
Abstract
The transition from acute kidney injury (AKI) to chronic kidney disease (CKD) remains a critical clinical challenge with limited therapeutic options. To our knowledge, this was the first study to investigate the renoprotective effects of silibinin, a flavonolignan from Silybum marianum, in mitigating AKI-to-CKD progression via modulation of MAPK and PI3K/AKT signaling pathways—addressing a critical unmet need, as no targeted therapies currently exist to block this transition. For in vivo evaluation, a mice renal ischemia-reperfusion injury (IRI) model was established, where 45 min of ischemia was followed by reperfusion for 1 day (AKI) or 14 days (CKD) across experimental groups. Human renal proximal tubular (HK-2) cells were utilized for in vitro modeling, including hypoxia/reoxygenation (H/R) model and TGF-β1-induced fibrosis. Bioinformatics analysis was employed for target prediction of silibinin. Results showed that silibinin significantly decreased serum creatinine, blood urea nitrogen, oxidative stress, inflammation, and apoptosis induced by IRI. The HE, Masson and Sirius Red staining showed that silibinin decreased the kidney damage and collagen deposition dramatically. Mechanistically, silibinin enhanced PI3K/AKT phosphorylation, suppressed phosphorylation of MAPK components (p38, ERK1/2, JNK), elevated antioxidant enzyme activity, reduced ROS/malondialdehyde levels, inhibited pro-inflammatory cytokine release, downregulated Bax/cleaved caspase-3 expression, and upregulated the anti-apoptotic factor Bcl-2. These effects were recapitulated in vitro, where silibinin mitigated H/R and TGF-β1-driven cellular injury by restoring redox balance and modulating dual signaling axes. Collectively, our findings demonstrated silibinin as a multi-target therapeutic candidate that impedes AKI-CKD transition via coordinated regulation of PI3K/AKT and MAPK pathways. These findings position silibinin as a promising agent for clinical intervention, potentially improving long-term renal outcomes in AKI patients at risk of progressing to CKD.
Similar content being viewed by others


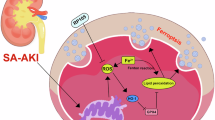
Introduction
The kidneys maintain homeostasis by filtering waste, regulating fluids, and producing essential hormones. Acute kidney injury (AKI), a critical clinical condition often triggered by sepsis, ischemia, nephrotoxic drugs, or chronic diseases such as diabetes, leads to rapid renal function decline and is associated with high morbidity and mortality worldwide1,2. Approximately 13.3 million people suffer from AKI annually, with up to 2 million deaths in developing countries3. In intensive care units (ICU), AKI affected nearly 50% of patients4. AKI has emerged as a pressing global health concern, with sharply escalating incidence and mortality rates over the past decade. Its strong association with a spectrum of adverse health outcomes positions it as a major public health challenge5. Patients with AKI face a higher risk of progression to chronic kidney disease (CKD) and end-stage renal disease (ESRD)6. Notably, approximately 29% of hospitalized AKI patients develop CKD or ESRD7. Renal interstitial fibrosis, characterized by myofibroblast accumulation and excessive extracellular matrix deposition, serves as a hallmark of AKI-to-CKD progression8,9. The pathogenic mechanisms underlying the AKI-to-CKD transition remain incompletely understood; however, emerging evidence suggests that failed tubular repair-potentially due to the severity of the initial AKI episode, incomplete functional recovery, or persistent renal injury (e.g., tubular atrophy or interstitial fibrosis)—predisposes the kidneys to subsequent damage and potential progression to CKD10,11. The transitional phase features a complex pathophysiological microenvironment involving oxidative stress, inflammation, fibrosis, and programmed cell death, collectively impairing renal function during disease progression12. Despite these insights, no targeted therapies currently exist to halt renal fibrosis. The AKI-to-CKD transition has emerged as a critical public health challenge, imposing substantial societal and economic burdens worldwide.
Oxidative stress and inflammation play pivotal roles in the AKI-to-CKD transition. AKI, particularly ischemic injury, induced mitochondrial dysfunction due to hypoxia-induced disruption of the electron transport chain (ETC), resulting in excessive reactive oxygen species (ROS) generation. These harmful molecules exacerbate renal tubular cell damage13. During subsequent CKD progression, elevated oxidative stress manifests as a consequence of dysregulated ROS production and impaired antioxidant clearance mechanisms14. The complexity of oxidative stress in CKD was closely linked to compromised mitochondrial function, establishing a self-perpetuating cycle that amplifies mitochondrial ROS production, ultimately driving apoptosis and contributing to renal injury and functional decline14. Lan et al.15demonstrated that mitochondrial metabolic dysfunction and its associated oxidative stress at 14 days post-IRI play pivotal roles in tubular atrophy development and the AKI-to-CKD transition. Following acute injury, stressed cells and damaged tissues activate innate immune pathways, triggering the release of pro-inflammatory cytokines, chemokines, and ROS, which collectively exacerbate cellular necrosis and tissue damage16,17. Furthermore, sustained release of fibrosis-associated cytokines such as transforming growth factor-β (TGF-β) and interleukin-13 (IL-13) triggers epithelial-mesenchymal transition (EMT), potentially driving renal fibrosis and chronic renal insufficiency18. Previous studies showed that mitigated the inflammatory response alleviated kidney injury and renal fibrosis. Sirtuin 1 (SIRT1) has been shown to mitigate renal injury by attenuating inflammatory responses in AKI19. Diosmin, a glycosylated polyphenolic flavonoid found in Citrus aurantium, alleviates renal fibrosis primarily through SIRT3-mediated suppression of NF-κB p65 nuclear translocation, exerting anti-inflammatory effects20. As a key regulator of inflammation-related gene expression, the NF-κB signaling pathway can be modulated through miRNA-mediated regulation and specific inhibition of NF-κB/NLRP3 activity, offering therapeutic potential for mitigating renal injury21. The long-acting thioredoxin (Trx) conjugate human serum albumin-Trx (HSA-Trx) confers renal protection during AKI-to-CKD transition by modulating oxidative stress and inflammatory responses22.
Silibinin, a naturally occurring flavonolignan isolated from the seeds of Silybum marianum, has a molecular formula of C25H22O10 and a molecular weight of 482.44 g/mol23. This compound exhibits diverse and significant biological activities, garnering considerable attention in medical research. Its most prominent feature was its potent antioxidant capacity, enabling efficient scavenging of free radicals and mitigation of oxidative stress-induced cellular damage. Additionally, silibinin demonstrated anti-inflammatory, antifibrotic, cell cycle-regulating, and pro-apoptotic properties, which collectively underpin its therapeutic potential across multiple disease contexts24,25,26. Traditionally, silibinin has been employed in the management of hepatic disorders. In alcoholic liver disease (ALD) and non-alcoholic fatty liver disease (NAFLD), it ameliorated pathological conditions by modulating fatty acid metabolism pathways. Studies revealed that silibinin down-regulated the expression of fatty acid synthase (FAS) and acetyl-CoA carboxylase (ACC), while upregulating carnitine palmitoyltransferase 1 A (CPT1A), thereby reducing hepatic lipid accumulation and improving lipid metabolism27,28. In viral hepatitis and drug-induced liver injury, silibinin primarily attenuated hepatic inflammation by suppressing pro-inflammatory cytokine release. Concurrently, it enhanced the antioxidant defense system in hepatocytes, reduced free radical-mediated damage, and promoted hepatocyte repair and regeneration24,29. Beyond hepatic diseases, silibinin demonstrated therapeutic potential across diverse pathological conditions. In oncology, it exerted multi-faceted anticancer mechanisms. In breast cancer cells, silibinin differentially modulated estrogen receptor (ER) subtypes (ERα and ERβ), thereby regulating RAS/ERK signaling pathways to suppress cancer cell proliferation and induce apoptosis30. In renal cancers, silibinin inhibits EMT, effectively blocking tumor cell migration and invasion31. In renal pathologies, silibinin attenuated fibrosis by inhibiting TGF-β1 signaling and downregulating fibrotic markers (collagen I, fibronectin, α-SMA)32,33,34. It also alleviated inflammatory lung injury via TXNIP/MAPKs/AP-1 pathway inhibition, reducing cytokine production35. Despite these advances, the role of silibinin in AKI-to-CKD progression remains underexplored. Our study elucidated that silibinin mitigated oxidative stress, inflammation, and apoptosis during AKI-to-CKD transition by modulating MAPK and PI3K/AKT pathways, offering novel therapeutic strategies for clinical intervention.
Materials and methods
Antibodies and reagents
Active recombinant mature TGF-β 1 protein was obtained from ABclonal.
(Wuhan, RP01458), and Silibinin was purchased from MedChemExpress (Shanghai, 65666−07-1). The product source and product number of other kits are described accordingly below.
Experimental animals
Ten-week-old male C57BL/6 mice were obtained from the Experimental Animal Center at Wuhan University School of Medicine (Wuhan, China). The project was approved by the Wuhan University Laboratory Animal Committee and carried out in compliance with the ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines. All procedures were in accordance with the Guidelines for the Care and Use of Laboratory Animals. The animals were maintained under specific pathogen-free conditions with controlled temperature (25 ± 1 °C) and humidity (55 ± 5%), subjected to standardized 12 h/12 h light-dark photoperiod cycles. Food and water were provided ad libitum throughout the experimental period. Following a 7-day acclimatization period in the institutional vivarium, the subjects were randomly assigned to experimental groups for subsequent induction of renal ischemia-reperfusion (I/R) injury. Surgical procedures were performed under aseptic conditions with appropriate anesthetic protocols, as detailed in the following methodology section.
Renal Ischemia-Reperfusion injury model establishment
For AKI model, male C57BL/6 mice (10 weeks old, body weight 22–25 g) were randomly assigned to 4 groups: Sham, IRI, IRI + Oil (IRI + O), IRI + Silibinin (IRI + S). For CKD model, mice were randomly assigned to 4 groups: Sham, IRI 14 d, IRI 14 d + O, IRI 14 d + S. The 14-day timepoint for CKD modeling was selected based on established literature, as it captures key transitional events of AKI-to-CKD progression (resolution of acute tubular injury, onset of persistent interstitial fibrosis, and upregulation of chronic fibrotic markers36,37.
All experimental groups, with the exception of the Sham-operated controls, received daily oral gavage of either Silibinin (100 mg/kg body weight) suspended in vegetable oil or equivalent volumes of vehicle (vegetable oil alone) for 7 consecutive days preceding surgical intervention. This pre-treatment design mimics clinical scenarios of prophylactic intervention for patients at high risk of perioperative AKI (e.g., cardiac bypass, renal transplantation)38,39. The dosing regimen was established based on published studies showing optimal renoprotective effects without toxicity in murine IRI models40,41,42, and validated through preliminary dose-response studies conducted in our laboratory.
For IRI surgery, Mice were anesthetized via intraperitoneal injection of sodium pentobarbital (50 mg/kg). After confirming the loss of pedal reflex, the mice were placed on a heating pad to maintain body temperature at 37 °C. A midline laparotomy was performed under aseptic conditions to expose the bilateral renal pedicles. The left renal artery and vein were occluded using non-traumatic vascular clamps (Fine Science Tools, 18055-04) for 30 min to induce ischemia, while the right kidney was removed. During the ischemic period, the surgical area was covered with saline-moistened gauze to prevent tissue dehydration. Reperfusion was initiated by clamp removal, confirmed by restoration of renal blood flow under microscopic observation. The abdominal cavity was closed in layers with 6 − 0 silk sutures. Sham-operated mice underwent identical procedures except for vascular occlusion. Postoperatively, animals were administered subcutaneous buprenorphine (0.1 mg/kg) for analgesia and allowed free access to water and food. At 24 h or 14 days post-reperfusion, euthanasia was performed by intraperitoneal injection of pentobarbital sodium (150 mg/kg) followed by confirmation of absent corneal reflex and respiration. then, renal tissues and blood samples were collected for histopathological and biochemical analyses.
Cell culture and H/R injury model
Human renal proximal tubular (HK-2) cells were provided by China Center for Type Culture Collection (CCTCC, Wuhan, China), and were maintained in DMEM (Invitrogen, USA) supplemented with 10% FBS under standard culture conditions (37 °C, 5% CO₂).
Cells were pre-treated with Silibinin (20µM, dissolved in DMSO) 24 h prior to H/R induction. For hypoxic challenge, cultures were transferred to serum-free/glucose-depleted DMEM and exposed to controlled hypoxia (1% O₂/5% CO₂/94% N₂) in a tri-gas incubator at 37 °C for 12 h. Reoxygenation was initiated by replacing hypoxic medium with complete growth medium (10% FBS), followed by normoxic recovery (5% CO₂) for 4 h. Experimental timelines were established based on our previous optimization studies43.
To establish in vitro fibrosis models, cells were stimulated with TGF-β1 at graded concentrations (1, 3, 5 ng/mL), using untreated cells as controls. Cells were pre-treated with Silibinin (20µM, dissolved in DMSO) 24 h prior to TGF-β1 treatment.
Assessment of renal function
Blood samples were collected and centrifuged (1,500 g, 10 min, 4 °C) to obtain serum. Creatinine (Cr) and blood urea nitrogen (BUN) concentrations were quantified using spectrophotometric assay kits (C011-2-1/C013-2-1; Nanjing Jiancheng Bioengineering Institute, China) according to manufacturer’s protocols. Absorbance measurements were performed at 510 nm (Cr) and 640 nm (BUN) using a microplate reader (BioTek Synergy H1) with internal calibration standards. All samples were analyzed in triplicate with inter-assay CV < 5%.
Histological staining
Kidney tissue samples were fixed in 4% paraformaldehyde (24–48 h, 4 °C), dehydrated through graded ethanol series, and paraffin-embedded. Serial 4-µm-thick sections were stained with:
-
1.
Hematoxylin and eosin (H&E) for evaluation of tubular architecture.
-
2.
Masson’s trichrome for collagen deposition visualization.
-
3.
Picrosirius red for fibrillar collagen characterization.
Two board-certified renal pathologists independently evaluated histopathological alterations in a blinded manner, including: Tubular dilatation index (TDI), Brush border loss percentage, Tubular necrosis score, Cortico-medullary patterning integrity. Picrosirius red-positive areas (indicative of collagen deposition) and tubulointerstitial fibrosis index were independently evaluated by blinded investigators using a semi-quantitative scoring system. Digital quantification was performed using ImageJ software (v1.53) with color thresholding algorithms, following calibration with internal reference standards. Three non-overlapping cortical fields (400× magnification) per sample were analyzed by two independent observers.
Dihydroethidium (DHE) staining
Renal tissues from euthanized mice were snap-frozen and sectioned at 4 μm thickness. Sections were incubated with 5 µM dihydroethidium (DHE; Sigma-Aldrich, D7008) in PBS (pH 7.4) at 37 °C for 30 min in the dark. After three washes with PBS, nuclei were counterstained with DAPI (1 µg/mL, 5 min). Fluorescence imaging was performed using a confocal microscope (Zeiss LSM 880; Ex/Em = 518/605 nm) with consistent laser power (15%) and exposure time (200 ms).
Quantitative Real-Time PCR analysis
Total RNA was isolated from treated cells and renal tissues using the RNApure Kit (Servicebio, China; Cat# G3013) following manufacturer’s protocol, with RNA integrity verified by A260/A280 ratios (1.8–2.0.8.0). Reverse transcription was performed using the HiScript II cDNA Synthesis Kit (Servicebio; Cat# G3333-100) under thermal cycling conditions: 25 °C for 5 min, 50 °C for 30 min, and 85 °C for 5 min. SYBR Green-based qPCR reactions were conducted in triplicate using Universal Blue Mix (Servicebio; Cat# G3326-05) on a CFX96 Touch™ system (Bio-Rad, USA) with the following parameters: 95 °C for 3 min, 40 cycles of 95 °C for 15 s and 60 °C for 30 s. Gene-specific primers (Sangon Biotech, China; sequences in Tables S1-S2) were designed to span exon-exon junctions, with amplification efficiencies (90–110%) validated through standard curves. Relative mRNA expression was calculated using the 2^(-ΔΔCt) method, normalized to β-actin as endogenous controls. Reference gene stability was confirmed using geNorm algorithm (M-value < 0.5). Melt curve analysis verified reaction specificity.
Western blot analysis
Protein extraction was performed using ice-cold RIPA lysis buffer (Beyotime Biotechnology, Cat# P0013B) supplemented with PMSF (Servicebio; Cat# G2008-1ML). Renal tissues and HK-2 cells were homogenized on ice, followed by centrifugation at 12,000 ×g for 20 min at 4 °C to remove insoluble debris. Supernatant protein concentrations were determined via BCA assay (Thermo Fisher Scientific, Cat# 23225) with bovine serum albumin (BSA) standards.Equal amounts of protein (30 µg/lane) were resolved on 10% SDS-polyacrylamide gels under denaturing conditions (100 V for 120 min) and subsequently transferred to PVDF membranes (Millipore, Cat# IPVH00010) using semi-dry electroblotting (25 V, 30 min). Membranes were blocked with 5% non-fat dry milk in TBST (Tris-buffered saline with 0.1% Tween-20) for 1 h at room temperature, then incubated overnight at 4 °C with primary antibodies diluted in blocking buffer: β-actin, anti-TNF-α (ab183218, abcam), anti-IL-1β (ab254360), anti-IL-6 (ab259341, abcam), anti-collagen I (ab138492, abcam), anti-collagen IV (ab6586, abcam), anti-α-SMA (ab7817, abcam), Kim-1 (AF1817, R&D Systems), NGAL (#DF6816, Affinity), Bax (50599-2-Ig, Proteintech Group), Bcl-2 (T40056, Abmart), Cleaved caspase-3(TA7022, Abmart), p-PI3K(T40116, Abmart), PI3K(T0115, Abmart), p-AKT(T40067, Abmart), AKT(T55561, Abmart), p38(T55600, Abmart), p-p38(TA4001, Abmart), p-ERK1/2(TA1015, Abmart), ERK1/2(T40071, Abmart), p-JNK1/2/3(T40074, Abmart), JNK1/2/3(T40073, Abmart), Caspase-3 (TA6311, Abmart), catalase (21260-1-AP, Proteintech Group), SOD1 (10269-1-AP, Proteintech Group), SOD2 (24127-1-AP, Proteintech Group), Then, the membranes were washed and incubated with the secondary antibody for 2 h. Protein levels were quantified using the Image J software.
Hydrogen peroxide (H₂O₂) quantification
H₂O₂ levels in mouse serum were measured using a peroxidase-mediated colorimetric assay (Jiancheng Bioengineering, A064-1). Briefly, 50 µL serum was mixed with 100 µL working reagent (0.1 M phosphate buffer containing 0.4 mM 4-aminoantipyrine and 2 mM 3,5-dichloro-2-hydroxybenzenesulfonic acid) and incubated at 37 °C for 10 min. Absorbance at 505 nm was recorded using a microplate reader (BioTek Synergy H1). Concentrations were calculated against an H₂O₂ standard curve (0–100 µM) and normalized to total protein content (BCA assay).
Antioxidant enzymatic activity profiling
Catalase activity was determined via ammonium molybdate colorimetry (Jiancheng Bioengineering, Cat# A007-1). Superoxide dismutase (SOD) activity was assessed using the hydroxylamine method (Jiancheng, Cat# A001-3). Reduced glutathione (GSH) content and malondialdehyde (MDA) levels were quantified fluorometrically (Beyotime, Cat# S0053) and spectrophotometrically (Beyotime, Cat# S0131S) respectively. All assays followed manufacturer protocols.
TUNEL staining
Apoptotic cells in renal tissues were identified using the In Situ Cell Death Detection Kit (Beyotime Biotechnology, Cat# C1088) per established protocols24. Paraffin-embedded kidney Sect. (4 μm) underwent sequential deparaffinization in xylene, rehydration through graded ethanol series (100%−70%), and antigen retrieval via proteinase K digestion (20 µg/mL, 37 °C, 20 min). Tissue sections were incubated with TdT enzyme reaction mixture (dUTP: FITC = 1:40 in labeling buffer) for 60 min at 37 °C protected from light, followed by DAPI counterstaining (5 µg/mL, 10 min).
Visualization was performed using a fluorescence microscope (Olympus BX53; 200× magnification) with consistent exposure settings. Ten randomly selected cortical fields per section were imaged.
Bioinformatics analysis
Drug target prediction
The predicted targets of Silibinin were acquired from SwissTargetPredition (http://www.swisstargetprediction.ch/) and HERB website (http://herb.ac.cn/). Gene names of all targets were matched and normalized through the UniProt database (https://www.uniprot.org/).
Acquisition of disease target
Candidate genes associated with renal ischemia-reperfusion injury (IRI) were systematically retrieved from two authoritative biomedical databases: Online Mendelian Inheritance in Man (OMIM, https://www.omim.org) and GeneCards (https://www.genecards.org).
Functional enrichment analysis
Gene ontology and KEGG pathway analysis were carried out through Metascape analysis resource (https://metascape.org/) and visualized by the Bioinformatics website (https://www.bioinformatics.com.cn/). Heatmap was plotted by the.
Bioinformatics website as well.
Protein − Protein interaction (PPI) network
Intersected gene were entered into GENEMANIA (http://genemania.org/) to determine the PPI network, and map the PPI network using Cytoscape3.7.1 software.
Statistical analysis
All quantitative data were presented as mean ± SD and analyzed using GraphPad Prism 5.0 (GraphPad Software, San Diego, CA). All data were first tested for normality using the Shapiro-Wilk test (appropriate for small sample sizes: n = 3 for cell experiments, n = 6 for animal experiments), and results showed conformity to a normal distribution (Shapiro-Wilk P > 0.05), justifying the use of parametric tests.
Two-group comparisons were evaluated by two-tailed unpaired Student’s t-test with Welch’s correction for unequal variances. Multiple-group comparisons utilized one-way ANOVA with Tukey’s post hoc test following verification of homogeneity of variances (Bartlett’s test, p > 0.1). Statistical significance was defined as p < 0.05 (FDR-adjusted for multiple comparisons).
Results
Silibinin attenuated Ischemia-Reperfusion injury (IRI)-Induced AKI
To investigate the role of silibinin in AKI, we first established a murine renal IRI model (Fig. 1A), with kidneys harvested after 24 h (IRI) or 14 days (IRI 14 d) of reperfusion for injury assessment. Compared to the Sham group, IRI mice exhibited significantly elevated serum creatinine (Cr) and blood urea nitrogen (BUN) levels (Fig. 1B, C). Histopathological analysis via hematoxylin and eosin (H&E) staining revealed severe renal damage in the IRI group, characterized by cellular swelling, tubular dilation, partial tubular epithelial cell detachment, protein casts, and marked interstitial inflammatory infiltration (Fig. 1D). Additionally, qRT-PCR and Western blot analyses demonstrated significant upregulation of renal injury markers, kidney injury molecule-1 (Kim-1) and neutrophil gelatinase-associated lipocalin (NGAL), at both mRNA and protein levels post-IRI (Fig. 1F-H).

Silibinin Attenuated Ischemia-Reperfusion Injury (IRI)-Induced AKI. (A). Schematic of Silibinin administration in the renal ischemia-reperfusion (I/R) model.(Created by Dr. Jian with Biorender.com, Agreement number: RY28DVQ026). (B-C). Serum creatinine (Cr) and blood urea nitrogen (BUN) levels in Sham and IRI groups. (D). H&E staining of renal tissues in each groups. (E). Chemical structure of silibinin. (F-G). Western blot and quantitative analysis of renal injury markers (Kim-1, NGAL) at protein levels. (H). qRT-PCR analysis of renal injury markers (Kim-1, NGAL) at mRNA levels. (I-J). Silibinin (IRI + S) reversed IRI-induced elevations in Cr and BUN (#P < 0.05,). (K). H&E staining of renal tissues in each groups. (L, M). Western blot and quantitative analysis of renal injury markers (Kim-1, NGAL) at protein levels. (N, O). qRT-PCR analysis of mRNA expression of Kim-1 and NGAL in Sham, IRI, IRI + O, and IRI + S groups. All the IRI in Fig. 1 stand for ischemia-reperfusion 1 day. Each experiment was repeated independently for a minimum three times. Data: mean ± SEM (n = 6). *P < 0.05, **P < 0.01, ***P < 0.001 vs. Sham; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. IRI.
Oral administration of silibinin (Fig. 1E) significantly reversed the IRI-induced elevations in serum Cr and BUN (Silibinin-treated group vs. IRI and vehicle-treated groups; Fig. 1I, J). H&E staining further confirmed that silibinin pretreatment markedly ameliorated renal structural damage, whereas the vehicle group showed no protective effects (Fig. 1K). Furthermore, qRT-PCR and WB results indicated that silibinin effectively suppressed the IRI-induced upregulation of Kim-1 and NGAL at transcriptional and translational levels (Fig. 1L-O). Collectively, these findings indicated that silibinin attenuated IRI-induced AKI in mice.
IRI induced AKI-to-CKD transition in mice
To investigate the role of silibinin in AKI-to-CKD progression, we subjected mice to renal IRI and collected kidney tissues and serum at 1 day (AKI phase) and 14 days (CKD phase) post-reperfusion. Serum Cr and BUN levels were significantly elevated at 1 day post-IRI (P < 0.05 vs. Sham). Although these levels slightly decreased by day 14, they remained significantly higher than those in the Sham group (Fig. 2A, B). qRT-PCR and Western blot analyses revealed time-dependent upregulation of fibrosis markers, including α-SMA, collagen I, and collagen IV, at both mRNA and protein levels in IRI mice (Fig. 2C-F, J).

IRI Induced AKI-to-CKD Transition in Mice. (A-B). Measurement of serum BUN, Cr production in each groups. (C-E). qRT-PCR analysis of mRNA expression of α-SMA, collagen I/IV in each groups. (F, G), (J-K). Western blot and quantitative analysis of α-SMA, collagen I/IV in each groups. (H, I, L). qRT-PCR analysis of mRNA expression of α-SMA, collagen I/IV in each groups. (M). H&E, Masson’s trichrome, and Sirius red staining revealing tubular injury and interstitial fibrosis in each groups. Each experiment was repeated independently for a minimum three times. Data: mean ± SEM (n = 6). *P < 0.05, **P < 0.01, ***P < 0.001 vs. control/Sham; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. IRI 1d.
To validate these findings in vitro, we treated HK-2 cells with TGF-β1 to establish a fibrotic model. Consistent with in vivo results, TGF-β1 dose-dependently increased the expression of α-SMA, collagen I, and collagen IV in HK-2 cells (Fig. 2G-I, K-L). Histological assessment via H&E staining confirmed significant renal damage in IRI mice compared to the Sham group, with the most severe injury observed at 1 day post-IRI and partial recovery by day 14 (Fig. 2M). Masson’s trichrome and Sirius red staining further demonstrated pronounced renal fibrosis in IRI mice, particularly in the 14-day group (Fig. 2M). These results collectively indicated that IRI triggers a transition from AKI to CKD in surviving mice.
Bioinformatics analysis of target prediction of renal I/RI
Although our previous findings demonstrated that silibinin alleviated renal injury in murine IRI models, its precise mechanisms remained unclear. To elucidate these mechanisms, we constructed an “ingredient-target-pathway-disease” interactive network to delineate the pathological processes and signaling pathways regulated by silibinin during renal IRI.A total of 240 non-redundant silibinin targets were identified by integrating 101 predicted genes from SwissTargetPrediction and 145 potential targets from the HERB database. For renal IRI-associated genes, 1,786 unique targets were compiled from OMIM (212 genes) and GeneCards (1,586 genes). Intersection analysis using Venny 2.1 software revealed 110 overlapping genes between silibinin and renal IRI targets (Fig. 3A). The top 50 silibinin targets predicted by SwissTargetPrediction were categorized by target class (Fig. 3B).

Bioinformatics Analysis of Target Prediction of Renal I/RI. (A). Venn diagram of overlapping genes between silibinin targets (240) and renal IRI-associated genes (1,786), highlighting 110 shared genes. (B). Functional classification of top 50 silibinin targets. (C, D). GO functional annotation of intersecting genes from drug and disease targets. (E, F). KEGG pathway analysis of enriched genes. (G). PPI network of shared genes, with node size/color reflecting interaction degree. (H). Degree distribution of PPI nodes, indicating hub genes critical to silibinin’s mechanism.
Gene Ontology (GO) enrichment analysis of the 110 shared genes highlighted significant enrichment in biological processes (BP), including response to decreased oxygen levels, response to hypoxia, response to oxidative stress, reactive oxygen species metabolic process, and regulation of apoptotic signaling pathway (Fig. 3C, D). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis further demonstrated enrichment in hepatitis B, Kaposi sarcoma-associated herpesvirus infection, PI3K-Akt signaling pathway, and MAPK signaling pathway (Fig. 3E, F). Protein-protein interaction (PPI) network analysis using GENEMANIA and Cytoscape 3.7.1 identified key hub genes among the 110 overlapping targets, with the top 20 genes visualized in Fig. 3F.
Silibinin attenuated renal IRI by modulating PI3K/AKT Signaling, oxidative Stress, Inflammation, and apoptosis
Among the predicted pathways, PI3K/AKT and MAPK signaling drew particular interest. We first investigated the role of PI3K/AKT in renal IRI. Silibinin significantly reduced IRI-induced elevations in serum Cr, BUN, MDA, and hydrogen peroxide (H₂O₂) (Fig. 4A, B,I, J). qRT-PCR and Western blot analyses demonstrated that silibinin reversed the IRI-induced upregulation of pro-inflammatory cytokines, including tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and interleukin-6 (IL-6), at both mRNA and protein levels (Fig. 4C-F, K).

Silibinin Attenuated Renal IRI by Modulating PI3K/AKT Signaling, Oxidative Stress, Inflammation, and Apoptosis. (A, B). Measurement of serum BUN, Cr production in each groups. (C-E). qRT-PCR analysis of mRNA expression of TNF-α, IL-1β, IL-6 in each groups. (F-H, K-M). Western blot and quantitative analysis of TNF-α, IL-1β, IL-6, Bax, cleaved caspase-3, Caspase-3 Bcl-2, p-PI3K, PI3K, p-AKT, AKT in each groups. (I, J). Measurement of oxidative stress markers (H₂O₂, MDA) production in each groups. (N, O). TUNEL staining and quantitative analysis showed apoptosis in kidneys. All the IRI in Fig. 4 stand for ischemia-reperfusion 1 day. Each experiment was repeated independently for a minimum three times. Data: mean ± SEM (n = 6). *P < 0.05, **P < 0.01, ***P < 0.001 vs. Sham; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. IRI. (one-way ANOVA with Tukey’s post hoc test).
Western blot further revealed that IRI significantly increased the expression of pro-apoptotic proteins Bax and cleaved caspase-3 while suppressing anti-apoptotic Bcl-2. These effects were markedly counteracted by silibinin pretreatment (Fig. 4G, L). Notably, IRI suppressed PI3K and AKT phosphorylation, a phenomenon reversed by silibinin (Fig. 4H, M). TUNEL staining confirmed that silibinin significantly attenuated IRI-induced renal cellular apoptosis (Fig. 4N, O). These results suggested that silibinin mitigated renal IRI by enhancing PI3K/AKT phosphorylation, thereby alleviating oxidative stress, inflammation, and apoptosis. Notably, silibinin simultaneously enhanced PI3K/AKT phosphorylation and suppressed MAPK (p38, ERK1/2, JNK) phosphorylation. This coordinated regulation aligns with published evidence of reciprocal crosstalk between PI3K/AKT and MAPK pathways—where PI3K/AKT activation negatively regulates MAPK via inhibiting upstream kinases, and excessive MAPK activation suppresses PI3K/AKT via feedback mechanisms44,45,46,47.
Silibinin mitigated H/R-Induced HK-2 cell injury by regulating PI3K/AKT Signaling, oxidative Stress, Inflammation, and apoptosis
To validate the role of silibinin in vitro, we established a hypoxia/reoxygenation (H/R) model in HK-2 cells (Fig. 5A). Consistent with in vivo findings, qRT-PCR and Western blot analyses revealed that silibinin significantly attenuated H/R-induced upregulation of pro-inflammatory cytokines TNF-α, IL-1β, and IL-6 at both mRNA and protein levels (Fig. 5B-E, I). Western blot further demonstrated that silibinin reversed H/R-mediated alterations in apoptosis-related proteins, suppressing Bax and cleaved caspase-3 while restoring Bcl-2 expression (Fig. 5G, J).

Silibinin Mitigated H/R-Induced HK-2 Cell Injury by Regulating PI3K/AKT Signaling, Oxidative Stress, Inflammation, and Apoptosis. (A). H/R model schematic.(Created by Dr. Jian with Biorender.com, Agreement number: LP28DVQ66J). (B-D). qRT-PCR analysis of mRNA expression of TNF-α, IL-1β, IL-6 in each groups. (F). Measurement of MDA production in each groups. (E, G, H, I-K). Western blot and quantitative analysis of TNF-α, IL-1β, IL-6, Bax, cleaved caspase-3, Caspase-3 Bcl-2, p-PI3K, PI3K, p-AKT, AKT in each groups. Each experiment was repeated independently for a minimum three times. Data: mean ± SEM (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001 vs. control; #P < 0.05, ##P < 0.01 vs. H/R. (one-way ANOVA with Tukey’s post hoc test).
Notably, H/R-induced reductions in PI3K and AKT phosphorylation were effectively rescued by silibinin treatment (Fig. 5H, K). Additionally, silibinin markedly decreased H/R-elevated MDA levels, indicating alleviation of oxidative stress (Fig. 5F). These results suggested that silibinin protected HK-2 cells against H/R injury by enhancing PI3K/AKT phosphorylation, thereby mitigating oxidative stress, inflammation, and apoptosis.
Silibinin delayed AKI-to-CKD progression by modulating PI3K/AKT Signaling, oxidative Stress, Inflammation, and apoptosis
Building on prior findings that silibinin mitigated renal IRI via PI3K/AKT regulation, we further investigated its role in AKI-to-CKD progression using mice subjected to 14 days of post-ischemic reperfusion (IRI 14 d). Silibinin significantly reduced IRI 14d-induced elevations in serum Cr, BUN, MDA, and H2O2 (Fig. 6A-D). qRT-PCR and Western blot analyses confirmed that silibinin suppressed the upregulation of pro-inflammatory cytokines (TNF-α, IL-1β, IL-6) and fibrosis markers (α-SMA, collagen I, collagen IV) at both mRNA and protein levels (Fig. 6E-G, J,L, M). Western blot further revealed that silibinin reversed IRI 14d-induced alterations in apoptosis-related proteins, decreasing Bax and cleaved caspase-3 while restoring Bcl-2 expression (Fig. 6H, N). Additionally, silibinin rescued the IRI 14d-mediated suppression of PI3K and AKT phosphorylation (Fig. 6I, O). Histological assessments via H&E, Masson’s trichrome, and Sirius red staining demonstrated that silibinin attenuated renal injury and interstitial fibrosis in IRI 14 d mice (Fig. 6P). TUNEL staining corroborated its anti-apoptotic effects, showing significantly reduced apoptotic cells in silibinin-treated kidneys (Fig. 6K, P).

3.6 Silibinin Delayed AKI-to-CKD Progression by Modulating PI3K/AKT Signaling, Oxidative Stress, Inflammation, and Apoptosis. (A-D). Measurement of Cr, BUN, H2O2, MDA production in each groups. (E, F, J). qRT-PCR analysis of mRNA expression of TNF-α, IL-1β, IL-6 in each groups. (G-I, L-O). Western blot and quantitative analysis of TNF-α, IL-1β, IL-6, Bax, cleaved caspase-3, Caspase-3 Bcl-2, p-PI3K, PI3K, p-AKT, AKT in each groups. (O, P). H&E, Masson’s trichrome, Sirius red, TUNELstaining(and its quantitative analysis) in each groups. Each experiment was repeated independently for a minimum three times. Data: mean ± SEM (n = 6). *P < 0.05, **P < 0.01, ***P < 0.001 vs. Sham; #P < 0.05, ##P < 0.01, ###P < 0.001vs. IRI 14d. (one-way ANOVA with Tukey’s post hoc test).
These results indicated that silibinin delayed AKI-to-CKD progression by enhancing PI3K/AKT phosphorylation, thereby alleviating oxidative stress, inflammation, apoptosis, and fibrosis. It is noted that the 14-day timepoint reflects the early stage of AKI-to-CKD transition; longer timepoints (e.g., 28–56 days) would be required to model established CKD (e.g., severe tubular atrophy, extensive fibrosis). However, our data highlight silibinin’s efficacy in targeting the early intervention window, which is critical for blocking irreversible CKD progression.
Silibinin attenuated TGF-β1-Induced HK-2 cell fibrosis by regulating PI3K/AKT Signaling, oxidative Stress, Inflammation, and apoptosis
To further validate these findings in vitro, we established a TGF-β1-induced fibrotic model in HK-2 cells (Fig. 7A). Silibinin significantly reduced TGF-β1-elevated MDA levels, indicating suppression of oxidative stress (Fig. 7B). qRT-PCR analysis demonstrated that silibinin downregulated TGF-β1-induced mRNA expression of pro-inflammatory cytokines TNF-α, IL-1β, and IL-6 (Fig. 7C-E). Western blot revealed that silibinin reversed TGF-β1-induced upregulation of inflammatory cytokines (TNF-α, IL-1β, IL-6), fibrosis markers (α-SMA, collagen I, collagen IV), and pro-apoptotic proteins (Bax, cleaved caspase-3), while restoring anti-apoptotic Bcl-2 expression. Concurrently, silibinin rescued the TGF-β1-mediated suppression of PI3K and AKT phosphorylation (Fig. 7F-L). These in vitro results corroborated that silibinin mitigated TGF-β1-driven fibrosis by modulating PI3K/AKT signaling, oxidative stress, inflammation, and apoptosis.

Silibinin Attenuated TGF-β1-Induced HK-2 Cell Fibrosis by Regulating PI3K/AKT Signaling, Oxidative Stress, Inflammation, and Apoptosis. (A). TGF-β1 fibrosis model schematic. (Created by Dr. Jian with Biorender.com, Agreement number: LP28DVQ66J). (B). Measurement of MDA production in each groups. (C-E).qRT-PCR analysis of mRNA expression of TNF-α, IL-1β, IL-6 in each groups. (F-L). Western blot and quantitative analysis of TNF-α, IL-1β, IL-6, Bax, cleaved caspase-3, Caspase-3 Bcl-2, p-PI3K, PI3K, p-AKT, AKT in each groups. Each experiment was repeated independently for a minimum three times. Data: mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 vs. control; #P < 0.05, ##P < 0.01, ###P < 0.001vs. TGF-β1. (one-way ANOVA with Tukey’s post hoc test).
Silibinin mitigated acute kidney injury via MAPK pathway and oxidative stress
Based on network pharmacology predictions implicating MAPK signaling in post-IRI renal injury, we validated this mechanism using in vivo and in vitro AKI models. In IRI mice, serum levels of catalase, glutathione (GSH), and superoxide dismutase (SOD) were significantly reduced, indicating severe oxidative stress. Silibinin treatment restored catalase, GSH, and SOD expression (Fig. 8A-C) and decreased renal ROS production, as confirmed by dihydroethidium (DHE) staining (Fig. 8D, I). Western blot analyses further demonstrated that silibinin rescued IRI-induced downregulation of catalase, SOD1, and SOD2 (Fig. 8E, F). Silibinin also significantly attenuated IRI-induced phosphorylation of MAPK pathway components, including p38, ERK1/2, and JNK1/2/3 (Fig. 8G, H). These findings were corroborated in vitro: silibinin partially restored H/R-suppressed catalase, SOD1, and SOD2 expression in HK-2 cells (Fig. 8J, K) and reduced H/R-induced phosphorylation of p38, ERK1/2, and JNK1/2/3 (Fig. 8L, M). Collectively, these results demonstrate that silibinin alleviated acute kidney injury by suppressing MAPK pathway activation and mitigating oxidative stress in both mice and cellular models.

Silibinin Mitigated Acute Kidney Injury via MAPK Pathway and Oxidative Stress. (A-C). Measurement of MDA, GSH, SOD production in each groups. (E-H). Western blot and quantitative analysis of p-p38, p38, p-ERK1/2, ERK1/2, p-JNK1/2/3, JNK1/2/3 in each groups. (D, I). DHE staining and quantitative analysis confirmed reduced ROS levels in silibinin-treated kidneys. (J-M). Western blot and quantitative analysis of p-p38, p38, p-ERK1/2, ERK1/2, p-JNK1/2/3, JNK1/2/3 in each groups. All the IRI in Fig. 8 stand for ischemia-reperfusion 1 day. Each experiment was repeated independently for a minimum three times. Data: mean ± SEM (n = 6). *P < 0.05, **P < 0.01, ***P < 0.001 vs. Control/Sham; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. IRI/H/R. (one-way ANOVA with Tukey’s post hoc test).
Silibinin delayed AKI-to-CKD transition via modulated MAPK pathway and oxidative stress
To further explore the role of MAPK signaling and oxidative stress in silibinin-mediated attenuation of AKI-to-CKD progression, in vivo and in vitro models were established. In IRI 14 d mice, serum catalase, GSH, and SOD levels were significantly reduced, while silibinin treatment partially restored their expression (Fig. 9A-C). DHE staining confirmed that silibinin markedly reduced renal ROS production in IRI 14 d kidneys (Fig. 9D, I), suggesting its potential to mitigate oxidative stress during AKI-to-CKD transition. Western blot analyses revealed that silibinin partially rescued IRI 14d-induced downregulation of catalase, SOD1, and SOD2 (Fig. 9E, F). Additionally, silibinin significantly attenuated IRI 14d-mediated phosphorylation of MAPK pathway components, including p38, ERK1/2, and JNK1/2/3 (Fig. 9G, H). These findings were recapitulated in vitro: silibinin restored TGF-β1-suppressed catalase, SOD1, and SOD2 expression in HK-2 cells (Fig. 9J, K) and reduced TGF-β1-induced phosphorylation of p38, ERK1/2, and JNK1/2/3 (Fig. 9L, M). These results suggested that silibinin delayed AKI-to-CKD progression by suppressing MAPK pathway activation and oxidative stress.

Silibinin Delayed AKI-to-CKD Transition via modulated MAPK Pathway and Oxidative Stress. (A-C). Measurement of MDA, GSH, SOD production in each groups. (E-H). Western blot and quantitative analysis of p-p38, p38, p-ERK1/2, ERK1/2, p-JNK1/2/3, JNK1/2/3 in each groups. (D, I). DHE staining and quantitative analysis showed reduced renal ROS in silibinin-treated CKD kidneys. (J-M). Western blot and quantitative analysis of p-p38, p38, p-ERK1/2, ERK1/2, p-JNK1/2/3, JNK1/2/3 in each groups. Each experiment was repeated independently for a minimum three times. Data: mean ± SEM (n = 6). *P < 0.05, **P < 0.01, ***P < 0.001 vs. Control/Sham; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. IRI 14d/TGF-β1. (one-way ANOVA with Tukey’s post hoc test).
Discussion
The transition from AKI to CKD represents a complex and highly detrimental pathological process, posing a significant threat to global public health41. AKI affects approximately 13.3 million individuals annually worldwide, with high mortality rates closely associated with its progression41. A substantial proportion of AKI patients progress to CKD, imposing severe health burdens and substantial socioeconomic costs7. Renal interstitial fibrosis, characterized by myofibroblast accumulation and excessive extracellular matrix deposition, serves as a hallmark of AKI-to-CKD progression. This transition involves a multifaceted pathophysiological microenvironment, where oxidative stress, inflammation, and apoptosis play central roles. In this study, we demonstrated that silibinin mitigates the AKI-to-CKD transition by suppressing phosphorylation of the MAPK and PI3K/AKT signaling pathways, thereby alleviating IRI-induced oxidative stress, inflammation, and apoptosis.
The IRI model was a classical model for AKI-to-CKD transition study. Following temporary renal blood flow occlusion and subsequent reperfusion, a cascade of detrimental cellular responses was triggered, leading to tubular cell injury, death, and inflammation. Beyond these acute changes, post-IRI kidneys might develop chronic pathologies over weeks to months48. In this study, we established a unilateral renal IRI model with contralateral nephrectomy, as this approach not only facilitated chronic renal pathology but also enhanced ischemic tolerance, ensuring long-term model stability48,49. Our 14-day timepoint captures the early transition phase of AKI-to-CKD, characterized by partial recovery of acute markers (Cr, BUN) and onset of chronic fibrosis—consistent with literature36,37. While longer timepoints (e.g., 28 days) would model established CKD, our data highlight silibinin’s ability to target the early intervention window, which is clinically critical for preventing irreversible renal damage. The results demonstrated that mice subjected to IRI for 1 day exhibited significant AKI characteristics, including elevated Cr, BUN, and renal injury markers (Kim-1 and NGAL). In contrast, IRI 14-day mice showed partial resolution of acute injury markers but marked upregulation of chronic fibrosis indicators. Masson’s trichrome and Sirius red staining revealed aggravated collagen deposition in renal tissues. These findings collectively validate the successful induction of AKI-to-CKD transition in the IRI model, recapitulating the pathophysiological progression from acute tubular damage to chronic fibrotic remodeling.
Oxidative stress, inflammation, and apoptosis play pivotal roles in the AKI-to-CKD transition12. Previous studies have demonstrated that ischemic injury triggers mitochondrial dysfunction, leading to excessive ROS generation and subsequent oxidative stress13, this finding consistent with our experimental results, our findings demonstrated that renal IRI in mice induced a marked increase in oxidative stress markers, including MDA and H₂O₂, accompanied by significant reductions in key antioxidant enzymes: catalase, SOD1, and SOD2. These findings indicated a pronounced imbalance in redox homeostasis, where elevated lipid peroxidation and impaired antioxidant defense mechanisms synergistically exacerbate oxidative damage during renal IRI. The suppression of catalase and SOD isoforms further underscores the disruption of enzymatic scavenging systems, which may contribute to sustained ROS accumulation and tubular injury progression. These observations align with established mechanisms of IRI pathophysiology and highlight potential therapeutic targets for restoring oxidative balance in acute-to-chronic kidney disease transitions. Oxidative stress not only directly damages renal tubular cells but also activates inflammatory cascades, promoting the release of pro-inflammatory cytokines such as tumor necrosis TNF-α, IL-1β, and IL-613. These cytokines exacerbate renal tissue damage by amplifying inflammatory responses, the mechanism corroborated in our study. Our work was consistent with these early insights, indicated that the renal tissues of IRI mice show a significant upregulation of the inflammatory cytokines TNF-α, IL-1β, and IL-6. As a crucial type of cell death, apoptosis induced by mitochondrial dysfunction played an important role in the progression of AKI-to-CKD50. The activation of CD36 in renal cells has been demonstrated to initiate the inflammatory pathway, leading to the production of pro-inflammatory cytokines and ROS, and ultimately resulting in cell damage and apoptosis51. Moreover, fucosyltransferase 8 (FUT8) promoted the transdifferentiation of pericytes by regulating mitochondrial function and the apoptotic pathway, contributing to the transition of AKI-to-CKD52. Our results also showed that the expression of apoptotic factors Bax and cleaved caspase-3 was increased in mice with IRI, while the expression of the anti-apoptotic protein Bcl-2 was decreased.
Silibinin, a natural flavonolignan isolated from Silybum marianum seeds, has demonstrated broad therapeutic potential across diverse pathologies53. In hepatic disorders, it ameliorates alcoholic liver disease and non-alcoholic fatty liver disease by modulating oxidative stress and fatty acid metabolism29,54. In oncology, silibinin exerts multi-pronged anticancer mechanisms, suppressing proliferation, inducing apoptosis, and inhibiting metastasis through pathways such as EMT blockade and RAS/ERK signaling regulation55,56,57. Additionally, silibinin exhibits neuroprotective and cardioprotective properties, attenuating neurodegenerative and ischemic injuries via antioxidant and anti-inflammatory actions25,58,59. In this study, we identified silibinin’s potent inhibitory effect on AKI-to-CKD progression. Experimental data revealed that oral administration of silibinin (100 mg/kg) significantly reversed IRI-induced elevations in Cr and BUN, while suppressing renal injury markers Kim-1 and NGAL at both transcriptional and translational levels. These findings align with prior reports of silibinin’s renoprotective effects. For instance, silibinin ameliorates cisplatin-induced nephrotoxicity by enhancing antioxidant defenses42, and mitigates IRI-AKI through ferroptosis inhibition via FTH1 targeting41. Our work extends these observations, suggesting that silibinin preserves renal cellular homeostasis by regulating metabolic and functional pathways, potentially through shared mechanisms involving oxidative stress mitigation. In our investigation of AKI-to-CKD transition, silibinin significantly reduced serum creatinine, BUN, MDA and H₂O₂ levels in IRI 14 d mice, while reversing the upregulation of pro-inflammatory cytokines (TNF-α, IL-1β, IL-6) and fibrosis markers (α-SMA, collagen I/IV). These effects collectively attenuated renal injury and interstitial fibrosis. These findings are consistent with silibinin’s documented anti-inflammatory and anti-fibrotic actions in other pathologies. For example, silibinin suppresses collagen I/III synthesis in keloid fibroblasts by inhibiting mTOR signaling60, and mitigates pulmonary inflammation through TLR4/MAPK/NF-κB pathway inhibition and NLRP3 inflammasome downregulation61. Similarly, in renal disease, silibinin likely interrupts AKI-CKD progression by targeting analogous pathways—modulating oxidative stress, suppressing inflammatory cascades, and blocking fibrotic remodeling. This mechanistic conservation across tissues underscores its potential as a multi-organ protective agent.
To elucidate the mechanisms underlying silibinin’s regulation of oxidative stress and inflammation during AKI-to-CKD transition, we employed network pharmacology to identify key downstream targets. The PPI network analysis identified top hub genes including AKT1, TNF, IL-1β, TP53 and Bcl-2. Our experimental data directly validate four critical hubs. This alignment supports the reliability of our bioinformatics-guided mechanistic exploration. From thousands of candidate targets, MAPK and PI3K/AKT signaling pathways were prioritized as critical mediators. In both in vivo and in vitro renal fibrosis models, silibinin elevated phosphorylation of PI3K/AKT components, a finding paralleling its previously reported inhibition of TGF-β2-induced JAK2/STAT3 activation and downstream PI3K/AKT signaling in fibrotic contexts62. Our data show silibinin coordinately enhances PI3K/AKT phosphorylation and suppresses MAPK activation. This aligns with published evidence of reciprocal crosstalk: PI3K/AKT inhibits MAPK via MEK/RAF, while MAPK suppresses PI3K/AKT via feedback44,45,46,47. Though dedicated inhibitor experiments are needed for direct validation, this coordinated modulation suggests silibinin targets a core signaling node of AKI-to-CKD progression. Notably, silibinin’s anti-tumor effects have also been linked to PI3K/AKT pathway modulation55, suggesting its role as a multi-target agent capable of disrupting disease progression across pathologies through conserved signaling nodes. Our findings align with prior evidence highlighting the critical involvement of these pathways in renal fibrosis and functional decline12,63. For instance, oxidative stress-driven mitochondrial dysfunction perpetuates ROS overproduction, exacerbating tubular cell damage and inflammatory responses13,14. Similarly, MAPK activation amplifies pro-fibrotic signaling, while PI3K/AKT dysregulation impairs cellular repair mechanisms18,21. Silibinin’s dual inhibition of these pathways not only attenuates acute injury but also disrupts the self-reinforcing cycle of fibrosis, offering a novel therapeutic strategy to decelerate CKD progression.
However, this study has several limitations. While murine models partially recapitulate human disease pathophysiology, interspecies differences in renal repair mechanisms and comorbidity profiles may affect translational relevance. Although MAPK and PI3K/AKT pathways were identified as central mediators of silibinin’s effects, additional molecular pathways (e.g., epigenetic regulators, mitochondrial quality control systems) may contribute to its renoprotective effects, warranting comprehensive multi-omics investigations. Furthermore, silibinin’s pharmacokinetic profile in humans remains poorly characterized—its absorption, distribution, metabolism, and excretion properties may diverge from preclinical observations, potentially limiting therapeutic efficacy in clinical settings54. Future studies may prioritize optimizing silibinin formulations (e.g., nanoparticle-based drug delivery systems) to enhance bioavailability and renal target specificity. Additionally, exploring synergistic combinations with established nephroprotective agents could amplify therapeutic outcomes while mitigating dose-dependent toxicity.
In conclusion, this study was the first to demonstrate that silibinin mitigated IRI-induced AKI-to-CKD progression by attenuating oxidative stress, inflammation, and apoptosis through suppression of MAPK signaling pathway phosphorylation and enhanced PI3K/AKT signaling pathway phosphorylation. These findings might provide novel insights into silibinin’s renoprotective mechanisms and highlight its potential as a therapeutic agent for interrupting AKI-CKD transition.
Data availability
The data that support the findings of this study are available from the correspondingauthor (drwanglei@whu.edu.cn) upon reasonable request.
References
Kale, A., Shelke, V., Sankrityayan, H., Dagar, N. & Gaikwad, A. B. Klotho restoration via ACE2 activation: A potential therapeutic strategy against acute kidney injury-diabetes comorbidity. Biochim. Biophys. Acta Mol. Basis Dis. 1868, 166532. https://doi.org/10.1016/j.bbadis.2022.166532 (2022).
See, E. J. et al. Long-term risk of adverse outcomes after acute kidney injury: A systematic review and meta-analysis of cohort studies using consensus definitions of exposure. Kidney Int. 95, 160–172. https://doi.org/10.1016/j.kint.2018.08.036 (2019).
Sawhney, S. et al. Harmonization of epidemiology of acute kidney injury and acute kidney disease produces comparable findings across four geographic populations. Kidney Int. 101, 1271–1281. https://doi.org/10.1016/j.kint.2022.02.033 (2022).
Hoste, E. A. J. et al. Epidemiology of acute kidney injury in critically ill patients: the multinational AKI-EPI study. Intensive Care Med. 41, 1411–1423. https://doi.org/10.1007/s00134-015-3934-7 (2015).
Sawhney, S., Mitchell, M., Marks, A., Fluck, N. & Black, C. Long-term prognosis after acute kidney injury (AKI): what is the role of baseline kidney function and recovery? A systematic review. BMJ Open. 5, e6497. https://doi.org/10.1136/bmjopen-2014-006497 (2015).
Su, C. et al. Outcomes associated with acute kidney disease: A systematic review and meta-analysis. EClinicalMedicine 55, 101760. https://doi.org/10.1016/j.eclinm.2022.101760 (2023).
Chawla, L. S. & Kimmel, P. L. Acute kidney injury and chronic kidney disease: an integrated clinical syndrome. Kidney Int. 82, 516–524. https://doi.org/10.1038/ki.2012.208 (2012).
Ferenbach, D. A. & Bonventre, J. V. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat. Rev. Nephrol. 11, 264–276. https://doi.org/10.1038/nrneph.2015.3 (2015).
Yu, S. M. & Bonventre, J. V. Acute kidney injury and maladaptive tubular repair leading to renal fibrosis. Curr. Opin. Nephrol. Hypertens. 29, 310–318. https://doi.org/10.1097/MNH.0000000000000605 (2020).
Zhang, X., Agborbesong, E. & Li, X. The role of mitochondria in acute kidney injury and chronic kidney disease and its therapeutic potential. Int. J. Mol. Sci. 22, https://doi.org/10.3390/ijms222011253 (2021).
Venkatachalam, M. A., Weinberg, J. M., Kriz, W. & Bidani, A. K. Failed tubule recovery, AKI-CKD transition, and kidney disease progression. J. Am. Soc. Nephrol. 26, 1765–1776. https://doi.org/10.1681/ASN.2015010006 (2015).
Sanz, A. B., Sanchez-Niño, M. D., Ramos, A. M. & Ortiz, A. Regulated cell death pathways in kidney disease. Nat. Rev. Nephrol. 19, 281–299. https://doi.org/10.1038/s41581-023-00694-0 (2023).
Bhargava, P. & Schnellmann, R. G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 13, 629–646. https://doi.org/10.1038/nrneph.2017.107 (2017).
Granata, S. et al. A new therapeutic target in chronic kidney disease. Nutr. Metab. (Lond). 12, 49. https://doi.org/10.1186/s12986-015-0044-z (2015).
Lan, R. et al. Mitochondrial pathology and glycolytic shift during proximal tubule atrophy after ischemic AKI. J. Am. Soc. Nephrol. 27, 3356–3367. https://doi.org/10.1681/ASN.2015020177 (2016).
Roh, J. S. & Sohn, D. H. Damage-Associated molecular patterns in inflammatory diseases. Immune Netw. 18, e27. https://doi.org/10.4110/in.2018.18.e27 (2018).
Zhao, M. et al. Mitochondrial ROS promote mitochondrial dysfunction and inflammation in ischemic acute kidney injury by disrupting TFAM-mediated MtDNA maintenance. Theranostics 11, 1845–1863. https://doi.org/10.7150/thno.50905 (2021).
Sato, Y. & Yanagita, M. Immune cells and inflammation in AKI to CKD progression. Am. J. Physiol. Ren. Physiol. 315, F1501–F1512. https://doi.org/10.1152/ajprenal.00195.2018 (2018).
Peasley, K., Chiba, T., Goetzman, E. & Sims-Lucas, S. Sirtuins play critical and diverse roles in acute kidney injury. Pediatr. Nephrol. 36, 3539–3546. https://doi.org/10.1007/s00467-020-04866-z (2021).
Zhao, W. et al. Diosmin ameliorates renal fibrosis through Inhibition of inflammation by regulating SIRT3-mediated NF-κB p65 nuclear translocation. BMC Complement. Med. Ther. 24, 29. https://doi.org/10.1186/s12906-023-04330-z (2024).
Song, N., Thaiss, F. & Guo, L. NFκB and kidney injury. Front. Immunol. 10, https://doi.org/10.3389/fimmu.2019.00815 (2019).
Nishida, K. et al. Recombinant Long-Acting thioredoxin ameliorates AKI to CKD transition via modulating renal oxidative stress and inflammation. Int. J. Mol. Sci. 22, https://doi.org/10.3390/ijms22115600 (2021).
Zare Mehrjerdi, P., Asadi, S., Ehsani, E., Askari, V. R. & Baradaran Rahimi, V. Silibinin as a major component of milk Thistle seed provides promising influences against diabetes and its complications: A systematic review. Naunyn Schmiedebergs Arch. Pharmacol. 397, 7531–7549. https://doi.org/10.1007/s00210-024-03172-x (2024).
Abenavoli, L. et al. Milk Thistle (Silybum marianum): A concise overview on its chemistry, pharmacological, and nutraceutical uses in liver diseases. Phytother Res. 32, 2202–2213. https://doi.org/10.1002/ptr.6171 (2018).
Ashique, S. et al. Unlocking the possibilities of therapeutic potential of Silymarin and Silibinin against neurodegenerative Diseases-A mechanistic overview. Eur. J. Pharmacol. 981, 176906. https://doi.org/10.1016/j.ejphar.2024.176906 (2024).
Polachi, N. et al. Modulatory effects of Silibinin in various cell signaling pathways against liver disorders and cancer - a comprehensive review. Eur. J. Med. Chem. 123, 577–595. https://doi.org/10.1016/j.ejmech.2016.07.070 (2016).
Yang, L. et al. Silibinin improves nonalcoholic fatty liver by regulating the expression of miR–122: An in vitro and in vivo study. Mol. Med. Rep. 23, https://doi.org/10.3892/mmr.2021.11974 (2021).
Chu, C. et al. Role of Silibinin in the management of diabetes mellitus and its complications. Arch. Pharm. Res. 41, 785–796. https://doi.org/10.1007/s12272-018-1047-x (2018).
Selc, M., Macova, R. & Babelova, A. Novel strategies enhancing bioavailability and therapeutical potential of Silibinin for treatment of liver disorders. Drug Des. Devel Ther. 18, 4629–4659. https://doi.org/10.2147/DDDT.S483140 (2024).
Binienda, A., Ziolkowska, S. & Pluciennik, E. The anticancer properties of silibinin: its molecular mechanism and therapeutic effect in breast cancer. Anticancer Agents Med. Chem. 20, 1787–1796. https://doi.org/10.2174/1871520620666191220142741 (2020).
Fan, Y. et al. Silibinin inhibits epithelial–mesenchymal transition of renal cell carcinoma through autophagy–dependent Wnt/β–catenin signaling. Int. J. Mol. Med. 45, 1341–1350. https://doi.org/10.3892/ijmm.2020.4521 (2020).
Liu, K. et al. Silibinin attenuates high-fat diet-induced renal fibrosis of diabetic nephropathy. Drug Des. Devel Ther. 13, 3117–3126. https://doi.org/10.2147/DDDT.S209981 (2019).
Liu, R. et al. Silibinin augments the antifibrotic effect of Valsartan through inactivation of TGF-β1 signaling in kidney. Drug Des. Devel Ther. 14, 603–611. https://doi.org/10.2147/DDDT.S224308 (2020).
Ma, Z., Zang, W., Wang, H. & Wei, X. Silibinin enhances anti-renal fibrosis effect of MK-521 via downregulation of TGF-β signaling pathway. Hum. Cell. 33, 330–336. https://doi.org/10.1007/s13577-019-00314-9 (2020).
Lim, J. O. et al. Silibinin attenuates silica dioxide Nanoparticles-Induced inflammation by suppressing TXNIP/MAPKs/AP-1 signaling. Cells 9, https://doi.org/10.3390/cells9030678 (2020).
Jiang, M. et al. Mitochondrial dysfunction and the AKI-to-CKD transition. Am. J. Physiol. Ren. Physiol. 319, F1105–F1116. https://doi.org/10.1152/ajprenal.00285.2020 (2020).
Chang, L., Chao, Y., Chiu, C., Chen, P. & Lin, H. Y. Mitochondrial signaling, the mechanisms of AKI-to-CKD transition and potential treatment targets. Int. J. Mol. Sci. 25, https://doi.org/10.3390/ijms25031518 (2024).
Mcilroy, D. R., Lopez, M. G. & Billings, F. T. T. Perioperative clinical trials in AKI. Semin Nephrol. 40, 173–187. https://doi.org/10.1016/j.semnephrol.2020.01.008 (2020).
Saadat-Gilani, K. & Zarbock, A. Perioperative renal protection. Curr. Opin. Crit. Care. 27, 676–685. https://doi.org/10.1097/MCC.0000000000000881 (2021).
Khoshnoodi, M., Fakhraei, N. & Dehpour, A. R. Possible involvement of nitric oxide in antidepressant-like effect of Silymarin in male mice. Pharm. Biol. 53, 739–745. https://doi.org/10.3109/13880209.2014.942787 (2015).
Deng, Y. et al. Silibinin attenuates ferroptosis in acute kidney injury by targeting FTH1. Redox Biol. 77, 103360. https://doi.org/10.1016/j.redox.2024.103360 (2024).
Yang, F. et al. Silibinin ameliorates cisplatin-induced acute kidney injury via activating Nfe2l1-mediated antioxidative response to suppress the ROS/MAPK signaling pathway. J. Mol. Histol. 53, 729–740. https://doi.org/10.1007/s10735-022-10089-3 (2022).
Zhang, B. et al. Nephroprotective effects of Cardamonin on renal ischemia reperfusion Injury/UUO-Induced renal fibrosis. J. Agric. Food Chem. 71, 13284–13303. https://doi.org/10.1021/acs.jafc.3c01880 (2023).
Zhang, Y. et al. Signaling pathways involved in diabetic renal fibrosis. Front. Cell. Dev. Biol. 9, 696542. https://doi.org/10.3389/fcell.2021.696542 (2021).
Schultze, S. M., Hemmings, B. A., Niessen, M. & Tschopp, O. PI3K/AKT, MAPK and AMPK signalling: protein kinases in glucose homeostasis. Expert Rev. Mol. Med. 14, e1. https://doi.org/10.1017/S1462399411002109 (2012).
Cui, X. et al. Scutellariae radix and coptidis rhizoma improve glucose and lipid metabolism in T2DM rats via regulation of the metabolic profiling and MAPK/PI3K/Akt signaling pathway. Int. J. Mol. Sci. 19, https://doi.org/10.3390/ijms19113634 (2018).
Shorning, B. Y., Dass, M. S., Smalley, M. J. & Pearson, H. B. The PI3K-AKT-mTOR pathway and prostate cancer: At the crossroads of AR, MAPK, and WNT signaling. Int. J. Mol. Sci. 21, https://doi.org/10.3390/ijms21124507 (2020).
Fu, Y. et al. Rodent models of AKI-CKD transition. Am. J. Physiol. Ren. Physiol. 315, F1098–F1106. https://doi.org/10.1152/ajprenal.00199.2018 (2018).
Yang, L., Besschetnova, T. Y., Brooks, C. R., Shah, J. V. & Bonventre, J. V. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat. Med. 16, 535–543. https://doi.org/10.1038/nm.2144 (2010) (1p-143p).
Aranda-Rivera, A. K., Cruz-Gregorio, A., Aparicio-Trejo, O. E. & Pedraza-Chaverri, J. Mitochondrial redox signaling and oxidative stress in kidney diseases. Biomolecules 11, https://doi.org/10.3390/biom11081144 (2021).
Yang, X. et al. CD36 in chronic kidney disease: novel insights and therapeutic opportunities. Nat. Rev. Nephrol. 13, 769–781. https://doi.org/10.1038/nrneph.2017.126 (2017).
Shang, Y. et al. FUT8 upregulates CD36 and its core fucosylation to accelerate pericyte-myofibroblast transition through the mitochondrial-dependent apoptosis pathway during AKI-CKD. Mol. Med. 30, 222. https://doi.org/10.1186/s10020-024-00994-6 (2024).
Tuli, H. S. et al. Path of Silibinin from diet to medicine: A dietary polyphenolic flavonoid having potential anti-cancer therapeutic significance. Semin Cancer Biol. 73, 196–218. https://doi.org/10.1016/j.semcancer.2020.09.014 (2021).
Bai, Y. et al. Silibinin, a commonly used therapeutic agent for non-alcohol fatty liver disease, functions through upregulating intestinal expression of fibroblast growth factor 15/19. Br. J. Pharmacol. 181, 3663–3684. https://doi.org/10.1111/bph.16431 (2024).
Li, P. et al. Anti-Tumor activity and mechanism of silibinin based on network pharmacology and experimental verification. Molecules 29, https://doi.org/10.3390/molecules29081901 (2024).
Pecori Giraldi, F., Cassarino, M. F., Sesta, A., Lasio, G. & Losa, M. Silibinin, an HSP90 Inhibitor, on human ACTH-Secreting adenomas. Neuroendocrinology 113, 606–614. https://doi.org/10.1159/000529710 (2023).
Jo, A. I. & Kim, M. Silibinin inhibits cell invasion through the Inhibition of MMPs, p-p38, and IL-1β in human fibrosarcoma cells. Front. Biosci. (Landmark Ed). 28, 64. https://doi.org/10.31083/j.fbl2804064 (2023).
Liu, P. et al. Silibinin ameliorates STING-mediated neuroinflammation via downregulation of ferroptotic damage in a sporadic alzheimer’s disease model. Arch. Biochem. Biophys. 744, 109691. https://doi.org/10.1016/j.abb.2023.109691 (2023).
Li, W. et al. Silibinin exerts neuroprotective effects against cerebral hypoxia/reoxygenation injury by activating the GAS6/Axl pathway. Toxicology 495, 153598. https://doi.org/10.1016/j.tox.2023.153598 (2023).
Choi, S. et al. Silibinin downregulates types i and III collagen expression via suppression of the mTOR signaling pathway. Int. J. Mol. Sci. 24, https://doi.org/10.3390/ijms241814386 (2023).
Im, H. et al. Silibinin mitigates vanadium-induced lung injury via the TLR4/MAPK/NF-κB pathway in mice. Vivo 38, 2179–2189. https://doi.org/10.21873/invivo.13681 (2024).
Wu, X. et al. Silibinin attenuates TGF-β2-induced fibrogenic changes in human trabecular meshwork cells by targeting JAK2/STAT3 and PI3K/AKT signaling pathways. Exp. Eye Res. 244, 109939. https://doi.org/10.1016/j.exer.2024.109939 (2024).
Sun, J. et al. Mitochondria in Sepsis-Induced AKI. J. Am. Soc. Nephrol. 30, 1151–1161. https://doi.org/10.1681/ASN.2018111126 (2019).
Acknowledgements
This work was supported by The research project of Health Commission of Hubei Province(WJ2025Q020) and Natural Science Foundation of Hubei Province (No.2025AFB803).
Funding
Natural Science Foundation of Hubei Province (No.2025AFB803). The research project of Health Commission of Hubei Province(WJ2025Q020).
Author information
Authors and Affiliations
Contributions
Sijin Dong: Writing-review & editing, Writing-original draft, Methodology, Formal analysis, Data curation, Software, Conceptualization. Jun Jian: Writing-review & editing, Writing-original draft, Methodology, Formal analysis, Data curation, Software, Conceptualization. Yufeng xiong: Formal analysis, Data curation, Methodology, Software, Investigation. Jingsong Wang: Conceptualization, Formal analysis. Qianxue Lu: Methodology, Formal analysis. Wei Li: Conceptualization, Formal analysis, Data curation. Shanshan Wan: Supervision, Formal analysis, Data curation. Lei Wang: Validation, Supervision, Project administration, Investigation, Funding acquisition.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Dong, S., Jian, J., Xiong, Y. et al. Silibinin mitigates AKI-to-CKD transition via MAPK and PI3K/AKT signaling pathways in Ischemia-Reperfusion injury. Sci Rep 15, 40609 (2025). https://doi.org/10.1038/s41598-025-24433-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-24433-6
