function. Front. Endocrinol. (Lausanne. 10, 187. https://doi.org/10.3389/fendo.2019.00187 (2019).
van Ginkel, P. R. et al. Resveratrol inhibits tumor growth of human neuroblastoma and mediates apoptosis by directly targeting mitochondria. Clin. Cancer Res. 13, 5162–5169. https://doi.org/10.1158/1078-0432.CCR-07-0347 (2007).
Kilkenny, C., Browne, W. J., Cuthill, I. C., Emerson, M. & Altman, D. G. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol. 8, e1000412. https://doi.org/10.1371/journal.pbio.1000412 (2010).
Kaech, S. & Banker, G. Culturing hippocampal neurons. Nat. Protoc. 1, 2406–2415. https://doi.org/10.1038/nprot.2006.356 (2006).
Klimaschewski, L., Nindl, W., Pimpl, M., Waltinger, P. & Pfaller, K. Biolistic transfection and morphological analysis of cultured sympathetic neurons. J. Neurosci. Methods. 113, 63–71. https://doi.org/10.1016/S0165-0270(01)00473-3 (2002).
Acknowledgements
This work was supported by the 2024 Inje University research grant.
Author information
Authors and Affiliations
Contributions
Conceptualization: SWP, JGL, and MKS; Formal analysis: MKS, JHK, and JYL; Funding acquisition: SWP; Investigation: JGL, SWP, MKS, and JHK; Methodology: MKS, JHK, DHS, SWP, and JGL; Resources: MKS, DHS, and SHK; Supervision: SWP; Writing-original draft: SWP; Editing: MKS, DHS, SHK and SHJ. All authors have read and agreed to the published version of the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics statement
The animal study was reviewed and approved by Inje Medical College Committee for Animal Experimentation and the Institutional Animal Laboratory Review Board (Approval No. 2023-001).
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Seo, M.K., Lee, J.G., Kim, J.H. et al. SRT2104 enhances dendritic outgrowth and spine formation through Sirtuin 1-mediated mTORC1 signaling. Sci Rep 15, 21283 (2025). https://doi.org/10.1038/s41598-025-06203-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-06203-6
