
Abstract
Human RIPK4 mutation leads to Bartsocas-Papas syndrome (BPS), characterized by severe skin, craniofacial and limb abnormalities. Currently, our understanding of RIPK4’s function has focused on epidermal differentiation and development, whether RIPK4 regulates skeletal homeostasis remains largely elusive. Herein, through global RIPK4 ablation in adult mice, we demonstrate that RIPK4 deficiency leads to osteoporosis, promotes myeloid-biased hematopoiesis and osteolineage RIPK4 plays a crucial role in bone formation and myeloid hematopoiesis. Further detailed investigation pinpoints that RIPK4 interacts with mitochondrial fusion protein MFN2 in a kinase-dependent manner. RIPK4 facilitates the phosphorylation of MFN2, which subsequently undergoes degradation through the proteasome pathway and disrupts the dynamic equilibrium of mitochondrial fission and fusion. Additionally, we also show that osteolineage RIPK4 maintains bone marrow myelopoiesis by MFN2-mediated mitochondrial transfer. More interestingly, while osteocytic RIPK4 could modestly influence the osteogenesis, it is insufficient to sustain bone marrow myelopoiesis owing to the limited amount of mitochondria transfer. These findings decipher the essential role of RIPK4 in maintaining skeletal homeostasis and unveil an unappreciated mechanism of RIPK4-MFN2 axis in regulating osteogenesis and bone marrow myelopoiesis.
Similar content being viewed by others


Introduction
Receptor-interacting protein kinase 4 (RIPK4) is a receptor-interacting protein family kinase with ankyrin repeats that plays a critical role in normal epidermal differentiation and maintenance of the barrier function of the skin1,2. Mutations in human RIPK4 result in Bartsocas-Papas syndrome (BPS), a severe autosomal recessive disorder that is characterized by skin, craniofacial, and limb abnormalities3,4, indicating a close relationship between RIPK4 and skin and the skeletal system. While there were studies investigating the role of RIPK4 in skin development using mouse models2,5,6, the effects of RIPK4 mutation on skeletal system development remain largely elusive. Also, research has demonstrated that RIPK4 interferes with B cell development and proliferation7,8,9, implying a potential involvement of RIPK4 in the regulation of bone marrow hematopoiesis. However, by which RIPK4 regulates bone marrow hematopoiesis is not fully elucidated.
Stem cells persist throughout an organism’s life, sustaining organogenesis, maintaining tissue homeostasis, and facilitating repair upon damage. The skeletal system hosts two stem cell lineages, skeletal stem cells (SSCs) and hematopoietic stem cells (HSCs)10,11,12, which give rise to a series of bone and blood lineage cells that perform essential functions including movement, structural support, hematopoiesis, immune responses, and endocrine regulations13,14,15. During hematopoiesis, HSCs give rise to lymphoid and myeloid lineage cells, including B cells, neutrophils, and monocytes as well as osteoclasts. Meanwhile, SSCs differentiate into osteoblastic (OB) lineage cells, bone marrow adipocytes, and form fibroconnective tissues. The sophisticated processes of differentiation and dynamic interactions among these cell lineages are critical not only to skeletal development, but also to the integrity of hematopoietic, immune, and endocrine systems10,16,17. Osteolineage cells, including osteoprogenitors, OB cells, and osteocytes, are primarily responsible for bone formation, and have increasingly been thought as important regulators of both bone and bone marrow. They have been reported to regulate hematopoietic niche within bone marrow and influence the commitment of myeloid and lymphoid lineage and their progeny10,18,19,20. While the intricate communications among these cell lineages have been documented, the underlying regulatory mechanisms are not fully understood.
Emerging evidence indicates that mitochondria are actively involved in the regulation of stem cell behaviors11,21,22,23. During stem cell self-renewal, proliferation, and differentiation, mitochondria undergo a series of specific dynamic processes (biogenesis, fission, fusion, and mitophagy). The alteration of mitochondrial dynamics, mediated by niche factors and stress signals, plays an important role in stem cell behavior23,24,25. However, whether mitochondrial dynamics participate in the regulation of bone and bone marrow homeostasis remains deciphered. Herein, we generated RIPK4 global and conditional knockout mice and demonstrated that RIPK4 regulates stem cell lineage commitments, including osteogenesis and myelopoiesis via regulating mitochondrial dynamics. We unveiled the role of osteolineage RIPK4 in regulating bone and bone marrow homeostasis via the RIPK4-MFN2 axis, which may provide insights into the function of RIPK4-mediated mitochondrial dynamics in maintaining skeletal metabolic homeostasis.
Results
Global RIPK4 deficiency in adult mice induces osteoporosis and myeloid-biased hematopoiesis
RIPK4 global knockout mice die perinatally because of fusions of all external orifices, as well as barrier and epidermal differentiation defects2. To study the function of RIPK4 in the skeletal system in vivo, we first examined the expression pattern of RIPK4. We have established RIPK4 reporter mice (The EUCOMM RIPK4tm1a/tm1a mice containing a LacZ cassette5) to investigate its expression in bone and bone marrow across different time points, including postnatal day 0 (P0), 4 W, 12 W, and 12 months. We found that during prenatal period, expression of RIPK4 could be detected in both bone and bone marrow (Supplementary Fig. 1a), and its expression in bone and bone marrow become increasingly high in 12-week-old mice, inferring its important role in postnatal skeletal development (Supplementary Fig. 1a). Interestingly, RIPK4 expression decreases slightly after 12 months (Supplementary Fig. 1a). We then performed Alizarin red/Alcian blue staining of whole mount skeleton at P0, which showed no apparent differences in craniofacial, long bones or spines between WT and RIPK4 homozygous knockout mice (RIPK4ho) (Supplementary Fig. 1b), indicating that RIPK4 did not perform a significant role in the prenatal skeletal development.
To further investigate whether global deletion of RIPK4 has an impact on postnatal maturation of bone tissue, we crossed Ripk4fl/fl mice with UBCCre/ERT2 mice to obtain inducible deletion of RIPK4 during adult life (8-week-old). Eight weeks after tamoxifen administration, micro-computed tomography (μCT) examination of the appendicular skeleton revealed a significant trabecular bone loss with the decrease of bone mineral density (BMD), bone volume fraction (BV/TV), trabecular number (Tb.N), trabecular thickness (Tb.Th), as well as greater trabecular separation (Tb.Sp) in UbcCre/ERT2 Ripk4fl/fl mice compared to Ripk4fl/fl mice (Fig. 1a, b). Moreover, deletion of RIPK4 also led to the cortical bone loss as indicated by decreased cortical thickness (Ct.Th) and increased cortical porosity (Ct.Po) (Fig. 1a, c). Similarly, μCT observation of axial skeleton also revealed the significant bone loss in vertebral bodies in UbcCre/ERT2 Ripk4fl/fl mice (Fig. 1d, e). Interestingly, obvious kyphosis was observed in UbcCre/ERT2 Ripk4fl/fl mice with significant loss of hair and body weight (Supplementary Fig. 1c, d). Whole body μCT scan revealed that there was an increase of lumbar curvature of UbcCre/ERT2 Ripk4fl/fl mice with higher kyphosis index (Supplementary Fig. 1e, f), which may be caused by spine bone loss. Together, these data demonstrated that global RIPK4 deficiency causes severe osteoporosis.

a–c Representative μCT reconstructive images of 16-week-old male Ripk4fl/fl and UbcCre/ERT2 Ripk4fl/fl mice femur (a) and trabecular (BMD, BV/TV, Tb.N, Tb.Sp and Tb.Th) (b) and cortical microstructural parameters (Ct.Th and Ct.Po) (c) derived from μCT analysis, demonstrating obvious bone loss in UbcCre/ERT2 Ripk4fl/fl Rhot1fl/fl mice (n = 4 per group). d, e Representative μCT reconstructive images of 16-week-old male male Ripk4fl/fl and UbcCre/ERT2 Ripk4fl/fl mice spine (d) and trabecular microstructural parameters (e) derived from μCT analysis (n = 4 per group). f, g Representative image of flow cytometry (f) and analysis of proportions of myeloid progenitors (CMP, GMP and MEP) (g) (n = 5 in Ripk4fl/fl group and n = 6 in UbcCre/ERT2 Ripk4fl/fl group). h, i Representative image of flow cytometry (h) and analysis of proportions of CD11b+ myeloid cells (i) of 16-week-old Ripk4fl/fl and UbcCre/ERT2 Ripk4fl/fl mice (n = 5 in Ripk4fl/fl group and n = 6 in UbcCre/ERT2 Ripk4fl/fl group). j, k Representative image of flow cytometry (j) and analysis of proportions of B220+ lymphoid cells (k) of 16-week-old Ripk4fl/fl and UbcCre/ERT2 Ripk4fl/fl mice (n = 5 in Ripk4fl/fl group and n = 6 in UbcCre/ERT2 Ripk4fl/fl group). l, m Representative image of flow cytometry (l) and analysis of proportions of monocyte, Ly-6Cint and Ly-6Chi monocytes (m) of 16-week-old Ripk4fl/fl and UbcCre/ERT2 Ripk4fl/fl mice (n = 5 in Ripk4fl/fl group and n = 6 in UbcCre/ERT2 Ripk4fl/fl group). Data are presented as mean ± s.d., with biologically individual data points shown. P values were determined by unpaired two-tailed Student’s t-test (BV/TV, Tb.N, Tb.Sp, Tb.Th of b, c, e, i, k, m), unpaired two-tailed Student’s t test with Welch’s correction (g), and two-tailed Mann-Whitney U test (BMD of b). Source data are provided as a Source Data file.
As a part of the skeletal system, bone marrow has vital functions in maintaining bone homeostasis26,27,28. To gain a full insight of bone marrow changes after RIPK4 deletion, flow cytometry and further analysis were performed after gating single live cells (Supplementary Fig. 2a). Although there was no significant change of HSC (Lin−c-Kit+Sca1+, LSK+ cell) numbers between Ripk4fl/fl and UbcCre/ERT2 Ripk4fl/fl mice (Supplementary Fig. 2b, c), UbcCre/ERT2 Ripk4fl/fl mice demonstrated significantly increased number of short-term HSC (ST-HSC) but decreased number of long-term HSC (LT-HSC), indicating that HSC in UbcCre/ERT2 Ripk4fl/fl mice bone marrow was mobilized (Supplementary Fig. 2d, e). Further flow cytometry analysis revealed that the number of myeloid progenitors, including common myeloid progenitors (CMP), granulocyte-monocyte progenitors (GMP) was substantially increased after RIPK4 deletion (Fig. 1f, g), but megakaryocyte erythroid progenitors (MEP) numbers were decreased (Fig. 1f, g). Furthermore, the total number of CD11b+ myeloid cells was also increased (Fig. 1h, i) at the expense of a reduction in total B220+ lymphoid cells (Fig. 1j, k) in UbcCre/ERT2 Ripk4fl/fl mice, in which both monocytes (Fig. 1l, m) and neutrophils (Supplementary Fig. 2f, g) significantly expanded. Together, these results demonstrated that RIPK4 deletion alters hematopoietic lineage, characterized by a shift from lymphopoiesis to myelopoiesis.
Global RIPK4 deficiency in adult mice impairs bone formation
The imbalance between bone formation and resorption contributes to the development of osteoporosis. To explore the potential effect of the deletion of RIPK4 on bone formation and resorption, RNA sequencing was performed on whole bone with bone marrow flushed out from Ripk4fl/fl mice and UbcCre/ERT2 Ripk4fl/fl mice. Principal component analysis (PCA) demonstrated significant separation between Ripk4fl/fl mice and UbcCre/ERT2 Ripk4fl/fl mice (Fig. 2a). A total of 1503 significantly regulated genes (358 significantly upregulated genes and 1145 significantly downregulated genes, fold change >2.0-fold and false discovery rate <0.05) were identified in UbcCre/ERT2 Ripk4fl/fl mice compared with Ripk4fl/fl mice. Among these, a volcano plot of top 20 significantly differentiated genes revealed that genes critical for osteogenesis including Bglap, Bglap2, Col11a1, Col11a2, Col5a2, Col1a2 and Col2a1, were affected by the RIPK4 deficiency (Fig. 2b). Similarly, top 20 Gene Ontology (GO) analysis revealed that significantly downregulated genes in RIPK4 deficiency mice were enriched in ossification, skeletal system development, bone development, OB differentiation, bone mineralization, skeletal system morphogenesis and regulation of bone mineralization (Fig. 2c). Gene set enrichment analysis (GSEA) revealed that osteogenesis related pathways including Wnt signaling pathway, Bmp signaling pathway, Notch signaling pathway and Hedgehog signaling pathway were downregulated (Fig. 2d–g). These data indicated that RIPK4 deficiency impaires osteogenesis.

a PCA of RNA sequencing of bone of Ripk4fl/fl and UbcCre/ERT2 Ripk4fl/fl mice. b Volcano plot of the top 20 significantly differentially expressed genes between Ripk4fl/fl and UbcCre/ERT2 Ripk4fl/fl mice. c Top 20 GO analysis of downregulated genes by RIPK4 deficiency. d–g GSEA of osteogenesis related pathways after RIPK4 ablation. h–j Goldner trichrome staining of 16-week-old male Ripk4fl/fl and UbcCre/ERT2 Ripk4fl/fl mice femur (h) and histomorphometry analysis of BV/TV, Tb.N (i), N.Ob/BS and OS/BS (j) (n = 3 per group). Scale bar, 500 and 20 µm. k, l Representative images of calcein double labeling of the mineral layers of 16-week-old male Ripk4fl/fl and UbcCre/ERT2 Ripk4fl/fl mice femur (k) and histomorphometry analysis of the BFR/BS, MAR, and MS/BS (l) (n = 4 per group). Scale bar, 50 µm. m Hematoxylin-eosin staining of 16-week-old male Ripk4fl/fl and UbcCre/ERT2 Ripk4fl/fl mice femur, Scale bar, 20 µm. n qPCR analysis of osteoblast signature genes expression at the mRNA levels (n = 4 per group from three independent experiments), indicating impaired osteogenesis in UbcCre/ERT2 Ripk4fl/fl mice. o, p TRAP staining of 16-week-old male Ripk4fl/fl and UbcCre/ERT2 Ripk4fl/fl mice femur (o) and histomorphometry analysis of Oc.S/BS and N.Oc/BS (p) (n = 3 per group). Scale bar, 20 µm. Data are presented as mean ± s.d., with biologically individual data points shown. P values were determined by two-tailed wald test (b), hypergeometric test (c), two-tailed permutation test (d–g), unpaired two-tailed Student’s t-test (i, j), MS/BS, MAR of (l), Sp7, Dmp1, Sost of (n), Oc.S/BS of (p), unpaired two-tailed Student’s t test with Welch’s correction (Col1a1, Bglap, Spp1of (n), N.Oc/BS of (p)), and two-tailed Mann–Whitney U test (Alpl, Runx2 of (n)). Source data are provided as a Source Data file.
To further evaluate the bone formation after RIPK4 deletion, we performed histomorphometry analysis. Consistent with the μCT data, histomorphometric analysis showed a significant decrease in bone mass with reduced BV/TV and Tb.N in UbcCre/ERT2 Ripk4fl/fl mice compared to Ripk4fl/fl mice (Fig. 2h, i). Meanwhile, the number of osteoblasts (N.Ob/BS) and osteoid-covered surface (OS/BS) were decreased in UbcCre/ERT2 Ripk4fl/fl mice compared to Ripk4fl/fl mice (Fig. 2h, j). Moreover, dynamic histomorphometric analysis using calcein labeling demonstrated that the bone formation rate (BFR), mineral apposition rate (MAR) and mineralized surface (MS) were significantly decreased in UbcCre/ERT2 Ripk4fl/fl mice (Fig. 2k, l). Furthermore, H&E staining showed that the cortical bone in the femurs of UbcCre/ERT2 Ripk4fl/fl mice was significantly thinner compared to that of Ripk4fl/fl mice (Fig. 2m). This observation was consistent with the GO analysis, which revealed the enrichment of extracellular matrix, extracellular region, collagen-containing extracellular matrix, extracellular matrix organization, and extracellular matrix structural constituent among the downregulated genes in RIPK4 deficiency mice (Fig. 2c). qPCR analysis confirmed that mRNA levels of osteogenic markers including Col1a1, Bglap, Alpl, Sp7, Runx2, Spp1, Dmp1, Sost were downregulated after RIPK4 deletion (Fig. 2n).
In the aspect of osteoclastogenesis, histomorphometric analysis revealed that neither the osteoclast number (N.Oc/BS) nor the osteoclast surface (Oc.S/BS) was altered following ablation of RIPK4 (Fig. 2o, p), indicating that knockout of RIPK4 has no effect on osteoclast activity. Together, all these data demonstrated that global RIPK4 deficiency impaires bone formation.
Osteolineage RIPK4 deficiency impairs osteogenesis
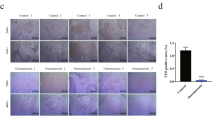
As we have confirmed that the underlying cause of osteoporosis observed in global RIPK4 deficiency mice is impaired bone formation, we then reasoned whether osteolineage RIPK4 regulated bone and bone marrow phenotype. We first examined RIPK4 expression during osteogenesis. Consistent with the temporal expression pattern of osteogenic markers (Alpl and Bglap) from day 0, day 7, and day 14, the expression of RIPK4 became increasingly higher during osteogenesis (Supplementary Fig. 3a), indicating the essential role of osteolineage RIPK4 in bone homeostasis. We then generated osteolineage RIPK4 ablation mice by crossing Prrx1Cre, Dmp1Cre mice with Ripk4fl/fl mice (Supplementary Fig. 3b, c). Whole-mount skeletal staining of P0 pubs demonstrated no significant differences in craniofacial structures, long bones or spines between Ripk4fl/fl and Prrx1Cre Ripk4fl/fl mice (Supplementary Fig. 3d), as well as Ripk4fl/fl and Dmp1Cre Ripk4fl/fl mice (Supplementary Fig. 3e), suggesting that RIPK4 does not play a significant role in prenatal skeletal development. However, μCT analysis of 8-week mice femur revealed that both trabecular and cortical bone mass of Prrx1Cre Ripk4fl/fl mice were both decreased compared to Ripk4fl/fl mice (Fig. 3a–c). Consistently, histomorphometry analysis also showed decreased bone mass with decreased BV/TV and Tb.N in Prrx1Cre Ripk4fl/fl mice in comparison to Ripk4fl/fl mice (Fig. 3d, e). Moreover, N.Ob/BS and OS/BS were also decreased in Prrx1Cre Ripk4fl/fl mice (Fig. 3d, f). Dynamic bone formation analysis revealed that the bone formation index, including BFR/BS, MAR, and MS/BS were impaired following osteolineage RIPK4 ablation (Fig. 3g, h). We then extracted SSCs and induced in vitro osteogenic differentiation. Alkaline phosphatase (ALP) staining (Fig. 3i) and alizarin red staining (Fig. 3j) showed decreased osteogenesis and mineralization. And mRNA levels of osteogenic markers including Alpl, Bglap, Col1a1, Sp7 and Runx2 were also decreased (Fig. 3k).

a–c Representative μCT reconstructive images of male Ripk4fl/fl and Prrx1Cre Ripk4fl/fl mice femur at 16 weeks (a) and trabecular microstructural parameters (BMD, BV/TV, Tb.N, Tb.Sp and Tb.Th) (b) and cortical microstructural parameters (Ct.Th and Ct.Po) (c) derived from μCT analysis (n = 6 per group), demonstrating significant bone loss after osteolineage RIPK4 ablation. d–f Goldner trichrome staining of male Ripk4fl/fl and Prrx1Cre Ripk4fl/fl mice femur at 16 weeks (d) and histomorphometry analysis of BV/TV, Tb.N (e), N.Ob/BS and OS/BS (f) (n = 3 per group). Scale bar, 20 µm. g, h Representative images of calcein double labeling of the mineral layers of male Ripk4fl/fl and Prrx1Cre Ripk4fl/fl mice femur at 16 weeks (g) and histomorphometry analysis of the BFR/BS, MAR, and MS/BS (h) (n = 4 per group). Scale bar, 50 µm. i, j ALP staining (i) and alizarin red staining (j) of in vitro osteogenesis from Ripk4fl/fl and Prrx1Cre Ripk4fl/fl mice. Scale bar, 250 µm. k qPCR analysis of the expression of osteoblast signature genes at the mRNA level between Ripk4fl/fl and Prrx1Cre Ripk4fl/fl mice (n = 3 per group). Data are presented as mean ± s.d., with biologically individual data points shown. P values were determined by unpaired two-tailed Student’s t-test (BMD, BV/TV, Tb.N, Tb.Sp of (b, c), BV/TV of (e, f), MAR of (h), Alpl, Bglap, Col1a1, Sp7, Runx2 of (k)), unpaired two-tailed Student’s t test with Welch’s correction (Tb.N of (e), MS/BS, BFR/BS of (h)), and Mann–Whitney U test (Tb.Th of (b), Spp1 of (k)). Source data are provided as a Source Data file.
We also further investigated whether osteocytic RIPK4 could regulate bone mass. Similarly, Dmp1Cre Ripk4fl/fl mice revealed significant bone loss as indicated by the μCT analysis of trabecular bone but with no significant difference of cortical bone (Supplementary Fig. 3f–h). Furthermore, we compared the degree of bone loss among UbcCre/ERT2 Ripk4fl/fl mice, Prrx1Cre Ripk4fl/fl mice, and Dmp1Cre Ripk4fl/fl mice. We observed no significant difference in the degree of trabecular bone loss between UbcCre/ERT2 Ripk4fl/fl mice and Prrx1Cre Ripk4fl/fl mice (Supplementary Fig. 3i), indicating that osteolineage RIPK4 contributed most to the osteoporosis phenotype. However, the trabecular bone loss degree of Dmp1Cre Ripk4fl/fl mice is significantly less than that of Prrx1Cre Ripk4fl/fl mice (Supplementary Fig. 3i), indicating a less contribution of osteocytic RIPK4 to the osteoporosis phenotype. Interestingly, in cortical bone, the bone loss degree of both Prrx1Cre Ripk4fl/fl mice and Dmp1Cre Ripk4fl/fl mice was less than that of UbcCre/ERT2 Ripk4fl/fl mice (Supplementary Fig. 3i), suggesting that other factors may participate in the regulation of the cortical bone formation. Together, all these data indicated that osteolineage RIPK4 ablation induces bone loss which recapitulated the bone loss phenotype after global RIPK4 deficiency.
Osteolineage RIPK4 deficiency results in myeloid-biased hematopoiesis
Previous studies have demonstrated the close relationship between bone and bone marrow18,20. Considering that osteolineage cells could support bone marrow hematopoietic niche and influence myeloid and lymphoid lineage and their progeny commitments10,18,19,20, we then investigated whether osteolineage RIPK4 contributed to the regulation of myelopoiesis. We examined bone marrow myelopoiesis and lymphopoiesis following osteolineage RIPK4 ablation, respectively. Intriguingly, while the total number of HSCs was not significantly altered (Supplementary Fig. 4a), HSCs were mobilized with increased ST-HSC and decreased LT-HSC in Prrx1Cre Ripk4fl/fl mice compared to Ripk4fl/fl mice (Fig. 4a). Simultaneously, we revealed that the myelopoiesis was enhanced at the expense of lymphopoiesis with increased myeloid progenitors (Fig. 4b and Supplementary Fig. 4a), subsequent myeloid cells (Fig. 4c and Supplementary Fig. 4a) and decreased lymphoid cells in Prrx1Cre Ripk4fl/fl mice compared to Ripk4fl/fl mice (Fig. 4c). These data confirmed the critical role of osteolineage RIPK4 in sustaining bone marrow myelopoiesis, while the absence of osteolineage RIPK4 leads to myeloid-biased hematopoiesis process.

a Flow cytometry analysis of proportions of LT-HSC and ST-HSC in 16-week-old male Ripk4fl/fl and Prrx1Cre Ripk4fl/fl mice femur (n = 3 per group). b Flow cytometry analysis of proportions of CMP, GMP, and MEP in 16-week-old male Ripk4fl/fl and Prrx1Cre Ripk4fl/fl mice femur (n = 3 per group). c Flow cytometry analysis of proportions of CD11b+ myeloid cells, monocytes and B220+ lymphoid cells in male Ripk4fl/fl and Prrx1Cre Ripk4fl/fl mice femur (n = 3 per group). d Flow cytometry analysis of proportions of LT-HSC and ST-HSC in16-week-old male Ripk4fl/fl and Dmp1Cre Ripk4fl/fl mice femur (n = 5 per group). e Flow cytometry analysis of proportions of CMP, GMP and MEP in16-week-old male Ripk4fl/fl and Dmp1Cre Ripk4fl/fl mice femur (n = 5 per group). f Flow cytometry analysis of proportions of CD11b+ myeloid cells, monocytes and B220+ lymphoid cells in 16-week-old male Ripk4fl/fl and Dmp1Cre Ripk4fl/fl mice femur (n = 5 per group). g Flow cytometry analysis of proportions of CD11b+ myeloid cells, monocytes, neutrophils and B220+ lymphoid cells in16-week-old male Ripk4fl/fl and Lyz2Cre Ripk4fl/fl mice femur (n = 3 per group). h Flow cytometry analysis of proportions of CD11b+ myeloid cells, monocytes, neutrophils and B220+ lymphoid cells in 16-week-old male Ripk4fl/fl and Cd4Cre Ripk4fl/fl mice femur s (n = 4 in Ripk4fl/fl group and n = 3 in Cd4Cre Ripk4fl/fl group). Data are presented as mean ± s.d., with biologically individual data points shown. P values were determined by unpaired two-tailed Student’s t-test (a, b, c, CMP, MEP of e, f, g), unpaired two-tailed Student’s t test with Welch’s correction (GMP of e, h), and Mann-Whitney U test (d, g). Source data are provided as a Source Data file.
Osteocytes, as the terminal cell type of osteolineage cells, have also been shown to regulate bone marrow myelopoiesis12. We have discovered that osteocytic RIPK4 regulates the bone mass, consequently, we hypothesized whether osteocytic RIPK4 also influences myelopoiesis. However, bone marrow analysis revealed no significant difference in HSC mobilization, myelopoiesis, and lymphopoiesis between Ripk4fl/fl and Dmp1Cre Ripk4fl/fl mice (Fig. 4d and Supplementary Fig. 4b). These data implied that osteocytic RIPK4 exerts minimal influence on myelopoiesis in the bone marrow. Meanwhile, to eliminate the potential influence of myeloid and lymphoid RIPK4 on regulating bone marrow myelopoiesis, we established a myeloid RIPK4 ablation mouse model by crossing Lyz2Cre mice with Ripk4fl/fl mice (Supplementary Fig. 4c), as well as a lymphoid RIPK4 ablation mouse model by crossing Cd4Cre mice with Ripk4fl/fl mice (Supplementary Fig. 4d). Bone marrow myelopoiesis and lymphopoiesis also showed no significant differences between Ripk4fl/fl and Lyz2Cre Ripk4fl/fl mice (Fig. 4g), as well as between Ripk4fl/fl and Cd4Cre Ripk4fl/fl mice (Fig. 4h). Together, all these data confirmed that myeloid or lymphoid RIPK4 is dispensable for regulating myelopoiesis and lymphopoiesis in the bone marrow.
RIPK4 regulates osteogenesis via MFN2 degradation
Stem cell specification is controlled by the concerted actions of signaling pathways, among which metabolic programs mediated by mitochondrial regulation are intensely involved in this process11,22,29,30,31. Previous studies have reported the association between RIPK3 and mitochondria. For example, RIPK3 could induce diabetic podocytopathy by regulating mitochondrial fission via PGAM5-DRP1 signaling through MLKL32. RIPK3 induces mitochondrial apoptosis via inhibition of FUNDC1-mediated mitophagy in cardiac ischemia-reperfusion (IR) injury33. RIPK4, as a member of RIPK protein kinase family, has been reported to regulate keratinocyte differentiation1 and B cell development8. Furthermore, our studies also demonstrated that RIPK4 promotes oxidative stress34, indicating the close relationship between RIPK4 and mitochondria. We thus hypothesized that RIPK4 may regulate osteogenesis and bone marrow myelopoiesis through mitochondrial regulation. We initially performed a metabolic cage test comparing Ripk4fl/fl and UbcCre/ERT2 Ripk4fl/fl mice. However, there was no significant difference of metabolic characteristics, including the rate of O2 consumption (Supplementary Fig. 5a), the rate of CO2 consumption (Supplementary Fig. 5b), the respiration exchange rate (RER) (Supplementary Fig. 5c) and energy expenditure (Supplementary Fig. 5d) in both light and dark cycles after RIPK4 ablation. Additionally, food intake remained unchanged (Supplementary Fig. 5e). Consistent with these findings, the mitochondrial DNA (mtDNA) levels of bone were not affected by RIPK4 depletion (Supplementary Fig. 5f). Furthermore, mitochondrial function in isolated bone cells was evaluated, revealing no significant differences in mitochondrial mass, membrane potential and mitochondrial ROS levels between Ripk4fl/fl and UbcCre/ERT2 Ripk4fl/fl mice (Supplementary Fig. 5g). These data indicate that RIPK4 does not regulate mitochondrial metabolic functions. However, the transmission electron microscope (TEM) detected elongated tubular structures of mitochondria in both tissues and cells (Fig. 5a, b), indicating abnormal dynamics of mitochondrial fusion and fission following RIPK4 ablation.

a, b TEM detected long extended tubules of mitochondria in muscle tissues (a) and osteoprogenitor cells (b) in 16-week-old Ripk4fl/fl, UbcCre/ERT2 Ripk4fl/fl and UbcCre/ERT2 Ripk4fl/flMfn2fl/fl mice (n = 5 per group). Scale bar, 500 nm. The right arrow defines the mitochondria. c Co-immunoprecipitation of endogenous MFN2 and FLAG-RIPK4 in HEK293T cells. d Endogenous immunoprecipitation of RIPK4 and MFN2 was performed in hBMSC cells. e Representative images PLA signal (in situ close association between FLAG and MFN2) in U2OS cells. The nuclear marker DAPI is in blue, and the PLA signal is in red. Scale bar, 10 μm. f HEK293T cells were transfected with plasmids expressing 3×FLAG-RIPK4 (WT, 3×FLAG-RIPK4; K51R, 3×FLAG-RIPK4-K51R, kinase-dead mutant; T184I, 3×FLAG RIPK4-T184I, ATP binding mutant), RIPK4 was immunoprecipitated using FLAG-M2 affinity gel. g Myc-MFN2 and 3×FLAG-RIPK4 expressing plasmids (WT, 3×FLAG-RIPK4; K51R, 3×FLAG-RIPK4-K51R) were transfected in HEK293T cells. Cells were harvested and cell lysates were incubated with or without lambda protein phosphatase (λ-PPase). Phosphorylated and non-phosphorylated MFN2 proteins were separated by Phos-Tag SDS-PAGE. h HA-MFN2 and 3×FLAG-RIPK4 or 3×FLAG-RIPK4 K51R expressing plasmids were co-transfected in HEK293T cells. HA-MFN2 was immunoprecipitated using anti-HA-agarose. i Immunoblots analysis of MFN2 and RIPK4 proteins in RIPK4 knockout HBMSC cells. j, k WT and K51R RIPK4 were inducibly expressed in U2OS cells with 50 ng/ml doxycycline for 24 h. MFN2, MFN1, DRP1, and OPA1 proteins were detected by immunoblots. l WT RIPK4 was inducibly expressed in U2OS cells with 50 ng/ml doxycycline for 24 h, and mitochondria were extracted according to the manufacturer’s protocol. MFN2, MFN1, and DRP1 proteins were detected by immunoblots and quantified by ImageJ. m RIPK4 was inducibly expressed in U2OS cells with 50 ng/ml doxycycline for 24 h, then cells were treated with MG132 or Bafilomycin A1 (BafA) for 4 h. MFN2 protein was detected by immunoblots. n FLAG-RIPK4 or FLAG-RIPK4-K51R with dsRed-mito expressing plasmids were co-transfected in U2OS cells for 24 h, then immune-stained for RIPK4 (green). dsRed-mito (red) was utilized to visualize the morphology of mitochondria. DAPI staining indicated the nuclei. Scale bar, 10 μm. Source data were provided as a Source Data file.
We subsequently investigated the interaction of RIPK4 with proteins involved in mitochondrial fusion/fission. Our results demonstrated that RIPK4 specifically interacted with MFN2, a mitochondrial membrane protein primarily involved in mediating the fusion of mitochondria, rather than OPA1, DRP1 or MFN1 (Fig. 5c). We conducted endogenous immunoprecipitation in human BMSC (hBMSC) cells and observed that endogenous RIPK4 pulled down MFN2 (Fig. 5d). Proximity ligation assay (PLA) in RIPK4 overexpressed U2OS cells also demonstrated remarkable interaction between RIPK4 and endogenous MFN2 (Fig. 5e). These data confirmed the direct interaction between RIPK4 and MFN2. Interestingly, wildtype RIPK4 exhibited significantly stronger MFN2 interaction compared to its kinase-dead mutant K51R and ATP-binding mutant T184I (Fig. 5f), suggesting the importance of RIPK4’s kinase activity. Therefore, we further investigated whether RIPK4 could phosphorylate MFN2. Utilizing phos-tag gels, we demonstrated that WT RIPK4 but not its kinase-dead mutant induced the shifted band of MFN2 in the phos-tag gel, which was eliminated by λ-phosphatase treatment, suggesting that the shifted band of MFN2 was indeed the phosphorylated form (Fig. 5g). Moreover, the phosphorylation of MFN2 by WT RIPK4 was confirmed using a pan-phospho-threonine antibody (Fig. 5h).
Subsequently, we aimed to elucidate the effect of the phosphorylation of MFN2. Upon knockout of RIPK4 using specific sgRNA, MFN2 protein accumulated in human BMSC cells with RIPK4 deficiency, while its mRNA level remained unchanged (Fig. 5i and Supplementary Fig. 6a). On the other hand, we examined both mRNA and protein levels of MFN2 in RIPK4 inducible osteosarcoma U2OS cells. Although RIPK4 overexpression did not affect MFN2 mRNA levels (Supplementary Fig. 6b), MFN2 protein levels were decreased in WT RIPK4 inducible cells but not in cells expressing its kinase-dead mutant (Fig. 5j). It should be noticed that other mitochondria fusion and fission proteins including MFN1, OPA1 and DRP1 were not changed (Fig. 5j, k). To further exclude the contribution of the cytosolic compartment, we have extracted mitochondria from RIPK4-inducible U2OS cells. Consistently, we observed a decrease in MFN2 protein levels, but not in MFN1 or DRP1 protein levels within the mitochondria (Fig. 5l). Moreover, the decrease of MFN2 was blocked by the proteasome inhibitor MG132 but not by the V-ATPase inhibitor Bafilomycin A1, which blocks late-phase autophagy (Fig. 5m), suggesting that the phosphorylation of MFN2 by RIPK4 leads to its proteasomal degradation. Meanwhile, WT RIPK4 induced obvious mitochondrial fragmentation but not its kinase-dead mutant (Fig. 5n), suggesting the important role of its kinase activity in this process. Given that MFN2 is critical for mitochondria-ER tethering, mitophagy and mitochondrial calcium uptake, we further checked these activities in cells-overexpressed RIPK4. However, RIPK4 overexpression did not affect ER tethering, as indicated by PLA dots between VDAC1 and IP3R3 (Supplementary Fig. 6c). At the same time, neither mtDNA levels nor the expression of LC3ІІ was induced by RIPK4 overexpression (Supplementary Fig. 6d, e). Additionally, the mitochondrial Ca2+ concentration, measured using a mitochondrial-targeted calcium probe (Rhod-2, AM staining), was comparable between WT and RIPK4 overexpressed cells (Supplementary Fig. 6f).
We then downregulated RIPK4 expression in OBs, an impaired osteogenesis was observed as revealed by the ALP and alizarin red staining (Fig. 6a–c). Meanwhile, MFN2 downregulation was able to reverse the impairment in osteogenesis (Fig. 6a–c). To further confirm the role of RIPK4-MFN2 axis in regulating osteogenesis in vivo, we established double knockout mice of RIPK4 and MFN2 by crossing UbcCre/ERT2 Ripk4fl/fl mice with Mfn2fl/fl mice, namely UbcCre/ERT2 Ripk4fl/fl Mfn2fl/fl mice, in which MFN2 was ablated (Supplementary Fig. 7a). We found that elongated tubular structures of mitochondria in both tissues and cells were reversed to normal in UbcCre/ERT2 Ripk4fl/fl Mfn2fl/fl mice (Fig. 5a, b) while its mitochondrial mass, membrane potential and mitochondrial ROS were not altered (Supplementary Fig. 5g). Consistently, while UbcCre/ERT2 Ripk4fl/fl Mfn2fl/fl mice still displayed hair loss, their trabecular bone mass was significantly higher than that of UbcCre/ERT2 Ripk4fl/fl mice with increased BMD, Tb.N and decreased Tb.Sp of trabecular bone (Fig. 6d, e). There was also an increased tendency of cortical bone mass in UbcCre/ERT2 Ripk4fl/fl Mfn2fl/fl mice compared to UbcCre/ERT2 Ripk4fl/fl mice (Fig. 6d, f). Combined with literatures reporting that knockdown of MFN2 leads to accelerated osteogenesis35 and depletion of MFN2 in osteolineage cells enhances bone formation36, we could conclude that osteolineage RIPK4 regulates its osteogenesis via MFN2 degradation.

a qPCR analysis demonstrating the downregulation of Ripk4 and Mfn2 (n = 3 per group). b, c ALP and alizarin red staining of in vitro osteogenesis following RIPK4 knockdown and dual knockdown of RIPK4 and MFN2. Scale bar, 250 µm. d–f Representative μCT reconstructive images of male Ripk4fl/fl, UbcCre/ERT2 Ripk4fl/fl and UbcCre/ERT2 Ripk4fl/fl Mfn2fl/fl mice femur at 16 weeks (d) and trabecular microstructural parameters (BMD, BV/TV, Tb.N, Tb.Sp and Tb.Th) (e) and cortical microstructural parameters (Ct.Th and Ct.Po) (f) derived from μCT analysis (n = 8 in Ripk4fl/fl group, n = 7 in UbcCre/ERT2 Ripk4fl/fl group and n = 6 in UbcCre/ERT2 Ripk4fl/fl Mfn2fl/fl group), demonstrating that knockout of MFN2 rescued RIPK4 ablation induced bone loss. Data are presented as mean ± s.d., with biologically individual data points shown. P values were determined by ordinary one-way ANOVA test with Tukey’s multiple comparisons (a, BMD, Tb.N, Tb.Sp of e), Brown-Forsythe and Welch ANOVA tests with Dunnett’s T3 multiple comparisons (Tb.Th of e) and non-parametric one-way ANOVA with Dunn’s multiple comparisons (BV/TV of e, f). Source data are provided as a Source Data file.
Osteolineage RIPK4 maintains bone marrow myelopoiesis via MFN2-mediated mitochondrial transfer
Mitochondria play pivotal roles in the regulation of cell lineage preservation and specification11,37,38,39. Cell-to-cell transferred mitochondria, as dynamic organelles involved in energy-transforming and signaling, have been increasingly recognized as important information processing systems40 that could modulate neighboring and distant cells41,42,43,44,45,46,47,48. We previously reported the mitochondrial transfer network within the bone marrow and osteolineage cells enables the transfer of mitochondria to myeloid cells, thereby regulating their commitments49. Our group also showed that osteocytes, as terminal osteolineage cells, transfer mitochondria among one another within their network via MFN250. Thus, we hypothesized that osteolineage RIPK4 regulates bone marrow myelopoiesis via MFN2-mediated mitochondrial transfer.
Based on our established osteolineage-specific mitochondria reporter mice, we previously reported that osteolineage cells are capable of transferring mitochondria to myeloid cells in vivo49. And we found that the number of monocytes/macrophages and neutrophils in the Dmp1Cre MitoDendra+/+ reporter mice that received transferred mitochondria was significantly lower than that in the Prrx1Cre MitoDendra+/+ reporter mice49. We then extracted OBs from Prrx1Cre MitoDendra+/+ mice and bone marrow mononuclear cells (BMNCs) from mT/mG mice as previously reported12,51,52. BMNCs were induced to differentiate into myeloid cells through stimulation with M-CSF (20 ng/mL). Concurrently, OBs were added and co-incubated for three days, after which flow cytometry was performed. Flow cytometry analysis showed that downregulation of RIPK4 expression in OBs promoted mitochondrial transfer (Fig. 7a, b) and increased in vitro myelopoiesis as revealed by an elevation in CD11b+ myeloid cells (Fig. 7c, d). However, simultaneous downregulation of MFN2 expression abrogated the enhanced mitochondrial transfer and myelopoiesis (Fig. 7c, d).

a, b Representative image of flow cytometry (a) and analysis (b) demonstrating that RIPK4-MFN2 axis regulated mitochondrial transfer from OBs to BMNCs (n = 4 per group). c, d Representative image of flow cytometry (c) and analysis (d) demonstrating that RIPK4-MFN2 axis regulated in vitro myelopoiesis (n = 4 per group). e, f Representative image of flow cytometry (e) and analysis (f) showing that in vitro mitochondrial transfer was reduced in osteocytic cells (MLO-Y4) compared to OBs (n = 4 per group). g Representative image of flow cytometry and analysis showing that in vitro myelopoiesis was lower in osteocytic cells (MLO-Y4) than OBs (n = 4 per group). h, i Flow cytometry analysis of proportions of HSCs, myeloid progenitors (CMP, GMP and MEP) (h), myeloid cells (CD11b+, monocytes) and B220+ cells (i) in male Ripk4fl/fl, UbcCre/ERT2 Ripk4fl/fl and UbcCre/ERT2 Ripk4fl/fl Mfn2fl/fl mice femur at 16 weeks (n = 6 per group). Data are presented as mean ± s.d., with biologically individual data points shown. P values were determined by ordinary one-way ANOVA test with Tukey’s multiple comparisons (b, d, h, i), unpaired two-tailed Student’s t-test (f, g). Source data are provided as a Source Data file.
Meanwhile, we performed a coculture of the osteocytic cell line (MLO-Y4 cells, labeled with dendra2 signal) and BMNCs derived from mT/mG mice. Consistent with the in vivo mitochondrial transfer, the number of BMNCs that received mitochondria from MLO-Y4 cells was significantly lower than that of BMNCs that received mitochondria from OBs (Fig. 7e, f). And in vitro myelopoiesis also showed significantly lower than that coincubated with OBs (Fig. 7g). These data indicated that the quantity of transferred mitochondria is a critical determinant in the promoting potential of myelopoiesis, thereby maintaining the bone marrow myelopoiesis. These findings may elucidate the underlying mechanism by which osteocytic RIPK4 knockout fails to elicit significant myeloid-biased hematopoiesis. Furthermore, we characterized bone marrow myelopoiesis and found that the number of myeloid progenitors and subsequent myeloid cells in UbcCre/ERT2 Ripk4fl/fl Mfn2fl/fl mice was markedly reduced compared to UbcCre/ERT2 Ripk4fl/fl mice (Fig. 7h, i, Supplementary Fig. 7b), indicating that myeloid-biased hematopoiesis was reversed following MFN2 ablation. Moreover, the lymphopoiesis was also rescued in UbcCre/ERT2 Ripk4fl/fl Mfn2fl/fl mice compared to UbcCre/ERT2 Ripk4fl/fl mice (Fig. 7i). Together, all these data demonstrated that osteolineage RIPK4 inhibits bone marrow myelopoiesis by suppressing MFN2-mediated mitochondrial transfer.
Discussion
In this study, we deciphered the important role of RIPK4 in regulating stem cell lineage commitments to maintain skeletal homeostasis. By means of a global and conditional mouse model, we uncovered that osteolineage RIPK4, rather than myeloid or lymphoid RIPK4, regulated bone mass and bone marrow myelopoiesis. Mechanically, by interacting with MFN2 to regulate mitochondrial dynamics, osteolineage RIPK4 regulated the osteogenesis and myeloid-biased hematopoiesis, thus preserving skeletal homeostasis (Fig. 8).

In normal conditions, RIPK4 in osteolineage cells regulates osteogenesis and hematopoiesis by phosphorylating mitochondrial fusion protein MFN2 to regulate mitochondrial dynamics. When RIPK4 is ablated, mitochondrial dynamics was disturbed due to the interruption of RIPK4-MFN2 axis, leading to the impaired osteogenesis and promoted myeloid-biased hematopoiesis, thus resulting in severe osteoporosis and myeloid-biased hematopoiesis. This figure was created by P.D. and cartoonized by Mr. Zihao Li using Procreate software.
The mutation of RIPK4 in humans leading to limb abnormalities indicated a close relationship between RIPK4 and the skeletal system4. We first utilized RIPK4ho mice which showed no significant developmental difference in the skeletal system at P0 compared to WT mice. Similarly, whole mount skeletal staining of P0 pubs showed that there were no significant differences of craniofacial, long bones or spines between Ripk4fl/fl and Prrx1Cre Ripk4fl/fl mice, Ripk4fl/fl and Dmp1Cre Ripk4fl/fl mice. Also, there was a study reporting no significant difference in limb development at E18.5 between Ripk4fl/fl and K14Cre Ripk4fl/fl mice5. These data confirmed that RIPK4 did not play a significant role in prenatal skeletal system development. We then investigated the role of RIPK4 in postnatal skeletal system development. Using an inducible global RIPK4 ablation mouse model, we found that there was significant bone loss after RIPK4 deficiency, which was recapitulated by our osteolineage and osteocytic RIPK4 ablation mice model, demonstrating the essential role of osteolineage RIPK4 in regulating osteogenesis and subsequent bone formation. Further, we uncovered that RIPK4 interacted with MFN2, a mitochondrial membrane protein, dynamically regulating mitochondrial shape and structure53, to regulate osteogenesis. Moreover, RIPK4 phosphorylated MFN2 to regulate mitochondrial dynamics, and this process was dependent on RIPK4 kinase activity. Knockdown of RIPK4 led to the upregulation of MFN2, and overexpression of RIPK4 resulted in the degradation of MFN2, thus leading to the fragmentation of mitochondria, which could be antagonized by MFN2 co-expression. These data demonstrated the importance of mitochondria dynamics in regulating bone metabolism. Previous studies have reported that increased fragmentation of mitochondria and donut formation could promote osteogenesis54 and MFN2 knockdown promotes osteogenic differentiation through aerobic glycolysis mediated by the Wnt/β-catenin signaling pathway35. Our RNA sequencing also demonstrated the enrichment of osteogenic pathways including Wnt signaling pathway. Besides, osteolineage depletion of MFN2 enhanced cortical bone formation36 and our RIPK4 and MFN2 double knockout mice also revealed enhanced osteogenesis. Together, all these data demonstrated the essential role of RIPK4-MFN2 axis in regulating osteogenesis.
SSCs and HSCs form a specific bone marrow niche10, in which different lineage cells communicate with each other to influence their commitments. Osteolineage cells were increasingly thought as the important regulator to maintain bone marrow niche homeostasis and the process of osteogenesis and hematopoiesis are closely correlated. For example, aged osteolineage cells generated an inflammatory niche to promote myeloid skewing hematopoiesis55. And myelodysplastic syndrome (MDS) cells impair osteolineage differentiation to suppress hematopoiesis56. In our study, we uncovered the mechanism of RIPK4-MFN2 axis in regulating osteogenesis and subsequent myeloid-biased hematopoiesis via regulating mitochondrial dynamics, which provided insights of osteolineage cells regulating bone and bone marrow homeostasis. Different cell types transferring mitochondria to regulate neighboring or remote cells has been observed41,42,43,44,45,46,57,58 and our group has demonstrated that osteocytes transfer mitochondria within dendrite network via MFN250. Recently, we also reported the mitochondrial transfer network within the bone marrow and detected the mitochondrial transfer from osteolineage cells to myeloid cells49. Thus, we inferred that osteolineage RIPK4 regulated bone marrow myelopoiesis via MFN2-mediated mitochondria transfer. Further detailed investigation pinpointed that the number of transferred mitochondria determined the myelopoiesis-promoting potential of osteolineage RIPK4, which may be the underlying cause that osteocytic RIPK4 knockout cannot induce significant myeloid-biased hematopoiesis. Combined with a previous report showing that bone marrow regeneration requires hematopoietic lineage mitochondrial transfer to the mesenchymal environment59, these data indicate that mitochondria transfer between hematopoietic lineage and osteolineage cells may be a bilateral interactive process that is important for bone marrow regeneration.
In summary, our findings depict the unappreciated role of RIPK4 in regulating the skeletal system homeostasis and reveal the RIPK4-MFN2 axis to regulate osteogenesis and myeloid-biased hematopoiesis via MFN2. Therapeutically targeting the RIPK4-MFN2 axis may represent a potential way to ameliorate skeletal metabolic diseases.
Methods
Study approval
Our research complies with all relevant ethical regulations according to protocols approved by the Animal Care and Use Committee of Shanghai Jiao Tong University affiliated with Sixth People’s Hospital (No: 2021-0934). All of the mice were sacrificed and all of the animal experiments were performed under isoflurane anesthesia. Every effort was made to minimize and avoid suffering.
Mice
UbcCre/ERT2 mice (stock number, 007001), Prrx1Cre mice (stock number, 005584) MitoDendra+/+ mice (stock number, 018385) and mT/mG mice (stock number, 007676) were acquired from Jackson Laboratory. RIPK4tm1a(EUCOMM)Wtsi (C57BL6/N) mice were purchased from EUCOMM (Helmholtz Zentrum, München, Germany). Lyz2Cre mice (strain ID, T003822) were acquired from GemPharmatech. Dmp1Cre mice were provided by J. Q. (Jerry) Feng from Texas A&M College of Dentistry, USA (Jackson Laboratory stock number, 023047). Cd4Cre mice were a gift from Dr. Qiang Zou (Shanghai Jiao Tong University School of Medicine). Global inducible RIPK4 ablation mice were established by crossing UbcCre/ERT2 mice with Ripk4fl/fl mice to generate UbcCre/ERT2 Ripk4fl/fl mice. Osteolineage Ripk4 knockout mice were established by crossing Prrx1Cre mice with Ripk4fl/fl mice to obtain Prrx1cre Ripk4fl/fl mice. Osteocytic Ripk4 knockout mice was established by crossing Dmp1Cre mice with Ripk4fl/fl mice to obtain Dmp1Cre Ripk4fl/fl mice. Myeloid Ripk4 knockout mice were established by crossing Lyz2cre mice with Ripk4fl/fl mice to obtain Lyz2Cre Ripk4fl/fl mice. T cell Ripk4 knockout mice were established by crossing Cd4Cre mice with Ripk4fl/fl mice to obtain Cd4Cre Ripk4fl/fl mice. Osteolineage mitochondria reporter mice were established as previously reported49. For tamoxifen depletion, tamoxifen (~75 mg/kg body weight) is given by intraperitoneal injection (using an ACUC approved injection procedure) for a total of 5 consecutive days. All mice were maintained with normal chow (1010082) at the animal facility of Shanghai Jiao Tong University affiliated to Sixth People’s Hospital under 12-h light/dark cycle at 20–26 °C and 40–70% humidity. The data for key experiments are in both sexes.
Flow cytometry
To obtain bone marrow cells, mice femurs were flushed with bone marrow then filtered through 70 μm nylon mesh. After removing red blood cells with erythrocyte lysis solution, the isolated cells were blocked for 15 min with an anti-mouse CD16/32 antibody (Biolegend, 101302). Fluorescence-conjugated antibodies were used for staining. Staining was performed at 4 °C in the dark for 30 min. All the antibodies were purchased from Biolegend and listed as below: anti-CD45R-PE/Cy5 (103209), anti-CD45R-APC (103212), anti-CD117-PE (105808), anti-CD117-APC/Cy7 (105825), anti-Ly-6G-PE/Cy7 (127617), anti-Ly-6A/E-APC (108111), anti-Ly-6A/E-Alexa Fluor®700 (108142), anti-Ly-6C-Pacific BlueTM (128013), anti-Ly-6C-PE (128007), anti-Ly-6G-Pacific BlueTM (127611), anti-lineage cocktail-Pacific BlueTM (133305), anti-CD11b-Alexa Fluor®700 (101202), anti-CD11b-APC/Cy7 (101225), 7-AAD viability staining solution (420404). The cells were then washed and run on the Cytometer CytoFlex (Beckman Coulter). The samples were analyzed using FlowJo version 10.4 after gating single live cells. 50000 events were collected for each sample.
Fluorescence-activated cell sorting (FACS)
For BMSCs sorting, bone marrow cells were flushed and stained with anti-CD45- Pacific BlueTM (Biolegend, 157212), anti-CD11b-FITC (Biolegend, 101205), anti-Ly-6A/E-APC (Biolegend, 108111) and anti-CD29-PE (Biolegend, 102208). Stained cells were sorted on the Sony platform. CD11b−CD45−CD29+Sca1+ cells were gated as targeted cells in purifying mode. Sorted cells were cultured in mouse mesenchymal stem cell culture medium (Cyagen) and then induced for osteogenesis. Similarly, splenic were stained with anti-CD4-APC/Cy7 (Biolegend, 100413) and CD4+ cells were sorted. Bone marrow cells were stained with anti-CD11b-FITC (Biolegend, 101205) and CD11b+ cells were sorted.
Bone histomorphometry analysis
The dissected femurs were fixed in 4% paraformaldehyde (PFA) for 2 days, followed by immersion in 10% EDTA (pH = 7.2) at 4 °C for about 2 weeks for decalcification. We embedded decalcified femurs in paraffin and sectioned them at a thickness of 4 μm to identify osteoclasts using TRAP staining. We stained OB with Goldner trichrome staining after embedding undecalcified femurs in plastic and sectioning them at a thickness of 5 μm. Double calcein-labeling was used for dynamic histomorphometry analysis. On days 1 and 7, mice were injected intraperitoneally with 30 μg/gram body weight calcein (Sigma-Aldrich). At day 9, mice were sacrificed, and their femurs were dissected, fixed in 4% PFA for 2 days, embedded in plastic, and sectioned at 5 μm thickness. Bioquant Osteo software (Bioquant) was used for bone histomorphometry analysis.
Bone density measurements
We fixed the femurs and L3 lumbar in 4% PFA for 2 days. After removing soft tissues from femurs and limbs, 70% ethanol was soaked in them, and the CT scanner (SkyScan 1176) was used to scan them. Relevant structure parameters of the μCT instrument were as previously reported51. The scanning voxel size was 9 × 9 × 9 μm3. The X-ray tube potential was 50 kV and 450 μA. Integration time was 520 ms and rotation step was 0.4° for 180° scanning. CTAn micro-CT software version 1.13 (Bruker) was used for analysis. All femurs and lumbar trabecular bone analyses were thresholded at 75 (grayscale index) and all cortical bone analyses were thresholded at 110. The femurs and L3 lumbar were analyzed at a resolution of 9 μm. For cortical bone analysis, the volumetric regions include 600 μm long at mid-diaphysis of the femur (300 μm extending proximally and distally from the diaphyseal midpoint between the proximal and distal growth plates). For vertebrae, the volumetric regions exclude the primary spongiosa (300 μm below the cranial and above the caudal growth plate). Morphometric data included BMD, BV/TV, Tb.N, Tb.Th, Tb.Sp, Ct.Th and Ct.Po.
Whole mount alcian blue/alizarin red staining
We fixed and dehydrated mouse embryos (P0) overnight in 95% ethanol with gentle agitation after removing their skin, viscera, and muscles. After degreasing with absolute acetone overnight, the embryos were stained overnight with 0.015% alcian blue (Sigma-Aldrich) and 0.005% alizarin red (Sigma-Aldrich) in 70% ethanol with gentle agitation. A 1% KOH solution was used to digest the embryos after staining them in 70% ethanol for 30 min three times. Embryos were stored in a 75% (vol/vol) KOH/glycerol solution after most of their soft tissue was digested and gradually replaced with glycerol. Pictures of the embryos were captured using a microscope (Leica).
Whole-body µCT scan
With a dedicated cradle and holder, 14-week-old Ripk4fl/fl and UbcCre/ERT2 Ripk4fl/fl mice were deeply anesthetized and carefully placed into the CT instrument (SkyScan 1176) to capture a 35 µm resolution whole-body radiograph (excluding the tail). Scanning details were listed as following: X-ray tube potential, 65 kV and 375 µA; exposure time, 150 ms; rotation step, 0.5° for 180° scanning. CTAn micro-CT software version 1.13 (Bruker) was used to reconstruct pictures.
X-Gal staining
For X-Gal staining, mice femur was dissected and fixed in 4% PFA for 2 days and further decalcified with 10% EDTA (pH = 7.2) at 4 °C for about 2 weeks. Then, specimens were dehydrated in 30% sucrose and embedded in OCT and frozen sectioned at 10 μm thickness. Then, X-Gal staining was performed according to the manufacturer’s instructions (Beyotime). For P0 pubs, the whole tibia was stained with X-Gal staining.
qPCR
RNeasy® Mini Kit (Qiagen) was used to isolate total RNA. PrimeScriptTM RT Master Mix (Takara, RR036A) was used to reverse transcribe 500 ng of total RNA into cDNA. Applied Biosystems’ ABI HT7900 platform was used to perform qPCR analysis using Takara’s SYBR Premix Ex TaqTM II (RR820L). SYBR Green PCR conditions were 1 cycle of 95 °C for 30 s (s), and 40 cycles of 95 °C for 5 s and 34 °C for 60 s. Melting curve stage was added to check primer specificity. Relative gene expression levels were calculated using the threshold cycle (2–ΔΔCT) method. Relevant primers were used as previously reported12.
Isolation of mitochondria
To obtain dissociative mitochondria from cells, a Mitochondria Isolation Kit (Thermo Fisher) was used according to the manufacturer’s instructions. Briefly, RIPK4 inducible U2OS cells were collected in a 2.0 ml microcentrifuge tube and centrifuged at 850 × g for 5 min. The supernatant was removed and 800 µl Mitochondria Isolation Reagent A was added on ice. The tube was vortexed at medium speed for about 5 s and incubated on ice for 2 min. The cell suspension was then transferred to Dounce Tissue Grinder and homogenized on ice. Cell lysates were returned to a new tube and 1 mL of Mitochondria Isolation Reagent C was added. The tube was inverted several times gently to mix and centrifuged at 700 × g for 10 min at 4 °C. The supernatant was transferred to a new 2.0 ml tube and then centrifuged at 12,000 × g for 15 min at 4 °C. Remove the supernatant and the pellet contained the isolated mitochondria. Finally, 500 µl Mitochondria Isolation Reagent C was added to the pellet and centrifuged at 12,000 × g for 5 min. Discard the supernatant and resuspend the pellet by using complete medium.
Western blotting analysis and immunoprecipitation
Cell pellets were collected and lysed in RIPA buffer (50 mM Tris-HCl, PH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Nonidet P-40, 0.5% sodium deoxycholate and APExBio protease inhibitor cocktail) on ice for 30 min and subsequently centrifuged at 20,000 × g for 30 min at 4 °C. The supernatants were collected for western-blot analysis or immunoprecipitation. For FLAG-RIPK4 immunoprecipitation, the supernatants were incubated with anti-FLAG M2 Affinity Gel (Sigma-Aldrich, A2220) overnight at 4 °C. The next day the beads were washed with RIPA buffer 3 times and then eluted with 0.25 mg/ml 3× FLAG peptide in RIPA buffer for 6 h at 4 °C. For HA-MFN2 immunoprecipitation, the supernatants were incubated with anti-HA Affinity Gel (Sigma-Aldrich, A2095) overnight at 4 °C. The next day the beads were washed with RIPA buffer 3 times and then eluted with 0.25 mg/ml HA peptide in RIPA buffer for 6 h at 4 °C. For endogenous RIPK4 immunoprecipitation, the supernatants were pre-cleared with protein A/G agarose (Thermo Fisher, 20333) at 4 °C for 4 h. At the same time, anti-RIPK4 antibody (Cell Signaling Technology, 126236) or control IgG was coupled to protein A/G agarose (Thermo Fisher, 20333) in PBS (containing 5 mg/ml BSA) for 2 h at room temperature. Then the pre-cleared supernatants were incubated with protein A/G agarose coupled with RIPK4 antibody or control IgG overnight at 4 °C. The next day, the beads were washed with RIPA buffer three times and the immunoprecipitants were eluted with low pH elution buffer (Thermo Fisher, 21004).
In vitro kinase assay
RIPK4 kinase assays were performed according to the detailed protocol previously reported60 with modifications. Briefly, RIPK4 kinase was incubated with MFN2 protein in 30 µL kinase reaction system containing kinase buffer (20 mM HEPES, pH7.4, 1 mM EGTA, 5 mM MgCl2), 50 μM DTT and 0.1 mM ATPγS (Abcam, ab138911) at 37 °C for 30 min. To terminate kinase reaction and alkylate thiophosphorylated proteins, EDTA (20 mM) and p-Nitrobenzyl mesylate (2.5 mM, 5% DMSO) were added. After 1 h at room temperature, the samples were analyzed by western blotting using anti-Thiophosphate Ester antibody (Abcam, ab133473).
Lentivirus package and generation of stable cell lines
HEK293T, HBMSC, and U2OS cells were purchased from ATCC and were cultured in DMEM (Sigma-Aldrich, D6429) supplemented with 10% FBS (ExCell Bio, FSP500) and 100 U/ml Penicillin-Streptomycin (GIBCO, 15140122). RIPK4 sgRNA sequences were cloned into lentiCRISPR-v2 (Addgene) with Cas9 expression, and RIPK4 KO hBMSC cells were established as previously described in ref. 34.
Harvest of calvaria osteoblasts and osteogenic differentiation
Neonatal mice were decapitated and calvaria were separated from their skulls. Soft tissue was removed from the calvaria and then calvaria was digested using α-MEM which contains 0.1% collagenase (Roche) and 0.2% dispase (Roche) in a 37 °C constant temperature shaking table set at 10 × g for 10 min. Calvaria were digested for five times in total, the first four times for 10 min and the last time for 30 min, and the digestive production which contains calvaria OB from the last four times was collected and cultured in α-MEM containing 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin. Calvaria OB was then re-plated in 96-well plates (5000 cells per well) for staining or in 6-well plates (2 × 105 cells per well) for RNA isolation. After culturing the cells to 70–80% confluence, osteogenic differentiation medium (Cyagen) was replaced for further differentiation. The medium was changed every 2 days. ALP staining, alizarin red staining or RNA isolation was performed for further research.
Co-culture
For coculture to investigate the mitochondria transfer, BMNCs were extracted and induced towards myelopoiesis with M-CSF (20 ng/mL) stimulation. Meanwhile, OBs were extracted and coincubated for two days and then flow cytometry was performed. For flow cytometry analysis, 12-well dish was used to coculture BMNCs (1 × 105/ well) and OBs (1 × 105/ well). On the indicated day, cells were first digested with trypsin-EDTA and then detached with a cell scraper. For direct coculture, OBs extracted from Prrx1cre MitoDendra+/+ mice were added and coincubated for 2 days. After coculture for 2 days, flow cytometry examining the mitochondrial transfer and myelopoiesis was performed.
Analysis of mitochondrial mass, membrane potential and ROS
To stain mitochondria and mitochondrial ROS, BMSCs were stained with MitoTracker Green (Thermo Fisher, M7514) or MitoSOX (Thermo Fisher, M36008) for 1 h. For mitochondria membrane potential analysis, BMSCs were stained with JC-1 dye (Thermo Fisher, T3168). Flow cytometry analysis was performed using Cytometer CytoFlex (Beckman Coulter) and analyzed using FlowJo software version 10.4.
Immunofluorescence staining
Cells were cultured on coverslips and transfected with indicated plasmids using Lipofectamine 3000 Transfection Reagent (Invitrogen, L3000015) according to the manufacturer’s protocol for 48 h. Subsequently, cells were fixed with ice-cold 4% PFA for 15 min and washed with ice-cold PBS 3 times. Then cells were incubated with PBS containing 0.1% Triton X for 10 min and blocked with blocking buffer (4% BSA in PBS). Cells were incubated with primary antibodies overnight at 4 °C, washed 3 times with PBS, and then incubated with appropriate secondary antibodies for 1 h at room temperature. Primary antibodies used in this paper were mouse monoclonal anti-Flag-M2 (Sigma-Aldrich, F1804), and rabbit monoclonal anti-myc-tag antibody (Cell Signaling Technology, 2278). Secondary antibodies were goat anti-mouse Alexa Fluor 488 (Invitrogen, A32723) and goat anti-rabbit Alexa Fluor 647 (Invitrogen, A21244). Cells were imaged by the Olympus IX83 laser scanning confocal microscope.
Proximal ligation assay
The Duolink Proximity Ligation In Situ Assay (Sigma-Aldrich, DUO92008) was used to identify the in vivo interaction between VDAC1/IP3R3 or FLAG(RIPK4)/MFN2. WT and K51R RIPK4 were inducibly expressed in osteosarcoma U2OS cells with 50 ng/ml doxycycline in a 6-well plate with coverslips for 24 h. The cells were then washed with PBS at room temperature and fixed with 4% PFA for 20 min. Subsequently, the cells were permeabilized with ice-cold methanol at 20 °C for 15 min. The remaining steps of the assay were conducted according to the manufacturer’s instructions, using rabbit anti-VDAC1 antibody (1:200, Abcam, ab15895), and mouse anti-IP3R3 antibody (1:200, BD Biosciences, 610312)/mouse anti-FLAG antibody (1:500, Sigma-Aldrich, F1804) and rabbit anti-MFN2 antibody (1:200, Cell Signaling Technology, 9482) to detect protein interactions. The PLA signal was visualized using an Olympus FV 3000 microscope (×40), and the obtained images were processed using Fiji software.
TEM
TEM was used to observe mitochondrial morphology. We collected suspended cells and tissues and then fixed them at 4 °C with glutaraldehyde (2.5%, electron microscope grade). After dehydrating in 70%, 90%, and 100% alcohol, samples were embedded in fresh Epon overnight. The mitochondria were photographed using a Tecnai G2 Spirit 120 kv microscope (FEI, Hong Kong).
RNA sequencing
Total RNA of whole bone with bone marrow flushed out from Ripk4fl/fl and UbcCre/ERT2 Ripk4fl/fl mice was extracted using Trizol reagent (Thermo Fisher), quantified and purified using Bioanalyzer 2100 and RNA 6000 Nano LabChip Kit (Agilent). RNA was extracted using Trizol reagent (Thermo Fisher), quantified using Bioanalyzer 2100 and purified with RNA 6000 Nano LabChip Kit (Agilent). After RNA purification, mRNA library was constructed, fragmented and amplified according to the manufacturer’s instructions. The samples were then loaded into the nanoarray, and Illumina NovaseqTM 6000 platform was used for RNA-sequencing following the vendor’s recommended protocol. Generated reads were filtered and mapped to the reference genome using HISAT2 (v2.0.4) and assembled using StringTie (v1.3.4 d) with default parameters. Gffcompare software (v0.9.8) was used to merge all transcriptomes from all samples to reconstruct a comprehensive transcriptome. After merging, the expression levels of all transcripts were calculated by Stringtie and ballgown. DESeq2 software was performed to analyze different gene expression and GO functions were performed for enrichment analysis. The data were deposited into the GEO repository (accession no. GSE243610).
Statistics and reproducibility
GraphPad Prism (v8.2.1) software was used for statistical analysis of all data, and the results were presented as mean ± s.d. A Shapiro–Wilk test was used to test normality. F-tests were used to verify homogeneity of error variances (P > 0.05). In order to determine significant differences between two groups of normally distributed data, we used the unpaired two-tailed Student’s t-test. For groups with unequal variances, Welch’s correction was used. For comparisons between two groups without a normal distribution, a Mann–Whitney U test was used without a paired sample. An ordinary one-way ANOVA test with Tukey’s multiple comparisons was used for comparisons between multiple groups with normally distributed data. For groups with unequal variances, Brown-Forsythe and Welch ANOVA tests with Dunnett’s T3 multiple comparisons were used. Non-parametric one-way ANOVA with Dunn’s multiple comparisons was used to compare multiple groups without normally distributed data. All inclusion and exclusion criteria were predetermined and no samples or animals were excluded from analysis. Experiments were randomized and investigators were blinded to allocation throughout the experiments and when analyzing the results and each experiment was repeated independently at least three times with similar results.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
RNA sequencing data have been deposited into GEO repository with accession codes GSE243610. Source data generated in this study are provided in Figshare (https://doi.org/10.6084/m9.figshare.29108861).
References
Lee, P. et al. Phosphorylation of Pkp1 by RIPK4 regulates epidermal differentiation and skin tumorigenesis. EMBO J. 36, 1963–1980 (2017).
Oberbeck, N. et al. The RIPK4-IRF6 signalling axis safeguards epidermal differentiation and barrier function. Nature 574, 249–253 (2019).
Huang, X. et al. Phosphorylation of dishevelled by protein kinase RIPK4 regulates Wnt signaling. Science 339, 1441–1445 (2013).
Erturan, G., Holton, J., Wall, S. & Giele, H. Bartsocas-Papas syndrome. Ann. Plast. Surg. 76, 459–462 (2016).
Urwyler-Rosselet, C. et al. Keratinocyte-specific ablation of RIPK4 allows epidermal cornification but impairs skin barrier formation. J. Investig. Dermatol. 138, 1268–1278 (2018).
De Groote, P. et al. A novel RIPK4-IRF6 connection is required to prevent epithelial fusions characteristic for popliteal pterygium syndromes. Cell Death Differ. 22, 1012–1024 (2015).
Cariappa, A. et al. A catalytically inactive form of protein kinase C-associated kinase/receptor interacting protein 4, a protein kinase C beta-associated kinase that mediates NF-kappa B activation, interferes with early B cell development. J. Immunol. 171, 1875–1880 (2003).
Oleksyn, D. et al. PKK deficiency in B cells prevents lupus development in Sle lupus mice. Immunol. Lett. 185, 1–11 (2017).
Chen, L. et al. A critical role for the protein kinase PKK in the maintenance of recirculating mature B cells and the development of B1 cells. Immunol. Lett. 172, 67–78 (2016).
Mendez-Ferrer, S. et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 466, 829–834 (2010).
Zheng, C. X., Sui, B. D., Qiu, X. Y., Hu, C. H. & Jin, Y. Mitochondrial regulation of stem cells in bone homeostasis. Trends Mol. Med. 26, 89–104 (2020).
Ding, P. et al. Osteocytes regulate senescence of bone and bone marrow. Elife 11, e81480 (2022).
Karsenty, G. & Ferron, M. The contribution of bone to whole-organism physiology. Nature 481, 314–320 (2012).
Katsnelson, A. Physiology: the bones of contention. Nature 466, 914–915 (2010).
Quarles, L. D. Dem bones’ are made for more than walking. Nat. Med. 17, 428–430 (2011).
Le, P. M., Andreeff, M. & Battula, V. L. Osteogenic niche in the regulation of normal hematopoiesis and leukemogenesis. Haematologica 103, 1945–1955 (2018).
Yu, V. W. & Scadden, D. T. Heterogeneity of the bone marrow niche. Curr. Opin. Hematol. 23, 331–338 (2016).
Kim, M. J., Valderrabano, R. J. & Wu, J. Y. Osteoblast lineage support of hematopoiesis in health and disease. J. Bone Min. Res. 37, 1823–1842 (2022).
Coskun, S. et al. Development of the fetal bone marrow niche and regulation of HSC quiescence and homing ability by emerging osteolineage cells. Cell Rep. 9, 581–590 (2014).
Emoto, T. et al. Colony stimulating factor-1 producing endothelial cells and mesenchymal stromal cells maintain monocytes within a perivascular bone marrow niche. Immunity 55, 862–878.e868 (2022).
Seo, B. J., Yoon, S. H. & Do, J. T. Mitochondrial dynamics in stem cells and differentiation. Int. J. Mol. Sci. 19, 3893 (2018).
Wang, Y., Barthez, M. & Chen, D. Mitochondrial regulation in stem cells. Trends Cell Biol. 34, 685–694 (2023).
Chen, H. & Chan, D. C. Mitochondrial dynamics in regulating the unique phenotypes of cancer and stem cells. Cell Metab. 26, 39–48 (2017).
Hong, X. et al. Mitochondrial dynamics maintain muscle stem cell regenerative competence throughout adult life by regulating metabolism and mitophagy. Cell Stem Cell 29, 1298–1314.e1210 (2022).
Khacho, M. et al. Mitochondrial dynamics impacts stem cell identity and fate decisions by regulating a nuclear transcriptional program. Cell Stem Cell 19, 232–247 (2016).
Divieti Pajevic, P. & Krause, D. S. Osteocyte regulation of bone and blood. Bone 119, 13–18 (2019).
Fulzele, K. et al. Myelopoiesis is regulated by osteocytes through Gsalpha-dependent signaling. Blood 121, 930–939 (2013).
Asada, N. et al. Matrix-embedded osteocytes regulate mobilization of hematopoietic stem/progenitor cells. Cell Stem Cell 12, 737–747 (2013).
Guo, B., Huang, X., Lee, M. R., Lee, S. A. & Broxmeyer, H. E. Antagonism of PPAR-gamma signaling expands human hematopoietic stem and progenitor cells by enhancing glycolysis. Nat. Med. 24, 360–367 (2018).
Gu, W. et al. Glycolytic metabolism plays a functional role in regulating human pluripotent stem cell state. Cell Stem Cell 19, 476–490 (2016).
Mihaylova, M. M. et al. Fasting activates fatty acid oxidation to enhance intestinal stem cell function during homeostasis and aging. Cell Stem Cell 22, 769–778.e764 (2018).
Kang, J. S. et al. RIPK3 causes mitochondrial dysfunction and albuminuria in diabetic podocytopathy through PGAM5-Drp1 signaling. Metabolism 159, 155982 (2024).
Zhou, H. et al. Ripk3 induces mitochondrial apoptosis via inhibition of FUNDC1 mitophagy in cardiac IR injury. Redox Biol. 13, 498–507 (2017).
Zhang, J. et al. RIPK4 promotes oxidative stress and ferroptotic death through the downregulation of ACSM1. Proc. Natl. Acad. Sci. USA 121, e2410628121 (2024).
Deng, L., Yi, S., Yin, X., Li, Y. & Luan, Q. MFN2 knockdown promotes osteogenic differentiation of iPSC-MSCs through aerobic glycolysis mediated by the Wnt/beta-catenin signaling pathway. Stem Cell Res. Ther. 13, 162 (2022).
Zarei, A. et al. Osteolineage depletion of mitofusin2 enhances cortical bone formation in female mice. Bone 148, 115941 (2021).
Hsu, Y. C., Wu, Y. T., Yu, T. H. & Wei, Y. H. Mitochondria in mesenchymal stem cell biology and cell therapy: from cellular differentiation to mitochondrial transfer. Semin. Cell Dev. Biol. 52, 119–131 (2016).
Wei, P., Dove, K. K., Bensard, C., Schell, J. C. & Rutter, J. The force is strong with this one: metabolism (over)powers stem cell fate. Trends Cell Biol. 28, 551–559 (2018).
Snoeck, H. W. Mitochondrial regulation of hematopoietic stem cells. Curr. Opin. Cell Biol. 49, 91–98 (2017).
Picard, M. & Shirihai, O. S. Mitochondrial signal transduction. Cell Metab. 34, 1620–1653 (2022).
Nicolas-Avila, J. A. et al. A network of macrophages supports mitochondrial homeostasis in the heart. Cell 183, 94–109.e123 (2020).
Rosina, M. et al. Ejection of damaged mitochondria and their removal by macrophages ensure efficient thermogenesis in brown adipose tissue. Cell Metab. 34, 533–548.e512 (2022).
Borcherding, N. et al. Dietary lipids inhibit mitochondria transfer to macrophages to divert adipocyte-derived mitochondria into the blood. Cell Metab. 34, 1499–1513.e1498 (2022).
Zhang, K. et al. TREM2(hi) resident macrophages protect the septic heart by maintaining cardiomyocyte homeostasis. Nat. Metab. 5, 129–146 (2023).
Yang, C. et al. Mitochondria transfer mediates stress erythropoiesis by altering the bioenergetic profiles of early erythroblasts through CD47. J. Exp. Med. 219, e20220685 (2022).
van der Vlist, M. et al. Macrophages transfer mitochondria to sensory neurons to resolve inflammatory pain. Neuron 110, 613–626.e619 (2022).
Baldwin, J. G. et al. Intercellular nanotube-mediated mitochondrial transfer enhances T cell metabolic fitness and antitumor efficacy. Cell 187, 6614–6630.e6621 (2024).
Ikeda, H. et al. Immune evasion through mitochondrial transfer in the tumour microenvironment. Nature 638, 225–236 (2025).
Ding, P. et al. Mitochondria from osteolineage cells regulate myeloid cell-mediated bone resorption. Nat. Commun. 15, 5094 (2024).
Gao, J. et al. Endoplasmic reticulum mediates mitochondrial transfer within the osteocyte dendritic network. Sci. Adv. 5, eaaw7215 (2019).
Ding, P. et al. Toll-like receptor 9 deficiency induces osteoclastic bone loss via gut microbiota-associated systemic chronic inflammation. Bone Res. 10, 42 (2022).
Bakker, A. D. & Klein-Nulend, J. Osteoblast isolation from murine calvaria and long bones. Methods Mol. Biol. 816, 19–29 (2012).
Chandhok, G., Lazarou, M. & Neumann, B. Structure, function, and regulation of mitofusin-2 in health and disease. Biol. Rev. Camb. Philos. Soc. 93, 933–949 (2018).
Suh, J. et al. Mitochondrial fragmentation and donut formation enhance mitochondrial secretion to promote osteogenesis. Cell Metab. 35, 345–360.e347 (2023).
Ambrosi, T. H. et al. Aged skeletal stem cells generate an inflammatory degenerative niche. Nature 597, 256–262 (2021).
Hayashi, Y. et al. MDS cells impair osteolineage differentiation of MSCs via extracellular vesicles to suppress normal hematopoiesis. Cell Rep. 39, 110805 (2022).
Brestoff, J. R. et al. Intercellular mitochondria transfer to macrophages regulates white adipose tissue homeostasis and is impaired in obesity. Cell Metab. 33, 270–282.e278 (2021).
Levoux, J. et al. Platelets facilitate the wound-healing capability of mesenchymal stem cells by mitochondrial transfer and metabolic reprogramming. Cell Metab. 33, 283–299.e289 (2021).
Golan, K. et al. Bone marrow regeneration requires mitochondrial transfer from donor Cx43-expressing hematopoietic progenitors to stroma. Blood 136, 2607–2619 (2020).
Allen, J. J. et al. A semisynthetic epitope for kinase substrates. Nat. Methods 4, 511–516 (2007).
Acknowledgements
We thank Dr. Qiang Zou (Shanghai Jiao Tong University School of Medicine) for kindly providing Cd4Cre mice. We acknowledge Mr. Zihao Li for his assistance in the cartonization of the schematic diagrams. This work was supported by National Natural Science Foundation of China (82402849 to P.D., 82002339 to J.J.G., 32370794 to J.Z., 92254307 to Q.Z.), the Ministry of Science and Technology of the People’s Republic of China (2023YFA0914900), Shanghai Municipal Health Commission Key Priority Discipline Project, Shanghai Spinal Disease and Trauma Orthopedics Research Center (2022ZZ01014), and Shanghai Municipal Hospital Orthopedic Specialist Alliance (SHDC22021308), the National Key Research and Development Program of China (Grant No. 2023YFC2509600), the Shanghai Frontiers Science Center of Cellular Homeostasis and Human Diseases; and innovative research team of high-level local universities in Shanghai (SHSMU-ZDCX20212000 and SHSMU-ZDCX20211202).
Author information
Authors and Affiliations
Contributions
C.Q.Z., Q.Z., J.J.G. and J.Z. conceived, designed, and supervised the study. P.D., X.W., C.A.G., Y.H.W. performed the experiment and analyzed the data. W.L. Z., N. W., W.F., G.L., Q.H.F., M.Y., Y.G.H., C.Y.J. and Y.S.G. provided suggestions. P.D., X.W., C.A.G., Y.H.W. wrote the manuscript, C.Q.Z., Q.Z., J.J.G. and J.Z. revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Noriaki Ono and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ding, P., Wang, X., Gao, C. et al. RIPK4-mediated MFN2 degradation drives osteogenesis through mitochondrial fragmentation and restricts myelopoiesis by blocking mitochondrial transfer. Nat Commun 16, 6666 (2025). https://doi.org/10.1038/s41467-025-61808-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-61808-9
