
Abstract
This study conducted exploratory analyses of the effects of BIIB080, a MAPT (microtubule-associated protein tau)-targeting antisense oligonucleotide, in participants with mild Alzheimer’s disease. A multicenter, randomized, double-blind, phase 1b trial was conducted as a placebo-controlled, multiple-ascending dose (MAD) study followed by an open-label, long-term extension (LTE). During the MAD study, participants were randomized and received either intrathecal placebo or BIIB080 10 mg (n = 6), 30 mg (n = 6) or 60 mg (n = 9) every 4 weeks or 115 mg (n = 13) every 12 weeks (Q12W). During the LTE, participants received high-dose BIIB080 (60 mg (n = 7) or 115 mg (n = 9) Q12W). BIIB080 was generally well tolerated. Here we present findings from exploratory analyses, which showed a consistent trend of slowed decline on cognitive, functional and global measures favoring BIIB080 high-dose groups at the end of both study periods. This favorable trend is supported by reported reductions from baseline in brain neurofibrillary tangles measured with tau positron emission tomography. Trial registration: ClinicalTrials.gov identifier: NCT03186989.
Similar content being viewed by others

Main
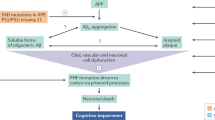
Alzheimer’s disease (AD) is a progressive neurogenerative disorder characterized by pathological accumulation of extracellular amyloid plaques and intracellular aggregates of hyperphosphorylated tau known as neurofibrillary tangles (NFTs)1. Tau aggregates, measured by tau positron emission tomography (PET), are closely linked to symptom severity and the clinical progression of AD2, and it has been hypothesized that reduction of these aggregates may lead to slowing or halting of disease progression in this amyloid-driven secondary tauopathy3,4. First-generation disease-modifying therapies for AD target amyloid and have mixed effects on NFT accumulation5,6; presently, there are no approved treatments that specifically target tau. Although progress has been made, there remains a considerable unmet need for the treatment of AD.
BIIB080 is a MAPT-targeting antisense oligonucleotide (ASO) that selectively reduces MAPT premessenger RNA, decreasing de novo production of all isoforms of tau, including posttranslationally modified and toxic species of tau, regardless of location in or outside of the cell3,4. Although it remains unclear how or what forms of tau contribute to pathogenesis4,7,8, BIIB080 may provide a potential therapeutic advantage because of its ability to reduce overall production of tau.
A multiple-ascending dose (MAD) phase 1b study (Fig. 1) was completed in participants with mild AD, with the primary objective of evaluating the safety and tolerability of intrathecally administered BIIB080 (ref. 4). BIIB080 was generally well tolerated4. Adverse events, reported in 94% of BIIB080-treated patients and in 75% of participants who received placebo, were all mild (88%) or moderate (12%) in severity4. The most commonly reported adverse events were post–lumbar puncture syndrome and headache (approximately 44%); most of these cases were considered mild4. Analyses of exploratory biomarkers of cerebrospinal fluid (CSF) demonstrated robust, dose-dependent and sustained BIIB080-induced reductions in total tau (t-tau) and phosphorylated tau 181 (p-tau181) levels in the MAD period4; this effect was maintained in the open-label LTE period3.

For cohorts A and B, week 64 was counted from LTE registration. In cohorts C and D, week 100 was counted from MAD screening. Cohorts A and B had a gap (5−19 months) between completion of the MAD treatment period and commencement of the LTE treatment period; cohorts C and D had a seamless transition between the MAD and LTE. CDR, MMSE, FAQ and RBANS were also assessed at the safety follow-up visit at week 72 for the low-dose groups and at week 100 for the high-dose groups. a FAQ was not collected at week 9. mo., months; Post-Rx, posttreatment period; Rx, treatment period; wk, week.
In the exploratory tau PET substudy, BIIB080 impacted parenchymal tau pathology at week 25 during the MAD period3. After the LTE period was completed (week 100), BIIB080 was shown to reduce parenchymal tau pathology from baseline in all brain composites assessed3. This result represents, to our knowledge, the first time that a reduction in aggregated tau has been observed across multiple brain regions in response to a disease-modifying therapy. As tau pathology is strongly correlated with clinical decline9, the outcomes of the BIIB080 phase 1b study are of special interest. The concurrent observation of favorable trends in tau biomarkers as well as clinical outcomes, if confirmed in subsequent clinical trials in AD, would be an important milestone for the field and potentially of great benefit for patients with AD.
Here we report the exploratory analyses of clinical outcomes collected in the MAD and LTE periods of the BIIB080 phase 1b study. Recognizing that the study was not powered for the statistical inferences of the effect on the clinical outcomes, we emphasize the exploratory nature of these findings.
Results
Exploratory MAD analysis
Characteristics of participants at MAD baseline are summarized in Table 1. This is a younger study population than in other recent AD trials6,10. Differences in MAD baseline clinical scores were observed between treatment groups (Table 1). The mean Clinical Dementia Rating-Sum of Boxes (CDR-SB) scores at baseline were numerically lower for BIIB080 high-dose groups (cohort C, 60 mg every 4 weeks (Q4W): 2.9 (s.d. 0.6); cohort D, 115 mg every 12 weeks (Q12W): 3.3 (s.d. 1.1)) compared to low-dose groups (cohort A, 10 mg Q4W: 4.8 (s.d. 0.5); cohort B, 30 mg Q4W: 4.7 (s.d. 1.0)) due to an amendment during enrollment of cohort B that allowed inclusion of participants with a CDR-Global Score (CDR-GS) of 0.5 in addition to the original criterion of 1.0. This amendment resulted in more participants with a CDR-GS of 1.0 in cohorts A and B compared to cohorts C and D and could potentially explain the differences observed in mean CDR-SB scores at baseline. In addition, the placebo group included participants from all cohorts, resulting in more participants with a CDR-GS of 1.0 and higher CDR-SB scores at baseline compared to cohorts C and D. Moreover, Functional Activities Questionnaire (FAQ) and Repeatable Battery for the Assessment of Neuropsychological Status (RBANS) Delayed Memory scores were much higher (suggestive of more advanced disease) in cohort B versus the other groups.
The low-dose groups showed mixed trends for the Mini-Mental State Examination (MMSE), FAQ and RBANS Delayed Memory scores relative to placebo (Fig. 2), which may be partially attributed to the differences in baseline characteristics among groups. In the high-dose groups, numerically smaller decline on MMSE was seen compared to placebo beginning at week 9 and across all postbaseline visits (Fig. 2a). Numerically smaller decline was also observed on FAQ at weeks 25 and 37 in the high-dose groups compared to placebo (Fig. 2b). For RBANS Delayed Memory scale, cohort D showed slightly less decline at week 37 compared to placebo, and cohort C exhibited improvement from baseline across all postbaseline visits (Fig. 2c). Spaghetti plots of MMSE, RBANS Delayed Memory scale and FAQ change from baseline scores and scores at each visit for individual participants during the MAD period are shown in Extended Data Figs. 1 and 2. Although the interpretation of the BIIB080 high-dose group performance was admittedly limited given the differences in the baseline characteristics among groups, it is still encouraging to observe numerical differences favoring the BIIB080 high-dose groups at weeks 25 and 37 in the context of the trend suggesting a pharmacodynamic effect on tau PET at week 25 that was previously reported3.

Gray shading indicates the 13-week treatment period. Results at each visit were based on an ANCOVA model, fitted with change from baseline as a dependent variable, and with treatment group, baseline value and baseline CDR-GS as independent variables. Error bars reflect s.e. No statistical testing or multiple comparison adjustments were performed. a Number of participants by study week based on MMSE. Small differences in n were seen for RBANS and FAQ. FAQ was not collected at week 9. In a, at baseline, the number of participants with MMSE scores in the placebo group was n = 12; BIIB080 10 mg Q4W had an n = 6; BIIB080 30 mg Q4W had an n = 6; BIIB080 60 mg Q4W had an n = 9; and BIIB080 115 mg Q12W had an n = 13. In b, at baseline, the number of participants with RBANS scores in the placebo group was n = 12; BIIB080 10 mg Q4W had an n = 6; BIIB080 30 mg Q4W had an n = 6; BIIB080 60 mg Q4W had an n = 9; and BIIB080 115 mg Q12W had an n = 13. In c, at baseline, the number of participants with FAQ scores in the placebo group was n = 12; BIIB080 10 mg Q4W had an n = 6; BIIB080 30 mg Q4W had an n = 6; BIIB080 60 mg Q4W had an n = 9; and BIIB080 115 mg Q12W had an n = 13.
Exploratory LTE analysis
Of 33 participants who enrolled in the LTE3, a total of 16 (nine in cohort D and seven in cohort C) received high-dose BIIB080 throughout the MAD and LTE periods and had evaluable clinical data at both MAD baseline and week 100. These 16 LTE participants consisted of the high → high groups, and their changes in outcomes were analyzed from MAD baseline to week 100. Participants who received BIIB080 high doses in the LTE only were not the focus of this analysis because they either had a variable gap period that would complicate the analysis and data interpretation or had no baseline data collected at LTE entry due to seamless transition between the MAD to the LTE (see Methods for additional details).
For the open-label portion of the trial, a pooled placebo was no longer available, and, therefore, an external control comparator was selected. TANGO, a phase 2 clinical trial of the anti-tau monoclonal antibody gosuranemab, was selected as a contemporaneous study with similar population characteristics as the BIIB080 phase 1b study. BIIB080-treated groups were compared to TANGO participants who were selected with propensity score matching (PSM)11. Baseline characteristics for the high → high groups were well matched with the TANGO PSM external controls for age, sex and disease severity (Table 2). The change on CDR-SB in the TANGO PSM dataset was estimated to be 3.1 at week 100, which is within the range estimated for mild AD (Fig. 3a)12. A numerical difference favoring BIIB080 was seen on CDR-SB, MMSE and FAQ change from baseline for the high → high groups at week 100 versus matched external controls (Fig. 3a). Numerical differences favoring BIIB080 were also observed in all CDR box scores at week 100 (Fig. 3b). Spaghetti plots of MMSE, CDR-SB and FAQ change from baseline scores and scores at each visit during the combined MAD and LTE periods are shown for the 16 participants in the high → high groups in Extended Data Figs. 3 and 4.

For high → high groups, TANGO PSM used 1:1 match and adjusted for seven covariates: CDR-GS (exact match), CDR-SB, MMSE, FAQ, APOE, age and sex. In a and b, changes in clinical outcomes from the MAD baseline to the end of LTE (week 100) were analyzed, and results were based on an ANCOVA model, with treatment group, baseline value and baseline CDR-GS as independent variables. No statistical testing or multiple comparison adjustments were performed. In a, nine participants in cohort D, seven participants in cohort C and 16 participants in the TANGO PSM group had CDR-SB, MMSE and FAQ scores. In b, nine participants in cohort D, seven participants in cohort C and 16 participants in the TANGO PSM group had CDR Memory Box, Orientation, Judgment and Problem Solving, Community Affairs, Home and Hobbies and Personal Care scores. CI, confidence interval; Chg Ctrl., change in control; Diff vs Ctrl., difference versus control.
Supplementary analyses were performed for the high → high groups using additional external control groups selected from TANGO with alternative methods (that is, the subset of TANGO participants selected with key inclusion/exclusion (I/E) criteria of the BIIB080 phase 1b study (TANGO I/E criteria) and a subset of TANGO participants selected by resampling from the TANGO I/E criteria (TANGO resample)) and additional external control groups selected from the Alzheimer’s Disease Neuroimaging Initiative (that is, ADNI PSM, ADNI and ADNI resample). Despite varying magnitude of numerical differences between these analyses and the analysis using TANGO PSM, all group comparisons consistently favored BIIB080 across all clinical scales (Fig. 4).

At week 100, TANGO I/E criteria had an n = 101 for MMSE scores and an n = 99 for FAQ scores. At week 100, ADNI I/E criteria had an n = 40 for MMSE scores. TANGO PSM used 1:1 match and adjusted for seven covariates: CDR-GS (exact match), CDR-SB, MMSE, FAQ, APOE, age and sex. ADNI PSM used 1:1 match and adjusted for five covariates: CDR-GS (exact match), CDR-SB, MMSE, FAQ and APOE. The I/E criteria method selected participants by key inclusion criteria of the BIIB080 phase 1b study. The resample method randomly selected 16 participants from the I/E criteria set, and the results were summarized from 1,000 rounds of resampling. In a and b, changes in clinical outcomes from the MAD baseline to the end of the LTE (week 100) were analyzed, and results were based on an ANCOVA model, with treatment group, baseline value and baseline CDR-GS as independent variables. No statistical testing or multiple comparison adjustments were performed. CI, confidence interval.
Primary endpoint of safety
As previously reported, BIIB080 was generally well tolerated during the MAD4. An overall summary of treatment-emergent adverse events (TEAEs) among the safety population during the LTE for all dose groups (N = 33) is presented in Table 3. For TEAEs, the relationship to study treatment or procedure was determined by the investigator. During the LTE, 31 participants (93.9%) reported one or more TEAEs, and 14 participants (42.4%) reported one or more treatment-related TEAEs. Most TEAEs were mild or moderate in severity; however, one participant reported a severe TEAE (pain in extremity). Twenty-four (72.7%) participants reported at least one study procedure-related TEAE, which were all mild or moderate in severity and non-serious. The most common TEAEs (occurring in four or more participants) during the LTE included back pain (39.4%), pain in extremity (27.3%), headache (24.2%), dizziness (15.2%), procedural pain (15.2%), confusional state (12.1%), fatigue (12.1%) and post–lumbar puncture syndrome (12.1%). A total of four participants (12.1%) reported one or more serious TEAEs, which were assessed as not related to study treatment or procedure. Three participants (9.1%) reported a total of four TEAEs that led to treatment discontinuation: one participant with two serious adverse events (paranoia and aggression) and two participants with dementia Alzheimer’s type. All events that led to discontinuation were moderate in severity. One of the four TEAEs (dementia Alzheimer’s type) was possibly related to study treatment and not related to study procedure. No deaths were reported during the LTE. BIIB080 was generally well tolerated in the LTE.
Discussion
BIIB080 was generally well tolerated during the MAD and LTE4. At the end of the MAD in the BIIB080 phase 1b study, exploratory analyses demonstrated a numerical difference of slowed clinical decline favoring the BIIB080 high-dose groups compared to the placebo group, based on change from baseline in MMSE, FAQ and RBANS Delayed Memory scale at week 25 and/or week 37. Similarly, a favorable numerical difference of slowed clinical decline was seen at the end of the LTE in participants who received high-dose BIIB080 in both the MAD and the LTE versus the external comparator on CDR-SB, MMSE and FAQ scales at week 100.
In the low-dose groups versus placebo, mixed trends were observed at the end of the MAD period. As previously noted, the low-dose groups had more advanced disease at the start of the MAD. In addition, there were no tau PET data for these early cohorts because tau PET was not available at the beginning of the study; thus, it is not presently clear whether these low doses had an impact on aggregated tau pathology. Due to these limitations, it is difficult to make any conclusions based on the observations from the low-dose groups.
Consistent trends favoring the BIIB080 high-dose groups on multiple cognitive and functional scales at the end of the MAD and LTE periods in the BIIB080 phase 1b study align with mechanistic evidence from preclinical studies and tau PET results from the phase 1b study3.
In preclinical studies of transgenic mice expressing mutant P301S human tau, antisense-mediated knockdown of MAPT premessenger RNA led to lowered tau protein expression and reduction of preexisting tau pathology assessed histologically by the phospho-tau epitope AT8 (ref. 13). The reduction of tau pathology was accompanied by reduction of the severity of toxin-induced seizures, reduction of neuronal loss, improvement in behavioral phenotypes and lengthening of survival in these adult tau-transgenic mouse models13,14. Consistent with the preclinical data, the BIIB080 phase 1b study demonstrated reduction from baseline in parenchymal tau pathology (measured by tau PET) across all brain regions assessed at the end of the LTE, indicating an impact of BIIB080 on this pathologic hallmark of AD. These data, taken together, provide evidence that BIIB080 may alter the underlying pathology of AD and potentially impact clinical progression.
Ongoing investigation of BIIB080 in the phase 2 CELIA study (NCT05399888)15 will generate additional data regarding the efficacy and safety of BIIB080 as well as insights into the dynamics of tau PET effects and their relationship with clinical outcomes.
Study limitations
Interpretation of the impact of BIIB080 on clinical progression in this study focused on numerical differences or trends rather than the magnitude or statistical inference of group differences given the exploratory nature of this analysis, the small sample size and the use of external controls in the LTE analyses. Due to the small sample size, data were analyzed at each postbaseline visit by an analysis of covariance (ANCOVA) model, not accounting for the within-participant correlation between visits. Recognizing that the exploratory analyses described in this study were not powered for the statistical inferences of the effect on the clinical outcomes, we do not report P values; however, standard errors and/or confidence intervals are reported when applicable.
Several other factors should be noted when interpreting the results of these exploratory analyses. Distribution of APOE (apolipoprotein E) alleles varied across the cohorts; cohorts A and D had higher percentages of APOE4 carriers compared to the placebo and other cohort groups. Additionally, this study was conducted in relatively younger participants than those in contemporaneous trials who were screened for genetic AD mutations but were not excluded based on this screening. Indeed, one participant in the early-start cohort D who rolled over into the LTE had a mutation in PSEN2 (encoding presenilin 2).
Of note, in the open-label portion of the trial, a placebo group was no longer a part of the study, and, therefore, an external control comparator was selected using PSM. Despite best efforts to match key baseline characteristics between the treated participants and the external control comparator, residual confounding variables may still exist. This could be due to some important clinical, biological or functional characteristics that influence clinical outcomes of AD which were not collected and, therefore, not accounted for in the matching process. In addition, the open-label design in the LTE may have affected participants’ behavior and/or expectations as well as clinicians’ assessments, potentially introducing bias into the clinical outcomes. These limitations highlight the importance of confirming these preliminary observations in a randomized controlled clinical trial that is designed to rigorously evaluate the efficacy of BIIB080.
The results for the exploratory analyses of clinical outcomes from the BIIB080 phase 1b MAD and LTE study in mild AD revealed numerical differences favoring high-dose BIIB080 treatment across multiple clinical endpoints, and BIIB080 was generally well tolerated. Although the limited sample sizes and wide confidence interval ranges warrant cautious interpretation, the consistency of the favorable trends across clinical endpoints for patients receiving high doses of BIIB080 aligns with preclinical and biomarker (for example, tau PET) data. These data support further investigation of the clinical efficacy and safety of BIIB080 in patients with mild cognitive impairment (MCI) due to AD or mild AD in the ongoing CELIA phase 2 trial15.
Methods
Study design
This phase 1b, multicenter, randomized, double-blind clinical trial evaluated the safety, tolerability and optimal dosing of BIIB080. The study was conducted from October 2017 to May 2022 in the following countries: Germany, United Kingdom, Netherlands, Sweden, Finland and Canada. Details of clinical study NCT03186989 were reported previously3,4. Investigators, participants and the sponsor were blinded to trial group assignments for the trial duration. Part 1 was a placebo-controlled MAD period with a 13-week treatment and a 24-week posttreatment observation period, followed by an open-label LTE with a 48-week treatment and an up to 23-week posttreatment observation period. The study design, dosing schema and timing of clinical and tau PET measurements are shown in Fig. 1. The full trial protocol was published previously3.
Eligibility criteria
Eligible participants were between the ages of 50 years and 74 years and had mild AD as defined by a CDR-GS of 1.0 or 0.5 with a Memory Score of 1 and MMSE of 20–27 inclusive (scores range from 0 to 30; 20–27 may represent mild AD)4; a CSF pattern of low Aβ42 and elevated t-tau and p-tau consistent with diagnosis of mild AD; and a diagnosis of probable AD based on National Institute of Aging-Alzheimer’s Association criteria16. An amendment during enrollment of cohort B allowed inclusion of participants with a CDR-GS of 0.5 in addition to the original criterion of 1.0. This amendment resulted in more participants with a CDR-GS of 1.0 in cohorts A and B compared to cohorts C and D.
During the MAD period, participants were randomized 3:1 to receive intrathecal bolus administration of BIIB080 or placebo in four dose cohorts during the 13-week treatment period: cohort A (low dose: 10 mg Q4W; total of four doses each); cohort B (low dose: 30 mg Q4W; total of four doses each); cohort C (high dose: 60 mg Q4W; total of four doses each); or cohort D (high dose: 115 mg Q12W; total of two doses each)3.
After the 13-week MAD treatment period, eligible participants in cohorts C and D seamlessly transitioned to the LTE, after a 24-week posttreatment observation period, whereas participants in cohorts A and B had a variable gap period ranging from 5 months to 19 months between completion of the MAD treatment period and commencement of the LTE treatment period. Participants in the LTE treatment period were dosed for 48 weeks with 60 mg Q12W (MAD cohorts A, B or C) or 115 mg Q12W (MAD cohort D).
The trial was conducted in accordance with the Declaration of Helsinki and all applicable International Council for Harmonisation and Good Clinical Practice guidelines. Investigators were required to obtain institutional review board or ethics committee approval before beginning the study. The list of ethics committees that approved the study was reported previously4. All participants provided written informed consent before participating in any study-related activities. No financial compensation was provided to participants in this study.
Clinical outcomes
The primary outcome was safety and tolerability of BIIB080 treatment assessed by the incidence and severity of AEs. Exploratory analyses of clinical outcomes included MMSE, FAQ, RBANS and CDR scale. MMSE, FAQ and RBANS were assessed longitudinally over the MAD and LTE. CDR was assessed only at baseline and at the end of the LTE (Fig. 1).
The MMSE is used to quantify cognitive function and to screen for cognitive impairment. It is a severity scale, not a diagnostic scale, and can be confounded by level of education. MMSE has a maximum possible score of 30 points17, with lower scores indicating greater impairment18. CDR is a global scale used at screening to categorize the severity of Alzheimer’s type dementia19,20. It utilizes a semi-structured test administrator interview with the patient and the study partner to obtain information necessary to rate the patient’s performance in six domains: Memory, Orientation, Judgment and Problem Solving, Community Affairs, Home and Hobbies and Personal Care. Categorical scores for each domain are 0 (none), 0.5 (questionable), 1 (mild), 2 (moderate) and 3 (severe). A summed total score (sum of boxes) is produced, and a global score (using the same five grades of dementia) is derived from the category scores according to the practice described by Morris20. The FAQ is a widely used scale to assess activities of daily living in patients with mild AD21,22. It is a brief questionnaire in which the trial partner rates the patient’s abilities in 10 areas, such as keeping track of current events and preparing a balanced meal, on a scale of 0 (normal) to 3 (dependent). A score of 30 represents maximal dependence, and score of 0 represents complete independence23. A score of 9 or higher is often used as a potential indicator of mild AD, suggesting that the individual may need assistance with several daily living tasks. The RBANS is a neurological assessment designed to identify abnormal cognitive decline in older adults and to differentiate between dementia etiologies24. The assessment yields five index scores, one each for the domains tested: Attention, Visuospatial/Constructional Abilities, Language, Immediate Memory and Delayed Memory. An index score is calculated by summing the age-corrected standardized total scores of specific subtests within a particular cognitive domain, with each index score having a mean of 100 and an s.d. of 15 (refs. 24,25,26).
The scales used in this study are commonly used in trials of early-stage AD.
Statistics and reproducibility
Although no statistical methods were used to predetermine sample size, the sample sizes were selected based on previous publications assessing ASOs4. The exploratory analyses reported in this paper were post hoc and were not included in the previously published statistical analysis plan4. Statistical analyses were performed using Statistical Analysis System (SAS) v.9.4 software. For the MAD, clinical outcomes were compared between the groups that received BIIB080 versus the pooled placebo group. Changes from MAD baseline on MMSE total score, FAQ total score and RBANS Delayed Memory Index were analyzed using an ANCOVA model adjusting for treatment group, baseline clinical scale score and baseline CDR-GS. ANCOVA models were used to analyze the change from baseline scores of clinical scales assuming that these change scores were normally distributed, but this assumption was not formally tested. No statistical tests were performed for the group differences. CDR Memory Box scores were explored in greater depth as memory loss is a prominent symptom in AD; exploration of CDR Memory Box scores was also data driven as a result of the findings from the phase 1b study.
For the LTE, analyses were performed for participants who transitioned seamlessly from the MAD to the LTE (referred to as the high → high groups). The high → high groups included participants in cohorts D (115 mg Q12W → 115 mg Q12W) and C (60 mg Q4W → 60 mg Q12W) who received high doses throughout the MAD and LTE. The analysis period for the high → high groups was change from MAD baseline to week 100 (at the end of the LTE). An ANCOVA model was fitted for each clinical scale (CDR-SB, CDR box scores, MMSE total score and FAQ total score), adjusting for treatment group, baseline CDR-GS and baseline clinical scale score (the same model used for the MAD). Participants who received BIIB080 high doses in the LTE only were not the focus of this study because they either had a variable gap period of 5−19 months between the end of the MAD and the start of the LTE (participants from cohorts A and B), which would complicate the analysis and data interpretation, or had no baseline data collected at LTE entry due to seamless transition between the MAD to the LTE (placebo participants from cohorts C and D). The LTE safety population included all participants who were randomized and received one or more doses of BIIB080 (including participants who received placebo in the MAD and BIIB080 in the LTE).
No data were excluded from the analyses for the placebo-controlled MAD. For the LTE, analyses were performed for participants who transitioned seamlessly from the MAD to the LTE (these participants received BIIB080 high dose in both MAD and LTE and are referred to as the high → high groups). No data were excluded from the analyses of the high → high groups.
External controls and PSM
Given the lack of a placebo group in the LTE, the high → high groups were compared to external controls selected with PSM, a methodology that identified participants in the external control data source with characteristics similar to those of participants in the BIIB080-treated groups. The following baseline covariates were included in the PSM model: baseline CDR-GS, baseline CDR-SB, baseline MMSE, APOE carrier status, baseline FAQ, age and sex. The primary data source for external controls was the TANGO study (NCT03352557), an 18-month placebo-controlled, randomized, phase 2 study with a dose-blinded LTE period of anti-tau monoclonal antibody gosuranemab in early AD (MCI due to AD or mild AD) that demonstrated no efficacy signal on disease biomarkers or clinical endpoints27. Data up to week 104 across the placebo-controlled and LTE periods from participants with mild AD from the pooled placebo and active treatment groups of the TANGO study were included as the data source for external controls. TANGO was chosen because it had similar inclusion criteria as the BIIB080 phase 1b study (for example, amyloid positivity, CDR-GS scores of 0.5 or 1 in both studies, ages 50−80 years in TANGO and 50−74 years in the phase 1b study and MMSE scores of 22−30 in TANGO and 20−27 in the phase 1b study), except that TANGO enrolled participants with either MCI due to AD or mild AD, whereas the phase 1b study only enrolled participants with mild AD4,27,28. Additionally, TANGO (May 2018 to August 2021) was a contemporaneous study with rigorous and extensive data collection. The decision to use TANGO participants as primary external controls was made prior to conducting any exploratory analyses for the LTE. Supplementary analyses were performed using ADNI as an alternative data source for external controls and additional matching methods to select external control comparators. External controls were not used in the safety analysis.
As noted previously, the primary external control comparator was selected using PSM from the TANGO study (that is, TANGO PSM). The following important demographic and prognostic factors were included in the PSM model: baseline CDR-GS, baseline CDR-SB, baseline MMSE, APOE carrier status, baseline FAQ, age and sex. Matching was exact for the CDR-GS. These factors were selected based on previous experience with clinical trials in the early AD population, in addition to consideration of data availability in both the phase 1b and TANGO studies. For each baseline covariate, a standardized difference of ≤0.2 between the BIIB080-treated group and the external control comparator was considered a good matching performance (that is, balanced baseline characteristics after matching). For the high → high groups, the TANGO PSM external comparator was selected using optimal 1:1 matching with all seven baseline covariates listed above. All covariates achieved a standardized difference of ≤0.2 after matching.
Supplementary analysis was performed using the ADNI as an alternative data source for external controls. ADNI is a large, open-access database that contains longitudinal clinical, imaging, genetic and biochemical biomarker data from participants at different AD stages29,30. ADNI was not selected as the primary choice for the external control because it enrolled participants aged 55−90 years, resulting in an older cohort than those enrolled in the BIIB080 phase 1b study (aged 50−74 years)29,31. As a consequence of the relatively small overlap in age range, the PSM model could not achieve a standardized difference of ≤0.2 for all covariates for the high → high groups. A modified PSM algorithm was implemented to prioritize the matching for baseline CDR-SB and MMSE. Specifically, the PSM model was updated to exclude sex, age and baseline FAQ sequentially until the standardized difference was ≤0.2 for CDR-SB and MMSE or until sex, age and baseline FAQ were all dropped. In the end, the ADNI PSM external comparator was selected using optimal 1:1 matching with the following five baseline covariates: CDR-GS (exact match), CDR-SB, MMSE, FAQ and APOE.
In addition to PSM, additional methods were used to select external control comparators from TANGO and ADNI. The external controls were selected using the key inclusion criteria of the phase 1b study (amyloid positivity, mild AD, CDR-GS 0.5 with CDR Memory Box score of 1.0 or a CDR-GS of 1.0, MMSE 20–27 and ages 50–74 years) and are referred to as TANGO I/E criteria (n = 102) and ADNI I/E criteria (n = 41), respectively. At week 100, TANGO I/E criteria comparators had an n = 101 for MMSE scores and an n = 99 for FAQ scores; at week 100, the ADNI I/E criteria comparators had an n = 40 for MMSE scores. In addition, a resampling approach was performed, referred to as TANGO resample and ADNI resample, respectively. For each of the resampling steps, 16 participants were randomly selected from the ‘TANGO I/E criteria’ set and the ‘ADNI I/E criteria’ set, respectively, to compare to the BIIB080 high → high groups using the ANCOVA model. For each resampling set, an ANCOVA model was fitted for each clinical scale (CDR-SB, MMSE total score and FAQ total score) and adjusted for treatment group, baseline CDR-GS and baseline clinical scale score. This process was repeated 1,000 times, and the results of 1,000 resamplings were summarized.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The trial information is publicly available at ClinicalTrials.gov (https://clinicaltrials.gov/study/NCT03186989). Individual participant data collected during the trial may be shared after anonymization and upon approval of the research proposal in accordance with internal policies and procedures. Biogen commits to sharing patient-level data, study-level data, clinical study reports and protocols with qualified scientific researchers who provide a methodologically sound proposal. Biogen reviews all data requests internally based on the review criteria and in accordance with its Clinical Trial Transparency and Data Sharing Policy, which is available at https://www.biogentrialtransparency.com. Deidentified data and documents will be shared under agreements that further protect against participant reidentification. To request access to data, please visit https://vivli.org/.
References
DeTure, M. A. & Dickson, D. W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 14, 32 (2019).
Coomans, E. M. et al. A head-to-head comparison between plasma ptau181 and tau PET along the Alzheimer’s disease continuum. J. Nucl. Med. 64, 437–443 (2023).
Edwards, A. L. et al. Exploratory tau biomarker results from a multiple ascending-dose study of BIIB080 in Alzheimer disease: a randomized clinical trial. JAMA Neurol. 80, 1344–1352 (2023).
Mummery, C. J. et al. Tau-targeting antisense oligonucleotide MAPTRX in mild Alzheimer’s disease: a phase 1b, randomized, placebo-controlled trial. Nat. Med. 29, 1437–1447 (2023).
Budd Haeberlein, S. et al. Two randomized phase 3 studies of aducanumab in early Alzheimer’s disease. J. Prev. Alzheimers Dis. 9, 197–210 (2022).
Sims, J. R. et al. Donanemab in early symptomatic Alzheimer disease: the TRAILBLAZER-ALZ 2 randomized clinical trial. JAMA 330, 512–527 (2023).
Corsi, A. et al. Tau isoforms: gaining insight into MAPT alternative splicing. Int. J. Mol. Sci. 23, 15383 (2022).
Yang, J. et al. Complicated role of post-translational modification and protease-cleaved fragments of tau in Alzheimer’s disease and other tauopathies. Mol. Neurobiol. 61, 4712–4731 (2024).
Malpetti, M., Joie, R. & Rabinovici, G. D. Tau beats amyloid in predicting brain atrophy in Alzheimer disease: implications for prognosis and clinical trials. J. Nucl. Med. 63, 830–832 (2022).
van Dyck, C. H. et al. Lecanemab in early Alzheimer’s disease. N. Engl. J. Med. 388, 9–21 (2023).
Austin, P. C. An introduction to propensity score methods for reducing the effects of confounding in observational studies. Multivar. Behav. Res. 46, 399–424 (2011).
O’Bryant, S. E. et al. Staging dementia using Clinical Dementia Rating Scale Sum of Boxes scores: a Texas Alzheimer’s Research Consortium study. Arch. Neurol. 65, 1091–1095 (2008).
DeVos, S. L. et al. Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci. Transl. Med. 9, eaag0481 (2017).
DeVos, S. L. et al. Antisense reduction of tau in adult mice protects against seizures. J. Neurosci. 33, 12887–12897 (2013).
A Study to Learn About the Safety of BIIB080 Injections and Whether They Can Improve Symptoms of Participants With Mild Cognitive Impairment Due to Alzheimer's Disease (AD) or Mild AD Dementia Between 50 to 80 Years of Age (CELIA). https://www.clinicaltrials.gov/ct2/show/NCT05399888 (2025).
Albert, M. S. et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7, 270–279 (2011).
Folstein, M. F., Folstein, S. E. & McHugh, P. R. ‘Mini-mental state’. A practical method for grading the cognitive state of patients for the clinician. J. Psychiatr. Res. 12, 189–198 (1975).
Kennedy, R. E. et al. Using baseline cognitive severity for enriching Alzheimer’s disease clinical trials: how does Mini-Mental State Examination predict rate of change? Alzheimers Dement. (N Y) 1, 46–52 (2015).
Morris, J. C. Clinical Dementia Rating: a reliable and valid diagnostic and staging measure for dementia of the Alzheimer type. Int. Psychogeriatr. 9, 173–176 (1997).
Morris, J. C. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 43, 2412–2414 (1993).
Marshall, G. A. et al. Executive function and instrumental activities of daily living in mild cognitive impairment and Alzheimer’s disease. Alzheimers Dement. 7, 300–308 (2011).
Brown, P. J. et al. Functional impairment in elderly patients with mild cognitive impairment and mild Alzheimer disease. Arch. Gen. Psychiatry 68, 617–626 (2011).
Pfeffer, R. I. et al. Measurement of functional activities in older adults in the community. J. Gerontol. 37, 323–329 (1982).
Randolph, C. et al. The Repeatable Battery for the Assessment of Neuropsychological Status (RBANS): preliminary clinical validity. J. Clin. Exp. Neuropsychol. 20, 310–319 (1998).
Duff, K. et al. Diagnostic accuracy of the RBANS in mild cognitive impairment: limitations on assessing milder impairments. Arch. Clin. Neuropsychol. 25, 429–441 (2010).
Randolph, C. RBANS: Repeatable Battery for the Assessment of Neuropsychological Status: Manual (The Psychological Corporation, 1998).
Shulman, M. et al. TANGO: a placebo-controlled randomized phase 2 study of efficacy and safety of the anti-tau monoclonal antibody gosuranemab in early Alzheimer’s disease. Nat. Aging 3, 1591–1601 (2023).
Phase 2 Study of BIIB092 in Participants With Early Alzheimer's Disease (TANGO). https://www.clinicaltrials.gov/ct2/show/NCT03352557 (2022).
Petersen, R. C. et al. Alzheimer’s Disease Neuroimaging Initiative (ADNI): clinical characterization. Neurology 74, 201–209 (2010).
Alzheimer’s Disease Neuroimaging Initiative. https://adni.loni.usc.edu/
Ziogas, N. et al. Exploratory clinical outcomes from the BIIB080 phase 1b multiple ascending dose and long term extension study in mild Alzheimer’s disease. In 16th Clinical Trials on Alzheimerʼs Disease. https://investors.biogen.com/static-files/75a32d30-01ce-4816-87a6-20735ff25b3c (2023).
Acknowledgements
We thank all the participants and their caregivers for participating in the BIIB080 phase 1b and TANGO studies as well as the investigators and their staff for conducting these studies. We thank ADNI and the ADNI participants and their families who made this analysis possible. This study was sponsored and funded by Ionis Pharmaceuticals and Biogen, Inc. The TANGO study was sponsored by Biogen. Biogen authors contributed to the conceptualization, design, data collection, analysis, decision to publish and preparation of the manuscript. Medical writing and editorial support were provided by Nucleus Global, in accordance with Good Publication Practice guidelines (http://www.ismpp.org/gpp-2022), and were funded by Biogen.
Author information
Authors and Affiliations
Contributions
M.S., S.W., N.Z., Y.T. and Y.L. had full access to all study data and take responsibility for the integrity of the data and accuracy of the data analysis. Concept and design of this study: R.L. and J.B. Study supervision and oversight: D.G., C.M., C.J., J.B., J.L. and S.B. Acquisition, analysis or interpretation of data: M.S., S.W., N.Z., C.J., J.B., J.L. and Y.L. All authors contributed to the drafting of the manuscript.
Corresponding author
Ethics declarations
Competing interests
M.S., S.W., A.E., J.C., G.C., S.B. and D.G. are employees of and may hold stock in Biogen, Inc. N.Z., L.L., I.T., Y.L., J.B., Y.T. and J.L. are former employees of and may hold stock in Biogen, Inc. R.L. and C.J. are employees of Ionis Pharmaceuticals and may hold stock. C.M. is a BIIB080 trial site investigator and is supported by the National Institute for Health and Care Research Biomedical Research Centre at University College London Hospitals and has acted as a consultant to Biogen, Roche, Eli Lilly, Prevail, Alnylam, Alector, Eisai, WAVE, PeerView and Ionis Pharmaceuticals.
Peer review
Peer review information
Nature Aging thanks Marwan N. Sabbagh and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Spaghetti plots of MMSE (A), RBANS DMI (B), and FAQ (C) change from baseline during the MAD period.
FAQ, Functional Activities Questionnaire; MAD, multiple–ascending dose; MMSE, Mini-Mental State Examination; Q4W, every 4 weeks; RBANS DMI, Repeatable Battery for the Assessment of Neuropsychological Status delayed memory index. FAQ was not collected at week 9. Grey shading indicates the 13-week treatment period.
Extended Data Fig. 2 Spaghetti plots of MMSE (A), RBANS DMI (B), and FAQ (C) scores at each visit during the MAD period.
FAQ, Functional Activities Questionnaire; MAD, multiple–ascending dose; MMSE, Mini-Mental State Examination; Q4W, every 4 weeks; RBANS DMI, Repeatable Battery for the Assessment of Neuropsychological Status delayed memory index. FAQ was not collected at week 9. Grey shading indicates the 13-week treatment period.
Extended Data Fig. 3 Spaghetti plots of MMSE (A), CDR-SB (B), and FAQ (C) change from baseline during the MAD and LTE periods.
CDR-SB, Clinical Dementia Rating Scale–Sum of Boxes; FAQ, Functional Activities Questionnaire; LTE, long-term extension; MAD, multiple–ascending dose; MMSE, Mini-Mental State Examination; Q12W, every 12 weeks. Data from the MAD period up to Week 100 for CDR-SB, MMSE, and FAQ, is included for the 16 participants from the early C and D cohorts (n = 16 participants in the high→high groups). Grey shading indicates the MAD period.
Extended Data Fig. 4 Spaghetti plots of MMSE (A), CDR-SB (B), and FAQ (C) scores at each visit during the MAD and LTE periods.
CDR-SB, Clinical Dementia Rating Scale–Sum of Boxes; FAQ, Functional Activities Questionnaire; LTE, long-term extension; MAD, multiple–ascending dose; MMSE, Mini-Mental State Examination; Q12W, every 12 weeks. Data from the MAD period up to Week 100 for CDR-SB, MMSE, and FAQ, is included for the 16 participants from the early C and D cohorts (n = 16 participants in the high→high groups). Grey shading indicates the MAD period.
Supplementary information
Supplementary Information (download PDF )
Study protocol and statistical analysis plan for the study
Source data
Source Data Fig. 2 (download ZIP )
Statistical source data for Fig. 2
Source Data Fig. 3 (download ZIP )
Statistical source data for Fig. 3
Source Data Fig. 4 (download ZIP )
Statistical source data for Fig. 4
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Shulman, M., Wu, S., Ziogas, N. et al. Exploratory analyses of clinical outcomes from the BIIB080 phase 1b study in mild Alzheimer’s disease. Nat Aging 6, 445–453 (2026). https://doi.org/10.1038/s43587-025-01031-9
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s43587-025-01031-9
